

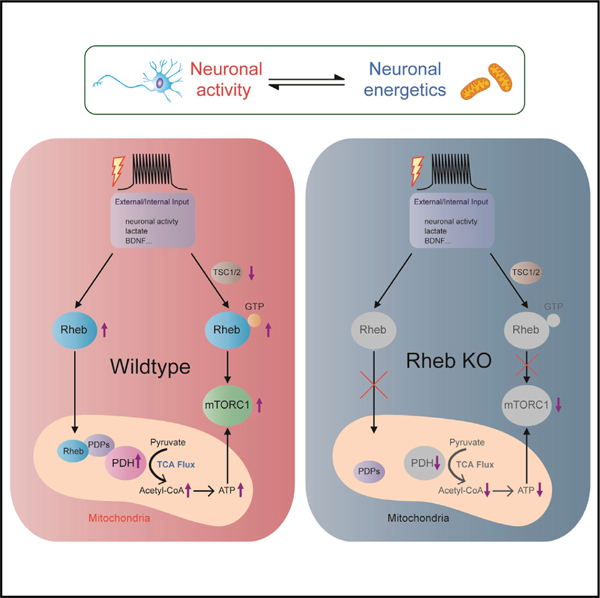
SUMMARY
Neuronal activity increases energy consumption and requires balanced production to maintain neuronal function. How activity is coupled to energy production remains incompletely understood. Here, we report that Rheb regulates mitochondrial tricarboxylic acid cycle flux of acetyl-CoA by activating pyruvate dehydrogenase (PDH) to increase ATP production. Rheb is induced by synaptic activity and lactate and dynamically trafficked to the mitochondrial matrix through its interaction with Tom20. Mitochondria-localized Rheb protein is required for activity-induced PDH activation and ATP production. Cell-type-specific gain- and loss-of-function genetic models for Rheb reveal reciprocal changes in PDH phosphorylation/activity, acetyl-CoA, and ATP that are not evident with genetic or pharmacological manipulations of mTORC1. Mechanistically, Rheb physically associates with PDH phosphatase (PDP), enhancing its activity and association with the catalytic E1α-subunit of PDH to reduce PDH phosphorylation and increase its activity. Findings identify Rheb as a nodal point that balances neuronal activity and neuroenergetics via Rheb-PDH axis.
Graphical abstract
In brief
Neuronal activity is coordinated with neuroenergetics. Yang et al. demonstrate that dynamic translocation of Rheb to mitochondrial matrix activates pyruvate dehydrogenase (PDH) independent of mTORC1 and mediates neuronal-activity-regulated mitochondrial metabolism. Their findings suggest a mechanism by which Rheb integrates PDH-dependent energy production and mTORC1-dependent energy consumption.
INTRODUCTION
Neurons coordinate activity-regulated energetics and synaptic plasticity (Butterfield and Halliwell, 2019; Rangaraju et al., 2014; Harris et al., 2012). Activity-regulated neuroenergetics is essential for maintaining energy homeostasis that supports neuronal functions (Zilberter and Zilberter, 2017; Le Masson et al., 2014; Lutas et al., 2014; Rangaraju et al., 2014). Increased synaptic signaling/transmission between neurons leads to higher energy consumption (Sibson et al., 1998), a finding that has been exploited in brain imaging experiments (Vanzetta and Grinvald, 1999). Although neuronal activity is a high-energy demanding process and neurons lack energy stores, neurons engage multiple cellular and molecular mechanisms to balance energy consumption and production, maintaining ATP levels stable (Du et al., 2008).
Neurons use both glucose and lactate as energy source in the resting and activated states (Baeza-Lehnert et al., 2019; Wyss et al., 2011). In response to activity, neurons not only elevate glucose uptake and glycolysis (Díaz-García et al., 2017; Lundgaard et al., 2015) but also outsource glycolysis to glia for lactate supply to maintain synaptic function and long-term viability (Liu et al., 2017; Suzuki et al., 2011; Rouach et al., 2008). Ultimately, neurons rely on mitochondria for the majority of activity-induced ATP production (Rangaraju et al., 2014; Hall et al., 2012). While being a signaling molecule (Yang et al., 2014), glia-derived lactate as an energy substrate for neurons is converted to pyruvate for ATP production through tricarboxylic acid (TCA) cycle and oxidative phosphorylation. This lactate-fueled neuroenergetics may be essential for sustaining synaptic function during the period of heightened activity (Magistretti and Allaman, 2018; Machler et al., 2016; Nagase et al., 2014; Wyss et al., 2011). Interruption of glial-derived lactate results in motor neuron degeneration (Lee et al., 2012). Consistent with neuronal-activity-induced mitochondrial activity, neuronal pyruvate consumption in mitochondria is increased when neurons are in activated state (Baeza-Lehnert et al., 2019). The outstanding question is how neuronal mitochondria adapt to increased supply of energy source (activity-induced lactate and pyruvate consumption in this case), to increase mitochondrial activity and ATP production. This activity-regulated neuronal energetics is essential for maintaining synaptic function and survival because neurons are susceptible to an even brief disruption of ATP homeostasis (Rangaraju et al., 2014), with possible consequences for neurodegeneration (Zilberter and Zilberter, 2017; Le Masson et al., 2014). Pursuant to the stimulation patterns, neuronal-activity-induced events associated with neuron ensembles can last for hours (Tyssowski et al., 2018). This sustained neuronal activation imposes a significant energetic challenge to neurons.
During our study of the roles of the activity-induced gene Rheb in synaptic signaling and cell differentiation (Kang et al., 2015; Delgoffe et al., 2011; Zou et al., 2011; Yamagata et al., 1994), we identified a mitochondrial role of Rheb in regulating energetics involving pyruvate metabolism. Rheb is a small H-Ras-like GTPase originally identified as an immediate early gene based on its rapid de novo transcription in 3T3 fibroblasts induced by serum stimulation and brain neurons by electroconvulsive seizure (Yamagata et al., 1994). In its GTP state (controlled by tuberous sclerosis complex [TSC]), Rheb functions as a direct and essential activator of mechanistic target of rapamycin complex 1 (mTORC1) on the surface of lysosomes (Menon et al., 2014). mTORC1 is a serine/threonine kinase complex that is known to regulate protein synthesis and protein synthesis–dependent synaptic plasticity (Bockaert and Marin, 2015; Buffington et al., 2014).
Previous studies suggest Rheb’s localization to mitochondrial outer membrane (Ma et al., 2008), where Rheb seems to regulate mitochondrial autophagy/mitophagy (Melser et al., 2013). Using genetically modified mouse models, we examined how Rheb might regulate mitochondrial activity beyond previously described mTORC1-dependent mechanisms and linked Rheb to activity-induced mitochondrial energetics by activating pyruvate dehydrogenase (PDH) independently of mTORC1.
RESULTS
Dynamic trafficking of Rheb to mitochondria involves Tom20
To examine whether Rheb directly regulates mitochondrial function, we validated mitochondrial localization of Rheb (Melser et al., 2013; Ma et al., 2008). Immunostaining showed myc-Rheb colocalization with a mitochondrial marker (Mitotracker) in HeLa cells, as reported previously (Figure 1A). The related protein myc-Rheb2 did not colocalize with Mitotracker, and glutamine did not alter its subcellular localization (Figure S1A). We further performed biochemical isolation and subfractionation of mitochondria and demonstrated the presence of Rheb in mitochondrial matrix (Mx) (Figures 1B–1D). Furthermore, the presence of Rheb in mitochondria matrix was enhanced by neuronal activity and metabolic needs (Figures 1C and 1D).
Figure 1. Dynamic trafficking of Rheb to mitochondria.
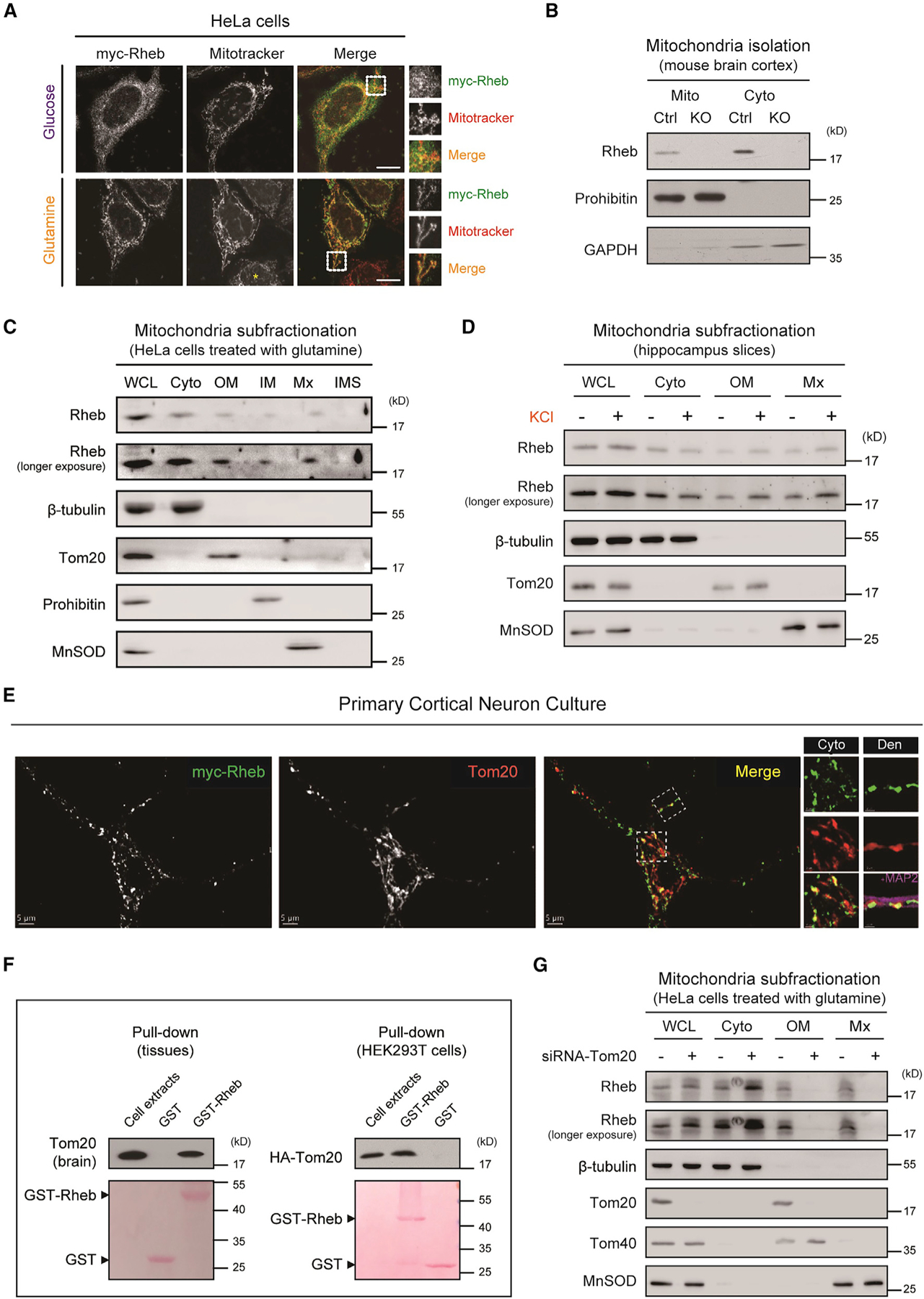
(A) Immunofluorescence showing that myc-Rheb colocalized with Mitotracker (red) in mitochondria of HeLa cells. Note enhanced colocalization of Rheb with Mitotracker by glutamine treatment. Asterisk denotes nontransfected cells. Scale bar, 10 µm.
(B) Subcellular fractionation of brain cortex showing mitochondrial localization of Rheb, compared with controls Ctrl (Rhebf/+ or Rhebf/f) and Rheb KO (Rhebf/f;CaMKII-cre). Cyto, cytosol; Mito, mitochondria.
(C) Subfractionation of highly purified mitochondria from glutamine-treated HeLa cells showing the presence of Rheb protein in the mitochondrial matrix. WCL, whole cell lysate; cyto, cytosol; OM, mitochondrial outer membrane; IM, mitochondrial inner membrane; Mx, mitochondrial matrix; IMS, mitochondrial inter-membrane space.
(D) Western blots showing increased Rheb mitochondrial localization by KCl treatment (40 mM, 1 h) in hippocampal slices of wildtype mice.
(E) Immunofluorescence showing myc-Rheb colocalization with Tom20 in the soma (cyto) and dendrites (Den) of cortical neuronal culture derived from myc-Rheb transgenic mice (Rosa mycRhebK/+;Nestin-Cre). Scale bar, 5 µm.
(F) GST pull-down demonstrating that purified GST-Rheb binds endogenous Tom20 in the brain (left panel), and HA-tagged Tom20 expressed in HEK293T cells (right panel).
(G) Western blots showing reduced Rheb presence in mitochondrial outer membrane (OM) and matrix (Mx) by Tom20 knockdown. See also Figure S1.
Mitochondrial protein import and sorting is a complex process. The best-characterized mitochondrial protein import begins with the recognition of matrix-targeted proteins by the translocase of the outer membrane (TOM) receptors, followed by translocation through the outer membrane to the translocase of the inner membrane and into the matrix (Chacinska et al., 2009; Abe et al., 2000). We found that Rheb targeting to mitochondrial matrix is dependent on the mitochondrial translocase protein Tom20. Rheb binds Tom20 indicated by GST pull-down assay and they colocalize in neurons revealed by immunostaining (Figures 1E and 1F). Furthermore, knocking down Tom20 significantly reduced the amount of Rheb in the outer membrane and matrix (Figure 1G). These results indicate that Rheb is dynamically trafficked to mitochondrial matrix by action of Tom20, which is consistent with the general mode of protein trafficking to mitochondria (Chacinska et al., 2009; Abe et al., 2000).
Rheb regulates mitochondrial PDH activity and energy production
The presence of Rheb in mitochondrial matrix (further validated by proximity ligation assay [PLA] assay shown later) suggests a role for Rheb to regulate metabolic events. To assess the role of Rheb in mitochondria, we first examined how Rheb knockout (KO) in neural cells could affect the central metabolites of mitochondrial energy metabolism—acetyl coenzyme A (acetyl-CoA) and ATP—and found significantly lower levels of acetyl-CoA and ATP levels in the cortex of Rheb KO mice (Rhebf/f;Nestin-cre) (Figure 2A). In Rhebf/f;Nestin-cre mice, Rheb is deleted in all neural cells (neurons, astrocytes, and oligodendrocytes) (Zou et al., 2011). Mice were maintained with ad libitum access to food and water and were not otherwise manipulated. Reduced acetyl-CoA levels suggest that the conversion of pyruvate to acetyl-CoA was suppressed. Pyruvate is oxidized by PDH to acetyl-CoA, an irreversible reaction in mitochondria, and therefore, PDH is considered a mitochondrial gatekeeper. In this way, PDH controls the TCA cycle input of acetyl-CoA (Kaplon et al., 2013). Therefore, the dynamic regulation of PDH activity in neurons might play a role in activity-regulated mitochondrial energetics.
Figure 2. Rheb regulates neuronal PDH activity and energy production.
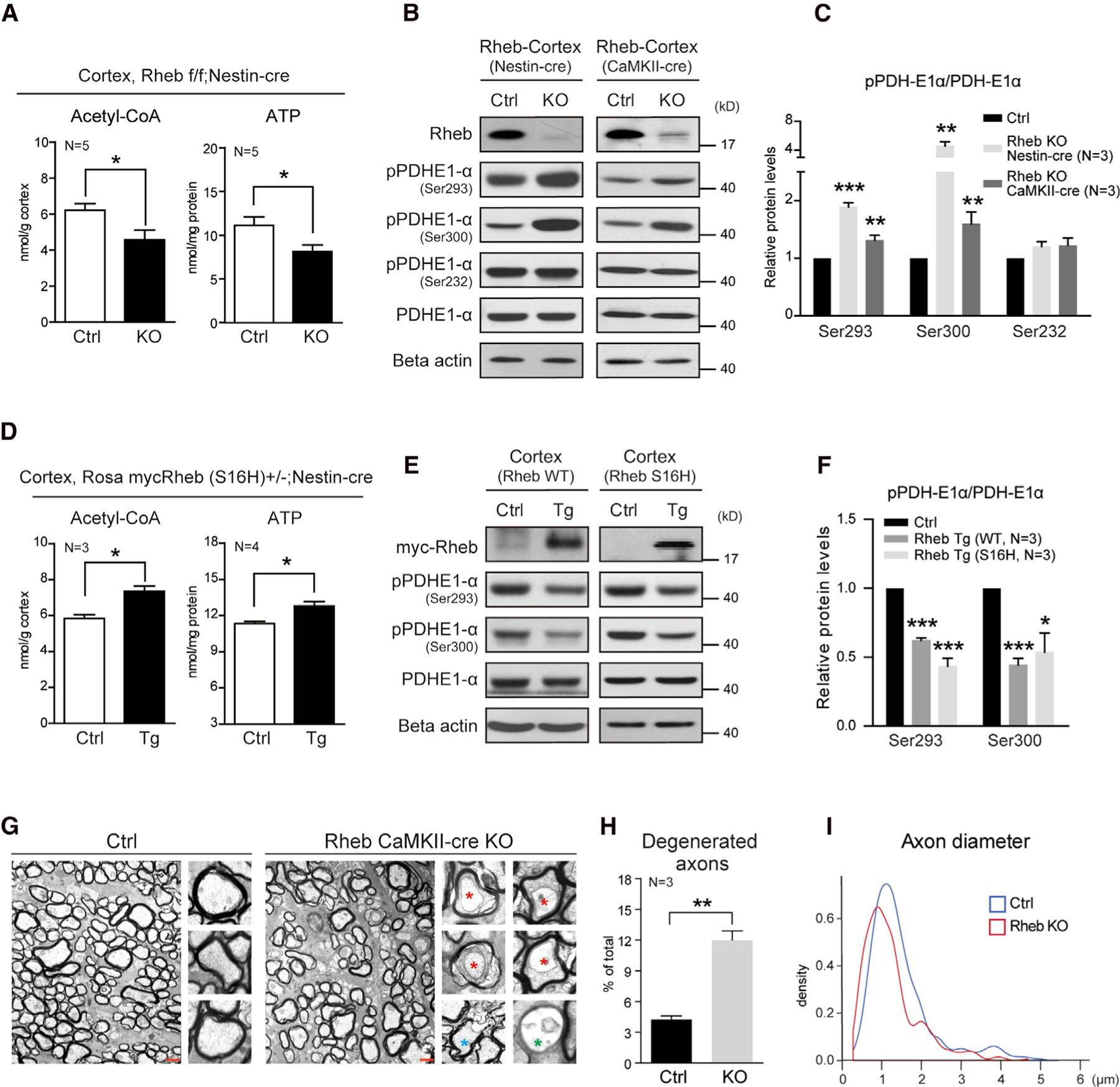
(A) Acetyl-CoA and ATP reductions in the cerebral cortex of Rheb Nestin-cre KO mice. N = 5 pairs of mice. Acetyl-CoA, p = 0.0342; ATP, p = 0.0392.
(B and C) Western blots (B) and quantification (C) indicating increase of PDH phosphorylation in the cortex of Rheb Nestin-cre and CaMKII-cre KO mice. N = 3 pairs of mice. Ser293, Rheb Nestin-cre, p = 0.0002; Rheb CaMKII-cre, N = 3, p = 0.0040; Ser300, Rheb Nestin-cre, p = 0.0037; Rheb CaMKII-cre, p = 0.0096; Ser232, Rheb Nestin-cre, p = 0.0708; Rheb CaMKII-cre, p = 0.0404.
(D) Increase of acetyl-CoA and ATP in the cerebral cortex of Rheb (S16H) Nestin-cre Tg mice. Acetyl-CoA, N = 3 pairs of mice, p = 0.0121; ATP, N = 4 pairs of mice, p = 0.0116.
(E and F) Western blots (E) and quantification (F) indicating decrease of PDH phosphorylation in the cortex of Rheb WT (wildtype) and S16H Nestin-cre Tg (Rosa mycRheb+/−; Nestin-cre and Rosa mycRheb(S16H)+/−;Nestin-cre) mice. N = 3 pairs of mice. Ser293, Rheb Tg-WT cortex, p < 0.0001; Rheb Tg-S16H cortex, p = 0.0006; Ser300, Rheb Tg-WT cortex, p = 0.0003; Rheb Tg-S16H cortex, p = 0.0277.
(G) Cross-sectional electron micrographs showing axonal degeneration in optic nerves of Rheb CaMKII-cre KO mice. Scale bar, 1 µm.
(H and I) Quantifications of electron micrographs showing the degenerated axons (H), axonal diameters (I) in optic nerves of Rheb CaMKII-cre KO mice. All data represent mean ± SEM. Statistical analysis was performed by using 2-tailed Student’s t test (A, C, D, F, H), and *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figures S1–S3.
In eukaryotes, PDH consists of >100 proteins assembled in 3 enzymatic complexes termed PDH (E1), dihydrolipoyl transacetylase (E2), and dihydrolipoyl dehydrogenase (E3). PDH activity is inhibited by phosphorylation of the enzymatic subunit of E1 (PDH-E1α) at Ser293, Ser300, and Ser232 that prevents access of pyruvate to the catalytic site (Rardin et al., 2009; Kolobova et al., 2001). We found that deletion of Rheb in neurons and glia cells increased phosphorylated PDH-E1α levels (Figures 2B, 2C, S1B, and S1C). To examine the specific contribution of Rheb in neurons to PDH activity, we deleted Rheb in excitatory neurons using CaMKII-cre (Rhebf/f;CaMKII-cre). Deletion of Rheb in excitatory neurons likewise increased phosphorylated PDH-E1α, particularly at sites 1 and 2 (Ser293 and Ser300) (Figures 2B, 2C, S1B, and S1C). During postnatal brain development, PDH-E1α phosphorylation was increased in close temporal association with Rheb deletion (assayed in Rhebf/f;CaMKII-cre mice at P4 and P6), suggesting a direct dependence upon Rheb in the control of PDH phosphorylation/activity (Figures S1D and S1E). As a consequence of reduced PDH activity indicated by increased phosphorylated PDH, we found that acetyl-CoA and ATP were reduced by 22.0% and 22.7%, respectively, in cerebral cortex of Rhebf/f; CaMKII-cre mice (Figure S1F). Pyruvate level was lower than the control in the cortex (Figure S1G), which could be caused by reduced glycolysis consequential to mTORC1 reduction (Düvel et al., 2010). Consistent with the finding of reduced ATP production in Rheb KO neurons, adenosine diphosphate (ADP) level was increased, suggesting less phosphorylation of ADP to ATP. To compensate for reduced ATP production, neurons could use more phospho-creatine to generate ATP, as indicated by decreased phospho-creatine level in the brain tissue (Figures S2A–S2D). Moreover, neuronal ATP reduction by Rheb KO did not require metabolic stress (naive mice euthanized from home cage) and was not due to impairment in mitochondrial machinery (Figures S2E and S2F).
In another mouse model that reciprocally increases Rheb expression in the brain, phosphorylation of PDH-E1α was reduced in the cortex by wild-type (WT) Rheb transgene expression in Rosa mycRheb+/−; Nestin-cre to an extent similar to Rheb mutant Rosa mycRheb(S16H)+/−;Nestin-cre (Figures 2E and 2F). Accordingly, acetyl-CoA and ATP were increased in the cerebral cortex by Rheb transgene (Figure 2D). Unlike mycRheb(S16H), which increases mTORC1, mycRheb WT transgene does not cause an increase of mTORC1 and their similar effect on PDH phosphorylation suggests that Rheb-regulated PDH activity may not be dependent on its role of activating mTORC1 (Figures S1B and S1C).
PDH activity and acetyl-CoA/ATP levels are essential for neuronal function and maintenance of axon integrity (Currais et al., 2019; Le Masson et al., 2014; Sorbi et al., 1983). Therefore, we examined whether neuronal Rheb plays a role in the maintenance of axon integrity. Results showed that neuronal Rheb KO mice manifested age-dependent axon degeneration. Electron microscopy revealed age-dependent axon degeneration of optic nerves in neuronal Rheb KO mice. Notably, 12% of axons of optic nerves exhibited structural changes characteristic of axon degeneration by 9–14 months, including separations of axonglial adhesion (separated), collapsed myelinated axons (collapsed), and production of empty myelin sheaths (empty) (Figures 2G and 2H). We also noted that the number of axons with smaller diameter was increased, which is consistent with the notion that neuronal Rheb KO leads to axon degeneration (Figure 2I).
Because PDH is fundamental to mitochondrial metabolism with implications for multiple synthetic pathways, we wondered whether Rheb-regulated PDH is a general mechanism cells use to regulate mitochondrial metabolism. Toward this goal, we examined the effect of Rheb KO on liver hepatocytes using Albumin-cre (hereafter Alb-cre) (Postic et al., 1999), in comparison with mTOR deletion. Rheb and mTOR deletion reduced mTORC1 activity to similar level; Rheb deletion increased phosphorylated PDH-E1α in the liver, whereas mTOR deletion did not alter phosphorylated PDH (Figures S3A and S3B), suggesting that Rheb activates PDH independently of its role of activating mTORC1. Biochemical measurement of PDH enzymatic activity also revealed a reduction selectively in Rheb KO liver (Figure S3C). As a result, Rheb KO reduced acetyl-CoA (Figure S3D) and produced a more profound ATP reduction in the liver (Figure S3E). The bulk of cellular acetyl-CoA is derived from pyruvate, although beta-oxidation of fatty acids also contributes to some extent. We reasoned that increased PDH phosphorylation in Rheb KO samples was not due to changes in pyruvate levels, because reduced pyruvate levels were noted in both Rheb and mTOR KO mice (Figure S3F), but changes to phosphorylated PDH were noted only in Rheb KO mice. Lactate was not reduced in the liver of Rheb KO mice, suggesting enhanced pyruvate-to-lactate conversion (Figure S3G). These results indicate that Rheb plays a general role in regulating mitochondrial energy production by activating PDH.
Rheb regulates PDH activity independently of mTORC1
The comparison of the effects of mTOR and Rheb KO on metabolites and phosphorylated PDH suggests that Rheb-activated mTORC1 may not play a role in PDH activation (Figure S3). To further examine this notion in neurons, we evaluated PDH-E1α phosphorylation in Raptorf/f;CaMKII-cre mice. Raptor encodes an essential component of mTORC1 complex (Peterson et al., 2011). Deletion of Raptor reduced mTORC1 activity in the brain but did not alter PDH phosphorylation (Figure 3A). To examine the effect of in vitro inhibition of mTOR on PDH phosphorylation, we monitored the time course of pharmacological inhibition of mTORC1 by rapamycin or Torin1 and found that these treatments produced a profound and sustained reduction of mTORC1 without a change in PDH phosphorylation (Figure 3B). Consistent with this finding, we found that inhibition of mTORC1 did not alter acetyl-CoA level (Figure S4A).
Figure 3. Rheb regulates PDH activity independently of mTORC1.
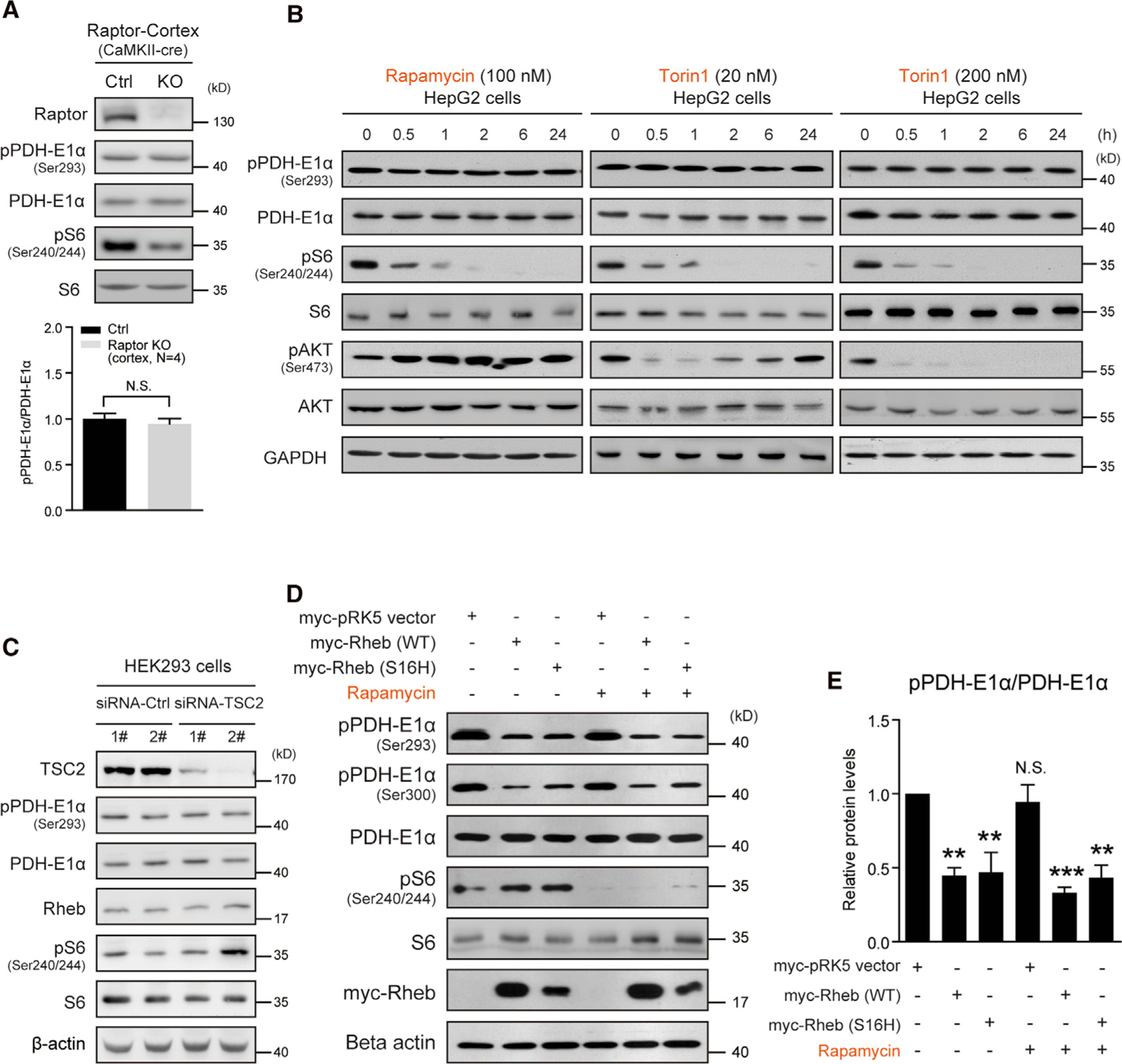
(A) Western blots and quantification indicating PDH phosphorylation not altered in the cortex of Raptor CaMKII-cre KO (Raptorf/f;CaMKII-cre) mice. Ctrl (raptorf/+ or raptorf/f). Ser293, cortex, N = 4 pairs of mice, p = 0.53.
(B) Western blots showing that rapamycin (100 nM) or Torin1 (20 and 200 nM) treatment for 24 h does not alter PDH phosphorylation in HepG2 cells. Note that mTORC1 activity was persistently inhibited, but mTORC2 activity assayed as pAKT (Ser473) initially decreased and then gradually recovered (Torin1 20 nM).
(C) Western blot showing that TSC2 knockdown activates mTORC1 activity, but does not alter PDH phosphorylation.
(D and E) Western blots (D) and quantification (E) showing that transient overexpression of Rheb (WT or S16H) decreases PDH phosphorylation in HepG2 cells and rapamycin (100 nM, 6 h) treatment does not prevent the decrease. pPDH-E1α (Ser293), N = 3 independent experiments, myc-pRK5 versus Rheb WT, p < 0.01; Rheb-S16H, p < 0.01; rapamycin, N.S.; Rheb WT + rapamycin, p < 0.001; Rheb-S16H+Rapamycin, p < 0.01. All data represent mean ± SEM. Statistical analysis was performed by using 2-tailed Student’s t test (A) or one-way ANOVA with Dunnett post hoc test (E), **p < 0.01 and ***p < 0.001. See also Figure S4.
The canonical mTORC1 signaling cascade inhibits TSC (TSC1/TSC2) to reduce its GTPase activity for Rheb and thereby increase Rheb-GTP and activate mTORC1 (Menon et al., 2014; Garami et al., 2003). Consistent with the separation of this pathway from PDH activation, knockdown of TSC2 protein increased mTORC1 activity, but phosphorylated PDH level was not altered (Figure 3C), suggesting that TSC-mTORC1 axis is not involved in the regulation of PDH phosphorylation.
To further distinguish Rheb’s role in activating PDH versus mTORC1, we examined PDH-E1α phosphorylation in cell cultures transfected with Rheb WT or Rheb(S16H) expression constructs and then treated cells with rapamycin and Torin1. Similar to the expression of Rheb transgene in vivo, transfection of Rheb (WT or S16H mutant) reduced PDH-E1α phosphorylation while maintaining the total protein level of PDH-E1α. Rapamycin or Torin1 treatment did not prevent the reduction of PDH-E1α phosphorylation in Rheb transfected cells, despite prominent reduction of pS6 (Ser240/244) (Figures 3D, 3E, and S4B).
Finally, to assess the specificity of Rheb-PDH, we compared its action to Rheb2. Rheb2 is not required for mTORC1 activation in vivo (Zou et al., 2011); however, overexpression of Rheb2 transgene increases mTORC1 activity in HepG2 cells, and Rheb2 overexpression did not alter PDH phosphorylation (Figures S4C and S4D). Therefore, we conclude that Rheb specifically regulates PDH phosphorylation, and this action is independent of its role in activation of mTORC1.
Rheb binding to PDPs enhances its activity to PDH
PDH is activated by dephosphorylation mediated by 2 isoforms of PDH phosphatases (PDPs). PDP1 is predominate in brain and its activity exhibits Ca2+ sensitivity (Lawson et al., 1993). PDP2 is enriched in liver and is Ca2+ insensitive (Huang et al., 1998). PDPs form a stable complex with PDH to maintain reduced phosphorylation and increased PDH activity. PDP dissociation from PDH leads to PDH phosphorylation (Fan et al., 2014). In biochemical assays using recombinant proteins, we did not detect direct binding of GST-Rheb with PDH-E1α subunit (Figure S4E). However, GST-Rheb binds PDP1 from brain and PDP2 from liver (Figure 4A). The association of Rheb with PDP1 was also corroborated by an in situ PLA (Bagchi et al., 2015), in which antibodies for 2 different proteins are labeled with DNA sequences that can be ligated and amplified if the antibodies are within ~40 nm. HeLa cells transfected with GFP-Rheb and processed with antibodies for GFP and PDP1 to detect native protein produced distinct PLA signals (Figure 4B). GST-Rheb could also interact with recombinant HA-tagged PDP2 expressed in HEK293T cells (Figure 4C). The specificity of Rheb-PDP interaction is supported by experiments showing that GST-Rheb does not bind the PDP2-related phosphatase PP2C (Figure 4C). GST-PDP2 pulls down Rheb but not Rheb2 or Rac1 (Figure S4F). myc-Rheb and HA-PDP2 transgenes expressed in HEK293T cells coimmunoprecipitated in both directions (Figure 4D), and binding was reconstituted in vitro using purified recombinant Rheb and PDP2 proteins from E. coli (Figures 4E and 4F). Rheb-PDP binding was not dependent on the GTP/GDP state of Rheb (Figures 4G and 4H). Deletion analysis reveals that aa 168–196 of PDP2 and N-terminal region (aa 1–93) of Rheb are required for their interaction (Figures S4G and S4H).
Figure 4. Rheb interacts with PDH phosphatases (PDPs).
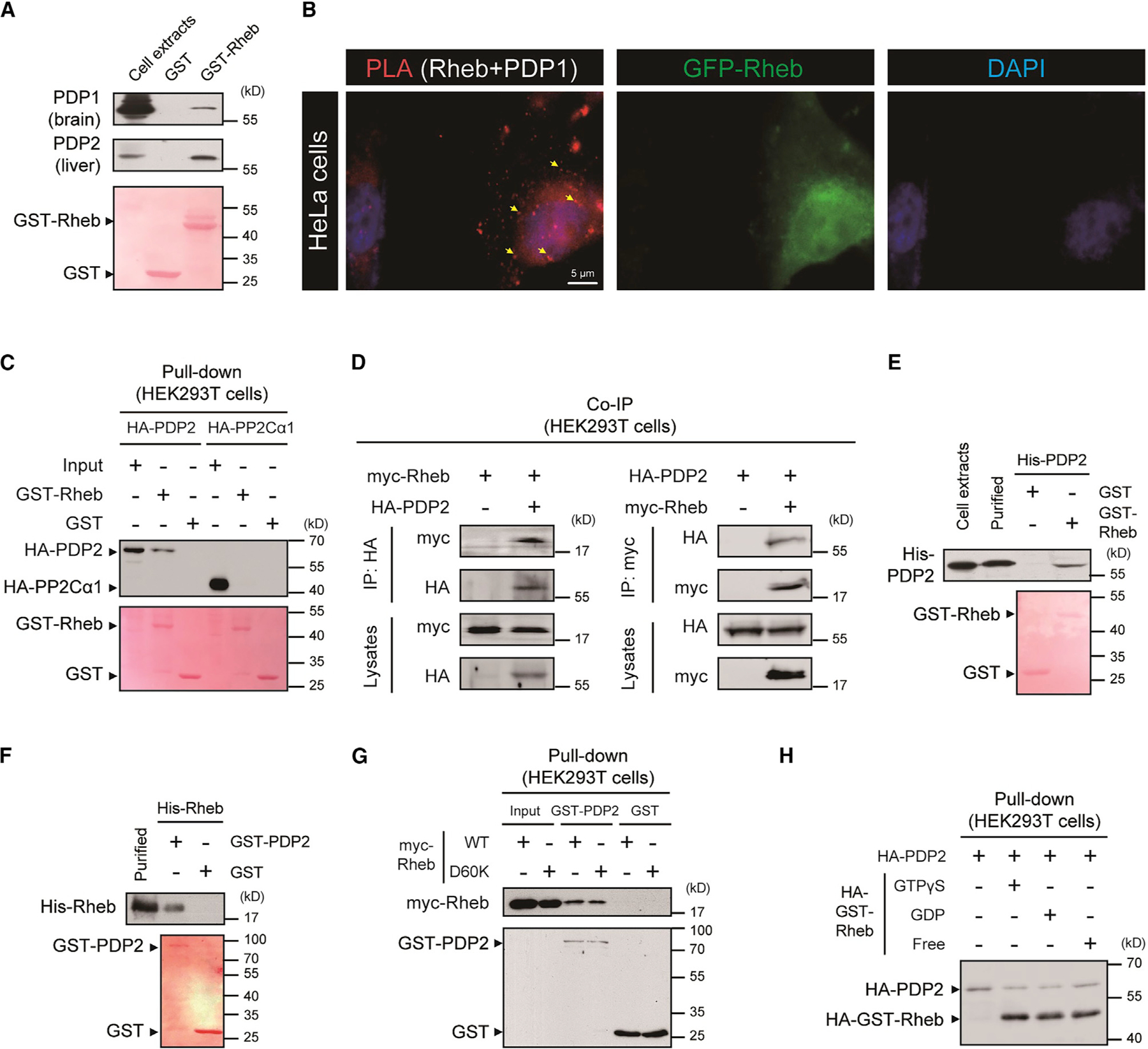
(A) GST pull-down demonstrating that purified GST-Rheb binds endogenous PDP1 in the brain (A, top panel) and PDP2 in the liver (A, middle panel).
(B) Images showing PLA signals (indicated by arrow heads) in HeLa cells transfected with GFP-Rheb using GFP and PDP1 antibody.
(C) GST pull-down demonstrating that GST-Rheb binds HA-tagged PDP2 transiently expressed in HEK293T cells. Note that GST-Rheb does not pull down HA-PP2Cα1 transiently expressed in HEK293T cells.
(D) Coimmunoprecipitation of myc-Rheb and HA-PDP2 in HEK293T cells demonstrating the interaction of Rheb and PDP2.
(E) In vitro reconstitution of direct binding between bacterially expressed purified GST-Rheb and recombinant His-PDP2 purified from HEK293T cells.
(F) In vitro reconstitution of direct binding between bacterially expressed recombinant His-Rheb and GST-PDP2.
(G) GST pull-down demonstrating that purified GST-PDP2 binds myc-tagged Rheb (wild type) and D60K mutant transiently expressed in HEK293T cells.
(H) GST pull-down demonstrating that purified HA-GST-Rheb loaded with GTPγS, GDP, or nucleotide-free (free) binds HA-tagged PDP2 transiently expressed in HEK293T cells. See also Figure S4.
Building on the observation that Rheb interacts with PDPs, we tested whether Rheb can enhance complex formation of PDPs with PDH. Using GST-Rheb, we pulled down PDP2 from lysates of WT liver and monitored PDH-E1α (Figure 5A, 2 leftmost lanes). The same pull-down experiment with liver extracts from Rhebf/f; Alb-cre mice revealed comparable pull-down of PDP2 compared with WT liver; however, the amount of PDH-E1α was less (Figures 5A and 5B). PDP2 and PDH-E1α protein expression were not altered in Rhebf/f;Alb-cre liver (Figure 5C). GST-Rheb pull-down from the liver of mTorf/f;Alb-cre mice revealed PDP2 and PDH-E1α amounts similar to WT (Figure 5A, rightmost lane). Findings are consistent with the notion that Rheb enhances PDP-PDH-E1α complex formation and that this assembly is important for the reduced phosphorylation of PDH in WT compared with Rheb deleted liver. Moreover, we examined the effect of Rheb on PDP1 activity using purified recombinant proteins. The incubation of Rheb with PDP1 increased its phosphatase activity toward a synthetic peptide substrate by 24% (Figure 5D). By contrast, Rheb2 protein had no effect on the phosphatase activity of PDP1 (Figure S4I). Results support the model that Rheb facilitates the assembly of PDPs-PDH complex and enhances the phosphatase activity of PDP toward PDH-E1α (Figure 5E).
Figure 5. Rheb binding to PDPs enhances its activity to PDH.
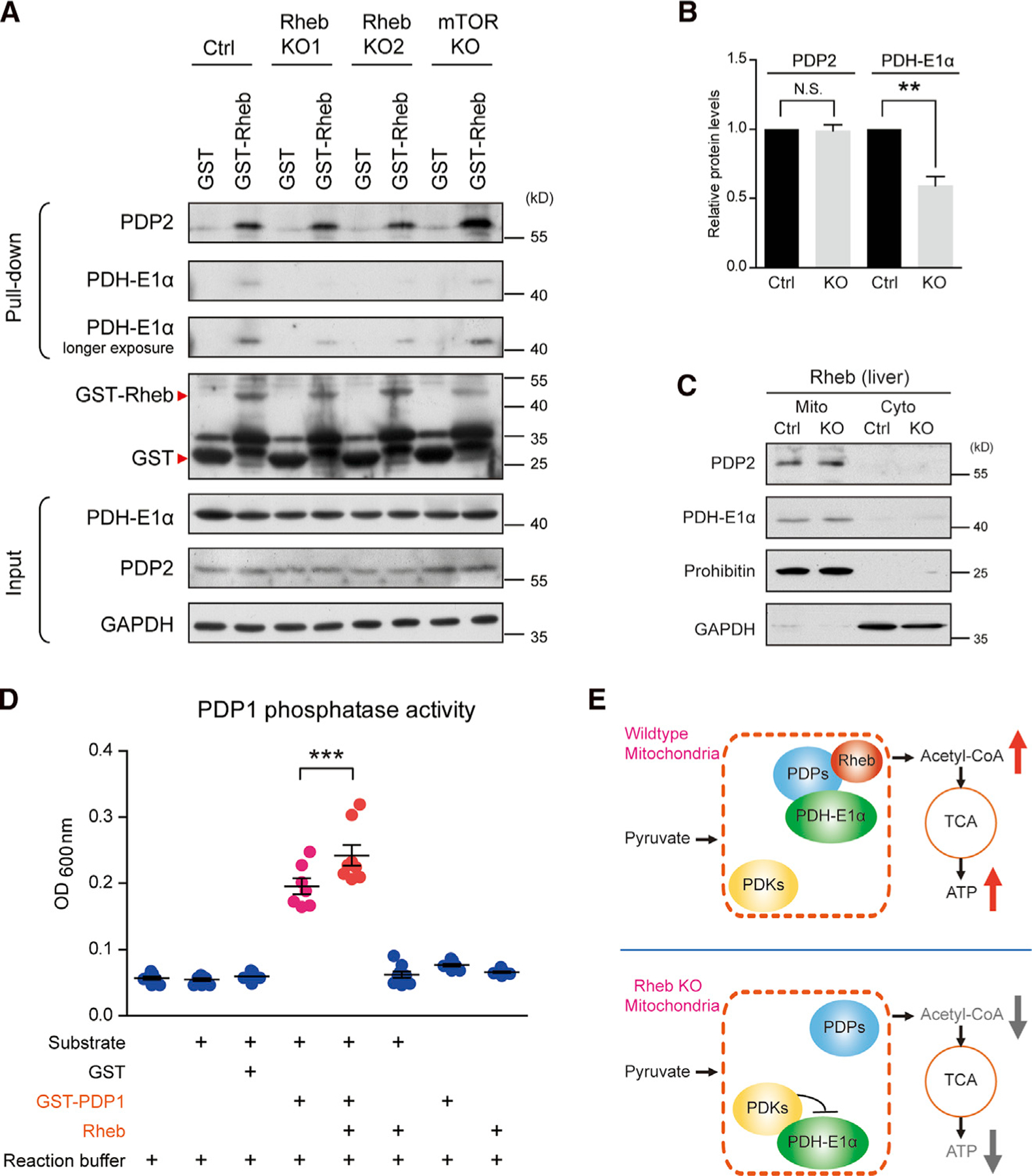
(A and B) GST pull-down (A) and quantification (B) comparing GST-Rheb pull-down of PDP2 and PDH-E1α from liver of Rheb KO versus mTor KO mice. Despite equivalent amounts of PDP2, less PDH-E1α is pulled down from Rheb KO. N = 3 independent experiments, PDP2, p = 0.8183; PDH-E1α, p = 0.0034.
(C) Subcellular fractionation showing comparable amounts of PDP2 and PDH-E1α in liver mitochondria of Ctrl and Rheb KO mice. Prohibition, the mitochondria marker; cyto, cytosol; and Mito, mitochondria.
(D) Incubation of Rheb protein with PDP1 increases the phosphatase activity of PDP1. N = 8 independent experiments.
(E) Diagram illustrating that Rheb activates PDH consequent to binding PDH phosphatases (PDP) and enhancing the association of PDH with PDP. All data represent mean ± SEM. Statistical analysis was performed by using two-tailed Student’s t test (B) or one-way ANOVA with Tukey post hoc test (D), and *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figure S4.
Coordinated induction of Rheb expression and PDH activity by neuronal activity
To elucidate the hypothesis that neuronal activity is linked to PDH activation through Rheb expression, we examined the coordinated expression of Rheb and PDH activation. Based on the raw data of single-cell sequencing analysis (GSE60361) (Lake et al., 2017; Zeisel et al., 2015), we found that the expressions of Rheb and PDH-E1α are enriched in mouse neurons, compared with glial cells (Figures 6A and 6B). In addition, their expression levels are highly correlated, but less so for Rheb and PDP1 (Figures 6C and S5A)., To examine the functional interactions among neuronal activity Rheb, and PDH activity, we cultured cortical neurons (DIV10) and treated them with bicuculline (50 µM), which blocks inhibition and increases neuronal activity (Ueno et al., 1997). Bicuculline induced a parallel increase of Rheb expression and decrease of PDH-E1α phosphorylation (Figures 6D and 6E). Potassium chloride (KCl) has long been used to stimulate neuronal activation in vitro, and this paradigm is useful for studying neuronal transcription under various stimulation patterns (Tyssowski et al., 2018). Under KCl stimulation (40 mM) for 30 to 60 min, cortical neuron cultures increased Rheb expression and reduced PDH-E1α phosphorylation (Figures 6D, 6E, and S5B). Similar observations on Rheb expression and PDH-E1α dephosphorylation were made with hippocampal slices treated with KCl (Figures S5C and S5D), and in addition, PDP-PDH interaction was enhanced (Figures S5E and S5F).
Figure 6. Coordinated induction of Rheb expression and PDH activation by neuronal activity.
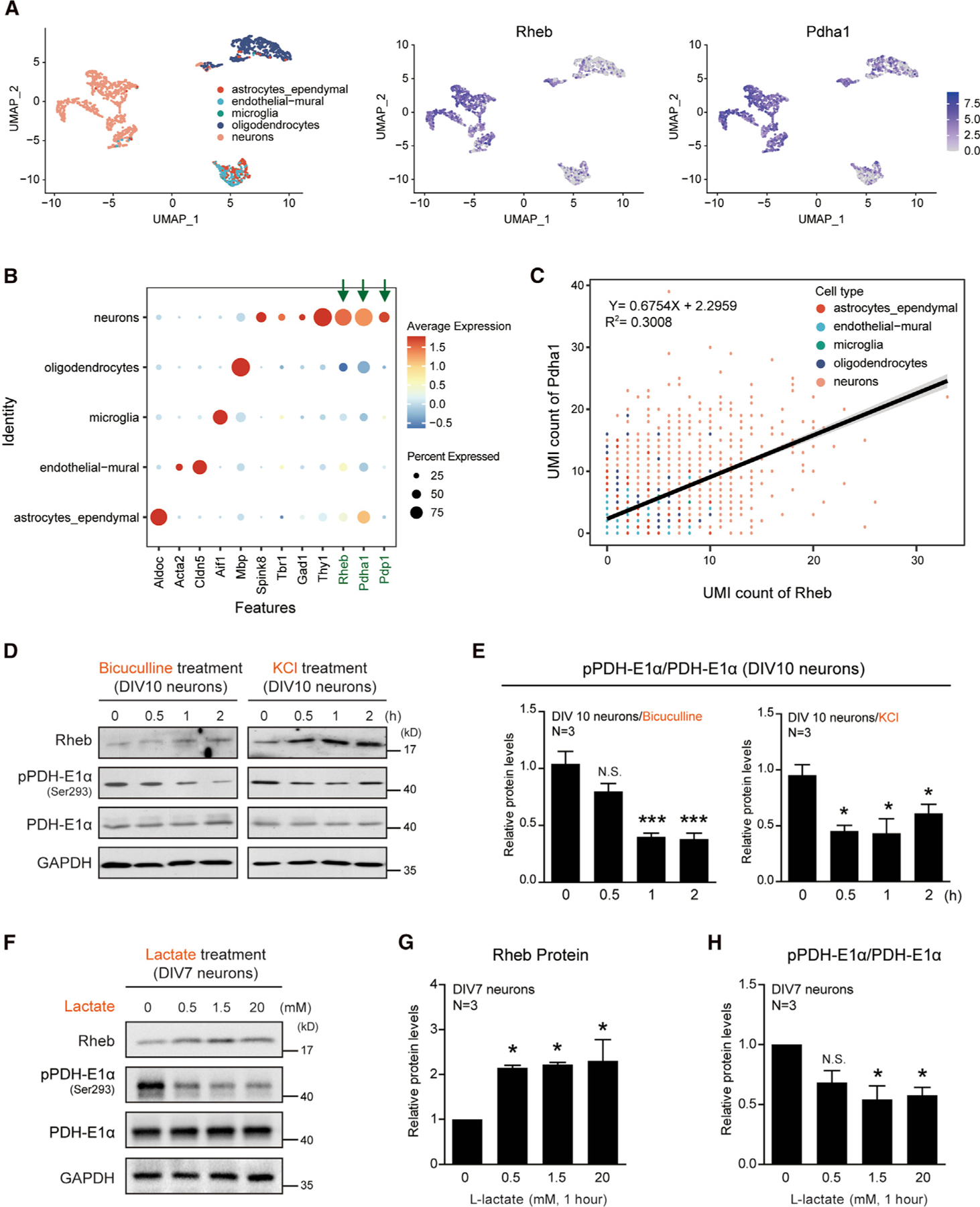
(A) scRNA-seq analysis showing that Rheb and Pdha1 coordinately expressed in brain neurons of mouse (data: GSE60361).
(B) Dot plot data showing that the expressions of Rheb and Pdha1 were enriched in neurons.
(C) Diagram showing that the expressions of Rheb and Pdha1 were highly correlated in mouse brain.
(D and E) Western blots (D) and quantification (E) demonstrating time-dependent increase of Rheb and decrease of PDH-E1α phosphorylation in primary cultured neurons (DIV 10) after bicuculline (50 µM) and KCl treatment (40 mM). N = 3 independent experiments.
(F–H) Western blots (F) and quantification (G and H) showing induction of Rheb expression and lower pPDH-E1α in DIV7 primary cortical neurons by L-lactate treatment (0.5, 1.5, and 20 mM). N = 3 independent experiments. All data represent mean ± SEM. Statistical analysis was performed by using one-way ANOVA with Dunnett (E, G, H) post hoc test, and *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figure S5.
In response to activity, neuronal uptake of lactate released by astrocytes could be increased (Bélanger et al., 2011) and lactate functions as both a signaling molecule [that induces expression of plasticity genes (Yang et al., 2014)] and an energy source for neurons. We applied L-lactate to hippocampal slices and found that L-lactate (0.5– to 20 mM) induced Rheb expression and PDH-E1α dephosphorylation (Figures 6F–6H and S6A–S6D). These studies establish that activity induces mitochondrial PDH activation through Rheb expression in neurons.
Rheb mediates neuronal-activity-regulated PDH activation and energy production
Because Rheb regulates neuronal PDH activation and Rheb and PDH activity were concomitantly induced by neuronal activity, we examined if the activity-induced PDH activation is dependent on Rheb expression in neurons. First, we applied KCl treatment to WT and Rheb KO hippocampal slices and assessed how Rheb KO affected KCl-induced PDH activation indicated by its phosphorylation status. Although KCl treatment lowered phosphorylated PDH-E1α in slices from Rhebf/f controls, KCl failed to produce similar effect on PDH-E1α in slices from Rhebf/f;CaMKII-cre mice (Figures 7A, 7B, and S6E), supporting the notion that Rheb is required for activity-dependent PDH activation.
Figure 7. Rheb mediates activity-regulated PDH activation and energy production.
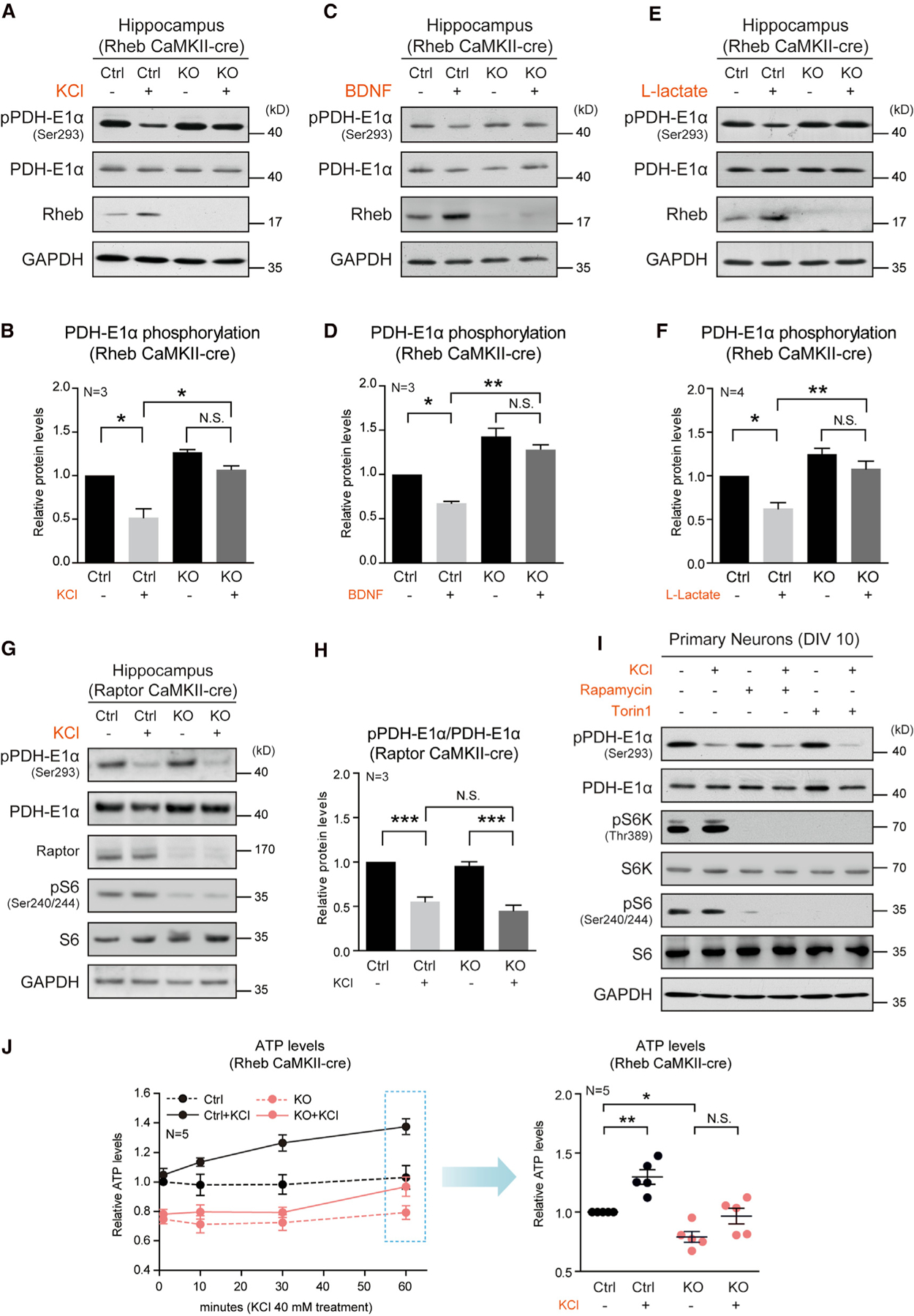
(A and B) Western blots (A) and quantification (B) demonstrating absence of change of PDH-E1α phosphorylation following KCl treatment (40 mM, 1 h) in acute hippocampal slices of Rheb CaMKII-cre KO mice. N = 3 pairs of mice, Ctrl versus Ctrl+KCl, p < 0.05; KO versus KO+KCl, N.S.; Ctrl+KCl versus KO+KCl, p < 0.05.
(C and D) Western blots (C) and quantification (D) demonstrating absence of change of PDH-E1α phosphorylation following BDNF treatment (50 ng/mL, 1 h) in acute hippocampal slices of Rheb CaMKII-cre KO mice. pPDH-E1α (Ser293), N = 3 pairs of mice, Ctrl versus Ctrl+BDNF, p < 0.05; KO versus KO+BDNF, p < 0.05; Ctrl+BDNF versus KO+BDNF, p < 0.01.
(E and F) Western blots (E) and quantification (F) showing the impairment of pPDH-E1α decrease by L-lactate treatment (20 mM, 2 h) in acute hippocampal slices of Rheb CaMKII-cre KO mice. pPDH-E1α (Ser293), Ctrl versus Ctrl+lactate, N = 4 pairs of mice, p < 0.05; KO versus KO+lactate, N = 4, N.S.; Ctrl+lactate versus KO+lactate, N = 4, p < 0.01.
(G and H) Western blots (G) and quantification (H) showing a decrease of PDH-E1α phosphorylation in acute hippocampal slices (Raptor CaMKII-cre KO and control mice) by KCl treatment (40 mM, 1 h). N = 3 pairs of mice, Ctrl versus Ctrl+KCl, p < 0.001; KO versus KO+KCl, p < 0.001; Ctrl+KCl versus KO+KCl, N.S.
(I) Western blots showing a decrease of PDH-E1α phosphorylation in cortical neurons (WT mice) by KCl treatment (40 mM) with or without rapamycin (100 nM) and Torin1 (200 nM).
(J) Impairment of KCl-induced (40 mM, 1 h) ATP production of acute hippocampal slices derived from Rheb CaMKII-cre KO mice N = 5 pairs of mice. Ctrl versus KO, p < 0.05; Ctrl versus Ctrl+KCl, p < 0.01; KO versus KO+KCl, N.S.. All data represent mean ± SEM. Statistical analysis was performed by using one-way ANOVA with Tukey (B, D, F, H, J) post hoc test, and *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figure S6.
Next, we examined the role of Rheb in PDH activation under BDNF-induced physiological activation of the brain (Korb et al., 2015; Messaoudi et al., 2002). Brain-derived neurotrophic factor (BDNF) is induced by neuronal activity and plays a role in multiple forms of plasticity. We found that BDNF treatment induces Rheb expression and reduces phosphorylated PDH, and this effect is impaired by Rheb deletion (Figures 7C and 7D). Finally, we examined whether Rheb is required for L-lactate-induced PDH activation and found that L-lactate failed to induce PDH dephosphorylation in slices from Rhebf/f;CaMKII-cre mice (Figures 7E and 7F). These results support the conclusion that activity-induced PDH activation is mediated by Rheb in neurons under sustained stimulation, with direct impact on activity-induced ATP production.
In neuronal-activity-induced PDH activation, Rheb’s role is clearly separable from its role in activating mTORC1 because activity-regulated PDH phosphorylation status is intact under conditions where mTORC1 activation is blunted by Raptor KO (Figures 7G and 7H) or pharmacological inhibition by rapamycin or Torin1 (Figure 7I).
To validate the impact of Rheb deletion on neuronal-activity-induced ATP production, we used KCl to activate ATP production in hippocampal slices from WT and Rheb KO mice. WT slices showed significant increases in ATP levels at 30 and 60 min after the application of KCl (Figure 7J, left panel)—the time course correlating with the induction of Rheb protein expression and change to PDH phosphorylation status (Figure S5D). The extent of ATP increase is similar to what was reported (Alavian et al., 2011). At every time point tested, we found ATP levels in Rheb KO slices were lower than in WT slices. Particularly, in Rheb KO slices, KCl-induced ATP elevation was blunted, particularly at 60 min (Figure 7J, right panel). These results link activity-induced Rheb expression/PDH activation to neuronal ATP production.
DISCUSSION
Rheb and activity-regulated neuroenergetics
This study identifies Rheb as a major regulator of mitochondrial energy production by activating PDH. Activity/energy demand–induced expression of Rheb enhances lactate/pyruvate metabolism in activated neurons. Disruption of Rheb-PDH axis by Rheb deletion significantly reduced cellular acetyl-CoA and ATP levels. Rheb and PDH activities are coordinately regulated by neuronal activation. We found that neuronal-activity-induced ATP production was dependent on induced Rheb expression and PDH activation.
Dynamic regulation of neuroenergetics involves multiple mechanisms. Previous work shows that Rheb could activate AMPK activity (Lacher et al., 2010), and AMPK enhances PDH activity (Cai et al., 2020). However, AMPK is not involved in Rheb-regulated PDH activation, because Rheb KO does not reduce AMPK activity in brain tissues (Figures S7A and S7B). In the short term, AMPK might be responsible for neuronal-activity-induced energy boost by increasing the localization of glucose transporter GLUT4 at presynaptic terminals and glycolysis (Ashrafi et al., 2017). For sustained neuronal activation, neurons may also depend on glial cells that release lactate to fuel neuronal mitochondrial oxidative phosphorylation, as the astrocyte-neuron lactate shuttle model indicates (Suzuki et al., 2011; Chuquet et al., 2010; Kasischke et al., 2004; Pellerin and Magistretti, 1994). In this case, Rheb could play a role in enhancing neuronal mitochondrial metabolism of increased energy substrates—lactate and pyruvate. In neurons, Rheb is prominently enriched and activity-and lactate-induced Rheb expression increases mitochondrial function on a time scale that parallels Rheb expression. We also found that neuronal deletion of Rheb blunted activity-induced ATP production in brain slices.
Neuronal ATP production is critical for sustained firing (Lutas et al., 2014) and ATP-and mTORC1-regulated synaptic function (Bockaert and Marin, 2015). The role of Rheb in regulation of neuroenergetics is consistent with its role in the formation of synapses and spatial memory (Shahani et al., 2017; Yasuda et al., 2014). Neuronal acetyl-CoA production is directly relevant to neurodegeneration through ATP production and acetylation of regulatory proteins such as histones (Currais et al., 2019). We found that neuronal deletion of Rheb causes neurodegeneration, which is consistent with the finding that, in human brains, Rheb protein level and PDH activity in the frontal cortex of patients with Alzheimer’s disease were significantly lower than those in non-Alzheimer’s controls (Shahani et al., 2014; Sorbi et al., 1983). All this effect may be related to reduced acetyl-CoA production (Currais et al., 2019), but not mTORC1 inactivation, because inhibiting mTORC1 activity often produces neuroprotective effect in neurodegenerative disease models (Zheng et al., 2016; Bové et al., 2011). PDH deficiency is linked to neuro-developmental and neurodegenerative disorders and touted as a therapeutic target (Jakkamsetti et al., 2019; Naia et al., 2017; Stacpoole, 2012; Sorbi et al., 1983). Taken together, our work provides a critical missing link between neuronal activity and mitochondrial metabolism and supports the notion that disruption of activity-regulated Rheb-PDH function in mitochondrial metabolism could have serious clinical implications. Therefore, Rheb-dependent mitochondrial function in disease deserves future attention.
The interplay between Rheb, mTORC1, and mitochondrial activity
Rheb-regulated PDH activation seems to be a general mechanism cells use to control mitochondrial acetyl-CoA and ATP production. We show that PDH activities in neural cells and hepatocytes were similarly affected by Rheb KO. The role for Rheb to activate PDH is not dependent on Rheb’s role in activating mTORC1 via TSC. We found that TSC knockdown did not alter phosphorylated PDH. Rheb WT transgene (expressed at a level that does not activate mTORC1) and Rheb S16H transgene (resistant to TSC inhibition and activating mTORC1) activate PDH to similar extent. Deletion of Raptor and mTOR, respectively, did not affect PDH phosphorylation like Rheb deletion does. In addition, pharmacological inhibition of mTORC1 did not affect the effect of Rheb on PDH phosphorylation/activity. The efficacy of Rheb in activating PDH may depend on the amount of Rheb protein trafficking to mitochondria. This notion is consistent with the finding that stimulating mitochondrial metabolism promotes Rheb trafficking to mitochondria. The comparison of hepatic Rheb and mTOR deletion reveals that Rheb deletion had a more profound effect on ATP production, highlighting the significance of Rheb-regulated PDH activation and acetyl-CoA synthesis in mitochondrial energy metabolism.
Rheb-regulated PDH activity involves Rheb’s stabilizing PDP-PDH interactions and enhancing PDP phosphatase activity. A previous study suggests that PDP-PDH complex formation is regulated by PDP acetylation (Fan et al., 2014). When PDP is acetylated (e.g., K202), PDP will dissociate from PDH, leading to PDH phosphorylation and inactivation by PDKs. Because aa 168–196 of PDP is required for Rheb-PDP interaction, we speculate that Rheb binding with PDP might affect PDP’s acetyltransferase (e.g., ACAT1) accessing PDP. Moreover, we show that Rheb enhances the phosphatase activity of PDP toward PDH. Thus, Rheb has a dual role in PDP regulation; one is to facilitate the assembly of PDP-PDH complex, and the other to enhance PDP phosphatase activity.
Earlier work shows that Rheb responds to cellular energetic status and promotes mitophagy (Melser et al., 2013). This role for Rheb might be a result of increased mitochondrial activity as we demonstrate in this study. Increased mitochondrial activity renders mitochondria vulnerable for damage and thus more mitophagy for maintaining the content of healthy, productive mitochondria. Rheb’s role in promoting mitophagy involves its binding with nix at the outer membrane of mitochondria, independent of mTOR. In this model, mitophagy regulated by outer membrane-bound Rheb might be aligned with mitochondrial activity-regulated matrix-localized Rheb, for optimizing mitochondrial ATP production. These observations highlight Rheb as a multi-faced regulator of mitochondrial function.
Rheb is known to bind mTOR, also an ATP sensor (Dennis et al., 2001), and activate mTORC1 activity (Yang et al., 2017; Zou et al., 2011). Activation of mTORC1 is dependent on cellular ATP levels (Dennis et al., 2001). Our study provides new insight to the coordinated regulation of acetyl-CoA/ATP production and mTORC1 activation by Rheb, consistent with the recent discovery that acetyl-CoA could activate mTORC1 (Son et al., 2019). In parallel, Rheb-activated mTORC1 could also facilitate mitochondrial energy production, through regulating mitochondrial gene expressions (Cunningham et al., 2007) and mitochondrial dynamics (Yuan et al., 2021; Morita et al., 2017). However, mTORC1-regulated mitochondrial dynamics is mediated by acetyl-CoA–dependent acetylation of multiple GTPases including MFN1/2, OPA1, and DNM1L that collectively control mitochondrial fission and fusion (Samant et al., 2014). Therefore, the effect of mTORC1 activation on mitochondrial dynamics might be consequential to Rheb-regulated acetyl-CoA production and overall mitochondrial activity. These findings support a model where Rheb-regulated ATP production through mitochondrial activity is coordinated with mTORC1 activation and mitochondrial dynamics (Figure S7C).
In conclusion, our work identifies a mitochondrial function of activity-regulated gene Rheb, which is independent of its well-known function to activate mTORC1. Rheb positively regulates neuronal mitochondrial energy production by activating PDH. Findings indicate that Rheb functions in a dual role as both an activator of mTORC1 and as an activator of mitochondrial energy production and suggest that neurons utilize this shared mechanism to balance energy consumption and production.
STAR☆METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bo Xiao (xiaob@sustech.edu.cn).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse
All mouse work was done in accordance with the Animal Care and Use Committee guidelines of West China Hospital, Sichuan University and Johns Hopkins University School of Medicine. The generation and characterization of floxed Rheb and Rosa26-Rheb(S16H) mice has been described previously (Zou et al., 2011). To delete or overexpress Rheb in neural cells of the brain, floxed Rheb, Rosa26-Rheb(wildtype) and Rosa26-Rheb(S16H) mice were crossed respectively with mice harboring the Cre recombinase under control of the Nestin promoter. To delete Rheb and Raptor in excitatory neurons of the brain, floxed Rheb and Raptor mice were crossed respectively with the CaMKII-cre mice (Casanova et al., 2001). To delete Rheb and mTOR in hepatocytes of the liver, floxed Rheb and mTOR mice were crossed respectively with Albumin-cre mice (Postic et al., 1999).
All the mice were kept in SPF conditions with standard housing conditions in a temperature-controlled environment with 12-hour light/dark cycles and received normal diet and water ad libitum. Littermates were used for comparison, and mice of both sexes were used for all experiments described, because no obvious differences were found between the two sexes.
METHOD DETAILS
Mitochondrial isolation and subfractionation
Mitochondria and cytosol fractions from mouse liver or glutamine-treated HeLa cells or hippocampal slices were isolated using a mitochondria fractionation kit from Beyotime Biotechnology (China) according to manufacturer’s instructions. Mitochondrial proteins and cytosolic proteins were used for Western blotting. Mitochondrial subfractionation assay was performed as previously described (Li et al., 2016; She et al., 2011). Isolated mitochondria were resuspended in 10 µM KH2PO4 (pH 7.4) for 20 min on ice. An equal volume of iso-osmotic solution (32% sucrose, 30% glycerol, 10 mM MgCl2) was added and spun at 10,000 g and 4°C for 10 min. The supernatant was centrifuged at 15,000 g and 4°C for 1 h; the pellet and supernatant contained outer membrane (OM) and intermembrane space (IMS) proteins. Then, the pellet was resuspended in 10 µM KH2PO4 (pH 7.4) for 20 min on ice, and iso-osmotic solution was added, followed by centrifugation at 15,000 g and 4°C for 1 h; the pellet and supernatant contained inner membrane (IM) and matrix (Mx) proteins.
Immunofluorescence assay
To examine the colocalization of Rheb with mitochondrial marker Mitotracker, HeLa cells transfected with myc-Rheb or myc-Rheb2 for ~10 hours and stained with rabbit monoclonal anti-Myc antibody using standard immunofluorescence combined with Mitotracker, as follows. Cells were fixed with 3% glyoxal (Richter et al., 2018) and then quenched in 100 mM NH4Cl. Next, cell permeabilization and blocking were performed in PBS containing 0.5% Triton X-100 and 2.5% BSA. Cells were then incubated with anti-Myc (dilution, 1:500) and fluorescence secondary antibody-Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen). The coverslips were mounted onto glass slides with antifade reagent and DAPI to stain nuclei. Images were acquired using a confocal laser-scanning microscope (ZEISS 880+ Airyscan and Nikon N-SIM).
Metabolite assays
Extracts from mouse tissues or cell cultures were used for the assessment of metabolites, including ATP, pyruvate, acetyl-CoA, lactate, ADP, AMP, creatine and phospho-creatine contents using commercial kits. The ATP measurement was based on Luciferase assay. Pyruvate was measured based on pyruvate oxidase catalyzed reaction to generate a colorimetric product at OD=570 nm in the microplate reader. To assay acetyl-CoA content in tissues and cell cultures, acetyl-CoA in supernatant was converted to CoA and incubated with PicoProbe to generate fluorescence at Ex/Em=535/589 nm. Lactate level was indicated by the fluorescence intensity (at Ex/Em=535/589 nm) emitted through the enzymatic reaction of lactate catalyzed by an enzyme mix. To assay ADP level, ADP was converted to ATP and pyruvate, and the generated pyruvate was quantified by fluorometric method. To assess AMP level, AMP was converted to pyruvate to generate a colored product with a strong absorbance at 570 nm. To measure creatine in the extracts of tissues, creatine was enzymatically converted to a product that was able to convert a colorless probe to highly fluorescent one (Ex/Em=538/587 nm). Phospho-Creatine was measured by an enzyme-linked immunosorbent assay for metabolite detection. All metabolites were measured following manufacturer’s instructions.
Seahorse assay
Bioenergetic analysis of mitochondrial preparations was performed on Seahorse XF96 Extracellular Flux Analyzer. Synaptosomes were prepared as described (Choi et al., 2009) from the forebrains of 3-week Rheb KO and control mice. Forebrains were homogenized with Dounce glass homogenizer and centrifuged at 1,000 g for 10 minutes. The supernatant was then laid on top of discontinuous Percoll gradient made in sucrose medium, and centrifuged at 32,500 g for 10 minutes to separate the synaptosomes. The synaptosomal band was then isolated and diluted into ‘Ionic Medium’ (20 mM HEPES, 10 mM D-glucose, 1.2 mM Na2HPO4, 1 mM MgCl2, 5 mM NaHCO3, 5 mM KCl, 140 mM NaCl, pH 7.4). Diluted synaptosomes were centrifuged at 15,000 g for 15 minutes to remove the Percoll, and resuspended in the ionic medium. The total protein level of synaptosomes was measured with Lowry Protein Assay. Synaptosomes were then aliquoted into a polyehtyleneimine-coated Seahorse XF96 Cell Culture Microplate (10 µg protein each well) and the microplate was centrifuged at 3,400 g for 1 hour to allow attachment of synaptosomal aggregates. Ionic medium was replaced by pre-warmed ‘incubation medium’ (3.5 mM KCl, 120 mM NaCl, 1.3 mM CaCl2, 0.4 mM KH2PO4, 1.2 mM Na2SO4, 2 mM MgSO4, 15 mM D-glucose, 10 mM Pyruvate, 4 mg/mL bovine serum albumin, 37°C), and plates were used immediately for assays. Oligomycin (4 µg/mL) and FCCP (4 µM) were added to incubation medium and injected automatically to each well at pre-designated times. Oxygen consumption rate (OCR) were then determined and reported in pmoles/minute.
Western blotting
Extracts from mouse tissues or cultured cells were sonicated in lysis buffer (2% SDS with proteinase inhibitors and phosphatase inhibitors). The protein concentration of each extract was measured using the BCA Protein Assay kit (Thermo Scientific Pierce). Equal amounts of proteins from each extract were loaded into SDS-PAGE gel and blotted with respect primary (dilution, 1:500~1:1000) and secondary antibodies, according to standard procedures. Films were scanned, and optical densities were quantified using ImageJ (https://imagej.nih.gov/ij/).
Measurement of mRNA levels by real-time PCR
Total RNA was extracted from tissues using TRizol reagent (Invitrogen). RNA was subjected to reverse transcription with reverse transcriptase as manufacturer’s instructions (Thermo). Quantitative real-time PCR was performed using the Bio-Rad iQ5 system, and the relative gene expression was normalized to internal control as Beta actin. Primer sequences for SYBR Green probes of target genes are as list in the key resources table under the Oligonucleotide section.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal Rheb antibody | Lab made | N/A |
| Rabbit monoclonal pS6K (Thr389) antibody | Cell Signaling Technology | Cat#9234 |
| Rabbit monoclonal S6K antibody | Cell Signaling Technology | Cat#2708 |
| Rabbit polyclonal pS6 (Ser240/244) antibody | Cell Signaling Technology | Cat#2215 |
| Rabbit monoclonal S6 antibody | Cell Signaling Technology | Cat#2217 |
| Rabbit polyclonal p4EBP1 (Thr37/46) antibody | Cell Signaling Technology | Cat#9459 |
| Rabbit polyclonal 4EBP1 antibody | Cell Signaling Technology | Cat#9452 |
| Mouse monoclonal Beta actin antibody | Boster Biological Technology | Cat#BM0627 |
| Rabbit polyclonal mTOR antibody | Upstate | Cat#07–231 |
| Rabbit polyclonal pPDH-E1α (Ser293) antibody | Abcam | Cat#ab92696 |
| Rabbit polyclonal pPDH-E1α (Ser300) antibody | Millipore | Cat#AP1064 |
| Rabbit polyclonal pPDH-E1α (Ser232) antibody | Millipore | Cat#AP1063 |
| Mouse monoclonal PDH-E1α antibody | Abcam | Cat#ab110334 |
| Rabbit monoclonal Raptor antibody | Cell Signaling Technology | Cat#2280 |
| Rabbit polyclonal PDP1 antibody | Abcam | Cat#ab59706 |
| Rabbit polyclonal PDP2 antibody | Proteintech | Cat#13404–1-AP |
| Mouse monoclonal c-Myc antibody | Merck | Cat#OP10L |
| Rabbit monoclonal c-Myc antibody | Cell Signaling Technology | Cat#2278 |
| Rabbit His-Tag antibody | Cell Signaling Technology | Cat#2365 |
| Mouse HA-Tag antibody | Cell Signaling Technology | Cat#2367 |
| Rabbit monoclonal MnSOD antibody | Epitomics | Cat#2299–1 |
| Mouse monoclonal GAPDH antibody | Millipore | Cat#MAB374 |
| Mouse monoclonal Beta tubulin antibody | Millipore | Cat#05–661 |
| Mouse monoclonal ARC antibody | Lab made | N/A |
| Rabbit polyclonal Tom20 antibody | Santa Cruz Biotechnology | Cat#sc-136211 |
| Rabbit polyclonal Tom40 antibody | Proteintech | Cat#18409–1-AP |
| Rabbit polyclonal pAKT (Ser473) antibody | Cell Signaling Technology | Cat#9271 |
| Rabbit polyclonal AKT antibody | Cell Signaling Technology | Cat#9272 |
| Rabbit monoclonal TSC2 antibody | Cell Signaling Technology | Cat#3990 |
| Rabbit monoclonal pAMPKα (Thr172) antibody | Cell Signaling Technology | Cat#2535 |
| Rabbit polyclonal AMPKα antibody | Cell Signaling Technology | Cat#2532 |
|
Chemicals, peptides, and recombinant proteins | ||
| Rapamycin | Sigma-Aldrich | Cat#PHZ1233 |
| Torin1 | Tocris Bioscience | Cat#4247 |
| KCl | Sigma-Aldrich | Cat#P5405 |
| Bicuculline | Tocris Bioscience | Cat#0130 |
| L-Lactate | Sigma-Aldrich | Cat#L7022 |
| Recombinant Human/Murine/Rat BDNF | PeproTech | Cat#450–02 |
| GTPγS | Millipore | Cat#20–176 |
| GDP | Millipore | Cat#20–177 |
| ATP Assay Kit | Biovision, Inc, CA, USA | Cat#791–100 |
| Acetyl-CoA Assay Kit | Biovision, Inc, CA, USA | Cat#K317–100 |
| Mitochondria Isolation Kit | Beyotime, China | Cat#C3601/3606 |
| PDH Enzyme activity kit | Abcam | Cat#ab109902 |
| Serine/Threonine Phosphatase Assay kit | Promega | Cat#V2460 |
| Duolink In Situ Red Starter Kit Mouse/Rabbit | Sigma-Aldrich | Cat#DUO92101 |
| Pyruvate Assay Kit | Biovision, Inc, CA, USA | Cat#K609–100 |
| Lactate Assay Kit | Biovision, Inc, CA, USA | Cat#K607–100 |
| ADP Assay Kit | Abcam | Cat#ab83359 |
| AMP Assay Kit | Abcam | Cat#ab273275 |
| Creatine Assay Kit | Abcam | Cat# ab65339 |
| ELISA Kit for phospho-creatine | Cloud Clone | Cat#CEV808Ge |
|
Experimental models: organisms/strains | ||
| Floxed Rheb | Zou et al., 2011 | N/A |
| Rosa26-Rheb(S16H) | Zou et al., 2011 | N/A |
| Rosa26-Rheb(Wildtype) | This paper | N/A |
| Floxed mTOR | The Jackson Laboratory | JAX: 011009 |
| Floxed Raptor | The Jackson Laboratory | JAX: 013191 |
| Albumin-cre | The Jackson Laboratory | JAX: 003574 |
| Nestin-cre | Tronche, et al., 1999 | N/A |
| CaMKII-cre | Casanova, et al., 2001 | N/A |
|
Oligonucleotides | ||
| Primer: Pgc1α real-time PCR Forward: CTCCCTGTGGATGAAGACGG |
This paper | N/A |
| Primer: Pgc1α real-time PCR primer Reverse: ATCCTGTGATTGTGATTTGC |
This paper | N/A |
| Primer: Ndufs8 real-time PCR primer Forward: TGGCGGCAACGTACAAGTAT |
This paper | N/A |
| Primer: Ndufs8 real-time PCR primer Reverse: CCTCGGATGAGTTCTGTCCA |
This paper | N/A |
| Primer: Cox5 real-time PCR primer Forward: GGGTCACACGAGACAGATGA |
This paper | N/A |
| Primer: Cox5 real-time PCR primer Reverse: GGAACCAGATCATAGCCAACA |
This paper | N/A |
| Primer: Atp5g1 real-time PCR primer Forward: AGTTGGTGTGGCTGGATCA |
This paper | N/A |
| Primer: Atp5g1 real-time PCR primer Reverse: GCTGCTTGAGAGATGGGTTC |
This paper | N/A |
| Primer: Rheb real-time PCR primer Forward: AAGTCCCGGAAGATCGCCA |
This paper | N/A |
| Primer: Rheb real-time PCR primer Reverse: GGTTGGATCGTAGGAATCAACAA |
This paper | N/A |
| Primer: Arc real-time PCR primer Forward: AAGTGCCGAGCTGAGATGC |
This paper | N/A |
| Primer: Arc real-time PCR primer Reverse: CGACCTGTGCAACCCTTTC |
This paper | N/A |
| siRNA targeting sequence: TSC2 siRNA 1# sense: GCACCUCUACAGGAACUUUTT |
This paper | N/A |
| siRNA targeting sequence: TSC2 siRNA 1# anti-sense: AAAGUUCCUGUAGAGGUGCGG |
This paper | N/A |
| siRNA targeting sequence: TSC2 siRNA 2# sense: GCCUCACAGGUGCAUCAUATT |
This paper | N/A |
| siRNA targeting sequence: TSC2 siRNA 2# anti-sense: UAUGAUGCACCUGUGAGGCCA |
This paper | N/A |
| siRNA targeting sequence: Tom20 siRNA sense: GGUCUUACAGCAAACUCUUTT |
This paper | N/A |
| siRNA targeting sequence: Tom20 siRNA anti-sense: AAGAGUUUGCUGUAAGACCTG |
This paper | N/A |
|
Software and algorithms | ||
| ImageJ | Schneider, et al., 2012 | imagej.nih.gov/ij |
| GraphPad Prism ver 6.0 | GraphPad software Inc | www.graphpad.com |
Enzymatic assay of PDH activity
The PDH enzyme activity microplate assay kit (Abcam; #ab109902) was used to measure PDH activity of liver tissues and purified mitochondria from liver. Liver tissues were homogenized to determine protein concentrations by BCA Protein Assay kit (Thermo Scientific Pierce) and supernatants were used for PDH assay using manufacturer’s protocol.
Enzymatic assay of PDP1 phosphatase activity
PDP1 phosphatase activity was determined by measuring the release of phosphate from a phosphorylated polypeptide [Ac-Tyr-His-Gly-His-Ser(P)-Met-Ser-Asp-Pro-Gly-Val-Ser(P)-Tyr-Arg-NH2] (Mullinax et al., 1985) by the Serine/Threonine Phosphatase Assay kit (V2460) from Promega (Madison, WI, USA) (Huang et al., 2003; Caruso et al., 2001). The polypeptide substrate was synthesized according to the amino acid sequence surrounding phosphorylation sites 1 and 2 of the PDH-E1α component. In brief, recombinant GST-PDP1 (0.4 mg/mL) was incubated with substrate polypeptide in the reaction mixture for 30 min at 30 °C, with or without recombinant Rheb proteins (0.4 mg/mL). The reaction was stopped by adding the Molybdate Dye and measured by spectrophotometric quantitation of released phosphate at 600 nm following manufacturer’s instructions and previous reports (Huang et al., 2003; Caruso et al., 2001).
GST pull-down and in vitro reconstitution of protein-protein interaction
To examine the protein interactions of Rheb and PDPs, GST pull-down assay was performed. Briefly, GST fusion proteins were expressed in BL21 (DE3) E. coli strains and soluble fractions of the recombinant proteins were purified as follows. Bacteria were harvested and lysed in PBS (with 1% Triton X-100, and 2 mM phenylmethylsulfonyl fluoride (PMSF)). Lysates were incubated with glutathione–sepharose beads (GE Healthcare), and bound proteins were eluted with 10 mM glutathione and dialyzed against PBS at 4°C. Protein concentrations were determined by BCA Protein Assay kit.
Expression constructs were transiently transfected into HEK293T cells to express various tagged proteins. Cells were lysed 48 hours post-transfection with PBS (1% Triton X-100 with proteinase inhibitors (Merck) and phosphatase inhibitors (Biovision)). GST pull-down assays were performed by mixing 300 µL cell lysates (0.5–1 µg/µL) with beads charged with 300–500 µL GST fusion proteins (0.5–2 µg/µL) at 4°C overnight. Beads were then washed with PBS + 1% Triton X-100 and additionally with PBS three times. Bound proteins were eluted with 100 µL 4×SDS loading buffer and detected by SDS-PAGE and western blotting. GST pull-down assays of Rheb and PDPs from tissue lysates were performed by sonicating mouse brain cortex or liver with PBS (1% Triton X-100 with proteinase inhibitors (Merck)) and processed as above.
For Rheb nucleotide-loading experiment, recombinant Rheb protein doubly tagged with HA and GST was expressed in HEK293T cells by transient transfection. HEK293T cells were then harvested in lysis buffer (150 mM NaCl, 50 mM HEPES pH 7.4, 1% Triton X-100, 5 mM MgCl2, protease inhibitors). Cleared lysates were incubated with immobilized glutathione for 5 hours at 4°C. Beads were washed three times with lysis buffer and loaded with GTPγS by incubating with 10 mM EDTA and 0.1 mM GTPγS or 1 mM GDP at 30 °C for 30 min. MgCl2 was added to a final concentration of 20 mM to stop the reaction. GTP/GDP loaded HA-GST-Rheb, as well as nucleotide-free HA-GST-Rheb, was used for GST pull-down experiments.
For in vitro reconstitution of Rheb binding to PDP2, GST-Rheb, His-Rheb and GST-PDP2 were prepared as above. HEK293T cells were transfected with HA-PDP2-His plasmid for 48 hours, and cells were harvested and transferred to a new centrifuge tube with His Trap HP (GE Healthcare). Incubation was performed overnight at 4 °C, and then centrifuged at 4,000 rpm for 5 min to collect precipitates. Precipitates were washed with 4 °C wash buffer three times (PBS with 1% Triton X-100, 15 mM imidazole and proteinase inhibitors), and eluted by elution buffer (PBS with 250 mM imidazole, pH 7.4). Recombinant protein preparations were dialyzed with PBS. In vitro reconstitution assay was performed by mixing 500 µL His recombinant proteins (0.5 µg/µL) with beads charged with 300–500 µL GST fusion proteins (0.5–2 µg/µL) at 4°C overnight followed by washing with PBS + 1% Triton X-100 and then PBS three times. Bound proteins were eluted with 100 µL 4×SDS loading buffer and detected by SDS-PAGE and Western blots.
Proximity ligation assay (PLA)
The PLA was performed following the DUOLINK PLA Fluorescence Protocol. HeLa cells were plated on coverslips and transfected with GFP-Rheb using Lipofectamine 2000 (Invitrogen). After transfection for twenty-four hours, cells were washed with PBS for three times, then fixed with 4% paraformaldehyde (PFA) for 30 minutes at room temperature. Then fixed cells were washed with PBS, and added with one drop of DUOLINK blocking solution, and incubated in a wet chamber at 37°C for 1 hour. Next, cells were incubated with primary antibodies (GFP, sc9996 from Santa Cruz, 1:500; PDP1, A6332 from ABclonal, 1:100) in the DUOLINK Antibody Diluent at 4°C overnight. The next day, cells were washed twice with DUOLINK wash buffer A and incubated with PLA probes (1:5 in DUOLINK Antibody Diluent, 1 hour), the Ligase in Ligation buffer (1:40, 30 minutes) and the polymerase in Amplification buffer (1:80, 100 minutes) at 37°C. Finally, cells were washed with DUOLINK wash buffer B twice and wash buffer B. The coverslips were mounted onto glass slides with DAPI to stain nuclei. Images were acquired using Olympus BX63 microscope.
Co-immunoprecipitation
HEK293T cells were transfected with myc-tagged Rheb and HA-tagged PDP2. After ~18 hours cells were rinsed with ice-cold PBS and harvested in lysis buffer (1% Triton with protease inhibitors). Cell lysates were cleared by centrifugation at 13,000 r.p.m. at 4 °C for 10 min. Then anti-myc or anti-HA antibodies (2 µg for each Co-IP reaction) were added to lysates for incubation with rotation for overnight at 4 °C. 50 µL of washed affinity beads (50%) were added to lysates for 5 hours at 4 °C. Finally, immunoprecipitated proteins were eluted from beads by the addition of 50 µL SDS-PAGE sample buffer, boiled for 5 min, then resolved by SDS–PAGE and analyzed by Western blotting.
Induction of Rheb and PDH by neuronal activation and metabolites
To induce neuronal activity in cultured neurons and acute hippocampal slices KCl, bicuculline or BDNF were added to Neurobasal medium at a final concentration of KCl (40 mM), bicuculline (50 µM), or BDNF (50 ng/mL) for 30 min to 2 hours. To assess the effects of metabolites on Rheb expression and PDH activity, L-lactate was added to Neurobasal medium to a final concentration of 0.5 mM, 1.5 mM and 20 mM.
Gene knockdown
For small interference (si) RNA experiments, cells were transfected using Lipofectamine RNAimax (Invitrogen) with 20 nM siRNA (GenePharma) for 3 days. The siRNA sequences were list in the key resources table under the Oligonucleotide section.
QUANTIFICATION AND STATISTICAL ANALYSIS
In all graphs, the data is the mean value from at least three independent experiments. The error bars represent the standard error of the mean (S.E.M.). Statistical analysis was determined using GraphPad Prism 6.0 (https://www.graphpad.com/). Two-tailed Student’s t test and one-way ANOVA (followed by Tukey or Dunnett post hoc test) were performed for statistical significance analysis. A p value of > 0.05 is not considered statistically significant. * indicates p < 0.05, **p < 0.01 and ***p < 0.001.
Supplementary Material
Highlights.
Rheb activates mitochondrial pyruvate dehydrogenase (PDH) to produce energy
Rheb interacts with PDH phosphatases (PDPs) to activate PDH independent of mTORC1
Rheb mediates neuronal-activity-induced PDH activation
Rheb coordinates PDH-dependent neuroenergetics and mTORC1-dependent neuronal functions
ACKNOWLEDGMENTS
We thank the Transgenic Facility of Johns Hopkins University School of Medicine for the assistance with the generation of Rosa26 Rheb (wildtype) mouse, Dr. Chenghui Li of the Analytical & Testing Center of Sichuan University for the help with laser scanning confocal imaging, and Ms. Hui Zheng for the artistic work. This work was supported by grants from the National Natural Science Foundation of China (31501155 to W.Y., 31530042, 31371484 to B.X., and 81571195 to M.C.), Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions (2021SHIBS0002), and Shenzhen Innovation Committee of Science and Technology Grants.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.devcel.2021.02.022.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abe Y, Shodai T, Muto T, Mihara K, Torii H, Nishikawa S, Endo T, and Kohda D (2000). Structural basis of presequence recognition by the mitochondrial protein import receptor Tom20. Cell 100, 551–560. [DOI] [PubMed] [Google Scholar]
- Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, et al. (2011). Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat. Cell Biol 13, 1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Wu Z, Farrell RJ, and Ryan TA (2017). GLUT4 mobilization supports energetic demands of active synapses. Neuron 93, 606–615.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeza-Lehnert F, Saab AS, Gutiérrez R, Larenas V, Díaz E, Horn M, Vargas M, Hösli L, Stobart J, Hirrlinger J, et al. (2019). Non-canonical control of neuronal energy status by the Na(+) pump. Cell Metab 29, 668–680.e4. [DOI] [PubMed] [Google Scholar]
- Bagchi S, Fredriksson R, and Wallén-Mackenzie Å (2015). In situ proximity ligation assay (PLA). Methods Mol. Biol 1318, 149–159. [DOI] [PubMed] [Google Scholar]
- Bélanger M, Allaman I, and Magistretti PJ (2011). Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14, 724–738. [DOI] [PubMed] [Google Scholar]
- Bockaert J, and Marin P (2015). mTOR in brain physiology and pathologies. Physiol. Rev 95, 1157–1187. [DOI] [PubMed] [Google Scholar]
- Bové J, Martínez-Vicente M, and Vila M (2011). Fighting neurodegeneration with rapamycin: mechanistic insights. Nat. Rev. Neurosci 12, 437–452. [DOI] [PubMed] [Google Scholar]
- Buffington SA, Huang W, and Costa-Mattioli M (2014). Translational control in synaptic plasticity and cognitive dysfunction. Annu. Rev. Neurosci 37, 17–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, and Halliwell B (2019). Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci 20, 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Li CF, Han F, Liu C, Zhang A, Hsu CC, Peng D, Zhang X, Jin G, Rezaeian A-H, et al. (2020). Phosphorylation of PDHA by AMPK drives TCA cycle to promote cancer metastasis. Mol. Cell 80, 263–278.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso M, Maitan MA, Bifulco G, Miele C, Vigliotta G, Oriente F, Formisano P, and Beguinot F (2001). Activation and mitochondrial translocation of protein kinase Cdelta are necessary for insulin stimulation of pyruvate dehydrogenase complex activity in muscle and liver cells. J. Biol. Chem 276, 45088–45097. [DOI] [PubMed] [Google Scholar]
- Casanova E, Fehsenfeld S, Mantamadiotis T, Lemberger T, Greiner E, Stewart AF, and Schütz G (2001). A CamKIIalpha iCre BAC allows brainspecific gene inactivation. Genesis 31, 37–42. [DOI] [PubMed] [Google Scholar]
- Chacinska A, Koehler CM, Milenkovic D, Lithgow T, and Pfanner N (2009). Importing mitochondrial proteins: machineries and mechanisms. Cell 138, 628–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SW, Gerencser AA, and Nicholls DG (2009). Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: spare respiratory capacity and stochastic mitochondrial failure. J. Neurochem 109, 1179–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuquet J, Quilichini P, Nimchinsky EA, and Buzsáki G (2010). Predominant enhancement of glucose uptake in astrocytes versus neurons during activation of the somatosensory cortex. J. Neurosci 30, 15298–15303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, and Puigserver P (2007). mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 450, 736–740. [DOI] [PubMed] [Google Scholar]
- Currais A, Huang L, Goldberg J, Petrascheck M, Ates G, Pinto-Duarte A, Shokhirev MN, Schubert D, and Maher P (2019). Elevating acetyl-CoA levels reduces aspects of brain aging. eLife 8, e47866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, and Powell JD (2011). The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol 12, 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, and Thomas G (2001). Mammalian TOR: a homeostatic ATP sensor. Science 294, 1102–1105. [DOI] [PubMed] [Google Scholar]
- Díaz-García CM, Mongeon R, Lahmann C, Koveal D, Zucker H, and Yellen G (2017). Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab 26, 361–374.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du F, Zhu XH, Zhang Y, Friedman M, Zhang N, Ugurbil K, and Chen W (2008). Tightly coupled brain activity and cerebral ATP metabolic rate. Proc. Natl. Acad. Sci. USA 105, 6409–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Shan C, Kang HB, Elf S, Xie J, Tucker M, Gu TL, Aguiar M, Lonning S, Chen H, et al. (2014). Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol. Cell 53, 534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, and Thomas G (2003). Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457–1466. [DOI] [PubMed] [Google Scholar]
- Hall CN, Klein-Flügge MC, Howarth C, and Attwell D (2012). Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J. Neurosci 32, 8940–8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, and Attwell D (2012). Synaptic energy use and supply. Neuron 75, 762–777. [DOI] [PubMed] [Google Scholar]
- Huang B, Gudi R, Wu P, Harris RA, Hamilton J, and Popov KM (1998). Isoenzymes of pyruvate dehydrogenase phosphatase. DNA-derived amino acid sequences, expression, and regulation. J. Biol. Chem 273, 17680–17688. [DOI] [PubMed] [Google Scholar]
- Huang B, Wu P, Popov KM, and Harris RA (2003). Starvation and diabetes reduce the amount of pyruvate dehydrogenase phosphatase in rat heart and kidney. Diabetes 52, 1371–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakkamsetti V, Marin-Valencia I, Ma Q, Good LB, Terrill T, Rajasekaran K, Pichumani K, Khemtong C, Hooshyar MA, Sundarrajan C, et al. (2019). Brain metabolism modulates neuronal excitability in a mouse model of pyruvate dehydrogenase deficiency. Sci. Transl. Med 11, eaan0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang E, Kim JY, Liu CY, Xiao B, Chen PY, Christian KM, Worley PF, Song H, and Ming GL (2015). Rheb1 mediates DISC1-dependent regulation of new neuron development in the adult hippocampus. Neurogenesis (Austin) 2, e1081715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, et al. (2013). A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498, 109–112. [DOI] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, and Webb WW (2004). Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 305, 99–103. [DOI] [PubMed] [Google Scholar]
- Kolobova E, Tuganova A, Boulatnikov I, and Popov KM (2001). Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem. J 358, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb E, Herre M, Zucker-Scharff I, Darnell RB, and Allis CD (2015). BET protein Brd4 activates transcription in neurons and BET inhibitor Jq1 blocks memory in mice. Nat. Neurosci 18, 1464–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacher MD, Pincheira R, Zhu Z, Camoretti-Mercado B, Matli M, Warren RS, and Castro AF (2010). Rheb activates AMPK and reduces p27Kip1 levels in Tsc2-null cells via mTORC1-independent mechanisms: implications for cell proliferation and tumorigenesis. Oncogene 29, 6543–6556. [DOI] [PubMed] [Google Scholar]
- Lake BB, Codeluppi S, Yung YC, Gao D, Chun J, Kharchenko PV, Linnarsson S, and Zhang K (2017). A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Sci. Rep 7, 6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson JE, Niu XD, Browning KS, Trong HL, Yan J, and Reed LJ (1993). Molecular cloning and expression of the catalytic subunit of bovine pyruvate dehydrogenase phosphatase and sequence similarity with protein phosphatase 2C. Biochemistry 32, 8987–8993. [DOI] [PubMed] [Google Scholar]
- Le Masson G, Przedborski S, and Abbott LF (2014). A computational model of motor neuron degeneration. Neuron 83, 975–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang P-W, et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Jiang Y, Meisenhelder J, Yang W, Hawke DH, Zheng Y, Xia Y, Aldape K, He J, Hunter T, et al. (2016). Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol. Cell 61, 705–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, MacKenzie KR, Putluri N, Maletić-Savatić M, and Bellen HJ (2017). The glia-neuron lactate shuttle and elevated ROS Promote Lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab 26, 719–737.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgaard I, Li B, Xie L, Kang H, Sanggaard S, Haswell JD, Sun W, Goldman S, Blekot S, Nielsen M, et al. (2015). Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat. Commun 6, 6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutas A, Birnbaumer L, and Yellen G (2014). Metabolism regulates the spontaneous firing of substantia nigra pars reticulata neurons via KATP and nonselective cation channels. J. Neurosci 34, 16336–16347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Bai X, Guo S, and Jiang Y (2008). The switch I region of Rheb is critical for its interaction with FKBP38. J. Biol. Chem 283, 25963–25970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber-Castell A, Kaelin V, Zuend M, San Martin A, Romero-Gomez I, et al. (2016). In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab 23, 94–102. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, and Allaman I (2018). Lactate in the brain: from metabolic end-product to signalling molecule. Nat. Rev. Neurosci 19, 235–249. [DOI] [PubMed] [Google Scholar]
- Melser S, Chatelain EH, Lavie J, Mahfouf W, Jose C, Obre E, Goorden S, Priault M, Elgersma Y, Rezvani HR, et al. (2013). Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab 17, 719–730. [DOI] [PubMed] [Google Scholar]
- Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, and Manning BD (2014). Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156, 771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaoudi E, Ying SW, Kanhema T, Croll SD, and Bramham CR (2002). Brain-derived neurotrophic factor triggers transcription-dependent, late phase long-term potentiation in vivo. J. Neurosci 22, 7453–7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Prudent J, Basu K, Goyon V, Katsumura S, Hulea L, Pearl D, Siddiqui N, Strack S, McGuirk S, et al. (2017). mTOR controls mitochondrial dynamics and cell survival via MTFP1. Mol. Cell 67, 922–935.e5. [DOI] [PubMed] [Google Scholar]
- Mullinax TR, Stepp LR, Brown JR, and Reed LJ (1985). Synthetic peptide substrates for mammalian pyruvate dehydrogenase kinase and pyruvate dehydrogenase phosphatase. Arch. Biochem. Biophys 243, 655–659. [DOI] [PubMed] [Google Scholar]
- Nagase M, Takahashi Y, Watabe AM, Kubo Y, and Kato F (2014). On-site energy supply at synapses through monocarboxylate transporters maintains excitatory synaptic transmission. J. Neurosci 34, 2605–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naia L, Cunha-Oliveira T, Rodrigues J, Rosenstock TR, Oliveira A, Ribeiro M, Carmo C, Oliveira-Sousa SI, Duarte AI, Hayden MR, and Rego AC (2017). Histone deacetylase inhibitors protect against pyruvate dehydrogenase dysfunction in Huntington’s disease. J. Neurosci 37, 2776–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, and Magistretti PJ (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 91, 10625–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, and Sabatini DM (2011). mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, and Magnuson MA (1999). Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem 274, 305–315. [DOI] [PubMed] [Google Scholar]
- Rangaraju V, Calloway N, and Ryan TA (2014). Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, and Dixon JE (2009). Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal. Biochem 389, 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter KN, Revelo NH, Seitz KJ, Helm MS, Sarkar D, Saleeb RS, D’Este E, Eberle J, Wagner E, Vogl C, et al. (2018). Glyoxal as an alternative fixative to formaldehyde in immunostaining and super-resolution microscopy. EMBO J 37, 139–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K, and Giaume C (2008). Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 322, 1551–1555. [DOI] [PubMed] [Google Scholar]
- Samant SA, Zhang HJ, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D, Archer SL, Chan DC, and Gupta MP (2014). SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol. Cell. Biol 34, 807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahani N, Huang WC, Varnum M, Page DT, and Subramaniam S (2017). Forebrain depletion of Rheb GTPase elicits spatial memory deficits in mice. Neurobiol. Aging 50, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahani N, Pryor W, Swarnkar S, Kholodilov N, Thinakaran G, Burke RE, and Subramaniam S (2014). Rheb GTPase regulates beta-secretase levels and amyloid beta generation. J. Biol. Chem 289, 5799–5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She H, Yang Q, Shepherd K, Smith Y, Miller G, Testa C, and Mao Z (2011). Direct regulation of complex I by mitochondrial MEF2D is disrupted in a mouse model of Parkinson disease and in human patients. J. Clin. Invest 121, 930–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, and Shulman RG (1998). Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc. Natl. Acad. Sci. USA 95, 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son SM, Park SJ, Lee H, Siddiqi F, Lee JE, Menzies FM, and Rubinsztein DC (2019). Leucine signals to mTORC1 via its metabolite acetyl-coenzyme A. Cell Metab 29, 192–201.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorbi S, Bird ED, and Blass JP (1983). Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann. Neurol 13, 72–78. [DOI] [PubMed] [Google Scholar]
- Stacpoole PW (2012). The pyruvate dehydrogenase complex as a therapeutic target for age-related diseases. Aging Cell 11, 371–377. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, and Alberini CM (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, and Schütz G (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet 23, 99–103. [DOI] [PubMed] [Google Scholar]
- Tyssowski KM, DeStefino NR, Cho JH, Dunn CJ, Poston RG, Carty CE, Jones RD, Chang SM, Romeo P, Wurzelmann MK, et al. (2018). Different neuronal activity patterns induce different gene expression programs. Neuron 98, 530–546.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Bracamontes J, Zorumski C, Weiss DS, and Steinbach JH (1997). Bicuculline and gabazine are allosteric inhibitors of channel opening of the GABAA receptor. J. Neurosci 17, 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanzetta I, and Grinvald A (1999). Increased cortical oxidative metabolism due to sensory stimulation: implications for functional brain imaging. Science 286, 1555–1558. [DOI] [PubMed] [Google Scholar]
- Wyss MT, Jolivet R, Buck A, Magistretti PJ, and Weber B (2011). In vivo evidence for lactate as a neuronal energy source. J. Neurosci 31, 7477–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Sanders LK, Kaufmann WE, Yee W, Barnes CA, Nathans D, and Worley PF (1994). rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J. Biol. Chem 269, 16333–16339. [PubMed] [Google Scholar]
- Yang H, Jiang X, Li B, Yang HJ, Miller M, Yang A, Dhar A, and Pavletich NP (2017). Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 552, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Ruchti E, Petit JM, Jourdain P, Grenningloh G, Allaman I, and Magistretti PJ (2014). Lactate promotes plasticity gene expression by potentiating NMDA signaling in neurons. Proc. Natl. Acad. Sci. USA 111, 12228–12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda S, Sugiura H, Katsurabayashi S, Shimada T, Tanaka H, Takasaki K, Iwasaki K, Kobayashi T, Hino O, and Yamagata K (2014). Activation of Rheb, but not of mTORC1, impairs spine synapse morphogenesis in tuberous sclerosis complex. Sci. Rep 4, 5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q, Chen M, Yang W, and Xiao B (2021). Circadian Rheb oscillation alters the dynamics of hepatic mTORC1 activity and mitochondrial morphology. FEBS Lett 595, 360–369. [DOI] [PubMed] [Google Scholar]
- Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, Marques S, Munguba H, He L, Betsholtz C, et al. (2015). Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zheng X, Boyer L, Jin M, Kim Y, Fan W, Bardy C, Berggren T, Evans RM, Gage FH, and Hunter T (2016). Alleviation of neuronal energy deficiency by mTOR inhibition as a treatment for mitochondria-related neurodegeneration. eLife 5, e13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberter Y, and Zilberter M (2017). The vicious circle of hypometabolism in neurodegenerative diseases: ways and mechanisms of metabolic correction. J. Neurosci. Res 95, 2217–2235. [DOI] [PubMed] [Google Scholar]
- Zou J, Zhou L, Du XX, Ji Y, Xu J, Tian J, Jiang W, Zou Y, Yu S, Gan L, et al. (2011). Rheb1 is required for mTORC1 and myelination in postnatal brain development. Dev. Cell 20, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.