

Abstract
It has long been known (circa 1917) that environmental conditions, as well as speciation, can affect dramatically the frequency distribution of Spo11/Rec12-dependent meiotic recombination. Here, by analyzing DNA sequence-dependent meiotic recombination hotspots in the fission yeast Schizosaccharomyces pombe, we reveal a molecular basis for these phenomena. The impacts of changing environmental conditions (temperature, nutrients, and osmolarity) on local rates of recombination are mediated directly by DNA site-dependent hotspots (M26, CCAAT, and Oligo-C). This control is exerted through environmental condition-responsive signal transduction networks (involving Atf1, Pcr1, Php2, Php3, Php5, and Rst2). Strikingly, individual hotspots modulate rates of recombination over a very broad dynamic range in response to changing conditions. They can range from being quiescent to being highly proficient at promoting activity of the basal recombination machinery (Spo11/Rec12 complex). Moreover, each different class of hotspot functions as an independently controlled rheostat; a condition that increases the activity of one class can decrease the activity of another class. Together, the independent modulation of recombination rates by each different class of DNA site-dependent hotspots (of which there are many) provides a molecular mechanism for highly dynamic, large-scale changes in the global frequency distribution of meiotic recombination. Because hotspot-activating DNA sites discovered in fission yeast are conserved functionally in other species, this process can also explain the previously enigmatic, Prdm9-independent, evolutionarily rapid changes in hotspot usage between closely related species, subspecies, and isolated populations of the same species.
Keywords: recombination, meiosis, recombination hotspot, plasticity, evolution, Prdm9
Introduction
In meiosis cells express the broadly conserved Spo11/Rec12 protein which, along with other components of the basal meiotic recombination machinery, catalyzes the formation of double-strand DNA breaks (DSBs) that initiate homologous recombination (Lam and Keeney 2014; Robert et al. 2016; Jing et al. 2019). The broken chromosome uses its intact homolog as a template for repair, leading to gene conversion (recombination) on the initiating chromosome. A subset of these gene conversion events generate crossovers (reciprocal recombination) between the homologs. As is the case for transcription, meiotic recombination can occur anywhere in the genome, but its distribution is tightly regulated. In diverse species, distribution maps of recombination-initiating DSBs and/or recombination events (i.e., gene conversions, conversion-associated crossovers, or both) revealed that most recombination is tightly clustered at hotspots that regulate the frequency distribution of recombination across the genome (Wahls and Davidson 2012; Choi and Henderson 2015; Dluzewska et al. 2018; Ergoren 2018; Jing et al. 2019).
The global distribution of DSB hotspots can be defined with precision using several molecular tools (de Massy et al. 1995; Wu and Lichten 1995; Baudat and Nicolas 1997; Gerton et al. 2000; Blitzblau et al. 2007; Buhler et al. 2007; Pan et al. 2011). The concordance and reproducibility of the different approaches are exemplified by two, extensively studied, highly diverged organisms, fission yeast, and budding yeast. Experiments conducted using different DSB detection methods, at different times, and even in different laboratories have yielded similar patterns for the distribution of DSB hotspots within each of these species, differing primarily in the resolution and sensitivity of the assay employed (Gerton et al. 2000; Young et al. 2002; Blitzblau et al. 2007; Buhler et al. 2007; Cromie et al. 2007; Pan et al. 2011; Fowler et al. 2014). However, most of the intra-species comparative studies have used similar genetic backgrounds and experimental conditions, such as the media and temperature in which meiosis was induced. Remarkably, other experiments revealed that differences in metabolic states and environmental cues can trigger dramatic changes in the positioning of recombination at hotspots, even within isogenic or genetically identical strains.
Plasticity in the frequency distribution of meiotic recombination is a long-recognized, well-documented phenomenon (Plough 1917). For example, differences in sex or mating-type affect the distribution of recombination among hotspots of diverse species (e.g., fungi and mammals), as does the addition of an ectopic mating-type cassette (Parvanov et al. 2008; Kong et al. 2010; Hyppa et al. 2014; Brick et al. 2018). Both auxotrophies and nutritional states, such as the addition or removal of amino acids in the media, affect hotspot activity (Abdullah and Borts 2001; Cotton et al. 2009). Differences in parental mating type and the freezing of diploids each affect the activity of hotspots in subsequent meiosis, which suggests that there is epigenetic imprinting (Parvanov et al. 2008; Stahl et al. 2016). Such imprinting can also be inferred from the structure and composition of chromatin at hotspots (de Castro et al. 2011; Storey et al. 2018; Mukiza et al. 2019). Differences in temperature during meiosis affect patterns of recombination across the genomes of diverse species (Bomblies et al. 2015), and in two of these species the effects of temperature on the global distribution of recombination-initiating DSBs have been examined. In budding yeast only about 20% of DSB hotspots, as defined using a frequency threshold, occur at the same positions when meiosis is carried out at 14°C, 30°C, and 37°C (Zhang et al. 2017). In fission yeast differences in temperature affect the global distribution of DSBs (Hyppa et al. 2014) and rates of recombination (Brown et al. 2020) at the M26 class of DNA site-dependent hotspots (Kon et al. 1997; Steiner and Smith 2005). Hypothetically, such regulatory DNA sites and their binding/activator proteins might contribute to the plasticity of recombination positioning (Zhang et al. 2017; Mukiza et al. 2019). There are other, even more perplexing manifestations of plasticity. For example, while the transcription factor Prdm9 (which is present in a subset of metazoans) can modulate the initiation of recombination at its own DNA binding sites (Brick et al. 2018), the deletion of Prdm9 leads to the generation of new hotspots elsewhere in the genome (Brick et al. 2012; Mihola et al. 2021). A similar situation applies for transcription factors Bas1 and Ino4 of budding yeast, whose removal represses DSBs at some hotspots and induces DSBs at others (Mieczkowski et al. 2006; Zhu and Keeney 2015). In summary, environmental conditions and metabolic states can reshape—in some cases quite dramatically and by yet unknown mechanisms—the frequency distribution of meiotic recombination across the genomes of diverse taxa.
To gain insight into mechanisms for plasticity in the frequency distribution of meiotic recombination, we studied three exceptionally well-characterized classes of recombination hotspots in fission yeast. Each different class of hotspot is regulated by a discrete DNA sequence motif that has been defined functionally at single-nucleotide resolution by systematic, comprehensive, scanning base-pair substitutions in the genome (Table 1; Schuchert et al. 1991; Steiner et al. 2009, 2011). In each case, the binding of transcription factors to those DNA sites is essential for hotspot activity. The Atf1-Pcr1 heterodimer (of the ATF/CREB/AP-1 family) promotes recombination at M26 (CRE-like) DNA sites (Kon et al. 1997; Gao et al. 2008); Php2-Php3-Php5 complex activates recombination at CCAAT box DNA sites (Steiner et al. 2011); and Rst2 regulates recombination at Oligo-C DNA sites (Steiner et al. 2011). Notably, the removal of these proteins has little or no impact on the rates of recombination for well-matched, basal recombination controls that lack those DNA sites, demonstrating that the binding/activator proteins are hotspot-specific regulators of recombination. These disparate cis-acting regulatory modules each share a common downstream mechanism. Each protein-DNA complex triggers the displacement of nucleosomes to promote access of the basal recombination machinery (Spo11/Rec12 complex) to its DNA substrates within chromatin (Mukiza et al. 2019), thereby stimulating locally the frequency of recombination-initiating DSBs (Steiner et al. 2002; Wahls and Davidson 2010; Fowler et al. 2014). Here we reveal mechanisms by which these types of cis-acting regulatory modules can reshape the frequency distribution of meiotic recombination across the genome in response to environmental and metabolic cues.
Table 1.
DNA sequences of ade6 alleles
| ade6 allele | Relevant sequencea |
|---|---|
| 121 1461 | |
| ade6+ (wt) | AAACAAATTGA.TGGAGGACGTGAGCACAT… AGATGCCTCG |
| ade6-M26 | AAACAAATTGA.TGGAtGACGTGAGCACAT …AGATGCCTCG |
| ade6-CCAAT b | AAACAAATTGAtTGGAGGACGTGAGCACAT …AGATGCCTCG |
| ade6-Oligo-C | t AACAAATTGA . accccGcac TGAGCACAT …AGATGCCTCG |
| ade6-M375 | AAACAAATTGA.T t GAGGACGTGAGCACAT …AGATGCCTCG |
| ade6-M210 | AAACAAATTGA.TGGAGGACGTGAGCACAT … AGATGC t TCG |
The ade6 ORF is 1,659 bp in length and coordinates are numbered relative to the first nucleotide of the start codon (+1). The hotspot (M26, CCAAT, Oligo-C), basal recombination control (M375), and test-cross (M210) alleles differ from wild-type (wt) only by the indicated bp substitutions (lower case). The bp substitutions of the hotspot alleles create DNA binding sites (grey shading) for DNA sequence-specific, hotspot-activating binding proteins (Atf1-Pcr1 heterodimer, Php2-Php3-Php5 complex, and Rst2, respectively). Each allele also renders cells auxotrophic for adenine, allowing one to score for adenine prototrophic recombinants (ade6+) from heteroallelic crosses that contain a 5’ allele (M375, M26, CCAAT, or Oligo-C) and the 3’ allele (M210).
The consensus motif 5’-CCAATCA-3’ is on the complementary strand.
Materials and methods
Fission yeast husbandry
The DNA sequences of ade6 alleles used in this study are provided in Table 1 and the genotypes of all strains are listed in Supplementary Table S1. Strains were constructed using standard genetic techniques and were cultured in rich or minimal media supplemented as necessary with amino acids and bases at 100 µg/ml (Gutz et al. 1974; Forsburg and Rhind 2006; Gao et al. 2009). Mating and meiosis were carried out on sporulation agar (SPA) that contained 3% agar, 1% glucose, 0.1% K2HPO4, vitamins and any required supplements. SPA-glu contained 5% glucose and SPA-KCl contained 0.5M KCl. Spores were harvested when asci became abundant, which takes 3–4 days at our standard temperature of 25°C and up to 10 days at 15°C. Ectopic expression of rst2 was from the plasmid pREP3X-Rst2 (Takenaka et al. 2018), which uses the regulatable nmt1 promoter (Maundrell 1993); thiamine (0.5 µg/ml) was included in the media to maintain low expression levels (Forsburg 1993).
Measurements of meiotic recombination
Methods to determine rates of meiotic recombination were as described (Kon et al. 1997, 1998). In brief, haploid strains with different ade6 alleles (Table 1) were mated; then, haploid meiotic products (spores) were harvested and serial dilutions of spores were plated on minimal media that contained or lacked adenine. The titer of Ade+ recombinant spore colonies was divided by the titer of all viable spore colonies to yield the recombinant frequency of each cross. Frequencies reported in the figures are mean ± SD of values from three or more independent biological replicates of each cross.
Measurements of mRNA abundance
Quantitative, real-time, reverse-transcription PCR (qRT-PCR) was used to measure the abundance of mRNAs in cells (A595 = 0.5) that had been starved for nitrogen for 60 min to induce sexual differentiation. For each sample, total RNA from approximately 1 × 108 cells was extracted using the Quick-RNA Fungal/Bacterial Miniprep (Zymo Research, Irvine, CA, USA). For each sample, 1 µg of RNA was treated with TURBO DNase using the TURBO DNA-free™ Kit, which includes reagents for the digestion of DNA along with the removal of the enzyme and divalent cations post-digestion. Following DNase treatment, cDNA was synthesized from 400 ng RNA as template using the iScript cDNA Synthesis kit (BioRad, Hercules, CA, USA). The cDNA was used as template for qPCR using BlazeTaq SYBR Green qPCR Master Mix 2.0 (GeneCopeia, Rockville, MD, USA) and the PCR primers listed in Supplementary Table S2; reactions were carried out using a CFX96 Real Time System (BioRad, Hercules, CA, USA). Each qPCR reaction (10 µl) contained 1.0 µl of template and 200 nM of forward and reverse primers. Thermocycler parameters were: one cycle at 95°C for 10 min; followed by 40 cycles of 95°C for 10 s, 60°C for 20 s, and 72°C for 15 s. In each experiment, specificity was confirmed by melting point analyses from 65°C to 95°C at 0.5°C increments. For each transcript and experimental condition, fold change in mutant vs wild-type samples was calculated using the ΔΔCt method normalized to cam1 as the internal control (Livak and Schmittgen 2001; Schmittgen and Livak 2008). Calculations used the equation: ΔΔCt = [(Ct gene—Ct ref) in wild-type]—[(Ct gene—Ct ref) in mutant], where gene refers to transcript being measured (atf1, pcr1, php2, php3, php5, and rst2), and ref is the cam1 reference transcript. Values reported in the figures are mean ± SD of values from four independent biological replicates.
Measurements of protein-DNA binding
Reagents, methods and controls for chromatin immunoprecipitation (ChIP) of epitope-tagged Pcr1 (Pcr1-HA) were as previously described (Ekwall and Partridge 1999). Analyses included vegetative cells in log-phase growth and cells in meiotic prophase (Mukiza et al. 2019). Abundance of DNA from ChIP samples was determined by using quantitative PCR with the primers listed in Supplementary Table S2; relative enrichment within each sample (% ChIP signal vs input efficiency) was calculated by normalization to DNA abundance in aliquots of the same sample prior to immunoprecipitation. Values reported in the figures are mean ± SD of values from three independent biological replicates.
Statistics
Each experiment was conducted using three or more independent biological replicates. Two-sided t-test was used to calculate significance of differences between recombinant frequencies; linear least squares regression was used for population-level data on dose responses; 95% confidence intervals were used for gene expression values from the ΔΔCt approach. Differences with P ≤ 0.05 were judged to be significant statistically.
Results
All known DNA site-specific, recombination hotspot-binding/activating proteins of fission yeast (Atf1, Pcr1, Php2, Php3, Php5, and Rst2) are also transcription factors (Kon et al. 1997; Steiner et al. 2011). In their roles as environmental condition/stress-responsive transcription factors, each cis-acting regulatory module (protein-DNA complex) responds to positive and negative signals to control the rate of transcription of its target genes (e.g., Higuchi et al. 2002; Davidson et al. 2004; Mercier et al. 2008). Hypothetically, such signals could also modulate the rate of recombination at each class of DNA site-dependent hotspots. To test this hypothesis, we compared the effects of environmental and metabolic conditions on recombination at the DNA site-dependent hotspots. We included a well-matched basal recombination control (which lacks any hotspot-activating DNA sequence motif) to distinguish between hotspot-specific and nonspecific modes of regulation. To avoid any changes in the overall structure or spacing of elements in the genome, each allele used in this study was created by only one or a few base pair substitutions within the ade6 test locus (Table 1).
The M26, CCAAT, and Oligo-C DNA sites activate recombination hotspots under standard conditions
We compared the rates of meiotic recombination from crosses of strains bearing either a basal recombination control allele (M375) or one of three different hotspot DNA sequence motifs (M26, CCAAT, and Oligo-C) located near the 5′ end of the ade6 gene (Table 1). Haploid strains with these alleles, which are auxotrophic for adenine, were crossed to another adenine auxotroph that harbors a tester allele (M210) near the 3′ end of ade6 (Figure 1A). After mating and meiosis, haploid spores were plated and the spore colonies were scored for the frequency of Ade+ recombinants. Because meiosis is a single round event and the reversion rates of the alleles are negligible, the recombinant frequency provides a measure of recombination rate. In each case, the hotspot DNA sequence motifs (M26, CCAAT, and Oligo-C) promoted the rate of recombination substantially (by about 7-fold to 16-fold), relative to that of the basal recombination control (M375) (Figure 1B).
Figure 1.

DNA site-dependent recombination hotspots directly modulate rates of recombination in response to changing environmental conditions. (A) Diagram of genetic crosses showing relative positions of ade6 alleles (see Table 1 for sequences of alleles). (B) Rates of recombination under standard conditions for crosses with hotspot-activating DNA sequence motifs (M26, CCAAT, and Oligo-C) are compared to those of a basal recombination control (M375). (C) Effects of temperature (15°C, 20°C, 25°C, and 30°C) on recombination rates. Here and subsequently, rates under standard conditions are depicted for comparison (horizontal dashed lines). (D) Effects of increased carbon source (+Glu, 5% glucose) and hyperosmolarity (+KCl, 0.5 M) on rates are compared to those under standard conditions (std, 1% glucose and 0.0 M KCl). Data are mean ± SD from three independent biological replicates. In B and D, differences with P ≤ 0.05 are indicated (*) for hotspot vs control (B) and altered environmental conditions vs standard conditions (D). In C, all six possible pairwise comparisons within each data set were significantly different except for those indicated (ns, not significant).
These experiments were conducted using standard, widely employed conditions for mating and meiosis, which are incubation on SPA at 25°C until asci are abundant. We then changed one environmental variable at a time to determine their effects on the specific activities of the recombination hotspots. To facilitate visual comparisons of data in subsequent figure panels, values observed under standard conditions are shown using a horizontal, dashed line.
Effects of environmental conditions on local recombination rates are mediated directly and differentially by each class of hotspot
Changes in the environment that are encountered regularly by free-living, unicellular organisms (such as fission yeast) include those of temperature, energy source, and osmolarity. We therefore included each of these factors as variables in our experiments.
Effects of temperature
We varied the temperature of mating and meiosis, from 15°C to 30°C in 5°C increments, for crosses harboring the hotspot (M26, CCAAT, or Oligo-C) and basal recombination control (M375) alleles. For the basal recombination control, changing the temperature had no statistically significant impacts on recombination rates (Figure 1C). These findings indicate that the basal meiotic recombination machinery (Spo11/Rec12 complex) is proficient for initiating recombination in the face of, and is not affected appreciably by, changes in temperature from 15°C to 30°C. In sharp contrast, changing the temperature of meiosis triggered substantial changes in the rates of recombination for each of the DNA site-dependent hotspots (Figure 1C).
For the M26 hotspot, the differences in the temperature of meiosis led to an approximately 8.6-fold range in rates of recombination (Figure 1C). The rates were lowest at the coolest temperature (15°C) and, relative to that temperature, rose with sequential increases in temperature by about 5.2-fold (20°C), 6.9-fold (25°C), and 8.6-fold (30°C). Each of the differences between the six pair-wise comparisons was statistically significant. Because temperature had no significant impact on rates of recombination for the basal recombination control, we conclude that signals from environmental temperature are transduced though this DNA site-dependent hotspot to control local rates of recombination. We also conclude that individual DNA site-dependent hotspots can modulate recombination rates over a broad dynamic range.
For the CCAAT hotspot, varying the temperature led to an approximately 11-fold range in the rates of recombination (Figure 1C). Recombination was lowest at the coolest temperature (15°C) and, relative to that value, was stimulated at higher temperatures by about 2.9-fold (20°C), 8.9-fold (25°C), and 11-fold (30 C). Five of the six inter-sample differences were significant. We conclude that, like the M26 hotspot, the CCAAT hotspot controls local rates of recombination over a broad dynamic range in response to changes in temperature.
For the Oligo-C hotspot, the changes in temperature led to an approximately 4.1-fold range in recombination rates (Figure 1C). There was a statistically significant, biphasic response where the rates increased in the 15°C to 25°C range, then decreased between 25°C and 30°C. Thus, like the M26 and CCAAT hotspots, the Oligo-C hotspot directly mediates environmentally induced changes in local recombination rates. Like the other hotspots, this hotspot functions as a rheostat that modulates the activity of the basal recombination machinery. The lower dynamic range for control of recombination by this Oligo-C hotspot, relative to the others, is attributable to the fact that this hotspot retained substantial activity at the 15°C temperature, whereas the M26 and CCAAT hotspots behaved similarly to the basal control M375.
Although each class of DNA site-dependent recombination hotspots responded to changes in temperature, there were clear differences between the hotspots. There were statistically significant differences in minimum hotspot activity, maximum activity, dynamic range, optimal temperature, and temperature response profiles (Figure 1C). For example, at 15°C the Oligo-C hotspot promoted recombination substantially (3.5-fold higher recombination rate than for the basal recombination control M375), whereas the M26 and CCAAT hotspots did not (i.e., had rates equivalent to that of the basal recombination control). As another example, the Oligo-C hotspot had maximal activity at 25°C, whereas the M26 and CCAAT hotspots were most active at 30°C. Such differences in the data set revealed unambiguously that individual, discrete, seemingly modest changes in the environment can elicit substantially different (even opposite) dynamic responses among different classes of hotspots. For example, changing the temperature from 25°C to 30°C increased significantly the activity of the M26 hotspot while significantly reducing the activity of the Oligo-C hotspot. We conclude that each class of DNA site-dependent hotspots can respond independently and differentially, relative to other classes of hotspots, to the same environmental cues.
Effects of energy source
Our standard SPA contains 1% glucose as the energy source. We compared rates of recombination under these conditions to rates under conditions of elevated glucose (5%) (Figure 1D). This increase in glucose had no significant impacts on recombination rates for the basal recombination control. However, rates of recombination for the M26 hotspot were increased significantly, those for the CCAAT hotspot were unchanged, and there was a nonsignificant decrease for the Oligo-C hotspot. We conclude that signals from the environmental abundance of this key nutrient are transduced through at least one of these DNA site-dependent hotspots to control local rates of recombination. We do not mean to imply that the other hotspots are insensitive to carbon source availability; they might be affected by glucose concentrations or energy sources not tested in this study. Notwithstanding this possibility, the observed differential responses further support the idea that each class of hotspots can be regulated independently by its own constellation of signals.
Effects of osmolarity
Our standard SPA formulation contains no osmolytes or salts other than the 7 mM potassium phosphate buffer. As the variable, we supplemented the SPA with 500 mM KCl (Figure 1D). For the basal recombination control (M375), this increase in osmolarity led to a significant decrease in the rates of recombination. In contrast, there were significant increases in recombination rates for the M26 and CCAAT hotspots, as well as a significant reduction in rates for Oligo-C. Thus, as for changes in temperature and glucose, different classes of DNA site-dependent hotspots respond differentially to changes in osmolarity. However, in this case (osmolarity), the magnitude of the hotspot-specific effects is partially obscured by the fact that KCl also triggered a reduction in basal recombination. In such cases, the hotspot-specific component of control can be ascertained by comparing hotspot activity ratios, which are the rates of recombination for a given hotspot under a given condition divided by the rates for the basal recombination control under the same conditions. By these criteria (using M375 control-normalized data), the hotspot-specific impacts of adding KCl were about 3.1-, 3.6-, and 1.3-fold increases in recombination, respectively, for the M26, CCAAT, and Oligo-C hotspots. Both ways of considering the data, either by measuring absolute levels of recombination or by taking into account the hotspot-independent impacts on KCl on basal recombination, support the same conclusions: Signals from changes in osmolarity, like those from changes in temperature and nutrients, are transduced through DNA site-dependent hotspots to help control local rates of recombination. Moreover, as seen for the other environmental cues, the impacts of osmolarity on local rates of recombination are mediated differentially by distinct classes of hotspots.
Key conclusions and overarching model for the plasticity of meiotic recombination
Together these results support three fundamental conclusions. First, the impacts of environmental conditions on local rates of meiotic recombination are mediated directly and primarily by DNA site-dependent recombination hotspots. Second, individual DNA site-dependent hotspots can modulate rates of recombination over a very broad dynamic range—even in the same, genetically identical strain. They can, in response to changing conditions, range from not promoting recombination beyond basal levels to being highly active at promoting local functions of the basal meiotic recombination machinery (Spo11/Rec12 complex). Third, different classes of DNA site-dependent hotspots respond independently and differentially, relative to other classes of hotspots, to environmental cues. For example, discrete changes in the environment that increase the activity of one class of hotspots can decrease the activity of another class of hotspots. These striking discoveries revealed a molecular basis for the environmentally induced plasticity of meiotic recombination. Importantly, the independent modulation of recombination rates over a broad dynamic range by each different class of hotspots can trigger substantial, highly dynamic changes in the global frequency distribution of recombination (see model in Figure 7 of the Discussion section).
Figure 7.

Model for plasticity in the distribution of meiotic recombination. The magnitudes of dynamic changes in relative hotspot activity depicted here are based on those defined experimentally in this study. For the sake of illustration, rates of recombination (arbitrary units) were assigned at random for low (rate range of 1–5), medium (11–15), and high (21–25) activity of individual hotspots. (A) Conceptual distribution of three different types of hotspot-activating DNA sequence motifs (a, blue; b, orange; c, gray) along the chromosome. (B) Each different class of DNA site-dependent hotspots is regulated independently, through its own binding/activating proteins, in response to changes in environmental and metabolic conditions. (C–E) Contribution of each DNA site to the rate of recombination in its vicinity (blue, orange, and gray lines) under the experimental conditions listed in B. (F–H) Net impact of the three classes of regulatory DNA sites on the overall distribution of recombination (black lines) under the three different conditions. Note substantial differences in the frequency distribution of recombination events. Also note that if one applies a cutoff value for what is a hotspot (e.g., local recombination rate ≥ 10), as is often done in the literature, the number of annotated hotspots and their apparent positions change substantially from condition to condition.
Components of environmental condition-responsive signaling pathways control the rate of recombination at each class of hotspots
The impacts of environmental conditions on hotspot activity (Figure 1) must be transduced by intracellular signaling pathways. In support of this idea, the activation of M26-class hotspots is affected positively and negatively by multiple different signaling pathways, including those that respond to environmental and metabolic cues (Kon et al. 1998; Mizuno et al. 2001; Hirota et al. 2003, 2004, 2007, 2008; Yamada et al. 2004; Gao et al. 2009; Storey et al. 2018). Notably, each of the hotspot-binding/activating proteins (Atf1, Pcr1, Php2, Php3, Php5, and Rst2) is also a transcription factor that can affect the expression of target genes directly via binding to their promoters and indirectly via inter-pathway connections (e.g., Davidson et al. 2004). This raises an interesting question with regard to the activation of meiotic recombination hotspots. Do the hotspot-activating proteins control exclusively hotspot activity at their own DNA sites, or can they also help to control DNA site-dependent hotspots to which they do not bind? To address this question, we analyzed rates of recombination in null mutants lacking each of the hotspot-binding/activating proteins, and we included all possible pairwise combinations of the activator proteins and DNA sequence motifs.
Direct control of hotspots
Each of the hotspot DNA sequence motifs (M26, CCAAT, and Oligo-C) promoted substantially the rate of recombination, relative to a basal recombination control that lacks those DNA sites (Figure 1B). Removing either subunit of the Atf1-Pcr1 heterodimer, which binds to the M26 DNA site, greatly reduced the rate of recombination at the M26 hotspot (Figure 2A). The amount of recombination that remained was similar to that of the basal recombination control allele in wild-type cells. Likewise, subunits of the Php2-Php3-Php5 (CCAAT box-binding) complex were required for CCAAT motif-promoted recombination (Figure 2B) and Rst2 was required for hotspot activity of its DNA binding site, Oligo-C (Figure 2C). These results support a specific conclusion that is germane to the topic of this study: Known components of environmental condition/stress-responsive signal transduction pathways (transcription factors Atf1, Pcr1, Php2, Php3, Php5, and Rst2) control local rates of recombination directly via their own DNA binding sites. We next asked if these proteins could also help to regulate in trans other classes of DNA site-dependent hotspots.
Figure 2.

Components of environmental condition-responsive signaling pathways control DNA site-dependent recombination hotspots in cis and in trans. The effects of each indicated protein on the activation of each class hotspots was measured; proteins that bind directly to each hotspot DNA site are highlighted (bold). Relative rates of recombination in null mutant vs wild-type are grouped by class of hotspot: (A) M26 DNA site; (B) CCAAT DNA site; (C) Oligo-C DNA site. Data are mean ± SD from at least three independent biological replicates; differences with P ≤ 0.05 (*) are indicated for mutant vs wild-type.
Indirect control of hotspots
The removal of the Php2, Php3, or Php5 proteins (which bind to and promote recombination at the CCAAT motif) had no significant impact on rates of recombination at the M26 hotspot (Figure 2A). Similarly, the deletions of Php2 and Php3 did not affect appreciably the activation of the Oligo-C hotspot (Figure 2C). Likewise, the removal of the Rst2 protein (which binds to and promotes recombination at the Oligo-C motif) had no significant impact on the rates of recombination at the M26 hotspot (Figure 2A) or at the CCAAT hotspot (Figure 2B). These findings suggest that the Php2, Php3, and Rst2 proteins function with high specificity to promote recombination at their own DNA binding sites, although it is possible that these proteins help to control other, distinct classes of DNA site-dependent hotspots not examined in this study.
The binding of Atf1-Pcr1 heterodimer to M26 DNA sites directly activates this class of hotspots. Strikingly, the removal of Pcr1 strongly reduced (by about 74% and 70%, respectively) rates of recombination at the CCAAT and Oligo-C hotspots (Figure 2, B and C). Removing Atf1 also reduced recombination (by 46% and 30%, respectively) for CCAAT and Oligo-C (Figure 2, B and C). Similarly, the removal of Php5 significant reduced the activity of the Oligo-C hotspot (Figure 2C). We conclude that hotspot-activating transcription factors, which are also key components of environmental condition/stress-responsive regulatory networks, can control the activation of heterologous DNA sequence-dependent hotspots to which they do not bind. The possibility that these trans effects are mediated by the indirect recruitment of the activator proteins is explored in greater detail below.
Hotspot-activating proteins are rate-limiting for promoting, and thus can modulate, recombination at their own DNA binding sites
Results described in the previous two sections revealed that extracellular cues (Figure 1) and components of intracellular signal transduction networks (Figure 2) each modulate the activity of DNA site-dependent recombination hotspots. For this to occur, the cis-acting regulatory modules (protein-DNA complexes) must serve as variable-output effectors of those signals. A key prediction of this hypothesis is that each class of DNA sequence motif-dependent hotspots should be sensitive to the abundance of their binding/activating proteins, which we tested as follows.
To test for dose-dependent responses, we compared rates of recombination for each hotspot DNA sequence motif (M26, CCAAT, and Oligo-C) in meioses that were homozygous wild-type, heterozygous wild-type/null mutant, and homozygous null mutant for their respective binding proteins. Essentially identical results were observed for each of the six different hotspot-activating proteins (Atf1, Pcr1, Php2, Php3, Php5, and Rst2). In every case, the rate of recombination in the heterozygotes was intermediate between that of homozygous wild-type (full hotspot activity) and homozygous null mutant (no hotspot activity) (Figure 3). Linear regression analysis of the entire data set revealed a robust, positive correlation between dose and recombination rate (R2 = 0.89, P < 0.0001). We confirmed this dose-dependent response using second experimental approach that alters the expression of genes without changing their copy numbers (results presented in the next section). The dose-dependent responses, observed using two different approaches, support an important conclusion: Each hotspot-activating protein is rate-limiting for promoting recombination at its DNA binding site. Consequently, any factor (cellular or environmental) that affects the abundance or functionality of a particular, DNA sequence-specific, hotspot-binding/activating protein will affect rates of recombination at the corresponding class of DNA site-dependent hotspots in the genome.
Figure 3.

Hotspot-binding/activating proteins are rate-limiting for promoting recombination at their own DNA binding sites. Recombination was measured in crosses that were homozygous wild-type, heterozygous wild-type/null mutant, and homozygous null mutant for hotspot-activating proteins. The heterozygous crosses were in two configurations. In the first configuration (+/Δ), the parent with the hotspot allele was wild-type for the binding protein (e.g., genotype ade6-M26 atf1+) and the parent with the test-cross allele was null mutant for the binding protein (e.g., ade6-M210 atf1Δ). In the second configuration (Δ/+), the parent with the hotspot allele was null mutant for the binding protein (e.g., ade6-M26 atf1Δ) and the parent with the test-cross allele expressed the binding protein (e.g., ade6-M210 atf1). Rates of recombination are plotted relative to those in homozygous wild-type and are grouped by class of hotspot: (A) M26 DNA site; (B) CCAAT DNA site; (C) Oligo-C DNA site. Data are mean from three or more independent biological replicates; differences with P ≤ 0.05 are indicated for mutant vs wild-type (*).
The control of hotspots is mediated in part by altering expression of the rate-limiting binding/activator proteins
The Atf1 and Pcr1 proteins, which are environmental condition/stress-responsive transcription factors, each controlled the activity of DNA sequence-dependent hotspots that do not contain in their vicinity any binding sites for Atf1-Pcr1 (Figure 2). For example, the removal of Pcr1 reduced by 74% and 70%, respectively, rates of recombination at the CCAAT and Oligo-C hotspots. Hypothetically, such control could be mediated in trans by altering the expression or activity of corresponding binding/activator proteins. Alternatively, the M26 hotspot-binding/activating protein complex (Atf1-Pcr1 heterodimer) could be recruited by the heterologous hotspots and help to control them in cis. We therefore sought to distinguish between these modes of regulation.
Atf1-dependent, Pcr1-dependent control of the CCAAT and Oligo-C hotspots is not mediated by the recruitment of Atf1-Pcr1
ChIP of the Atf1-Pcr1 heterodimer revealed that it binds specifically at M26 DNA sequence motifs, but not at appreciable levels to our ade6 test locus in the absence of this DNA site (Eshaghi et al. 2010). We used the same approach to test whether presence of the CCAAT and Oligo-C DNA sites at ade6 can recruit Pcr1. As an internal, previously validated, positive control, we gauged binding at the hsp9 promoter; as an internal negative control, we used the cdc18 ORF (Sanso et al. 2008). We analyzed, and obtained equivalent results for, protein-DNA binding in vegetative cells and cells in meiotic prophase (Figure 4). There was about a 25-fold higher ChIP signal for the binding of Pcr1 to the positive control hsp9 than to the negative control cdc18. Under these conditions, we detected no appreciable binding of Pcr1 to either the CCAAT or the Oligo-C hotspots. We therefore reject the hypothesis that the recruitment of Pcr1 or the Atf1-Pcr1 heterodimer to these hotspots contributes substantially to their activation.
Figure 4.

Pcr1-dependent control of the CCAAT and Oligo-C hotspots is not mediated by the recruitment of Pcr1. Chromatin immunoprecipitation of Pcr1 was used to test whether it is recruited to the test locus (ade6) by the CCAAT and Oligo-C hotspots in vegetative cells (mitosis) and cells in meiotic prophase (meiosis). Signal intensities from ChIP vs the input material used for ChIP (ChIP signal %) were compared to those of internal positive (hsp9) and negative (cdc18) controls. Data are mean ± SD from three independent biological replicates.
Control of the CCAAT and Oligo-C hotspots is mediated in part by regulating expression of their binding/activator proteins
We next tested whether Atf1 and Pcr1 regulate the expression of genes encoding the proteins that bind to and activate the CCAAT box hotspot (Php2-Php3-Php5 complex) and the Oligo-C hotspot (Rst2). We used conditions for inducing sexual differentiation that are known to trigger Atf1-Pcr1 heterodimer-dependent changes in the expression of both its direct and indirect targets, such as cgs2 and ste11 (Davidson et al. 2004). Quantitative, real-time, reverse-transcription PCR (qRT-PCR) was used to compare the relative abundance of each mRNA in null mutant cells vs wild-type (Figure 5). It was reported previously, based on quantitative Northern blot analyses of gene expression under conditions similar to those used in this study, that Atf1 promotes the expression of pcr1 and that Pcr1 represses the expression of atf1 (Kon et al. 1998). We obtained concordant results using qRT-PCR: removing Atf1 reduced the expression of pcr1 by 40% (1.7-fold difference) (Figure 5A) and ablating Pcr1 increased by 2.9-fold the expression of atf1 (Figure 5B).
Figure 5.

Hotspot-activating proteins can regulate the expression of other hotspot-activating proteins. Quantitative, reverse-transcription PCR was used to determine the effects of Atf1 and Pcr1 on the expression of the six genes (atf1, pcr1, php2, php3, php5, and rst2) that encode hotspot-binding/activating proteins. Gene expression levels in mutants are plotted as % relative to wild-type for: (A) atf1Δ mutant; (B) pcr1Δ mutant. Data are mean ± SD from four independent biological replicates; differences with P ≤ 0.05 are indicated for mutant vs wild-type (*). (C) Conclusions from this and prior figures. The impacts of environmental conditions on meiotic recombination are mediated via cis-acting regulatory modules (site-specific protein-DNA complexes). Transcriptional regulation of hotspot-binding/activating proteins and cross-talk between regulatory networks each contribute to the control of hotspots. Arrows depict transcription-mediated pathway connections elucidated in this study (solid lines in upper half of panel), as well as connections where hotspot control in trans was also observed, but where there were no significant changes in expression of the genes encoding the respective activator proteins (dotted lines).
Overall, considering all six genes analyzed, the removal of Atf1 reduced significantly the expression of pcr1, php2, and php3 (Figure 5A); whereas the removal of Pcr1 significantly increased the expression of atf1 and reduced that of rst2 (Figure 5B). We conclude that some hotspot-activating proteins (which are also transcription factors) can regulate the expression of other hotspot-activating proteins. Our finding that deletions of atf1 and pcr1 affected differentially the expression of the other genes was not surprising, given that Atf1 and Pcr1 (like bZIP proteins in general) can form combinatorial homodimers and heterodimers, each of which regulates the expression of a different set of genes (Wahls and Smith 1994; Kon et al. 1998; Sanso et al. 2008; Eshaghi et al. 2010).
The regulated expression of genes encoding hotspot-binding/activating proteins, coupled with the finding that those activator proteins are rate-limiting for promoting recombination at their own DNA sites (Figure 3), provided insight into mechanisms by which hotspots can be controlled in trans. The removal of Pcr1 led to an 84% reduction in the expression of rst2 (Figure 5B) and, correspondingly, reduced by 70% the rate of recombination at the Oligo-C hotspot (Figure 2C), which is activated by the binding of Rst2. Similarly, the removal of Atf1 led to reductions in the expression of php2, php3, and php5 (reduced by 39%, 35%, and 19%, respectively) (Figure 5A). This led, correspondingly, to a 46% reduction in the rate of recombination at the CCAAT hotspot (Figure 2B), which is activated by the binding of the Php3-Php3-Php5 complex. These findings indicate that the regulation of the Oligo-C and CCAAT hotspots are each mediated, at least in part, via the transcriptional control of the respective hotspot-binding/activating proteins (pcr1 → rst2 → recombination at Oligo-C hotspot; atf1 → php2, php3 → recombination at CCAAT hotspot; see pathway diagram in Figure 5C). This type of signal transduction mechanism, exerted through modulating the expression of rate-limiting activator proteins, has broad implications for how diverse signaling networks can affect dynamically the frequency distribution of recombination across the genome. We do not mean to imply that this is the sole or even the primary mechanism for controlling hotspot activity; additional, transcription-independent modes of control could also impinge upon the rate-limiting, hotspot-binding/activating proteins.
Deficient activation of the Oligo-C hotspot in pcr1Δ cells is complemented by ectopic expression of rst2
Our conclusion that there are transcription-based pathway mechanisms for the control of meiotic recombination hotspots (e.g., pcr1 → rst2 → recombination at Oligo-C hotspot) stemmed from—and required—experiments in which the regulatory transcription factors were deleted. However, deleting transcription factors can have pleiotropic effects. We therefore sought confirmation that dysregulation of the rst2 mRNA in pcr1Δ mutants (Figure 5B) is a specific factor that compromises activation of the Oligo-C hotspot in the in pcr1Δ strains (Figure 2C). If so, then adding back some of the missing rst2 transcript should restore functionality. To test this, we measured rates of recombination at the Oligo-C hotspot in the presence and absence of Rst2 (its binding/activator protein), Pcr1, and a plasmid that expresses rst2 from a heterologous promoter (Figure 6). The removal of Rst2 abolished hotspot activity of its DNA binding site (Oligo-C) and ectopic expression of rst2 from the plasmid restored hotspot activation. Thus, the rst2 mRNA (and Rst2 protein) expressed from this plasmid is biologically active. The removal of Pcr1 strongly reduced (by about 80%) the rate of recombination at the Oligo-C hotspot, and this defect was complemented substantially by the ectopic expression of rst2. The ectopic expression of rst2 did not fully restore recombination to wild-type levels in the pcr1Δ strain, suggesting that the deletion of pcr1 has effects other than downregulation of rst2 expression, as expected for a transcription factor with several other roles. Nevertheless, the substantial complementation provides independent validation of our conclusion that the control of the Oligo-C hotspot in trans by Pcr1 is mediated in part by the Pcr1-dependent transcriptional regulation of rst2.
Figure 6.
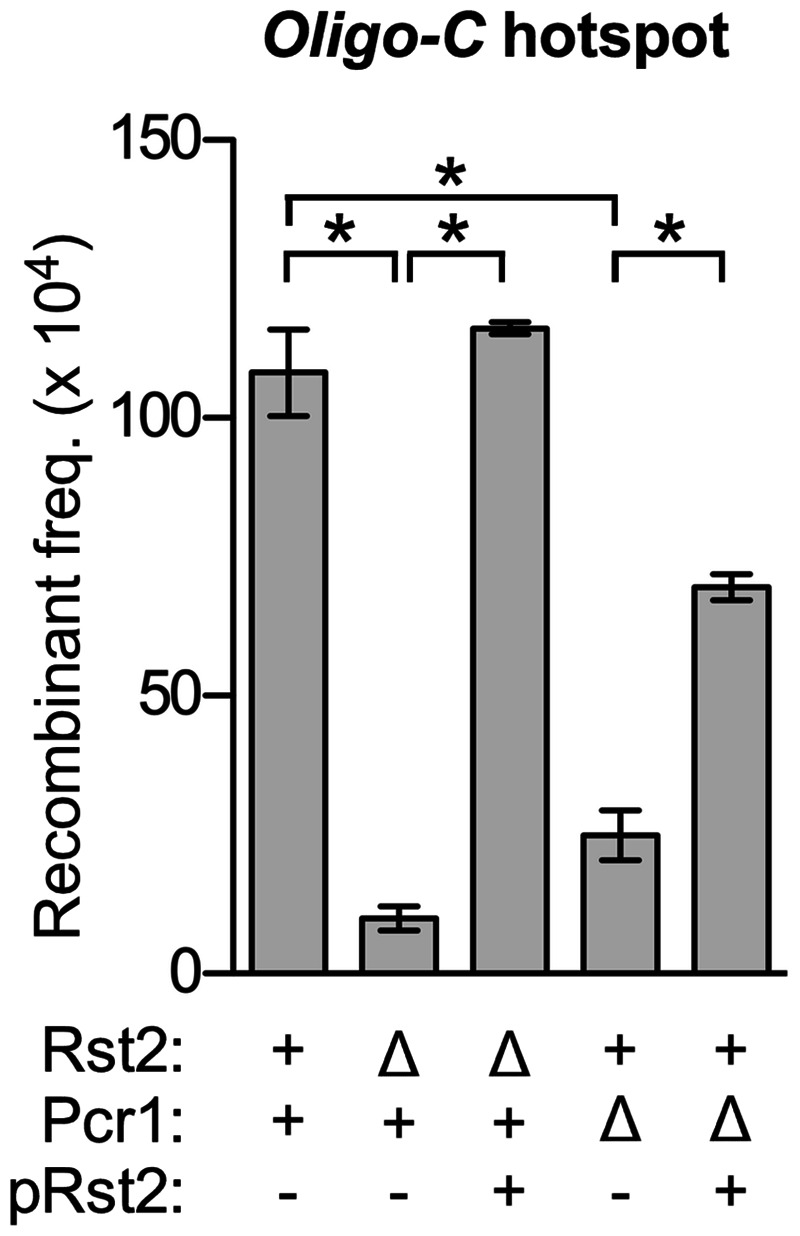
Ectopic expression of rst2 from a heterologous promoter complements deficient activation of the Oligo-C hotspot in pcr1Δ cells. Plot shows rates of recombination for the Oligo-C hotspot in the presence and absence of its binding/activating protein (Rst2), an environmental condition-responsive transcription factor that does not bind to the Oligo-C hotspot (Pcr1), and a plasmid that expresses rst2 ectopically from a heterologous promoter (pRst2). Data are mean ± SD from three independent biological replicates; relevant significant differences are indicated (*).
Evidence for additional, transcription-independent regulatory mechanisms
Interestingly, our qRT-PCR data also revealed that the regulated transcription of genes encoding hotspot-binding/activating proteins cannot be the sole mechanism for regulating hotspots in trans. For example, Pcr1 was strongly required for the activation of the CCAAT box hotspot: rates of recombination in the pcr1Δ mutant were only 26% of those in wild-type (Figure 2B). However, this removal of Pcr1 had no significant impact on the expression of php2, php3, and php5 (Figure 5B). The expression levels of these three genes in the pcr1Δ mutant were 115%, 109%, and 99% of those in wild-type cells. These findings are inconsistent with a model in which the Pcr1-dependent control of this hotspot is exerted via altering the expression of its binding/activating proteins, Php2, Php3, and Php5. We infer that there must be at least one additional mechanism for the control of DNA site-dependent hotspots in trans.
Discussion
In diverse taxa, environmental cues and metabolic states can reshape dramatically the frequency distribution of meiotic recombination. To explore potential mechanisms for these phenomena, we analyzed the control of recombination by each type of DNA site-dependent recombination hotspot in fission yeast whose binding/activator proteins are known. About 200 additional, distinct DNA sequence elements also activate recombination hotspots in fission yeast, but their binding proteins are unknown (Steiner et al. 2009). Processes that modulate the activities of hotspots uncovered in this study likely apply to many, if not most of those other DNA sequence-dependent hotspots. Moreover, because the regulation of hotspots by specific DNA sites and their binding proteins is conserved between species whose latest common ancestor occurred about 400 million years ago (Sipiczki 2000; Steiner and Steiner 2012; Wahls and Davidson 2012) and is implicated by association to be even more widely conserved (e.g., Mougel et al. 2014), the mechanisms revealed by this study likely apply broadly across taxa.
A molecular basis for environmentally and metabolically induced plasticity in the frequency distribution of meiotic recombination
Our experiments revealed the following. First, the impacts of differences in various environmental conditions (temperature, energy source, and osmolarity) on local rates of recombination are mediated directly and primarily by DNA site-dependent recombination hotspots (Figure 1). Second, and correspondingly, components of intracellular signal transduction pathways that respond to environmental conditions control local rates of recombination through DNA site-specific hotspots (Figures 2 and 3). Third, individual hotspots can modulate rates of recombination over a broad dynamic range—even in the same (i.e., genetically identical) strain (Figure 1). They can, in response to changing conditions, range from not promoting recombination beyond basal levels to being highly active at promoting local functions of the basal recombination machinery. Fourth, relative to each other, different classes of DNA site-dependent hotspots respond differentially to the constellation of environmental cues and metabolic conditions (Figures 1 and 2). For example, discrete changes in the environment that increase the activity of one class of hotspots can reduce the activity of another class of hotspots. These findings revealed—more than a century after environmentally induced plasticity was discovered (Plough 1917)—a molecular basis for dramatic changes in the recombination landscape in response to extracellular and intracellular conditions. Together, the independent modulation of recombination rates by each different class of DNA site-dependent hotspots can remodel dynamically and extensively the distribution of recombination across the genome (see model in Figure 7).
Multiple mechanisms for controlling recombination rates via hotspot-activating proteins—with implications for complex regulatory networks and the Butterfly Effect
In addition to revealing molecular processes by which extracellular and intracellular conditions can reshape the global frequency distribution of meiotic recombination, our results provide insight into discrete mechanisms that control protein-DNA complex-dependent recombination hotspots.
The first type of mechanism for controlling hotspot activity is exerted through controlling the abundance of the hotspot-binding/activating proteins, which are rate-limiting for promoting recombination at their own DNA sites (Figure 3). For example, removing the environmental condition/stress-responsive transcription factor Pcr1 led to an 84% reduction in the expression of rst2 (Figure 5B) and, correspondingly, led to a 70–80% reduction in recombination at the Oligo-C hotspot (Figures 2B and 6), which is activated by the binding of Rst2. We obtained evidence for the use of this same type of transcription-mediated pathway mechanism for controlling two different classes of hotspots (pcr1 → rst2 → recombination at Oligo-C hotspot; atf1 → php2, php3 → recombination at CCAAT hotspot; see pathway diagram in Figure 5C). Moreover, we confirmed that ectopic expression of the dysregulated gene from a heterologous promoter complements the hotspot-activation defect (Figure 6). We note that any other factors (cellular or environmental) whose signals modulate the expression of the rate-limiting, hotspot-binding/activating proteins would also modulate rates of recombination at their DNA sites of action.
A second type of mechanism for controlling hotspot activity can be exerted by changing the functionality of hotspot-binding/activating proteins without necessarily changing their abundance. Our finding that Pcr1 strongly controls activation of the CCAAT box hotspot (Figure 2B) without affecting significantly the expression of php2, php3, or php5 (Figure 5B) supports this idea. There are several ways to achieve robust, transcription-independent control of hotspots. For example, signals that affect the subcellular localization, DNA binding affinity, or specific activity of rate-limiting, hotspot-binding/activating proteins will control rates of recombination at their DNA sites of action. An excellent case in point is the hotspot-activating Atf1-Pcr1 heterodimer, whose functions are controlled by each of these three different processes (Gaits et al. 1998; Kon et al. 1998; Neely and Hoffman 2000; Lawrence et al. 2007; Gao et al. 2008, 2009).
A third, currently hypothetical type of mechanism for controlling hotspot activity lies in the fact that each of the cis-acting regulatory modules analyzed in this study promotes recombination through downstream chromatin remodeling pathways (Mukiza et al. 2019). Consequently, any signals that affect chromatin remodelers that help to activate a given class of hotspots would contribute to the regulation of those hotspots. A recent, proteomics-based approach revealed that many different chromatin remodeling enzymes and histone PTMs function in concert to activate individual hotspots (Storey et al. 2018), so there are many potential targets for the modulation of hotspot activity at this level.
The multiple different modes of hotspot control are not mutually exclusive and likely operate in concert with each other. Moreover, as with other biological processes, the control of recombination hotspots occurs within the context of, and is likely affected by, complex regulatory networks. Indeed, our discovery that hotspot-binding/activating proteins can help to control the activation of DNA site-dependent hotspots to which they do not bind (Figures 2, 4–6) provides a striking example of how inter-pathway cross-talk can help to shape the positioning of meiotic recombination.
In summary, there are many different classes of DNA sequence-dependent hotspots and multiple different ways to modulate rates of recombination at each class of hotspots via their own, rate-limiting, hotspot-binding/activating proteins. Each of these known and hypothetical mechanisms provides a nexus for the control of hotspot activity by other factors and signaling networks. As in the Butterfly Effect, even minor perturbations to the biological system can, through the diversity of hotspots, inter-pathway connections, and regulatory networks, have large impacts on the overall distribution of recombination (see model in Figure 7).
A molecular basis for the off-target effects of hotspot-activating proteins in other species
In mice and rats the sequence-specific, hotspot-activating protein Prdm9 represses the activity of some hotspots to which it does not bind (Brick et al. 2012; Mihola et al. 2021), although it is not yet known whether those other hotspots are controlled by their own DNA sites. Similarly, transcription factors Bas1 and Ino4 of budding yeast affect the frequency distribution of DSBs for hotspots both at their DNA binding sites and elsewhere (Mieczkowski et al. 2006; Zhu and Keeney 2015), although those studies did not test for DNA sequence dependence of the presumptively direct or indirect effects. In contrast, this study revealed dependent pathway relationships between multiple different proteins and DNA sites (e.g., Figure 2). Moreover, we identified molecular mechanisms for inter-pathway connections (Figures 4–6). Notably, Prdm9, Bas1, and Ino4 are each transcription factors—as is each currently known, hotspot-binding/activating protein in fission yeast. Thus, the previously enigmatic, off-target effects of Prdm9, Bas1, and Ino4 on the distribution of recombination (Mieczkowski et al. 2006; Brick et al. 2012; Zhu and Keeney 2015; Mihola et al. 2021) are readily explained by our findings and model. Deleting a given hotspot-binding/activating protein (transcription factor) will affect not only the rate of recombination at its own DNA binding sites. It will also affect, via transcription-dependent regulatory networks, all biological processes downstream of that transcription factor. Some of those signals will impinge (positively or negatively) on the binding/activator proteins for other classes of DNA site-dependent recombination hotspots (as documented in Figures 2, 5, and 6), thereby mediating the off-target changes in local recombination rates elsewhere in the genome.
A molecular basis for the evolutionarily rapid “repositioning” of Prdm9-independent hotspots
A subset of metazoans (e.g., mice, cattle, and primates) express the sequence-specific DNA binding protein Prdm9, whose binding sites activates meiotic recombination hotspots located in intergenic regions (reviewed by Grey et al. 2018; Paigen and Petkov 2018). Species that lack Prdm9 or one of its crucial domains (e.g., fungi, birds, amphibians, many fishes, canids, marsupials, and plants) still have hotspots which tend to cluster in a “yeast-like” fashion around functional elements such as promoters (e.g., Munoz-Fuentes et al. 2011; Axelsson et al. 2012; Kawakami et al. 2017). Moreover, Prdm9-expressing species can still activate hotspots that cluster at promoters (remote from Prdm9 binding sites) (Schield et al. 2020), as do mice and rats when Prdm9 is deleted (Brick et al. 2012; Mihola et al. 2021). There seems to be an evolutionarily ancient, Prdm9-independent mechanism for distributing recombination to hotspots. This mechanism likely involves a constellation of hotspot-activating DNA sites like those discovered in fission yeast, the majority of which are known to be conserved functionally between species whose latest common ancestor was about 400 million years ago (Steiner and Steiner 2012; Wahls and Davidson 2012) and that are implicated by association to be even more widely conserved (e.g., Mougel et al. 2014).
Prdm9 is among the most rapidly evolving proteins known and the changes in its zinc finger domain change its DNA site specificity, thereby relocating Prdm9-dependent hotspots over short evolutionary time scales. It has been assumed that this property is unique to Prdm9-class hotspots and that the positions of other types of hotspots are more stable evolutionarily, “even over tens of millions of years of evolution” (e.g., Grey et al. 2018). However, a growing body of evidence suggests that the positions of Prdm9-independent hotspots can change rapidly over evolutionary time scales. For example, in flycatcher birds there are substantial differences in the positions of annotated hotspots between pairs of closely related species (55–61% of hotspots in different locations) and between isolated populations of the same species (31–49% in different locations) (Kawakami et al. 2017). Similarly, about 80% of mapped hotspots are at different locations in subspecies of rice (Marand et al. 2019). Likewise, about 85% of inferred hotspots are in different positions in isolated populations of stickleback fish, which became separated from each other within the last 15 thousand years (Shanfelter et al. 2019). Such evolutionarily rapid changes in the mapped positions of Prdm9-independent recombination hotspots are perplexing because the low amounts of genomic DNA sequence divergence involved preclude the possibilities of large-scale changes in the DNA sequences of cis-acting regulatory elements or in the DNA binding site specificities of the various binding/activator proteins. A solution to this quandary lies in our discovery of mechanisms that can immediately affect local recombination rates, resulting in an overall redistribution of recombination (Figure 7). Importantly, these mechanisms do not require any changes in the distribution of the regulatory DNA sites themselves.
The inference that the positions of hotspots can change rapidly over evolutionary time scales and the implications of such movement have each been influenced strongly by the standard practice of defining recombination hotspots based on either/or cutoff frequency values. However, as shown directly in this study, and as can also be inferred indirectly from data in other studies (Pengelly et al. 2016; Kawakami et al. 2017; Shanfelter et al. 2019), individual, Prdm9-independent hotspots actually function as rheostats that can modulate rates of recombination over a very broad dynamic range (Figure 1). Moreover, as little as a single heterology in the genome can be sufficient to strongly adjust these rheostats, as shown directly by mutating individual components of signal transduction networks (Figure 2). We therefore posit that even minor genetic or metabolic differences between species, subspecies and isolated populations can; by affecting signal transduction networks that differentially control each class of DNA site-dependent hotspots; trigger substantial changes in the distribution of recombination across the genome (as in Figure 7). In short, the dynamic modulation of local recombination rates by different classes of DNA site-dependent hotspots—which can be induced at laboratory/experimental time scales—can explain why Prdm9-independent hotspots (as defined previously using either/or cutoff frequency values) appear to move rapidly over evolutionarily time scales. This postulate has substantial implications for the conservation of fundamental mechanisms that control the distribution of meiotic recombination in diverse taxa and for current thinking about its evolution.
Conclusions
The striking, previously enigmatic plasticity in the frequency distribution of meiotic recombination across genomes is readily explained by the fact that many, distinct classes of DNA sequence motifs each control rates of recombination locally. Each different type of cis-acting regulatory module (hotspot-activating protein-DNA complex) serves as an independently controlled rheostat that modulates rates of recombination over a broad dynamic range in response to environmental cues and metabolic states. Together, the independent modulation of recombination rates by each different class of DNA site-dependent hotspots can remodel dynamically and profoundly the distribution of recombination across the genome (as depicted in Figure 7). This process can also explain why the distribution of recombination among hotspots varies markedly between closely related species, subspecies, and isolated populations of the same species that lack Prdm9.
Data availability
All data necessary for confirming the conclusions presented in this article are represented fully within the tables and figures. Full genotypes of strains used are provided in Supplementary Table S1; DNA sequences and positional coordinates of the PCR primers employed are provided in Supplementary Table S2. Yeast strains and other materials generated by the study are available upon request.
Supplementary material is available at GENETICS online.
Supplementary Material
Acknowledgments
The authors thank Yasuhiro Matsuo’s laboratory for providing the plasmid pREP3X-Rst2 and Elena Hidalgo’s laboratory for providing a strain expressing Pcr1-HA (NG96).
Funding
This work was supported by a grant from the National Institutes of Health [grant number GM081766 to W.P.W.].
Conflicts of interest
The authors declare that there is no conflict of interest.
Literature cited
- Abdullah MF, Borts RH.. 2001. Meiotic recombination frequencies are affected by nutritional states in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 98:14524–14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson E, Webster MT, Ratnakumar A, Ponting CP, Lindblad-Toh K.. 2012. Death of PRDM9 coincides with stabilization of the recombination landscape in the dog genome. Genome Res. 22:51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Nicolas A.. 1997. Clustering of meiotic double-strand breaks on yeast chromosome III. Proc Natl Acad Sci USA. 94:5213–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzblau HG, Bell GW, Rodriguez J, Bell SP, Hochwagen A.. 2007. Mapping of meiotic single-stranded DNA reveals double-stranded-break hotspots near centromeres and telomeres. Curr Biol. 17:2003–2012. [DOI] [PubMed] [Google Scholar]
- Bomblies K, Higgins JD, Yant L.. 2015. Meiosis evolves: adaptation to external and internal environments. New Phytol. 208:306–323. [DOI] [PubMed] [Google Scholar]
- Brick K, Smagulova F, Khil P, Camerini-Otero RD, Petukhova GV.. 2012. Genetic recombination is directed away from functional genomic elements in mice. Nature. 485:642–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brick K, Thibault-Sennett S, Smagulova F, Lam KG, Pu Y, et al. 2018. Extensive sex differences at the initiation of genetic recombination. Nature. 561:338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SD, Audoynaud C, Lorenz A.. 2020. Intragenic meiotic recombination in Schizosaccharomyces pombe is sensitive to environmental temperature changes. Chromosome Res. 28:195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler C, Borde V, Lichten M.. 2007. Mapping meiotic single-strand DNA reveals a new landscape of DNA double-strand breaks in Saccharomyces cerevisiae. PLoS Biol. 5:e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K, Henderson IR.. 2015. Meiotic recombination hotspots—a comparative view. Plant J. 83:52–61. [DOI] [PubMed] [Google Scholar]
- Cotton VE, Hoffmann ER, Abdullah MF, Borts RH.. 2009. Interaction of genetic and environmental factors in Saccharomyces cerevisiae meiosis: the devil is in the details. Methods Mol Biol. 557:3–20. [DOI] [PubMed] [Google Scholar]
- Cromie GA, Hyppa RW, Cam HP, Farah JA, Grewal SI, et al. 2007. A discrete class of intergenic DNA dictates meiotic DNA break hotspots in fission yeast. PLoS Genet. 3:e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson MK, Shandilya HK, Hirota K, Ohta K, Wahls WP.. 2004. Atf1-Pcr1-M26 complex links stress-activated MAPK and cAMP-dependent protein kinase pathways via chromatin remodeling of cgs2+. J Biol Chem. 279:50857–50863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro E, Soriano I, Marin L, Serrano R, Quintales L, et al. 2011. Nucleosomal organization of replication origins and meiotic recombination hotspots in fission yeast. EMBO J. 31:124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Massy B, Rocco V, Nicolas A.. 1995. The nucleotide mapping of DNA double-strand breaks at the CYS3 initiation site of meiotic recombination in Saccharomyces cerevisiae. EMBO J. 14:4589–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dluzewska J, Szymanska M, Ziolkowski PA.. 2018. Where to cross over? Defining crossover sites in plants. Front Genet. 9:609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekwall K, Partridge JF.. 1999. Fission yeast chromosome analysis: fluorescenct in situ hybridization (FISH) and chromatin immunoprecipitation (ChIP). In: Bickmore W, editor. Chromosome Structural Analysis: a Practical Approach. New York, NY: Oxford University Press USA. p. 39–57. [Google Scholar]
- Ergoren MC. 2018. The control of meiotic recombination in the human genome. Crit Rev Eukaryot Gene Expr. 28:187–204. [DOI] [PubMed] [Google Scholar]
- Eshaghi M, Lee JH, Zhu L, Poon SY, Li J, et al. 2010. Genomic binding profiling of the fission yeast stress-activated MAPK Sty1 and the bZIP transcriptional activator Atf1 in response to H2O2. PLoS One. 5:e11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsburg SL. 1993. Comparison of Schizosaccharomyces pombe expression systems. Nucleic Acids Res. 21:2955–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsburg SL, Rhind N.. 2006. Basic methods for fission yeast. Yeast. 23:173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler KR, Sasaki M, Milman N, Keeney S, Smith GR.. 2014. Evolutionarily diverse determinants of meiotic DNA break and recombination landscapes across the genome. Genome Res. 24:1650–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaits F, Degols G, Shiozaki K, Russell P.. 1998. Phosphorylation and association with the transcription factor Atf1 regulate localization of Spc1/Sty1 stress-activated kinase in fission yeast. Genes Dev. 12:1464–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Davidson MK, Wahls WP.. 2008. Distinct regions of ATF/CREB proteins Atf1 and Pcr1 control recombination hotspot ade6-M26 and the osmotic stress response. Nucleic Acids Res. 36:2838–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Davidson MK, Wahls WP.. 2009. Phosphorylation-independent regulation of Atf1-promoted meiotic recombination by stress-activated, p38 kinase Spc1 of fission yeast. PLoS One. 4:e5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerton JL, DeRisi J, Shroff R, Lichten M, Brown PO, et al. 2000. Inaugural article: global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 97:11383–11390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey C, Baudat F, de Massy B.. 2018. PRDM9, a driver of the genetic map. PLoS Genet. 14:e1007479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutz H, Heslot H, Leupold U, Loprieno N.. 1974. Schizosaccharomyces pombe. In: King RC, editor. Handbook of Genetics. New York, NY: Plenum Press. p. 395–446. [Google Scholar]
- Higuchi T, Watanabe Y, Yamamoto M.. 2002. Protein kinase A regulates sexual development and gluconeogenesis through phosphorylation of the Zn finger transcriptional activator Rst2p in fission yeast. Mol Cell Biol. 22:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Hasemi T, Yamada T, Mizuno KI, Hoffman CS, et al. 2004. Fission yeast global repressors regulate the specificity of chromatin alteration in response to distinct environmental stresses. Nucleic Acids Res. 32:855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Hoffman CS, Shibata T, Ohta K.. 2003. Fission yeast Tup1-like repressors repress chromatin remodeling at the fbp1+ promoter and the ade6-M26 recombination hotspot. Genetics. 165:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Mizuno K, Shibata T, Ohta K.. 2008. Distinct chromatin modulators regulate the formation of accessible and repressive chromatin at the fission yeast recombination hotspot ade6-M26. Mol Biol Cell. 19:1162–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Steiner WW, Shibata T, Ohta K.. 2007. Multiple modes of chromatin configuration at natural meiotic recombination hot spots in fission yeast. Eukaryot Cell. 6:2072–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyppa RW, Fowler KR, Cipak L, Gregan J, Smith GR.. 2014. DNA intermediates of meiotic recombination in synchronous S. pombe at optimal temperature. Nucleic Acids Res. 42:359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing JL, Zhang T, Wang YZ, He Y.. 2019. Advances towards how meiotic recombination is initiated: a comparative view and perspectives for plant meiosis research. Int J Mol Sci. 20:4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T, Mugal CF, Suh A, Nater A, Burri R, et al. 2017. Whole-genome patterns of linkage disequilibrium across flycatcher populations clarify the causes and consequences of fine-scale recombination rate variation in birds. Mol Ecol. 26:4158–4172. [DOI] [PubMed] [Google Scholar]
- Kon N, Krawchuk MD, Warren BG, Smith GR, Wahls WP.. 1997. Transcription factor Mts1/Mts2 (Atf1/Pcr1, Gad7/Pcr1) activates the M26 meiotic recombination hotspot in Schizosaccharomyces pombe. Proc Natl Acad Sci USA. 94:13765–13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon N, Schroeder SC, Krawchuk MD, Wahls WP.. 1998. Regulation of the Mts1-Mts2-dependent ade6-M26 meiotic recombination hotspot and developmental decisions by the Spc1 mitogen-activated protein kinase of fission yeast. Mol Cell Biol. 18:7575–7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, et al. 2010. Fine-scale recombination rate differences between sexes, populations and individuals. Nature. 467:1099–1103. [DOI] [PubMed] [Google Scholar]
- Lam I, Keeney S.. 2014. Mechanism and regulation of meiotic recombination initiation. Cold Spring Harb Perspect Biol. 7:a016634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence CL, Maekawa H, Worthington JL, Reiter W, Wilkinson CR, et al. 2007. Regulation of S. pombe Atf1 protein levels by Sty1-mediated phosphorylation and heterodimerisation with Pcr1. J Biol Chem. 282:5160–5170. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD.. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25:402–408. [DOI] [PubMed] [Google Scholar]
- Marand AP, Zhao H, Zhang W, Zeng Z, Fang C, et al. 2019. Historical meiotic crossover hotspots fueled patterns of evolutionary divergence in rice. Plant Cell. 31:645–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maundrell K. 1993. Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene. 123:127–130. [DOI] [PubMed] [Google Scholar]
- Mercier A, Watt S, Bahler J, Labbe S.. 2008. Key function for the CCAAT-binding factor Php4 to regulate gene expression in response to iron deficiency in fission yeast. Eukaryot Cell. 7:493–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieczkowski PA, Dominska M, Buck MJ, Gerton JL, Lieb JD, et al. 2006. Global analysis of the relationship between the binding of the Bas1p transcription factor and meiosis-specific double-strand DNA breaks in Saccharomyces cerevisiae. Mol Cell Biol. 26:1014–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihola O, Landa V, Pratto F, Brick K, Kobets T, et al. 2021. Rat PRDM9 shapes recombination landscapes, duration of meiosis, gametogenesis, and age of fertility. BMC Biol. 19:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno K, Hasemi T, Ubukata T, Yamada T, Lehmann E, et al. 2001. Counteracting regulation of chromatin remodeling at a fission yeast cAMP response element-related recombination hotspot by stress-activated protein kinase, cAMP-dependent kinase and meiosis regulators. Genetics. 159:1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougel F, Poursat MA, Beaume N, Vautrin D, Solignac M.. 2014. High-resolution linkage map for two honeybee chromosomes: the hotspot quest. Mol Genet Genomics. 289:11–24. [DOI] [PubMed] [Google Scholar]
- Mukiza TO, Protacio RU, Davidson MK, Steiner WW, Wahls WP.. 2019. Diverse DNA sequence motifs activate meiotic recombination hotspots through a common chromatin remodeling pathway. Genetics. 213:789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Fuentes V, Di Rienzo A, Vila C.. 2011. Prdm9, a major determinant of meiotic recombination hotspots, is not functional in dogs and their wild relatives, wolves and coyotes. PLoS One. 6:e25498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely LA, Hoffman CS.. 2000. Protein kinase A and mitogen-activated protein kinase pathways antagonistically regulate fission yeast fbp1 transcription by employing different modes of action at two upstream activation sites. Mol Cell Biol. 20:6426–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paigen K, Petkov PM.. 2018. PRDM9 and its role in genetic recombination. Trends Genet. 34:291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Sasaki M, Kniewel R, Murakami H, Blitzblau HG, et al. 2011. A hierarchical combination of factors shapes the genome-wide topography of yeast meiotic recombination initiation. Cell. 144:719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvanov E, Kohli J, Ludin K.. 2008. The mating-type-related bias of gene conversion in Schizosaccharomyces pombe. Genetics. 180:1859–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pengelly RJ, Gheyas AA, Kuo R, Mossotto E, Seaby EG, et al. 2016. Commercial chicken breeds exhibit highly divergent patterns of linkage disequilibrium. Heredity (Edinb). 117:375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plough HH. 1917. The effect of temperature on linkage in the second chromosome of Drosophila. Proc Natl Acad Sci USA. 3:553–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert T, Vrielynck N, Mezard C, de Massy B, Grelon M.. 2016. A new light on the meiotic DSB catalytic complex. Semin Cell Dev Biol. 54:165–176. [DOI] [PubMed] [Google Scholar]
- Sanso M, Gogol M, Ayte J, Seidel C, Hidalgo E.. 2008. Transcription factors Pcr1 and Atf1 have distinct roles in stress- and Sty1-dependent gene regulation. Eukaryot Cell. 7:826–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schield DR, Pasquesi GIM, Perry BW, Adams RH, Nikolakis ZL, et al. 2020. Snake recombination landscapes are concentrated in functional regions despite PRDM9. Mol Biol Evol. 37:1272–1294. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ.. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 3:1101–1108. [DOI] [PubMed] [Google Scholar]
- Schuchert P, Langsford M, Kaslin E, Kohli J.. 1991. A specific DNA sequence is required for high frequency of recombination in the ade6 gene of fission yeast. EMBO J. 10:2157–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanfelter AF, Archambeault SL, White MA.. 2019. Divergent fine-scale recombination landscapes between a freshwater and marine population of threespine stickleback fish. Genome Biol Evol. 11:1573–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipiczki M. 2000. Where does fission yeast sit on the tree of life? Genome Biol. 1:reviews1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl FW, Rehan MB, Foss HM, Borts RH.. 2016. Apparent epigenetic meiotic double-strand-break disparity in Saccharomyces cerevisiae: a meta-analysis. Genetics. 204:129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner WW, Davidow PA, Bagshaw AT.. 2011. Important characteristics of sequence-specific recombination hotspots in Schizosaccharomyces pombe. Genetics. 187:385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner WW, Schreckhise RW, Smith GR.. 2002. Meiotic DNA breaks at the S. pombe recombination hot spot M26. Mol Cell. 9:847–855. [DOI] [PubMed] [Google Scholar]
- Steiner WW, Smith GR.. 2005. Natural meiotic recombination hot spots in the Schizosaccharomyces pombe genome successfully predicted from the simple sequence motif M26. Mol Cell Biol. 25:9054–9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner WW, Steiner EM.. 2012. Fission yeast hotspot sequence motifs are also active in budding yeast. PLoS One. 7:e53090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner WW, Steiner EM, Girvin AR, Plewik LE.. 2009. Novel nucleotide sequence motifs that produce hotspots of meiotic recombination in Schizosaccharomyces pombe. Genetics. 182:459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey AJ, Wang HP, Protacio RU, Davidson MK, Tackett AJ, et al. 2018. Chromatin-mediated regulators of meiotic recombination revealed by proteomics of a recombination hotspot. Epigenetics Chromatin. 11:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka K, Tanabe T, Kawamukai M, Matsuo Y.. 2018. Overexpression of the transcription factor Rst2 in Schizosaccharomyces pombe indicates growth defect, mitotic defects, and microtubule disorder. Biosci Biotechnol Biochem. 82:247–257. [DOI] [PubMed] [Google Scholar]
- Wahls WP, Davidson MK.. 2010. Discrete DNA sites regulate global distribution of meiotic recombination. Trends Genet. 26:202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahls WP, Davidson MK.. 2012. New paradigms for conserved, multifactorial, cis-acting regulation of meiotic recombination. Nucleic Acids Res. 40:9983–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahls WP, Smith GR.. 1994. A heteromeric protein that binds to a meiotic homologous recombination hot spot: correlation of binding and hot spot activity. Genes Dev. 8:1693–1702. [DOI] [PubMed] [Google Scholar]
- Wu TC, Lichten M.. 1995. Factors that affect the location and frequency of meiosis-induced double-strand breaks in Saccharomyces cerevisiae. Genetics. 140:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Mizuno K, Hirota K, Kon N, Wahls WP, et al. 2004. Roles of histone acetylation and chromatin remodeling factor in a meiotic recombination hotspot. EMBO J. 23:1792–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JA, Schreckhise RW, Steiner WW, Smith GR.. 2002. Meiotic recombination remote from prominent DNA break sites in S. pombe. Mol Cell. 9:253–263. [DOI] [PubMed] [Google Scholar]
- Zhang K, Wu XC, Zheng DQ, Petes TD.. 2017. Effects of temperature on the meiotic recombination landscape of the yeast Saccharomyces cerevisiae. mBio. 8:e02099–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Keeney S.. 2015. High-resolution global analysis of the influences of Bas1 and Ino4 transcription factors on meiotic DNA break distributions in Saccharomyces cerevisiae. Genetics. 201:525–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data necessary for confirming the conclusions presented in this article are represented fully within the tables and figures. Full genotypes of strains used are provided in Supplementary Table S1; DNA sequences and positional coordinates of the PCR primers employed are provided in Supplementary Table S2. Yeast strains and other materials generated by the study are available upon request.
Supplementary material is available at GENETICS online.