

Abstract
Cell division cycle-associated 8 (CDCA8) is a component of chromosomal passenger complex (CPC) that participates in mitotic regulation. Although cancer-related CDCA8 hyperactivation has been widely observed, its molecular mechanism remains elusive. Here, we report that CDCA8 overexpression maintains tumorigenicity and is associated with poor clinical outcome in patients with prostate cancer (PCa). Notably, enhancer of zeste homolog 2 (EZH2) is identified to be responsible for CDCA8 activation in PCa. Genome-wide assays revealed that EZH2-induced H3K27 trimethylation represses let-7b expression and thus protects the let-7b-targeting CDCA8 transcripts. More importantly, EZH2 facilitates the self-activation of E2F1 by recruiting E2F1 to its own promoter region in a methylation-independent manner. The high level of E2F1 further promotes transcription of CDCA8 along with the other CPC subunits. Taken together, our study suggests that EZH2-mediated cell cycle regulation in PCa relies on both its methyltransferase and non-methyltransferase activities.
INTRODUCTION
Cell cycle progression is a fine-tuned process that leads to the replication of cellular components and cell division [1, 2]. As a hallmark of cancer, dysregulation of cell cycle directly contributes to the overactivated proliferation and aggressive behaviors of cancer cell [3]. In prostate cancer (PCa), androgen receptor (AR) is currently recognized as a master regulator of cell cycle which accelerates this process through transcriptional regulation and direct interaction with cell cycle-related genes [4]. However, uncontrolled cell proliferation is still or even more active in AR-negative PCa types such as neuroendocrine prostate cancer (NEPC), suggesting that core regulators other than AR may play a more important role in governing cell cycle in PCa.
Cell division cycle-associated 8 (CDCA8), also known as Borealin or Dasra B, is a subunit of the chromosomal passenger complex (CPC) [5, 6]. CDCA8 forms CPC with three other members [Aurora Kinase B (AURKB), Survivin (BIRC5) and INCENP] and is involved in the regulation of mitosis [7]. In particular, CDCA8 is required for guiding the CPC to centromeres, correction of kinetochore attachment errors and stabilization of the bipolar spindle [5]. Elevated expression of CDCA8 has been reported in multiple cancer types and is strongly associated with cancer aggressiveness and poor clinical outcome [8–11]. Despite this fact, the underlying mechanism by which CDCA8 is overactivated remains to be investigated.
Enhancer of zeste homolog 2 (EZH2) is the catalytic subunit of polycomb repressive complex 2 (PRC2) that is responsible for conducting H3K27 trimethylation (H3K27me3) [12, 13]. As a master driver and robust biomarker in PCa, EZH2 mostly works as an epigenetic silencer to inhibit expression of tumor suppressor genes [14, 15]. However, evidence has accumulated recently suggesting that EZH2 is capable of mediating gene expression beyond PRC2 and H3K27me3 in aggressive PCa [16–18]. Notably, our recent study proved that interaction of EZH2 with RNA methyltransferase FBL enhances 2′-O methylation in rRNAs and thus accelerates ribosome functions, which further extends the function of EZH2 from the transcriptional level to the translational level [19].
Here, we observed aberrant upregulation of CDCA8 in PCa specimens which is closely associated with the clinical characteristics and prognosis of PCa patients. Depletion of CDCA8 led to diminished aggressive and tumorigenic capabilities in PCa cells. Remarkably, significantly positive correlation was found between EZH2 and CDCA8 expressions in PCa samples. RNA-seq and other data showed that EZH2 could modulate CDCA8 expression transcriptionally and thus play a role in cell cycle regulation. Mechanistically, EZH2-mediated CDCA8 upregulation is partially due to the PRC2-dependent transcriptional repression of micro-RNA (miRNA) let-7b, which targets the mRNA of CDCA8. More importantly, EZH2 could further promote CDCA8 transcription by upregulation of transcription factor E2F1 in a methylation-independent manner. The regulatory role of EZH2 towards other CPC members was also discussed in the present study.
RESULTS
CDCA8 is upregulated in PCa tissues
To investigate the role of CDCA8 in PCa, we firstly assessed its mRNA level using TCGA database. Expressions of the other CPC members were also determined in parallel. Compared with normal prostate controls, PCa specimens were characterized by increases of CDCA8 (Fig. 1A). In addition, elevated CDCA8 expression was observed in PCa tumors at the late clinical and pathological stages (Fig. 1B, C). Prostate-specific antigen (PSA) is a commonly used biomarker for PCa detection [20]. As shown in Fig. 1D, CDCA8 expression was positively correlated with PSA level in PCa patients. Consistently, the median disease-free survival time of patient group with a high CDCA8 expression was significantly shorter than that of the group with a low CDCA8 expression (Fig. 1E). With respect to other CPC members, upregulation of AURKB and BIRC5 were observed in PCa tissues while the expression of INCENP remained unchanged (Supplementary Fig. 1A). Intriguingly, associations between the rest CPC components and clinical factors and prognosis of PCa patients could also be observed (Supplementary Fig. 1B–E), indicating that the whole CPC may be activated in a similar manner during the progression of PCa.
Fig. 1. Elevated CDCA8 expression in PCa.
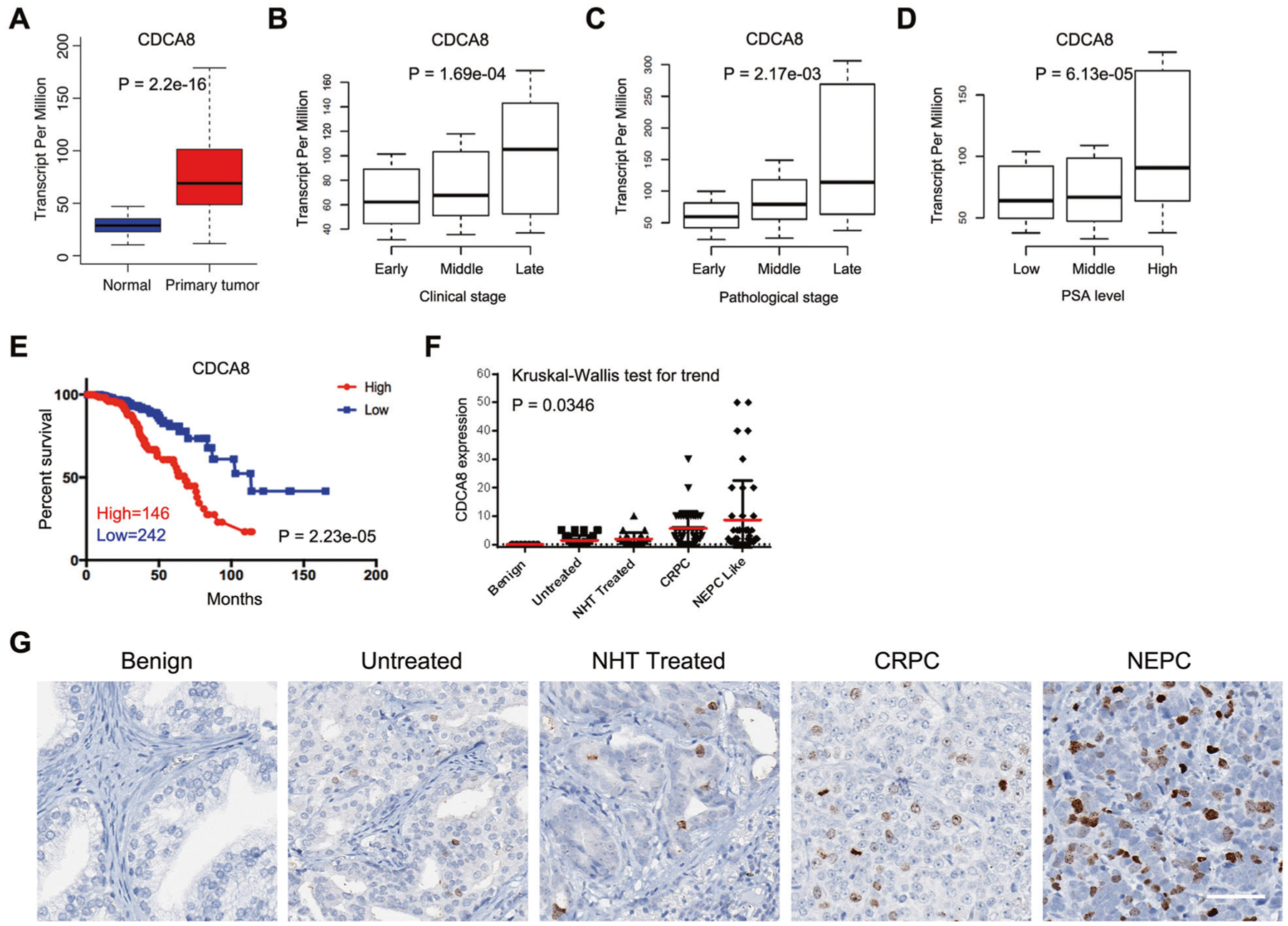
A Box plot showing the mRNA level of CDCA8 in normal (n = 52) and PCa (n = 497) specimens using data from TCGA. B Box plot showing the mRNA level of CDCA8 in PCa patients with different clinical stages (n(early) = 177, n(middle) = 173, n(late) = 53) using data from TCGA. C Box plot showing the mRNA level of CDCA8 in PCa patients with different pathological stages (n(early) = 186, n(middle) = 293, n(late) = 10) using data from TCGA. D Box plot showing the mRNA level of CDCA8 in PCa patients with different PSA levels (n(low) = 181, n(middle) = 209, n(high) = 48) using data from TCGA. E The association between CDCA8 expression and Disease-free survival time of PCa patients was analyzed by Kaplan–Meier analysis using data from TCGA. F Graph showing the CDCA8 protein levels based on the IHC results from benign prostate tissue (n = 7) and four PCa types including untreated (n = 35), NHT treated (n = 23), CRPC (n = 40) and NEPC (n = 41). NHT neoadjuvant hormonal therapy, CRPC castration-resistant prostate cancer, NEPC neuroendocrine prostate cancer. G Representative IHC images of CDCA8 expression in benign prostate tissue and four types of PCa as indicated. Scale bar = 70 μm.
To test whether the protein level of CDCA8 (Borealin) is also dysregulated in PCa tissues, immunohistochemistry (IHC) assay was next performed using serially sectioned PCa tissue microarray (TMA) slides. As shown in Fig. 1F, G, CDCA8 signals were barely detected in benign prostate tissues but gradually enriched with the advancement of PCa, further supporting the correlated pattern between CDCA8 expression and PCa stages.
CDCA8 potentiates tumorigenic capacity of PCa cells
The upregulation of CDCA8 in PCa prompted us to determine whether CDCA8 is essential for PCa progression. To this end, we specifically suppressed CDCA8 expression in PCa cell lines of C4–2 and PC-3 through shRNA-expressing lentiviruses (Fig. 2A and Supplementary Fig. 2A, upper panel). As expected, inhibition of CDCA8 led to significantly reduced proliferation in both PCa cell lines (Fig. 2A and Supplementary Fig. 2A, lower panel). In transwell migration assays, numbers of migratory cells presenting in the lower surface of filter were remarkably decreased upon CDCA8 knockdown (Fig. 2B and Supplementary Fig. 2B). Moreover, the Boyden chamber invasion assay showed that depletion of CDCA8 reduced invasive capacity of PCa cells through Matrigel (Fig. 2C and Supplementary Fig. 2C). Inversely, overexpression of CDCA8 in benign prostate cell line of BPH-1 promoted the cell growth and enhanced its migratory capacity (Fig. 2D, E). To extend our in vitro observations, a murine prostate tumor xenograft model was next used. As shown in Fig. 2F, G, mice grafted with CDCA8-deficient PCa cells exhibited much smaller tumors compared to control cells, suggesting that CDCA8 could execute its oncogenic functions both in vitro and in vivo.
Fig. 2. CDCA8 sustains tumorigenicity of PCa.
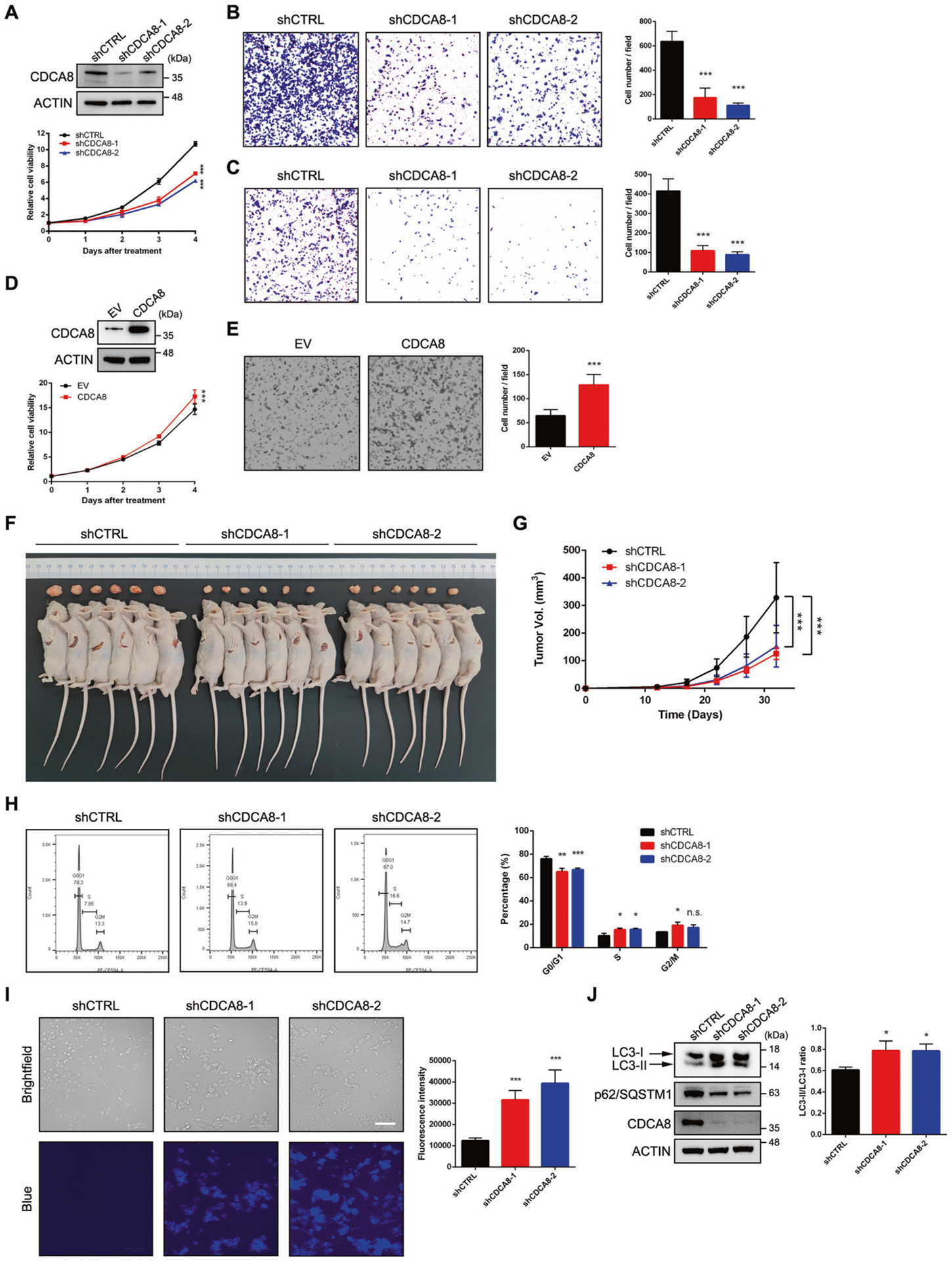
A Cell viability assay was used to assess the proliferative capacity of control and CDCA8-deficient C4–2 cells. The knockdown efficiency of CDCA8 was validated by western blot. B Boyden chamber migration assay was performed to determine the migratory capability of C4–2 cells after CDCA8 depletion. Graph showing the number of migrated cells in the lower surface of filter at 24 h. Data represent Mean ± SD from n = 5 random fields per filter. C Boyden chamber invasion assay was performed to determine the invasive capability of C4–2 cells after CDCA8 depletion. Graph showing the number of migrated cells passing through Matrigel at 24 h. Data represent Mean ± SD from n = 5 random fields per filter. D Cell viability assay was used to assess the proliferative capacity of control and CDCA8-overexpressing BPH-1 cells. The ectopic expression of CDCA8 was validated by western blot. EV empty vector. E Boyden chamber migration assay was performed to determine the migratory capability of BPH-1 cells upon CDCA8 overexpression. Graph showing the number of migrated cells in the lower surface of filter at 24 h. Data represent Mean ± SD from n = 5 random fields per filter. F, G Tumor formation in nude mice injected with control or CDCA8-deficient PC-3 cells. Xenograft tumors at the end point of measurement were presented in F. Tumors were measured by caliper every 5 days and plotted in G. Data represent Mean ± SD from n = 6 tumors in each group. Statistical significance was determined by two-way ANOVA. H Either control or CDCA8-deficient C4–2 cells were subjected to cell cycle analysis via flow cytometry with propidium iodide staining. Graph showing the percentage of cells at each stage of mitosis. I Either control or CDCA8-deficient C4–2 cells were subjected to autophagy analysis through staining of autophagic vacuoles. Graph showing the relative activity of autophagosomes in each group. Scale bar = 100 μm. J Western blot analysis of the proteins involved in autophagy from C4–2 cells upon CDCA8 knockdown. Graph showing the change of LC3-II/ LC3-I ratio upon CDCA8 knockdown in C4–2 cells. Data represent Mean ± SD from three replicates. *P < 0.05, **P < 0.01, ***P < 0.001 is based on the student’s t-test unless otherwise stated. Values are mean ± SD of at least three independent experiments.
Since CDCA8 mainly exerts its role during mitosis, we speculated that cell cycle arrest may be responsible for the abolished carcinogenic effects in CDCA8-deficient PCa cells. In line with our hypothesis, the proportion of C4–2 cells in G0/G1 phase was significantly decreased upon CDCA8 inhibition, whereas the proportion of cells in S phase was inversely increased (Fig. 2H). We then assessed whether apoptosis was induced along with cell cycle arrest in response to CDCA8 suppression. Notably, flow cytometry assay revealed no significant change of apoptotic rate in C4–2 cells undergoing CDCA8 inhibition (Supplementary Fig. 2D). Meanwhile, only minimal cleavage of PARP and Caspase-3 was detectable in CDCA8-deficient cells (Supplementary Fig. 2E), indicating that the reduced tumorigenic capacity of CDCA8-deficient PCa cells was not closely associated with the apoptotic cell death. Instead, autophagic vacuole staining demonstrated a profound induction of autophagy in CDCA8-deficient C4–2 cells (Fig. 2I). Consistently, increased LC3-II/LC3-I ratio and reduced p62/SQSTM1 protein level, which are two hallmarks of the autophagy activation, were clearly detected in CDCA8-deficient cells (Fig. 2J). In summary, these data suggest that CDCA8 maintains the tumorigenic effect of PCa cells by promoting cell cycle progression and evading the surveillance of autophagy.
Positive correlation between EZH2 and CDCA8 in PCa
We next sought to investigate the reason for the activation of CDCA8 in PCa. By searching for the TCGA database, we observed that expression of EZH2, a well-known prostate oncogene, was upregulated and positively correlated with that of CDCA8 in PCa (Fig. 3A and Supplementary Fig. 3A). Similarly, significantly positive correlation was also found between EZH2 and other CPC members (Supplementary Fig. 3B). In addition, PCa patients with high expressions of both EZH2 and CDCA8 predicted poorer survival rate as compared with double-low patients (Fig. 3B). To extend our observations to the protein level, we first checked the expressions of EZH2 and CDCA8 in a panel of prostate cells. As a result, both EZH2 and CDCA8 were expressed higher in PCa cells compared with benign BPH-1 cells (Fig. 3C). IHC Assay was next conducted in PCa TMA slides using anti-EZH2 and anti-CDCA8 antibodies. With the elevated expression of EZH2, the protein level of CDCA8 was increased coordinately in PCa tissues (Fig. 3D and Supplementary Table 1), further confirming the co-expression between EZH2 and CDCA8.
Fig. 3. Expression of CDCA8 is positively correlated with EZH2 in PCa.
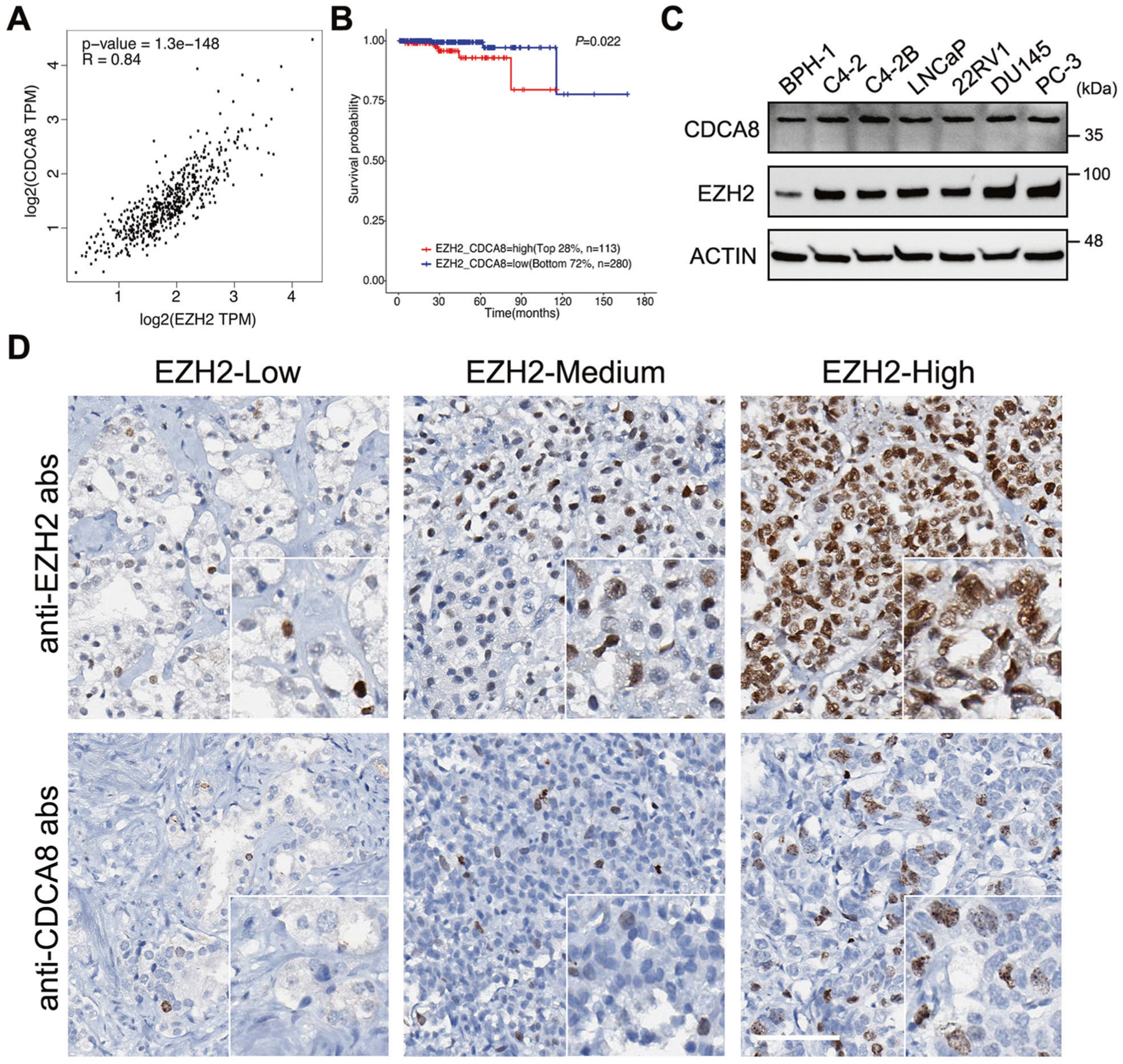
A Scatter plot showing the relationship between CDCA8 and EZH2 expressions using data from TCGA, with Spearman correlation coefficient (R) and P value as indicated. TPM transcript per million. B The association between EZH2/CDCA8 co-expression and overall survival time of PCa patients was analyzed by Kaplan–Meier analysis using data retrieved from TCGA. C Western blot analysis of CDCA8 and EZH2 expressions in six PCa cell lines and benign BPH-1 prostate cell line. D IHC staining of PCa TMA slides using the indicated antibodies. Scale bar = 80 μm.
EZH2 serves as an upstream regulator of CDCA8
The above findings suggested that EZH2 may play a role in activation of CDCA8. To validate, we knocked down EZH2 expression in two PCa cell lines using shRNAs, followed by RT-qPCR and western blot analyses. As shown in Fig. 4A and Supplementary Fig. 4A, suppression of EZH2 resulted in evident decrease of CDCA8 at both mRNA and protein levels, along with the other CPC members. Similar results could also be observed in PCa cells treated with EZH2 protein inhibitor DZNep or enzymatic inhibitor EPZ6438 (Fig. 4B and Supplementary Fig. 4B). In comparison, ectopic expression of EZH2 in PCa cells inversely increased the expression of all CPC members including CDCA8 (Fig. 4C and Supplementary Fig. 4C).
Fig. 4. EZH2 is a master regulator of cell cycle-related genes including CDCA8.
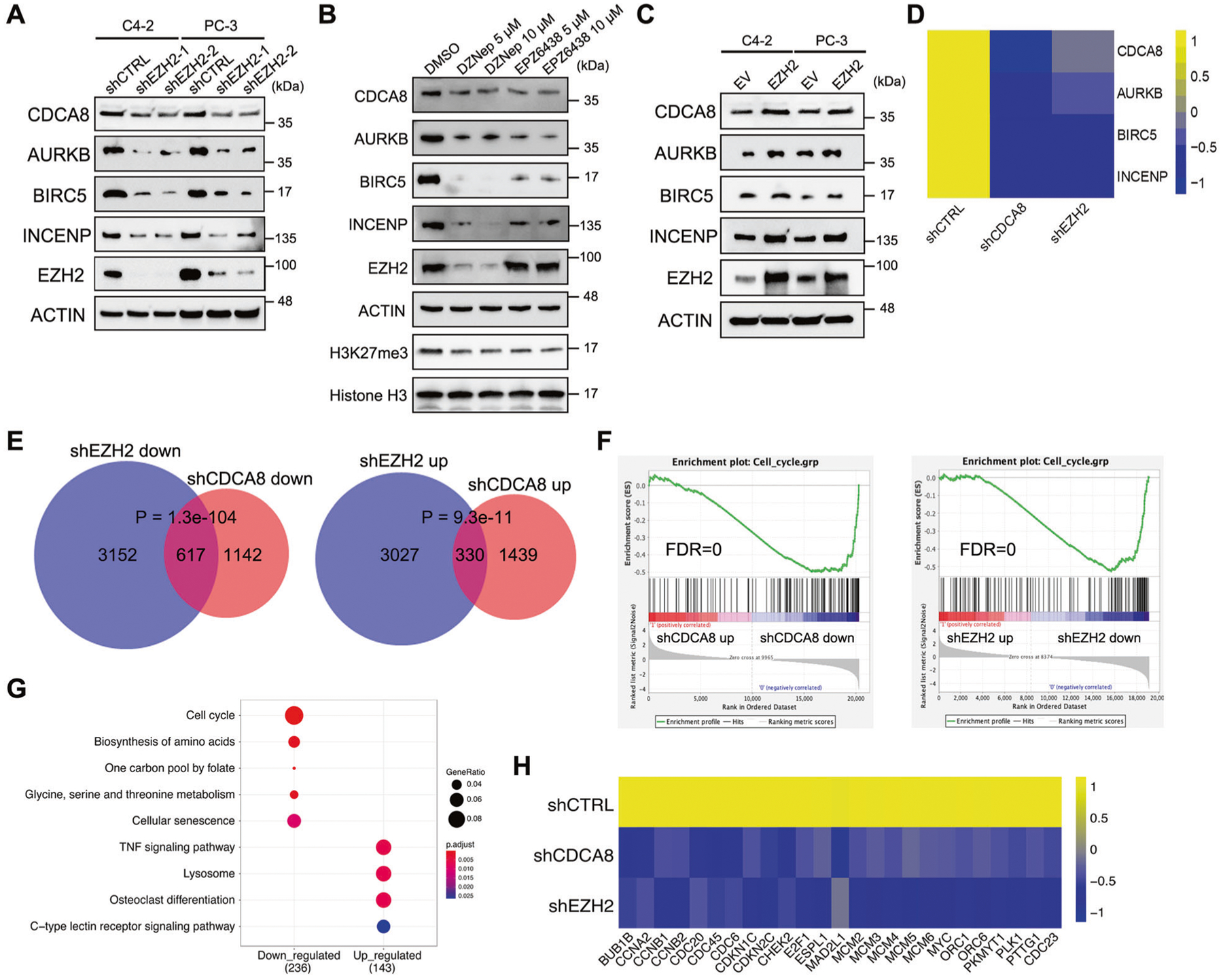
A Western blot analysis of the protein levels of CDCA8 and other CPC members in PCa cell lines upon EZH2 knockdown. B Western blot analysis of the protein levels of CDCA8 and other CPC members in C4–2 cells after treatment of EZH2 inhibitors as indicated. C Western blot analysis of the protein levels of CDCA8 and other CPC members in PCa cell lines upon EZH2 overexpression. EV empty vector. D Heatmap showing the transcriptional change of four CPC members upon CDCA8 or EZH2 inhibition in C4–2 cells, as revealed by RNA-seq data. E Venn diagram showing the overlap between genes that were downregulated or upregulated at mRNA level upon EZH2 deficiency and CDCA8 deficiency. P values were calculated by one-tailed Fisher’s exact test. F GSEA analysis showed a negative enrichment of cell cycle-related genes associated with the transcriptional changes after CDCA8 or EZH2 knockdown. FDR false discovery rate. G KEGG pathway analysis of genes which were co-regulated at mRNA level upon both EZH2 deficiency and CDCA8 deficiency. H Heatmap showing the transcriptional changes of 25 cell cycle-related genes upon CDCA8 or EZH2 inhibition in C4–2 cells, as revealed by RNA-seq data.
It is generally accepted that EZH2 exerts its role in cell cycle transition primarily by orchestrating the transcriptions of cell cycle-related genes [21–23]. To determine the significance of CDCA8 targeting in EZH2-mediated transcriptional programming, we performed RNA-seq transcriptome analysis in CDCA8-deficient C4–2 cells, and compared with our previously obtained RNA-seq data of EZH2-deficient C4–2 cells [19]. As depicted in Fig. 4D and Supplementary Table 2, depletion of either CDCA8 or EZH2 resulted in dramatic downregulation of all four CPC members. In addition, the differentially expressed genes (DEGs) upon CDCA8 or EZH2 inhibition were substantially overlapped with each other, especially for the downregulated genes (Fig. 4E). Gene set enrichment analysis (GSEA) showed a significant enrichment of cell cycle-related genes in genes downregulated by knockdown of CDCA8 or EZH2 (Fig. 4F). In line with these evidences, KEGG pathway analysis revealed that the transcripts co-downregulated in both CDCA8-deficient and EZH2-deficient cells were enriched in cell cycle pathway (Fig. 4G). To be specific, a total of 25 cell cycle-related genes showed reduced RNA level upon both CDCA8 inhibition and EZH2 inhibition (Fig. 4H). These co-affected genes spread over every single stage of cell cycle (Supplementary Fig. 4D). Collectively, these data suggest that EZH2 could target CDCA8 to mediate cell cycle.
EZH2 activates CDCA8 expression partially by repressing let-7b transcription
We then focused on the mechanism by which EZH2 promotes CDCA8 expression. Given that EZH2 normally functions as a transcriptional repressor, we wondered that EZH2 may increase CDCA8 expression indirectly by suppressing the transcription of miRNAs targeting CDCA8 [24]. To test this hypothesis, small RNA-seq was firstly performed in control and EZH2-deficient C4–2 cells to define the group of miRNAs affected by EZH2. As a result, a total of 74 miRNAs showed expression change upon EZH2 knockdown, with the majority of which (67/74) showed significant upregulation in EZH2-deficient cells (Fig. 5A and Supplementary Table 3). Meanwhile, miRNAs that target the transcripts of CPC members were predicted using miRTarBase [25] and listed in Fig. 5B. By evaluation of both lists, let-7b, a potential tumor suppressor [26], was selected as the candidate for further verification because its expression was strongly elevated upon EZH2 suppression [log2 (fold change) >0.7, P < 0.05] and it was identified to target multiple CPC members including CDCA8. In accordance with the small RNA-seq data, TaqMan qPCR result also revealed an increased let-7b level in EZH2-deficient C4–2 cells (Fig. 5C). In addition, the occupancy of H3K27me3 marks in the promoter region of let-7b was confirmed by ChIP-qPCR assay, and its enrichment was largely decreased upon EZH2 knockdown (Fig. 5D). The above data demonstrated that EZH2 could repress let-7b transcription by conferring H3K27me3 modification to its promoter.
Fig. 5. EZH2-mediated let-7b repression increases CDCA8 level in PCa.
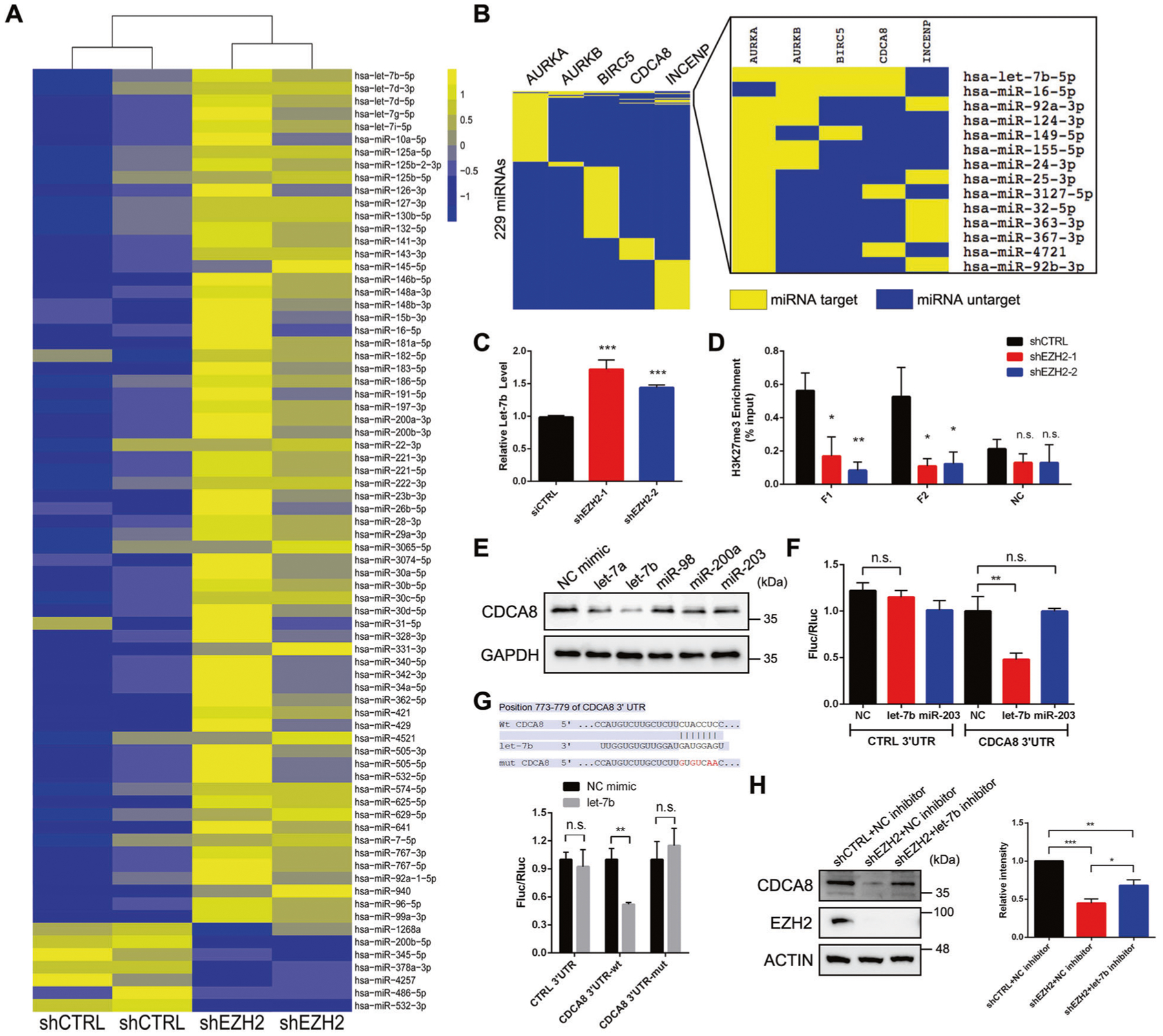
A Heatmap showing the expression levels of miRNAs that were dysregulated upon EZH2 inhibition in C4–2 cells. B List of CPC member mRNA-targeting miRNAs, as predicted by miRTarBase database. In addition to CPC members, the potential AURKA-targeting miRNAs were also presented. C TaqMan qPCR analysis to detect the change of let-7b expression upon EZH2 knockdown in C4–2 cells. D ChIP-qPCR assay to monitor the enrichment of H3K27me3 marks at the promoter region of let-7b in control and EZH2-deficient C4–2 cells. Two pairs of primers (F1 and F2) were used to amplify fragments inside let-7b promoter region while another pair of primers (NC) targeting nearby region was used as negative control. E Western blot analysis of the protein level of CDCA8 in C4–2 cells after treatment of various miRNA mimics as indicated. F Reporter plasmid containing control or CDCA8 3′-UTR sequence was transfected into C4–2 cells treated with different miRNA mimics, followed by detection of luciferase activities through dual-luciferase assay. NC negative control. G Reporter plasmid containing wild-type or mutant CDCA8 3′-UTR sequence was transfected into C4–2 cells treated with control or let-7b mimic, followed by detection of luciferase activities through dual-luciferase assay. The mutation sites were shown in the upper panel. H Rescue assay showing that treatment of let-7b inhibitor could not fully restore the downregulation of CDCA8 in EZH2-deficient C4–2 cells, as measured by western blot. Graph represents the relative CDCA8 protein level in each group. *P < 0.05, **P < 0.01, ***P < 0.001 is based on the student’s t-test unless otherwise stated. Values are mean ± SD of three independent experiments.
To confirm the inhibitory role of let-7b on CDCA8, a set of miRNA mimics were separately transfected into C4–2 cells, followed by western blot to detect CDCA8 expression. As shown in Fig. 5E, overexpression of let-7b mimics led to the most significant reduction of CDCA8 protein level as compared with other tested miRNAs. A dual-luciferase reporter containing a firefly luciferase (Fluc) fused with interested three prime untranslated region (3′-UTR) sequence and a stably expressed renilla luciferase (Rluc) was then introduced to measure the repression of RNA abundance via 3′-UTR regulation of genes. After overexpression of let-7b mimics, 3′-UTR of CDCA8, but not negative control, showed decreased luciferase activities compared to control mimic group (Fig. 5F). In comparison, overexpression of miR-203 mimics had no significant impact on the luciferase activity of CDCA8 3′-UTR (Fig. 5F). To further verify this finding, we constructed a CDCA8 3′-UTR mutant in which its complementary target sites with let-7b were fully replaced (Fig. 5G, upper panel). As expected, luciferase reporter with mutated CDCA8 3′-UTR became insensitive to let-7b mimics (Fig. 5G, lower panel), suggesting that the let-7b-CDCA8 3′-UTR interaction is essential for let-7b to target and affect CDCA8 mRNA. Moreover, forced expression of miRNA-resistant CDCA8 almost fully rescued the decreased proliferative, migratory and invasive capacities in C4–2 cells transfected with let-7b mimics (Supplementary Fig. 5A–C), reflecting the importance of CDCA8-targeting in let-7b-mediated tumor suppression.
We next wondered whether EZH2-mediated CDCA8 activation could be fully attributed to let-7b. Surprisingly, silencing of let-7b by specific inhibitor only partially rescued the downregulation of CDCA8 in EZH2-deificient C4–2 or PC-3 cells (Fig. 5H and Supplementary Fig. 5D), indicating that additional pathways are existed for EZH2 to modulate CDCA8 in PCa.
EZH2-induced self-activation of E2F1 promotes transcription of CDCA8 and other CPC members
To gain more insights into the EZH2-mediated CDCA8 activation, we looked back into the DEGs list in EZH2-deficient C4–2 cells (Supplementary Table 2). What is noteworthy is that transcription factor E2F1 ranked as the one of the top genes which showed sharp downregulation upon EZH2 knockdown (Supplementary Table 2 and Fig. 4H). This pattern of expression change was verified by both RT-qPCR and western blot assays (Fig. 6A and Supplementary Fig. 6A). E2F1 is a well-studied cell cycle regulator and PCa oncogene [27]. Correlation plots revealed significantly positive associations between E2F1 and all CPC members in PCa (Fig. 6B and Supplementary Fig. 6B), indicating that E2F1 may play a crucial role in control of CPC function. In line with this observation, both mRNA and protein levels of all CPC members showed remarkable downregulation in response to E2F1 inhibition in two PCa cell lines (Fig. 6C and Supplementary Fig. 6C). To determine whether all four CPC members are down-stream effectors of E2F1, we first checked the public ChIP-seq data from LNCaP-abl PCa cells [28] and observed clear E2F1 peaks at the promoter region of each CPC member (Fig. 6D). This observation was further confirmed by ChIP-qPCR assay in C4–2 cells (Fig. 6E and Supplementary Fig. 6D), proving that E2F1 could specifically bind to CPC promoters to activate transcription.
Fig. 6. EZH2 facilitates self-activation of E2F1 and thus promotes transcription of all CPC members.
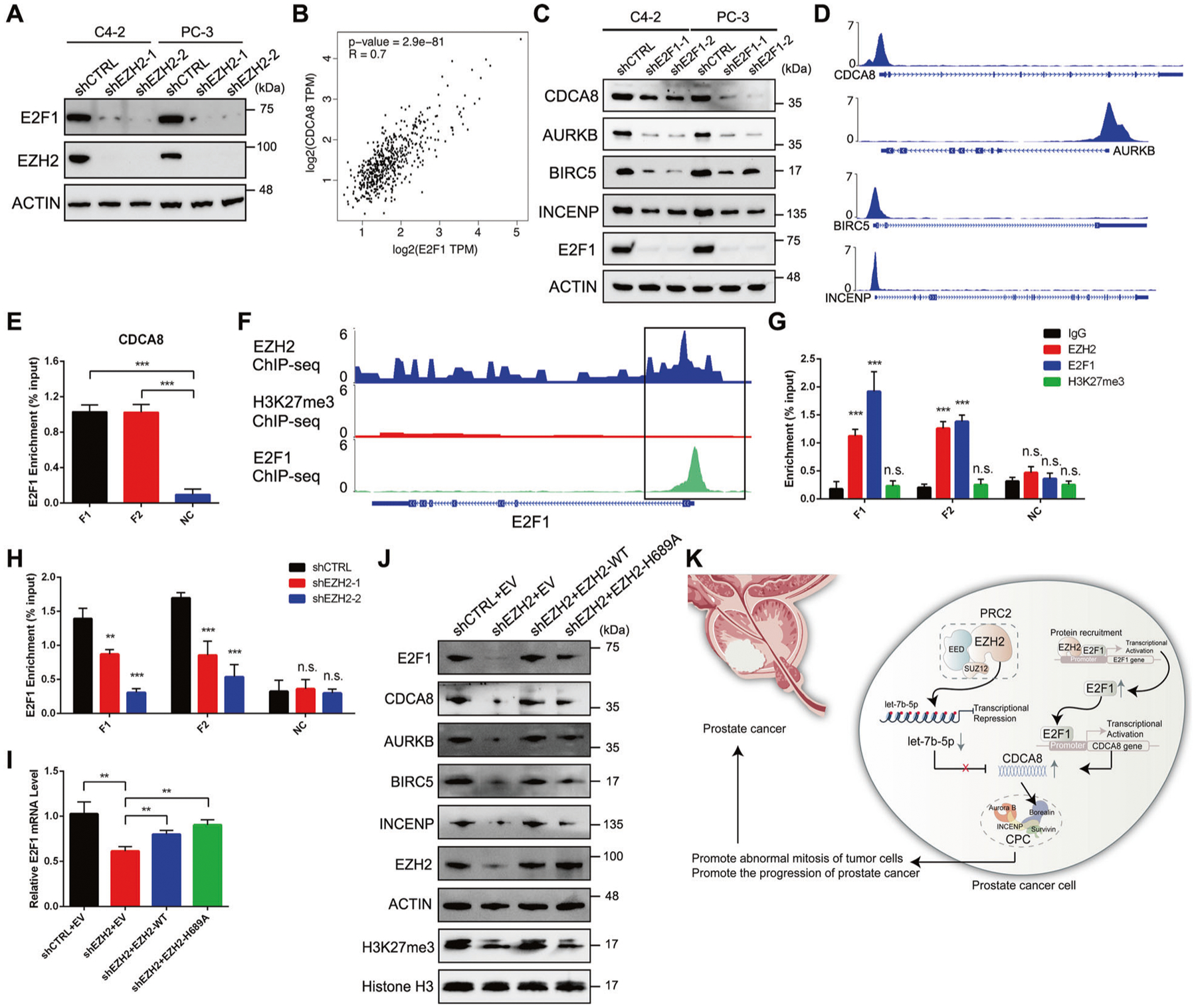
A Western blot analysis of the protein level of E2F1 in PCa cell lines upon EZH2 knockdown. B Scatter plot showing the relationship between CDCA8 and E2F1 expressions using data from TCGA, with Spearman correlation coefficient (R) and P value as indicated. C Western blot analysis of the protein levels of all CPC members in PCa cell lines upon E2F1 knockdown. D ChIP-seq profiles showing the E2F1 peaks at the promoter region of each CPC member in LNCaP-abl cells. E ChIP-qPCR assay to monitor the enrichment of E2F1 at the promoter region of CDCA8 in C4–2 cells. Two pairs of primers (F1 and F2) were used to amplify fragments inside CDCA8 promoter region while another pair of primers (NC) targeting nearby region was used as negative control. F ChIP-seq profiles showing the peaks of EZH2, H3K27me3 and E2F1 at the promoter region of E2F1 in PCa cells. G ChIP-qPCR assay to monitor the enrichment of EZH2, H3K27me3 and E2F1 at the promoter region of E2F1 in C4–2 cells. Two pairs of primers (F1 and F2) were used to amplify fragments inside E2F1 promoter region while another pair of primers (NC) targeting nearby region was used as negative control. H ChIP-qPCR assay to monitor the change of E2F1 occupancy at the promoter region of E2F1 upon EZH2 inhibition in C4–2 cells. I RT-qPCR analysis of E2F1 mRNA level in EZH2-deficient C4–2 cells overexpressed with either wild-type (WT) or H689A-mutant EZH2. J Rescue assay showing that ectopic expression of either wild-type (WT) or H689A-mutant EZH2 could restore the downregulation of all CPC members in EZH2-deficient cells, as measured by western blot. K The proposed model of this study. EZH2-mediated CDCA8 upregulation in PCa can be attributed to the combination of let-7b repression and E2F1 self-activation.
We then tried to unveil the mechanism by which EZH2 stimulates E2F1 expression. Previous reports demonstrated that EZH2 could cooperate with E2F1 to activate gene expression [28, 29]. In principle, EZH2 recruits E2F1 to a number of chromatin sites lacking H3K27me3 marks and thus activate genes that are critical for tumor progression [28]. Based on these findings, we speculated that E2F1 may self-promote its expression with the help of EZH2 in PCa cells. As expected, public ChIP-seq data in LNCaP and its derived cell lines [16, 28] showed co-localized EZH2 and E2F1 peaks at E2F1 promoter region, while no H3K27me3 peak was detected (Fig. 6F). By conducting ChIP-qPCR assay, we obtained similar results in C4–2 cells (Fig. 6G). More importantly, the enrichment of E2F1 in its own promoter was significantly reduced upon EZH2 knockdown (Fig. 6H), further supporting the idea that EZH2 could promote E2F1 transcription by facilitating the occupancy of E2F1 at the promoter site of itself.
Last, we attempted to determine the significance of E2F1 activation in EZH2-mediated CDCA8 regulation. Since EZH2 is also a direct transcriptional activation target of E2F1 [30], rescue of E2F1 expression in EZH2-deficient cells would significantly enhance endogenous EZH2 level and thereby prevent us from reaching accurate conclusion. As an alternative strategy, we sought to distinguish the methylation-dependent and -independent roles of EZH2 since our current findings suggested that the role of EZH2 in repressing let-7b expression is methylation-dependent, while its function in activating E2F1 does not rely on its lysine methyltransferase activity. To this end, we treated C4–2 cells with either control or EZH2 3′-UTR-targeting shRNA, followed by rescue expression using wild-type or catalytically dead H689A mutant of EZH2. Intriguingly, re-expression of either wild-type or H689A-mutant EZH2 rescued the E2F1 mRNA level to a great extent (Fig. 6I), supporting the methylation-independent transcriptional activation of E2F1 by EZH2. Western blot analysis further proved the re-upregulation of E2F1 protein level by both wild-type and H689A-mutant EZH2 in EZH2-deficient cells (Fig. 6J). Meanwhile, expression levels of CDCA8 as well as other CPC components were coordinately restored (Fig. 6J). Above all, our data provide strong evidence that, apart from its canonical role, EZH2 could directly induce E2F1 transcription in a methylation-independent manner, which subsequently contributes to the activation of the whole CPC in PCa.
DISCUSSION
More than one decade has passed since the beginning of EZH2 research in the aspect of cell cycle regulation [31]. Throughout these years, a number of key cell cycle-associated genes, such as BRCA1 [32], FBXO32 [33], BRG1 [34], and FOXA1 [35], have been reported to be modulated by EZH2 to accelerate cell division rates in cancer. In the present study, mitotic regulator CDCA8 is identified as a novel EZH2 target to maintain proliferation and malignant development in PCa cells. In addition to CDCA8, EZH2 also has a great impact on the expression of the other CPC members including AURKB, BIRC5 and INCENP. Indeed, our RNA-seq data revealed over 20 cell cycle-related genes which are transcriptionally activated by EZH2, further reflexing the importance and complexity of EZH2-mediated cell cycle regulation in cancer.
It is now clearly established that the interplay between EZH2 and miRNAs contributes to tumorigenesis in various cancer types [24, 36, 37]. Interestingly, double-negative feedback is frequently occurred between EZH2 and its targeting miRNAs. For instance, the EZH2/miR-138 feedback loop is associated with drug resistance in multiple myeloma [38], while the EZH2/miR-26a feedback loop regulates tumor cell growth in hepatocellular carcinoma [39]. Furthermore, multiple EZH2/miRNA feedback loops are identified in ovarian cancer which together promote the malignant behaviors of cancer cells [40]. In our study, let-7b expression is verified to be repressed by EZH2-induced H3K27me3 at its promoter region, which subsequently maintains the high expression of CDCA8 in PCa. Notably, a previous study showed that EZH2 mRNA is also a direct target of let-7b in PCa cells [41], suggesting the existence of a potential feedback between EZH2 and let-7b. It is anticipated that this double-negative feedback loop could facilitate PCa development by upregulating the expression of numerous oncogenes including CDCA8.
Likely, the most significant implication for our work is to unveil the non-canonical role of EZH2 in promoting E2F1 transcription. Although massive studies have reported the EZH2-targeting function of E2F1 [27], few investigations focus on the role of EZH2 in regulating E2F1. A previous work suggested that retinoblastoma protein (pRB) could bind to E2F1 and EZH2 simultaneously and thus establish H3K27me3-dependent repression at the genomic repeat sequences of somatic cells [42]. In comparison, other studies revealed that E2F1 alone could form a transcriptional complex with EZH2 to induce the transcription of a range of genes critical for tumor progression [28, 29]. In agreement with the latter opinion, our data further demonstrate that E2F1 itself is controlled by EZH2 through this methylation-independent pathway to mediate CPC member expression. In addition, disorder of E2F1 could also explain why such a large scale of co-affected DEGs were observed upon CDCA8/EZH2 inhibition (Fig. 4E). Since E2F1 expression is precisely controlled during mitosis, it is not surprising that the dramatic cell cycle arrest occurred in CDCA8-deficient cells were accompanied by a significant downregulation of E2F1 (Fig. 4H and Supplementary Table 2). As a result, transcriptions of numerous E2F1 target genes were co-altered in response to CDCA8/EZH2 suppression, which finally made these DEGs overlapped with each other.
In conclusion, our present work uncovers a dual-role of EZH2 in mediating CDCA8 expression in PCa (Fig. 6K). On one hand, EZH2 represses the transcription of CDCA8-targeting let-7b by conferring H3K27me3 marks at its promoter; on the other hand, EZH2 enhances the self-activation of E2F1 and thus promotes the E2F1-driven CDCA8 transcription.
METHODS AND MATERIALS
Cell culture
Human PCa cell lines PC-3, C4–2B, 22RV1, LNCaP and DU145 were purchased from ATCC, while C4–2 cell line was a generous gift from Dr. Leland Chung. Human benign prostatic hyperplasia cell line BPH-1 was a kind gift from XD. All prostate cell lines were grown in RPMI 1640 medium supplemented with 10% FBS. HEK293T cells were purchased from ATCC and cultured in DMEM medium supplemented with 10% FBS. All cell lines were maintained at 37 °C and 5% CO2 in a humidified atmosphere. All cell lines used were authenticated and routinely screened for Mycoplasma.
Lentivirus/Retrovirus construction
All lentiviral shRNA vectors were purchased from Sigma (shCDCA8–1: TRCN0000007898, shCDCA8–2: TRCN0000007900; shEZH2–1: TRCN0000286227, shEZH2–2: TRCN0000040077; shE2F1–1: TRCN0000000251, shE2F1–2: TRCN0 000000252). For lentivirus production, shRNA vectors and helper plasmids of pVSVG and psPAX2 were co-transfected into HEK293T cells using Lipofectamine 3000 (Invitrogen) following its protocol. The medium was renewed once at 24 h post-transfection. The supernatants containing viruses were collected at 48 h post-transfection and directly used for infection.
Retroviral vector of pBABE-puro is a gift from Hartmut Land & Jay Morgenstern & Bob Weinberg (Addgene plasmid # 1764). The coding sequence of CDCA8 was cloned into pBABE-puro vector to construct CDCA8-expressing plasmid. For retrovirus production, pBABE vectors and helper plasmids of pVSVG and pUMVC were co-transfected into HEK293T cells using Lipofectamine 3000 (Invitrogen) following its protocol. The medium was renewed once at 24 h post-transfection. The supernatants containing viruses were collected at 48 h post-transfection and directly used for infection.
EZH2 inhibitors treatment
DZNep and EPZ6438 were purchased from Selleck Chemicals and dissolved in DMSO. C4–2 cells were treated with the EZH2 inhibitors at the indicated concentration for 72 h until further use.
miRNA mimics and inhibitors treatment
All miRNA mimics and inhibitors were purchased from Thermo (let-7b mimic: MC12489, let-7a mimic: MC10050, miR-98 mimic: MC10426, miR-200a mimic: MC10250, miR-203 mimic: MC10152; let-7b inhibitor: MH11050). The mimics/inhibitors were delivered into PCa cells using Lipofectamine RNAiMAX (Invitrogen) following its protocol. Cells were collected at 48 h post-transfection for further use.
Western blot
To denature proteins, cell lysates were added to 4× loading buffer (Bio-Rad) and heated to 95 °C for 10 min. Protein samples were separated electrophoretically by SDS-PAGE, and semi-dry transferred to PVDF membranes (Roche). The membranes were blocked for 45 min in Tris-buffered saline-Tween 20 (TBST) with 5% nonfat milk. Thereafter, immunoblotting was performed by incubation with primary antibodies for 2 h at room temperature. After washing for three times, the membranes were incubated with goat anti-mouse/rabbit IgG (H + L)-HRP secondary antibody (GenDEPOT, 1:5000 dilution) for 1 h. The signals were developed using western ECL Substrate (Bio-Rad) and captured by a Bio-Rad imaging system. The relative protein level was evaluated using ImageJ software. The primary antibodies used in this paper were listed in Supplementary Table 4.
Real-time (RT)- qPCR analysis
To detect mRNA level, total RNA was extracted from cells using RNeasy Plus Mini Kit (Qiagen). The obtained RNA was then reverse transcribed into cDNA using Maxima H Minus First Strand cDNA Synthesis Kit (Thermo). Each cDNA sample was amplified using Universal SYBR Green Supermix (Bio-Rad) in the QuantStudio 6 Flex Real-time PCR System (GE Healthcare) following manufacturer’s instructions. The primers used for RT-qPCR analysis were summarized in Supplementary Table 5. The relative RNA level was calculated using the 2−ΔΔCt method with the Ct values normalized using GAPDH as an internal control.
To detect miRNA level, miRNeasy Mini Kit (Qiagen) was used to isolate miRNA and total RNA from cells. The obtained RNA was reverse transcribed into cDNA using TaqMan miRNA Reverse Transcription Kit (Thermo). TaqMan MicroRNA Assay was then performed following the guideline from Thermo. The relative let-7b level was calculated using the 2−ΔΔCt method with the Ct values normalized using U6 snRNA as an internal control. The probes used for TaqMan qPCR analysis were purchased from Thermo (let-7b: 002619, U6 snRNA: 001973).
Tissue microarrays (TMAs) and immunohistochemistry (IHC) staining
Prostate tumor biopsies retrieved from Vancouver Prostate Centre tissue bank were used to construct TMAs as published previously [43]. This protocol was approved by the office of research ethics in the University of British Columbia. IHC was performed using Ventana Discovery XT autostainer (Ventana) with anti-CDCA8 and anti-EZH2 antibodies as reported [44]. All stained slides were scanned by a Leica SCN400 scanner. Digital images were evaluated and scored by a pathologist, Dr. Ladan Fazli. EZH2 histology score (H-score) was calculated by the Aperio ImageScope software (Leica Biosystems) based on both intensity and percentage of the IHC signals. The primary antibodies used for IHC were listed in Supplementary Table 4.
Cell viability assay
The proliferation rate of prostate cells was determined using CellTiter-Glo Luminescent Cell Viability Assay (Promega) following the user’s manual. In brief, cells were seeded in 96-well plates at a density of 5 × 103 cells/well, and were incubated under 37 °C in a humidified 5% CO2 atmosphere. At each time-point, culture medium was discarded and 40 μL of CellTiter-Glo solution was added into each well, followed by incubation for 10 min on an orbital shaker at 37 °C to induce cell lysis. The bioluminescence was detected using a Tecan plate reader.
Boyden chamber invasion and migration assays
The invasiveness of PCa cells was assessed by their ability to pass through Matrigel (Corning)-coated Transwell inserts (Millipore). Briefly, the upper surface of the polycarbonic membranes (8.0-μm pore size) of the Transwell chambers was coated with Matrigel (1:20 diluted with 1640 medium). Cells (8 × 104) diluted in 300 μL of serum-free 1640 medium were seeded into the upper compartments of the chambers. Meanwhile, the lower compartments of the chambers were filled with 800 μL of 1640 medium containing 10% FBS. After 24 h, invasive cells that had migrated from Matrigel to the lower surface of the filters were fixed in methanol, stained with 0.1% crystal violet (Sigma) and subjected to microscopic inspection. The number of invasive cells was expressed as the average number of cells counted in 5 random fields per filter. The migration of prostate cells was assessed by their ability to pass through Transwell inserts without Matrigel coated, else is the same way with invasion assay as described above.
Cell cycle analysis
For cell cycle analysis, control or CDCA8-deficient C4–2 cells were harvested and fixed with absolute ethanol for at least 15 min at −20 °C. The fixed cells were rehydrated with PBS at room temperature for 5 min and then stained with 3 μM of propodium iodide solution (PI, Thermo) and subjected to flow cytometry analysis using LX200 Luminex Multiplexing Assay System (Thermo). Data were analyzed by FlowJo software.
Cell apoptosis analysis
The Annexin V-FITC Apoptosis Detection Kit (Sigma) was used for cell apoptosis analysis. Briefly, control or CDCA8-deficient C4–2 cells were harvested and resuspended by Annexin V-FITC binding buffer. Next, cells were incubated with 5 μl Annexin V and PI for 20 min. The apoptotic rate was detected by flow cytometry using LX200 Luminex Multiplexing Assay System (Thermo). Data were analyzed by FlowJo software.
Staining of autophagic vacuoles
Autophagy monitoring in PCa cells was achieved by Autophagy Assay Kit (Sigma) according to the manufacturer’s instructions. In short, control or CDCA8-deficient C4–2 cells without culturing medium were incubated with the autophagosome detection reagent under 37 °C in a humidified 5% CO2 atmosphere for 1 h. After washing with wash buffer for 3 times, cells were visualized using a fluorescence microscope (Bio-Rad) to detect fluorescence intensity. The bioluminescence was read on Synergy 2 Multi-Mode Reader (BioTek).
RNA sequencing (RNA-seq) analysis
For RNA-seq, total RNA (two for shCTRL and two for shCDCA8) was isolated from cells using RNeasy Plus Mini Kit (Qiagen) and subjected to BGI for library preparation and sequencing using the DNBseq platform. RNA-seq reads were mapped to the human reference genome hg19 using topHat (v2.1.1) [45]. Read counts of the annotated genes were obtained by the Python software HTSeq-count (v0.11.2) [46]. The edgeR package (v3.34.1) was used to identify DEGs from RNA-seq with two biological replicates [47]. Genes with at least 1.5-fold change in expression levels and false discovery rate (FDR) less than 0.05 were considered as differentially expressed. The heatmap plots were depicted using R plot function and pheatmap package (v1.0.12). The functional enrichment analysis was conducted by the R package clusterProfiler (v4.0.5) [48]. GSEA was performed with JAVA software.
Small RNA sequencing (RNA-seq) analysis
For small RNA-seq, small RNA (two for shCTRL and two for shEZH2) was extracted from cells using miRNeasy Mini Kit (Qiagen) and subjected to BGI for library preparation and sequencing using the DNBseq platform. The adaptor sequences in raw data were removed using cutadapt (v2.3), then an all-in-one small noncoding RNA annotation pipeline AASRA was applied to process the mapping and count the raw count for each small noncoding RNA [49]. DEGseq2 (v.1.34.0) was used to detect the differentially expressed miRNAs from raw counts matrix [50]. The adjusted P value less than 0.01 was considered as significant.
Chromatin immunoprecipitation (ChIP)-seq analysis
For analysis of public ChIP-seq data, the reads mapping and processing were achieved as described previously [51, 52]. Briefly, the reads were first mapped to the human genome hg19 by Bowtie (V1.3.1) [53]. The wig file was then generated using DANPOS (V.2.2.3) [54] and bigwig file was finally obtained using the tool WigToBigWig for visualization in genome browser.
ChIP-qPCR analysis
The ChIP experiment was performed using the EZ-Magna ChIP kit (Millipore) with the procedure provided by the manufacturer. Cells were cross-linked using paraformaldehyde solution (Invitrogen) and terminated by glycine solution. Chromatin fragment at an average size of 200 bp was obtained by cell lysis and sonication using a Diagenode bioruptor. DNA was isolated from samples by incubation with the antibody at 4 °C overnight followed by washing and reversal of cross-linking. To analyze the enrichment of DNA fragments at different regions, qPCR assay was conducted using the primers listed in Supplementary Table 5. Immunoprecipitated DNA was calculated as percentage of input DNA.
Dual-luciferase assay
All dual-luciferase vectors containing 3′-UTR sequence of interest were purchased from Genecopoeia using pEZX-MT06 vector as backbone. Dual-luciferase assays were performed at 24 h post-transfection of dual-luciferase vectors using the Dual-Glo luciferase reagent (Promega) according to the manufacturer’s instructions. A Tecan plate reader was used to measure the bioluminescence intensity.
EZH2 rescue assay
Both wild-type and H689A-mutant EZH2 plasmids were kind gifts from Dr. Jindan Yu. For rescue assay, the shEZH2–1 lentivirus was chosen since it targets the 3′-UTR region of endogenous EZH2 and will not affect the ectopic expression of EZH2. Samples were collected at 48 h post-transfection of EZH2 plasmids into EZH2-deficient C4–2 cells, followed by RT-qPCR and western blot assays to detect mRNA/protein levels.
In vivo animal experiment
Five-week-old male Balb/c athymic nude mice were purchased from Charles River. Animal care and use conditions were followed in accordance with institutional and National Institutes of Health protocols and guidelines, and all studies were approved by Houston Methodist Institution Animal Care and Use Committee. Nude mice were divided into three groups of 6 mice each.
For subcutaneous tumor model, each mouse was injected subcutaneously in the left flank with 1 × 107 of PC-3 cells (control or CDCA8-deficient) suspended in 100 μL of PBS. Tumor volumes were measured by length (a), width (b) and calculated as tumor volume = MIN(a)2 × MAX(b) × 0.5. Thirty-two days after the tumor cell injection, the mice were sacrificed and tumor xenografts were removed and photographed.
Statistics and reproducibility
Statistical analysis was performed using GraphPad Prism (version 6.0) or R and presented as means ± SD. Unless otherwise specified, the P values were obtained using two-tailed Student’s t-tests for comparison of two datasets. Statistical data were considered significant if P < 0.05. The results were reproducible and conducted with established internal controls. Experiments were repeated for at least three times and yielded similar results.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by American Cancer Society (RSG-15-192-01) and a startup funding provided by the Northwestern University. Part of effort for QC was supported by Northwestern University, U.S. Department of Defense (W81XWH-17-1-0357, W81XWH-19–1-0563 and W81XWH-20-1-0504), NIH/NCI (R01CA208257, R01CA256741 and Prostate SPORE P50CA180995 Development Research Program) and Polsky Urologic Cancer Institute of the Robert H. Lurie Comprehensive Cancer Center of Northwestern University at Northwestern Memorial Hospital.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41388-022-02208-x.
DATA AVAILABILITY
Next generation sequencing data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession code GSE184740. Our previously published RNA-seq data of EZH2-deficient C4–2 cells are available under accession code GSE143975. Previously published ChIP-seq data of LNCaP and its derived LNCaP-abl cells that were re-analyzed here are available under accession codes GSE107782 (EZH2 and H3K27me3) and GSE67809 (E2F1).
REFERENCES
- 1.Sullivan M, Morgan DO. Finishing mitosis, one step at a time. Nat Rev Mol Cell Biol. 2007;8:894–903. [DOI] [PubMed] [Google Scholar]
- 2.Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28:2925–39. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 4.Ben-Salem S, Venkadakrishnan VB, Heemers HV. Novel insights in cell cycle dysregulation during prostate cancer progression. Endocr-Relat cancer. 2021;28: R141–R155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gassmann R, Carvalho A, Henzing AJ, Ruchaud S, Hudson DF, Honda R, et al. Borealin: a novel chromosomal passenger required for stability of the bipolar mitotic spindle. J Cell Biol. 2004;166:179–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007;8:798–812. [DOI] [PubMed] [Google Scholar]
- 7.Carmena M, Wheelock M, Funabiki H, Earnshaw WC. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol. 2012;13:789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao X, Wen X, He H, Zheng L, Yang Y, Yang J, et al. Knockdown of CDCA8 inhibits the proliferation and enhances the apoptosis of bladder cancer cells. PeerJ. 2020;8:e9078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeon T, Ko MJ, Seo YR, Jung SJ, Seo D, Park SY, et al. Silencing CDCA8 Suppresses Hepatocellular Carcinoma Growth and Stemness via Restoration of ATF3 Tumor Suppressor and Inactivation of AKT/beta-Catenin Signaling. Cancers. 2021;13:1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qi G, Zhang C, Ma H, Li Y, Peng J, Chen J, et al. CDCA8, targeted by MYBL2, promotes malignant progression and olaparib insensitivity in ovarian cancer. Am J Cancer Res. 2021;11:389–415. [PMC free article] [PubMed] [Google Scholar]
- 11.Dai C, Miao CX, Xu XM, Liu LJ, Gu YF, Zhou D, et al. Transcriptional activation of human CDCA8 gene regulated by transcription factor NF-Y in embryonic stem cells and cancer cells. J Biol Chem. 2015;290:22423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. [DOI] [PubMed] [Google Scholar]
- 13.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–9. [DOI] [PubMed] [Google Scholar]
- 15.Yang YA, Yu J. EZH2, an epigenetic driver of prostate cancer. Protein Cell. 2013;4:331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J, Lee Y, Lu X, Song B, Fong KW, Cao Q, et al. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep. 2018;25:2808–20 e2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koyen AE, Madden MZ, Park D, Minten EV, Kapoor-Vazirani P, Werner E, et al. EZH2 has a non-catalytic and PRC2-independent role in stabilizing DDB2 to promote nucleotide excision repair. Oncogene. 2020;39:4798–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yi Y, Li Y, Meng Q, Li Q, Li F, Lu B, et al. A PRC2-independent function for EZH2 in regulating rRNA 2’-O methylation and IRES-dependent translation. Nat Cell Biol. 2021;23:341–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hernandez J, Thompson IM. Prostate-specific antigen: a review of the validation of the most commonly used cancer biomarker. Cancer. 2004;101:894–904. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez ME, Li X, Toy K, DuPrie M, Ventura AC, Banerjee M, et al. Downregulation of EZH2 decreases growth of estrogen receptor-negative invasive breast carcinoma and requires BRCA1. Oncogene. 2009;28:843–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, et al. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell. 2009;136:1122–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asenjo HG, Gallardo A, Lopez-Onieva L, Tejada I, Martorell-Marugan J, Carmona-Saez P, et al. Polycomb regulation is coupled to cell cycle transition in pluripotent stem cells. Sci Adv. 2020;6:eaay4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao Q, Mani RS, Ateeq B, Dhanasekaran SM, Asangani IA, Prensner JR, et al. Coordinated regulation of polycomb group complexes through microRNAs in cancer. Cancer Cell. 2011;20:187–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang HY, Lin YC, Li J, Huang KY, Shrestha S, Hong HC, et al. miRTarBase 2020: updates to the experimentally validated microRNA-target interaction database. Nucleic acids Res. 2020;48:D148–D154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chirshev E, Oberg KC, Ioffe YJ, Unternaehrer JJ. Let-7 as biomarker, prognostic indicator, and therapy for precision medicine in cancer. Clin Transl Med. 2019;8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chun JN, Cho M, Park S, So I, Jeon JH. The conflicting role of E2F1 in prostate cancer: A matter of cell context or interpretational flexibility? Biochim et Biophys Acta Rev Cancer. 2020;1873:188336. [DOI] [PubMed] [Google Scholar]
- 28.Xu H, Xu K, He HH, Zang C, Chen CH, Chen Y, et al. Integrative Analysis Reveals the Transcriptional Collaboration between EZH2 and E2F1 in the Regulation of Cancer-Related Gene Expression. Mol Cancer Res. 2016;14:163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tabbal H, Septier A, Mathieu M, Drelon C, Rodriguez S, Djari C, et al. EZH2 cooperates with E2F1 to stimulate expression of genes involved in adrenocortical carcinoma aggressiveness. Br J Cancer. 2019;121:384–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park SH, Fong KW, Mong E, Martin MC, Schiltz GE, Yu J. Going beyond Polycomb: EZH2 functions in prostate cancer. Oncogene. 2021;40:5788–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonzalez ME, DuPrie ML, Krueger H, Merajver SD, Ventura AC, Toy KA, et al. Histone methyltransferase EZH2 induces Akt-dependent genomic instability and BRCA1 inhibition in breast cancer. Cancer Res. 2011;71:2360–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Z, Lee ST, Qiao Y, Li Z, Lee PL, Lee YJ, et al. Polycomb protein EZH2 regulates cancer cell fate decision in response to DNA damage. Cell Death Differ. 2011;18:1771–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fillmore CM, Xu C, Desai PT, Berry JM, Rowbotham SP, Lin YJ, et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature. 2015;520:239–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park SH, Fong KW, Kim J, Wang F, Lu X, Lee Y, et al. Posttranslational regulation of FOXA1 by Polycomb and BUB3/USP7 deubiquitin complex in prostate cancer. Sci Adv. 2021;7:eabe2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322:1695–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yin H, Wang Y, Wu Y, Zhang X, Liu J, Wang T, et al. EZH2-mediated Epigenetic Silencing of miR-29/miR-30 targets LOXL4 and contributes to Tumorigenesis, Metastasis, and Immune Microenvironment Remodeling in Breast Cancer. Theranostics. 2020;10:8494–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rastgoo N, Pourabdollah M, Abdi J, Reece D, Chang H. Dysregulation of EZH2/ miR-138 axis contributes to drug resistance in multiple myeloma by down-regulating RBPMS. Leukemia. 2018;32:2471–82. [DOI] [PubMed] [Google Scholar]
- 39.Zhuang C, Wang P, Huang D, Xu L, Wang X, Wang L, et al. A double-negative feedback loop between EZH2 and miR-26a regulates tumor cell growth in hepatocellular carcinoma. Int J Oncol. 2016;48:1195–204. [DOI] [PubMed] [Google Scholar]
- 40.Liu T, Cai J, Wang Z, Cai L. EZH2-miRNA Positive Feedback Promotes Tumor Growth in Ovarian Cancer. Front Oncol. 2020;10:608393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong D, Heath E, Chen W, Cher ML, Powell I, Heilbrun L, et al. Loss of let-7 up-regulates EZH2 in prostate cancer consistent with the acquisition of cancer stem cell signatures that are attenuated by BR-DIM. PloS One. 2012;7:e33729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishak CA, Marshall AE, Passos DT, White CR, Kim SJ, Cecchini MJ, et al. An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol Cell. 2016;64:1074–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu Y, Yang O, Fazli L, Rennie PS, Gleave ME, Dong X. Progesterone receptor expression during prostate cancer progression suggests a role of this receptor in stromal cell differentiation. Prostate. 2015;75:1043–50. [DOI] [PubMed] [Google Scholar]
- 44.Xie N, Cheng H, Lin D, Liu L, Yang O, Jia L, et al. The expression of glucocorticoid receptor is negatively regulated by active androgen receptor signaling in prostate tumors. Int J Cancer. 2015;136:E27–38. [DOI] [PubMed] [Google Scholar]
- 45.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang C, Xie Y, Guo M, Yan W. AASRA: an anchor alignment-based small RNA annotation pipelinedagger. Biol Reprod. 2021;105:267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao D, Zhang L, Zhang M, Xia B, Lv J, Gao X, et al. Broad genic repression domains signify enhanced silencing of oncogenes. Nat Commun. 2020;11:5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xia B, Zhao D, Wang G, Zhang M, Lv J, Tomoiaga AS, et al. Machine learning uncovers cell identity regulator by histone code. Nat Commun. 2020;11:2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, et al. DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 2013;23:341–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Next generation sequencing data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession code GSE184740. Our previously published RNA-seq data of EZH2-deficient C4–2 cells are available under accession code GSE143975. Previously published ChIP-seq data of LNCaP and its derived LNCaP-abl cells that were re-analyzed here are available under accession codes GSE107782 (EZH2 and H3K27me3) and GSE67809 (E2F1).