

Abstract

Many snake venom toxins cause local tissue damage in prey and victims, which constitutes an important pathology that is challenging to treat with existing antivenoms. One of the notorious toxins that causes such effects is myotoxin II present in the venom of the Central and Northern South American viper, Bothrops asper. This Lys49 PLA2 homologue is devoid of enzymatic activity and causes myotoxicity by disrupting the cell membranes of muscle tissue. To improve envenoming therapy, novel approaches are needed, warranting the discovery and development of inhibitors that target key toxins that are currently difficult to neutralize. Here, we report the identification of a new peptide (JB006), discovered using phage display technology, that is capable of binding to and neutralizing the toxic effects of myotoxin II in vitro and in vivo. Through computational modeling, we further identify hypothetical binding interactions between the toxin and the peptide to enable further development of inhibitors that can neutralize myotoxin II.
1. Introduction
Snakebite envenoming is a neglected tropical disease of high impact in sub-Saharan Africa, Asia, and Latin America. There is an urgent need for the discovery and development of novel therapies that could complement antivenoms to reduce mortality and morbidity of this pathology on a global basis.1,2 One of the most serious consequences of snakebite envenoming is the local tissue damage inflicted by the venom of many species, which includes the necrosis of skeletal muscles.3 Muscle regeneration in these cases is often impaired, with consequent permanent tissue loss and dysfunction in these patients.4 The poor efficacy of many antivenoms against local tissue damage is a major medical concern, leaving many victims permanently maimed and disabled when antivenom is not administered rapidly after the bite.3 Part of the explanation for this may be that some of the toxins responsible for local pathology, such as phospholipases A2 (PLA2s), are only moderately immunogenic, which causes antivenoms derived via immunization processes to generally only have intermediate antibody titers against them.5
Among the viperid species that cause severe local tissue damage, Bothrops asper causes a high number of cases in Central America and northern South America, of which many result in severe envenoming characterized by prominent local tissue pathology.6 The venom of this viper is rich in myotoxic PLA2s and PLA2 homologues, which are responsible for local skeletal muscle necrosis.7−9 Among them, myotoxin II, a Lys49 PLA2 homologue devoid of enzymatic activity, is abundant in this venom and plays a key role in myonecrosis.7,8,10 Therefore, for effective treatment, myotoxin II is one of the key targets that must be neutralized.11 However, this toxin is only moderately immunogenic and fails to raise a strong antibody response during the animal immunization process of antivenom manufacture.12−14 Therefore, to improve therapy against B. asper envenoming, the development of myotoxin-II-neutralizing agents is greatly warranted.
One proposed solution for improving envenoming therapy includes the development of peptide-based inhibitors that target key medically relevant toxins.15,16 Very few peptides have been reported that neutralize the effect of snake venom toxins. However, peptides have a number of different therapeutic benefits, including low-cost synthesis, reproducibility, and engineerable pharmacokinetics,15,17,18 which position peptides as a relevant pharmaceutical scaffold. Here, we report the discovery of an anti-myotoxin II peptide (JB006) using phage display technology. This peptide was further assessed for its ability to selectively bind myotoxin II in vitro and functionally neutralize this toxin in both a cell-based assay and a rodent model.
2. Methods
2.1. Purification of Myotoxin II
Myotoxin II (Uniprot P24605) was purified from the venom of B. asper by cation-exchange chromatography followed by reversed-phase high-performance liquid chromatography (RP-HPLC), as described previously.9,10
2.2. Phage Display Selection and Assessment of Polyclonal and Monoclonal Output
For phage display selection, two random linear peptide libraries, TriCo-16 Phage Display Peptide Library and TriCo-20 Phage Display Peptide Library from Creative Biolabs, were employed, following previously described protocols.17,19 In short, five rounds of panning on directly coated myotoxin II were performed, followed by isolation of monoclonal phages and assessment of their ability to bind myotoxin II and two controls, human serum albumin and α-cobratoxin (≥99% purity, from Naja kaouthia, Latoxan), using ELISA.17 For phages that displayed specific binding, their ssDNA was isolated and sequenced, and based on an assessment of their solubility and isoelectric point using http://pepcalc.com/peptide-solubility-calculator.php, peptides were selected for further analysis.17
2.3. Synthetic Peptides
Synthetic JB001–JB006 peptides were purchased from Schafer-N (Copenhagen, Denmark), and JB006-free and biotinylated JB006 peptides were purchased from GenScript with purities >95% (Table 1). The 5(6)-carboxytetramethylrhodamine (TAMRA)-labeled JB006 peptide was synthesized using an automated peptide synthesizer and standard Fmoc (fluorenylmethoxycarbonyl)-based solid-phase peptide synthesis on Rink amide TentaGel resin (0.23 mmol/g) at a 0.02 mmol scale. Fmoc deprotection was performed in two steps: (1) piperidine in dimethylformamide (DMF; 2:3, v/v) for 3 min and (2) piperidine in DMF (1:4, v/v) for 12 min. Deprotection steps were followed by washing with DMF (2 × 45 s), CH2Cl2 (1 × 45 s), and DMF (2 × 45 s). Coupling steps were performed as double couplings with Fmoc-Xaa-OH (5.00 equiv to the resin loading), 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronoium hexafluorophosphate (4.90 equiv), and i-Pr2NEt in NMP (10.0 equiv, 2.0 M) in DMF (final concentration = 0.2 M) for 40 min for each coupling. TAMRA-acid (1.5 equiv) was coupled manually using 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronoium hexafluorphosphate (HATU; 1.50 equivalents) and i-Pr2NEt (3.00 equivalents) in DMF (final concentration = 0.06 M) for 18 h. Global deprotection and cleavage were conducted in the cleavage cocktail (TFA–i-Pr3SiH–water, 95:2.5:2.5, v/v/v) at room temperature (RT) for 2 h, followed by TFA evaporation and ether precipitation. The crude peptide was purified as a single isomer by preparative RP-HPLC on a C8 Phenomenex Luna column (5 μm, 100 Å, 250 × 20 mm) using an Agilent 1260 LC system. Fractions were analyzed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS), and pure fractions were pooled and lyophilized. Purity was determined using an Agilent 1100 system equipped with a C18 Phenomenex Luna column (2.6 μm, 100 Å, 150 mm × 4.60 mm). Analytical HPLC purity: 95% (λ = 210 nm; Table 1, Supporting Information).
Table 1. Overview of Sequenced Peptide Hits.
| ID | sequence | MW (theoretical) (Da) | MW (measured) (Da) | from panning round | no. of AAs | pI (calculated) |
|---|---|---|---|---|---|---|
| JB001 | Ac-VNRMLELKIMDYGGG-NH2 | 1737.06 | 1736.80 | 1 | 14 | 7.14 |
| JB002 | Ac-QSVTMGPGLITHSPIHTQSK-NH2 | 2160.39 | 2160.10 | 1 | 20 | 14.00 |
| JB003 | Ac-DYDRIPDIPMLGGGG-NH2 | 1616.76 | 1617.40 | 1 | 15 | 3.41 |
| JB004 | Ac-NGYWSSQQYMQQAPMPWRIP-NH2 | 2509.76 | 2509.60 | 1 | 20 | 10.09 |
| JB005 | Ac-SWEPYANPTRYKFHDW-NH2 | 2138.30 | 2137.80 | 1 | 16 | 7.89 |
| JB006 | Ac-DHWVWGWNYQYQPQEWHTES-NH2 | 2717.76 | 2717.40 | 1 | 20 | 4.30 |
| JB006-free | H-DHWVWGWNYQYQPQEWHTES-OH | 2676.80 | 2676.58 | 1 | 20 | 4.5 |
| biotin-JB006 | H-DHWVWGWNYQYQPQEWHTESGGG{LYS(BIOTIN)}-OH | 3202.40 | 3202.80 | 24 | ||
| TAMRA-JB006 | TAMRA-Peg2-DHWVWGWNYQYQPQEWHTES-NH2 | 3233.40 | 20 |
2.4. Fluorescence Polarization Binding Assay
Protein concentrations were determined by UV absorbance (NanoDrop One, Thermo Fisher) at 280 nm. The dimethyl sulfoxide stock concentration of TAMRA-JB006 was determined by the weight of the corresponding TFA salt (MW + 5 × TFA = 3689 g/mol). Binding affinities were determined in a 384-well plate format (Corning Life Science) using a Safire 2 plate reader (Tecan). The instrument G-factor was calibrated to give an initial millipolarization at 20 (excitation at 530 nm; emission at 580 nm), and the instrumental Z-factor was adjusted to maximum fluorescence. All measurements were conducted in phosphate-buffered saline (PBS; pH = 7.4) at 25 °C. The FP saturation assay was performed by mixing 50 nM of TAMRA-labeled JB006 peptide with increasing concentrations (0.125–233 μM) of myotoxin II. The resulting polarization was plotted as a function of the protein concentration and fitted to a one-site binding model using GraphPad Prism 8.4 software to estimate the dissociation constant (Kd).
2.5. Pulldown Experiments
Streptavidin–agarose resin (Pierce, Thermo Fisher; 100 μL of slurry) was placed in a centrifugal vial containing a filter and centrifuged at 500g for 1 min to remove excess liquid. The resin was then incubated with either biotinylated JB006 peptide in PBS (100 μL, 1.0 mg/mL) or biotin in PBS (100 μL, 1.0 mg/mL) for 1 h at RT followed by centrifugation at 500g for 1 min. The resin was washed twice by incubation with PBS (100 μL) for 2 min and subsequent centrifugation at 500g for 1 min. Next, crude B. asper venom in PBS (100 μL, 1.0 mg/mL) was added to the resin and incubated at 4 °C. The next day, the resin was centrifuged at 500g for 1 min, and the run-through (supernatant) was collected. The resin was then washed three times by incubation with PBS (100 μL) for 2 min at RT and subsequent centrifugation at 500g for 1 min, and the individual washing steps were collected. In the next step, the bound protein was eluted three times from the resin by incubation with glycine–HCl buffer (100 μL, 0.1 M, pH 2.8) for 2 min at RT and subsequent centrifugation at 500g for 1 min, and the individual elution steps were collected. The pulldown samples were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using NuPAGE (Thermo Fisher) 4–12% bis–tris gels and NuPAGE (Thermo Fisher) MES SDS running buffer (20×). Samples were loaded with NuPAGE (Thermo Fisher) LDS sample buffer (4×), and for reduced samples, a NuPAGE (Thermo Fisher) sample reducing agent (1×) was added, and the samples were heated to 85 °C for 5 min prior to loading. Gels were stained with Coomassie blue overnight.
2.6. Myogenic C2C12 Cells Experiments
The ATCC-CRL1772 murine myogenic cell line C2C12 was used to evaluate neutralization of the cytotoxic action of myotoxin II, as previously described.20 Cells were maintained as myoblasts at subconfluent density in 25 cm2 bottles using Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum (FCS) (DMEM with 10% FCS, penicillin, streptomycin, ciprofloxacin, pyruvate, and l-glutamine). Cells were detached using trypsin, seeded in 96-well plates, and allowed to differentiate to myotubes using DMEM, 1% FCS for 5–6 days. Myotoxin II (10 μg; 0.73 nmol), pre-incubated for 30 min at 37 °C with 900 μM of each inhibitory peptide in assay medium (DMEM, 1% FCS) or without peptides, was added in a total volume of 100 μL/well. Controls for 0% cytotoxicity consisted of the assay medium, while controls for 100% cytotoxicity consisted of 0.1% Triton X-100 diluted in the assay medium. After an incubation of 3 h at 37 °C, 55 μL of the supernatant was taken to determine the activity of lactic dehydrogenase (LDH) released by damaged cells, using a UV kinetic assay (LDH-BR Chromatest, Linear Chemicals, Montgat, Spain). Assays were performed in triplicate wells.
A neutralization curve using myotoxin II (10 μg; 0.73 nmol; 7.3 μM), pre-incubated for 30 min at 37 °C with 900, 225, 56.25, 14.06, 3.52, or 0 μM JB006 in assay medium (DMEM, 1% FCS), was obtained using the methodology described above. LDH release was plotted to estimate the half-maximal inhibitory concentrations (IC50) value by nonlinear regression with variable slope using GraphPad Prism 8.4 software.
2.7. In Vivo Mouse Assay
Mouse experiments followed ethical guidelines of the Institutional Committee for the Use and Care of Animals (CICUA, #084-17) of the University of Costa Rica. Groups of five mice (18–20 g body weight) received an intramuscular injection (total volume: 50 μL) in the right gastrocnemius of 50 μg (3.6 nmol; 73 μM) of myotoxin II, previously incubated for 30 min at 37 °C with 900, 100, or 20 μM of JB006. These concentrations were selected on the basis of the results observed in the cytotoxicity assay on C2C12 cells (Figure 5). The aim was to span a wide range by including a low, an intermediate, and a high amount of the peptide in the experiments. A control group of five mice received an injection of myotoxin II incubated with PBS alone. After 3 h, a blood sample from the tail was collected into heparinized capillaries and centrifuged. A plasma aliquot of 4 μL was utilized to determine the activity of creatine kinase (CK; E.C. 2.7.3.2) using a kinetic assay (CK-Nac, Biocon Diagnostik, Mönchberg, Germany), following the manufacturer’s instructions. Enzyme activity was expressed as a percentage, considering 0% activity the injection of PBS and as 100% activity the injection of the toxin in the absence of the peptide.
Figure 5.
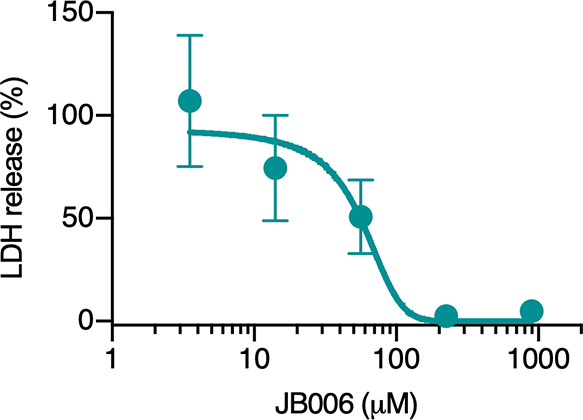
Dose–response curve for the determination of the IC50 value of JB006 for the inhibition of the cytotoxic activity of myotoxin II in C2C12 cells. Myotoxin II (10 μg; 0.73 nmol; 7.3 μM) was pre-incubated for 30 min at 37 °C with 900, 225, 56.25, 14.06, 3.52, or 0 μM JB006 in assay medium and added in triplicate wells. After an incubation of 3 h at 37 °C, the activity of LDH released by damaged cells was measured on the supernatant. 100% release corresponds to cells incubated with the toxin in the absence of the inhibitory peptide. Results are presented as mean ± SD (n = 3).
2.8. Statistical Analyses
All statistical analyses were performed using GraphPad Prism 8.4 software. The significance of the differences between the mean values of control groups and groups treated with synthetic peptides were determined using one-way analysis of variance and Tukey’s test. P values < 0.05 were considered significant. P < 0.05 (*), P < 0.01 (**), P < 0.001 (***), and P < 0.0001 (****).
2.9. Circular Dichroism Spectroscopy
Circular dichroism (CD) spectra were acquired using a JASCO J1500 spectrophotometer equipped with a water-circulating bath and a nitrogen gas flowmeter with a sensor. Measurements were carried out in 1 mm quartz cuvettes, and JB006 solutions with concentrations of 50 μM in aqueous buffers (acetate 10 mM, pH 4; phosphate 10 mM, pH 7, 7.4, and 8) were prepared based on the weight of the corresponding TFA salt (MW + 3 × TFA = 3018 g/mol). The CD data were obtained at 298 K with a bandwidth of 1.00 nm, a scanning speed of 50 nm/min, two accumulations, and a data integration time of 4 s. Spectra were recorded in millidegree units (m°) and normalized to molar ellipticity (θ) = 100 × m°/l × c × n, with c being the JB006 concentration in mM, l being the path length (0.1 cm), and n being the number of peptide amide bonds %.
2.10. Molecular Docking of JB006 and Myotoxin II
For the docking simulations, the structure of myotoxin II (1CLP; 2.80 Å; resolved via X-ray diffraction) was retrieved from the RCSB PDB database. The structure of JB006 was predicted using PEP-Fold2.21 Thereafter, docking between JB006 and myotoxin II was performed using ClusPro2 using standard settings.22 ClusPro2 predicted 60 different models, with the highest scoring one being chosen for further evaluation in ChimeraX.23
3. Results
3.1. Identification of Peptide Binders to Myotoxin II
Following five rounds of panning, the accumulation of binders was confirmed by polyclonal ELISA (Figure 1). Monoclonal phages were isolated from the first and fourth panning rounds and assessed using monoclonal phage ELISA, which upon DNA sequencing yielded 12 unique peptide hits. Based on their signal intensities and concentration curves using myotoxin II as antigen in the monoclonal phage ELISA, six of these peptides (JB001–JB006) were custom-synthesized by and purchased from a commercial vendor and further analyzed (Table 1).
Figure 1.
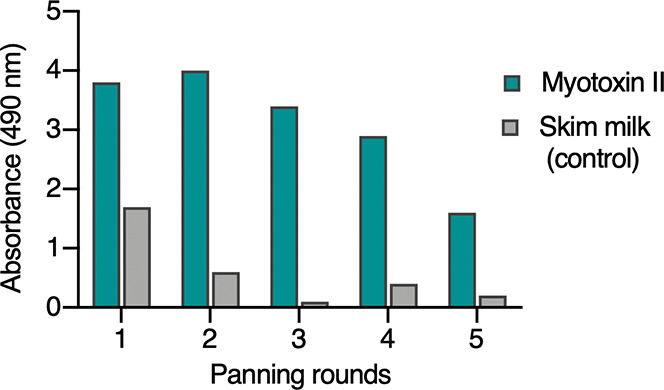
Polyclonal phage ELISA of the five different panning rounds performed in the phage display selection experiment against myotoxin II. Already from the first round, an accumulation of phages binding specifically to myotoxin II is observed.
3.2. Binding Properties of JB006
To investigate the affinity of JB006 toward myotoxin II, a TAMRA-labeled probe of JB006 was synthesized to conduct fluorescence polarization (FP) binding experiments (Figure S1). FP saturation experiments with a constant concentration of TAMRA-probe (50 nM) and increasing concentrations of myotoxin II (0.125–233 μM) gave an estimated Kd value for the TAMRA-probe of 130 ± 31 μM (Figure 2A). FP competition assays to determine the Ki of unlabeled JB006-free were not feasible as the presence of JB006-free peptide at low micromolar concentrations led to a significant increase in FP signal even without the addition of myotoxin II (Figure 2B). The observed FP signal in the absence of protein is likely to arise from peptide–peptide interactions, which would reduce the tumbling of the TAMRA-probe and therefore result in an increase of FP. The effect was concentration-dependent and rendered the determination of the Ki of JB006-free impossible.
Figure 2.

(A) Binding curve of TAMRA-JB006 to myotoxin II. FP saturation curve for the estimation of Kd between TAMRA-JB006 and myotoxin II, giving an estimated Kd value of 130 ± 30 μM. (B) FP increase in the absence of myotoxin II. Artificial increase in the FP signal in the absence of myotoxin II, but with increasing concentrations of non-labeled JB006-free peptide, indicates interactions between the JB006 peptide and the TAMRA probe. Results are presented as mean ± SD.
As an alternative method for studying the interaction between JB006 and myotoxin II, affinity-based pulldown experiments of myotoxin II with biotinylated JB006 peptide were performed to confirm target engagement of JB006 (Figure 3). Streptavidin-coated agarose resin was loaded with biotinylated JB006 and incubated with the crude venom of B. asper. The run-through (supernatant) of crude venom as well as the washing fractions did not contain the strong band at 15 kDa, in contrast to the elution fractions, which showed a strong band at the expected mass of myotoxin II (Figure 3A,B). To ensure the affinity pulldown of myotoxin II was specific for biotinylated JB006, a control experiment was conducted, where the resin was loaded with biotin (Figure 3C). Here, the run-through (supernatant) and the first washing fraction contained the majority of myotoxin II, and no band was observed in the elution fractions. These experiments together with the FP binding curve for the TAMRA-probe of JB006 confirm that JB006 binds to myotoxin II, however, with poor affinity.
Figure 3.

Coomassie-stained SDS-PAGE images for myotoxin II pulldown experiments, depicting the section of the gel where myotoxin II migrates. (A) Myotoxin II and B. asper crude venom, reduced (red.) and non-reduced. (B) B. asper crude venom incubation at 4 °C overnight with streptavidin-agarose resin loaded with biotinylated JB006 peptide shows selective enrichment of myotoxin II. (C) B. asper crude venom incubation at 4 °C overnight with streptavidin-agarose resin loaded with biotin did not result in enrichment of myotoxin II. See Methods for details of the experimental protocols. The complete pictures of the gels are shown in Figure S2.
3.3. Myogenic C2C12 Cell Experiments
The six selected peptides (JB001–JB006), discovered through phage display, were tested in cell culture to evaluate the inhibition of the cytotoxic activity of myotoxin II (Figure 4). The JB006 peptide displayed the highest inhibition of this activity and inhibited almost all of the cytotoxic activity at the concentration tested, while the other peptides inhibited cytotoxicity to a lesser extent. To determine the degree of inhibition by JB006, a neutralization curve was prepared (Figure 5), from which the IC50 of JB006 was determined to be 56 μM.
Figure 4.
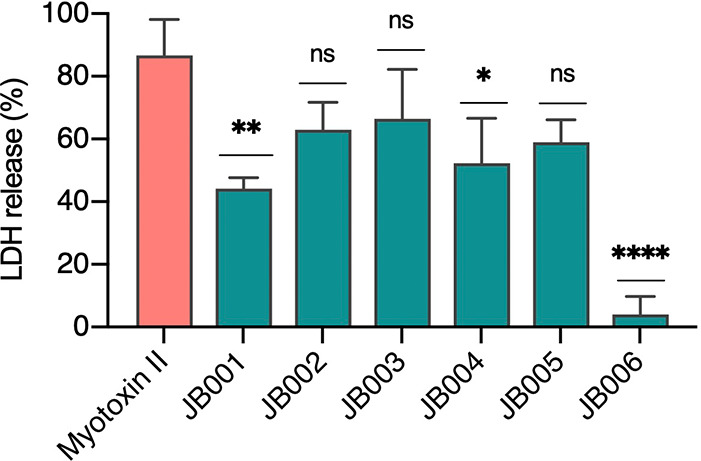
Inhibition of the cytotoxic activity of myotoxin II by different peptides (JB001–JB006). C2C12 myotubes were used. Myotoxin II (10 μg; 0.73 nmol; 7.3 μM) was pre-incubated for 30 min at 37 °C with 900 μM of each peptide in assay medium or without peptides and added in triplicate wells. After an incubation of 3 h at 37 °C, the activity of LDH released by damaged cells was measured on the supernatant. 100% release corresponds to cells incubated with 0.1% Triton X-100. Results are presented as mean ± SD (n = 3). Incubation of myotoxin II with JB006 caused a significant reduction of LDH release (4 ± 5%) compared to cells treated with myotoxin II alone (87 ± 11%). P < 0.05 (*), P < 0.01 (**), P < 0.001 (***), and P < 0.0001 (****).
3.4. In Vivo Mouse Assay
The intramuscular injection of mice with 50 μg of myotoxin II, pre-incubated with different concentrations of JB006, showed a dose-dependent inhibition of the myotoxic activity (Figure 6). A concentration of 900 μM of this peptide was able to completely inhibit CK release caused by toxin-induced damage.
Figure 6.
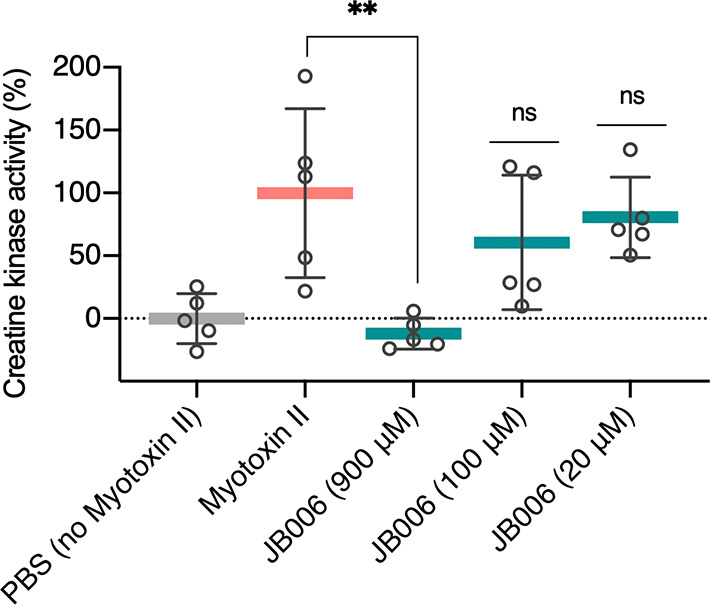
Inhibition of the in vivo myotoxic activity of myotoxin II by different concentrations of JB006. An intramuscular injection (50 μL) containing 50 μg (3.6 nmol; 73 μM) of myotoxin II, previously incubated for 30 min at 37 °C with different concentrations of JB006 (900, 100, or 20 μM) or PBS as the negative control, was administered to groups of five mice. After 3 h, a blood sample was collected from the tail. Plasma was obtained following centrifugation and used to determine the activity of CK, an enzyme released due to muscle damage. Results are presented as mean ± SD (n = 5). Mice injected with myotoxin II incubated with 900 μM of JB006 showed a statistically significant difference [P < 0.01 (**)] when compared with mice injected with myotoxin II incubated with PBS.
3.5. Investigation of the Modeling of the Molecular Interface between JB006 and Myotoxin II
In order to assess the formation of secondary structures for peptide JB006 in solution, CD spectroscopy was utilized (Figure 7).24 The spectra recorded at pH 7 and 7.4 indicate that the peptide possesses a secondary structure at neutral and physiological pH levels. The negative bands at 208 nm and 220 nm and the positive band at 193 nm indicate the presence of a characteristic helical signature at pH 7, while the spectrum recorded at pH 7.4 showed a significantly different shape and could indicate the presence of β-sheet structures. The folding properties of JB006 are highly pH-sensitive, and interestingly, no secondary structure was observed at slightly alkaline pH 8 and acidic pH 4.
Figure 7.
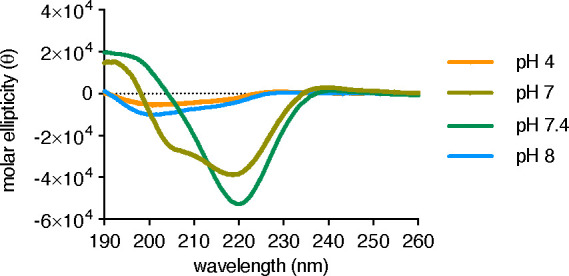
CD spectra of JB006 (50 μM) recorded at RT in acetate buffer (10 mM, pH 4) or phosphate buffer (10 mM, pH 7–8). The molar ellipticity (θ) is normalized with regard to the number of residues and peptide concentration.
Docking predictions between JB006-free and myotoxin II suggested that the interaction is primarily driven by two factors, that is, shape complementarity and electrostatic charge (Figure 8). The model suggests that JB006-free occupies an area of 2.1 Å2 and myotoxin II an area of 7.0 Å2. Notably, predictions also suggested that 28% (595 Å2) of JB006-free’s surface is buried in myotoxin II. The model also indicated a strong negative charge of JB006-free and a strong positive charge of myotoxin II. No hydrogen bonds were predicted at the interface. Furthermore, molecular lipophilicity potential was assessed, but no general patterns could be identified. Finally, specific residues involved in the peptide–toxin interface were identified and close interaction between Arg72 on myotoxin II and Glu19 on JB006-free was predicted (distance of 1.8 Å). Further interactions between Lys69, Phe3, and Asn16 (myotoxin II) and Trp9, Trp7, and Trp6 (JB006-free) (2.7, 4.0, 3.1 Å) were also predicted. Potentially, an interaction between Leu10 (myotoxin II) and Trp3 (JB006-free; 6.3 Å) could also exist. Finally, JB006-free might sterically block the hydrophobic channel of the toxin, with further interactions between the His48/Lys49 residues of myotoxin II and Trp5 (JB006-free) possibly being present.
Figure 8.

Molecular docking of JB006 (coral) and myotoxin II (lavender). (A) Shape complementarity and electrostatic charge driving the interactions (Coulombic color was used with red for the negative charge through white to blue for the positive charge). (B) Assessment of molecular lipophilicity potential (the surface coloring ranges from dark goldenrod for the most hydrophobic potentials, through white, to dark cyan for the most hydrophilic). (C) Depiction of the possible steric hindrance of the hydrophobic channel of myotoxin II by JB006. (D) Representation of residues involved in the toxin–peptide interface. “Green” sequence highlights indicate amino acids involved in the interface, “yellow” regions indicate protein structure helices, and “blue” indicates structure strands.
4. Discussion
Myotoxin II is a key toxin of significant medical importance in the context of B. asper envenomings in Central and Northern South America, as it contributes to local myotoxicity induced by this venom. However, due to its intermediate immunogenicity, it is difficult to ensure high antibody titers against this toxin in antivenoms derived from immunization processes.12−14 Thus, a need exists for the development of toxin-specific inhibitors, such as peptides or other small molecules, which can possibly be used as adjunct therapy or as fortification agents for improving existing antivenoms.19,25−28
Here, we report the discovery and assessment of a 20-mer peptide (JB006), which demonstrates the ability to bind and neutralize myotoxin II in vitro and in vivo, albeit at rather high concentrations, as shown by myogenic C2C12 cell experiments and in a rodent model involving preincubation of the peptide and toxin, followed by intramuscular injection. Based on the structural modeling data, it is speculated that electrostatic interactions could be largely responsible for the observed binding between the positively charged myotoxin II and the negatively charged JB006-free peptide. This might be supported by the previous observation that some binding to positively charged α-cobratoxin was observed, which however brings into question the specificity of the peptide.17 In comparison, it has also previously been reported that the negatively charged anti-trypanosomal drug, suramin, is also capable of inhibiting myotoxin II, further indicating that the charge of an inhibitor can play an important role in neutralizing this toxin.29 If this is indeed the case, it would have the implication that the binding enthalpy between the toxin and the peptide may need to be further optimized to improve selectivity. Possibly, this could be achieved via substitution of charged amino acids with amino acids capable of engaging in hydrogen bonding in JB006. Notably, the model mapped the primary interactions to the N-terminal helix of myotoxin II, which differs from the location of the neutralizing epitope identified previously.30 In this prior study, site-directed polyclonal antibodies that targeted the N-terminal helix of myotoxin II (residues 1–15), were found to be non-neutralizing in vivo. However, this discrepancy could potentially be explained by further residues playing an important role in neutralization (e.g., Asn16, Pro17, Ala18, Tyr22, Cys29, Lys69, and Arg72), many of which are predicted to interact with JB006-free. Additionally, it has been proposed that the structural regions that are essential for Lys49 PLA2s to exert toxicity mainly include residues near the C-terminal coil of the protein involved in the docking onto the cell membranes and in bilayer destabilization.28 Whilst the present docking results have not predicted these residues in its interface, it appears as if JB006-free might be sterically blocking the entrance of the so-called “hydrophobic channel” of the toxin. If so, such blockage has been suggested to prevent the allosteric activation of the toxin, as it would limit the orientation of the toxin, preventing it from docking onto and disrupting the cell membrane and thus inhibiting toxicity.28 Nevertheless, it is important to underline that further experiments, such as co-crystallization of JB006 and myotoxin II, would be necessary to confirm the hypotheses presented above.
The data presented here indicate that JB006 could potentially serve as a lead for further optimization using the main pharmacophore as a starting point. The benefits of such peptide scaffolds include the ease of introducing modifications that may improve potency, stability, bioavailability, and pharmacokinetics as peptide chemistry has been significantly standardized in the last many decades. In turn, the improvement of such properties (particularly shelf-life and bioavailability) may enable the application of other routes of administration (e.g., oral, subcutaneous, or intramuscular) instead of the routinely used intravenous route. Here, it could be hypothesized that smaller scaffolds combined with novel delivery methods that can be applied close to the bite site could find utility in treating local tissue damage, such as muscle necrosis induced by myotoxin II. In this relation, it is important to emphasize a limitation of this study, which is the lack of data on the half-life and bioavailability of JB006, which could be limiting factors for its further development. Moreover, the aggregation of JB006-free at low micromolar concentrations in FP competition experiments and the rather low solubility of the peptide in conjunction with the low binding affinity require more investigation, if such an approach is possible using JB006 as the lead peptide. Finally, it is important to further test such novel scaffolds in rescue experiments, as the in vivo experiments performed here utilize preincubation and do not allow for an accurate assessment of the potential of JB006 in a real life setting, where venom and inhibitor are introduced in separate anatomical sites.31 Nevertheless, the combined data presented here demonstrate the feasibility of using phage display technology to discover toxin-neutralizing peptide leads able to neutralize snake venom toxins in vivo.
Acknowledgments
This work was supported by the Department of Drug Design and Pharmacology, University of Copenhagen, and the Tech Transfer Office, University of Copenhagen (to B.L.). We also thank Professor Christian Adam Olsen from the University of Copenhagen for access to peptide synthesis facilities, Lorenzo Seneci from the Technical University of Denmark for help with formatting the text and figures, and Christoffer V. Sørensen from the Technical University of Denmark for help with proofreading.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00280.
Structure, formula, molecular mass, and HPLC trace of purified TAMRA-JB006; SDS-PAGE gels of B. asper crude venom and myotoxin II; and SDS-PAGE gels of pulldown experiments on B. asper crude venom with biotin- and JB006-coated streptavidin-agarose beads (PDF)
Author Contributions
A.H.L., B. L., and B. L. conceived the study; A.H.L., B.H.G., J.B., S.O., A.S.M., M.M.-M., B. L., J.F., J.M.G., and B. L. designed and performed experiments; A.H.L., B.H.G., J.B., S.O., M.M.-M., B. L., J.F., J.M.G., and B. L. analyzed the data. A.H.L., B. L., J.F., J.M.G., and B. L. provided supervision. A.H.L., B.H.G, B. L., J.F., and J.M.G. wrote the manuscript with input from all other authors.
The authors declare the following competing financial interest(s): B.Lohse is a founder of the company Serpentides ApS, which holds a patent (PCT/EP2019/057522) covering the use of the peptides described in this article.
Supplementary Material
References
- Chippaux J.-P.Snakebite envenomation turns again into a neglected tropical disease. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23. 10.1186/s40409-017-0127-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez J. M.Snakebite envenomation as a neglected tropical disease: new impetus for confronting an old scourge. In Handbook of Venoms and Toxins of Reptiles, 2nd ed.; CRC Press, 2021; pp 471–483. [Google Scholar]
- Gutiérrez J. M.; Calvete J. J.; Habib A. G.; Harrison R. A.; Williams D. J.; Warrell D. A. Snakebite envenoming. Nat. Rev. Dis. Prim. 2017, 3, 17063. [DOI] [PubMed] [Google Scholar]
- Gutiérrez J. M.; Escalante T.; Hernández R.; Gastaldello S.; Saravia-Otten P.; Rucavado A. Why is skeletal muscle regeneration impaired after myonecrosis induced by viperid snake venoms?. Toxins 2018, 10, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez JM; Lomonte B.; Sanz L.; Calvete JJ; Pla D. Immunological profile of antivenoms: Preclinical analysis of the efficacy of a polyspecific antivenom through antivenomics and neutralization assays. J. Proteomics 2014, 105, 340–50. 10.1016/j.jprot.2014.02.021. [DOI] [PubMed] [Google Scholar]
- Otero-Patiño R. Epidemiological, clinical and therapeutic aspects of Bothrops asper bites. Toxicon 2009, 54, 998–1011. [DOI] [PubMed] [Google Scholar]
- Lomonte B.; Gutiérrez J. A new muscle damaging toxin, myotoxin II, from the venom of the snake Bothrops asper (terciopelo). Toxicon 1989, 27, 725–733. 10.1016/0041-0101(89)90039-1. [DOI] [PubMed] [Google Scholar]
- Alape-Girón A.; Sanz L.; Escolano J.; Flores-Díaz M.; Madrigal M.; Sasa M.; Calvete J. J. Snake venomics of the lancehead pitviper Bothrops asper: Geographic, individual, and ontogenetic variations. J. Proteome Res. 2008, 7, 3556–3571. [DOI] [PubMed] [Google Scholar]
- Mora-Obando D.; Díaz C.; Angulo Y.; Gutiérrez J. M.; Lomonte B. Role of enzymatic activity in muscle damage and cytotoxicity induced by Bothrops asper Asp49 phospholipase A2 myotoxins: are there additional effector mechanisms involved?. PeerJ 2014, 2, e569 10.7717/peerj.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis B.; Gutierrez J. M.; Lomonte B.; Kaiser I. I. Myotoxin II from Bothrops asper (terciopelo) venom is a lysine-49 phospholipase A2. Arch. Biochem. Biophys. 1991, 284, 352–359. 10.1016/0003-9861(91)90307-5. [DOI] [PubMed] [Google Scholar]
- Lomonte B. Identification of linear B-cell epitopes on myotoxin II, a Lys49 phospholipase A2 homologue from Bothrops asper snake venom. Toxicon 2012, 60, 782–790. 10.1016/j.toxicon.2012.05.028. [DOI] [PubMed] [Google Scholar]
- Gutiérrez J. M.; Sanz L.; Flores-Díaz M.; Figueroa L.; Madrigal M.; Herrera M.; Villalta M.; León G.; Estrada R.; Borges A.; Alape-Girón A.; Calvete J. J. Impact of regional variation in Bothrops asper snake venom on the design of antivenoms: integrating antivenomics and neutralization approaches. J. Proteome Res. 2010, 9, 564–577. [DOI] [PubMed] [Google Scholar]
- Lomonte B.; Gutiérrez J.; Carmona E.; Rovira M. E. Equine antibodies to Bothrops asper myotoxin II: isolation from polyvalent antivenom and neutralizing ability. Toxicon 1990, 28, 379–384. 10.1016/0041-0101(90)90075-i. [DOI] [PubMed] [Google Scholar]
- Lomonte B.; Gutiérrez J.; Rojas G.; Calderón L. Quantitation by enzyme-immunoassay of antibodies against Bothrops myotoxins in four commercially-available antivenoms. Toxicon 1991, 29, 695–702. 10.1016/0041-0101(91)90061-u. [DOI] [PubMed] [Google Scholar]
- Laustsen A.; Engmark M.; Milbo C.; Johannesen J.; Lomonte B.; Gutiérrez J.; Lohse B. From fangs to pharmacology: The future of snakebite envenoming therapy. Curr. Pharm. Des. 2016, 22, 5270–5293. 10.2174/1381612822666160623073438. [DOI] [PubMed] [Google Scholar]
- Laustsen A. H.; Lohse B.; Lomonte B.; Engmark M.; Gutiérrez J. M. Selecting key toxins for focused development of elapid snake antivenoms and inhibitors guided by a Toxicity Score. Toxicon 2015, 104, 43–45. 10.1016/j.toxicon.2015.07.334. [DOI] [PubMed] [Google Scholar]
- Laustsen A. H.Recombinant antivenoms; University of Copenhagen, 2016. [Google Scholar]
- Knudsen C.; Laustsen A. Recent advances in next generation snakebite antivenoms. Trop. Med. Infect. Dis. 2018, 3, 42. 10.3390/tropicalmed3020042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynagh T.; Kiontke S.; Meyhoff-Madsen M.; Gless B. H.; Johannesen J.; Kattelmann S.; Christiansen A.; Dufva M.; Laustsen A. H.; Devkota K.; Olsen C. A.; Kümmel D.; Pless S. A.; Lohse B. Peptide inhibitors of the α-cobratoxin-nicotinic acetylcholine receptor interaction. J. Med. Chem. 2020, 63, 13709–13718. 10.1021/acs.jmedchem.0c01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo Y.; Lomonte B. Differential susceptibility of C2C12 myoblasts and myotubes to group II phospholipase A2 myotoxins from crotalid snake venoms. Cell Biochem. Funct. 2005, 23, 307–313. 10.1002/cbf.1208. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Maupetit J.; Derreumaux P.; Tufféry P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J. Chem. Theory Comput. 2014, 10, 4745–4758. 10.1021/ct500592m. [DOI] [PubMed] [Google Scholar]
- Kozakov D.; Hall D. R.; Xia B.; Porter K. A.; Padhorny D.; Yueh C.; Beglov D.; Vajda S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Meng E. C.; Couch G. S.; Croll T. I.; Morris J. H.; Ferrin T. E. UCSF ChimeraX : Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield N. J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kini R.; Sidhu S.; Laustsen A. Biosynthetic oligoclonal antivenom (BOA) for snakebite and next-generation treatments for snakebite victims. Toxins 2018, 10, 534. 10.3390/toxins10120534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albulescu L.-O.; Xie C.; Ainsworth S.; Alsolaiss J.; Crittenden E.; Dawson C. A.; Softley R.; Bartlett K. E.; Harrison R. A.; Kool J.; Casewell N. R. A therapeutic combination of two small molecule toxin inhibitors provides broad preclinical efficacy against viper snakebite. Nat. Commun. 2020, 11, 6094. 10.1038/s41467-020-19981-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador G. H. M.; Borges R. J.; Lomonte B.; Lewin M. R.; Fontes M. R. M. The synthetic varespladib molecule is a multi-functional inhibitor for PLA2 and PLA2-like ophidic toxins. Biochim. Biophys. Acta, Gen. Subj. 2021, 1865, 129913. 10.1016/j.bbagen.2021.129913. [DOI] [PubMed] [Google Scholar]
- Salvador G. H. M.; Gomes A. A. S.; Bryan-Quirós W.; Fernández J.; Lewin M. R.; Gutiérrez J. M.; Lomonte B.; Fontes M. R. M. Structural basis for phospholipase A2-like toxin inhibition by the synthetic compound Varespladib (LY315920). Sci. Rep. 2019, 9, 17203. 10.1038/s41598-019-53755-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M. T.; Arruda E. Z.; Melo P. A.; Martinez A. B.; Calil-Eliás S.; Tomaz M. A.; Lomonte B.; Gutiérrez J. M.; Arni R. K. Inhibition of myotoxic activity of Bothrops asper myotoxin II by the anti-trypanosomal drug suramin. J. Mol. Biol. 2005, 350, 416–426. 10.1016/j.jmb.2005.04.072. [DOI] [PubMed] [Google Scholar]
- Angulo Y.; Núñez C. E.; Lizano S.; Soares A. M.; Lomonte B. Immunochemical properties of the N-terminal helix of myotoxin II, a lysine-49 phospholipase A2 from Bothrops asper snake venom. Toxicon 2001, 39, 879–887. 10.1016/s0041-0101(00)00227-0. [DOI] [PubMed] [Google Scholar]
- Knudsen C.; Casewell N. R.; Lomonte B.; Gutiérrez J. M.; Vaiyapuri S.; Laustsen A. H. Novel snakebite therapeutics must be tested in appropriate rescue models to robustly assess their preclinical efficacy. Toxins 2020, 12, 528. 10.3390/toxins12090528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.