

Abstract
Genome‐wide association studies (GWAS) have successfully identified thousands of single nucleotide polymorphisms (SNPs) associated with complex traits; however, the identified SNPs account for a fraction of trait heritability, and identifying the functional elements through which genetic variants exert their effects remains a challenge. Recent evidence suggests that SNPs associated with complex traits are more likely to be expression quantitative trait loci (eQTL). Thus, incorporating eQTL information can potentially improve power to detect causal variants missed by traditional GWAS approaches. Using genomic, transcriptomic, and platelet phenotype data from the Genetic Study of Atherosclerosis Risk family‐based study, we investigated the potential to detect novel genomic risk loci by incorporating information from eQTL in the relevant target tissues (i.e., platelets and megakaryocytes) using established statistical principles in a novel way. Permutation analyses were performed to obtain family‐wise error rates for eQTL associations, substantially lowering the genome‐wide significance threshold for SNP‐phenotype associations. In addition to confirming the well known association between PEAR1 and platelet aggregation, our eQTL‐focused approach identified a novel locus (rs1354034) and gene (ARHGEF3) not previously identified in a GWAS of platelet aggregation phenotypes. A colocalization analysis showed strong evidence for a functional role of this eQTL.
Keywords: expression quantitative trait loci, family wise error rate, genome‐wide association studies, permutations, platelet aggregation, whole‐genome sequencing
1. INTRODUCTION
Platelet aggregation is critical for normal hemostasis and pathologic thrombus formation (Jackson, 2007). Platelets are known to play an important role in the pathogenesis of atherosclerosis and in the acute thrombotic events that characterize acute coronary syndromes (Freedman & Loscalzo, 2013; Libby, 2001). High residual levels of platelet reactivity despite antiplatelet therapy is also associated with increased likelihood of major adverse cardiovascular events after percutaneous coronary intervention (de Prado et al., 2006). Several large cohorts have documented the highly variable interindividual platelet responsiveness to a variety of agonists (Kunicki & Nugent, 2010). Furthermore, a number of genetic and environmental factors contribute to substantial variation in platelet function seen among normal persons.
Genome‐wide association studies (GWAS) have successfully identified several single nucleotide polymorphisms (SNPs) that are associated with platelet aggregation phenotype (Johnson et al., 2010; Keramati et al., 2021, 2019; Kim et al., 2013; Lewis & Ryan, 2013; Mathias et al., 2010; Qayyum et al., 2015). Previous family‐based studies have shown that the majority of these platelet traits are heritable, with estimates up to 70% in African Americans (AAs) and almost 60% in European Americans (EAs) (Bray et al., 2007; Faraday et al., 2007). But even in aggregate, the SNPs identified from prior GWAS explain only a small proportion of this heritability. This phenomenon is observed in most complex traits, because the effect size of most SNPs is small providing limited power to pass the GWAS significance threshold (He et al., 2013; Manolio et al., 2009). With the implementation of stringent thresholds, variants that confer small disease risks are likely to be missed among the millions of SNPs that are tested. Hence additional analytical approaches that exploit genetic information beyond SNP association are useful to uncover additional important genetic variants.
Establishing connections between genetic variants identified in GWAS and their biological mechanisms has been challenging (Gupta & Musunuru, 2013). Some studies have looked at the overlap between complex trait‐associated variants and expression quantitative trait loci (eQTL) variants as evidence of common causal molecular mechanisms (Dubois et al., 2010; Nica et al., 2010). The concept is that a GWAS variant, in some tissues, may affect expression at a nearby gene and that both the gene and the tissue might play a role in the disease mechanism (Huang et al., 2015). Others have also explored approaches that integrate summary‐level data from GWAS with eQTL data in a Mendelian randomization style to identify genes whose expression levels are associated with a complex trait because of pleiotropy (Zhu et al., 2016). There is also increasing evidence that SNPs associated with complex traits are more likely to be eQTL and that a substantial proportion of these GWAS risk variants influence complex trait by regulating gene expression levels of their target genes (Albert & Kruglyak, 2015; Emilsson et al., 2008; Nica et al., 2010; Nicolae et al., 2010). Integrating this information in GWAS can enhance the discovery of trait‐associated SNPs for complex phenotypes, as gene expression analyses can yield important information about genetic architecture and can point to mechanisms that link genetics and disease (Gupta & Musunuru, 2013). Annotating SNPs with information on expression can certainly improve our understanding of variants that underlie biological control of gene expression and genes involved in platelet aggregation.
Our goal in this study was to investigate the potential to leverage eQTL from a target tissue to identify novel loci associated with phenotype from prior GWAS. In this example, we leverage eQTL information from platelets (PLTs) and megakaryocytes (MKs) to identify novel loci associated with platelet aggregation phenotypes using Whole Genome Sequencing (WGS) data from EAs and AAs from the GeneSTAR family‐based study, generated as part of the NHLBI's Trans‐Omics for Precision Medicine (TOPMed) program. We incorporate eQTL information from RNA‐seq data on PLTs and induced pluripotent stem cell (iPSC) derived MKs (Kammers, Taub, Rodriguez, et al., 2021) to uncover novel genetic variants that determine platelet aggregation, using permutation tests to assess statistical significance.
2. MATERIALS AND METHODS
2.1. Genetic study of atherosclerosis risk cohort
GeneSTAR is an ongoing prospective study begun in 1983 designed to determine environmental, phenotypic, and genetic causes of premature cardiovascular disease. Participants came from EA and AA families identified from 1983 to 2006 from probands with a premature coronary disease event before 60 years of age who were identified at the time of hospitalization in any of 10 Baltimore area hospitals. Their apparently healthy 30–59 year old siblings without known coronary artery disease (CAD) were recruited and underwent initial phenotypic measurement and characterization between 1983 and 2007 (Vaidya et al., 2007; Yanek et al., 2013). Adult offspring (over 21 years of age) of siblings and probands along with the coparents of the offspring were recruited and underwent initial phenotypic measurement and characterization between 2003 and 2006. Participants for the current study took part in a 2‐week trial of aspirin from 2003 to 2006, and were apparently healthy, free of CAD, and had not used aspirin or antiplatelet medications for 2 weeks before the baseline visit (Becker et al., 2006). Platelet function was assessed before and after 2 weeks of aspirin in whole blood and platelet‐rich plasma (PRP) with multiple agonists such as collagen, ADP, and epinephrine (EPI) as described previously (Becker et al., 2006). Maximal aggregation (%) of PRP to 2 µM ADP was the phenotype we examined as proof of concept in this study.
2.2. Whole genome sequencing data
We used the sequencing data available through the NHLBI's Trans‐Omics for Precision Medicine (TOPMed) program (https://nhlbiwgs.org). WGS was performed to an average depth of 38X using DNA isolated from blood, PCR‐free library construction, and Illumina HiSeq X technology. Details for variant calling and quality control are described in detail in Taliun et al. (2021). In brief, variant discovery and genotype calling was performed jointly across all the available TOPMed studies using the GotCloud 6 pipeline, resulting in a single, multistudy, genotype call set. Sample‐level quality control was performed to check for pedigree errors, discrepancies between self‐reported and genetic sex, and concordance with prior genotyping array data. Among the GeneSTAR samples in TOPMed Freeze 6, 806 EAs in 196 families and 661 AAs in 190 families had complete phenotype data.
2.3. RNA sequencing data
Details on the iPSC derived MK and PLT samples used in the RNA sequencing are described in detail elsewhere (Kammers et al., 2017; Kammers, Taub, Mathias, et al., 2021; Kammers, Taub, Rodriguez, et al., 2021). Briefly, for 185 iPSC‐derived MK cell lines and for 290 PLT samples with WGS data we also obtained RNA‐seq data from extracted nonribosomal RNA. This included iPSC‐derived MKs on 84 AA and 101 EA subjects as well as platelets on 110 AA and 180 EA subjects. Details on data processing are provided in Kammers, Taub, Rodriguez, et al. (2021). In brief, we used the HISAT‐StringTie suite (Pertea et al., 2016) for alignment and assembly of RNA‐seq data and the Ballgown package (Frazee et al., 2015) for efficient data storage, processing, and analysis. Gene expression was quantified as fragments per kilobase per million reads mapped (FPKM), log‐transformed, and genes with median FPKM across all samples less than or equal to 1 (for MKs) or 0.3 (for PLTs) were excluded.
2.4. Genome‐wide association studies
A linear mixed effects model for genetic association was applied to the WGS data using the GENESIS Package (Conomos & Thornton, 2016), and analysis was first performed separately in each ethnic group (EA and AA). A genetic relationship matrix (GRM) was created using the PC‐Relate function to account for phenotype correlations due to the family structure of the GeneSTAR samples. GWAS WGS‐based association analysis was conducted using age and sex adjusted inverse normalized transformation of the platelet phenotypes. In each group, SNP quality control filtering was carried out family‐aware using PLINK (http://zzz.bwh.harvard.edu/plink/). Only SNPs with minor allele frequency (MAF) greater than 1% in the respective group, Hardy–Weinberg equilibrium test p value larger than 10−6 and missing genotype frequency less than 5% were tested for association, and reported. Further, SNPs with inflated estimated standard errors (larger than 10) due to collinearity were omitted.
2.5. Meta‐analysis
SNPs with MAF larger than 1% in both groups were then included in a meta‐analysis. Inverse variance weighted fixed effects meta‐analyses based on the slope and standard error estimates were conducted using the metagen function implemented in the R package meta, combining the stratified EA and AA results. Quantile–quantile (qq) plots of observed versus expected p values were examined to assess potential type I error inflation. Manhattan plots and regional association plots of the GWAS results using LocusZoom (Pruim et al., 2010) were created based on the Human Genome version 38 (hg38) build. Conditional analyses to potentially identify multiple causal variants in all regions identified using the GWAS WGS meta‐analysis approach were performed by conditioning on the most significant SNP in the regions of interest, and re‐assessing the strength of association in the respective regions.
2.6. eQTL analysis
Details of the eQTL analyses are provided in Kammers, Taub, Rodriguez, et al. (2021). In brief, eQTL analyses were carried out for both MK and PLT at the gene level stratified by ancestry (AA and EA), focusing on a 1 Mb window around each SNP and adjusting for sex, age, percent CD41+ CD42a+ MK pellets (MKs only), RNA‐seq batch, and 15 principal components (PCs) of the filtered and log‐transformed gene expression matrix. Only SNPs with at least two samples for each genotype and a call rate greater than 80% were tested, using the R package MatrixEQTL (Shabalin, 2012).
2.7. Permutation analysis
To simulate null distributions for tests of association between the set of eQTL identified SNPs and the trait, residuals (obtained after regressing the phenotype on the covariates) were randomly shuffled while SNP genotypes were kept the same, to preserve the SNP correlation structure (Churchill & Doerge, 1994). The 396 GeneSTAR families ranged from 1 to 15 members in size. For multiple‐member families, residuals were shuffled within families to also maintain within family phenotype correlation structure. Residuals were randomly swapped between singletons. To estimate the threshold for the 5% family‐wise error rate (FWER) under the global null of no association across all eQTL identified SNPs, we permuted each set of residuals 1000 times as described above, carried out 1000 separate GENESIS association analyses on the set of all eQTL identified SNPs, recorded the minimum p value for each of these 1000 analyses, and selected the 5th percentile of these 1000 minimum p values.
2.8. Colocalization
We performed a Bayesian colocalization analysis to investigate whether an observed association signal in the GWAS and eQTL analysis is consistent with a shared causal variant, using the framework described by Giambartolomei et al. (2014). In brief, for two separate traits (here, the phenotypes in the GWAS and the gene expression for the gene of interest in the eQTL analyses) five different hypothesis are considered under the assumption of a single causal variant for each trait: H0: no association with either trait; H1: association with trait 1, not with trait 2; H2: association with trait 2, not with trait 1; H3: association with trait 1 and trait 2, two independent SNPs; H4: association with trait 1 and trait 2, one shared SNP. Colocalization under the assumption of a single causal variant for each trait is inferred by support of hypothesis H5 calculating Bayes factors using the approximation proposed by Wakefield (2009). Prior probabilities for association with one or both traits were chosen as the default parameters in the coloc.abf function from the coloc R package ( that a SNP is associated with either of the two traits, and that a SNP is associated with both).
3. RESULTS
A total of 9,769,070 SNPs in the EA families and 16,415,214 SNPs in the AA families met the QC filtering criteria (described in Section 2). In the stratified association analysis, one SNP in gene GTF2IRD1 on chromosome 7 (rs13221023) exceeded the GWAS p value threshold in the EA families. In the AAs, one SNP (rs12041331) located in the PEAR1 gene met this GWAS threshold (Table 1A and Figure S1). The meta‐analysis of the 8,242,287 SNPs with a MAF of 1% or larger in both groups only yielded SNP rs12041331 in the PEAR1 gene (also identified in the stratified AA analysis) meeting the GWAS threshold (Table 1A and Figure 1A). The test statistics in the meta‐analysis and the stratified analyses were well‐calibrated, with genomic control parameters (Devlin & Roeder, 1999) of 1.011 in the meta‐analysis, and 1.014 and 1.012 in the EA and AA stratified analyses, respectively (Figure S2).
Table 1.
Loci identified using the standard genome‐wide significance level of through the WGS‐based GWAS meta‐analysis (A), and the eQTL PLTs (B) and MKs based (C) permutation tests using the respective FWER permutation thresholds
| (A) Loci identified through the WGS‐based GWAS meta‐analysis | ||||||||
|---|---|---|---|---|---|---|---|---|
| SNP | Model | CHR | Position | MEA | MAA | p | Gene | |
| rs12041331 | META | 1 | 156,899,922 | 0.09 | 0.35 |
|
PEAR1 | |
| rs12041331 | AA | 1 | 156,899,922 | 0.09 | 0.35 |
|
PEAR1 | |
| rs13221023 | EA | 7 | 74,528,803 | 0.04 | 0.07 |
|
GTF2IRD1 | |
| (B) Loci identified through the eQTL PLTs based permutation test | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Model | CHR | Position | MEA | MAA | p | Gene | eGEA | eGAA | |
| rs2182760 | META | 1 | 156,898,198 | 0.09 | 0.17 |
|
PEAR1 | PEAR1 | PEAR1 | |
| rs12041331 | META | 1 | 156,899,922 | 0.09 | 0.35 |
|
PEAR1 | PEAR1 | PEAR1 | |
| rs12041331 | AA | 1 | 156,899,922 | 0.09 | 0.35 |
|
PEAR1 | PEAR1 | PEAR1 | |
| rs1354034 | META | 3 | 56,815,721 | 0.40 | 0.25 |
|
ARHGEF3/SPATA12 | ARHGEF3 | ARHGEF3 | |
| (C) Loci identified through the eQTL MKs based permutation test | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Model | CHR | Position | MEA | MAA | p | Gene | eGEA | eGAA | |
| rs234103 | AA | 1 | 184,969,377 | 0.49 | 0.43 |
|
NIBAN1 | FAM129A | SWT1 | |
| rs85671 | AA | 1 | 184,970,425 | 0.49 | 0.44 |
|
NIBAN1 | FAM129A | SWT1 | |
| rs234104 | AA | 1 | 184,971,007 | 0.49 | 0.44 |
|
NIBAN1 | FAM129A | SWT1 | |
| rs234107 | AA | 1 | 184,973,263 | 0.49 | 0.44 |
|
NIBAN1 | FAM129A | SWT1 | |
| rs234111 | AA | 1 | 184,976,103 | 0.49 | 0.44 |
|
NIBAN1/RNF2 | FAM129A | SWT1 | |
| rs1354034 | META | 3 | 56,815,721 | 0.40 | 0.25 |
|
ARHGEF3/SPATA12 | ARHGEF3 | ARHGEF3 | |
Note: Column names as follows. SNP: the locus rs number when available. Model: the model used to identify the locus (EA/AA stratified, or META analysis). CHR: chromosome of the identified locus. Position: genomic position of the locus identified. Gene: gene the locus resides in. If intergenic, the flanking genes are reported. MEA/MAA: minor allele frequencies of the EA and AA families. P: statistical significance (p‐value) from the hypothesis test of no association based on a standard Gaussian null distribution. eGEA/eGAA: gene for which the reported SNP is an eQTL in the EA and AA families. An italicized MAF in column MAA indicates that the reference allele was switched.
Figure 1.
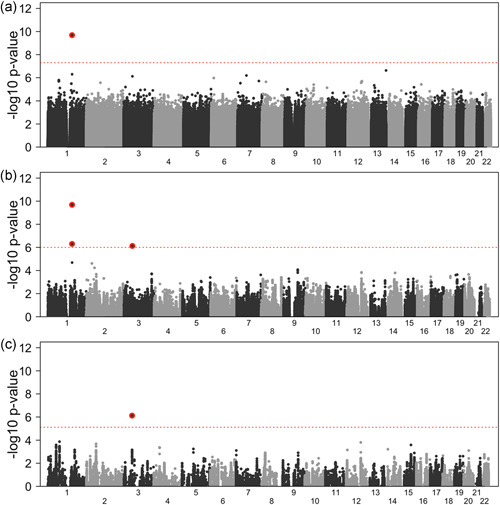
GWAS meta‐analysis results. (a) Manhattan plot of the GWAS for all 8,242,287 SNPs passing quality control. The dashed horizontal line is at p = , representing the standard GWAS cut‐off for significance. (b) Manhattan plot of the GWAS for the 229,674 eQTL in platelet. The dashed horizontal line is at 6.00 (p = ), representing the cut‐off for a 5% FWER derived using permutations. (c) Manhattan plot of the GWAS for the 55,088 eQTL in megakaryocytes. The dashed horizontal line is at 5.12 (p = ), representing the cut‐off for a 5% FWER derived using permutations. SNPs passing the respective significance threshold at the PEAR1 (chromosome 1) and ARHGEF3 (chromosome 3) loci are highlighted with a red background. eQTL, expression quantitative trait loci; FWER, family‐wise error rate; GWAS, genome‐wide association studies; SNP, single nucleotide polymorphism
In the eQTL analysis, a total of 16,641,225 SNP‐gene pairs were tested in the EA families and 20,101,156 pairs were tested in the AA families for PLTs, as previously described. Among those, 208,230 PLT eQTL SNP associations in the EA families met a false discovery rate of 5%, and 54,085 PLT eQTL SNP associations met the same threshold in the AA families. A combined total of 229,674 unique SNPs were common in both the EA and AA platelet eQTL analysis; these were used for the permutation approach applied to the GWAS meta‐analysis results. The MK data had a total of 30,802,119 SNP‐gene pairs tested in the EA families and 34,673,581 in the AA families for eQTL analysis. A total of 50,255 MK eQTL SNP associations in the EA families met a false discovery rate of 5%, and 9046 in the AA families, respectively. A combined total of 55,088 unique MK eQTL SNPs, found to be overlapping in EA and AA eQTL results, were then used for the permutation approach applied to the meta‐analysis of the GWAS signals.
In the GWAS meta‐analysis based on the 229,674 platelet‐identified eQTL, three SNPs met the PLT eQTL permutation FWER threshold of p = in two genes, PEAR1 on chromosome 1, and ARHGEF3 on chromosome 3 (Table 1B and Figure 1B). In the GWAS meta‐analysis based on the 55,088 MK‐identified eQTL, only the intron SNP rs1354034 in the ARHGEF3 gene met the permutation threshold of p = (Table 1C and Figures 1C, S3, and S4). While PEAR1 has been firmly established as a gene modifying platelet aggregation in response to agonists (Johnson et al., 2010; Kim et al., 2013; Lewis & Ryan, 2013; Mathias et al., 2010; Qayyum et al., 2015), the exchange factor ARHGEF3 found in platelets has largely gone unnoticed in that particular role. Associations of ARHGEF3, and in particular its intronic variant rs1354034, have been reported in the GWAS catalogue for many platelet and blood related phenotypes, such as platelet count, mean platelet volume, reticulocyte fraction of red cells, reticulocyte count, red blood cell count, blood protein levels, lymphocyte counts, hematocrit, hemoglobin concentration, mean corpuscular hemoglobin, and plateletcrit (https://www.ebi.ac.uk/gwas/). However, to our knowledge, ARHGEF3 has not been previously identified in a genome‐wide analysis as modifying platelet aggregation in response to agonists. The intronic ARHGEF3 SNP rs12485738, reported by Meisinger et al. (2009) as strongly associated with mean platelet volume, was considered by Johnson et al. (2010) as a platelet aggregation candidate SNP, and achieved a p value of when tested for association in a meta‐analysis with response to lower ADP levels (table S5a in Johnson et al., 2010). When ARHGEF3 was considered as a candidate gene (table S5b in Johnson et al., 2010), no SNPs were significant after multiple comparisons correction, but low p values were reported for SNPs rs4455300 (ADP, p = 0.0006), rs9851853 (epinephrine, p = 0.0029) and rs11716680 (collagen, p = 0.016). Also noteworthy, another exchange factor (ARHGEF11) was highlighted as a gene within proximity (60 kb) of the PEAR1 peak SNP (Johnson et al., 2010, table 4).
A Bayesian colocalization analysis using the platelet aggregation phenotype and gene expressions strongly supported the notion of a single shared common genetic causal variant in the newly detected gene ARHGEF3. Meta‐analysis p values for the association of the 7598 SNPs within 1 MB of rs1354034 with the platelet aggregation trait were considered, of which 4128 were PLT eQTL for ARHGEF3 gene expression, and 3809 were MK eQTL. The posterior probability of one common causal variant for association with the trait and ARHGEF3 gene expression (Hypothesis 4 as described in Giambartolomei et al., 2014) was 65.4% in the PLT and 99.8% in the MK. SNP rs1354034 had the strongest association with the phenotype (p = ) and the 8th smallest PLT eQTL p value (p = ), resulting in a posterior probability of 96.3% being the causal variant under the COLOC assumptions (Table 2 and Figure 2, PLT). However, because several SNPs had a stronger association with ARHGEF3 expression in the PLT than rs1354034 and the GWAS p value did not pass the traditional threshold of genome‐wide association, the posterior probabilities that the causal variant is only associated with gene expression (Hypothesis 2) or that two independent SNPs underly the associations (Hypothesis 3) also have appreciable support from the observed data (posterior probabilities of 14.8% and 19.8%, respectively). Among the ARHGEF3 MK eQTL on the other hand, rs1354034 also had the smallest eQTL p value (p = ), resulting in a posterior probability of virtually 100% being the causal variant (Table 2 and Figure 2, MK). A conditional analysis in this region supported the notion of a single independent variant affecting this platelet aggregation trait (Figure S5).
Table 2.
Bayesian colocalization results for the PLT and MK ARHGEF3 eQTL
| Bayesian colocalization results for PLT ARHGEF3 eQTL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PPH0 = 0.000, PPH1 = 0.000, PPH2 = 0.148, PPH3 = 0.198, PPH4 = 0.654 | ||||||||||||
| SNP | CHR | Position | MEA | MAA | P/GWAS | P/eQTL | BF/G | BF/E | BF | PP | ||
| rs1354034 | 3 | 56,815,721 | 0.40 | 0.25 |
|
|
9.12 | 16.46 | 25.58 | 0.963 | ||
| rs12488986 | 3 | 56,816,160 | 0.18 | 0.14 |
|
|
1.58 | 19.65 | 21.23 | 0.012 | ||
| rs1039383 | 3 | 56,815,027 | 0.23 | 0.16 |
|
|
0.27 | 20.87 | 21.14 | 0.011 | ||
| rs1039384 | 3 | 56,815,161 | 0.23 | 0.18 |
|
|
0.01 | 20.87 | 20.88 | 0.009 | ||
| rs17288922 | 3 | 56,817,359 | 0.17 | 0.13 |
|
|
1.67 | 18.36 | 20.03 | 0.004 | ||
| Bayesian colocalization results for MK ARHGEF3 eQTL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PPH0 = 0.000, PPH1 = 0.000, PPH2 = 0.001, PPH3 = 0.001, PPH4 = 0.998 | ||||||||||||
| SNP | CHR | Position | MEA | MAA | P/GWAS | P/eQTL | BF/G | BF/E | BF | PP | ||
| rs1354034 | 3 | 56,815,721 | 0.40 | 0.25 |
|
|
9.12 | 50.89 | 60.01 | 1.000 | ||
| rs6445826 | 3 | 56,814,971 | 0.50 | 0.13 |
|
|
1.84 | 24.17 | 26.01 | 0.000 | ||
| rs13085861 | 3 | 56,825,269 | 0.46 | 0.46 |
|
|
3.58 | 10.92 | 14.50 | 0.000 | ||
| rs13074522 | 3 | 56,826,855 | 0.48 | 0.12 |
|
|
2.27 | 12.23 | 14.50 | 0.000 | ||
| rs13062174 | 3 | 56,824,658 | 0.46 | 0.49 |
|
|
2.66 | 10.86 | 13.52 | 0.000 | ||
Note: PPH0–PPH4: posterior probabilities for Hypotheses 0–4 as described in Section 2 and Giambartolomei et al. (2014). Column names as in Table 1, and as follows. P/GWAS: p value from WGS GWAS. P/eQTL: p value from eQTL analysis. Bayes factors as described in Giambartolomei et al. (2014). BF/G: log10 Bayes factor for the SNP‐phenotype association. BF/E: log10 Bayes factor for the SNP‐gene association. BF: log10 Bayes factor for the joint association of the SNP with phenotype and gene expression.
Abbreviations: BF, Bayes factor; eQTL, expression quantitative trait loci; GWAS, Genome‐wide association studies; MK, megakaryocytes; PLT, platelets; PP, posterior probability of colocalization.
Figure 2.
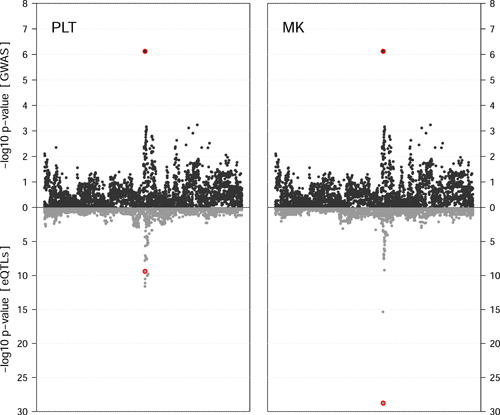
Colocalization using meta‐analysis p values (dark grey) and eQTL p values for association with ARHGEF3 (light grey), separately for platelets (PLT) and megakaryocytes (MK) eQTL. For clarity of display, the x‐axis represent the index in the SNP set, not the genomic locations. The respective p values for SNP rs1354034 are highlighted with a red background. eQTL, expression quantitative trait loci; SNP, single nucleotide polymorphism
4. DISCUSSION
GWAS have successfully identified tens of thousands of SNPs associated with complex traits, including genetic variants that affect platelet function by modifying platelet parameters such as platelet aggregation, platelet count, mean platelet volume and altering the expression of key platelet receptors. In general, SNPs that influence gene expression (eQTL) are significantly enriched (Nicolae et al., 2010), and consequently, researchers have explored various ways of incorporating eQTL into GWAS.
Using the ENCODE data base, Nicolae et al. (2010) constructed a score quantifying the likelihood that a SNP has a function in regulating transcript levels. They concluded that annotating SNPs with a score reflecting the strength of evidence that a SNP is an eQTL can improve ability to discover true associations. Gupta and Musunuru (2013) discussed the use of eQTL databases in the study of noncoding variants in cardiovascular and metabolic phenotypes, and reviewed successes in using eQTL to link variants with functional candidate genes. Zhu et al. (2016) proposed a new method called SMR that integrates summary‐level data from GWAS with expression data from eQTL to identify genes whose expression levels are associated with complex traits due to pleiotropy. The authors adopt a Mendelian randomization approach to estimate and test for the causative effect of an exposure variable on an outcome. J. Li et al. (2013) used eQTL weights as prior information in SNP‐based association tests to improve test power while maintaining control of the family‐wise error rate or false‐discovery rate. Some SNPs that were insignificant without eQTL weighting became significant using eQTL‐weighted Bonferroni or Benjamini–Hochberg procedures. The authors concluded that using informative weights may improve power, and little power is killed when uninformative weights are used. Saccone et al. (2010) developed an online prioritization tool (SPOT), which systematically combines multiple biological databases to prioritize SNPs by genomic information network. SNPs are assigned a prioritization score based on pathway information, comparative genomics, a linkage scan, and results from other independent GWAS. Wu et al. exploit the fact that complex traits are often affected by multiple genes in annotated gene pathways, and extend TWAS from a gene to a pathway based analysis (Wu & Pan, 2018). Integrating KEGG and GO pathways with GWAS and eQTL information, the authors were able to identify several novel pathways associated with schizophrenia. Zeng et al. (2019) investigated the prevalence and role of secondary cis‐eQTLs regulating gene expression in peripheral blood in two large cohort studies. A colocalization analysis of eQTL signals with GWAS hits detected 1349 genes whose expression in peripheral blood was associated with a total of 591 phenotypes, with more than 10% of the colocalized signals due to nonprimary cis‐eQTLs. After conducting GWAS on 12 complex eye diseases or traits, Strunz et al. (2020) examined regulation of gene expression in healthy retina as a disease relevant tissue, and identified more than 400,000 significant eQTL variants regulating more than 3000 genes. The authors found that expression of 10 of those genes was regulated by significant eQTLs associated with multiple eye diseases or traits, providing evidence for their role in the etiology of these conditions. These studies demonstrate that integrating eQTL information in GWAS can potentially improve power in highlighting causal genes.
In our study we presented an approach to improve power to detect GWAS signals when shared among eQTL by substantially lowering the genome‐wide significance threshold compared to the standard Bonferroni procedure using permutation analyses. In addition to improving power, focusing on eQTL also is more likely to yield functional variants. We also stress the importance of using relevant target tissues for eQTL analyses, particularly for the study of platelet related phenotypes. We previously investigated the transcriptional profile of platelets and iPSC‐derived megakaryocytes, and compared those with peak‐associated SNP‐expressed gene pairs of 48 other tissue types from the GTEx catalogue (GTEx Consortium, 2020). One of our key findings was that the eQTLs we detected were largely unique to MKs and PLs, with a somewhat large fraction not seen among any of the 48 GTEx tissues (Kammers, Taub, Rodriguez, et al., 2021).
Our here presented analysis strategy shares some of the characteristics of transcriptome‐wide association studies (TWAS). For example, both TWAS and our approach exploit the fact that gene expression may be a molecular mediator between genotype and phenotype. However, there are also important distinctions. TWAS were initially motivated by the fact that the majority of SNPs in the GWAS catalogue (https://www.ebi.ac.uk/gwas/) were in noncoding regions of the human genome, rendering functionality unclear. Thus, TWAS were introduced as an approach to directly investigate associations between genetically regulated gene expression and the phenotype of interest (Gamazon et al., 2015; Gusev et al., 2016). In general, a TWAS is a two‐step procedure where genetically regulated gene expression is first assessed in a reference data set for which transcriptomic data are available, and a prediction model is delineated based on these findings. The prediction model is then used in the second step to impute gene expression for the GWAS cohort, and the imputed gene expressions are then correlated with the phenotype of interest. Since its introduction, a variety of other TWAS‐like methods have been put forward (Barbeira et al., 2018, 2019; Hu et al., 2019; Luningham et al., 2020; Pividori et al., 2020; Xu et al., 2017; Zhou et al., 2020) that differ in a variety of aspects, such as the type of GWAS input, the statistical approaches to generate the eQTLs underlying the predictors, among others (see B. Li & Ritchie, 2021 for a review). A TWAS can be considered as a multimarker association approach that allows for the identification of genes underlying the etiology of the phenotype, while our approach identifies single variants that have potential to be causal. A TWAS is a more complicated procedure than our approach, and in particular the development of prediction models used in Step 1 is an active area of research (B. Li & Ritchie, 2021). Our approach for the main part is simply based on a GWAS and an eQTL analysis, with the latter being used to select SNPs from the former. The association p values from the GWAS do not change in our approach, only the significance threshold derived by the permutation test, does. In that sense it could also be reasoned that both TWAS and our methods improve power in part by reducing the multiple hypothesis burden.
In addition to confirming the well known PEAR1 platelet aggregation locus, we also identified a novel platelet and megakaryocyte eQTL rs1354034 (ARHGEF3) associated with aggregation to ADP after exposure to aspirin. The SNP rs1354034 falls within the protein coding gene ARHGEF3 (Rho guanine nucleotide exchange factor 3, RhoGEF3), which activates RhoGTPases and plays an important role in the regulation of cell morphology, cell aggregation, cytoskeletal rearrangements, and transcriptional activation. It regulates the switch of RhoGTPase from the inactive GDP‐bound state to the active GTP‐bound state and is one of the most abundant GEFs found in human megakaryocyte lineage and platelets (Astle et al., 2016; Eicher et al., 2016). ARHGEF3 has been shown in previous GWAS to be associated with platelet count and mean platelet volume (Gieger et al., 2011; J. Li et al., 2013; Lin et al., 2017; Read et al., 2019; Schick et al., 2016; Shameer et al., 2014). The silencing of ARHGEF3 has been shown to completely ablate erythropoiesis and thrombocyte formation in a zebrafish model (Serbanovic‐Canic et al., 2011). Serbanovic‐Canic et al. (2011) also reported that the disruption of the ARHGEF3 target, RhoA, produced severe anemia, which was corrected by iron injection. Zou et al. (2017) reported that rs1354034, which is located in a DNase I hypersensitive region in human megakaryocytes, is an eQTL associated with ARHGEF3 expression level in human platelets (Zou et al., 2017). They also suggested that it may be the causal SNP that accounts for the variations observed in human platelet traits and ARHGEF3 expression. They further reported that in vitro human platelet activation assays revealed rs1354034 is highly correlated with human platelet activation by ADP, and concluded that modulation of ARHGEF3 gene expression in humans with a promoter‐localized SNP may play a role in human megakaryocytes and human platelets. Our Bayesian colocalization analysis showed compelling evidence for a functional role of this eQTL.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supplementary Information
ACKNOWLEDGMENTS
Funding for methodological work was provided through NIH NHLBI R01 HL141944 (Rasika Mathias and Ingo Ruczinski, co‐PIs). GeneSTAR was supported through NIH NHLBI U01s HL072518, HL087698, HL112064, and M01‐RR000052 to the Johns Hopkins General Clinical Research Center from the NIH National Center for Research Resources.
Ngwa, J. S. , Yanek, L. R. , Kammers, K. , Kanchan, K. , Taub, M. A. , Scharpf, R. B. , Faraday, N. , Becker, L. C. , Mathias, R. A. , & Ruczinski, I. (2022). Secondary analyses for genome‐wide association studies using expression quantitative trait loci. Genetic Epidemiology, 46, 170–181. 10.1002/gepi.22448
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in the database of Genotypes and Phenotypes (dbGaP, www.ncbi.nlm.nih.gov/gap/) under accession number phs001218.v2.p1.
REFERENCES
- Albert, F. W. , & Kruglyak, L. (2015). The role of regulatory variation in complex traits and disease. Nature Reviews Genetics, 16(4), 197. [DOI] [PubMed] [Google Scholar]
- Astle, W. J. , Elding, H. , Jiang, T. , Allen, D. , Ruklisa, D. , Mann, A. L. , Mead, D. , Bouman, H. , Riveros‐Mckay, F. , Kostadima, M. A. , Lambourne, J. J. , Sivapalaratnam, S. , Downe, K. , Kundu, K. , Bomba, L. , Berentsen, K. , Bradley, J. R. , Daugherty, L. C. , Delaneau, O. , … Soranzo, N. (2016). The allelic landscape of human blood cell trait variation and links to common complex disease. Cell, 167(5), 1415–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeira, A. N. , Dickinson, S. P. , Bonazzola, R. , Zheng, J. , Wheeler, H. E. , Torres, J. M. , Torstenson, E. S. , Shah, K. P. , Garcia, T. , Edwards, T. L. , Stahl, E. A. , Huckins, L. M. , Nicolae, D. L. , Cox, N. J. , & Im, H. K. (2018). Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nature Communications, 9(1), 1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeira, A. N. , Pividori, M. , Zheng, J. , Wheeler, H. E. , Nicolae, D. L. , & Im, H. K. (2019). Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genetics, 15(1), e1007889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, D. M. , Segal, J. , Vaidya, D. , Yanek, L. R. , Herrera‐Galeano, J. E. , Bray, P. F. , Moy, T. F. , Becker, L. C. , & Faraday, N. (2006). Sex differences in platelet reactivity and response to low‐dose aspirin therapy. JAMA, 295(12), 1420–1427. [DOI] [PubMed] [Google Scholar]
- Bray, P. F. , Mathias, R. A. , Faraday, N. , Yanek, L. R. , Fallin, M. D. , Herrera‐Galeano, J. E , Wilson, A. F. , Becker, L. C. , & Becker, D. M. (2007). Heritability of platelet function in families with premature coronary artery disease. Journal of Thrombosis and Haemostasis, 5(8), 1617–1623. [DOI] [PubMed] [Google Scholar]
- Churchill, G. A. , & Doerge, R. W. (1994). Empirical threshold values for quantitative trait mapping. Genetics, 138(3), 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conomos, M. P. , & Thornton, T. (2016). Genetic estimation and inference in structured samples (genesis): Statistical methods for analyzing genetic data from samples with population structure and/or relatedness. R package version, 2(0.1).
- Devlin, B. , & Roeder, K. (1999). Genomic control for association studies. Biometrics, 55, 997–1004. [DOI] [PubMed] [Google Scholar]
- Dubois, P. C. , Trynka, G. , Franke, L. , Hunt, K. A. , Romanos, J. , Curtotti, A. , Zhernakova, A. , Heap, G. A. , Adány, R. , Aromaa, A. , Bardella, M. T. , van den Berg, L. H. , Bockett, N. A. , de la Concha, E. G. , Dema, B. , Fehrmann, R. S. , Fernández‐Arquero, M. , Fiatal, S. , Grandone, E. , … van Heel, D. A. (2010). Multiple common variants for celiac disease influencing immune gene expression. Nature Genetiics, 42(4), 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicher, J. D. , Chami, N. , Kacprowski, T. , Nomura, A. , Chen, M. H. , Yanek, L. R. , Tajuddin, S. M. , Schick, U. M. , Slater, A. J. , Pankratz, N. , Polfus, L. , Schurmann, C. , Giri, A. , Brody, J. A. , Lange, L. A. , Manichaikul, A. , Hill, W. D. , Pazoki, R. , Elliot, P. , … Johnson, A. D. (2016). Platelet‐related variants identified by exomechip meta‐analysis in 157,293 individuals. American Journal of Human Genetics, 99(1), 40–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emilsson, V. , Thorleifsson, G. , Zhang, B. , Leonardson, A. S. , Zink, F. , Zhu, J. , Carlson, S. , Helgason, A. , Walters, G. B. , Gunnarsdottir, S. , Mouy, M. , Steinthorsdottir, V. , Eiriksdottir, G. H. , Bjornsdottir, G. , Reynisdottir, I. , Gudbjartsson, D. , Helgadottir, A. , Jonasdottir, A. , Jonasdottir, A. , … Stefansson, K. (2008). Genetics of gene expression and its effect on disease. Nature, 452(7186), 423. [DOI] [PubMed] [Google Scholar]
- Faraday, N. , Yanek, L. R. , Mathias, R. , Herrera‐Galeano, J. E. , Vaidya, D. , Moy, T. F. , Fallin, M. D. , Wilson, A. F. , Bray, P. F. , Becker, L. C. , & Becker, D. M. (2007). Heritability of platelet responsiveness to aspirin in activation pathways directly and indirectly related to cyclooxygenase‐1. Circulation, 115(19), 2490–2496. [DOI] [PubMed] [Google Scholar]
- Frazee, A. C. , Pertea, G. , Jaffe, A. E. , Langmead, B. , Salzberg, S. L. , & Leek, J. T. (2015). Ballgown bridges the gap between transcriptome assembly and expression analysis. Nature Biotechnology, 33(3), 243–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman, J. , & Loscalzo, J. (2013). The role of platelets in coronary heart disease.
- Gamazon, E. R. , Wheeler, H. E. , Shah, K. P. , Mozaffari, S. V. , Aquino‐Michaels, K. , Carroll, R. J. , Eyler, A. E. , Denny, J. C. , Nicolae, D. L. , Cox, N. J. , & Im, H. K. (2015). A gene‐based association method for mapping traits using reference transcriptome data. Nature Genetics, 47(9), 1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giambartolomei, C , Vukcevic, D , Schadt, EE , Franke, L , Hingorani, AD , Wallace, C , & Plagnol, V (2014). Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genetics, 10(5), e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieger, C. , Radhakrishnan, A. , Cvejic, A. , Tang, W. , Porcu, E. , Pistis, G. , Serbanovic‐Canic, J. , Elling, U. , Goodall, A. H. , Labrune, Y. , Lopez, L. M. , Mägi, R. , Meacham, S. , Okada, Y. , Pirastu, N. , Sorice, R. , Teumer, A. , Voss, K. , Zhang, W. , … Soranzo, N. (2011). New gene functions in megakaryopoiesis and platelet formation. Nature, 480(7376), 201–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium . (2020). The gtex consortium atlas of genetic regulatory effects across human tissues. Science, 369, 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, R. M. , & Musunuru, K. (2013). Mapping novel pathways in cardiovascular disease using eqtl data: The past, present, and future of gene expression analysis. Frontiers in Genetics, 3, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev, A , Ko, A , Shi, H , Bhatia, G , Chung, W , Penninx, BW , Jansen, R , de Geus, EJ , Boomsma, DI , Wright, FA , Sullivan, PF , Nikkola, E , Alvarez, M , Civelek, M , Lusis, AJ , Lehtimäki, T , Raitoharju, E , Kähönen, M , Seppälä, I , … Pasaniuc, B (2016). Integrative approaches for large‐scale transcriptome‐wide association studies. Nature Genetics, 48(3), 245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, X. , Fuller, C. K. , Song, Y. , Meng, Q. , Zhang, B. , Yang, X. , & Li, H. (2013). Sherlock: Detecting gene‐disease associations by matching patterns of expression QTL and GWAS. American Journal of Human Genetics, 92(5), 667–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Y. , Li, M. , Lu, Q. , Weng, H. , Wang, J. , Zekavat, S. M. , Yu, Z. , Li, B. , Gu, J. , Muchnik, S. , Shi, Y. , Kunkle, B. W. , Mukherjee, S. , Natarajan, P. , Naj, A. , Kuzma, A. , Zhao, Y. , Crane, P. K. , Lu, H. , & Zhao, H. (2019). A statistical framework for cross‐tissue transcriptome‐wide association analysis. Nature Genetics, 51(3), 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y.‐T. , Liang, L. , Moffatt, M. F. , Cookson, W. O. , & Lin, X. (2015). IGWAS: Integrative genome‐wide association studies of genetic and genomic data for disease susceptibility using mediation analysis. Genetic Epidemiology, 39(5), 347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, S. P. (2007). The growing complexity of platelet aggregation. Blood, The Journal of the American Society of Hematology, 109(12), 5087–5095. [DOI] [PubMed] [Google Scholar]
- Johnson, A.D. , Yanek, L.R. , Chen, M.H. , Faraday, N. , Larson, M.G. , Tofler, G. , Lin, S.J. , Kraja, A.T. , Province, M.A. , Yang, Q. , Becker, D.M. , O'Donnell, C.J. , & Becker, L.C. (2010). Genome‐wide meta‐analyses identifies seven loci associated with platelet aggregation in response to agonists. Nature Genetics, 42(7), 608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammers, K. , Taub, M. A. , Mathias, R. A. , Yanek, L. R. , Kanchan, K. , Venkatraman, V. , Sundararaman, N. , Martin, J. , Liu, S. , Hoyle, D. , Raedschelders, K. , Holewinski, R. , Parker, S. , Dardov, V. , Faraday, N. , Becker, D. M. , Cheng, C. , Wang, Z. Z. , Leek, J. T. , Van Eyk, J. E. , & Becker, L.C. (2021). Gene and protein expression in human megakaryocytes derived from induced pluripotent stem cells. Journal of Thrombosis and Haemostasis, 19(7), 1783–1799. [DOI] [PubMed] [Google Scholar]
- Kammers, K. , Taub, M. A. , Rodriguez, B. , Yanek, L. R. , Ruczinski, I. , Martin, J. , Kanchan, K. , Battle, A. , Cheng, L. , Wang, Z. Z , Johnson, A. D. , Leek, J. T. , Faraday, N. , Becker, L. C. , & Mathias, R. A. (2021). Transcriptional profile of platelets and iPSC‐derived megakaryocytes from whole‐genome and RNA sequencing. Blood, 137, 959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammers, K. , Taub, M. A. , Ruczinski, I. , Martin, J. , Yanek, L. R. , Frazee, A. , Gao, Y. , Hoyle, D. , Faraday, N. , Becker, D. M. , Cheng, L. , Wang, Z. Z. , Leek, J. T. , Becker, L. C. , & Mathias, R. A. (2017). Integrity of induced pluripotent stem cell (iPSC) derived megakaryocytes as assessed by genetic and transcriptomic analysis. PLoS One, 12(1), e0167794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keramati, A. R. , Chen, M. H. , Rodriguez, B. A. T. , Yanek, L. R. , Bhan, A , Gaynor, B. J. , Ryan, K. , Brody, J. A. , Zhong, X. , Wei, Q. , Kammers, K. , Kanchan, K. , Iyer, K. , Kowalski, M. H. , Pitsillides, A. N. , Cupples, L. A. , Li, B. , Schlaeger, T. M. , Shuldiner, A. R. , … Johnson, A. D. (2021). Genome sequencing unveils a regulatory landscape of platelet reactivity. Nature Communication, 12(1), 3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keramati, A. R. , Yanek, L. R. , Iyer, K. , Taub, M. A. , Ruczinski, I. , Becker, D. M. , Becker, L. C. , Farada, N. , & Mathias, R. A. (2019). Targeted deep sequencing of the PEAR1 locus for platelet aggregation in European and African American families. Platelets, 30(3), 380–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. , Suktitipat, B. , Yanek, L. R. , Faraday, N. , Wilson, A. F. , Becker, D. M. , Becker, L. C. , & Mathias, R. A. (2013). Targeted deep resequencing identifies coding variants in the PEAR1 gene that play a role in platelet aggregation. PLoS One, 8(5), e64179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunicki, T. J. , & Nugent, D. J. (2010). The genetics of normal platelet reactivity. Blood, The Journal of the American Society of Hematology, 116(15), 2627–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, J. P. , Ryan, K. , O'Connell, J. R. , Horenstein, R. B. , Damcott, C. M. , Gibson, Q. , Pollin, T. I. , Mitchell, B. D. , Beitelshees, A. L. , Pakzy, R. , Tanner, K. , Parsa, A. , Tantry, U. S. , Bliden, K. P. , Post, W. S. , Faraday, N. , Herzog, W. , Gong, Y. , Pepine, C. J. , … Shuldiner, A. R. . (2013). Genetic variation in pear1 is associated with platelet aggregation and cardiovascular outcomes. Circulation: Cardiovascular Genetics, 6(2), 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B. , & Ritchie, M. D. (2021). From gwas to gene: Transcriptome‐wide association studies and other methods to functionally understand GWAS discoveries. Frontiers in Genetics, 12, 713230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Glessner, J.T. , Zhang, H. , Hou, C. , Wei, Z. , Bradfield, J.P. , Mentch, F.D. , Guo, Y. , Kim, C. , Xia, Q. , Chiavacci, R.M. , Thomas, K.A. , Qiu, H. , Grant, S.F. , Furth, S.L. , Hakonarson, H. , & Sleiman, P.M. (2013). GWAS of blood cell traits identifies novel associated loci and epistatic interactions in Caucasian and African‐American children. Human Molecular Genetics, 22(7), 1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby, P. (2001). Current concepts of the pathogenesis of the acute coronary syndromes. Circulation, 104(3), 365–372. [DOI] [PubMed] [Google Scholar]
- Lin, B. D. , Carnero‐Montoro, E. , Bell, J. T. , Boomsma, D. I. , de Geus, E. J. , Jansen, R. , Kluft, C. , Mangino, M. , Penninx, B. , Spector, T. D. , Willemsen, G. , & Hottenga, J. J. (2017). 2SNP heritability and effects of genetic variants for neutrophil‐to‐lymphocyte and platelet‐to‐lymphocyte ratio. Journal of Human Genetics, 62(11), 979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luningham, J. M. , Chen, J. , Tang, S. , De Jager, P. L. , Bennett, D. A. , Buchman, A. S. , & Yang, J. (2020). Bayesian genome‐wide TWAS method to leverage both cis‐ and trans‐eQTL information through summary statistics. American Journal of Human Genetics, 107(4), 714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio, T. A. , Collins, F. S. , Cox, N. J. , Goldstein, D. B. , Hindorff, L. A. , Hunter, D. J. , McCarthy, M. I. , Ramos, E. M. , Cardon, L. R. , Chakravarti, A. , Cho, J. H. , Guttmacher, A. E. , Kong, A. , Kruglyak, L. , Mardis, E. , Rotimi, C. N. , Slatkin, M. , Valle, D. , Whittemore, A. S. , … Visscher, P. M. (2009). Finding the missing heritability of complex diseases. Nature, 461(7265), 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias, R. A. , Kim, Y. , Sung, H. , Yanek, L. R. , Mantese, V. J. , Hererra‐Galeano, J. E. , Ruczinski, I. , Wilson, A. F. , Faraday, N. , Becker, L. C. , & Becker, D. M. (2010). A combined genome‐wide linkage and association approach to find susceptibility loci for platelet function phenotypes in European American and African American families with coronary artery disease. BMC Medical Genomics, 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisinger, C. , Prokisch, H. , Gieger, C. , Soranzo, N. , Mehta, D. , Rosskopf, D. , Lichtner, P. , Klopp, N. , Stephens, J. , Watkins, N. A. , Deloukas, P. , Greinacher, A. , Koenig, W. , Nauck, M. , Rimmbach, C. , Völzke, H. , Peters, A. , Illig, T. , Ouwehand, W. H. , … Döring, A. (2009). A genome‐wide association study identifies three loci associated with mean platelet volume. American Journal of Human Genetics, 84(1), 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nica, A.C. , Montgomery, S.B. , Dimas, A.S. , Stranger, B.E. , Beazley, C. , Barroso, I. , & Dermitzakis, E.T. (2010). Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genetics, 6(4), e1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolae, D.L. , Gamazon, E. , Zhang, W. , Duan, S. , Dolan, M.E. , & Cox, N.J. (2010). Trait‐associated SNPs are more likely to be eQTLs: Annotation to enhance discovery from GWAS. PLoS Genetics, 6(4), e1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez de Prado, A , ernández‐Vázquez, F , Cuellas, J. C. , Alonso‐Orcajo, N. , Carbonell, R. , Pascual, C. , Olalla, C. , Diego, A. , de Miguel, A. , & Calabozo, R. G. (2006). Association between level of platelet inhibition after early use of abciximab and myocardial reperfusion in ST‐elevation acute myocardial Infarction treated by primary percutaneous coronary intervention. American Journal of Cardiology, 97(6), 798–803. [DOI] [PubMed] [Google Scholar]
- Pertea, M. , Kim, D. , Pertea, G. M. , Leek, J. T. , & Salzberg, S. L. (2016). Transcript‐level expression analysis of RNA‐seq experiments with hisat, stringtie and ballgown. Nature Protocols, 11(9), 1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pividori, M. , Rajagopal, P. S. , Barbeira, A. , Liang, Y. , Melia, O. , Bastarache, L. , Park, Y. S. , GTEx, Consortium , Wen, X. , & Im, H. K. (2020). Phenomexcan: Mapping the genome to the phenome through the transcriptome. Science Advances, 6, eaba2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruim, R.J. , Welch, R.P. , Sanna, S. , Teslovich, T.M. , Chines, P.S. , Gliedt, T.P. , Boehnke, M. , Abecasis, G.R. , & Willer, C.J. (2010). LocusZoom: Regional visualization of genome‐wide association scan results. Bioinformatics, 26(18), 2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qayyum, R. , Becker, L.C. , Becker, D.M. , Faraday, N. , Yanek, L.R. , Leal, S.M. , Shaw, C. , Mathias, R. , Suktitipat, B. , & Bray, P.F. (2015). Genome‐wide association study of platelet aggregation in African Americans. BMC Genetics, 16, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read, R. W. , Schlauch, K. A. , Elhanan, G. , Metcalf, W. J. , Slonim, A. D. , Aweti, R. , Borkowski, R. , & Grzymski, J. J. (2019). GWAS and PHEWAS of red blood cell components in a northern Nevadan cohort. PLoS One, 14, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone, S. F. , Bolze, R. , Thomas, P. , Quan, J. , Mehta, G. , Deelman, E. , Tischfield, J. A. , & Rice, J. P. (2010). SPOT: A web‐based tool for using biological databases to prioritize SNPs after a genome‐wide association study. Nucleic Acids Research, 38(Web Server issue), W201–W209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schick, U.M. , Jain, D. , Hodonsky, C.J. , Morrison, J.V. , Davis, J.P. , Brown, L. , Sofer, T. , Conomos, M.P. , Schurmann, C. , McHugh, C.P. , Nelson, S.C. , Vadlamudi, S. , Stilp, A. , Plantinga, A. , Baier, L. , Bien, S.A. , Gogarten, S.M. , Laurie, C.A. , Taylor, K.D. , … Reiner, A.P. (2016). Genome‐wide association study of platelet count identifies ancestry‐specific loci in Hispanic/Latino Americans. American Journal of Human Genetics, 98(2), 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbanovic‐Canic, J. , Cvejic, A. , Soranzo, N. , Stemple, D. L. , Ouwehand, W. H. , & Freson, K. (2011). Silencing of rhoa nucleotide exchange factor, arhgef3, reveals its unexpected role in iron uptake. Blood, The Journal of the American Society of Hematology, 118(18), 4967–4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalin, A. A. (2012). Matrix eqtl: Ultra fast EQTL analysis via large matrix operations. Bioinformatics, 28(10), 1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shameer, K. , Denny, J. C. , Ding, K. , Jouni, H. , Crosslin, D. R. , de Andrade, M. , Chute, C. G. , Peissig, P. , Pacheco, J. A. , Li, R. , Bastarache, L. , Kho, A. N. , Ritchie, M. D. , Masys, D. R. , Chisholm, R. L. , Larson, E. B. , McCarty, C. A. , Roden, D. M. , Jarvik, G. P. , & Kullo, I. J. (2014). A genome‐ and phenome‐wide association study to identify genetic variants influencing platelet count and volume and their pleiotropic effects. Human Genetics, 133(1), 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strunz, T. , Kiel, C. , Grassmann, F. , Ratnapriya, R. , Kwicklis, M. , Karlstetter, M. , Fauser, S. , Arend, N. , Swaroop, A. , Langmann, T. , Wolf, A. , & Weber, B. H. F. (2020). A mega‐analysis of expression quantitative trait loci in retinal tissue. PLoS Genetics, 16(9), e1008934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliun, D. , Harris, D. N. , Kessler, M. D. , Carlson, J. , Szpiech, Z. A. , Torres, R. , Taliun, S. A. G. , Corvelo, A. , Gogarten, S. M. , Kang, H. M. , Pitsillides, A. N. , LeFaive, J. , Lee, S. B. , Tian, X. , Browning, B. L. , Das, S. , Emde, A. K. , Clarke, W. E. , Loesch, D. P. , … Abecasis, G. R. (2021). Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature, 590(7845), 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya, D. , Yanek, L. R. , Moy, T. F. , Pearson, T. A. , Becker, L. C. , & Becker, D. M. (2007). Incidence of coronary artery disease in siblings of patients with premature coronary artery disease: 10 years of follow‐up. American Journal of Cardiology, 100(9), 1410–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield, J. (2009). Bayes factors for genome‐wide association studies: Comparison with p‐values. Genetic Epidemiology, 33, 79–86. [DOI] [PubMed] [Google Scholar]
- Wu, C. , & Pan, W. (2018). Integrating EQTL data with gwas summary statistics in pathway‐based analysis with application to schizophrenia. Genetic Epidemiology, 42, 303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Z. , Wu, C. , Wei, P. , & Pan, W. (2017). A powerful framework for integrating eqtl and gwas summary data. Genetics, 207, 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanek, L. R. , Kral, B. G. , Moy, T. F. , Vaidya, D. , Lazo, M. , Becker, L. C. , & Becker, D. M. (2013). Effect of positive well‐being on incidence of symptomatic coronary artery disease. American Journal of Cardiology, 112(8), 1120–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, B. , Lloyd‐Jones, L. R. , Montgomery, G. W. , Metspalu, A. , Esko, T. , Franke, L. , Vosa, U. , Claringbould, A. , Brigham, K. L. , Quyyumi, A. A. , Idaghdour, Y. , Yang, J. , Visscher, P. M. , Powell, J. E. , & Gibson, G. (2019). Comprehensive multiple eQTL detection and its application to GWAS interpretation. Genetics, 212(3), 905–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, D. , Jiang, Y. , Zhong, X. , Cox, N. J. , Liu, C. , & Gamazon, E. R. (2020). A unified framework for joint‐tissue transcriptome‐wide association and Mendelian randomization analysis. Nature Genetics, 52(11), 1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Z. , Zhang, F. , Hu, H. , Bakshi, A. , Robinson, M. R. , Powell, J. E. , Montgomery, G. W. , Goddard, M. E. , Wray, N. R. , Visscher, P. M. , & Yang, J. (2016). Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nature Genetics, 48(5), 481–487. [DOI] [PubMed] [Google Scholar]
- Zou, S. , Teixeira, A. M. , Kostadima, M. , Astle, W. J. , Radhakrishnan, A. , Simon, L. M. , Truman, L. , Fang, J.S. , Hwa, J. , Zhang, P. X. , van der Harst, P. , Bray, P. F. , Ouwehand, W. H. , Frontini, M. , & Krause, D. S. (2017). SNP in human ARHGEF3 promoter is associated with DNase hypersensitivity, transcript level and platelet function, and Arhgef3 KO mice have increased mean platelet volume. PLoS One, 12(5), e0178095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Data Availability Statement
The data that support the findings of this study are openly available in the database of Genotypes and Phenotypes (dbGaP, www.ncbi.nlm.nih.gov/gap/) under accession number phs001218.v2.p1.