

Abstract
The formation of native disulfide bonds in complex eukaryotic proteins expressed in Escherichia coli is extremely inefficient. Tissue plasminogen activator (tPA) is a very important thrombolytic agent with 17 disulfides, and despite numerous attempts, its expression in an active form in bacteria has not been reported. To achieve the production of active tPA in E. coli, we have investigated the effect of cooverexpressing native (DsbA and DsbC) or heterologous (rat and yeast protein disulfide isomerases) cysteine oxidoreductases in the bacterial periplasm. Coexpression of DsbC, an enzyme which catalyzes disulfide bond isomerization in the periplasm, was found to dramatically increase the formation of active tPA both in shake flasks and in fermentors. The active protein was purified with an overall yield of 25% by using three affinity steps with, in sequence, lysine-Sepharose, immobilized Erythrina caffra inhibitor, and Zn-Sepharose resins. After purification, approximately 180 μg of tPA with a specific activity nearly identical to that of the authentic protein can be obtained per liter of culture in a high-cell-density fermentation. Thus, heterologous proteins as complex as tPA may be produced in an active form in bacteria in amounts suitable for structure-function studies. In addition, these results suggest the feasibility of commercial production of extremely complex proteins in E. coli without the need for in vitro refolding.
In Escherichia coli and other gram-negative bacteria, disulfide bonds form in the periplasmic space, a compartment topologically equivalent to the endoplasmic reticulum but much more oxidizing (35, 36). The formation of disulfide bonds in E. coli is catalyzed by a complex machinery involving at least two soluble, periplasmic cysteine oxidoreductases (DsbA and DsbC), two membrane-bound enzymes (DsbB and DsbD), and cytoplasmic proteins (3, 20, 24, 25, 30, 38, 39). In vitro, DsbA is a potent catalyst of protein cysteine oxidation, whereas DsbC exhibits disulfide isomerase activity (30, 39). The membrane proteins DsbB and DsbD appear to be responsible for maintaining DsbA and DsbC, respectively, in the proper oxidative state for optimal function.
Extensive studies over the last 15 years have demonstrated that multidisulfide proteins generally do not fold correctly in bacteria and accumulate largely in a misfolded form. Examples of commercially important proteins that cannot be produced in active form when secreted in the periplasm include enzymes such as tissue plasminogen activator (tPA) and kallikreins, the protease inhibitors, and various growth factors (3, 8, 10, 14, 18, 22, 26, 29, 32, 37). The production of these and other proteins with three or more disulfides is complicated and has to rely on either expression in higher eukaryotes that provide a favorable environment for the formation of disulfide bonds or refolding from inclusion bodies (8, 14).
Human tPA best exemplifies the challenges associated with the production of complex proteins in E. coli. It is a 527-amino-acid serine protease with 35 cysteine residues that participate in the formation of 17 disulfide bonds. tPA is comprised of five distinct structural domains: a finger region, an epidermal growth factor-like subdomain, two kringle domains, and finally, the catalytic domain. The function of tPA is to convert the zymogen plasminogen to plasmin, a serine protease of broad specificity that degrades the fibrin network in thrombi (34). The activity of tPA is markedly enhanced by binding to fibrin, a property of great physiological importance, as tPA is less likely than other proteases to cause inadvertent plasmin activation and internal bleeding. Binding to fibrin and modulation of the proteolytic activity are primarily mediated by the finger domain and the kringle 2 domain, respectively (21).
tPA secreted in the periplasm of E. coli is misfolded and completely inactive. Attempts to produce active tPA in Saccharomyces cerevisiae or in insect cells have been frustrated by problems due to hyperglycosylation, poor export, and improper folding (7, 27). Here we demonstrate that engineering the disulfide bond machinery of the cell through the high-level expression of DsbC allows the production of active full-length tPA. After purification from a high-cell-density fermentation, 180 μg of protein per liter with a specific activity nearly identical to the authentic tPA can be obtained, with a yield corresponding to 25% of the active material in cell lysates. To the best of our knowledge this is the first time full-length tPA has been expressed in active form in bacteria in significant amounts, and it bodes well for the production of other complex multidisulfide proteins in bacteria.
MATERIALS AND METHODS
Vector construction.
pAP-stII-tPA is a pBR322 derivative containing the tPA gene fused in frame to the heat-stable enterotoxin (stII) leader peptide and placed downstream from the phoA promoter (Genentech plasmid collection). pBAD-stII-tPA was constructed by amplifying the tPA gene from pAP-stII-tPA with the primers 5′-CGCGCGATATCATGAAAAAGAATATCGCATTTCTTCTT-3′ and 5′-TCTACGCAAAGCTTTCACGCTGGTCGCATGTTGTCA-3′. The PCR product was digested with EcoRV and HindIII and subcloned into pBAD33 (11) (kindly provided by Jon Beckwith, Harvard Medical School). pTrc-stII-tPA184 is a pACYC184 derivative in which the tPA gene with the stII leader peptide is placed downstream from the strong trc promoter from pTrc-99A (Pharmacia, Uppsala, Sweden).
pLppsOmpArPDI expressing the rat protein disulfide isomerase (rPDI) gene from the lpp-lac promoter was provided by Kristine De Sutter, University of Gent. pSE380dsbA and pSE420dsbC are derivatives of pSE380 and pSE420 (Invitrogen, Carlsbad, Calif.) expressing the dsbA and dsbC genes, respectively, and were provided by Satish Raina, Centre Medical Universitaire, Geneva, Switzerland. The vector pSE380dsbAC was constructed as follows. pSE420dsbC was digested with MluI, blunt ended by using mung bean nuclease, and further digested with HindIII. Subsequently, the fragment containing the dsbC gene downstream from the trc promoter was isolated and ligated with StuI/HindIII-treated pSE380dsbA to yield pSE380dsbAC.
Expression of tPA in shaker flasks.
To evaluate tPA expression in shaker flasks, E. coli SF110 (F− ΔlacX74 galE galK thi rpsL ΔphoA degP41 ΔompT) (2) cells transformed with the appropriate plasmids were grown in Luria-Bertani medium at 37°C supplemented with ampicillin (100 μg/ml), chloramphenicol (20 μg/ml), or kanamycin (40 μg/ml) as necessary. Synthesis of DsbA, DsbC, or DsbA-DsbC in cells bearing pSE380dsbA, pSE420dsbC, or pSE380dsbAC, respectively, was induced by the addition of IPTG (isopropyl-β-d-galactopyranoside; 2 mM, final concentration) when the culture optical density at 600 nm (OD600) reached between 0.8 and 1.0. Synthesis of rPDI in cells transformed with pLppsOmpArPDI was induced by the addition of IPTG to 0.5 mM at a culture OD600 of ca. 0.6. Expression of tPA from the PBAD promoter was induced 30 min after the addition of IPTG by adding arabinose to a final concentration of 0.2% (wt/vol).
After induction with arabinose, cultures were grown for an additional 3 h and harvested by centrifugation. The cells were then resuspended in 0.1 M Tris-HCl (pH 8.5) and lysed with a French pressure cell operated at 2,000 lb/in2. Subsequently, the cell lysates were centrifuged at 12,000 × g for 10 min at 4°C to separate the soluble and insoluble fractions.
Expression of tPA in fermentors.
For inoculum preparation, 1.0 ml of frozen SF110(pBAD-stII-tPA/pSE420dsbC) cells was added to 500 ml of Luria-Bertani medium containing 40 μg of ampicillin and 30 μg of chloramphenicol per ml. The culture was grown in a 2-liter flask for 10 h, reaching an OD550 of ca. 3.0. The inoculum culture was added to approximately 6.5 liters of mineral salts medium containing 1.2% digested casein, 1.2% yeast extract, and 1.5 g of isoleucine and 1 g of glucose per liter in a 15-liter Biolafite fermentor. The fermentor was operated at 37°C and 1,000 rpm, with 10 standard liters per min of aeration and a 0.3-bar back pressure to deliver an oxygen transfer rate of approximately 3.0 mmol/liter-min. When the initial glucose was depleted, a concentrated glucose solution was added to maintain a growth rate of 0.32 h−1 until the dissolved oxygen concentration (DO2) reached 30% of air saturation. At that point glucose feeding was adjusted to maintain a DO2 of 30%. At an OD550 of 25, a feed consisting of 13.5% digested casein and 6.5% yeast extract was added at 0.5 ml/min. When the OD550 reached 80, IPTG was added at a concentration of 0.05 mM, and 30 min later arabinose was added to 0.1% (wt/vol). When respiration poisoning caused the DO2 to rise, the glucose feed rate was lowered to avoid excessive acetate accumulation.
tPA purification.
tPA was purified from cell extracts by sequential l-lysine–Sepharose and Erythrina inhibitor-Sepharose affinity chromatography (26) as described below. Cell paste was resuspended in buffer A (50 mM Tris-HCl, pH 7.5; 5mM EDTA; 0.1% Tween 80). The cells were lysed by sonication on ice. The cell lysates were centrifuged at 12,000 × g for 15 min at 4°C, and the supernatant was loaded onto an l-lysine–Sepharose column (Pharmacia) preequilibrated with buffer A. The column was washed with 8 column volumes of buffer A, followed by 8 volumes of buffer A containing 0.1 M NaCl. tPA was eluted with buffer A containing 0.5 M NaCl and 0.2 M lysine. The eluant from the l-lysine–Sepharose column was loaded onto an Erythrina caffra inhibitor-Sepharose column prepared by coupling E. caffra inhibitor (ETI; American Diagnostica, Greenwich, Conn.) to cyanogen bromide-activated Sepharose 4B (Pharmacia). The column was washed with 4 column volumes of buffer B (0.5 M NH4HCO3, 0.1% Triton X-100), 4 volumes of buffer C (0.05 M NaH2PO4, pH 7.3), and 4 volumes of buffer C with 0.1 M KSCN. The column was eluted with buffer C containing 0.9 M KSCN. Then, 1-ml aliquots of the eluant from the ETI-Sepharose column were incubated with 100 μl of iminodiacetic acid-Sepharose (Pharmacia) that had been preequilibrated with ZnCl2 at 4°C for 30 min. Incubation of the tPA with Zn-Sepharose was carried out at 4°C for 2 h. The Sepharose was precipitated by centrifugation, washed with buffer E (0.05 M NaH2PO4, 0.5 M NaCl, 0.05% Tween 80 [pH 7.3]), and eluted with buffer E containing 0.05 M imidazole.
General methods.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 12% polyacrylamide gels and Western blotting were performed according to standard techniques (1). tPA quantitation was performed by the Imubind total tPA stripwell enzyme-linked immunosorbent assay (American Diagnotica). Glycosylated, single-chain tPA (Sigma Chemical Co., St. Louis, Mo.) was used as a standard.
Assays of tPA activity.
Fibrin plates were prepared essentially as described previously (23) except that 25 μg of tetracycline per ml was added to prevent bacterial growth. The rate of plasminogen activation was determined by using the Spectrolyse tPA/PAI activity assay kit from American Diagnostica and a total assay volume of 295 μl. Zymography was performed as described by Heussen and Dowdle (12) with the following modifications: SDS–12% polyacrylamide gels were copolymerized with 0.1% (wt/vol) α-casein and 10 μg of plasminogen per ml (Calbiochem, La Jolla, Calif.). After electrophoresis at 4°C, the gels were washed with 2.5% Triton X-100 for 1 h to remove the SDS, washed with distilled water exhaustively to remove the Triton X-100, incubated in 0.1 M glycine buffer (pH 8.3) for 5 h, and then stained.
RESULTS
Optimization of tPA expression in shaker flasks.
A cDNA encoding the complete amino acid sequence (amino acids 1 to 527) of the human tPA was fused in frame to the stII leader peptide, which had been shown earlier to be useful for the periplasmic expression of a variety of proteins (5). The stII-tPA gene was placed downstream from three different promoters: the arabinose-inducible promoter PBAD(11), the phoA promoter under which the tPA gene is transcribed constitutively at a moderate level in phoT mutant cells grown in high-phosphate medium, and, finally, the IPTG-inducible trc promoter. The respective expression vectors were transformed into several different E. coli strains. In every case, the expression level of tPA was low and could not be visualized by SDS-PAGE; a band corresponding to full-length tPA could only be detected by Western blotting, although, as might be expected, the intensity of the tPA band varied depending on the promoter and strain background. However, no fibrinolytic activity could be detected on the fibrin plates. It should be noted that less than 10 pg of purified tPA can be detected with this assay. Cell fractionation demonstrated that the majority (>70%) of the total tPA accumulates in a soluble yet inactive form.
Under certain conditions, the coexpression of the rPDI secreted in the periplasmic space increased the expression of bovine pancreatic trypsin inhibitor (BPTI), a small eukaryotic protein with three disulfide bonds, by up to 15-fold (28). To test the effect of rPDI coexpression on the formation of active tPA, E. coli SF110 (ompT degP) was transformed with the plasmid pLppsOmpArPDI, which contains the rPDI fused to the OmpA leader peptide downstream from the strong lpp-lac promoter. rPDI is secreted efficiently into the periplasmic space, and upon addition of IPTG it becomes the most abundant protein in the soluble fraction (see Fig. 2). A small yet detectable fibrin clearance was observed when stII-tPA was expressed from the PBAD promoter after induction of rPDI expression (Fig. 1A). However, active tPA was barely detectable in a quantitative assay that measures the rate of activation of plasminogen with a chromogenic substrate (Fig. 1B). The low levels of tPA activity were not due to poor expression of the stII-tPA. Even though coexpression of rPDI reduced the accumulation of stII-tPA relative to control cells (pBAD-stII-tPA plasmid alone), a band corresponding to mature tPA was readily visible by Western blotting (Fig. 1D). In other experiments, coexpression of yeast PDI as a secreted protein in E. coli did not facilitate the formation of active tPA either, even though it was shown to be functional in protein oxidation (data not shown).
FIG. 2.

SDS-PAGE of total cell extracts from cultures coexpressing both tPA and DsbA, DsbC, or DsbA-DsbC. Cells were harvested 3 h after induction and lysed, and equal amounts of total protein from each sample were loaded on a 12% SDS–PAGE gel as described in Materials and Methods. Lanes: A, pBAD-stII-tPA + pLppsOmpArPDI; B, pBAD-stII-tPA + pSE380dsbA; C, pBAD-stII-tPA + pSE420dsbC; D, pBAD-stII-tPA + pSE380dsbAC.
FIG. 1.



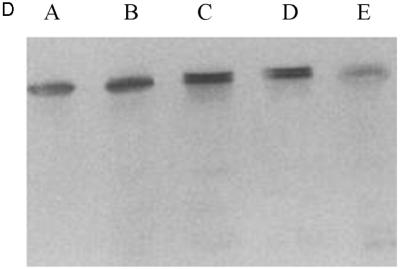
Expression of tPA in E. coli. (A) Fibrin plate analysis of tPA activity. Equal amounts of soluble protein from each culture were spotted on the fibrin plate. Lanes: A, pBAD-stII-tPA alone; B, pBAD-stII-tPA + pLppsOmpArPDI; C, pBAD-stII-tPA + pSE380dsbA; D, pBAD-stII-tPA + pSE420dsbC; E, pBAD-stII-tPA + pSE380dsbAC. (B) The specific rate of plasminogen activation in soluble fractions from cells harvested 3 h after induction was measured by the indirect chromogenic assay. tPA activities are reported as milliunits (mU) per microgram of total cell protein. Columns: 1, pBAD-stII-tPA alone; 2, pBAD-stII-tPA + pLppsOmpArPDI; 3, pBAD-stII-tPA + pSE380dsbA; 4, pBAD-stII-tPA + pSE420dsbC; 5, pBAD-stII-tPA + pSE380dsbAC. (C) Zymography of E. coli soluble fractions. DsbC is evident as an intense Coomassie blue-stained band. Lanes: A, single-chain tPA standard; B, pBAD-stII-tPA alone; C, pBAD-stII-tPA/pSE380dsbA; D, pBAD-stII-tPA/pSE420dsbC; E, pBAD-stII-tPA/pSE380dsbAC; F, pBAD-stII-tPA/pLppsOmpArPDI. (D) Western blot of tPA expression in different strains. Lanes: A, pBAD-stII-tPA alone; B, pBAD-stII-tPA + pSE380dsbA; C, pBAD-stII-tPA + pSE420dsbC; D, pBAD-stII-tPA + pSE380dsbAC; E, pBAD-stII-tPA + pLppsOmpArPDI.
In contrast, the coexpression of high levels of DsbC in cells grown in either rich or minimal medium resulted in a dramatic increase in plasminogen activation (Fig. 1). Zymography revealed that this activity arose from a band with a electrophoretic mobility slightly less than that of the single-chain tPA standard (Fig. 1C). This is consistent with the fact that the tPA standard is glycosylated whereas the bacterially expressed protein is not. No other bands were detectable in zymography gels (Fig. 1C), indicating that the activity observed on fibrin plates and with the Spectrolyse assay arises from the full-length protein and not from degradation products. In these experiments, DsbC expression was induced with 2 mM IPTG. However, because the trc promoter is leaky, DsbC synthesis occurred even in uninduced cultures, resulting in low but detectable tPA activities. Manipulation of the redox environment through the addition of GSH (glutathione, reduced form) and/or GSSG (glutathione, oxidized form) was not beneficial in cultures coexpressing DsbC and stII-tPA. In fact, the addition of as little as 0.5 mM GSSG resulted in a more than 40-fold-lower specific activity.
DsbA also stimulated the formation of active tPA, but the specific activity obtained was 25-fold lower than that of cultures expressing DsbC. The addition of 2 mM GSH, 2.0 mM GSSG, or 2.0 mM GSH–0.5 mM GSSG at the time of induction resulted in reduced tPA activities relative to control cultures that received no additives (data not shown). In vitro, DsbA and DsbC have been shown to act synergistically in the oxidative folding of BPTI (39). However, concomitant overexpression of DsbA and DsbC gave a lower specific activity of tPA than did DsbC alone. This may be because the level of DsbC accumulation in cells coexpressing DsbA-DsbC is slightly reduced (Fig. 2).
DsbC exhibits disulfide isomerase activity in vitro, and there is strong evidence that it has a similar role in vivo (19, 24, 30). DsbC can function as an isomerase only when its active-site cysteine thiols are reduced, presumably by the membrane protein DsbD (DipZ) (25, 30). In vitro, DsbC is oxidized readily by DsbA, the primary catalyst of cysteine oxidation in the periplasmic space (39). Bardwell and coworkers showed that substitution of the residues within the Cys-X-X-Cys active-site motif of DsbA modulates the redox potential of the protein (9). Several DsbA active-site mutants with a range of redox potentials were coexpressed together with tPA in a dsbA null mutant strain background. However, no improvement in tPA activity was observed relative to cells transformed with a control plasmid containing the wild-type dsbA gene. Similarly, coexpression of DsbC in a set of strains containing chromosomally integrated dsbA mutants did not result in any further increase in active tPA (3a).
tPA expression in high-cell-density fermentations.
SF110 (pBAD-stII-tPA+pSE420dsbC) cells were grown in a 10-liter fermentor in a synthetic medium supplemented with casein amino acids. The growth rate was maintained at 0.32 h−1 by controlling the addition of glucose until a DO2 level of 30% was reached. Preliminary experiments revealed that vigorous induction (with 2 mM IPTG) of DsbC expression, followed by induction of tPA expression, led to a dramatic reduction in the oxygen uptake rate after about 1.5 h (Fig. 3). Growth ceased, and a slow decline in the OD of the culture soon followed. Therefore, a lower concentration of IPTG (0.05 mM) was used to minimize the detrimental effects of DsbC overexpression. IPTG was added when the culture reached an OD550 of around 80; this was followed by a bolus of arabinose 30 min later. Under these conditions, the oxygen uptake rate remained constant for 3.5 h and then began to decline. The maximum specific tPA activity was attained 2.5 h after induction (Fig. 3) and then started to decrease, in part because the inducer, arabinose, is catabolized by strain SF110. The peak specific activity obtained in the fermentor was essentially identical to that in shaker flasks. Approximately 25 g of cell protein was obtained per liter of culture.
FIG. 3.

Production of tPA in a high-cell-density fermentation. The cell density, reported as OD550, and the oxygen uptake rate (OUR) are shown together with the tPA specific activity data from samples taken 0.5 h before and 0.5, 1.5, 2.5, 3.5, 4.5, and 5.5 h after the addition of arabinose. The tPA activity was determined by the indirect chromogenic assay. Comparable levels of activity were obtained in different runs (n = 3).
tPA purification.
Clarified cell lysates were loaded onto an l-lysine–Sepharose column. l-Lysine binds tightly and specifically to the kringle 2 domain of tPA assuming, of course, that it is correctly folded. About 50% of the total tPA activity that was loaded onto the l-lysine–Sepharose column was retained even after it was washed with 8 bed volumes of NaCl-containing buffer. The bound tPA was eluted with 0.2 M l-lysine. The eluant from the l-lysine–Sepharose affinity chromatography step was loaded onto a second column containing immobilized E. caffra inhibitor, a protein that binds to the tPA protease domain with a very high affinity (13). Active tPA was eluted with 0.9 M KSCN and was shown to have a specific activity of 373 IU/μg, a value nearly identical to that of the authentic glycosylated protein from mammalian cells (400 IU/μg). The bacterial tPA became bound irreversibly to ultrafiltration membranes and therefore could not be concentrated in this manner from the ETI column eluant. For this purpose and also to remove two contaminating E. coli proteins of 35 kDa, the active fraction from the second column was mixed with Zn-Sepharose beads. tPA was bound to Zn-Sepharose quantitatively and could be eluted with buffer containing 50 mM imidazole. The resulting protein was more than 90% pure, as determined by SDS-PAGE and silver staining (Fig. 4).
FIG. 4.
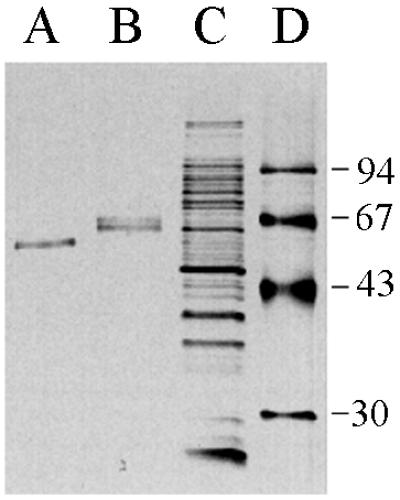
tPA purification. Shown is a silver-stained SDS-PAGE gel on which were loaded tPA purified from E. coli (A), the single-chain tPA standard from American Diagnostica (B), the total cell lysate from cells overproducing tPA and DsbC (C), and molecular size markers (D [in kilodaltons]).
The yield of purified tPA was 25% of the total activity in the starting material. Interestingly, although the bacterial tPA bound quantitatively to ETI, it could be eluted from the resin with a lower concentration of KSCN relative to the glycosylated, two-chain protein (0.9 and 1.6 M KSCN, respectively). Consequently, it appears that glycosylation affects the equilibrium dissociation constant for ETI. After ETI chromatography, 14 μg of high-specific-activity tPA was obtained from 2 g of cell protein. Thus, approximately 180 μg of purified tPA per liter can be obtained on the basis of the amount of cell protein produced per liter of culture by high-cell-density fermentation (25 g/liter).
DISCUSSION
We have identified conditions that allow the production of significant amounts of active, full-length tPA in E. coli. Normally, the formation of disulfide bonds takes place in the periplasmic space. In this study tPA was fused to the stII leader peptide, which was efficient in directing the mature protein into the periplasmic space. The stII leader peptide does not appear to be unique, and a similar yield of proteolytically active tPA was obtained when the tPA gene was fused to the OmpA leader peptide (29a). As discussed in Results and as shown in Fig. 1, in the absence of cysteine oxidoreductase overexpression, active tPA is virtually undetectable. However, a >100-fold increase in the specific rate of fibrinolysis in cell extracts is observed in cultures coexpressing DsbC. To the best of our knowledge, this dramatic increase in the production of active tPA represents by far the most striking improvement in the folding of a foreign protein ever obtained via the coexpression of foldases (8).
The rate-limiting step in the oxidative folding of eukaryotic proteins in the periplasmic space appears to be the isomerization of mismatched disulfides (29). Reduced DsbC has been shown to be an efficient catalyst of disulfide bond isomerization in vitro (6). Moreover, recent studies have shown that DsbC is maintained primarily in a reduced state in vivo, suggesting that its primary role in the cell is the catalysis of disulfide isomerization (17, 31, 33). The dramatic increase in the folding of tPA in cells coexpressing DsbC is consistent with this hypothesis. We believe that the high level of DsbC increased the disulfide isomerization capacity of the periplasmic space, thus facilitating the rearrangement of incorrect disulfides in nascent tPA. Indeed, Joly et al. (16) have shown that DsbC accumulated mostly in its partially reduced form when coexpressed in a fermentor together with insulin-like growth factor 1 (IGF-1). In this case, overexpression of DsbC increased the total yield of IGF-1 but not the amount of soluble active protein. However, this result may be a consequence of the very high level of IGF-1 overexpression (7.3 g of IGF-1 per liter of fermentation broth).
An alternative explanation for our results is that DsbC did not participate directly in the folding of tPA. Instead, its overexpression simply resulted in a higher concentration of protein thiols, thereby altering the redox potential of the periplasm to favor the formation of correct disulfide bonds. However, this hypothesis can be ruled out since neither the overexpression of other cysteine oxidoreductases nor the manipulation of the redox potential of the periplasm through the addition of GSH or GSSG had an effect similar to that of DsbC.
A low level of active tPA was detected in cells coexpressing DsbA, but this effect could not be further enhanced by the addition of reduced or oxidized glutathione. Chromosomally expressed DsbA is found almost exclusively in the oxidized form and is a potent catalyst of disulfide bond formation (19). It is possible that when DsbA is overexpressed, a fraction of the protein fails to be oxidized by DsbB and, instead, is present in the reduced form that can catalyze disulfide bond isomerization (16, 18). This activity of the reduced DsbA may in turn be responsible for the small levels of active tPA. Overexpression of rPDI has been shown to increase the yield of small heterologous proteins (15, 28), but it had a very minor effect on tPA. This may be because rPDI functions as a protein thiol oxidase in the bacterial periplasmic space but was shown to be rather ineffective in catalyzing the rate-limiting isomerization in BPTI in the periplasmic space of E. coli (28).
The highest level of active tPA was observed when the synthesis of DsbC from a trc promoter was induced first, followed by the induction of tPA expression 30 min later. Interestingly, high-level induction of DsbC, but not DsbA, rPDI, or tPA alone, was found to be particularly toxic, resulting in cessation of growth within 3 to 4 h after induction. The precursor form of DsbC was found to accumulate in the cell, raising the possibility that the observed toxicity is linked to the saturation of the protein translocation machinery. To obtain the maximum tPA specific activity in a high-cell-density fermentation, it was necessary to use a low concentration of IPTG, which allowed the production phase to be prolonged. Also, maintaining a low growth rate through the slow feeding of glucose was found to be essential in order to attain a high cell density in the fermentor. SF110 was found to be a prolific producer of acetate, which accumulated to inhibitory levels when glucose feeding was adjusted to control the growth rate above 0.65 h−1.
The above observations suggest a number of ways in which the expression of active, full-length tPA can be increased further. First, in the present study the level of accumulation of tPA was low regardless of the promoter used. A higher level of tPA synthesis may be beneficial and could be obtained by optimizing translation initiation and by substitution of rare codons in the tPA gene (4). Second, since DsbC overexpression has been found to inhibit growth, conditions that inhibit DsbC toxicity will have to be identified to prolong the production phase and help achieve even higher cell densities. Finally, it may be possible to further enhance the effect of DsbC by manipulating its interaction with DsbD (DipZ) or perhaps by isolating DsbC mutants with higher activity towards tPA.
The ability to produce substantial amounts of a heterologous protein as complex as tPA in E. coli bodes well for the expression of other complex eukaryotic proteins both for commercial purposes and for structure-function studies. When protein glycosylation is not essential, expression in bacteria is clearly advantageous in terms of both cost and simplicity.
ACKNOWLEDGMENTS
We are grateful to S. Raina, J. Beckwith, J. Bardwell, and K. De Sutter for gifts of plasmids. We also thank Susan Leung and the Genentech Fermentation Operations Department for assistance with the fermentation experiments and, finally, Paul Bessette, Jose Cotto, and John Joly for reading the manuscript.
Financial support was provided by Genentech and by NSF grant BES-9634036 to J.R.S. and G.G. and by NIH grant GM47520 to G.G.
REFERENCES
- 1.Ausubel F M. Current protocols in molecular biology. New York, N.Y: John Wiley & Sons; 1987. [Google Scholar]
- 2.Baneyx F, Georgiou G. In vivo degradation of secreted fusion proteins by the Escherichia coli outer membrane protease OmpT. J Bacteriol. 1990;172:491–494. doi: 10.1128/jb.172.1.491-494.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardwell J C. Building bridges: disulphide bond formation in the cell. Mol Microbiol. 1994;14:199–205. doi: 10.1111/j.1365-2958.1994.tb01281.x. [DOI] [PubMed] [Google Scholar]
- 3a.Bessette, P., J. Qiu, J. R. Swartz, J. Bardwell, and G. Georgiou. Unpublished data.
- 4.Brinkmann U, Mattes R E, Buckel P. High-level expression of recombinant genes in Escherichia coli is dependent on the availability of the dnaY gene product. Gene. 1989;85:109–114. doi: 10.1016/0378-1119(89)90470-8. [DOI] [PubMed] [Google Scholar]
- 5.Chang C N, Rey M, Bochner B, Heyneker H, Gray G. High-level secretion of human growth hormone by Escherichia coli. Gene. 1987;55:189–196. doi: 10.1016/0378-1119(87)90279-4. [DOI] [PubMed] [Google Scholar]
- 6.Darby N J, Raina S, Creighton T E. Contributions of substrate binding to the catalytic activity of DsbC. Biochemistry. 1998;37:783–791. doi: 10.1021/bi971888f. [DOI] [PubMed] [Google Scholar]
- 7.Furlong A M, Thomsen D R, Marotti K R, Post L E, Sharma S K. Active human tissue plasminogen activator secreted from insect cells using a baculovirus vector. Biotechnol Appl Biochem. 1988;10:454–464. [PubMed] [Google Scholar]
- 8.Georgiou G, Valax P. Expression of correctly folded proteins in Escherichia coli. Curr Opin Biotechnol. 1996;7:190–197. doi: 10.1016/s0958-1669(96)80012-7. [DOI] [PubMed] [Google Scholar]
- 9.Grauschopf U, Winther J R, Korber P, Zander T, Dallinger P, Bardwell J C. Why is DsbA such an oxidizing disulfide catalyst? Cell. 1995;83:947–955. doi: 10.1016/0092-8674(95)90210-4. [DOI] [PubMed] [Google Scholar]
- 10.Gustafson M E, Junger K D, Wun T C, Foy B A, Diaz-Collier J A, Welsch D J, Obukowicz M G, Bishop B F, Bild G S, Leimgruber R M, Palmier M O, Matthews B K, Joy W D, Frazier R B, Galluppi G R, Grabner R W. Renaturation and purification of human tissue factor pathway inhibitor expressed in recombinant E. coli. Protein Expr Purif. 1994;5:233–241. doi: 10.1006/prep.1994.1036. [DOI] [PubMed] [Google Scholar]
- 11.Guzman L M, Belin D, Carson M J, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heussen C, Dowdle E B. Electrophoretic analysis of plasminogen activators in polyacrylamide gels containing sodium dodecyl sulfate and copolymerized substrates. Anal Biochem. 1980;102:196–202. doi: 10.1016/0003-2697(80)90338-3. [DOI] [PubMed] [Google Scholar]
- 13.Heussen C, Joubert F, Dowdle E B. Purification of human tissue plasminogen activator with Erythrina trypsin inhibitor. J Biol Chem. 1984;259:11635–11638. [PubMed] [Google Scholar]
- 14.Hockney R C. Recent developments in heterologous protein production in Escherichia coli. Trends Biotechnol. 1994;12:456–463. doi: 10.1016/0167-7799(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 15.Humphreys D P, Weir N, Mountain A, Lund P A. Human protein disulfide isomerase functionally complements a dsbA mutation and enhances the yield of pectate lyase C in Escherichia coli. J Biol Chem. 1995;270:28210–28215. doi: 10.1074/jbc.270.47.28210. [DOI] [PubMed] [Google Scholar]
- 16.Joly J C, Leung W S, Swartz J R. Overexpression of Escherichia coli oxidoreductases increases recombinant insulin-like growth factor-1 accumulation. Proc Natl Acad Sci USA. 1998;95:2773–2777. doi: 10.1073/pnas.95.6.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joly J C, Swartz J R. In vitro and in vivo redox states of the Escherichia coli periplasmic oxidoreductase DsbA and DsbC. Biochemistry. 1997;36:10067–10072. doi: 10.1021/bi9707739. [DOI] [PubMed] [Google Scholar]
- 18.Joly J C, Swartz J R. Protein folding activities of Escherichia coli protein disulfide isomerase. Biochemistry. 1994;33:4231–4236. doi: 10.1021/bi00180a017. [DOI] [PubMed] [Google Scholar]
- 19.Kishigami S, Akiyama Y, Ito K. Redox states of DsbA in the periplasm of Escherichia coli. FEBS Lett. 1995;364:55–58. doi: 10.1016/0014-5793(95)00354-c. [DOI] [PubMed] [Google Scholar]
- 20.Kishigami S, Kanaya E, Kikuchi M, Ito K. DsbA-DsbB interaction through their active site cysteines. Evidence from an odd cysteine mutant of DsbA. J Biol Chem. 1995;270:17072–17074. doi: 10.1074/jbc.270.29.17072. [DOI] [PubMed] [Google Scholar]
- 21.Kohnert U, Rudolph R, Verheijen J H, Weening-Verhoeff E J, Stern A, Opitz U, Martin U, Lill H, Prinz H, Lechner M, Kresse G B, Buckel P, Fischer S. Biochemical properties of the kringle 2 and protease domains are maintained in the refolded t-PA deletion variant BM 06.022. Protein Eng. 1992;5:93–100. doi: 10.1093/protein/5.1.93. [DOI] [PubMed] [Google Scholar]
- 22.Lu H S, Hsu Y R, Lu L I, Ruff D, Lyons D, Lin F K. Isolation and characterization of human tissue kallikrein produced in Escherichia coli: biochemical comparison to the enzymatically inactive prokallikrein and methionyl kallikrein. Protein Exp Purif. 1996;8:215–226. doi: 10.1006/prep.1996.0094. [DOI] [PubMed] [Google Scholar]
- 23.Margareta W, Erik H, Anneli A, Christina K, Bjorn L, Benny R, Lennart H, Gunnar P. Synthesis and secretion of a fibrinolytically active tissue-type plasminogen activator variant in Escherichia coli. Gene. 1991;99:243–248. doi: 10.1016/0378-1119(91)90133-v. [DOI] [PubMed] [Google Scholar]
- 24.Missiakas D, Georgopoulos C, Raina S. The Escherichia coli dsbC (xprA) gene encodes a periplasmic protein involved in disulfide bond formation. EMBO J. 1994;13:2013–2020. doi: 10.1002/j.1460-2075.1994.tb06471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Missiakas D, Schwager F, Raina S. Identification and characterization of a new disulfide isomerase-like protein (DsbD) in Escherichia coli. EMBO J. 1995;14:3415–3424. doi: 10.1002/j.1460-2075.1995.tb07347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Obukowicz M G, Gustafson M E, Junger K D, Leimgruber R M, Wittwer A J, Wun T C, Warren T G, Bishop B F, Mathis K J, McPherson D T, Siegel N R, Jennings M G, Brightwell B B, Diaz-Collier J A, Bell L D, Craik C S, Tacon W C. Secretion of active kringle-2-serine protease in Escherichia coli. Biochemistry. 1990;29:9737–9745. doi: 10.1021/bi00493a033. [DOI] [PubMed] [Google Scholar]
- 27.Ogrydziak D M. Yeast extracellular proteases. Crit Rev Biotechnol. 1993;13:1–55. doi: 10.3109/07388559309069197. [DOI] [PubMed] [Google Scholar]
- 28.Ostermeier M, De Sutter K, Georgiou G. Eukaryotic protein disulfide isomerase complements Escherichia coli dsbA mutants and increases the yield of a heterologous secreted protein with disulfide bonds. J Biol Chem. 1996;271:10616–10622. doi: 10.1074/jbc.271.18.10616. [DOI] [PubMed] [Google Scholar]
- 29.Ostermeier M, Georgiou G. The folding of bovine pancreatic trypsin inhibitor in the Escherichia coli periplasm. J Biol Chem. 1994;269:21072–21077. [PubMed] [Google Scholar]
- 29a.Qiu, J., and G. Georgiou. Unpublished data.
- 30.Rietsch A, Belin D, Martin N, Beckwith J. An in vivo pathway for disulphide bond isomeration in Escherichia coli. Proc Natl Acad Sci USA. 1996;93:13048. doi: 10.1073/pnas.93.23.13048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rietsch A, Bessette P, Georgiou G, Beckwith J. Reduction of the periplasmic disulfide bond isomerase, DsbC, occurs by passage of electrons from cytoplasmic thioredoxin. J Bacteriol. 1997;179:6602–6608. doi: 10.1128/jb.179.21.6602-6608.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rudolph R, Lilie H. In vitro folding of inclusion body proteins. FASEB J. 1996;10:49–56. [PubMed] [Google Scholar]
- 33.Sone M, Akiyama Y, Ito K. Differential in vivo roles played by DsbA and DsbC in the formation of protein disulfide bonds. J Biol Chem. 1997;272:10349–10352. doi: 10.1074/jbc.272.16.10349. [DOI] [PubMed] [Google Scholar]
- 34.van Zonneveld A J, Veerman H, MacDonald M E, van Mourik J A, Pannekoek H. Structure and function of human tissue-type plasminogen activator (t-PA) J Cell Biochem. 1986;32:169–178. doi: 10.1002/jcb.240320302. [DOI] [PubMed] [Google Scholar]
- 35.Walker K W, Gilbert H F. Effect of redox environment on the in vitro and in vivo folding of RTEM-1 beta-lactamase and Escherichia coli alkaline phosphatase. J Biol Chem. 1994;269:28487–28493. [PubMed] [Google Scholar]
- 36.Wulfing C, Pluckthun A. Protein folding in the periplasm of Escherichia coli. Mol Microbiol. 1994;12:685–692. doi: 10.1111/j.1365-2958.1994.tb01056.x. [DOI] [PubMed] [Google Scholar]
- 37.Wunderlich M, Glockshuber R. In vivo control of redox potential during protein folding catalyzed by bacterial protein disulfide-isomerase (DsbA) J Biol Chem. 1993;268:24547–24550. [PubMed] [Google Scholar]
- 38.Wunderlich M, Glockshuber R. Redox properties of protein disulfide isomerase (DsbA) from Escherichia coli. Protein Sci. 1993;2:717–726. doi: 10.1002/pro.5560020503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zapun A, Missiakas D, Raina S, Creighton T E. Structural and functional characterization of DsbC, a protein involved in disulfide bond formation in Escherichia coli. Biochemistry. 1995;34:5075–5089. doi: 10.1021/bi00015a019. [DOI] [PubMed] [Google Scholar]