

Abstract
Novel spiropyrrolidine heterocyclic hybrids were synthesized for the first time in a sustainable fashion employing a 1,3-dipolar cycloaddition strategy to form a new class of azomethine ylides generated from tyrosine and acenaphthenequinone. Following their synthesis and characterization, these heterocyclic hybrids were tested for their anticancer activities by incubation at different concentrations and durations with different cancer and non-cancer cell cultures, and the results indicated a potential therapeutic activity. Further analysis of cancer cell death revealed that it occurred through a caspase-related apoptotic pathway, specifically mediated by caspase-3. These results demonstrated that the obtained spiropyrrolidine heterocyclic hybrids may be good hit compounds for the development of potential therapeutic agents for the treatment of malignant tumors.
Spiropyrrolidines were synthesized employing a new class of azomethine ylide for the first time and were tested for their anticancer activity, where the cell death mechanism revealed that it is occurring through the caspase-3 dependent pathway.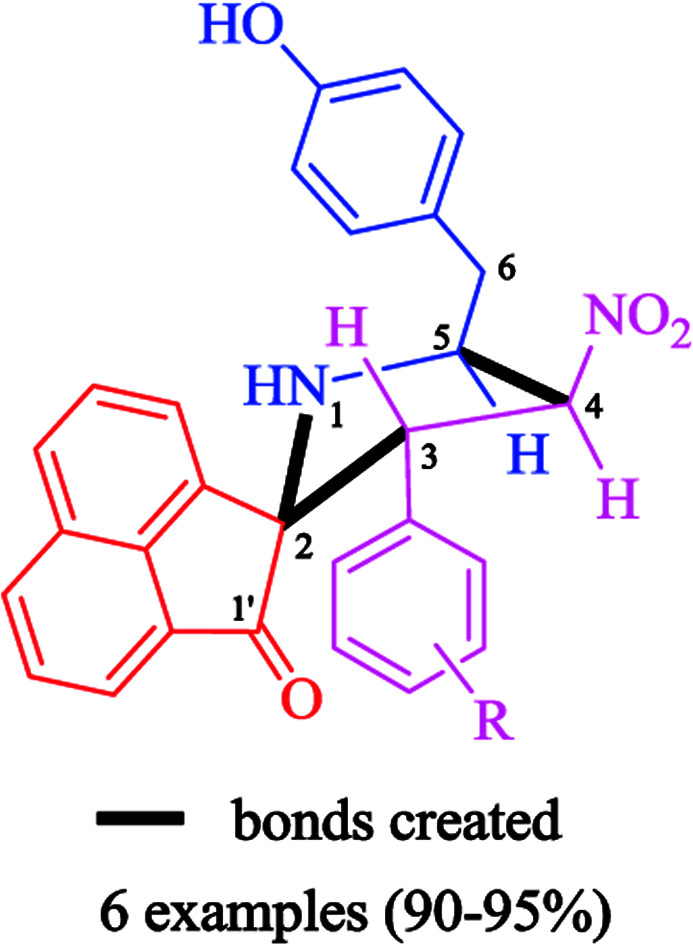
1. Introduction
Hybrid multifunctional molecules carrying more than one pharmacophoric unit, wherein each individual active unit exerts diverse modes of action, can be useful in the treatment of complex, multifactorial diseases such as cancer.1 Among various such compounds belonging to the nitrogen heterocycle class, spiropyrrolidine derivatives have attracted much interest as they constitute a central skeleton for a variety of alkaloids and pharmacologically active compounds.2–4 In particular, highly substituted pyrrolidines are common structures in natural products and are also prevalent in a variety of biologically active compounds,5,6 including compounds potentially useful for the treatment of diseases such as diabetes,7 cancer8 and viral infections.9,10 On the other hand, acenaphthenequinone derivatives have strong antioxidant properties and free radical scavenging activity that explains their ability to reduce lipid peroxidation11–14
The word “cancer” designs a potentially lethal group of diseases characterized by unregulated proliferation and a deregulation of apoptotic mechanisms. Apoptosis, an orchestrated event in which cells are programmed to die in response to specific stimuli, is crucial for sustaining the physiological balance between cell growth and cell death.15 Apoptosis can be caused by two major pathways: the extrinsic pathway, that involves the binding of “death ligands” to “death receptors”, and the intrinsic mitochondrial pathway,16 initiated by cytotoxicity. Both pathways are regulated by a group of proteases known as caspases.17 Targeting these pathways by chemotherapeutic drugs is a proven therapeutic strategy to control tumor growth and cancer progression.18 The side effects associated to the use of current chemotherapeutic agents and the development of resistance are major obstacles to treat cancer effectively.19–21 Thus, it is necessary to identify and develop new anti-cancer drug candidates with improved efficacy and reduced side effects to complement the present chemotherapeutic strategies.
The transcription factor p53 has been described as the “guardian of the human genome” and controls the cellular response to stress through the induction of apoptosis, among other processes. One of the mechanisms that regulate the levels of p53 is based on its interaction with mdm2, an ubiquitin ligase that targets p53 for degradation by the proteasome. Antagonists of the p53/mdm2 interaction are potentially very interesting as potential anticancer agents, although small molecules showing this activity are scarce. Among them, we will mention some synthetic spiropyrrolidine derivatives including MI-219, MI-773 and M-888. The low stability in solution of these compounds22 prompted us to attempt the generation of related, more stable compounds, and in this connection we have recently reported the synthesis of compounds I (Fig. 1), which caused apoptotic cell death but unfortunately displayed only moderate anticancer activity.23 In an effort to improve the activity of these compounds, we decided to investigate the replacement of the oxindole moiety by acenaphthene (compounds II).
Fig. 1. Some spiropyrrolidine derivatives that inhibit the mdm2–p53 interaction and compounds of our interest I and II.

The diversity of the biological functions of pyrrolidines has stimulated efforts for the expedient and controlled synthesis of this class of compounds.24,25 1,3-Dipolar cycloaddition reactions provides a direct and straightforward entry for the construction of pyrrolidine heterocyclic units in a highly regio- and stereoselective manner.26–30 In particular, the cycloaddition of non-stabilized azomethine ylides with olefins represents one of the most convergent approaches for the construction of five-membered hybrid heterocycles.31–33 These strategies permit the construction of complex molecules from the easily available starting materials in a single synthetic step. In view of their great synthetic potential, this cycloaddition reaction is a subject of intensive research in organic synthesis.34–36
The interesting properties of the above-mentioned structural motifs, in conjunction with our continued interest in the synthesis and/or biological activities of novel heterocyclic hybrids,37–43 prompted us to investigate the cycloaddition reactions of β-nitrostyrenes and non-stabilized azomethine ylides derived from acenaphthenequinone and tyrosine, a combination that has not been employed previously for the in situ generation of azomethine ylides. Tyrosine, in particular, has not, to our knowledge, been employed for the generation of azomethine ylides.
Our synthetic strategy for the formation of functionalized novel pyrrolidine analogues is summarized in Scheme 1. Furthermore, the synthesized analogues were tested for their therapeutic efficacy by means of in vitro cell culture systems, where the compounds were assayed for their effect of toxicity onto different cancer and non-cancer cell lines. The viability and proliferation of the cultured cells were measured followed by the in depth analysis of cell death mechanism.
Scheme 1. Synthetic strategy employed in this work.

2. Results and discussion
2.1. Chemistry
The synthetic approach was based on a three-component strategy involving the 1,3-dipolar cycloaddition reaction between β-nitrostyrenes 1(a–f) and azomethine ylide generated in situ from acenaphthenequinone 2 and tyrosine 3 as outlined in Scheme 2. The synthesis of required β-nitrostyrenes was performed following the previously reported procedure.44 The cycloaddition reactions were effected by refluxing equimolar amounts of the reactants 1, 2 and 3 in methanol, and the products obtained were purified by column chromatography using hexane and ethyl acetate as eluent. This reaction proceeded in a highly regio- and stereocontrolled fashion, as only one diastereoisomer of the cycloadduct 4(a–f) was formed in excellent yields (90–95%) despite the presence of four stereocenters. The spiropyrrolidine was obtained through a process which involved the formation of three C–C bonds and four contiguous stereocenters, one of which is quaternary.
Scheme 2. Synthesis of highly functionalized pyrrolidines 4(a–f).

A careful structural elucidation of the pyrrolidine 4 was accomplished with the help of FT-IR, NMR spectroscopy and mass spectrometric data as discussed for a representative example 4f (ESI of Fig. S1–S8†). In the IR spectrum of 4f, the main infrared absorption peaks at υmax 3357 and 1711 cm−1 is referred to N–H/O–H and C O respectively while the NO2 group appears at 1539 and 1351 cm−1. In the 1H NMR spectrum of 4f, H-3 proton appears as a doublet at 4.72 ppm (J = 8.5 Hz), this proton is further confirmed by its HMBCs with the carbonyl group of acenaphthenequinone at 205.85 ppm. Further H-3 shows H,H–COSY correlation with a triplet at 6.17 ppm (J = 9.0 Hz) assignable to H-4. H-5 proton appears as a multiplet at 4.85–4.89 ppm and the two doublet of doublets at 2.69 ppm (J = 13.5, 10.5 Hz) and 2.92 ppm (J = 13.5, 4.0 Hz) is due to the benzylic protons (6-CH2). The aromatic protons appear as doublets and multiplets around 6.71–8.00 ppm. From Dept 135 and HMQC spectra, the carbon chemical shifts at 56.97, 90.81, 59.48 and 37.11 ppm is assigned to C-3, C-4, C-5 and C-6 (benzylic carbon) respectively. The structure of other pyrrolidine derivatives 4(a–e) were also assigned similarly and were in good agreement with their NMR spectroscopic data. The selected 1H and 13C chemical shifts of 4f is shown in Fig. 2.
Fig. 2. Selected 1H and 13C chemical shifts of 4f.

A plausible mechanism for the regio- and stereoselective construction of spiropyrrolidine heterocyclic hybrids 4(a–f) is proposed in Scheme 3. The rationale for the regioselectivity observed is explicable on the basis that the transition state (TS1) leading to 4 is likely to be more stable than the other possible one (TS2), which would be destabilized by interactions between the aryl ring from the styrene derivative and the amino acid chain. The observed stereoselectivity is also explicable based on the fact that with the aryl ring attached to the pyrrolidine ring adjacent to the spiro carbon being cis to the carbonyl of the acenaphthenequinone moiety and trans to the nitro group. This stereoselectivity suggests that the transition state TS3 leading to the unobserved stereoisomers suffers from unfavourable interactions between the phenyl ring and the acenaphthlene group of the acenaphthenequinone moiety (Scheme 3). The selectivity observed is also supported by our earlier reports.45,46
Scheme 3. Explanation for the observed selectivity of 4(a–f).

2.2. Biology
The MTT assay dependent cell viability studies following the treatment of testing samples, 4(a–f) with varying concentrations (5, 25, 50, 75 and 100 μM) and time periods (24 and 48 h) on different cell types: non-cancer (PCS-130-010 and BRL-3A) and cancer cell lines (A549 and Jurkat cell types) are compared and are shown in Fig. 3 and 4 respectively. It can be observed from Fig. 3 that all the tested compounds up to 100 μM concentration onto the two non-cancer cell types of PCS-130-010 and BRL-3A are exhibiting no significant losses to the viability, revealing that the compounds are not toxic to the healthy-normal cells. This can be possibly due to the fact the healthy-normal cells in general possess resistance against xenobiotic impact until certain concentration levels, since oxidative stress or any other toxic mechanisms generated by the xenobiotics are easily nullified by the intracellular proteins having antioxidative properties.47 However, from Fig. 4, it can be observed that all compounds exhibit some level of toxicity to both cancer cell types as indicated by a reduction in the viability of cells treated with the compounds as compared with the cells without any treatment. We also observed from the results that the reduction of viability is getting increased upon an increase in sample concentration and incubation time. The most active compound was 4b while the least effective compound being the 4e as compared against the positive control of the anticancer drug camptothecin (CPT) (30 μM). The highest activity corresponded to 4b, with Br as a substituent, followed by Cl, H, CH3, CH3O and nitro derivatives. The two more active compounds were those with a higher lipophilicity (see the clog P values in ESI†), suggesting that cell penetration is of critical importance for activity. From the results, we calculated the IC50 values for the tested samples using both cancer cell lines and the values are shown in Table 1. In addition to the MTT assay-dependent absorption assay, the physical appearance and cell growth status for the Jurkat cells treated compounds over a 24 h and 48 h period were visualized by optical microscopy and are shown in the ESI of Fig. S19 and S20† respectively. From the figure, it can be observed that the healthy normal cells without any treatment are linked with each other while those treated with the positive control of CPT and our compounds are isolated due to loss of neighboring cells, and this gap increases upon increase of incubation time to 48 h.
Fig. 3. Comparison of non-cancer cell viability studies following the incubation of PCS-130-010 (A), and BRL-3A (B) cells at 24 h (1) and 48 h (2) time periods. For these studies, a positive control of CPT (30 μM) and the cells of without any treatment as negative control were applied. The tested samples, 4(a–f) with varying concentrations in the range of 5–100 μM were applied by dissolving the compounds in their respective media.

Fig. 4. Comparison of the cancer cell viability studies following the incubation of A549 cells (A) and Jurkat cells (B) at 24 h (1) and 48 h (2) time periods. Similar to the non-cancer cell viability studies, the testing samples 4(a–f) with a broad concentration range (5–100 μM), positive control (CPT, 30 μM), and negative control (cells of without any treatment) were selected for the cancer cell studies too. From the figure, * corresponds to the significant (p < 0.05) and ** corresponds to the highly significant (p < 0.01) values.

IC50 values of 4(a–f).
| Comp | IC50 μM (24 h) | IC50 μM (48 h) | ||
|---|---|---|---|---|
| Jurkat | A549 | Jurkat | A549 | |
| 4a (R = H) | 77.39 ± 2.46 | 81.99 ± 3.45 | 29.37 ± 1.57 | 40.03 ± 4.25 |
| 4b (R = 4-Br) | 65.68 ± 3.24 | 40.20 ± 4.34 | 33.09 ± 3.25 | 33.61 ± 2.50 |
| 4c (R = 4-Cl) | 65.68 ± 2.45 | 59.20 ± 3.25 | 38.39 ± 1.56 | 54.76 ± 4.14 |
| 4d (R = 4-Me) | 71.21 ± 2.50 | 52.98 ± 4.56 | 37.80 ± 4.47 | 35.83 ± 3.94 |
| 4e (R = 4-OMe) | NA | 96.52 ± 1.75 | 60.08 ± 3.54 | 68.25 ± 3.25 |
| 4f (R = 3-NO2) | 66.51 ± 3.25 | 81.25 ± 2.54 | 31.90 ± 3.65 | 65.42 ± 2.89 |
Cells undergoing apoptosis are generally characterized by distorted cell membrane architectures that allow the externalization/translocation of phosphatidylserine from the cytosol outward into the plasma membrane.47 Phosphatidylserine is the basic component of the phospholipid bilayer that helps to maintain the shape and control selective permeability across the cell membrane48 and is found outside the cell plasma membrane only when the cell membrane architecture gets compromised. Thus, phosphatidylserine is often used as a marker for the detection of early apoptotic cells owing to its strong binding affinity to fluorescence emitting calcium-dependent phospholipid-binding proteins (Annexin V).49,50 Simultaneous staining using AnnexinV-FITC and PI is included for the dye exclusion to differentiate between cells undergoing apoptosis (AnnexinV-FITC+ve PI−ve) and necrosis (AnnexinV-FITC+ve PI+ve).51,52 In this study, Annexin V-FITC assay was employed to differentiate the cells undergoing early and late apoptosis, following the treatment with the positive control (CPT) and 4a respectively in Fig. 5 (for a 24 h period). As shown in figure, 95% of the cells in the untreated groups are viable (Fig. 5A-1, upper left). However, for the groups treated with CPT, the number of viable cells were drastically reduced to 16% (Fig. 5B-1, lower left) and about 58% cells migrated into early stage apoptosis (Fig. 5B-1, upper right) and 14% into late apoptosis (Fig. 5B-1, lower right); the results further showed some of the cells (about 10%) becoming necrotic (Fig. 5B-1, upper left). Similarly, in the case of 4a treated groups, 53% of the cells remain viable (Fig. 5C-1, lower left), with almost 13% (Fig. 5C-1, lower right) in early stage of apoptosis and 27% (Fig. 5C-1, upper right) in late stage of apoptosis. From the results, it is obvious that the toxicity of CPT was greatly reduced in its combination form as 4a, owing to the fact that about 52% of the cells treated with 4a remain viable compared to the 16% cell viability for CPT treated groups. Similarly, for this group, compound 4a was able to regulate/inhibit the progression of the cells going into necrosis (6%) compared to 10% for the groups treated with CPT. Furthermore, the corresponding fluorescence intensities for the Annexin-V-FITC treated cells, i.e. untreated, CPT and 4a treated samples are shown in Fig. 5 of A-2, B-2, and C-2 respectively. From each section of figure, M1 corresponds to the fluorescence intensity from the live cells while the M2 represents the fluorescence from the nearly dying apoptotic cells. Hence from this analysis, it is clearly evident that 4a treated cells have the highest number (>89%) of apoptotic dying cells as compared with the CPT treated cells (68%) and cells without treatment (4%). The observation of similar findings even for the 48 h period sample incubated systems (ESI of Fig. S11†) were observed and all these analyses thereby confirming the apoptotic pathway of cell death in the 4a treated cells.
Fig. 5. Representative flow cytometric data for apoptosis in cells of without any treatment (A-1 and A-2), CPT treated (B-1 and B-2), and 4a sample treated (C-1 and C-2) in Jurkat cells measured by flow cytometry following the incubation of Annexin V-FITC and PI. Induction of apoptosis was significant in both the CPT and 4a treated cells compared to the untreated.

During the onset of apoptosis, complementary cascades were also observed to be activated; initiating caspase activation and inducing apoptotic cell execution.53,54 The activation of these cascades implies either the occurrence of intrinsic mitochondria-mediated apoptotic pathway triggered by ionizing radiation or cytotoxic metabolites or extrinsic apoptotic pathway, which is receptor-mediated.55 Specifically, caspase-3 and -9, terminal factor of apoptosis, exists as an inactive precursor in the cytoplasm, which is activated during the apoptosis and takes part in apoptosis induced by multiple factors.56 Caspase-3 is activated by the upstream caspase-8 and caspase-9, and since it serves as a convergence point for different signaling pathways, it is widely used as a monitor-device in an apoptosis assay. We explored the use of the active form of caspase-3 for the detection of apoptotic events. This protease has been implicated as an “effector” caspase associated with the initiation of the “death cascade” and is therefore an important marker to detect the onset/transformation cells into the apoptotic signaling pathway.57 The representative flow cytometric data of caspase-3 activity in cells treated with the CPT and 4a and that of the normal control over a 24 h period is shown in Fig. 6. The flow cytometric data for the untreated, CPT and 4a treated samples are shown in Fig. 6 of A-1, B-1, and C-1 respectively. Also, the fluorescence intensity data presented in the figure, the treatment of the cells with CPT showed an increase (>54%) in active caspase-3 expression (Fig. 6B-2) compared to the untreated cells (1.22% in Fig. 6A-2). Similarly, there was an increase in active caspase-3 staining (Fig. 6C-2) by >85% following the treatment with the test compound (4a) as compared against the negative control (Fig. 6A-2) and positive control (Fig. 6B-2). The treatment with 4a demonstrated an increase in active caspase-3 expression as compared to the CPT groups. The results revealed that the overexpression of caspase-3 improved the apoptosis of cells treated by CPT and 4a, meanwhile, the 4a in a more aggressive way strengthened the pro-apoptotic ability of caspase-3. In addition, the increased incubation period to a 48 h for the same samples shown in the ESI of Fig. S12† also revealing the similar information and from the analysis, these findings demonstrated that compound 4a played a synergistic role towards the induction of apoptosis in the cell lines.
Fig. 6. Representative flow cytometric data of caspase-3 activity in cells of untreated (A-1 and A-2), CPT treated (B-1 and B-2), and 4a treated (C-1 and C-2) Jurkat cells. The caspase-3 activity of the cells increased significantly in the CPT and 4a treated cells compared to the untreated. The M1 and M2 signified viable and apoptotic cells respectively.

3. Conclusion
An efficient synthesis of novel highly functionalized pyrrolidine derivatives has been achieved in excellent yields via a one pot three component 1,3-dipolar cycloaddition reaction of β-nitrostyrenes and non-stabilized azomethine ylides generated in situ from acenaphthenequinone and tyrosine. In addition to the synthesis protocol, the in vitro cytotoxicity assays shown that all the synthesized compounds have good therapeutic efficacy against both the cancer cell lines. However, this in vitro cytotoxic effect seems to be significantly affected by the substituent type, i.e. the halogen containing derivatives proved to be highly active as against the other derivatives. Further, the cell death in both the cancer cell lines indicated to be apoptotic way mediated by an increased activation of caspase-3 proteins. The highly functionalized pyrrolidine derivatives obtained in the present study can be employed for the sustainable treatment of cancerous tumors while simultaneously protecting the non-cancer cells during the cancer chemotherapy which in most cases is associated with many side effects. Furthermore, the presence of multiple functional groups in these hybrid heterocycles, enriches the budding for further alteration of these compounds into therapeutically useful candidates.
4. Materials and methods
4.1. Chemistry
All nitrostyrenes were prepared according to literature procedure44 and all other reagents and solvents were purchased from commercial suppliers and used without further purification. Reactions were monitored by thin-layer chromatography (TLC) on silica gel. Column chromatography was done on silica gel (230–400 mesh) using hexane-ethyl acetate as eluent. Melting points were recorded using open capillary tubes and are uncorrected. The 1H, 13C and 2-D NMR spectra were recorded on a Jeol 500 MHz instruments in CDCl3 using TMS as internal standard. Standard Jeol software was used throughout. Chemical shifts are given in parts per million (δ-scale) and the coupling constants are given in Hertz. IR spectra were recorded on a Perkin Elmer system 2000 FT IR instrument (KBr). Mass spectra were recorded on a Triple quadrupole mass spectrometer, Quattro Premier equipped with an electrospray ionization source (Z-spray) coupled with an Acquity UPLC system. Elemental analyses were performed on a Perkin Elmer 2400 Series II Elemental CHNS analyzer.
4.2. Biology
Fetal bovine serum (FBS), Dulbecco's phosphate buffered saline (DPBS), cell culture mediums of F-12K, RPMI-1640 were purchased from HiMedia (Mumbai, India). Propidium iodide (PI), APO-DIRECT™ Kit, FITC Rabbit Anti-Active Caspase-3, FITC Annexin V Apoptosis Detection Kit I, MitoScreen JC-1 Kit were obtained from BD Biosciences (San Jose, CA, USA) and CPT was procured from Sigma-Aldrich (St. Louis, MO, USA).
4.2.1. Preparation of stock solutions
The stock solutions with different concentrations of the testing compounds (5, 25, 50, 75, and 100 μM) and CPT (40 μM) were prepared in DMSO media. Further dilutions were made in either of smooth muscle cell medium (for PCS-130-010 cells), Eagle's minimum essential medium (BRL-3A cells), F-12K (A549 cells), or RPMI-1640 (Jurkat cells) mediums to the required concentrations of testing compounds for the treatment of in vitro cell culture systems.
4.2.2. Cell culture and maintenance
We tested the samples labelled in the range of 4(a–f) towards their cell viability and proliferation effects and further associated cell death mechanisms on different cell culture systems. For the testing, two different cancer (A549, human adenocarcinoma alveolar basal epithelial cells and Jurkat cells, human acute T-cell lymphoma) and two non-cancer cell lines (PCS-130-010, primary human lung smooth muscle cells and BRL-3A, rat fibroblast liver cells) obtained from the National Centre for Cell Science (NCCS, Pune, India) were used. The obtained cells were cultured in respective culture mediums supplemented with 10% FBS, penicillin (100 I.U. mL−1), streptomycin (100 μg mL−1) at 37 °C in a humidified atmosphere of 5% CO2 in air. The cultured cells when reaches to their 75–80% confluency, were used for the testing and for that the adherent cells were trypsinized first, counted and seeded in 96-well plates containing the complete growth medium. The cells were allowed to adhere overnight before the final treatment where the sample treatment was performed in a growth medium containing 10% FBS.
4.2.3. Cell viability and proliferation assay
In order to test the effects of cell viability and proliferation, each cells type was incubated with different concentrations and time periods where the assay is based on the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) agent. For that, the cells were seeded along with different concentrations of samples in the range of 4(a–f) at different concentrations of 5, 25, 50, 75, and 100 μM in 96 wells plate at the density of 2 × 104 cells per well and incubated for 24 and 48 h at 37 °C in a CO2 incubator. CPT of 30 μM concentration was used as a positive control and the cells of without any treatment were applied as the negative control. The assay was followed as per the manufacturer instructions and the absorbance of the treated and untreated wells was measured in a microplate reader at 570 nm wavelength. The results were represented as percentage of viability = {[A570 (treated cells) − background]/[A570 (untreated cells) − background]} × 100. Each treatment was performed in triplicate (N = 3). Further, the IC50 values, i.e. the concentration required to have 50% cell viability loss were determined for all the samples depending upon their incubation time and cell type.
4.2.4. Apoptosis assay
The apoptosis assay was performed using Annexin V/FITC kit. The fluorescence intensities of FITC-conjugated Annexin-V and PI in cells were analyzed using the flow cytometry. Briefly, Jurkat cells (1 × 106 cells per well) were seeded in a 6-well plate. After overnight growth, the cells were treated with 4a compound at IC50 concentration as determined from the cell viability assay. Following the trypsinization, the cells were centrifuged at 1000 × g for 10 min, and the pellet was gently re-suspended in 100 μL AnnexinV-FITC binding buffer and then incubated with 5 μL of AnnexinV-FITC in the dark for 10 min at 25 °C. The cells were then centrifuged at 2000 rpm for 5 min, gently re-suspended in 500 μL of AnnexinV-FITC binding buffer and 5 μL of PI was added in an ice bath, followed by flow cytometry analysis. From the result, the apoptotic cells were analyzed using the Cell Quest software.
4.2.5. Cell cycle analysis
Jurkat cells were plated at 1 × 106 cells per well in a six-well plate for 12 h and then cells were exposed with compound 4a at IC50 concentrations in serum-free media and incubated for an additional 24 or 48 h periods. Following the trypsinization, the cells were centrifuged at 1000 × g for 10 min, and the pellet was re-suspended in PBS. Fixation was completed by adding 70% cold ethanol for at least 2 h. The fixed cells were centrifuged at 1000g for 10 min, and the pellet suspended in PBS. After 60 s, the cells were centrifuged as before, and the pellet re-suspended in 1 mL of PI staining solution. Immediately after 15 min of incubation, the cells were analyzed to determine the cell cycle stage using flow cytometry with an excitation wavelength of 488 nm and an emission at 670 nm using the Cell Quest software. The data presented is of at least three independent experiments conducted in triplicates.
4.2.6. Caspase-3 expression
Briefly, Jurkat cells (1 × 106 cells per well) were seeded in a 6-well plate. After the adherence of cells for 12 h, the cells were treated with compound 4a (at IC50 concentrations) for another 24 or 48 h period. The cells were harvested and then fixed with 70% ice cold ethanol followed by incubation at −20 °C for 30 min. The cells were washed twice with PBS and treated with 20 μL of caspase-3 FITC followed by incubation for 60 min at room temperature in dark. The cells were washed with PBS and re-suspended in 0.5 mL of PBS and then analyzed for caspase-3 expression using BD FACS Calibur.
4.2.7. Statistical analysis
All the in vitro molecular biology experimental results presented are the mean ± SD of at least three replicate analyses. For the results, the performed statistical analysis was based on student's t-test with the significant value set at p < 0.05 (represented by *) and highly significant value set at p < 0.01 (represented by **) against the control ran with the aid of Graphpad prism software (version 6).
5. Experimental
5.1. General procedure for the synthesis of 3-(aryl)-4-nitro-5-(4-hydroxybenzyl)spiro[acenaphthene-2′.2-pyrrolidin]-1′-one 4(a–f)
A mixture of β-nitrostyrenes (1 mmol), acenaphthenequinone (1 mmol) and tyrosine (1 mmol) in methanol (10 mL) was stirred at reflux for 6 h. After completion of the reaction, as indicated by TLC, the excess solvent was evaporated under reduced pressure. The resultant solid was extracted with CH2Cl2 (3 × 20 mL). The product was further purified by column chromatography using hexane: ethyl acetate (3 : 2 v/v) as eluent.
5.1.1. 3-(Phenyl)-4-nitro-5-(4-hydroxybenzyl)spiro[acenaphthene-2′.2-pyrrolidin]-1′-one (4a)
Pale yellow solid; 93% yield; mp 105–107 °C; IR (KBr) νmax 3359, 1715, 1532, 1350 cm−1; 1H NMR (500 MHz, CDCl3): δH 2.72 (dd, 1H, J = 14.0, 10.5 Hz, 6-CH2), 2.92 (dd, 1H, J = 13.5, 4.0 Hz, 6-CH2), 4.77 (d, 1H, J = 9.0 Hz, H-3), 4.88–4.93 (m, 1H, H-5), 6.22 (t, 1H, J = 8.5 Hz, H-4), 6.71 (d, 2H, J = 7.0 Hz, ArH), 6.87 (dd, 2H, J = 8.0, 2.0 Hz, ArH), 6.97 (d, 2H, J = 7.5 Hz, ArH), 7.14 (d, 2H, J = 8.0 Hz, ArH), 7.54 (d, 1H, J = 8.0 Hz, ArH), 7.60 (d, 1H, J = 7.0 Hz, ArH), 7.75–7.78 (m, 2H, Ar–H), 7.85 (d, 1H, J = 8.0 Hz, ArH), 7.95 (d, 1H, J = 7.0 Hz, ArH), 7.98 (d, 1H, J = 8.0 Hz, ArH). 13C NMR (125 MHz, CDCl3): δC 37.09, 57.93, 59.76, 75.67, 91.42, 115.46, 120.12, 121.43, 125.49, 127.79, 127.94, 128.27, 128.35, 128.46, 128.83, 129.36, 130.22, 130.34, 130.58, 131.68, 132.78, 142.15, 154.39, 206.50. 450 (M+) Anal. calcd for C28H22N2O4: C, 74.65; H, 4.92; N, 6.22%; found: C, 74.87; H, 4.70; N, 6.35%.
5.1.2. 3-(4-Bromophenyl)-4-nitro-5-(4-hydroxybenzyl)spiro[acenaphthene-2′.2-pyrrolidin]-1′-one (4b)
Pale yellow solid; 92% yield; mp 135–137 °C; IR (KBr) νmax 3346, 1711, 1549, 1363 cm−1; 1H NMR (500 MHz, CDCl3): δH 2.69 (dd, 1H, J = 13.5, 10.5 Hz, 6-CH2), 2.92 (dd, 1H, J = 13.5, 4.0 Hz, 6-CH2), 4.72 (d, 1H, J = 8.5 Hz, H-3), 4.85–4.89 (m, 1H, H-5), 6.17 (t, 1H, J = 9.0 Hz, H-4), 6.72 (d, 2H, J = 8.0 Hz, ArH), 6.77 (d, 2H, J = 8.5 Hz, ArH), 7.09 (d, 2H, J = 8.5 Hz, ArH), 7.13 (d, 2H, J = 8.0 Hz, ArH), 7.59 (d, 1H, J = 7.5 Hz, Ar–H), 7.64 (d, 1H, J = 7.0 Hz, ArH), 7.75–7.78 (m, 1H, ArH), 7.87 (d, 1H, J = 8.5 Hz, ArH), 7.93 (d, 1H, J = 7.0 Hz, ArH), 8.00 (d, 1H, J = 8.0 Hz, ArH). 13C NMR (125 MHz, CDCl3): δC 37.12, 57.13, 59.45, 75.41, 91.18, 115.48, 120.16, 121.70, 122.02, 125.69, 128.45, 128.84, 129.25, 129.67, 130.37, 130.91, 131.55, 131.93, 134.41, 135.73, 137.80, 142.19, 154.42, 206.32. 529 (M+) Anal. calcd for C28H21BrN2O4: C, 63.53; H, 4.00; N, 5.29%; found: C, 63.40; H, 4.17; N, 5.38%.
5.1.3. 3-(4-Chlorophenyl)-4-nitro-5-(4-hydroxybenzyl)spiro[acenaphthene-2′.2-pyrrolidin]-1′-one (4c)
Pale yellow solid; 93% yield; mp 107–109 °C; IR (KBr) νmax 3455, 1710, 1538, 1357 cm−1; 1H NMR (500 MHz, CDCl3): δH 2.70 (dd, 1H, J = 13.5, 10.5 Hz, 6-CH2), 2.91 (dd, 1H, J = 13.5, 4.0 Hz, 6-CH2), 4.73 (d, 1H, J = 9.0 Hz, H-3), 4.87 (td, 1H, J = 10.0, 4.0 Hz, H-5), 6.17 (t, 1H, J = 8.5 Hz, H-4), 6.73 (d, 2H, J = 9.0 Hz, ArH), 6.83 (d, 2H, J = 8.5 Hz, ArH), 6.94 (d, 2H, J = 8.5 Hz, ArH), 7.14 (d, 2H, J = 8.5 Hz, ArH), 7.74–7.77 (m, 3H, Ar–H), 7.81 (d, 1H, J = 8.0 Hz, ArH), 7.92 (d, 1H, J = 7.0 Hz, ArH), 7.99 (d, 1H, J = 8.0 Hz, ArH). 13C NMR (125 MHz, CDCl3): δC 37.13, 57.07, 59.44, 75.46, 91.20, 115.48, 120.15, 121.64, 122.09, 125.67, 127.48, 128.45, 128.58, 128.84, 129.32, 130.36, 131.90, 132.64, 133.44, 135.33, 137.83, 142.16, 154.43, 206.26. 485 (M+) Anal. calcd for C28H21ClN2O4: C, 69.35; H, 4.36; N, 5.78%; found: C, 69.56; H, 4.48; N, 5.65%.
5.1.4. 3-(4-Methylphenyl)-4-nitro-5-(4-hydroxybenzyl)spiro[acenaphthene-2′.2-pyrrolidin]-1′-one (4d)
Yellow solid; 91% yield; mp 116–118 °C; IR (KBr) νmax 3545, 1711, 1540, 1356 cm−1; 1H NMR (500 MHz, CDCl3): δH 2.08 (s, 3H, CH3), 2.71 (dd, 1H, J = 13.0, 10.5 Hz, 6-CH2), 2.91 (dd, 1H, J = 13.5, 3.5 Hz, 6-CH2), 4.74 (d, 1H, J = 8.5 Hz, H-3), 4.86–4.91 (m, 1H, H-5), 6.19 (t, 1H, J = 9.0 Hz, H-4), 6.71 (d, 2H, J = 8.5 Hz, ArH), 6.72–6.80 (m, 4H, ArH), 7.14 (d, 2H, J = 8.5 Hz, ArH), 7.53–7.56 (m, 1H, ArH), 7.62 (d, 1H, J = 7.0 Hz, ArH), 7.75–7.78 (m, 1H, ArH), 7.85 (d, 1H, J = 8.5 Hz, ArH), 7.94 (d, 1H, J = 7.0 Hz, ArH), 7.98 (d, 1H, J = 7.5 Hz, ArH). 13C NMR (125 MHz, CDCl3): δC 20.82, 37.07, 57.60, 59.67, 75.58, 91.68, 115.45, 120.12, 121.45, 125.44, 127.82, 128.24, 128.83, 129.06, 129.38, 129.70, 130.24, 130.34, 131.17, 131.66, 137.47, 138.36, 142.19, 154.38, 206.53. 464 (M+) Anal. calcd for C29H24N2O4: C, 74.98; H, 5.21; N, 6.03%; found: C, 74.85; H, 5.40; N, 6.16%.
5.1.5. 3-(4-Methoxyphenyl)-4-nitro-5-(4-hydroxybenzyl)spiro[acenaphthene-2′.2-pyrrolidin]-1′-one (4e)
Yellow solid; 90% yield; mp 95–97 °C; IR (KBr) νmax 3341, 1714, 1545, 1351 cm−1; 1H NMR (500 MHz, CDCl3): δH 2.69 (dd, 1H, J = 13.5, 10.5 Hz, 6-CH2), 2.89 (dd, 1H, J = 13.5, 3.5 Hz, 6-CH2), 4.70 (d, 1H, J = 8.5 Hz, H-3), 4.86 (td, 1H, J = 9.5, 3.5 Hz, H-5), 6.16 (t, 1H, J = 8.5 Hz, H-4), 6.47 (d, 2H, J = 8.0 Hz, ArH), 6.69 (d, 2H, J = 8.5 Hz, ArH), 6.79 (d, 2H, J = 8.0 Hz, ArH), 7.12 (d, 2H, J = 7.5 Hz, ArH), 7.53 (t, 1H, J = 8.0 Hz, ArH), 7.60 (d, 1H, J = 6.5 Hz, ArH), 7.73–7.84 (m, 2H, Ar–H), 7.91 (d, 1H, J = 7.0 Hz, ArH), 7.96 (d, 1H, J = 8.0 Hz, ArH). 13C NMR (125 MHz, CDCl3): δC 37.16, 54.99, 57.30, 59.53, 75.57, 91.69, 113.75, 115.45, 120.08, 121.42, 125.42, 128.28, 128.81, 129.10, 129.38, 130.35, 131.14, 131.67, 133.45, 135.33, 138.35, 142.20, 154.40, 158.95, 206.85. 480 (M+) Anal. calcd for C29H24N2O5: C, 72.49; H, 5.03; N, 5.83%; found: C, 72.35; H, 5.19; N, 5.97%.
5.1.6. 3-(3-Nitrophenyl)-4-nitro-5-(4-hydroxybenzyl)spiro[acenaphthene-2′.2-pyrrolidin]-1′-one (4f)
Yellow solid; 95% yield; mp 121–123 °C; IR (KBr) νmax 3357, 1711, 1539, 1351 cm−1; 1H NMR (500 MHz, CDCl3): δH 2.69 (dd, 1H, J = 13.5, 10.5 Hz, 6-CH2), 2.92 (dd, 1H, J = 13.5, 4.0 Hz, 6-CH2), 4.72 (d, 1H, J = 8.5 Hz, H-3), 4.85–4.89 (m, 1H, H-5), 6.17 (t, 1H, J = 9.0 Hz, H-4), 6.72 (d, 2H, J = 8.5 Hz, ArH), 7.13 (d, 2H, J = 9.0 Hz, ArH), 7.18 (t, 1H, J = 7.5 Hz, ArH), 7.27 (d, 1H, J = 8.0 Hz, ArH), 7.55 (t, 1H, J = 7.5 Hz, Ar–H), 7.61 (d, 1H, J = 7.0 Hz, ArH), 7.70–7.72 (m, 1H, ArH), 7.78–7.81 (m, 1H, ArH), 7.83–7.89 (m, 2H, ArH), 7.97–8.00 (m, 2H, ArH). 13C NMR (125 MHz, CDCl3): δC 37.11, 56.97, 59.48, 75.50, 90.81, 115.53, 120.34, 121.82, 122.96, 123.19, 126.01, 128.47, 129.01, 129.47, 130.32, 130.37, 130.46, 130.64, 132.17, 133.80, 135.18, 137.19, 142.12, 495 (M+) 147.20, 154.53, 205.85. Anal. calcd for C28H21N3O6: C, 67.87; H, 4.27; N, 8.48%; found: C, 67.71; H, 4.42; N, 8.59%.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group No. RGP-026.
Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07985d
References
- Petrelli A. Valabrega G. Expert Opin. Pharmacother. 2009;10:589–600. doi: 10.1517/14656560902781907. [DOI] [PubMed] [Google Scholar]
- Monlineux R. J., in Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, Wiley, New York, 1987, ch. 1 [Google Scholar]
- Marti C. Carreira E. M. Eur. J. Org. Chem. 2003:2209–2219. doi: 10.1002/ejoc.200300050. [DOI] [Google Scholar]
- Galliford C. V. Scheidt K. A. Angew. Chem., Int. Ed. 2007;46:8748–8758. doi: 10.1002/anie.200701342. [DOI] [PubMed] [Google Scholar]
- Watson A. A. Fleet G. W. J. Asano N. Molyneux R. J. Nash R. J. Phytochemistry. 2001;56:265–295. doi: 10.1016/S0031-9422(00)00451-9. [DOI] [PubMed] [Google Scholar]
- O'Hagan D. Nat. Prod. Rep. 1997;14:637–651. doi: 10.1039/NP9971400637. [DOI] [Google Scholar]
- Horri S. Fukase H. Matsuo T. Kameda Y. Asano N. Matsui K. J. Med. Chem. 1986;29:1038–1046. doi: 10.1021/jm00156a023. [DOI] [PubMed] [Google Scholar]
- Spearman M. A. Jamieson J. C. Wright J. A. Exp. Cell Res. 1987;168:116–126. doi: 10.1016/0014-4827(87)90421-6. [DOI] [PubMed] [Google Scholar]
- Karpas A. Fleet G. W. J. Dwek R. A. Petursson S. Mamgoong S. K. Ramsden N. G. Jacob G. S. Rademacher T. W. Proc. Natl. Acad. Sci. U. S. A. 1988;85:9229–9233. doi: 10.1073/pnas.85.23.9229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleet G. W. J. Karpas A. Dwek R. A. Fellows L. E. Tyms A. S. Petursson S. Namgoong S. K. Ramsden N. G. Smith P. W. Son J. C. Wilson F. Witty D. R. Jacob G. S. Rademacher T. W. FEBS Lett. 1988;237:128–132. doi: 10.1016/0014-5793(88)80185-6. [DOI] [PubMed] [Google Scholar]
- Zhu Y. Z. Huang S. H. Tan K. H. Sun J. Whiteman M. Zhu Y. C. Nat. Prod. Rep. 2004;21:478–489. doi: 10.1039/B304821G. [DOI] [PubMed] [Google Scholar]
- Cao E. H. Liu X. Q. Wang J. J. Xu N. F. Free Radical Biol. Med. 1996;20:801–806. doi: 10.1016/0891-5849(95)02211-2. [DOI] [PubMed] [Google Scholar]
- Jiang W. Zhao Y. Zhao B. Wan Q. Xin W. Acta Biophys. Sin. 1994;10:685–689. [Google Scholar]
- Wu T. W. Zeng L. H. Fung K. P. Wu J. Pang H. Grey A. A. Weisel R. D. Wang J. Y. Biochem. Pharmacol. 1993;46:2327–2332. doi: 10.1016/0006-2952(93)90624-6. [DOI] [PubMed] [Google Scholar]
- Koff J. L. Ramachandiran S. Bernal-Mizrachi L. Int. J. Mol. Sci. 2015;16:2942–2955. doi: 10.3390/ijms16022942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomei L. D. and Cope F. O., Apoptosis: The Molecular Basis of Cell Death, Books on Demand, Stoughton, WI, USA, 1991 [Google Scholar]
- Parrish A. B. Freel C. D. Kornbluth S. Cold Spring Harbor Perspect. Biol. 2013;5:a008672. doi: 10.1101/cshperspect.a008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D. Xiao B. Han F. Wang E. Shi Y. BMC Psychiatry. 2012;12:211. doi: 10.1186/1471-244X-12-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J. J. J. Med. Chem. 2014;57:4427–4453. doi: 10.1021/jm401427c. [DOI] [PubMed] [Google Scholar]
- Chen Q. Yang Y. Lin X. Ma W. Chen G. Li W. Wang X. Yu Z. Chem. Commun. 2018;54:5369–5372. doi: 10.1039/C8CC02791A. [DOI] [PubMed] [Google Scholar]
- Wang H. Lu Z. Wang L. Guo T. Wu J. Wan J. Zhou L. Li H. Li Z. Jiang D. Song P. Xie H. Zhou L. Xu X. Zheng S. Cancer Res. 2017;77:6963–6974. doi: 10.1158/0008-5472.CAN-17-0984. [DOI] [PubMed] [Google Scholar]
- Zhao Y. Liu L. Sun W. et al. . J. Am. Chem. Soc. 2013;135:7223–7234. doi: 10.1021/ja3125417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam N. Almansour A. I. Suresh Kumar R. Periasamy V. S. Athinarayanan J. Alshatwi A. A. Govindasami P. Altaf M. Menéndez J. C. Tetrahedron. 2018;74:5358–5366. doi: 10.1016/j.tet.2018.04.032. [DOI] [Google Scholar]
- Daly J. W. Spande T. W. Whittaker N. Highet R. J. Feigl D. Noshimori N. Tokuyama T. Meyers C. W. J. Nat. Prod. 1986;49:265–280. doi: 10.1021/np50044a012. [DOI] [PubMed] [Google Scholar]
- Waldmann H. Synlett. 1995:133–141. doi: 10.1055/s-1995-4882. [DOI] [Google Scholar]
- Lown J. W., in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley-Interscience, New York, 1984, vol. 1, pp. 653–670 [Google Scholar]
- Carruthers V. W., Cycloaddition Reactions in Organic Synthesis, Pergamon Press, Elmsford, NY, 1990 [Google Scholar]
- Padwa A., Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry toward Heterocycles and Natural Products, John Wiley & Sons, New York, 2002 [Google Scholar]
- Grigg R. Sarker M. A. B. Tetrahedron. 2006;62:10332–10343. doi: 10.1016/j.tet.2006.08.077. [DOI] [Google Scholar]
- Gomes P. J. S. Nunes C. M. Pais A. A. C. C. Melo T. M. V. D. P. Arnaut L. G. Tetrahedron Lett. 2006;47:5475–5479. doi: 10.1016/j.tetlet.2006.05.179. [DOI] [Google Scholar]
- Dondas H. A. Fishwick C. W. G. Grigg R. Kilner C. Tetrahedron. 2004;60:3473–3485. doi: 10.1016/j.tet.2004.02.028. [DOI] [Google Scholar]
- Boruah M. Konwar D. Sharma S. D. Tetrahedron Lett. 2007;48:4535–4537. doi: 10.1016/j.tetlet.2007.04.131. [DOI] [Google Scholar]
- Kawashima K. Kakehi A. Noguchi M. Tetrahedron. 2007;63:1630–1643. doi: 10.1016/j.tet.2006.12.004. [DOI] [Google Scholar]
- Karthikeyan K. Perumal P. T. Etti S. Shanmugam G. Tetrahedron. 2007;63:10581–10586. doi: 10.1016/j.tet.2007.08.037. [DOI] [Google Scholar]
- Bortolini O. Nino A. D. Maiuolo L. Russo B. Sindona G. Tocci A. Tetrahedron Lett. 2007;48:7125–7128. doi: 10.1016/j.tetlet.2007.07.219. [DOI] [Google Scholar]
- Hong X. France S. Padwa A. Tetrahedron. 2007;63:5962–5976. doi: 10.1016/j.tet.2007.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R. S. Almansour A. I. Arumugam N. Qaide-Althomili D. M. Altaf M. Basiri A. Kotresha D. Manohar T. Venketesh S. Bioorg. Chem. 2018;77:263–268. doi: 10.1016/j.bioorg.2018.01.019. [DOI] [PubMed] [Google Scholar]
- Kumar R. S. Almansour A. I. Arumugam N. Soliman S. M. Kumar R. R. Altaf M. Ghabbour H. A. Krishnamoorthy B. S. J. Mol. Struct. 2018;1152:266–275. doi: 10.1016/j.molstruc.2017.09.073. [DOI] [Google Scholar]
- Kumar R. S. Almansour A. I. Arumugam N. Basiri A. Kia Y. Kumar R. R. Aust. J. Chem. 2015;68:863–871. doi: 10.1071/CH14370. [DOI] [Google Scholar]
- Kumar R. S. Almansour A. I. Arumugam N. Menéndez J. C. Osman H. Kumar R. R. Synthesis. 2015;47:2721–2730. doi: 10.1055/s-0034-1379931. [DOI] [Google Scholar]
- Almansour A. I. Kumar R. S. Beevi F. Shirazi A. N. Osman H. Ismail R. Choon T. S. Sullivan B. McCaffrey K. Nahhas A. Molecules. 2014;19:10033–10055. doi: 10.3390/molecules190710033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kia Y. Osman H. Kumar R. S. Basiri A. Murugaiyah V. Bioorg. Med. Chem. Lett. 2014;24:1815–1819. doi: 10.1016/j.bmcl.2014.02.019. [DOI] [PubMed] [Google Scholar]
- Kia Y. Osman H. Kumar R. S. Basiri A. Murugaiyah V. Bioorg. Med. Chem. 2014;22:1318–1328. doi: 10.1016/j.bmc.2014.01.002. [DOI] [PubMed] [Google Scholar]
- Kawai Y. Inaba Y. Tokitoh N. Tetrahedron: Asymmetry. 2001;12:309–318. doi: 10.1016/S0957-4166(01)00029-5. [DOI] [Google Scholar]
- Arumugam N. Almansour A. I. Suresh Kumar R. Altaf M. Padmanaban R. Sureshbabu P. Angamuthu G. Kotresha D. Manohar T. S. Venketesh S. Bioorg. Chem. 2018;79:64–71. doi: 10.1016/j.bioorg.2018.04.025. [DOI] [PubMed] [Google Scholar]
- Rajesh S. M. Perumal S. Menendez J. C. Yogeeswari P. Sriram D. Med. Chem. Commun. 2011;2:626–630. doi: 10.1039/C0MD00239A. [DOI] [Google Scholar]
- Bwatanglang I. B. Mohammad F. Yusof N. A. Abdullah J. Alitheen N. B. Hussein M. Z. Abu N. Mohammed N. E. Nordin N. Zamberi N. R. Yeap S. K. J. Colloid Interface Sci. 2016;480:146–158. doi: 10.1016/j.jcis.2016.07.011. [DOI] [PubMed] [Google Scholar]
- Abu N. Mohamed N. E. Yeap S. K. Lim K. L. Akhtar M. N. Zulfadli A. J. Kee B. B. Abdullah M. P. Omar A. R. Alitheen N. B. Drug Des., Dev. Ther. 2015;9:1401–1417. doi: 10.2147/DDDT.S67976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermes I. Haanen C. Steffens-Nakken H. Reutellingsperger C. J. Immunol. Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-I. [DOI] [PubMed] [Google Scholar]
- Zhang G. Gurtu V. Kain S. R. Yan G. BioTechniques. 1997;23:525–531. doi: 10.2144/97233pf01. [DOI] [PubMed] [Google Scholar]
- Schutte B. Nuydens R. Geerts H. Ramaekers F. J. Neurosci. Methods. 1998;86:63–69. doi: 10.1016/S0165-0270(98)00147-2. [DOI] [PubMed] [Google Scholar]
- Lale S. V. Kumar A. Naz F. Bharti A. C. Koul V. Polym. Chem. 2015;6:2115–2132. doi: 10.1039/C4PY01698J. [DOI] [Google Scholar]
- Fulda S. Debatin K. M. Curr. Med. Chem.: Anti-Cancer Agents. 2003;3:253–262. doi: 10.2174/1568011033482404. [DOI] [PubMed] [Google Scholar]
- Mooney L. M. Al-Sakkaf K. A. Brown B. L. Dobson P. R. Br. J. Cancer. 2002;87:909–917. doi: 10.1038/sj.bjc.6600541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton C. Davies D. Balkwill F. Burke F. Eur. J. Cancer. 2005;41:1474–1486. doi: 10.1016/j.ejca.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Kim Se.-L. Kim S. H. Trang K. T. T. Kim I. H. Lee S.-O. Lee S. T. Cancer Lett. 2013;335:479–486. doi: 10.1016/j.canlet.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Nicholson D. W. Ali A. Thornberry N. A. Vaillancourt J. P. Ding C. K. Gallant M. Gareau Y. Griffin P. R. Labelle M. Lazebnik Y. A. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.