

Abstract
Atopic disorders, including atopic dermatitis, food and environmental allergies, and asthma, are increasingly prevalent diseases. Atopic disorders are often associated with eosinophilia, driven by T helper type 2 (Th2) immune responses, and triggered by disrupted barrier function leading to abnormal immune priming in a susceptible host. Immune deficiencies, in contrast, occur with a significantly lower incidence, but are associated with greater morbidity and mortality. A subset of atopic disorders with eosinophilia and elevated IgE are associated with monogenic inborn errors of immunity (IEI). In this review, we discuss current knowledge of IEI that are associated with atopy and the lessons these immunologic disorders provide regarding the fundamental mechanisms that regulate type 2 immunity in humans. We also discuss further mechanistic insights provided by animal models.
Keywords: atopy, inborn errors of immunity (IEI), barrier function, cytoskeletal, TCR, cytokine, T cell
Introduction
Atopic diseases are common and result from dysregulated type 2 immunity driven by T helper type 2 (Th2) cells. Th2 cells are protective to the host when coordinated in response to specific helminth pathogens or toxins but can also induce tissue pathology when over-active or misdirected toward innocuous stimuli. Th2 effector functions are mediated primarily through secretion of pro-inflammatory cytokines including interleukin (IL)-4, IL-5, and IL-13 to activate and recruit effector cells such as mast cells and eosinophils. A fraction of Th2 cells home to peripheral tissues where they permanently reside as tissue-resident memory cells (Th2-Trm) (1–6). T follicular helper (Tfh) cells formed in the context of type 2 immune responses reside primarily in secondary lymphoid tissues and promote B-cell class switching to immunoglobulin E (IgE) production (7).
There is polygenic heritable risk for development of atopic diseases, and the increasing prevalence of these disorders also points toward changing environmental factors significantly influencing susceptibility. Living on a farm is a strong protective factor against the development of asthma (8), and childhood microbial exposures measured by home endotoxin levels have demonstrated a similar inverse relationship with asthma and environmental allergy prevalence (9). Specific gene–environment interactions have been shown to affect atopic disease activity in humans. For example, individuals homozygous for the Q576R risk allele in the IL-4 receptor alpha chain gene (IL4RA) demonstrated increased asthma symptoms when exposed to higher school endotoxin levels, whereas individuals that did not harbor this allelic variant showed protective benefit (10). However, complex gene plus environment interactions that result in atopic dermatitis, allergies, asthma, and other immune-mediated diseases in humans are difficult to study and recapitulate in animal models. This is in part due to the large sample sizes needed for controlled human studies related to polygenic diseases and imperfect methods for sampling and quantifying environmental variables (11).
Mouse models have been instrumental in elucidating immune pathways of atopic disease given the advantage of controlled genetics and allergic sensitization protocols. This work has solidified the principle that dysregulated type 2 immune responses can result from both immune cell-intrinsic and -extrinsic defects. Initially described in wild-type BALB/c mice, epicutaneous exposure to foreign protein antigens through tape-stripped skin elicits type 2 inflammation resembling atopic dermatitis (12). The phenotype in mice becomes much more pronounced with gene-targeted alterations that directly promote type 2 inflammation or repress mechanisms critical for normal immune homeostasis. Thus, either disrupted barrier function or immune dysregulation appear central to atopic diatheses, and monogenic disorders in humans provide additional insights that will be reviewed here.
Primary immunodeficiencies (PIDs) have significantly contributed to our understanding of human immunity to infection. More recently, monogenic variants associated with early and severe autoinflammation, autoimmunity, and atopic disease have been described. Inborn errors of immunity (IEI) now encompass monogenic immune dysregulation disorders in addition to immune deficiencies. These comprise autoinflammatory conditions affecting the innate immune system and autoimmune and allergic disorders primarily affecting the adaptive immune system. At the time of this review, IEI with mutations in more than 400 genes have been reported (13), and this number continues to rise as awareness of IEI grows and genetic testing becomes more readily available. The shared and unique clinical phenotypes and immune pathways involved in these inborn errors offer opportunities to gain insight into the pathogenesis of immune dysregulation disorders.
Monogenic IEI associated with atopy, or primary atopic disorders (14, 15), have implicated interrelated pathways involving the actin cytoskeleton and immunological synapse formation, aberrant T-cell receptor (TCR) signaling, cytokine signaling pathways, T-cell repertoire diversity, balance of regulatory T cells (Tregs) and effector T cells, and innate immune cell effector mechanisms as critical factors influencing type 2 inflammation and atopic disease in humans. Contextualized with mechanistic paradigms developed largely in experimental animal models, IEI may also provide significant insight into common atopic disease mechanisms and lead to improved prevention and treatment strategies.
Disorders of Barrier Function
Gene defects associated with skin barrier function have been associated with early and severe atopic dermatitis and allergen sensitization. While not immunodeficiencies per se as they involve non-hematopoietic cells, these mutations may increase risk of infections secondary to disrupted barrier to microbial penetration and to the inhibitory effect of cutaneously expressed Th2 cytokines on the production of antimicrobial peptides by keratinocytes. Skin permeability also allows entry of allergens and other foreign antigens, toxins, and irritants. Subsequent production of pro-inflammatory type 2 cytokines leads to a feed-forward loop locally in the skin and also affects immune cells residing in other peripheral tissues. Clinically, atopic dermatitis has long been appreciated as the primary manifestation in individuals with multiple atopic diseases. The natural history of atopic dermatitis early in life transitioning to later development of food allergies, asthma, and allergic rhinitis is frequently described as the “atopic march” (16).
Ichthyosis vulgaris is an inherited disorder of skin keratinization presenting in the first months of life with skin scaling and palmar hyperlinearity, along with keratosis pilaris and atopic dermatitis. Originally discovered as homozygous and compound heterozygous individuals with ichthyosis vulgaris (17), loss-of-function (LOF) mutations in the FLG gene encoding filament aggregating protein (filaggrin) have been associated with early and severe atopy. Heterozygous carriers of FLG mutations are also at increased risk for atopic dermatitis, allergic rhinitis, asthma, and food allergy (18–20). Expressed as an inactive polymer by keratinocytes, profilaggrin is cleaved by serine proteases to generate filaggrin monomers, which are cross-linked to facilitate the impermeability of the stratum corneum, prevent water loss, and maintain the skin barrier (21). Lympho-epithelial Kazal-type-related inhibitor (LEKTI) is a serine protease inhibitor also expressed by keratinocytes. LOF mutations in SPINK5, the gene encoding LEKTI, lead to the Netherton syndrome with congenital ichthyosis, abnormal hair shafts, and severe atopy also extending beyond the skin (22–24). Loss of LEKTI function leads to increased protease activity affecting intercellular adhesions, which causes peeling of deeper layers of the skin (25). Disruption of keratinocyte cell junctions by mutations in genes encoding corneodesmosin (CDSN) (26, 27), desmoglein 1 (DSG1) (28), and desmoplakin (DSP) (29) involved in corneodesmosomes also cause skin barrier defects and severe atopy ( Figure 1A ).
Figure 1.

Disrupted barrier function and the atopic march. (A) Disrupted skin barrier through primary disorders of the skin or external factors enable allergen entry; uptake by dendritic cells in the presence of alarmin signals such as TSLP and IL-33 promotes dendritic cell maturation and migration to the draining LN. Genes noted in red indicate monogenic IEI affecting this pathway. (B) Dendritic cells present allergen-derived peptide:MHCII to naive CD4+ T cells to induce Th2 effector cells and Tfh cells that drive IgE responses to allergen. (C) A portion of Th2 cells traffic back to skin and permanently reside as tissue-resident memory cells to drive local inflammation through release of type 2 cytokines such as IL-4, -5, and -13 upon re-encountering allergen. (D) Systemic IL-33 from keratinocytes and IL-25 from intestinal tuft cells synergize to activate ILC2s, which in turn produce IL-4 and activate mast cell degranulation (anaphylaxis) in response to oral allergen exposure.
Murine studies have also provided mechanistic explanations for the relationship between allergic sensitization through the skin and multiple atopic comorbidities affecting different tissues. Indeed, mice sensitized to chicken ovalbumin (OVA) or peanut through mechanically disrupted skin that are subsequently challenged to the same antigen in the airways or gut result in phenotypes resembling asthma, food allergen induced anaphylaxis, and eosinophilic esophagitis (12, 30, 31). Damaged keratinocytes express high levels of alarmins such as TSLP, IL-33, and IL-25 that promote type 2 inflammatory responses (32). Dendritic cells in the skin become activated in response to alarmin signals, migrate to skin draining lymph nodes, and present phagocytosed and processed foreign antigens to naïve CD4+ T cells in the form of peptides bound to MHCII (peptide:MHCII) to drive the Th2 differentiation program (33) and T-cell-dependent IgE production. Th2 cells subsequently traffic back to the skin where they reside as Th2-Trm and drive hallmark features of atopic dermatitis—inflammatory cell recruitment and epidermal hyperplasia. Recent studies also suggest that timing may be important for skin sensitization, as early life skin inflammation in mice enables a Th2-Trm niche with stromal cells in the skin (34). In the model of food allergy, systemic IL-33 produced by damaged keratinocytes synergizes with IL-25 produced by intestinal tuft cells (35) to promote activation and expansion of ILC2s in the gut, which in turn produce IL-4 locally to activate and expand mast cells to cause anaphylaxis following oral rechallenge with allergen (30) ( Figures 1B–D ).
Cytoskeletal Abnormalities
Wiskott–Aldrich syndrome (WAS) is an X-linked recessive disease characterized by atopic dermatitis, thrombocytopenia, and combined immune deficiency (36). WAS results from mutations in the WAS protein (WASp), which is critical for actin polymerization affecting immunological synapse formation, mechanotransduction, and tissue migration in multiple hematopoietic cell lineages (37). WASp forms a complex with WASp-interacting protein (WIP) and dedicator of cytokinesis 8 (DOCK8), a guanine nucleotide exchange factor that is essential for WASp’s TCR-dependent function (38). In its GTP-bound form, the small Rho GTPase cell division cycle 42 (CDC42) binds and activates WASp (39). This causes a conformational change in the WASp C-terminus that activates the actin-related protein 2/3 (ARP2/3) complex (40) and leads to actin polymerization ( Figure 2A ). Less common bi-allelic LOF mutations in the genes encoding WIP (41, 42) and ARP2/3 subunit 1B (43, 44) have been described in individuals with similar clinical presentations to WAS, including elevated IgE and eosinophilia.
Figure 2.

IEI influencing CD4+ T-cell activation and differentiation. (A) Genes noted in red involved in the actin cytoskeleton and formation of the immunological synapse. (B) T-cell receptor (TCR) signaling genes influence the generation of Th2 cells and type 2 inflammation. (C) IEI associated with the IL-6-STAT3 cytokine signaling pathway.
Autosomal recessive DOCK8 deficiency leads to a combined immunodeficiency with many shared features with WAS including atopic dermatitis, food allergies with increased incidence of anaphylaxis, recurrent viral infections, and an increased risk for autoimmunity and malignancies (45–48). CD4+ T cells from DOCK8-deficient patients are biased toward a Th2 cell fate (49). Studies in DOCK8-deficient mice have recently identified a novel CD4+ T-cell subset, Tfh13, which drives high-affinity IgE responses following allergen exposure and promotes anaphylaxis (50). WAS and DOCK8 deficiencies are of particular interest to understanding atopic diatheses given their diverse functions in TCR-dependent actin assembly and the formation of the immunological synapse (38), IL-2-dependent STAT5 phosphorylation and T regulatory cell function (51), and composition of the microbiome (52). Rare defects in serine/threonine-protein kinase 4 (STK4), which is upstream of DOCK8, have also demonstrated combined immunodeficiencies with many of the expanded features of WAS and DOCK8 deficiency including poor viral control, malignancy, autoimmunity, severe atopic dermatitis, eosinophilia, and elevated IgE (53, 54). Whether similar mechanisms underlie STK4’s pleiotropic effects require further investigation.
Aberrant TCR Signaling
Human mutations affecting immunological synapse formation and TCR signaling molecules, resulting in aberrant or impaired TCR signal strength, have been associated with combined immunodeficiencies with severe atopic phenotypes ( Figure 2B ). The CARD11–BCL10–MALT1 (CBM) complex links T- and B-cell receptor engagement with the nuclear factor-kappa B (NF-κB) (55) and mTOR (56) signaling pathways. IEI involving the CBM complex can manifest in a broad range of clinical phenotypes. Complete LOF in Caspase Recruitment Domain Family Member 11 (CARD11, also called CARMA1) mutations result in severe combined immunodeficiency, while dominant negative mutations in CARD11 can cause combined immunodeficiency and atopy (57–59). Autosomal dominant gain-of-function (GOF) mutations have also been described in individuals with “B cell expansion with NF-κB and T cell anergy” (BENTA) disease and B-cell malignancies, highlighting differences in regulation of antigen receptor signaling pathways in T and B cells (60). Two different cases of complete LOF mutations B cell CLL/lymphoma 10 (BCL10) (61, 62) have been described with a combined immunodeficiency without atopic features, though some patients with IEI involving mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) paracaspase have presented with severe atopic dermatitis in infancy (63–65). Interestingly, in a genome-wide association study (GWAS) of the Learning Early About Peanut allergy (LEAP) study participants, the top gene associated with independent risk of developing peanut allergy in the peanut avoidance group was MALT1 (66). While the extent to which the atopic phenotypes observed in LOF mutations in the CBM complex are explained by disrupting downstream NF-κB signaling is not clear, a retrospective analysis of patients with IEI registered in the USIDNET revealed that more than half of patients with NF-κB Essential Modulator (NEMO, also known as IKBKG) deficiency were noted to have atopic dermatitis (67). A more recent analysis of eosinophilia and elevated IgE in the USIDNET revealed that 3 out of 3 patients with NFKB2 defects had at least one atopic manifestation and eosinophilia above the upper limit of normal in the reference population and an increased proportion of patients with IKBKG defects had both eosinophilia and elevated IgE (68).
Capping protein regulator and myosin 1 linker 2 (CARMIL2, also known as RLTPR) integrates TCR and CD28 co-stimulation signaling at the immunological synapse. CARMIL2 deficiency results in variable clinical presentations, including a combined immunodeficiency with atopic dermatitis and eosinophilic esophagitis (69–72). Diffuse warts resulting from uncontrolled human papillomavirus infection is another feature reported in many of these patients. Interestingly, CD28 deficiency has recently been described in humans and revealed susceptibility to human papillomavirus infection with severe warts or giant cutaneous horns, though only 1 out of 3 patients were noted to have atopic features of food allergy and asthma (73). Whether this association is significant will require larger patient cohorts and future investigation; however, autoantibodies against CD28 have been associated with atopic disease (74).
Deficiency in the zeta chain of T-cell receptor associated protein kinase 70 (ZAP70) (75) resulting in impaired but not absent TCR signaling can present with significant atopic manifestations before an immunodeficiency is evident. Transgenic mice expressing a homologous point mutation found in humans in Linker for Activation of T cells (LAT) develop Th2 lymphoproliferation, systemic inflammation, and autoimmunity (76, 77). Additional studies in mice taking advantage of a variety of experimental approaches including TCR transgenic systems and altered peptide ligands to control for individual TCR affinities, varying antigen loads, and peptide:MHCII tetramer selection to remove high-affinity polyclonal CD4+ cells have further supported a model where weak TCR stimulation favors a Th2 differentiation program, whereas strong TCR stimulation can suppress GATA3 transcription and Th2 differentiation (78–81). Low TCR signal strength appears to promote Th2 cell induction independent of adjuvant effects (82). The intrinsic capacity of TCR signaling to influence T-cell fate decisions was further demonstrated with adoptive transfer of single naive T-cell clones, each expressing a unique TCR, into normal murine hosts and immunizing under identical conditions. Individual clones produced different patterns of effector T-cell and Tfh subsets, predicted by the strength of TCR signaling (83). Thus, numerous studies now support a dominant role of TCR signal strength in shaping helper T-cell differentiation and function in vivo (7).
Disrupted Cytokine Signaling Pathways
Cytokines produced by innate cells following activation through pattern recognition receptors (PRRs) provide a third signal in addition to TCR and co-stimulatory molecules involved in T-cell activation and differentiation. LOF variants in cytokine and cytokine-induced signaling pathways that result in IEI with atopy reveal mechanisms that typically suppress Th2 in favor of other innate sensing-dependent responses ( Figure 2C ). IL-6 is an acute phase cytokine produced primarily by monocytes and macrophages with pleiotropic effects on hematopoietic and non-hematopoietic cell types. IL-6 binds its receptor (IL-6Rα) to form a hexameric complex with the co-receptor GP130 and activate the downstream JAK-STAT3 signaling pathway (84). In naïve T cells, this canonical pathway promotes the generation of Th17 and Th22 cells, which are important for clearance of extracellular infections. Supporting a simultaneous role of this pathway as a negative regulator of Th2 responses, two reports of different somatic LOF mutations in IL-6Rα in hematopoietic cells were recently identified in patients with atopic dermatitis and elevated IgE in addition to recurrent deep skin and lung infections (85, 86). IL-6R-deficient patients exhibit decreased STAT3 phosphorylation and increased frequencies of GATA3-expressing Th2 cells (85). LOF mutations in IL6ST, the gene encoding GP130, result in atopic dermatitis, elevated IgE, and thrombocytopenia, in addition to connective tissue, pulmonary, neurologic, and renal features (87), owing to the involvement of GP130 in multiple cytokine signaling pathways in numerous cell types (84). Autosomal dominant forms have been recently described with similar atopic features and increased Th2 cytokine production but less connective tissue defects, which may delineate IL-6- and IL-11-dependent GP130 functions (88).
Job’s syndrome was first described in 1966 in individuals with recurrent staphylococcal abscesses, pulmonary infections, and atopic dermatitis (89). This was later found to also be associated with elevated IgE and eosinophilia due to STAT3 mutations with an autosomal dominant (AD) inheritance pattern (90), subsequently referred to as autosomal dominant hyper IgE syndrome (AD-HIES). STAT3 phosphorylation leads to its homodimerization, translocation to the nucleus, and transcription of downstream genes, including cytokines promoting Th17 differentiation and repression of Th2 cells. One such transcription factor, ZNF341, is involved in basal and inducible STAT3 gene expression and patients with ZNF341 mutations have been described with a similar phenotype to AD-HIES (91, 92). STAT3 LOF also have reduced anaphylaxis responses despite highly elevated IgE levels, owing to effects of STAT3 influencing mast cell activation in response to IgE (93) as well as endothelial cell responsiveness to histamine (94).
Descriptions of monogenic IEI are also supported by GWAS studies that have identified IL-6R polymorphisms associated with atopic dermatitis, elevated IgE, and asthma (95–97). Interestingly, a patient with neutralizing anti-IL-6 autoantibodies was described with Staphylococcal skin infections similar to those that affect patients with atopic dermatitis, but otherwise lacked an atopic phenotype (98). Similarly, the treatment of various inflammatory diseases with anti-IL-6 monoclonal antibodies has not resulted in reports of significantly increased atopic manifestations (99). This reinforces that the timing of immune development is critical for the pathogenesis of atopic disease, perhaps through interactions with the microbiome or “early life imprinting” (100).
Decreased T-Cell Repertoire Diversity
Severe combined immunodeficiency (SCID) is defined by very low or absent T-cell number and function. Prior to the addition of T-cell receptor excision circles to the newborn screening program, patients with SCID typically presented in the first year of life with failure to thrive and life-threatening infections. Immune dysregulation and atopy are not features of classical SCID, owing to the lack of T cells necessary to cause immunopathology. However, it is now appreciated that there is a spectrum of SCID and combined immunodeficiency that includes at least 64 distinct genetic disorders (101). Along this spectrum, whereas null mutations in many of these genes result in SCID, numerous hypomorphic variants with partial function have now been described and lead to “leaky SCID” and Omenn syndrome. In addition to significantly increased risk of infections including opportunistic infections, individuals with Omenn syndrome develop a lymphoproliferative state with clonal expansion of a limited number of T cell clones that lead to damaging tissue infiltration of the skin and other organs ( Figure 3A ). Oligoclonal T-cell expansion in Omenn syndrome is associated with Th2 differentiation, secretion of type 2 cytokines, elevated total serum IgE, and eosinophilia (102). Histologically, the erythrodermic skin inflammation of Omenn syndrome very closely resembles that of severe atopic dermatitis.
Figure 3.
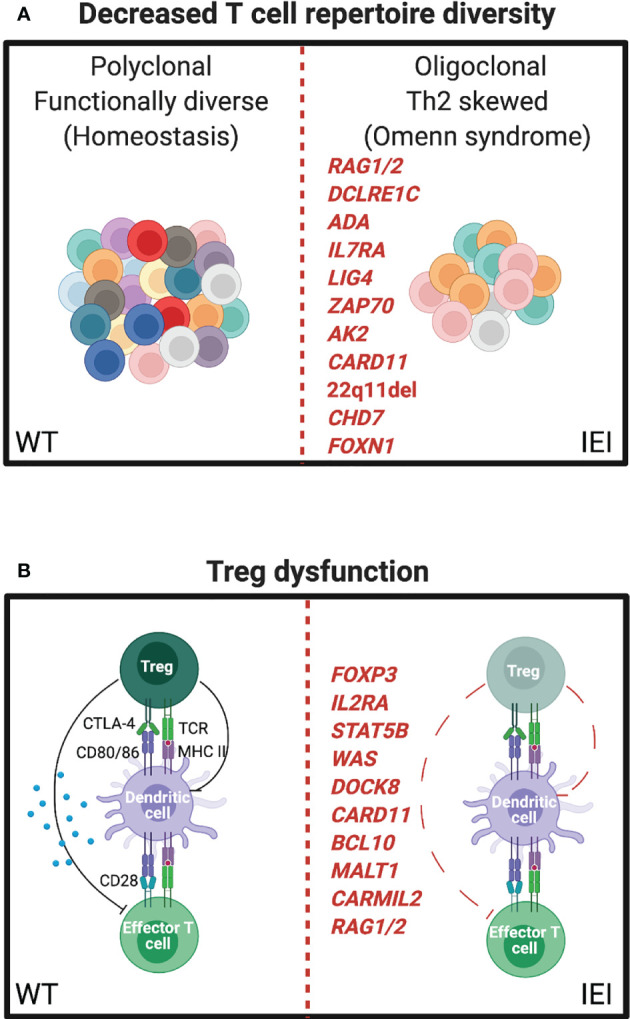
IEI influencing the T-cell repertoire can manifest as severe atopy. (A) Left, resting polyclonal T cells in a normal (WT) immune repertoire. Right, oligoclonal expansion of T cells in leaky SCID, often leading to Th2 skewing and eosinophilia (Omenn syndrome). (B) Left, normal balance between regulatory T cell (Treg) and effector T cell (Teff) functions. Right, mutations affecting Treg number or function leading to autoimmune and atopic phenotypes.
Omenn syndrome has been well described for a number of genes including recombination activating gene 1 and 2 (RAG1/RAG2) (103), ARTEMIS (DCLRE1C) (104), adenosine deaminase (ADA) (105, 106), IL-7 receptor alpha (IL7RA) (107), DNA ligase 4 (LIG4) (108), ZAP70 (75), adenylate kinase 2 (AK2) (109), and CARD11 (110). Thymic defects, in DiGeorge syndrome (22q11del) (111), CHARGE syndrome (CHD7) (112), and forkhead box protein N1 (FOXN1) (113), can also present with Omenn syndrome. While it is not entirely clear how these heterogenous disorders lead to the common atopic features associated with Omenn syndrome, lymphopenia-induced homeostatic proliferation, insufficient thymic deletion of autoreactive T-cell clones, insufficient Treg generation, uncontrolled viral infections, and stochastic variability in effector T-cell differentiation are possibilities.
Murine studies have shed further light on the impact of T-cell repertoire diversity. Milner and Paul et al. reconstituted lymphopenic RAG-/- mice with different numbers of polyclonal naïve CD4+ T cells from a C57BL/6 host with normal thymic development to demonstrate that a limited repertoire results in preferential Th2 skewing, IgE production, and multiorgan eosinophilic inflammation when compared to more complete immune repertoire reconstitution (114). Importantly, tissue pathology could be prevented by altering the balance of Tregs to conventional CD4+ T cells in this system, and IEI associated with Treg dysfunction provide further evidence for their role in limiting atopic disease.
Altered Balance of Conventional T cells and Regulatory T Cells
Inborn errors specifically associated with Treg dysfunction have clarified their important role in autoimmunity, atopy, and tissue homeostasis. Tregs are dependent on the expression of the master transcription factor forkhead box protein P3 (FoxP3). Early descriptions of FoxP3 deficiency in humans resulted in “X-linked autoimmunity-allergic dysregulation syndrome” and “Immunodysregulation polyendocrinopathy enteropathy X-linked” (IPEX) syndrome (115–117). Features of IPEX include multisystem autoimmunity and severe atopy including eczema, food allergy, and eosinophilic inflammation. FoxP3 deficiency in scurfy mice recapitulates many atopic and autoimmune features of IPEX in humans (118), and Treg-specific disruption of multiple immune pathways leads to systemic type 2 inflammation (119–122).
IL2RA, the high-affinity IL-2 receptor, is most highly expressed on Tregs and is critical for their survival and function through the actions of the transcription factor STAT5B. IL2RA- and STAT5B-deficient patients can also exhibit a combined immunodeficiency with autoimmunity, atopic dermatitis, and elevated IgE (123, 124) in addition to non-hematopoietic effects on growth found in STAT5B deficiency. Further evidence supporting a central role for Tregs as regulators of Th2 immunity and atopic disease is the multiple reports of IEI described above also affecting Treg diversity or function, including WAS (125–128), DOCK8 (45, 51), CBM complex components (129–131), CARMIL2 (132), and RAG (133) ( Figure 3B ).
Innate Cell Effector Mechanisms
Monogenic disorders primarily affecting innate immune cells and manifesting with unique atopic features have also been described. The PLCγ2-associated antibody deficiency and immune dysregulation (PLAID) results from temperature-sensitive spontaneous enzyme activity of PLCγ2 leading to mast cell degranulation. Individuals with PLAID have cold or evaporative cooling-induced urticaria (134–136). The cold-induced urticaria in individuals with PLAID can trigger burning sensations in the mouth following consumption of cold foods, which mimics symptoms of food allergy, but does not lead to anaphylaxis. Syncopal episodes have been described following prolonged exposures to cold swimming pools, however.
Summary
Here, we discussed human monogenic disorders associated with significant type 2 inflammation and atopic disease. This review is not a comprehensive list of all IEI associated with increased risk of atopic manifestations, but rather an attempt to illustrate key mechanistic pathways that drive unwanted type 2 immune responses in humans and are supported by experimental mouse models. Impaired barrier function or dysregulated Th2 responses are central drivers of common atopic disease and IEI offer considerable insight into disease pathogenesis (summarized in Table 1 ). Further awareness of the features and mechanisms of IEI will aid in improved diagnosis and inform development of more targeted therapeutics for atopic disease.
Table 1.
Monogenic IEI associated with atopic disease.
| Gene | Inheritance | Immune Manifestations | Atopic Manifestations |
|---|---|---|---|
| Disorders of barrier function | |||
| FLG | AR | Skin infections | A.D., FA, A.R., As, eos, high IgE |
| SPINK5 | AR | Skin infections | A.D., FA, A.R., As, eos, high IgE |
| CDSN | AR | Skin infections | A.D., FA, eos, high IgE |
| DSG1 | AR | Skin infections | A.D., FA, eos, high IgE |
| DSP | AR | Skin infections | A.D., FA, eos, high IgE |
| Cytoskeletal abnormalities | |||
| WAS | XL | CID | A.D., FA, eos, high IgE |
| WIP | AR | CID | A.D., FA, eos, high IgE |
| DOCK8 | AR | CID | A.D., FA, eos, high IgE |
| STK4 | AR | CID | A.D., FA, eos, high IgE |
| Aberrant TCR signaling | |||
| CARD11 | AR | CID, SCID | Eos, high IgE |
| BCL10 | AR | CID, SCID | Eos, high IgE |
| MALT1 | AR | CID, SCID | Eos, high IgE |
| CARMIL2 | AR | CID | Eos, high IgE |
| ZAP70 | AR | CID, SCID | Eos, high IgE |
| LAT | AR | CID, SCID | Eos, high IgE |
| Cytokine pathways | |||
| IL6RA | AR | Skin, lung infections | A.D., eos, high IgE |
| IL6ST | AR, AD | Skin, lung infections | A.D., eos, high IgE |
| STAT3 | AD | Skin, lung infections | A.D., eos, high IgE |
| ZNF341 | AR | Skin, lung infections | A.D., eos, high IgE |
| Decreased T-cell repertoire | |||
| Multiple genes presenting as Omenn syndrome | AR | Leaky SCID | Erythroderma, eos, high IgE |
| Regulatory T-cell defect | |||
| FOXP3 | XL | AI | A.D., FA, As, eos, high IgE |
| IL2RA | AR | CID, AI | A.D., FA, As, eos, high IgE |
| Innate cell effector mechanisms | |||
| PLCG2 | AD | CVID, AI, autoinflammatory | Temperature-sensitive mast cell degranulation |
A summary of causative genes, heritability, and major clinical features is presented. Table includes disorders of barrier function where infections and atopy overlap with IEI. Overlapping mechanisms are discussed further in the main text. AD, autosomal dominant; AR, autosomal recessive; XL, x-linked; CID, combined immune deficiency; SCID, severe combined immune deficiency; CVID, common variable immune deficiency; A.D., atopic dermatitis; eos, eosinophilia; AI, autoimmunity; A.R., allergic rhinitis; FA, food allergy; As, asthma.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by the National Institutes of Health grant NIAID T32 AI007512 (RWN) and AI139633 (RSG). Figures created with BioRender.com.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- 1. Thome JJ, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, et al. Spatial Map of Human T Cell Compartmentalization and Maintenance Over Decades of Life. Cell (2014) 159(4):814–28. doi: 10.1016/j.cell.2014.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rahimi RA, Nepal K, Cetinbas M, Sadreyev RI, Luster AD. Distinct Functions of Tissue-Resident and Circulating Memory Th2 Cells in Allergic Airway Disease. J Exp Med (2020) 217(9). doi: 10.1084/jem.20190865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hondowicz BD, An D, Schenkel JM, Kim KS, Steach HR, Krishnamurty AT, et al. Interleukin-2-Dependent Allergen-Specific Tissue-Resident Memory Cells Drive Asthma. Immunity (2016) 44(1):155–66. doi: 10.1016/j.immuni.2015.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bangert C, Rindler K, Krausgruber T, Alkon N, Thaler FM, Kurz H, et al. Persistence of Mature Dendritic Cells, TH2A, and Tc2 Cells Characterize Clinically Resolved Atopic Dermatitis Under IL-4Ralpha Blockade. Sci Immunol (2021) 6(55). doi: 10.1126/sciimmunol.abe2749 [DOI] [PubMed] [Google Scholar]
- 5. von Moltke J, Pepper M. Sentinels of the Type 2 Immune Response. Trends Immunol (2018) 39(2):99–111. doi: 10.1016/j.it.2017.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Masopust D, Soerens AG. Tissue-Resident T Cells and Other Resident Leukocytes. Annu Rev Immunol (2019) 37:521–46. doi: 10.1146/annurev-immunol-042617-053214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ruterbusch M, Pruner KB, Shehata L, Pepper M. In Vivo CD4(+) T Cell Differentiation and Function: Revisiting the Th1/Th2 Paradigm. Annu Rev Immunol (2020) 38:705–25. doi: 10.1146/annurev-immunol-103019-085803 [DOI] [PubMed] [Google Scholar]
- 8. Ege MJ, Mayer M, Normand AC, Genuneit J, Cookson WO, Braun-Fahrlander C, et al. Exposure to Environmental Microorganisms and Childhood Asthma. N Engl J Med (2011) 364(8):701–9. doi: 10.1056/NEJMoa1007302 [DOI] [PubMed] [Google Scholar]
- 9. Braun-Fahrlander C, Riedler J, Herz U, Eder W, Waser M, Grize L, et al. Environmental Exposure to Endotoxin and Its Relation to Asthma in School-Age Children. N Engl J Med (2002) 347(12):869–77. doi: 10.1056/NEJMoa020057 [DOI] [PubMed] [Google Scholar]
- 10. Lai PS, Massoud AH, Xia M, Petty CR, Cunningham A, Chatila TA, et al. Gene-Environment Interaction Between an IL4R Variant and School Endotoxin Exposure Contributes to Asthma Symptoms in Inner-City Children. J Allergy Clin Immunol (2018) 141(2):794–6.e3. doi: 10.1016/j.jaci.2017.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wolf M, Lai PS. Indoor Microbial Exposures and Chronic Lung Disease: From Microbial Toxins to the Microbiome. Clin Chest Med (2020) 41(4):777–96. doi: 10.1016/j.ccm.2020.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Spergel JM, Mizoguchi E, Brewer JP, Martin TR, Bhan AK, Geha RS. Epicutaneous Sensitization With Protein Antigen Induces Localized Allergic Dermatitis and Hyperresponsiveness to Methacholine After Single Exposure to Aerosolized Antigen in Mice. J Clin Invest (1998) 101(8):1614–22. doi: 10.1172/JCI1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Notarangelo LD, Bacchetta R, Casanova JL, Su HC. Human Inborn Errors of Immunity: An Expanding Universe. Sci Immunol (2020) 5(49). doi: 10.1126/sciimmunol.abb1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lyons JJ, Milner JD. Primary Atopic Disorders. J Exp Med (2018) 215(4):1009–22. doi: 10.1084/jem.20172306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Milner JD. Primary Atopic Disorders. Annu Rev Immunol (2020) 38:785–808. doi: 10.1146/annurev-immunol-042718-041553 [DOI] [PubMed] [Google Scholar]
- 16. Hill DA, Spergel JM. The Atopic March: Critical Evidence and Clinical Relevance. Ann Allergy Asthma Immunol (2018) 120(2):131–7. doi: 10.1016/j.anai.2017.10.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sandilands A, O’Regan GM, Liao H, Zhao Y, Terron-Kwiatkowski A, Watson RM, et al. Prevalent and Rare Mutations in the Gene Encoding Filaggrin Cause Ichthyosis Vulgaris and Predispose Individuals to Atopic Dermatitis. J Invest Dermatol (2006) 126(8):1770–5. doi: 10.1038/sj.jid.5700459 [DOI] [PubMed] [Google Scholar]
- 18. Venkataraman D, Soto-Ramirez N, Kurukulaaratchy RJ, Holloway JW, Karmaus W, Ewart SL, et al. Filaggrin Loss-of-Function Mutations are Associated With Food Allergy in Childhood and Adolescence. J Allergy Clin Immunol (2014) 134(4):876–82 e4. doi: 10.1016/j.jaci.2014.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brown SJ, Asai Y, Cordell HJ, Campbell LE, Zhao Y, Liao H, et al. Loss-Of-Function Variants in the Filaggrin Gene are a Significant Risk Factor for Peanut Allergy. J Allergy Clin Immunol (2011) 127(3):661–7. doi: 10.1016/j.jaci.2011.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thyssen JP, Tang L, Husemoen LL, Stender S, Szecsi PB, Menne T, et al. Filaggrin Gene Mutations Are Not Associated With Food and Aeroallergen Sensitization Without Concomitant Atopic Dermatitis in Adults. J Allergy Clin Immunol (2015) 135(5):1375–8.e1. doi: 10.1016/j.jaci.2015.01.001 [DOI] [PubMed] [Google Scholar]
- 21. Irvine AD, McLean WH, Leung DY. Filaggrin Mutations Associated With Skin and Allergic Diseases. N Engl J Med (2011) 365(14):1315–27. doi: 10.1056/NEJMra1011040 [DOI] [PubMed] [Google Scholar]
- 22. Chavanas S, Bodemer C, Rochat A, Hamel-Teillac D, Ali M, Irvine AD, et al. Mutations in SPINK5, Encoding a Serine Protease Inhibitor, Cause Netherton Syndrome. Nat Genet (2000) 25(2):141–2. doi: 10.1038/75977 [DOI] [PubMed] [Google Scholar]
- 23. Walley AJ, Chavanas S, Moffatt MF, Esnouf RM, Ubhi B, Lawrence R, et al. Gene Polymorphism in Netherton and Common Atopic Disease. Nat Genet (2001) 29(2):175–8. doi: 10.1038/ng728 [DOI] [PubMed] [Google Scholar]
- 24. Kusunoki T, Okafuji I, Yoshioka T, Saito M, Nishikomori R, Heike T, et al. SPINK5 Polymorphism Is Associated With Disease Severity and Food Allergy in Children With Atopic Dermatitis. J Allergy Clin Immunol (2005) 115(3):636–8. doi: 10.1016/j.jaci.2004.12.1114 [DOI] [PubMed] [Google Scholar]
- 25. Kubo A, Nagao K, Amagai M. Epidermal Barrier Dysfunction and Cutaneous Sensitization in Atopic Diseases. J Clin Invest (2012) 122(2):440–7. doi: 10.1172/JCI57416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Telem DF, Israeli S, Sarig O, Sprecher E. Inflammatory Peeling Skin Syndrome Caused a Novel Mutation in CDSN. Arch Dermatol Res (2012) 304(3):251–5. doi: 10.1007/s00403-011-1195-z [DOI] [PubMed] [Google Scholar]
- 27. Oji V, Eckl KM, Aufenvenne K, Natebus M, Tarinski T, Ackermann K, et al. Loss of Corneodesmosin Leads to Severe Skin Barrier Defect, Pruritus, and Atopy: Unraveling the Peeling Skin Disease. Am J Hum Genet (2010) 87(2):274–81. doi: 10.1016/j.ajhg.2010.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Samuelov L, Sarig O, Harmon RM, Rapaport D, Ishida-Yamamoto A, Isakov O, et al. Desmoglein 1 Deficiency Results in Severe Dermatitis, Multiple Allergies and Metabolic Wasting. Nat Genet (2013) 45(10):1244–8. doi: 10.1038/ng.2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McAleer MA, Pohler E, Smith FJ, Wilson NJ, Cole C, MacGowan S, et al. Severe Dermatitis, Multiple Allergies, and Metabolic Wasting Syndrome Caused by a Novel Mutation in the N-Terminal Plakin Domain of Desmoplakin. J Allergy Clin Immunol (2015) 136(5):1268–76. doi: 10.1016/j.jaci.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leyva-Castillo JM, Galand C, Kam C, Burton O, Gurish M, Musser MA, et al. Mechanical Skin Injury Promotes Food Anaphylaxis by Driving Intestinal Mast Cell Expansion. Immunity (2019) 50(5):1262–75.e4. doi: 10.1016/j.immuni.2019.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Venturelli N, Lexmond WS, Ohsaki A, Nurko S, Karasuyama H, Fiebiger E, et al. Allergic Skin Sensitization Promotes Eosinophilic Esophagitis Through the IL-33-Basophil Axis in Mice. J Allergy Clin Immunol (2016) 138(5):1367–80.e5. doi: 10.1016/j.jaci.2016.02.034 [DOI] [PubMed] [Google Scholar]
- 32. Ziegler SF. Thymic Stromal Lymphopoietin, Skin Barrier Dysfunction, and the Atopic March. Ann Allergy Asthma Immunol (2021) 127(3):306–11. doi: 10.1016/j.anai.2021.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sokol CL, Camire RB, Jones MC, Luster AD. The Chemokine Receptor CCR8 Promotes the Migration of Dendritic Cells Into the Lymph Node Parenchyma to Initiate the Allergic Immune Response. Immunity (2018) 49(3):449–63.e6. doi: 10.1016/j.immuni.2018.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boothby IC, Kinet MJ, Boda DP, Kwan EY, Clancy S, Cohen JN, et al. Early-Life Inflammation Primes a T Helper 2 Cell-Fibroblast Niche in Skin. Nature (2021) 599. doi: 10.1038/s41586-021-04044-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-Cell-Derived IL-25 Regulates an Intestinal ILC2-Epithelial Response Circuit. Nature (2016) 529(7585):221–5. doi: 10.1038/nature16161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ochs HD, Thrasher AJ. The Wiskott-Aldrich Syndrome. J Allergy Clin Immunol (2006) 117(4):725–38; quiz 39. doi: 10.1016/j.jaci.2006.02.005 [DOI] [PubMed] [Google Scholar]
- 37. Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich Syndrome: A Comprehensive Review. Ann NY Acad Sci (2013) 1285:26–43. doi: 10.1111/nyas.12049 [DOI] [PubMed] [Google Scholar]
- 38. Janssen E, Tohme M, Hedayat M, Leick M, Kumari S, Ramesh N, et al. A DOCK8-WIP-WASp Complex Links T Cell Receptors to the Actin Cytoskeleton. J Clin Invest (2016) 126(10):3837–51. doi: 10.1172/JCI85774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Abdul-Manan N, Aghazadeh B, Liu GA, Majumdar A, Ouerfelli O, Siminovitch KA, et al. Structure of Cdc42 in Complex With the GTPase-Binding Domain of the ‘Wiskott-Aldrich Syndrome’ Protein. Nature (1999) 399(6734):379–83. doi: 10.1038/20726 [DOI] [PubMed] [Google Scholar]
- 40. Carlier MF, Ducruix A, Pantaloni D. Signalling to Actin: The Cdc42-N-WASP-Arp2/3 Connection. Chem Biol (1999) 6(9):R235–40. doi: 10.1016/S1074-5521(99)80107-0 [DOI] [PubMed] [Google Scholar]
- 41. Lanzi G, Moratto D, Vairo D, Masneri S, Delmonte O, Paganini T, et al. A Novel Primary Human Immunodeficiency Due to Deficiency in the WASP-Interacting Protein WIP. J Exp Med (2012) 209(1):29–34. doi: 10.1084/jem.20110896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schwinger W, Urban C, Ulreich R, Sperl D, Karastaneva A, Strenger V, et al. The Phenotype and Treatment of WIP Deficiency: Literature Synopsis and Review of a Patient With Pre-Transplant Serial Donor Lymphocyte Infusions to Eliminate CMV. Front Immunol (2018) 9:2554. doi: 10.3389/fimmu.2018.02554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Somech R, Lev A, Lee YN, Simon AJ, Barel O, Schiby G, et al. Disruption of Thrombocyte and T Lymphocyte Development by a Mutation in ARPC1B. J Immunol (2017) 199(12):4036–45. doi: 10.4049/jimmunol.1700460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kahr WH, Pluthero FG, Elkadri A, Warner N, Drobac M, Chen CH, et al. Loss of the Arp2/3 Complex Component ARPC1B Causes Platelet Abnormalities and Predisposes to Inflammatory Disease. Nat Commun (2017) 8:14816. doi: 10.1038/ncomms14816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alroqi FJ, Charbonnier LM, Keles S, Ghandour F, Mouawad P, Sabouneh R, et al. DOCK8 Deficiency Presenting as an IPEX-Like Disorder. J Clin Immunol (2017) 37(8):811–9. doi: 10.1007/s10875-017-0451-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined Immunodeficiency Associated With DOCK8 Mutations. N Engl J Med (2009) 361(21):2046–55. doi: 10.1056/NEJMoa0905506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aydin SE, Kilic SS, Aytekin C, Kumar A, Porras O, Kainulainen L, et al. DOCK8 Deficiency: Clinical and Immunological Phenotype and Treatment Options - A Review of 136 Patients. J Clin Immunol (2015) 35(2):189–98. doi: 10.1007/s10875-014-0126-0 [DOI] [PubMed] [Google Scholar]
- 48. Engelhardt KR, Gertz ME, Keles S, Schaffer AA, Sigmund EC, Glocker C, et al. The Extended Clinical Phenotype of 64 Patients With Dedicator of Cytokinesis 8 Deficiency. J Allergy Clin Immunol (2015) 136(2):402–12. doi: 10.1016/j.jaci.2014.12.1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tangye SG, Pillay B, Randall KL, Avery DT, Phan TG, Gray P, et al. Dedicator of Cytokinesis 8-Deficient CD4(+) T Cells Are Biased to a TH2 Effector Fate at the Expense of TH1 and TH17 Cells. J Allergy Clin Immunol (2017) 139(3):933–49. doi: 10.1016/j.jaci.2016.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gowthaman U, Chen JS, Zhang B, Flynn WF, Lu Y, Song W, et al. Identification of a T Follicular Helper Cell Subset That Drives Anaphylactic IgE. Science (2019) 365(6456). doi: 10.1126/science.aaw6433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Janssen E, Kumari S, Tohme M, Ullas S, Barrera V, Tas JM, et al. DOCK8 Enforces Immunological Tolerance by Promoting IL-2 Signaling and Immune Synapse Formation in Tregs. JCI Insight (2017) 2(19). doi: 10.1172/jci.insight.94298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Abdel-Gadir A, Stephen-Victor E, Gerber GK, Noval Rivas M, Wang S, Harb H, et al. Microbiota Therapy Acts via a Regulatory T Cell MyD88/RORgammat Pathway to Suppress Food Allergy. Nat Med (2019) 25(7):1164–74. doi: 10.1038/s41591-019-0461-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Radwan N, El-Owaidy R, El-Sayed ZA, Abdel-Baky A, El-Haddad A, Rashad H, et al. A Case of STK4 Deficiency With Complications Evoking Mycobacterial Infection. J Clin Immunol (2020) 40(4):665–9. doi: 10.1007/s10875-020-00783-w [DOI] [PubMed] [Google Scholar]
- 54. Al-Saud B, Alajlan H, Sabar H, Anwar S, Alruwaili H, Al-Hussain T, et al. STK4 Deficiency in a Patient With Immune Complex Glomerulonephritis, Salt-Losing Tubulopathy, and Castleman’s-Like Disease. J Clin Immunol (2019) 39(8):823–6. doi: 10.1007/s10875-019-00682-9 [DOI] [PubMed] [Google Scholar]
- 55. Bertin J, Wang L, Guo Y, Jacobson MD, Poyet JL, Srinivasula SM, et al. CARD11 and CARD14 Are Novel Caspase Recruitment Domain (CARD)/membrane-Associated Guanylate Kinase (MAGUK) Family Members That Interact With BCL10 and Activate NF-Kappa B. J Biol Chem (2001) 276(15):11877–82. doi: 10.1074/jbc.M010512200 [DOI] [PubMed] [Google Scholar]
- 56. Banerjee A, El-Sayes N, Budylowski P, Jacob RA, Richard D, Maan H, et al. Experimental and Natural Evidence of SARS-CoV-2-Infection-Induced Activation of Type I Interferon Responses. iScience (2021) 24(5):102477. doi: 10.1016/j.isci.2021.102477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dadi H, Jones TA, Merico D, Sharfe N, Ovadia A, Schejter Y, et al. Combined Immunodeficiency and Atopy Caused by a Dominant Negative Mutation in Caspase Activation and Recruitment Domain Family Member 11 (CARD11). J Allergy Clin Immunol (2018) 141(5):1818–30.e2. doi: 10.1016/j.jaci.2017.06.047 [DOI] [PubMed] [Google Scholar]
- 58. Ma CA, Stinson JR, Zhang Y, Abbott JK, Weinreich MA, Hauk PJ, et al. Germline Hypomorphic CARD11 Mutations in Severe Atopic Disease. Nat Genet (2017) 49(8):1192–201. doi: 10.1038/ng.3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dorjbal B, Stinson JR, Ma CA, Weinreich MA, Miraghazadeh B, Hartberger JM, et al. Hypomorphic Caspase Activation and Recruitment Domain 11 (CARD11) Mutations Associated With Diverse Immunologic Phenotypes With or Without Atopic Disease. J Allergy Clin Immunol (2019) 143(4):1482–95. doi: 10.1016/j.jaci.2018.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Snow AL, Xiao W, Stinson JR, Lu W, Chaigne-Delalande B, Zheng L, et al. Congenital B Cell Lymphocytosis Explained by Novel Germline CARD11 Mutations. J Exp Med (2012) 209(12):2247–61. doi: 10.1084/jem.20120831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Torres JM, Martinez-Barricarte R, Garcia-Gomez S, Mazariegos MS, Itan Y, Boisson B, et al. Inherited BCL10 Deficiency Impairs Hematopoietic and Nonhematopoietic Immunity. J Clin Invest (2014) 124(12):5239–48. doi: 10.1172/JCI77493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Van Den Rym A, Taur P, Martinez-Barricarte R, Lorenzo L, Puel A, Gonzalez-Navarro P, et al. Human BCL10 Deficiency Due to Homozygosity for a Rare Allele. J Clin Immunol (2020) 40(2):388–98. doi: 10.1007/s10875-020-00760-3 [DOI] [PubMed] [Google Scholar]
- 63. Jabara HH, Ohsumi T, Chou J, Massaad MJ, Benson H, Megarbane A, et al. A Homozygous Mucosa-Associated Lymphoid Tissue 1 (MALT1) Mutation in a Family With Combined Immunodeficiency. J Allergy Clin Immunol (2013) 132(1):151–8. doi: 10.1016/j.jaci.2013.04.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Punwani D, Wang H, Chan AY, Cowan MJ, Mallott J, Sunderam U, et al. Combined Immunodeficiency Due to MALT1 Mutations, Treated by Hematopoietic Cell Transplantation. J Clin Immunol (2015) 35(2):135–46. doi: 10.1007/s10875-014-0125-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Frizinsky S, Rechavi E, Barel O, Najeeb RH, Greenberger S, Lee YN, et al. Novel MALT1 Mutation Linked to Immunodeficiency, Immune Dysregulation, and an Abnormal T Cell Receptor Repertoire. J Clin Immunol (2019) 39(4):401–13. doi: 10.1007/s10875-019-00629-0 [DOI] [PubMed] [Google Scholar]
- 66. Winters A, Bahnson HT, Ruczinski I, Boorgula MP, Malley C, Keramati AR, et al. The MALT1 Locus and Peanut Avoidance in the Risk for Peanut Allergy. J Allergy Clin Immunol (2019) 143(6):2326–9. doi: 10.1016/j.jaci.2019.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tuano KS, Orange JS, Sullivan K, Cunningham-Rundles C, Bonilla FA, Davis CM. Food Allergy in Patients With Primary Immunodeficiency Diseases: Prevalence Within the US Immunodeficiency Network (USIDNET). J Allergy Clin Immunol (2015) 135(1):273–5. doi: 10.1016/j.jaci.2014.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Smith KL, Dai D, Modi BP, Sara R, Garabedian E, Marsh RA, et al. Inborn Errors of Immunity Associated With Type 2 Inflammation in the USIDNET Registry. Front Immunol (2022) 13:831279. doi: 10.3389/fimmu.2022.831279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schober T, Magg T, Laschinger M, Rohlfs M, Linhares ND, Puchalka J, et al. A Human Immunodeficiency Syndrome Caused by Mutations in CARMIL2. Nat Commun (2017) 8:14209. doi: 10.1038/ncomms14209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Alazami AM, Al-Helale M, Alhissi S, Al-Saud B, Alajlan H, Monies D, et al. Novel CARMIL2 Mutations in Patients With Variable Clinical Dermatitis, Infections, and Combined Immunodeficiency. Front Immunol (2018) 9:203. doi: 10.3389/fimmu.2018.00203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sorte HS, Osnes LT, Fevang B, Aukrust P, Erichsen HC, Backe PH, et al. A Potential Founder Variant in CARMIL2/RLTPR in Three Norwegian Families With Warts, Molluscum Contagiosum, and T-Cell Dysfunction. Mol Genet Genomic Med (2016) 4(6):604–16. doi: 10.1002/mgg3.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang Y, Ma CS, Ling Y, Bousfiha A, Camcioglu Y, Jacquot S, et al. Dual T Cell- and B Cell-Intrinsic Deficiency in Humans With Biallelic RLTPR Mutations. J Exp Med (2016) 213(11):2413–35. doi: 10.1084/jem.20160576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Beziat V, Rapaport F, Hu J, Titeux M, Bonnet des Claustres M, Bourgey M, et al. Humans With Inherited T Cell CD28 Deficiency are Susceptible to Skin Papillomaviruses But are Otherwise Healthy. Cell (2021) 184(14):3812–28.e30. doi: 10.1016/j.cell.2021.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Neuber K, Mahnss B, Hubner C, Gergely H, Weichenthal M. Autoantibodies Against CD28 are Associated With Atopic Diseases. Clin Exp Immunol (2006) 146(2):262–9. doi: 10.1111/j.1365-2249.2006.03218.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Turul T, Tezcan I, Artac H, de Bruin-Versteeg S, Barendregt BH, Reisli I, et al. Clinical Heterogeneity can Hamper the Diagnosis of Patients With ZAP70 Deficiency. Eur J Pediatr (2009) 168(1):87–93. doi: 10.1007/s00431-008-0718-x [DOI] [PubMed] [Google Scholar]
- 76. Aguado E, Richelme S, Nunez-Cruz S, Miazek A, Mura AM, Richelme M, et al. Induction of T Helper Type 2 Immunity by a Point Mutation in the LAT Adaptor. Science (2002) 296(5575):2036–40. doi: 10.1126/science.1069057 [DOI] [PubMed] [Google Scholar]
- 77. Mingueneau M, Roncagalli R, Gregoire C, Kissenpfennig A, Miazek A, Archambaud C, et al. Loss of the LAT Adaptor Converts Antigen-Responsive T Cells Into Pathogenic Effectors That Function Independently of the T Cell Receptor. Immunity (2009) 31(2):197–208. doi: 10.1016/j.immuni.2009.05.013 [DOI] [PubMed] [Google Scholar]
- 78. Milner JD, Fazilleau N, McHeyzer-Williams M, Paul W. Cutting Edge: Lack of High Affinity Competition for Peptide in Polyclonal CD4+ Responses Unmasks IL-4 Production. J Immunol (2010) 184(12):6569–73. doi: 10.4049/jimmunol.1000674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tao X, Grant C, Constant S, Bottomly K. Induction of IL-4-Producing CD4+ T Cells by Antigenic Peptides Altered for TCR Binding. J Immunol (1997) 158(9):4237–44. [PubMed] [Google Scholar]
- 80. Jorritsma PJ, Brogdon JL, Bottomly K. Role of TCR-Induced Extracellular Signal-Regulated Kinase Activation in the Regulation of Early IL-4 Expression in Naive CD4+ T Cells. J Immunol (2003) 170(5):2427–34. doi: 10.4049/jimmunol.170.5.2427 [DOI] [PubMed] [Google Scholar]
- 81. Pfeiffer C, Stein J, Southwood S, Ketelaar H, Sette A, Bottomly K. Altered Peptide Ligands can Control CD4 T Lymphocyte Differentiation In Vivo. J Exp Med (1995) 181(4):1569–74. doi: 10.1084/jem.181.4.1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. van Panhuys N, Klauschen F, Germain RN. T-Cell-Receptor-Dependent Signal Intensity Dominantly Controls CD4(+) T Cell Polarization In Vivo . Immunity (2014) 41(1):63–74. doi: 10.1016/j.immuni.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, et al. Single Naive CD4+ T Cells From a Diverse Repertoire Produce Different Effector Cell Types During Infection. Cell (2013) 153(4):785–96. doi: 10.1016/j.cell.2013.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chen YH, Spencer S, Laurence A, Thaventhiran JE, Uhlig HH. Inborn Errors of IL-6 Family Cytokine Responses. Curr Opin Immunol (2021) 72:135–45. doi: 10.1016/j.coi.2021.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Spencer S, Kostel Bal S, Egner W, Lango Allen H, Raza SI, Ma CA, et al. Loss of the Interleukin-6 Receptor Causes Immunodeficiency, Atopy, and Abnormal Inflammatory Responses. J Exp Med (2019) 216(9):1986–98. doi: 10.1084/jem.20190344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Nahum A, Sharfe N, Broides A, Dadi H, Naghdi Z, Mandola AB, et al. Defining the Biological Responses of IL-6 by the Study of a Novel IL-6 Receptor Chain Immunodeficiency. J Allergy Clin Immunol (2020) 145(3):1011–5 e6. doi: 10.1016/j.jaci.2019.11.015 [DOI] [PubMed] [Google Scholar]
- 87. Chen YH, Grigelioniene G, Newton PT, Gullander J, Elfving M, Hammarsjo A, et al. Absence of GP130 Cytokine Receptor Signaling Causes Extended Stuve-Wiedemann Syndrome. J Exp Med (2020) 217(3). doi: 10.1084/jem.20191306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Beziat V, Tavernier SJ, Chen YH, Ma CS, Materna M, Laurence A, et al. Dominant-Negative Mutations in Human IL6ST Underlie Hyper-IgE Syndrome. J Exp Med (2020) 217(6). doi: 10.1084/jem.20191804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Davis SD, Schaller J, Wedgwood RJ. Job’s Syndrome. Recurrent, “cold”, staphylococcal abscesses. Lancet (1966) 1(7445):1013–5. doi: 10.1016/S0140-6736(66)90119-X [DOI] [PubMed] [Google Scholar]
- 90. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-Negative Mutations in the DNA-Binding Domain of STAT3 Cause Hyper-IgE Syndrome. Nature (2007) 448(7157):1058–62. doi: 10.1038/nature06096 [DOI] [PubMed] [Google Scholar]
- 91. Beziat V, Li J, Lin JX, Ma CS, Li P, Bousfiha A, et al. A Recessive Form of Hyper-IgE Syndrome by Disruption of ZNF341-Dependent STAT3 Transcription and Activity. Sci Immunol (2018) 3(24). doi: 10.1126/sciimmunol.aat4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Frey-Jakobs S, Hartberger JM, Fliegauf M, Bossen C, Wehmeyer ML, Neubauer JC, et al. ZNF341 Controls STAT3 Expression and Thereby Immunocompetence. Sci Immunol (2018) 3(24). doi: 10.1126/sciimmunol.aat4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Siegel AM, Stone KD, Cruse G, Lawrence MG, Olivera A, Jung MY, et al. Diminished Allergic Disease in Patients With STAT3 Mutations Reveals a Role for STAT3 Signaling in Mast Cell Degranulation. J Allergy Clin Immunol (2013) 132(6):1388–96. doi: 10.1016/j.jaci.2013.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hox V, O’Connell MP, Lyons JJ, Sackstein P, Dimaggio T, Jones N, et al. Diminution of Signal Transducer and Activator of Transcription 3 Signaling Inhibits Vascular Permeability and Anaphylaxis. J Allergy Clin Immunol (2016) 138(1):187–99. doi: 10.1016/j.jaci.2015.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wang Y, Hu H, Wu J, Zhao X, Zhen Y, Wang S, et al. The IL6R Gene Polymorphisms are Associated With sIL-6r, IgE and Lung Function in Chinese Patients With Asthma. Gene (2016) 585(1):51–7. doi: 10.1016/j.gene.2016.03.026 [DOI] [PubMed] [Google Scholar]
- 96. Ferreira RC, Freitag DF, Cutler AJ, Howson JM, Rainbow DB, Smyth DJ, et al. Functional IL6R 358Ala Allele Impairs Classical IL-6 Receptor Signaling and Influences Risk of Diverse Inflammatory Diseases. PloS Genet (2013) 9(4):e1003444. doi: 10.1371/journal.pgen.1003444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Esparza-Gordillo J, Schaarschmidt H, Liang L, Cookson W, Bauerfeind A, Lee-Kirsch MA, et al. A Functional IL-6 Receptor (IL6R) Variant is a Risk Factor for Persistent Atopic Dermatitis. J Allergy Clin Immunol (2013) 132(2):371–7. doi: 10.1016/j.jaci.2013.01.057 [DOI] [PubMed] [Google Scholar]
- 98. Puel A, Picard C, Lorrot M, Pons C, Chrabieh M, Lorenzo L, et al. Recurrent Staphylococcal Cellulitis and Subcutaneous Abscesses in a Child With Autoantibodies Against IL-6. J Immunol (2008) 180(1):647–54. doi: 10.4049/jimmunol.180.1.647 [DOI] [PubMed] [Google Scholar]
- 99. Choy EH, De Benedetti F, Takeuchi T, Hashizume M, John MR, Kishimoto T. Translating IL-6 Biology Into Effective Treatments. Nat Rev Rheumatol (2020) 16(6):335–45. doi: 10.1038/s41584-020-0419-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Al Nabhani Z, Eberl G. Imprinting of the Immune System by the Microbiota Early in Life. Mucosal Immunol (2020) 13(2):183–9. doi: 10.1038/s41385-020-0257-y [DOI] [PubMed] [Google Scholar]
- 101. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification From the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Villa A, Notarangelo LD, Roifman CM. Omenn Syndrome: Inflammation in Leaky Severe Combined Immunodeficiency. J Allergy Clin Immunol (2008) 122(6):1082–6. doi: 10.1016/j.jaci.2008.09.037 [DOI] [PubMed] [Google Scholar]
- 103. Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, et al. Partial V(D)J Recombination Activity Leads to Omenn Syndrome. Cell (1998) 93(5):885–96. doi: 10.1016/S0092-8674(00)81448-8 [DOI] [PubMed] [Google Scholar]
- 104. Ege M, Ma Y, Manfras B, Kalwak K, Lu H, Lieber MR, et al. Omenn Syndrome Due to ARTEMIS Mutations. Blood (2005) 105(11):4179–86. doi: 10.1182/blood-2004-12-4861 [DOI] [PubMed] [Google Scholar]
- 105. Roifman CM, Zhang J, Atkinson A, Grunebaum E, Mandel K. Adenosine Deaminase Deficiency can Present With Features of Omenn Syndrome. J Allergy Clin Immunol (2008) 121(4):1056–8. doi: 10.1016/j.jaci.2007.12.1148 [DOI] [PubMed] [Google Scholar]
- 106. Joshi AY, Ham EK, Shah NB, Dong X, Khan SP, Abraham RS. Atypical Omenn Syndrome Due to Adenosine Deaminase Deficiency. Case Rep Immunol (2012) 2012:919241. doi: 10.1155/2012/919241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Giliani S, Bonfim C, de Saint Basile G, Lanzi G, Brousse N, Koliski A, et al. Omenn Syndrome in an Infant With IL7RA Gene Mutation. J Pediatr (2006) 148(2):272–4. doi: 10.1016/j.jpeds.2005.10.004 [DOI] [PubMed] [Google Scholar]
- 108. Grunebaum E, Bates A, Roifman CM. Omenn Syndrome is Associated With Mutations in DNA Ligase IV. J Allergy Clin Immunol (2008) 122(6):1219–20. doi: 10.1016/j.jaci.2008.08.031 [DOI] [PubMed] [Google Scholar]
- 109. Henderson LA, Frugoni F, Hopkins G, Al-Herz W, Weinacht K, Comeau AM, et al. First Reported Case of Omenn Syndrome in a Patient With Reticular Dysgenesis. J Allergy Clin Immunol (2013) 131(4):1227–30.e3. doi: 10.1016/j.jaci.2012.07.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Fuchs S, Rensing-Ehl A, Pannicke U, Lorenz MR, Fisch P, Jeelall Y, et al. Omenn Syndrome Associated With a Functional Reversion Due to a Somatic Second-Site Mutation in CARD11 Deficiency. Blood (2015) 126(14):1658–69. doi: 10.1182/blood-2015-03-631374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Markert ML, Alexieff MJ, Li J, Sarzotti M, Ozaki DA, Devlin BH, et al. Complete DiGeorge Syndrome: Development of Rash, Lymphadenopathy, and Oligoclonal T Cells in 5 Cases. J Allergy Clin Immunol (2004) 113(4):734–41. doi: 10.1016/j.jaci.2004.01.766 [DOI] [PubMed] [Google Scholar]
- 112. Gennery AR, Slatter MA, Rice J, Hoefsloot LH, Barge D, McLean-Tooke A, et al. Mutations in CHD7 in Patients With CHARGE Syndrome Cause T-B + Natural Killer Cell + Severe Combined Immune Deficiency and may Cause Omenn-Like Syndrome. Clin Exp Immunol (2008) 153(1):75–80. doi: 10.1111/j.1365-2249.2008.03681.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Chou J, Massaad MJ, Wakim RH, Bainter W, Dbaibo G, Geha RS. A Novel Mutation in FOXN1 Resulting in SCID: A Case Report and Literature Review. Clin Immunol (2014) 155(1):30–2. doi: 10.1016/j.clim.2014.08.005 [DOI] [PubMed] [Google Scholar]
- 114. Milner JD, Ward JM, Keane-Myers A, Paul WE. Lymphopenic Mice Reconstituted With Limited Repertoire T Cells Develop Severe, Multiorgan, Th2-Associated Inflammatory Disease. Proc Natl Acad Sci USA (2007) 104(2):576–81. doi: 10.1073/pnas.0610289104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, et al. JM2, Encoding a Fork Head-Related Protein, is Mutated in X-Linked Autoimmunity-Allergic Disregulation Syndrome. J Clin Invest (2000) 106(12):R75–81. doi: 10.1172/JCI11679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-Linked Neonatal Diabetes Mellitus, Enteropathy and Endocrinopathy Syndrome is the Human Equivalent of Mouse Scurfy. Nat Genet (2001) 27(1):18–20. doi: 10.1038/83707 [DOI] [PubMed] [Google Scholar]
- 117. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome (IPEX) is Caused by Mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713 [DOI] [PubMed] [Google Scholar]
- 118. Ochs HD, Ziegler SF, Torgerson TR. FOXP3 Acts as a Rheostat of the Immune Response. Immunol Rev (2005) 203:156–64. doi: 10.1111/j.0105-2896.2005.00231.x [DOI] [PubMed] [Google Scholar]
- 119. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-Cell Suppressor Program Co-Opts Transcription Factor IRF4 to Control T(H)2 Responses. Nature (2009) 458(7236):351–6. doi: 10.1038/nature07674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. Regulatory T Cell-Derived Interleukin-10 Limits Inflammation at Environmental Interfaces. Immunity (2008) 28(4):546–58. doi: 10.1016/j.immuni.2008.02.017 [DOI] [PubMed] [Google Scholar]
- 121. Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, et al. Extrathymically Generated Regulatory T Cells Control Mucosal TH2 Inflammation. Nature (2012) 482(7385):395–9. doi: 10.1038/nature10772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 Control Over Foxp3+ Regulatory T Cell Function. Science (2008) 322(5899):271–5. doi: 10.1126/science.1160062 [DOI] [PubMed] [Google Scholar]
- 123. Belaid B, Lamara Mahammed L, Mohand Oussaid A, Migaud M, Khadri Y, Casanova JL, et al. Case Report: Interleukin-2 Receptor Common Gamma Chain Defect Presented as a Hyper-IgE Syndrome. Front Immunol (2021) 12:696350. doi: 10.3389/fimmu.2021.696350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Nadeau K, Hwa V, Rosenfeld RG. STAT5b Deficiency: An Unsuspected Cause of Growth Failure, Immunodeficiency, and Severe Pulmonary Disease. J Pediatr (2011) 158(5):701–8. doi: 10.1016/j.jpeds.2010.12.042 [DOI] [PubMed] [Google Scholar]
- 125. Maillard MH, Cotta-de-Almeida V, Takeshima F, Nguyen DD, Michetti P, Nagler C, et al. The Wiskott-Aldrich Syndrome Protein Is Required for the Function of CD4(+)CD25(+)Foxp3(+) Regulatory T Cells. J Exp Med (2007) 204(2):381–91. doi: 10.1084/jem.20061338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Humblet-Baron S, Sather B, Anover S, Becker-Herman S, Kasprowicz DJ, Khim S, et al. Wiskott-Aldrich Syndrome Protein is Required for Regulatory T Cell Homeostasis. J Clin Invest (2007) 117(2):407–18. doi: 10.1172/JCI29539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Marangoni F, Trifari S, Scaramuzza S, Panaroni C, Martino S, Notarangelo LD, et al. WASP Regulates Suppressor Activity of Human and Murine CD4(+)CD25(+)FOXP3(+) Natural Regulatory T Cells. J Exp Med (2007) 204(2):369–80. doi: 10.1084/jem.20061334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lexmond WS, Goettel JA, Lyons JJ, Jacobse J, Deken MM, Lawrence MG, et al. FOXP3+ Tregs Require WASP to Restrain Th2-Mediated Food Allergy. J Clin Invest (2016) 126(10):4030–44. doi: 10.1172/JCI85129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Policheni A, Horikawa K, Milla L, Kofler J, Bouillet P, Belz GT, et al. CARD11 is Dispensable for Homeostatic Responses and Suppressive Activity of Peripherally Induced FOXP3(+) Regulatory T Cells. Immunol Cell Biol (2019) 97(8):740–52. doi: 10.1111/imcb.12268 [DOI] [PubMed] [Google Scholar]
- 130. Altin JA, Tian L, Liston A, Bertram EM, Goodnow CC, Cook MC. Decreased T-Cell Receptor Signaling Through CARD11 Differentially Compromises Forkhead Box Protein 3-Positive Regulatory Versus T(H)2 Effector Cells to Cause Allergy. J Allergy Clin Immunol (2011) 127(5):1277–85 e5. doi: 10.1016/j.jaci.2010.12.1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Bornancin F, Renner F, Touil R, Sic H, Kolb Y, Touil-Allaoui I, et al. Deficiency of MALT1 Paracaspase Activity Results in Unbalanced Regulatory and Effector T and B Cell Responses Leading to Multiorgan Inflammation. J Immunol (2015) 194(8):3723–34. doi: 10.4049/jimmunol.1402254 [DOI] [PubMed] [Google Scholar]
- 132. Yonkof JR, Gupta A, Rueda CM, Mangray S, Prince BT, Rangarajan HG, et al. A Novel Pathogenic Variant in CARMIL2 (RLTPR) Causing CARMIL2 Deficiency and EBV-Associated Smooth Muscle Tumors. Front Immunol (2020) 11:884. doi: 10.3389/fimmu.2020.00884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Rowe JH, Stadinski BD, Henderson LA, Ott de Bruin L, Delmonte O, Lee YN, et al. Abnormalities of T-Cell Receptor Repertoire in CD4(+) Regulatory and Conventional T Cells in Patients With RAG Mutations: Implications for Autoimmunity. J Allergy Clin Immunol (2017) 140(6):1739–43 e7. doi: 10.1016/j.jaci.2017.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P, et al. Cold Urticaria, Immunodeficiency, and Autoimmunity Related to PLCG2 Deletions. N Engl J Med (2012) 366(4):330–8. doi: 10.1056/NEJMoa1102140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Gandhi C, Healy C, Wanderer AA, Hoffman HM. Familial Atypical Cold Urticaria: Description of a New Hereditary Disease. J Allergy Clin Immunol (2009) 124(6):1245–50. doi: 10.1016/j.jaci.2009.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Schade A, Walliser C, Wist M, Haas J, Vatter P, Kraus JM, et al. Cool-Temperature-Mediated Activation of Phospholipase C-Gamma2 in the Human Hereditary Disease PLAID. Cell Signal (2016) 28(9):1237–51. doi: 10.1016/j.cellsig.2016.05.010 [DOI] [PubMed] [Google Scholar]