

Abstract
Since its first detection in 2019, the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS‐CoV‐2) has been the cause of millions of deaths worldwide. Despite the development and administration of different vaccines, the situation is still worrisome as the virus is constantly mutating to produce newer variants some of which are highly infectious. This raises an urgent requirement to understand the infection mechanism and thereby design therapeutic‐based treatment for COVID‐19. The gateway of the virus to the host cell is mediated by the binding of the receptor binding domain (RBD) of the virus spike protein to the angiotensin‐converting enzyme 2 (ACE2) of the human cell. Therefore, the RBD of SARS‐CoV‐2 can be used as a target to design therapeutics. The α1 helix of ACE2, which forms direct contact with the RBD surface, has been used as a template in the current study to design stapled peptide therapeutics. Using computer simulation, the mechanism and thermodynamics of the binding of six stapled peptides with RBD have been estimated. Among these, the one with two lactam stapling agents has shown binding affinity, sufficient to overcome RBD‐ACE2 binding. Analyses of the mechanistic detail reveal that a reorganization of amino acids at the RBD‐ACE2 interface produces favorable enthalpy of binding whereas conformational restriction of the free peptide reduces the loss in entropy to result higher binding affinity. The understanding of the relation of the nature of the stapling agent with their binding affinity opens up the avenue to explore stapled peptides as therapeutic against SARS‐CoV‐2.
Keywords: binding free energy, RBD‐ACE2 interaction, SARS‐CoV‐2, stapled peptide
1. INTRODUCTION
The global pandemic caused by COVID‐19 has been the cause of more than 5.5 million deaths as of January 2022 and has massively affected the global economy.[ 1 ] Several respiratory syndromes, pneumonia, etc. are caused by the infection of the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) and these are highly contagious in nature.[ 2 , 3 ] Although SARS‐CoV‐2 belongs to the same coronavirus family as MERS‐CoV, SARS‐CoV, etc.[ 4 ] the mutations have made the virus more resistant to neutralizing antibodies.[ 5 ] In addition to that, several new variants of the virus have evolved which can escape the binding of antibodies, effective against the native form of the virus. The process of viral infection comprises several stages and different components of the virus are involved in these stages. In the very first step, the Spike protein of SARS‐CoV‐2 binds to the human ACE2 receptor.[ 6 , 7 , 8 , 9 ] The spike protein has a trimeric structure and each monomer is formed by several structural domains (Figure 1a).[ 10 ] During the virus attachment stage, the receptor binding domain (RBD) of one monomer gets activated and attaches to the human ACE2 receptor followed by membrane fusion and internalization of genetic material.[ 11 , 12 , 13 ] A closer look at the RBD‐ACE2 complex shows that the complexation is guided by the interaction of the α1 helix of ACE2 with the surface residues of the RBD.[ 14 ] The complexation eventually leads to the internalization of the virus and host membrane and the subsequent transfer of viral genetic material into the host cell. The attempts made to find the cure for this includes designing vaccines, antibodies, and drugs that can inhibit the crucial stages of the infection process. The continuous effort to design suitable inhibitor molecules, which can be, used as drugs to target different proteins of the virus, for example, the main protease (MP), non‐structural proteins (NSP) domains, receptor‐binding domain (RBD), etc. Among these, RBD has been targeted most of the time because of its direct interaction with the human ACE2 receptor. Unlike the other targets, the protein–protein interaction interface between ACE2 and RBD is quite extended and flexible,[ 15 ] thereby designing an inhibitor is challenging.[ 16 ]
FIGURE 1.
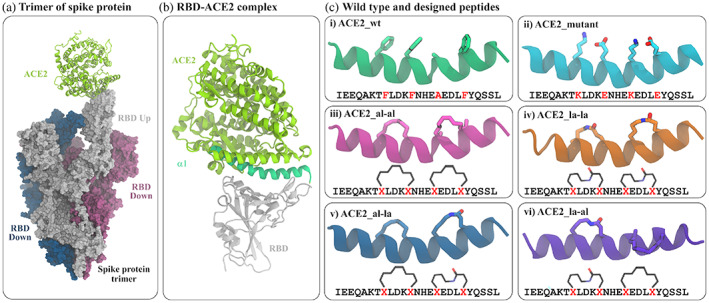
Modeling of the RBD‐ACE2 interaction. (a) The trimeric assembly of SARS‐CoV‐2 spike protein in complex with human ACE2 protein. Out of three spike proteins, two (blue and red surface) have their RBD buried (RBD down) and one (white surface) is with an exposed RBD (RBD up) conformation which binds at one end of human ACE2 (light green cartoon). (b) ACE2 interacts with amino acids of RBD (white ribbon) through its α1 helix (green ribbon). (c) The structures and sequences of the truncated α1 and five modified peptides. The amino acids at positions 28, 32, 36, and 40 are shown in sticks. The details of mutation and attachment of stapling agents are listed in Table 1
The two most widely used classes of inhibitors are small molecules and peptides.[ 17 , 18 ] Due to the extended shape of the binding pocket, small molecule inhibitors often lack specificity against wider binding pockets like that of RBD.[ 19 ] Rather a peptide inhibitor is more suitable to target such kind of interfaces.[ 20 ] There are few efforts to design peptide inhibitors against RBD. The basic working principle is to design a peptide that can mimic the binding mode of ACE2 with RBD. The simplest possible solution is to use the alpha helix of ACE2, which binds to RBD as a potential inhibitor. However, the wildtype α1 helix of ACE2 has been reported to lack proper binding with RBD in previous studies.[ 21 ] This has led to a modification of the wild‐type peptide yielding new peptides. These modifications mainly include mutation of some nonessential amino acids, use of crosslinkers to enhance binding affinity. Amino acids at suitable positions along the peptide backbone can be replaced with a specific functional group, which can be further crosslinked to generate stapled peptides.[ 22 , 23 ] Stapled peptides have shown promising results to inhibit different protein–protein interactions. By altering the length, attachment point, and chemical nature of the stapling agents, the design of stapled peptides can be tuned to achieve desirable affinity and target specificity.[ 18 , 24 , 25 ] Few experimental and computational reports have checked the stability of stapled ACE2 and their binding with RBD.[ 21 , 26 , 27 ] However, a rationale to combine stapling agents chemical nature and length with their binding affinity is still lacking which is crucial for the future design of stapled peptides.
In the current work, we have considered aliphatic and lactam stapling agents and used a combination of them to design four stapled peptides. Binding free energy calculation from extensive molecular dynamics simulation for the binding of stapled and unstapled peptides reveals that the binding is guided by both gain in enthalpic interaction and loss in entropic penalty. The staple ACE2 peptide, with two i − i + 4 lactam staples, is found to exhibit the most favorable binding.
2. METHODS
2.1. Computational design of peptide inhibitor
The structure of RBD bound to the α1 helical domain of ACE2 is modeled starting from the complex of SARS‐CoV‐2 RBD and full‐length human ACE2 (PDB ID: 6M0J).[ 7 ] In the structure, the length of the α1 helix of ACE2 is about ~30 residue (~21–56). The residues of α1 helix of ACE2 which interact favorably with RBD binding pocket are Gln24, Tyr27, Asp30, Lys31, His34, Glu35, Glu37, Asp38, Tyr41, and Gln42.[ 28 , 29 ] A careful inspection of the structure shows that the entire α1 does not interact with RBD, rather the 4–5 residues at the C‐terminal end are not in direct contact with RBD instead interact with the remaining part of full length ACE2. Therefore, we have considered a smaller section of the α1 helix as a template to increase the effectiveness of the peptide. The first 25 residues of α1 is found sufficient to span over the entire binding surface of RBD and it also accommodates the residues of ACE2 (Gln24, Tyr27, Asp30, Lys31, His34, Glu35, Glu37, Asp38, Tyr41, Gln42) involved in important ACE2‐RBD interactions. Thus, from the crystal structure, this 25‐residue domain (I21EEQAKTFLDKFNHEAEDLFYQSSL45) is chosen as our starting peptide (termed as ACE2_wt [wild type ACE2] in the rest of the manuscript) and modified further to design other peptides. This truncated form of α1 has been found to effectively bind RBD in some previous studies.[ 30 , 31 , 32 , 33 ]
In this work, we have used a stapling approach where the side chains of two amino acids of ACE2_peptide are joined together to form a stapled peptide.[ 22 , 23 ] and their binding with RBD is investigated. Since α1 of ACE2 binds in helical conformation, the helical propensity of the designed peptide will be crucial in binding with its target partner RBD of SARS‐CoV‐2.[ 28 ] Hence, for a large peptide of 25 residues, two i − i + 4 staples will be more useful than a single i − i + 7 staple in maintaining its helical structure. The stapling positions are selected based on the conditions that replaced residues should not be among those residues, which have favorable interactions with RBD as previously reported[ 28 , 29 ] and the newly designed peptides should have the proper orientation to gain a stronger binding affinity to RBD.[ 32 ] In the ACE2‐RBD complex, the ACE2 residues (Gln24, Tyr27, Asp30, Lys31, His34, Glu35, Glu37, Asp38, Tyr41, and Gln42) having favorable interaction with RBD are present at the ACE2‐RBD contact surface. These important residues were kept unaltered and residues on the opposite face of the ACE2 α1 helix, those are not directly interacting with RBD were available to attach a stapling agent. Considering all these above facts, we have selected our stapling positions by replacing Phe28, Phe32 to form an i − i + 4 staple and another i − i + 4 staple in the positions of Ala36 and Phe40. Two stapling agents are considered as crosslinkers between the two pairs of residues one of which is an all‐hydrocarbon staple and the other is a lactam crosslinker. A combination of these two stapling agents is used to design four stapled peptides. A peptide with mutations at the aforementioned four positions (F28K, F32E, A36K, and F40E) is also designed. The details of these peptides and their complexes with RBD are listed in Table 1.
TABLE 1.
Details of the system compositions and simulation lengths for different systems
| System identifier | Protein | Peptide | Simulation length | ||
|---|---|---|---|---|---|
| 1 | ACE2_wt | — | Truncated α1 helix of ACE2 (21 to 45) |
|
|
| 2 | ACE2_mutant | — | ACE2_wt with F28K, F32E, A36K, and F40E mutations |
|
|
| 3 | ACE2_al‐al | — | ACE2_wt with two i‐i + 4 aliphatic staples between residue pairs 28, 32 and 36, 40 |
|
|
| 4 | ACE2_la‐la | — | ACE2_wt with two i‐i + 4 lactam staples between residue pairs 28, 32 and 36, 40 |
|
|
| 5 | ACE2_al‐la | — | ACE2_wt with one i‐i + 4 aliphatic staples between residue pairs 28, 32 and one lactam staple between residue pairs 36, 40 |
|
|
| 6 | ACE2_la‐al | — | ACE2_wt with one i‐i + 4 lactam staples between residue pairs 28, 32 and one aliphatic staple between residue pairs 36, 40 |
|
|
| 7 | RBD | SARS‐Cov‐2 Receptor Binding domain (Residues 333 to 526) | — |
|
|
| 8 | RBD + ACE2_wt | RBD | ACE2_wt |
|
|
| 9 | RBD + ACE2_mutant | RBD | ACE2_mutant |
|
|
| 10 | RBD + ACE2_al‐al | RBD | ACE2_al‐al |
|
|
| 11 | RBD + ACE2_la‐la | RBD | ACE2_la‐la |
|
|
| 12 | RBD + ACE2_al‐la | RBD | ACE2_al‐la |
|
|
| 13 | RBD + ACE2_la‐al | RBD | ACE2_la‐al |
|
|
|
|
2.2. Simulation details
The CHARMM36[ 34 ] forcefield parameters are employed to design the protein‐peptide complex and a combination of CHARMM parameters of proteins and small molecules (CGENFF) are used to model the two stapling agents. For designing an aliphatic crosslinker at targeted i and i + 4 positions, first, the amino acids are replaced by lysine and then s‐NH2 groups of lysine are replaced by an aliphatic chain having an olefinic bond, and end of the two aliphatic side chains are joined together.[ 35 ] For ACE2_mutant, i and i + 4 residues are mutated to lysine (K) and glutamic acid (E) and for lactam crosslinker designing, these mutated residues are patched together to form a —C(O)—NH linkage.[ 27 ] This strategy has been successfully used to model stapled peptides in other studies as well. Taking all these cross‐linker designs into account, we have modeled four stapled versions of ACE2_wt, first one is ACE2 with a double aliphatic crosslinker (abbreviated as ACE2_al‐al) where one staple is at 28–32 residue and another at 36–40 position, the second one is ACE2_wt with double lactam crosslinker (abbreviated as ACE2_la‐la) where two staples are at 28–32 and 36–40 positions, the third one is ACE2_wt with one aliphatic and one lactam staple (abbreviated as ACE2_al‐la) where aliphatic liker is at 28–32 and lactam linker is at 36–40 and the last one is vice versa of the previous one that is lactam is at 28–32 and aliphatic is at 36–40 position (abbreviated as ACE2_la‐al). Along with all these designed peptides, 25‐mer ACE2 (abbreviated as ACE2_wt) and a mutated version of ACE2_wt (ACE2_mutant) are considered for a better structural comparison and their binding with RBD. All the six peptides are simulated in the free state and in complex with RBD. In addition, only the RBD is simulated for the calculation of binding energy following the multiple trajectory approach.
The N‐terminal and C‐terminal amino acids of RBD and ACE2‐peptides in the free and bound states are capped with acetyl and amide groups respectively to avoid unwanted interaction of the bare terminal charges with the rest of the system. Each of the systems is neutralized by adding the required numbers of K+ and Cl− ions and solvated in a cubic water box made of TIP3P water,[ 36 ] where the size of the box is determined by maintaining a distance of at least 1 nm between the water box edge and protein/peptide atoms to satisfy the periodic boundary condition. For the removal of initial steric clashes, a 5000‐step energy minimization is performed for each system using the steepest descent method.[ 37 ] Subsequently, a 500 ps equilibration in the NVT ensemble is performed to equilibrate each system at 310 K to avoid void formation in the box followed by a 20 ns equilibration at isothermal‐ isobaric (NPT) ensemble to attain a steady pressure of 1 atm considering a pressure relaxation of 1 ps. The temperature is kept constant at 310 K by applying the V–rescale thermostat[ 38 ] and the pressure was maintained to be at 1 atm using Parrinello‐Rahman barostat[ 39 ] with a pressure relaxation time of 1 ps, used for the attainment of desired pressure for all simulations. Finally, the production runs for 1000 ns with a time step of 2 fs are performed. All the simulations are performed in GROMACS.[ 40 ] Short‐ranged Lennard‐Jones interactions are calculated using the minimum image convention.[ 41 ] For estimating nonbonding interactions including electrostatic as well as van der Waals interactions, a spherical cut‐off distance of 1 nm is chosen. Periodic boundary conditions have been used in all three directions to remove edge effects. SHAKE algorithm[ 42 ] is applied to constrain bonds involving the hydrogen atoms of the water molecules. Long‐range electrostatic interactions are calculated using the particle mesh Ewald (PME) method.[ 43 ] The frames in the trajectory are saved at a frequency of 2 ps for analyses. To extract different structural properties and for visualization, in‐built modules of GROMACS,[ 40 ] VMD,[ 44 ] and some in‐house scripts are used.
2.3. Binding energy calculation
The most popular and comparably less computationally expensive method MM‐GBSA (molecular mechanics with the generalized bond surface area) is used to calculate the standard free energy of binding (∆G). Generally, the binding free energy of a protein–peptide complexation is calculated by subtracting the free energy of both protein and peptide in their free state from the free energy of the protein‐peptide complex.[ 45 ]
| (1) |
where ∆Gsolvated can be represented as,
| (2) |
which can further can be simplified as ‐
| (3) |
The first three terms of Equation (3) describe the gas‐phase energy which arises due to molecular motions such as bond vibration, angle bending, dihedral rotation, etc. and van der Waals and electrostatic interaction respectively. The fourth and fifth terms represent the solvation free energy calculated considering implicit solvent environments in two separate parts, that is, polar and non‐polar components. The polar solvation energy is estimated considering generalized born (GB) solvent model and the nonpolar component is assumed to be proportional to the solvent accessible surface area (SASA).[ 30 , 46 ] The last term is solute entropy, where T represents the absolute temperature and S is the entropy of the solute, whereas the solvation entropy is already included in solvation energy terms.[ 47 ] The solute entropy is calculated by considering quasi‐harmonic approximation.[ 48 ] In short, the sum of the first five terms is considered as enthalpy, H although it includes the solvation entropy term and simply can be written as,
| (4) |
In practice, all the simulations of the free and complex state are performed in explicit water to get the conformational microstates of the solute, and then the free energy of solvation of these conformations are estimated using an implicit environment after the removal of explicit water molecules. In this work, we have followed multiple trajectory protocols (MTP) and performed three independent simulations of RBD + ACE2_wt, modified RBD + ACE2_wt systems, RBD and ACE2, and their mutant and stapled versions. All of the free energy of solvation and entropy calculation is performed in CHARMM[ 49 ] and averaged over 50,000 conformations of each system over the last 500 ns of the simulations. The values calculated for the three simulations are summarized in Supporting information tables (Table S1–S19) and an average of the three simulations is provided in Table 2. Similar stepwise procedures were followed in an earlier work to successfully reproduce the favorable binding of stapled p53 peptide with mdm2 compared to wild type p53.[ 35 ]
TABLE 2.
Components of binding free energy of RBD‐ACE2‐peptide binding
| Energy components | RBD + ACE2_wt | RBD + ACE2_mutant | RBD + ACE2_al‐al | RBD + ACE2_la‐la | RBD + ACE2_al‐la | RBD + ACE2_la‐al |
|---|---|---|---|---|---|---|
| ∆EElec | −273.05 | −272.86 | −294.54 | −285.80 | −215.85 | −349.18 |
| ∆Evdw | −30.74 | −38.03 | −45.65 | −47.43 | −30.50 | −41.44 |
| ∆Einternal | 19.77 | −0.98 | −0.36 | −0.57 | −2.23 | 3.05 |
| ∆Esolv(polar) | 245.30 | 262.66 | 288.16 | 272.07 | 209.51 | 336.51 |
| ∆Esolv(non−polar) | −1.60 | −2.28 | −2.89 | −3.25 | −1.89 | −2.65 |
| ∆EElec+solv(polar) | −27.75 | −10.20 | −6.38 | −13.73 | −6.34 | −12.67 |
| ∆H | −40.31 | −51.50 | −55.28 | −64.98 | −40.96 | −53.72 |
| −T∆S | 21.97 | 27.83 | 28.67 | 19.08 | 6.08 | 31.74 |
| ∆G | −18.34 | −23.67 | −26.61 | −45.90 | −34.88 | −21.98 |
Note: All energy values are in kcal/mol.
3. RESULTS
3.1. The dynamics of the N‐terminal loop of RBD is regulated by ACE2‐peptide binding
The binding of ACE2 and RBD is governed by mainly electrostatic interaction between the residues of the binding pocket of RBD and the α1 helix of ACE2. To compare the structural changes in RBD induced by ACE2_wt peptide and other designed peptides, the root mean square fluctuations of the residues of RBD were calculated. The RMSFs of the amino acids of RBD in the presence of different peptides and the absence of them are plotted in Figure 2. The fluctuations in different regions of RBD vary when different peptides interact with the binding pocket residues. Maximum fluctuation is observed in the residue range 470–490, which belongs to the loop region close to the ACE2 binding pocket of RBD. Higher fluctuation of this loop at the interface of RBD and ACE2 have been reported in a previous study.[ 50 ] The higher fluctuations in that region in the presence of peptides are reflections of the direct interaction of the loop residues with the peptides. The interaction of the loop residues with ACE2 peptides was quantified by using a contact map between the residues of the two regions (Figure S1). From the plot, it is clear that there is a significant enhancement of the contacts between the two structural regions when the ACE2‐peptide is modified by mutation or by adding stapling agents. Since the stapling agents modify the peptide conformations, the interaction of ACE2‐peptides with the binding pocket residues of RBD will also be altered. A qualitative description of that has been provided as a representative snapshot of the three ACE2‐peptides in Figure 3.
FIGURE 2.
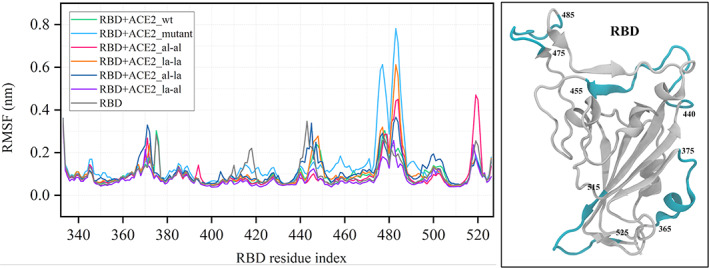
Root mean square fluctuation (RMSF) of the receptor‐binding domain (RBD) in a bound state with different designed peptides. RBD in apo state is shown on the right in white ribbon. The regions with higher fluctuation in the RMSF plot are marked in cyan
FIGURE 3.
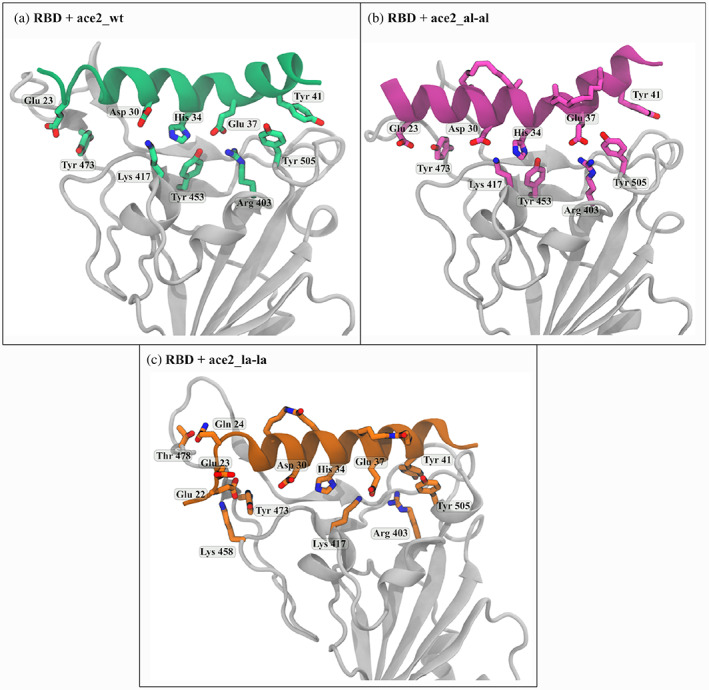
Residue level interaction between receptor binding domain (RBD) (white ribbon) and (a) 25‐mer ACE2_wt, (b) ACE2 with double aliphatic crosslinker (ACE2_al‐al) and (c) ACE2 with double lactam crosslinker (ACE2_la‐la)
The ACE2 peptide is stabilized on the binding pocket of RBD mainly by polar interaction between the amino acids (Glu23, Asp30, His34, Glu37, Tyr41) of RBD facing side of ACE2 peptide and the amino acids (Tyr473, Lys 417, Tyr453, Arg403 and Tyr505) at the surface of RBD. The interacting amino acids of ACE2_wt and RBD are shown in Figure 3a. The interacting residues get reorganized and that modifies the interaction between the peptide and RBD upon introduction of stapling agents. The extent of this alteration is different in the presence of different kinds of stapling agents. For example, the interactions are slightly modified between RBD and ACE2_al‐al (Figure 3b) whereas there is a significant change in the peptide conformation in RBD + ACE2_la‐la and that involves more RBD residues in interaction (Figure 3c).
The structural rearrangements in both ACE2‐peptide and RBD will impact the ACE2‐peptide‐RBD binding process and this can be quantified by calculating their binding free energies. The binding free energy is calculated using MMGBSA protocol as it is found to be a considerably accurate yet computationally less expensive method for binding energy estimation.[ 35 , 51 , 52 ]
3.2. Binding is governed by both enthalpy and entropy
The binding free energies are estimated for RBD bound to six ACE2‐peptides. The average binding free energy was quantified considering both enthalpy and entropy. The enthalpy was calculated using the MMGBSA protocol, a formalism used to find the solvation energy using a continuum model of implicit solvent. The conformational entropy is estimated considering quasi‐harmonic approximation on the ensemble of conformations. These values are calculated over the last 500 ns of 1 μs trajectory and further averaged over the three independent simulations for each system. The results are summarized in Table 2.
From the binding energy values, it is clear that most of the ACE2 peptides with the stapling agent or with mutation show favorable binding affinity to RBD of SARS‐CoV‐2. Out of six ACE2 peptides and their stapled versions, ACE2 with lactam double crosslinker (ACE2_la‐la) shows the best binding affinity value (~−46 kcal/mol). A direct comparison of the binding energy values with the experimental binding energy is not possible as that is not available for a peptide with the exact same sequence. However, a 30‐residue peptide derived from ACE2 (TIEEQAKTFLDKFNHEAEDLFYQSSLASWN) has shown a binding affinity of −5.71 kcal/mol and a stapled version of it with two aliphatic staples has affinity of −7.81 kcal/mol.[ 21 ] A comparison of the binding of another ACE2‐α1‐derived peptide (IEEQAKTFLDKFNHEAEDLFYQSSLASWNYNTNIT) and its variant with one lactam staple has shown significantly improved binding for the stapled one.[ 27 ] Although a quantitative comparison of the experimental binding energy and that calculated from this study is not possible, the relative binding affinity of an unstapled and stapled peptide with RBD calculated here agrees well with the experimental findings. A comparison of both enthalpy and entropy values for different peptides reveals that the stapling agents not only reduce the entropic cost of complexation but also enhance the enthalpy of binding. Interestingly, both enthalpy and entropy contribute to the binding energy value as for enthalpy, polar interactions play a very important role whereas conformational flexibility of peptide both in free state and complex bound state contributes to the entropy.
By mutating some hydrophobic and non‐interacting residues of ACE2_wt with polar residues such as lysine and glutamic acid, a sharp increase in the value of polar solvation energy is noticed for RBD + ACE2_mutant system. Since, for RBD + ACE2_la‐la these mutated polar residues are patched together to form lactam crosslinker, additional protein‐peptide and peptide‐solvent polar interactions are developed which is reflected in their electrostatic and polar solvation energy values. In addition, this effect is observed in other peptides designed with lactam groups such as RBD + ACE2_la‐al but not in the case of RBD + ACE2_al‐la. Exceptionally, in the case of RBD + ACE2_al‐al, despite using a hydrophobic chain as a crosslinker, polar interactions have developed. Generally, electrostatic and polar solvation energies are complementary to each other i. e. if there is a gain in the electrostatic energy upon binding it is usually associated with a loss in the polar solvation energy because the protein‐solvent polar interactions for the residues forming the binding interface are replaced by the protein–protein electrostatic interactions. However, due to the replacement of polar residues as a stapling agent, both the values increase simultaneously and result in a favorable enthalpy value. In addition to this, there is also a 15 to 17 kcal/mol increasing van der Waals energy particularly for RBD + ACE2_al‐al and RBD + ACE2_la‐la, which eventually contribute to the enthalpy value. All these changes in the enthalpy and entropy values can be correlated with the molecular level protein‐peptide interaction and conformational switching from the free to a bound state of the peptides. These two have been quantified in detail in the following sections.
3.3. Stapling induces polar amino acid‐mediated interaction
A sum of electrostatic and van der Waals interaction energies of each amino acid of ACE2‐peptides with the RBD residues has been calculated and plotted in Figure 4. For all of the modified peptides, there is a net increase in interaction energy value compared to that in the RBD + ACE2_wt. Interestingly, the addition of stapling agents to positions 28, 32, 36, and 40 enhances the interaction energy of residues facing the binding pocket of RBD, mainly Glu23, Asp30, and Glu37 (Figure 4a). These aspartates and glutamates with acidic side chains are well known to form several polar interactions with other amino acids containing polar side chains. The most common interaction is formation of a salt bridge interaction with amino acids having positively charged side chains i. e. Arginine and Lysine. RBD contains few such residues in its binding pocket, for example, Arg403, Lys417, and Lys458. The distances between the residue pairs forming effective salt‐bridge interaction are plotted throughout the simulation for different systems (Figure 4c). A typical salt bridge interaction is formed when the distance between any of the oxygen atoms (O) of an acidic amino acid side chain from the nitrogen (N) of any basic amino acid side chain is less than 0.4 nm.[ 53 ] For the RBD + ACE2_wt system, the distances for salt bridges Lys31‐Glu484, Asp38‐Lys403, and Gly37‐Lys417 are found to be greater than 0.4 nm during the simulation (Figure 4c). However, the residue pair Asp30‐Lys417 are found to be in a favorable distance to form stable salt bridge interaction. Interestingly in the RBD‐stapled ACE2‐peptide complexes, some of these residue pairs satisfy the distance criteria and form stable salt‐bridges interaction (Figure 4c). For example, Glu23 and Lys485 form strong salt bridges in RBD + ACE2_mutant and RBD + ACE2_al‐la system whereas Asp30 and Lys417 are at a favorable distance for salt bridge interaction in RBD + ACE2_al‐al and RBD + ACE2_al‐la systems.
FIGURE 4.
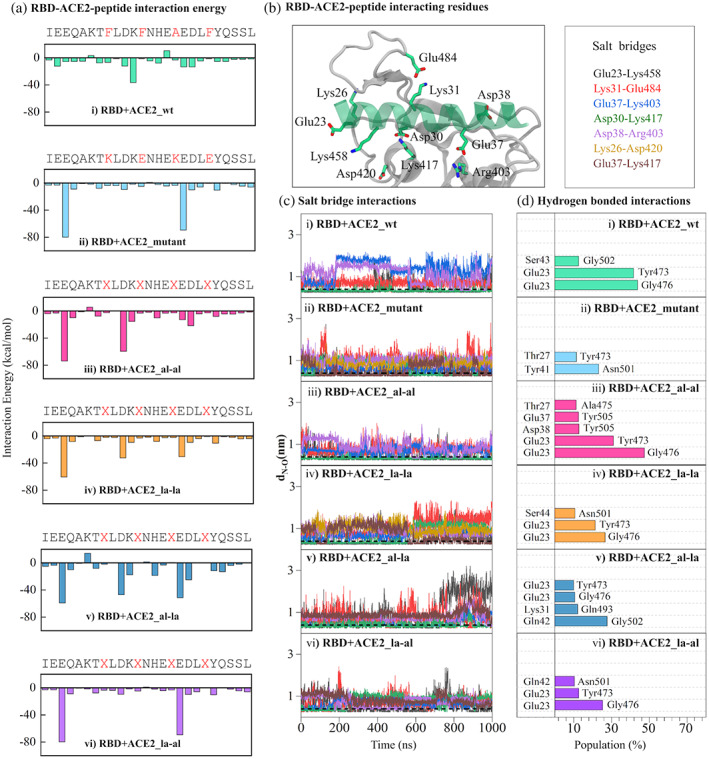
RBD‐ACE2 interactions: (a) the average interaction energy (as a sum of electrostatic and van der Waals energy) of each ACE2‐peptide residue with the binding pocket residues of RBD. The residues mutated or replaced by stapling agents are shown in red in the ACE2 sequence. (b) The amino acids of ACE2 (green transparent ribbon) and RBD (white ribbon) involved in salt‐bridge interaction are shown in stick representation. The carbon, nitrogen, and oxygen atoms are colored green, blue, and red respectively. The residue pairs forming effective salt‐bridge interaction are listed in the box on right. (c) the distances (dN—O) between any of the oxygen atoms of Glu/Asp with the nitrogen atom of respective Lys/Arg for the salt‐bridge pairs listed in (B) are plotted for different systems. The horizontal black dotted line at dN—O = 0.4 nm represents the cutoff for effective salt‐bridge formation. (d) the populations of hydrogen bonds formed between ACE2 and RBD residues are shown for different systems. The population is calculated as the percentage of simulation time the pairs form a successful hydrogen bond
In addition to these salt‐bridges, several new hydrogen‐bonded interactions have been developed between RBD and modified ACE2 peptides (Figure 4d). The occupancies of the significant hydrogen bonds are shown in the plot and the corresponding molecular level pictures showing these interactions are presented in Figure S2. Both these two types of polar interactions have contributed to the improved enthalpic component of the binding energies. Some of the residues involved in important interactions have been found to be crucial for RBD‐ACE2 binding and their mutation has influenced the RBD‐ACE2 binding.[ 54 ] The involvement of many of these residues has been reported in previous studies as well. For example, Asp30, Lys31, and Asp38 have been shown to be involved in hydrogen‐bonded interaction with the residues of RBD by Sitthiyotha et al.[ 32 ] Similarly, the salt bridge pairs found in our study, Lys31‐E484, E30‐Lys417, and Asp38‐Arg403 have been reported to play a crucial in the interaction of some designed ACE2 peptides with RBD.[ 26 ]
3.4. Conformational restriction by stapling agent contributes to entropic stabilization
The changes in entropy in the process of complex formation by combining two moieties depends on their entropy in the free and bound state. In the biomolecular complexation process like the one studied here, this entropy is usually the conformational entropy and is directly related to their conformational fluctuation. Since peptides are more flexible in their free state compared to in‐complex with protein, the entropy change due to protein‐peptide binding is usually negative. Stapled peptides are known to reduce this entropic penalty by inducing conformational restriction to the free peptide.[ 35 ] The fluctuations of the peptides in their free and the RBD‐bound state have been compared by calculating two properties, root mean square fluctuation (RMSF) of peptide residues and the helical propensity of the peptides.
The RMSFs of the 25‐residue peptides are plotted both in their free state and in their complex with RBD in Figure S3. From the values of RMSF it is evident that the fluctuation gets reduced in the complexed state (Figure S3B) compared to their free state (Figure S3A). In the free state, the introduction of two lactam staples (ACE2_la‐la) reduces the fluctuation quite significantly. For other stapling agents also, there are considerable decreases in RMSF values compared to ACE2_wt except for ACE2_al‐al and ACE2_mutant which shows higher RMSF especially towards the C‐terminal end of the peptide (residues 32–45) (Figure S3A). In the RBD‐bound state, all of the modified peptides show reduced RMSF compared to RBD + ACE2_wt representing a favorable binding in the RBD binding pocket. The only exception is the system where the aliphatic stapling agent was added (RBD + ACE2_al‐al) which shows higher fluctuation even in the complexed state. Although these comparisons provide an understanding of the relative stability of the peptides in the free and RBD‐bound state, the fluctuations of the same peptide in the free and RBD‐bound state have to be compared to find the difference in fluctuation which can be accounted for the entropy of binding. This comparison for each peptide is presented in Figure 5.
FIGURE 5.
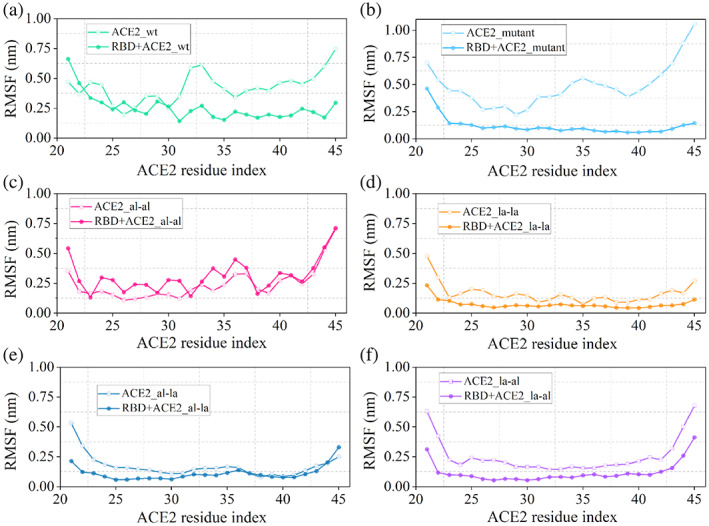
Comparison of RMSF values of wild‐type ACE2 and stapled peptides in the free and RBD‐bound states
If there is a considerable overlap between the RMSF of the residues of ACE2‐peptide in the free and RBD bound state that will require less conformational reorientation during binding hence the process will be associated with a smaller decrease in entropy. The two plots (free and complexed) deviate significantly for ACE2_mutant (Figure 5b) and ACE2_al‐al (Figure 5c) compared to that in ACE2_wt (Figure 5a). This leads to an increase in the entropic penalty to ~28 and ~29 kcal/mol compared to ~22 kcal/mol for ACE2_wt (Table 2). Since the overlap has improved in the case of ACE2_la‐la (Figure 5d) and ACE2_al‐la (Figure 5f) from ACE2_wt, the associated change in entropy has been reduced to ~19 and ~6 kcal/mol, respectively.
The ACE2 peptide forms complex with RBD in a helical conformation to fit into the binding pocket and also to maximize polar interactions with the RBD residues. Therefore, the helical propensity of the peptides will also play a key role in determining the binding thermodynamics. The helical fraction of peptide is calculated using a formula implemented in PLUMED.[ 55 ] First the number of six residue α‐helical stretches (S) in peptide is calculated using formula[ 56 ]:
| (5) |
| (6) |
where, is the coordinate of the set representing six‐residue peptide stretch and {R 0} is the same for an ideal α‐ helix. Finally the f H is obtained by dividing S by maximum no of possible six‐residue‐helical stretches in peptide, Smax.
| (7) |
The time evolution of this helical fraction is calculated for different peptides in both free state and bound states with RBD and represented in Figure 6.
FIGURE 6.
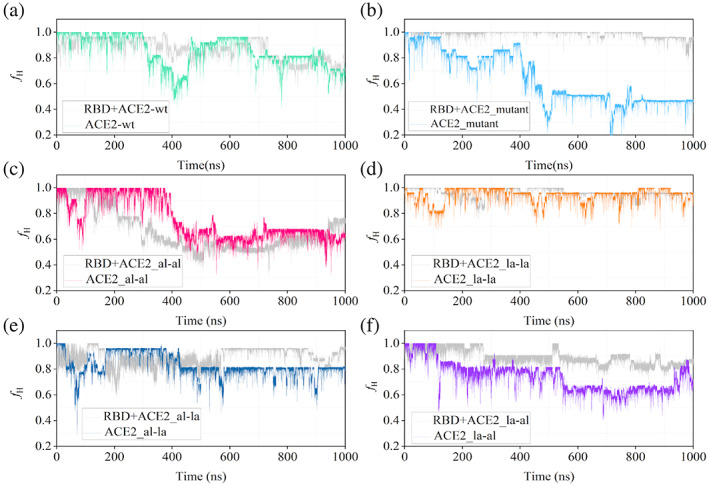
Comparison of time evolution of the helical fraction (f H ) of all ACE2 peptide variants in their free state (represented by their respective color codes same as in figure 5) and in complex with the receptor‐binding domain (RBD) the helical fraction of the RBD‐bound state is shown in gray in all the plots
The helical fraction is plotted for all the peptides throughout the 1000 ns simulation in both free and RBD‐bound states (Figure S4). The helicity of the free peptides decreases after ~500 ns for the majority of the peptides although the extent varies depending on the stapling agent (Figure S4A). The partial unfolding of the ACE2_wt has been observed by Das et al. in a recent experimental study.[ 57 ] The peptide with double lactam staple (ACE2_la‐la) is capable of maintaining maximum helicity (f H ~ 1.0) most of the time. For the ACE2_mutant system, the helicity is reduced to ~50% leading to the partial unfolding of the peptide. The ACE2_al‐al and ACE2_la‐al are also stabilized in a conformation with ~60% helicity. However, the ACE_wt and ACE2_al‐la maintain their helicity at around 75%. In the complex with RBD, most of the peptides maintain a higher degree of helicity (more than ~70%) in the confinement of RBD binding pocket throughout 1000 ns except RBD + ACE2_al‐al (Figure S4B). Similar to RMSF, a comparison of the helical fractions of the peptides in the uncomplexed state and in complex with RBD will provide a better understanding of conformational switching upon binding. The f H for each peptide in their apo and complexed states are plotted together in Figure 6.
From the plot of helical fraction, it is clear that f H of unstapled peptides decreases at earlier stages of the simulation. For example, for ACE2_wt, it is dropped at 200 ns and then it fluctuates throughout the simulation whereas, for ACE2_mutant, it decreases after 400 ns and peptide remains in a coiled conformation later on. Then, by adding lactam crosslinker as a stapling agent, the helical fraction of ACE2_la‐la is maintained around 0.8–0.9 throughout the simulation but in the case of aliphatic crosslinker, despite a stapling agent, helicity gets reduced after 400 ns. This exceptional behavior of ACE2_al‐al could be explained based on the chemical nature of the stapling agent of the hydrophobic aliphatic chain and also it supported by large fluctuation in their RMSF values (Figure 5). Similarly, for ACE2_al‐la and ACE2_la‐al, the helical fraction is maintained with a slight decrease in value during the simulation. Overall, lactam crosslinker is proved to be a good binder in maintaining the helicity of a long 25‐mer ACE2 peptide.
Furthermore, change in entropy of stapled designed peptides from complex to free state increases in comparison to hACE2_wt due to conformational restriction and rigidity of peptides by adding stapling agents both in the free and complex states. This is observed in the residue level fluctuation (RMSF) (Figure 5) and helicity (Figure 6) of peptides. Also, a helical conformation of peptide is more suitable for binding with its target partner. Therefore, a comparison of helicity of peptide in the free state and the bound state with RBD will give an idea of energy associated with conformational transition required for peptide to bind with RBD.
For ACE2_la‐la, there is a great overlap of helicity values for ACE2_la‐la and its bound state, thus the transition from unbound state to bound state will require less amount of energy whereas, for the mutant system, they are quite apart. While considering the rest of the systems, there is slight overlap of helical fraction between free and complex states even though their helicity decreases during the simulation. Considering all these facts, ACE2_la‐la is found to be a good binder with the RBD of SARS‐CoV‐2.
4. DISCUSSION
Since the emergence of the global pandemic, there have been several attempts to design vaccines, antibodies, to combat viral attacks. Alongside that, several efforts have been employed to design small molecule inhibitors which include both repurposing of clinically approved drugs or designing novel therapeutics.[ 16 , 58 , 59 , 60 , 61 , 62 , 63 ] At the same time, we have found the virus to mutate continuously to produce a large number of its variants, some of which are found to be many times infectious than the original virus.[ 64 , 65 , 66 ] In this circumstance, it is important to understand the infection mechanism with structural detail and use that knowledge to design a more potent inhibitor. The strategy of designing peptide inhibitors takes the advantage of using some native protein–protein interactions responsible for the disease progression and use an effective shorter version of one of the partners involved in the PPI with modifications that can enhance the binding affinity. In the present work, we have considered the interaction of spike protein of SARS‐CoV‐2 with human ACE2 receptor and designed some peptide inhibitors which can effectively bind to the receptor‐binding domain of SARS‐CoV‐2 to inhibit them from binding to ACE2. The human ACE2 effectively interacts with the viral RBD through its α1 helix, more precisely the residue ranging from 21 to 45. This 25‐residue domain has been considered as the starting structure (termed as ACE2_wt) and further modified to design a series of stapled peptides. A combination of two stapling agents (a hydrocarbon and a lactam stapling agent) has been used to design four stapled variants (Table 1). A detailed analyses of their binding mechanism to RBD in terms of different structural parameters and binding thermodynamics have revealed that the peptide with two lactam crosslinkers (ACE2_la‐la) is the most effective inhibitor. The protein‐peptide binding free energy is a sum of enthalpic and entropic components. The value of binding free energy provides a quantitative estimation of the energetic gain when the protein and the peptide combine to form a complex instead of being in their free state separately. Therefore, a proper understanding of the binding mechanism in terms of binding free energy requires consideration of the ensemble of conformations of RBD and the ACE2‐peptides in the free and bound state. There is a lack of these details in many protein‐peptide binding energy estimation studies including the few that consider SRS‐CoV‐2‐ACE2 binding. For example, the estimated free energies for some variants of ACE2‐peptide by de Campos et al. are in the range of −75 to −90 kcal/mol.[ 26 ] In their study they have not considered separate simulations for ACE2 and RBD and also the calculation of entropy is ignored. Similarly, Sitthiyota et al. have found MM‐GBSA binding energy for some 25‐residue ACE2‐peptides with RBD to be in the range −50 to −70 kcal/mol which does not include entropy of binding.[ 32 ] Unlike those studies, we have considered separate simulations for both ACE2‐peptides and RBD along with their complexes and entropy of binding has also been calculated. The outcome is a considerable range of binding free energy (−15 to −45 kcal/mol) and the change in enthalpy and entropy can be correlated with structural modifications in the peptides. Although there are studies that have measured the binding affinity of the ACE2 peptide and some of its modified forms to RBD, none of them have considered the exact same length of the peptide or the stapling agents. However, the relative order of affinity of an ACE2 peptide of different length and its stapled variant with two aliphatic stapling agents to RBD from an experimental measurement[ 21 ] and the same order of binding energy from the present simulation show reasonable agreement. The dynamics of the truncated α1 helix of ACE2 (ACE2_wt) at the binding pocket of RBD shows that the peptide does not bind in a fixed and stable orientation to the binding pocket, rather it undergoes large reorientation to explore different conformations (Video S1). During this process, it undergoes partial unfolding towards the C‐terminal and N‐terminal ends. The introduction of stapling agents to positions 28, 32, 36, and 40 improves the binding considerably. In the case of ACE2_al‐al, the fluctuation of the N‐terminal end of the peptide (21–30) has significantly decreased compared to ACE2_wt though the C‐terminal end (30–45) shows deviation from strong association with RBD surface (Video S2). This has been reflected in the RMSF (Figure 5c) and fraction of helicity (Figure 6c) as well. Changing the stapling agent from aliphatic to lactam has further reduced this fluctuation (Figures 5d and 6d) and the ACE2_la‐la anchors very well on the surface of RBD (Video S3). This differences in the dynamics of the ACE2_peptides in complex with RBD and their associations with binding pocket residues of RBD have been reflected in the binding free energies which follow the order: ΔGRBD−ACE2_la−la < ΔGRBD−ACE2_al−al < ΔGRBD−ACE2_wt (Table 2). A deconvolution of the free energies into enthalpy and entropy reveals that the binding is guided by changes in both enthalpy (ΔH) and entropy (ΔS). Both ACE2_al‐al and ACE2_la‐la have improved ΔH (−51.5 and −55.28 kcal/mol, respectively) than ACE2_wt (−40.31 kcal/mol). This enhancement in ΔH arises from the increase in salt‐bridge interaction and hydrogen‐bonded interaction of the polar amino acids of ACE2_al‐al and ACE2_la‐la with the amino acids at the RBD surface (Figure 4). A higher value of −TΔS indicates higher entropic penalty which disfavors the binding. A comparison of −TΔS of binding of ACE2_la‐la (19.08 kcal/mol) to RBD compared to RBD‐ACE2_wt (21.97 kcal/mol) and RBD‐ACE2_mutant (27.83 kcal/mol) shows that introduction of a crosslinker by replacing hydrophobic residues (F28, F32, A36, and F40) or just simple lactamization of two Lys‐Glu pairs reduces the entropic penalty. The reduction in the fluctuation of the free peptide and thereby a conformational similarity between the peptides in unbound and RBD‐bound states has been reflected in the overlaps between RMSFs and fractions of helicity. The plots of RMSF and helical fraction of free and RBD‐bound state together for different peptides shows considerable overlap between them for ACE2_la‐la (Figures 5d and 6d) while they differ significantly for ACE2_wt (Figures 5a and 6a) and ACE2_mutant (Figures 5b and 6b). The impact of these overlaps has been reflected in the −TΔS values (Table 2).
5. CONCLUSION
Employing computational modeling and molecular dynamics simulation, we have developed an approach to design stapled peptide inhibitors derived from human ACE2 against the receptor‐binding domain (RBD) of SARS‐CoV‐2. These peptides are designed to improve the binding affinity by forming new interactions with RBD in addition to the native RBD‐ACE2 interactions. Starting from a 25‐residue domain of the α1 helix of ACE2, a series of mutated and stapled ACE2 peptides have been designed using a combination of aliphatic and lactam stapling agents. From the estimation of binding free energy, the ACE2 peptide with two lactam staples connected in the positions 28, 32, 36, and 40 is found to be the most promising one. The benefit of using a lactam stapling agent is twofold. In the presence of the stapling agents, the RBD‐ACE2 interactions get reorganized to enhance salt‐bridge and hydrogen‐bonded interactions which contribute to a more negative change in the enthalpy of binding. In addition to that, the stapling agents restrict the conformational flexibility of the free peptide and thereby reduce the change in entropy on going from a free to an RBD‐bound state. To the best of our knowledge, this is the first study where the binding thermodynamics of RBD‐ACE2 interaction have been fine‐tuned by modulating the nature of the stapling agent to design a promising stapled peptide inhibitor against SARS‐CoV‐2. We believe that with experimental validation of the preferential binding of the designed peptide, this study will pioneer the rational designing of suitable inhibitors to combat the infection by SARS‐CoV‐2.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
Supporting information
Data S1. Supporting information.
Video S1. Supporting information.
Video S2. Supporting information.
Video S3. Supporting information.
ACKNOWLEDGMENTS
Rajarshi Chakrabarti acknowledges SERB (Grant CRG/2020/000279) for financial support. Asha Rani Choudhury thanks Department of Science and Technology INSPIRE for fellowship. Atanu Maity thanks IIT Bombay for the fellowship. Sayantani Chakraborty thanks SERB for the fellowship. The authors acknowledge the computational facility (HPC) provided by IIT Bombay. Authors thank XSEDE Covid‐19 HPC Consortium award and Microsoft Azure for providing computation time.
Choudhury A. R., Maity A., Chakraborty S., Chakrabarti R., Pept. Sci. 2022, 114(5), e24267. 10.1002/pep2.24267
Funding information IIT Bombay; Department of Science and Technology INSPIRE; SERB, Grant/Award Number: CRG/2020/000279
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Weekly epidemiological update on COVID‐19 . World Heal Organ, 2022. https://www.who.int/publications/m/item/weekly‐epidemiological‐update‐on‐covid‐19‐‐‐18‐january‐2022
- 2. Chan J. F. W., Yuan S., Kok K. H., To K. K. W., Chu H., Yang J., Xing F., Liu J., Yip C. C. Y., Poon R. W. S., Tsoi H. W., Lo S. K. F., Chan K. H., Poon V. K. M., Chan W. M., Ip J. D., Cai J. P., Cheng V. C. C., Chen H., Hui C. K. M., Yuen K. Y., Lancet 2020, 395, 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kupferschmidt K., Cohen J., Science 2020, 367, 610. [DOI] [PubMed] [Google Scholar]
- 4. Abdelrahman Z., Li M., Wang X., Front. Immunol. 2020, 11, 552909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shah M., Ahmad B., Choi S., Woo H. G., Comput. Struct. Biotechnol. J. 2020, 18, 3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fallon L., Belfon K. A. A., Raguette L., Wang Y., Stepanenko D., Cuomo A., Guerra J., Budhan S., Varghese S., Corbo C. P., Rizzo R. C., Simmerling C., J. Am. Chem. Soc. 2021, 143, 11349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X., Nature 2020, 581, 215. [DOI] [PubMed] [Google Scholar]
- 8. Acharya A., Lynch D. L., Pavlova A., Pang Y. T., Gumbart J. C., Chem. Commun. 2021, 57, 5949. [DOI] [PubMed] [Google Scholar]
- 9. Pavlova A., Zhang Z., Acharya A., Lynch D. L., Pang Y. T., Mou Z., Parks J. M., Chipot C., Gumbart J. C., J. Phys. Chem. Lett. 2021, 12, 5494. [DOI] [PubMed] [Google Scholar]
- 10. Astuti I., Ysrafil, Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang Y., Yang C., Xu X.‐F., Xu W., Liu S.‐W., Acta Pharmacol. Sin. 2020, 41, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zimmerman M. I., Porter J. R., Ward M. D., Singh S., Vithani N., Meller A., Mallimadugula U. L., Kuhn C. E., Borowsky J. H., Wiewiora R. P., Hurley M. F. D., Harbison A. M., Fogarty C. A., Coffland J. E., Fadda E., Voelz V. A., Chodera J. D., Bowman G. R., Nat. Chem. 2021, 13, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brotzakis Z. F., Löhr T., Vendruscolo M., Chem. Sci. 2021, 12, 9168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q., Science 2020, 367, 1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barros E. P., Casalino L., Gaieb Z., Dommer A. C., Wang Y., Fallon L., Raguette L., Belfon K., Simmerling C., Amaro R. E., Biophys. J. 2021, 120, 1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Muratov E. N., Amaro R., Andrade C. H., Brown N., Ekins S., Fourches D., Isayev O., Kozakov D., Medina‐Franco J. L., Merz K. M., Oprea T. I., Poroikov V., Schneider G., Todd M. H., Varnek A., Winkler D. A., Zakharov A. V., Cherkasov A., Tropsha A., Chem. Soc. Rev. 2021, 50, 9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scott D. E., Bayly A. R., Abell C., Skidmore J., Nat. Rev. Drug Discov. 2016, 15, 533. [DOI] [PubMed] [Google Scholar]
- 18. Pelay‐Gimeno M., Glas A., Koch O., Grossmann T. N., Angew. Chem. Int. Ed. 2015, 54, 8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smith M. C., Gestwicki J. E., Expert Rev. Mol. Med. 2012, 14, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wójcik P., Berlicki Ł., Bioorg. Med. Chem. Lett. 2016, 26, 707. [DOI] [PubMed] [Google Scholar]
- 21. Curreli F., Victor S. M. B., Ahmed S., Drelich A., Tong X., Tseng C. K., Hillyer C. D., Debnath A. K., MBio 2020, 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klein M., Expert Opin. Drug Discovery 2017, 12, 1117. [DOI] [PubMed] [Google Scholar]
- 23. Walensky L. D., Bird G. H., J. Med. Chem. 2014, 57, 6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tan Y. S., Lane D. P., Verma C. S., Drug Discov. Today 2016, 21, 1642. [DOI] [PubMed] [Google Scholar]
- 25. Das A., Yadav A., Gupta M., R P., Terse V. L., Vishvakarma V., Singh S., Nandi T., Banerjee A., Mandal K., Gosavi S., Das R., Ainavarapu S. R. K., Maiti S., J. Am. Chem. Soc. 2021, 143, 18766. [DOI] [PubMed] [Google Scholar]
- 26. de Campos L. J., Palermo N. Y., Conda‐Sheridan M., J. Phys. Chem. B 2021, 125, 6572. [DOI] [PubMed] [Google Scholar]
- 27. Maas M. N., Hintzen J. C. J., Löffler P. M. G., Mecinović J., Chem. Commun. 2021, 57, 3283. [DOI] [PubMed] [Google Scholar]
- 28. Han Y., Král P., ACS Nano 2020, 14, 5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Othman H., Bouslama Z., Brandenburg J. T., da Rocha J., Hamdi Y., Ghedira K., Srairi‐Abid N., Hazelhurst S., Biochem. Biophys. Res. Commun. 2020, 527, 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sitkoff D., Sharp K. A., Honig B., J. Phys. Chem. 1994, 98, 1978. [Google Scholar]
- 31. Zhang G., Pomplun S., Loftis A. R., Tan X., Loas A., Pentelute B. L.. The first‐in‐class peptide binder to the SARS‐CoV‐2 spike protein. bioarxiv, 2020. https://www.biorxiv.org/content/10.1101/2020.03.19.999318v1. Accessed March 17, 2021.
- 32. Sitthiyotha T., Chunsrivirot S., J. Phys. Chem. B 2020, 124, 10930. [DOI] [PubMed] [Google Scholar]
- 33. Chaturvedi P., Han Y., Král P., Vuković L., Adv. Theory Simul. 2020, 3, 2000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Best R. B., Zhu X., Shim J., Lopes P. E. M., Mittal J., Feig M., MacKerell A. D. Jr., J. Chem. Theory Comput. 2012, 8, 3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maity A., Choudhury A. R., Chakrabarti R., J. Chem. Inf. Model. 2021, 61, 1989. [DOI] [PubMed] [Google Scholar]
- 36. Mark P., Nilsson L., J. Phys. Chem. A 2001, 105, 9954. [Google Scholar]
- 37. Petrova S. S., Solov'Ev A. D., Hist. Math. 1997, 24, 361. [Google Scholar]
- 38. Bussi G., Donadio D., Parrinello M., J. Chem. Phys. 2007, 126, 014101. [DOI] [PubMed] [Google Scholar]
- 39. Parrinello M., Rahman A., J. Appl. Phys. 1981, 52, 7182. [Google Scholar]
- 40. Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., Berendsen H. J. C., J. Comput. Chem. 2005, 26, 1701. [DOI] [PubMed] [Google Scholar]
- 41. Allen M. P., Tildesley D. J., Computer Simulation of Liquids, 2nd ed., Oxford University Press, London 2017. [Google Scholar]
- 42. Andersen H. C., J. Comput. Phys. 1983, 52, 24. [Google Scholar]
- 43. Darden T., York D., Pedersen L., J. Chem. Phys. 1993, 98, 10089. [Google Scholar]
- 44. Humphrey W., Dalke A., Schulten K., J. Mol. Graph. 1996, 14, 33. [DOI] [PubMed] [Google Scholar]
- 45. Bill R., Miller I., Dwight McGee T. Jr., Swails J. M., Homeyer N., Gohlke H., Roitberg A. E., J. Chem. Theory Comput. 2012, 8, 3314. [DOI] [PubMed] [Google Scholar]
- 46. Tan C., Yu‐Hong Tan A., Luo R., J. Phys. Chem. B 2007, 111, 12263. [DOI] [PubMed] [Google Scholar]
- 47. Genheden S., Ryde U., Expert Opin. Drug Discovery 2015, 10, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brooks B. R., Janežič D., Karplus M., J. Comput. Chem. 1995, 16, 1522. [Google Scholar]
- 49. Mackerell A. D., Feig M., Brooks C. L., J. Comput. Chem. 2004, 25, 1400. [DOI] [PubMed] [Google Scholar]
- 50. Williams J. K., Wang B., Sam A., Hoop C. L., Case D. A., Baum J., Proteins 2021, 1‐10, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Greenidge P. A., Kramer C., Mozziconacci J. C., Wolf R. M., J. Chem. Inf. Model. 2013, 53, 201. [DOI] [PubMed] [Google Scholar]
- 52. Dastidar S. G., Lane D. P., Verma C. S., J. Am. Chem. Soc. 2008, 130, 13514. [DOI] [PubMed] [Google Scholar]
- 53. Kumar S., Nussinov R., Chembiochem 2002, 3, 604. [DOI] [PubMed] [Google Scholar]
- 54. Aggarwal A., Naskar S., Maroli N., Gorai B., Dixit N. M., Maiti P. K., Phys. Chem. Chem. Phys. 2021, 23, 26451. [DOI] [PubMed] [Google Scholar]
- 55. Bonomi M., Branduardi D., Bussi G., Camilloni C., Provasi D., Raiteri P., Donadio D., Marinelli F., Pietrucci F., Broglia R. A., Parrinello M., Comput. Phys. Commun. 2009, 180, 1961. [Google Scholar]
- 56. Pietrucci F., Laio A., J. Chem. Theory Comput. 2009, 5, 2197. [DOI] [PubMed] [Google Scholar]
- 57. Das A., Vishvakarma V., Dey A., Dey S., Gupta A., Das M., Vishwakarma K. K., Roy D. S., Yadav S., Kesarwani S., Venkatramani R., Maiti S., Biophys. J. 2021, 120, 2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vithani N., Ward M. D., Zimmerman M. I., Novak B., Borowsky J. H., Singh S., Bowman G. R., Biophys. J. 2021, 120, 2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Acharya A., Agarwal R., Baker M. B., Baudry J., Bhowmik D., Boehm S., Byler K. G., Chen S. Y., Coates L., Cooper C. J., Demerdash O., Daidone I., Eblen J. D., Ellingson S., Forli S., Glaser J., Gumbart J. C., Gunnels J., Hernandez O., Irle S., Kneller D. W., Kovalevsky A., Larkin J., Lawrence T. J., LeGrand S., Liu S. H., Mitchell J. C., Park G., Parks J. M., Pavlova A., Petridis L., Poole D., Pouchard L., Ramanathan A., Rogers D. M., Santos‐Martins D., Scheinberg A., Sedova A., Shen Y., Smith J. C., Smith M. D., Soto C., Tsaris A., Thavappiragasam M., Tillack A. F., Vermaas J. V., Vuong V. Q., Yin J., Yoo S., Zahran M., Zanetti‐Polzi L., J. Chem. Inf. Model. 2020, 60, 5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chan H. T. H., Moesser M. A., Walters R. K., Malla T. R., Twidale R. M., John T., Deeks H. M., Johnston‐Wood T., Mikhailov V., Sessions R. B., Dawson W., Salah E., Lukacik P., Strain‐Damerell C., Owen C. D., Nakajima T., Świderek K., Lodola A., Moliner V., Glowacki D. R., Spencer J., Walsh M. A., Schofield C. J., Genovese L., Shoemark D. K., Mulholland A. J., Duarte F., Morris G. M., Chem. Sci. 2021, 12, 13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mao R., Bie L., Xu M., Wang X., Gao J., Phys. Chem. Chem. Phys. 2021, 23, 12549. [DOI] [PubMed] [Google Scholar]
- 62. Abu‐Saleh A. A. A. A., Awad I. E., Yadav A., Poirier R. A., Phys. Chem. Chem. Phys. 2020, 22, 23099. [DOI] [PubMed] [Google Scholar]
- 63. Lokhande K. B., Banerjee T., Swamy K. V., Ghosh P., Deshpande M., Proteins 2021, 1‐15, 1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Triveri A., Serapian S. A., Marchetti F., Doria F., Pavoni S., Cinquini F., Moroni E., Rasola A., Frigerio F., Colombo G., J. Chem. Inf. Model. 2021, 61, 4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bai C., Wang J., Chen G., Zhang H., An K., Xu P., du Y., Ye R. D., Saha A., Zhang A., Warshel A., J. Am. Chem. Soc. 2021, 143, 17646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen J., Gao K., Wang R., Wei G. W., Chem. Sci. 2021, 12, 6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting information.
Video S1. Supporting information.
Video S2. Supporting information.
Video S3. Supporting information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.