

Abstract
Caspase-11 detection of intracellular lipopolysaccharide (LPS) from invasive Gram-negative bacteria mediates noncanonical activation of the NLRP3 inflammasome. While avirulent bacteria do not invade the cytosol, their presence in tissues necessitates clearance and immune system mobilization. Despite sharing LPS, only live avirulent Gram-negative bacteria activate the NLRP3 inflammasome. Here, we found that bacterial mRNA, which signals bacterial viability, was required alongside LPS for noncanonical activation of the NLRP3 inflammasome in macrophages. Concurrent detection of bacterial RNA by NLRP3 and binding of LPS by procaspase-11 mediated a procaspase-11-NLRP3 interaction prior to caspase-11 activation and inflammasome assembly. LPS binding to procaspase-11 augmented bacterial mRNA-dependent assembly of the NLRP3 inflammasome, while bacterial viability and an assembled NLRP3 inflammasome were necessary for activation of LPS-bound procaspase-11. Thus, the procaspase-11-NLRP3 interaction nucleated a scaffold for their interdependent activation explaining their functional reciprocal exclusivity. Our findings inform novel vaccine adjuvant combinations and sepsis therapy.
Murine caspase-11 and its human orthologues, caspases 4 and 5, are cytoplasmic receptors of LPS1,2,3. Caspase-11 orchestrates defense against Gram-negative bacteria and lethality in sepsis models through cleavage of gasdermin D (GSDMD), which executes pyroptosis of infected cells and damages tissue in endotoxic shock4,5,6. Caspase-11 also mediates noncanonical (NC)-activation of the highly inflammatory NLRP3 inflammasome to elicit caspase-1 cleavage and interleukin-1β (IL-1β) production7,8,9,10,11,12. Caspase-11 does not cleave IL-1β directly, while both caspases 1 and 11 cleave GSDMD6. Potential mechanisms of NC-activation of the NLRP3 inflammasome include caspase-11 heterodimerization with caspase-111,13 and caspase-11-mediated GSDMD-dependent cellular perturbations acting through a cell-intrinsic pathway4,5,14.
Live, but not dead, forms of virulent and avirulent Gram-negative bacteria elicit NC-activation of the NLRP3 inflammasome in macrophages8,9,10,11,12,15,16,17. A specific class of pathogen associated molecular patterns (PAMPs) called vita-PAMPs, such as bacterial messenger RNA (mRNAbac) and the cyclic dinucleotide c-di-AMP, signals microbial viability and potential infectivity, and augments the immune response15,17,18,19,20. Detection of the vita-PAMP mRNAbac from live bacteria triggers the activation of the NLRP3 inflammasome in response to Gram-negative, but not Gram-positive bacteria15,17,18,19,20, which do not have LPS, irrespective of bacterial virulence factors that activate inflammasomes21.
Here we report that coincident cytosolic detection of LPS with mRNAbac was required for NC-activation of the NLRP3 inflammasome in macrophages. Co-detection of LPS and bacterial RNA triggered an interaction of procaspase-11 with NLRP3 mediated by the NLRP3 LRR and PYD domains and the procaspase-11 scaffold domain. Biochemical interaction and interdependent activation of procaspase-11 and NLRP3 mechanistically underlie their functional reciprocal exclusivity.
Results
mRNAbac and LPS activate caspase-11 and the inflammasome
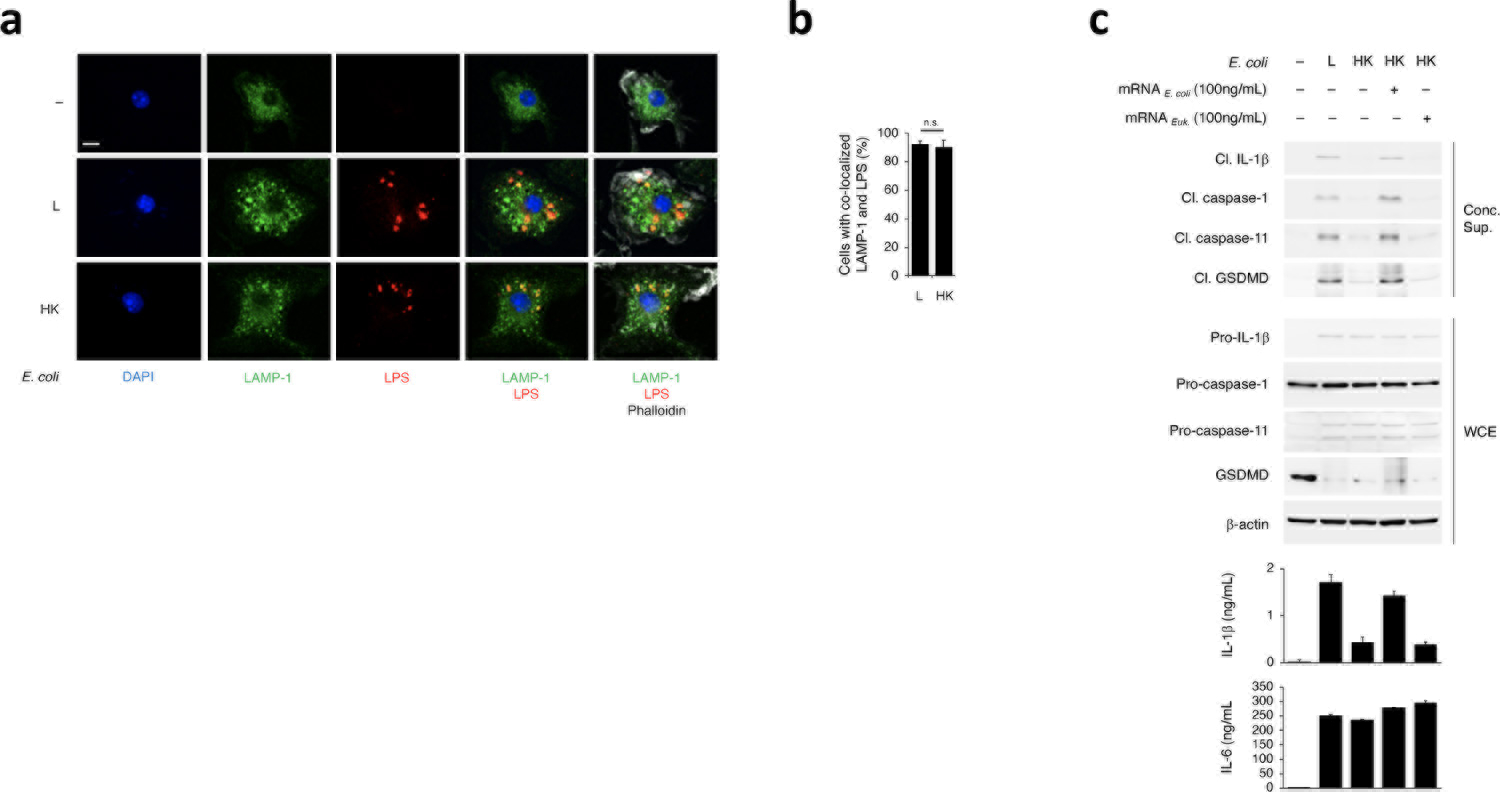
We investigated NC-activation of the NLRP3 inflammasome in bone marrow-derived macrophages (hereafter ‘macrophages’) following phagocytosis of Gram-negative bacteria. We observed similar lysosomal localization of live and dead E. coli (Extended Data Fig. 1a,b). To avoid the compounding effects of replication or virulence factors, we used thymidine auxotrophs of non-pathogenic Escherichia coli K12, strain DH5α17,19. Live E. coli elicited cleavage of caspase-11, caspase-1, GSDMD and IL-1β, and induced pyroptosis as visualized by electron microscopy and measured by lactate dehydrogenase (LDH) release (Fig. 1a,b,c and Extended Data Fig. 1c). These responses, which reflect inflammasome effector functions, were significantly impaired in response to killed E. coli, which have LPS but lack the vita-PAMP mRNAbac17, despite similar inflammasome priming3 (expression of NLRP3 and pro-forms of IL-1β, caspase-1 and caspase-11) by either live or killed E. coli (Fig. 1a,c and Extended Data Fig. 1c). Delivery of mRNAbac or bacterial RNA (RNAbac, which contains 1% mRNAbac) with phagocytosed killed E. coli, irrespective of RNAbac isolation from either Gram-positive or Gram-negative bacteria, restored caspase-11 and GSDMD cleavage (Fig. 1a,c and Extended Data Fig. 1c) and restored IL-1β secretion and pyroptosis to levels comparable to those elicited by live E. coli (Fig. 1a,c and Extended Data Fig. 1c). Eukaryotic mRNA was not able to do so (Extended Data Fig. 1c). Inflammasome-independent IL-6 and TNF production were similar for all stimulation conditions (Fig. 1a,c and Extended Data Fig. 1c). E. coli treatment with Rifampicin, which inhibits RNAbac synthesis, reduced caspase-11, caspase-1 and IL-1β cleavage, while retaining ~70% bacterial viability (Fig. 1d,e).
Figure 1. Bacterial mRNA and stimulatory LPS are both required for caspase-11 and noncanonical inflammasome activation.
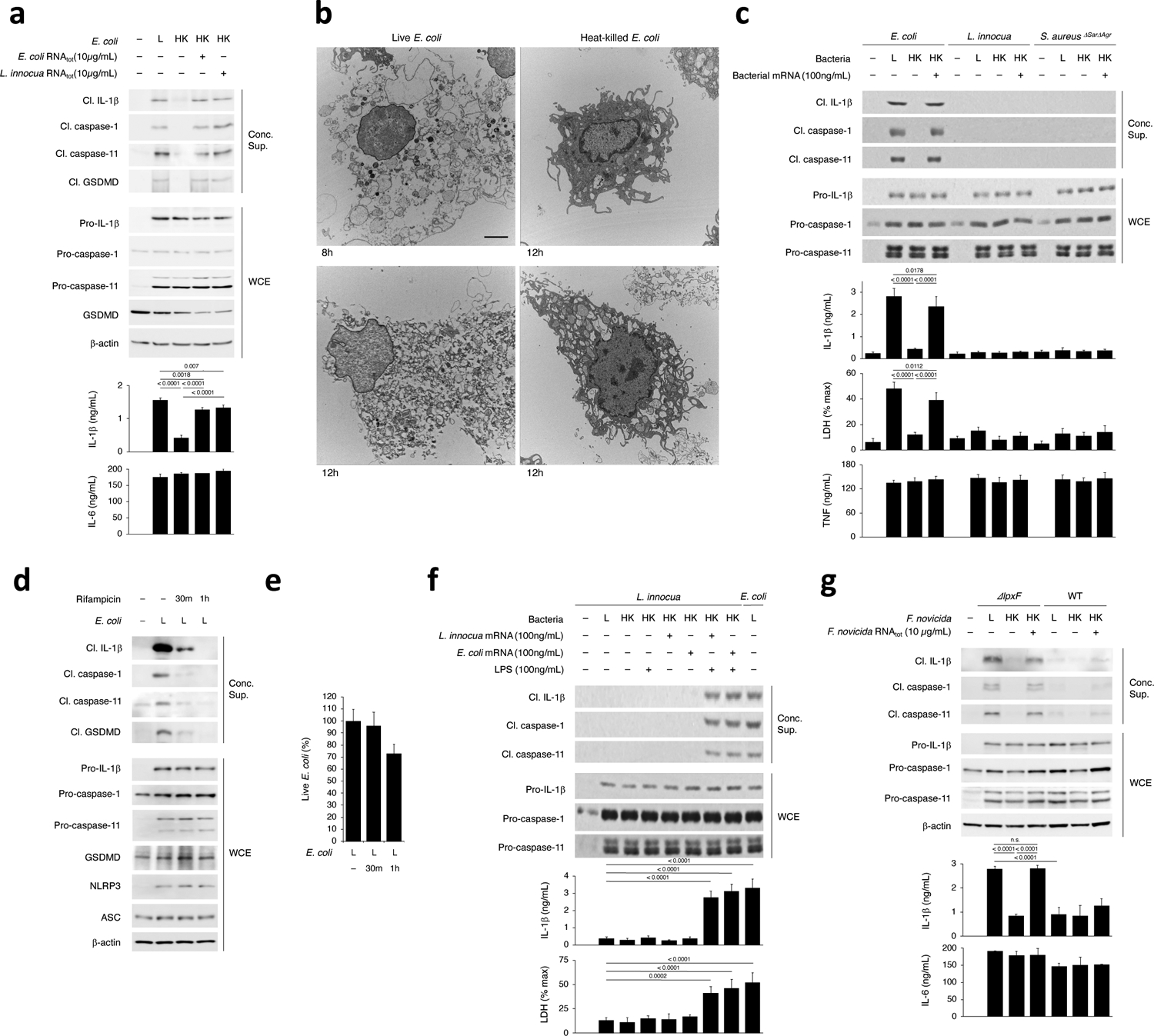
a, Immunoblots of macrophage concentrated supernatants (20hr) or whole cell extracts (WCE, 6hr), and cytokine concentrations in culture supernatants (20hr) post-stimulation with live (L), heat-killed (HK) or HK E. coli supplemented with 10μg/mL total bacterial RNA (RNAtot) from E. coli or L. innocua. b, Electron microscopy on macrophages stimulated with L or HK E. coli for 8 or 12 hrs. Scale bar=2μm. c, Immunoblots and cytokine concentrations as in a and lactate dehydrogenase (LDH) release (pyroptosis) post-stimulation with L or HK E. coli, L. innocua or S. aureusΔSarΔAgr supplemented with their respective 100ng/mL bacterial mRNA, d, Immunoblots as in a post-stimulation with L E. coli grown in 2μg/mL Rifampicin for indicated times. e, Percentage live E. coli following Rifampicin treatment. f, Immunoblots, cytokine concentrations and LDH release as in c post-stimulation with L E. coli or L, HK or HK L. innocua supplemented with LPS and/or mRNA from E. coli or L. innocua. g, Immunoblots and cytokine concentrations as in a post-stimulation with L, HK or HK F. novicida or F. novicida ΔlpxF with 10μg/mL RNAtot from each bacterium. LDH measured by cytotoxicity assay; IL-1β, TNF and IL-6 by ELISA. Error bars, mean ± s.e.m. One-way ANOVA followed by multiple comparisons Sidak tests and p values indicated in bar graphs in a,g (n=4), c,f (n=3). ns: non-significant. Cleaved caspase-11 corresponds to caspase-11 p30. Bacteria:macrophage=20:1. Results represent at least 3 independent experiments.
mRNAbac did not elicit NC-activation of the NLRP3 inflammasome when added to avirulent Gram-positive bacteria (L. innocua or Staphylococcus aureusΔSarΔAgr) that lack LPS, despite similar inflammasome priming (Fig. 1c,f). Live Gram-positive bacteria did not activate the inflammasome either (Fig. 1c,f). Only LPS with mRNAbac from either Gram-positive or Gram-negative bacteria elicited inflammasome activation when delivered to macrophages with killed Gram-positive bacteria (Fig. 1f). Delivery of LPS with dead L. innocua without mRNAbac did not elicit inflammasome activation (Fig. 1f). Conversely, L. innocua mRNAbac delivered to cells with killed E. coli restored inflammasome activation to levels similar to those elicited by live E. coli and killed E. coli plus mRNAbac (Fig. 1f).
RNAbac with LPS from the Gram-negative Francisella novicida bacteria, in which lipid A is modified to evade immune stimulation1, did not activate the inflammasome (Fig. 1g), indicating the stimulatory activity of LPS was required for inflammasome effector function. RNAbac with killed mutant bacteria F. novicidaΔlpxF, which express stimulatory penta-acylated LPS, led to inflammasome effector functions at levels comparable to those elicited by the live F. novicidaΔlpxF (Fig. 1g). Comparatively, delivery of RNAbac with killed F. novicida, which express non-stimulatory tetra-acylated LPS, induced significantly less activation (Fig. 1g), as were live F. novicida1 (Fig. 1g). The live or killed forms of all bacteria induced similar inflammasome priming (Fig. 1g). Similar results were observed with E. coliLPSmut bacteria that express tetra-acylated non-stimulatory LPS2,3. Thus, coincident detection of stimulatory LPS and the vita-PAMP mRNAbac significantly augmented NC-activation of the NLRP3 inflammasome in macrophages in response to Gram-negative bacteria.
mRNAbac with low LPS NC-activate the NLRP3 inflammasome
Next, we tested whether experimental cytoplasmic delivery of mRNAbac with LPS into macrophages augmented NC-activation of the NLRP3 inflammasome. We transfected 2ng/mL ultrapure LPS and 100ng/mL of mRNAbac free of LPS or LPS-stimulatory activity (from E. coliLPSmut, L. innocua, or transcribed in vitro (IVT)). These concentrations approximated endotoxin and mRNAbac amounts measured in the cytosolic fraction of macrophages following phagocytosis of avirulent E. coli (see Methods, Extended Data Fig. 2)17,22. Cytosolic LPS amounts were on average 7–8 fold lower (~2ng/mL) after macrophage stimulation with avirulent compared to virulent E. coli (Extended Data Fig. 2a), consistent with lack of specialized machineries used for cytosolic entry21, and possibly linked to phagosome-to-cytosol translocation mediating virulence factor-independent cytosolic innate immunity or the cytosolic pathway of antigen cross-presentation15,17,23,24,25.
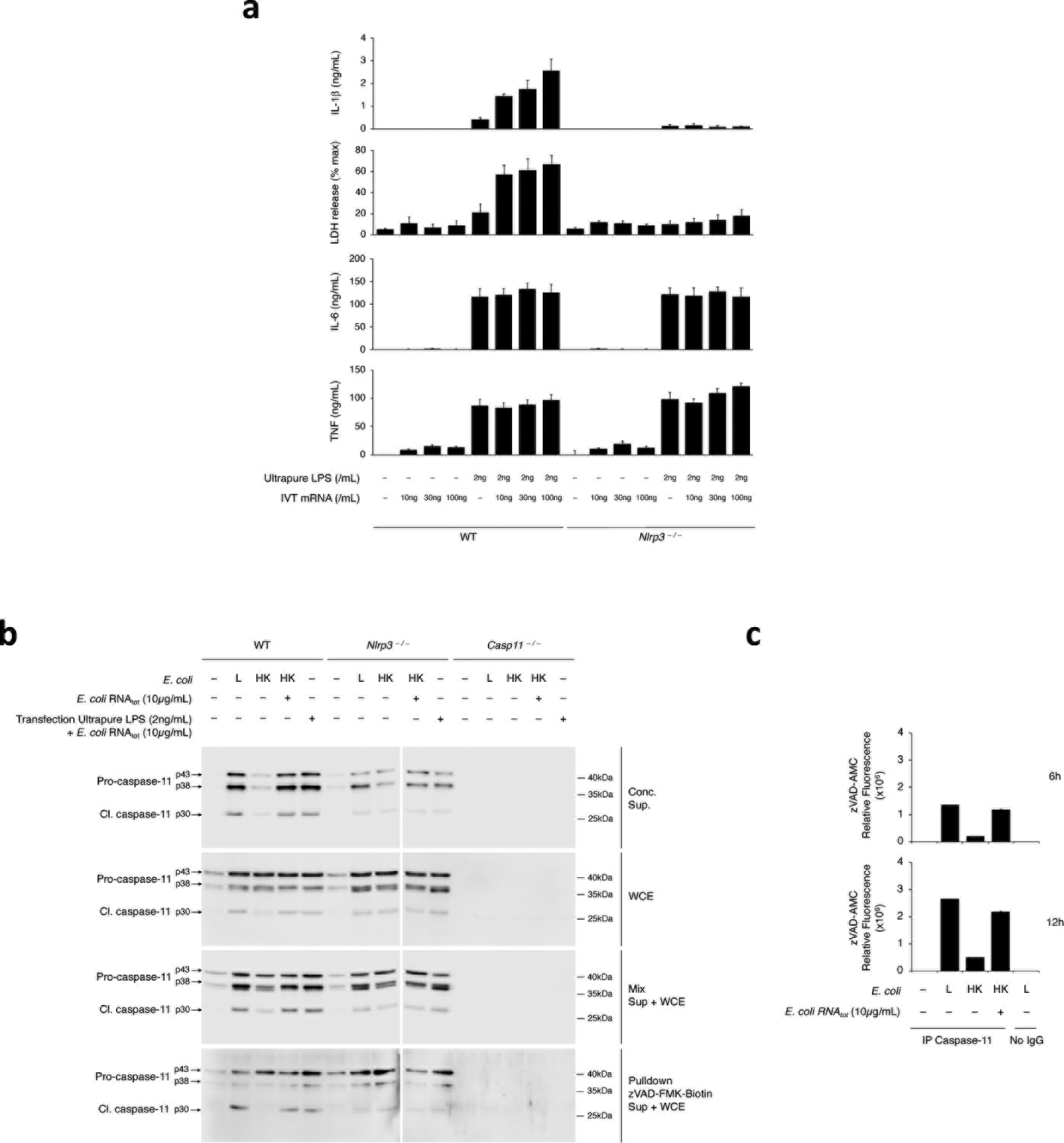
Transfection of 2ng/mL ultrapure LPS did not elicit inflammasome effector functions (Fig. 2a,b), despite intact inflammasome priming (Fig. 2a,b), while co-transfection with 100ng/mL mRNAbac irrespective of its source (E. coli, E. coliLPSmut, L. innocua, IVT mRNAbac) did elicit it (Fig. 2a,b), and in an NLRP3-dependent manner (Fig. 2b); transfected RNAbac alone did not (Fig. 2a,b), irrespective of dose (Extended Data Fig. 3a). Co-transfection of eukaryotic mRNA with 2ng/mL ultrapure LPS did not elicit NC-activation of the inflammasome (Fig. 2a). Co-transfection of 100ng/mL IVT mRNAbac and 2ng/mL ultrapure LPS into cells also elicited levels of IL-1β production at levels 2–3ng/mL that approximated those elicited by the supplementation of killed bacteria with 100ng/mL bacterial mRNA (Extended Data Fig. 3a). In contrast, transfection of a high concentration of LPS (1μg/mL) elicited inflammasome effector functions without mRNAbac (Fig. 2a), as previously reported1,2,3. Caspase-11 cleavage faithfully reflected caspase-11 activation (Extended Data Fig. 3b,c). Notably, cytosolic LPS after transfection of 1μg/mL LPS was higher than after phagocytosis of avirulent E. coli (on average 100-fold higher), virulent E. coli (10–11-fold higher) (Extended Data Fig. 2a), and Salmonella typhimurium infection (6-fold higher)3.
Figure 2. Low cytosolic concentration of LPS triggers noncanonical activation of the NLRP3 inflammasome when cytosolic bacterial mRNA is also present.
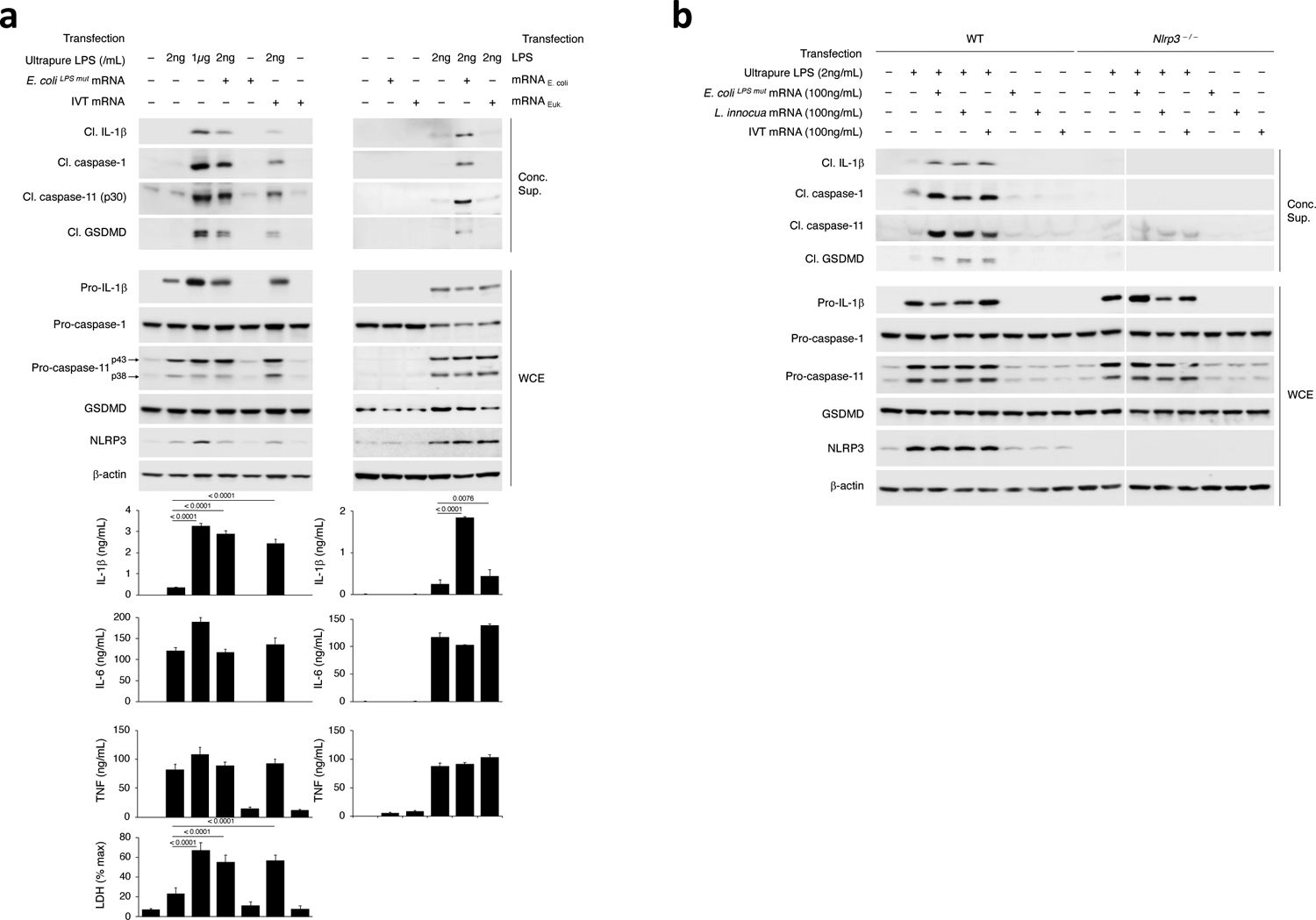
a-b, Immunoblots of macrophage concentrated supernatants (20hr) or WCE (6hr), and cytokine concentrations and LDH release in culture supernatants (20hr) as indicated following transfection of ultrapure LPS (low dose 2ng/mL or high dose 1μg/mL) +/− mRNA from E. coliLPSmut or in vitro transcribed (IVT) (100ng/mL), or transfection of LPS (2ng/mL) +/− mRNA from E. coli or eukaryotic cells (100ng/mL) (a), and transfection of wild-type (WT) or Nlrp3–/– macrophages with indicated doses of ultrapure LPS or mRNA from E. coliLPSmut, L. innocua, or IVT (b). Bacteria:macrophage ratio=20:1, except 50:1 for F. novicida. LDH measured by cytotoxicity assay; IL-1β, TNF and IL-6 by ELISA. Error bars, mean ± s.e.m. One-way ANOVA followed by multiple comparisons Sidak tests and p values indicated in bar graphs in a (IL-1β: n=4, LDH: n=3), and b (n=4). Cleaved caspase-11 corresponds to caspase-11 p30. Results represent at least 3 independent experiments.
mRNAbac (LPS-free RNA from L. innocua or IVT mRNAbac, and mRNAbac from E. coli or E. coliLPSmut) did not induce expression of NLRP3, procaspase-11 and pro-IL-1β protein above that in unstimulated macrophages (Fig. 2a), while ultrapure LPS, at either 2ng/mL or 1μg/mL, was sufficient to elicit inflammasome priming (Fig. 2a,b). Transfection of mRNAbac alone (either from E. coli, E. coliLPSmut or IVT mRNAbac) did not elicit appreciable IL-6 or TNF production, and did not increase their expression at any dose compared to transfection of LPS alone (Fig. 2a and Extended Data Fig. 3a). Contrarily, co-transfection of 2ng/mL LPS with mRNAbac elicited inflammasome-dependent pro-IL-1β cleavage and secretion of IL-1β (Fig. 2a,b), in a manner dependent on the dose of co-transfected mRNAbac (Extended Data Fig. 3a). Thus, the ability of mRNAbac to augment NC-activation of the NLRP3 inflammasome by transfected 2ng/mL LPS was not through inflammasome priming, which was mediated primarily by LPS. At cytosolic concentrations equivalent to those found in macrophages following stimulation with bacteria, cytoplasmic LPS and mRNAbac, which alone did not induce inflammasome activation, synergized in mediating NC-activation of the NLRP3 inflammasome.
LPS augments mRNAbac-driven NLRP3 inflammasome assembly
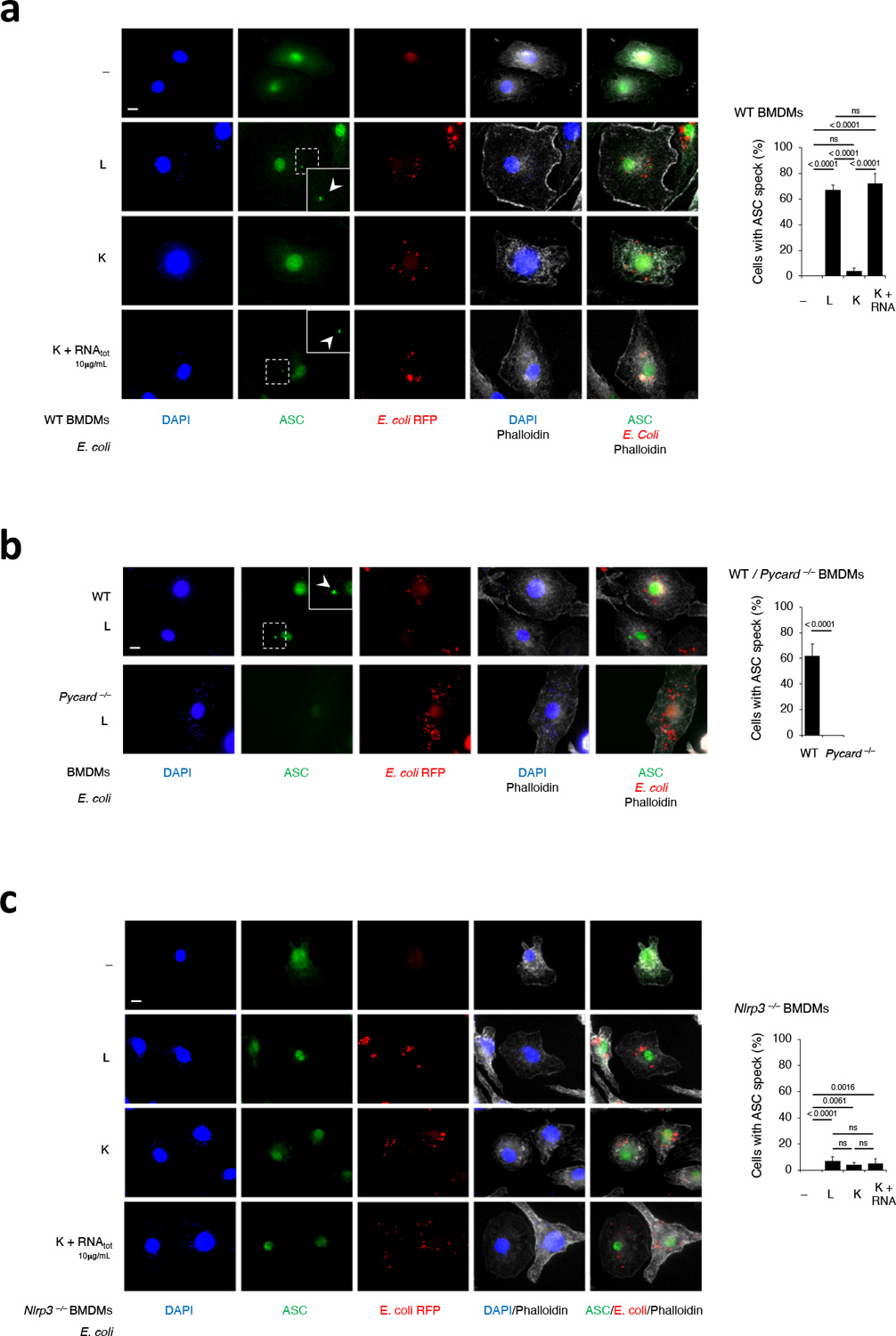
A prerequisite for NLRP3 activation is the oligomerization of the adaptor protein ASC (encoded by Pycard) into prion-like structures, which recruit and activate caspase-14,26,27. Killed Y. pestis, which expresses a stimulatory LPS when grown at 24°C1, and killed E. coli without mRNAbac or RNAbac did not induce detectable ASC oligomerization in macrophages (Fig. 3a–c), although killed and live bacteria similarly induced inflammasome priming (Fig. 3d,e). On the other hand, live bacteria or delivery of mRNAbac or RNAbac isolated from each strain, with killed bacteria, elicited ASC oligomerization (Fig. 3a–c), which was impaired in Nlrp3–/– macrophages (Fig. 3c)17,28. ASC oligomerization was triggered by various forms of RNAbac, mRNAbac from E. coli or L. innocua, or LPS-free IVT mRNAbac, but not eukaryotic mRNA, if delivered with killed E. coli (Fig. 3f,g) but not when added alone (Fig. 3f). Confocal microscopy showed that live, but not dead bacteria induced ASC specks29 (Fig. 3h,i and Extended Data Fig. 4a,b). RNAbac delivery with killed E. coli restored ASC speck formation (Fig. 3i and Extended Data Fig. 4a), and was dependent on NLRP3 (Extended Data Fig. 4c), consistent with previous observations17,28.
Figure 3. NLRP3 inflammasome assembly requires bacterial mRNA with LPS, irrespective of LPS stimulatory activity.
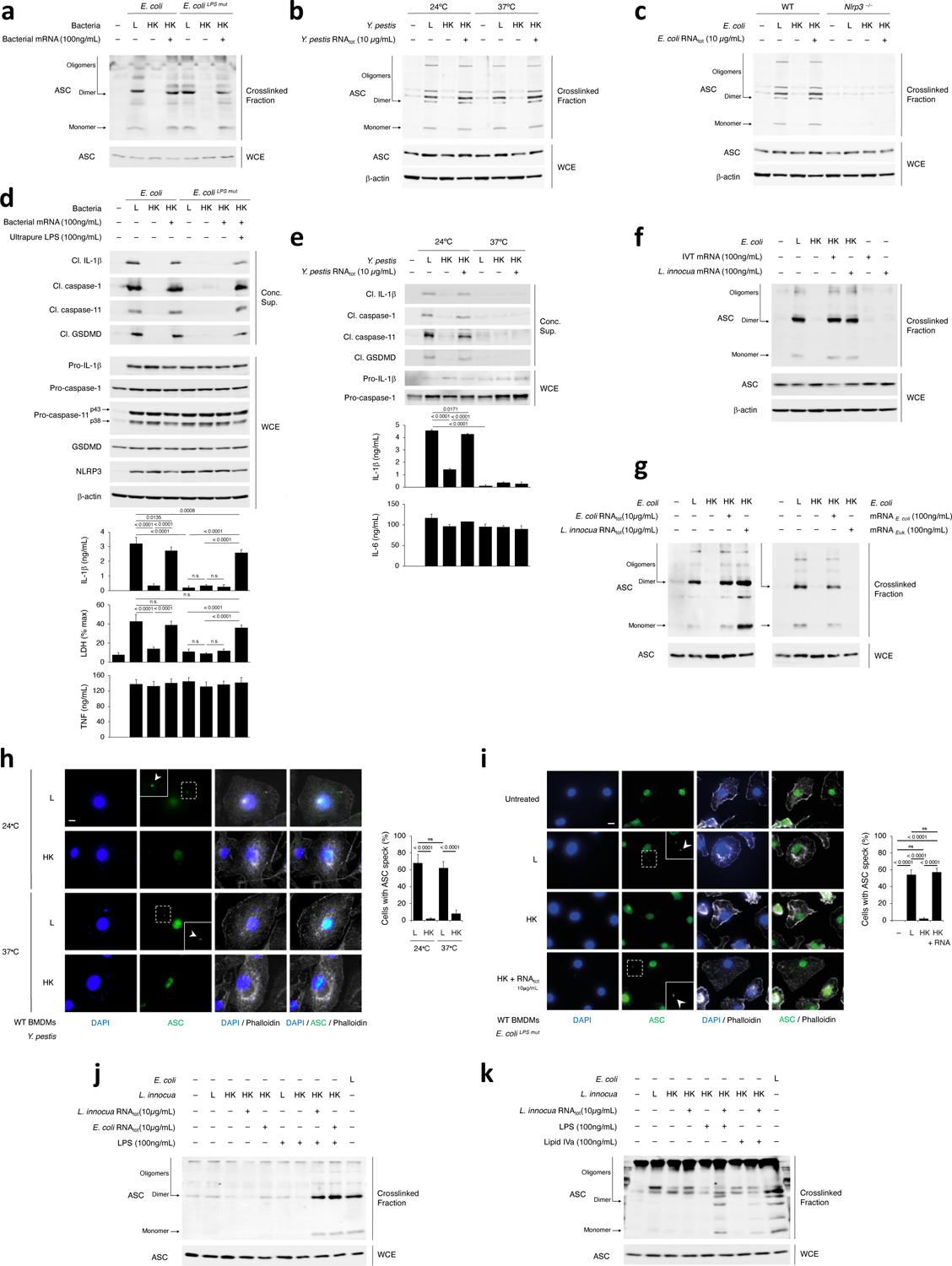
a-c, Immunoblots of macrophage cross-linked fractions and matching WCE (16hr), post-stimulation with L, HK or HK E. coli and E. coliLPSmut + mRNA from each bacterium (a), L, HK or HK Y. pestis grown at 24°C or 37°C + Y. pestis RNAtot (b), L, HK or HK E. coli + E. coli RNAtot (c). d,e, Immunoblots of macrophage concentrated supernatants (20hr) or WCE (6hr), and cytokine concentrations and LDH release in culture supernatants (20hr) post-stimulation with L, HK or HK E. coli or E. coliLPSmut +100ng/mL ultrapure LPS or 100ng/mL mRNA from each bacterium (d), and L, HK or HK Y. pestis grown at 24°C or 37°C +Y. pestis RNAtot (e). LDH measured by cytotoxicity assay; IL-1β, TNF and IL-6 by ELISA. f,g, Immunoblots of macrophage cross-linked fractions and matching WCE as in a, post-stimulation with L, HK or HK E. coli+IVT or L. innocua mRNA (f), and L, HK or HK E. coli+E. coli or L. innocua RNAtot, or E. coli mRNA or eukaryotic mRNA (g). h,i, Immunofluorescence confocal microscopy on macrophages stimulated for 16hr with L or HK Y. pestis grown at 24°C or 37°C (h), and L, HK or HK E. coliLPSmut + E. coliLPSmut RNAtot or untreated (i). Bar graphs in h,i, % cells exhibiting ASC speck. Inserts, magnification of indicated areas. White arrowheads, ASC specks. Phalloidin delineates the macrophage actin cytoskeleton. Scale bar=10μm. j,k, Immunoblots of macrophage cross-linked fractions and matching WCE as in a, post-stimulation with L, HK or HK E. coli or L. innocua + E. coli or L. innocua RNAtot with LPS or Lipid IVa as indicated. Error bars, mean ± s.e.m. One-way ANOVA followed by multiple comparisons Sidak tests and p values indicated in bar graphs in d (IL-1β: n=3, LDH: n=4), e (n=4), h (n=10) and i (Unst.: n=6, L: n=8, HK and HK+RNA: n=11). ns: non-significant. All macrophages are WT except as indicated in (c). Bacteria:macrophage=20:1. Results represent at least 3 independent experiments.
Live bacteria expressing a non-stimulatory tetra-acylated LPS (37°C Y. pestis and E. coliLPSmut)1,2,3 or delivery of RNAbac with killed forms of these bacteria elicited ASC oligomerization and ASC speck formation (Fig. 3a,b,h,i), indicating the stimulatory activity of LPS was dispensable for NLRP3 inflammasome ‘assembly’, although ASC oligomerization under these conditions was accompanied by inefficient activation of caspase-11 and inflammasome effector functions (Fig. 3d,e). L. innocua which lack LPS, had impaired ability to assemble ASC oligomers, regardless of bacterial viability or the presence of RNAbac from either Gram-positive or Gram-negative bacteria (Fig. 3j). Addition of LPS to killed L. innocua along with RNAbac from either Gram-positive or Gram-negative bacteria augmented ASC oligomerization (Fig. 3j). Supplementation of killed L. innocua with lipid IVa, which binds to but does not activate caspase-113, also augmented ASC oligomerization when RNAbac was co-supplemented (Fig. 3k). These data showed that viability of Gram-negative bacteria or co-detection of mRNAbac with LPS drove efficient ASC oligomerization irrespective of the ability of LPS to stimulate caspase-11, and indicated the uncoupling of inflammasome assembly from inflammasome activation.
NLRP3 and ASC are required for caspase-11 activation
We next tested whether the activation of NLRP3 and the activation of caspase-11 in macrophages were interdependent. Caspase-11 activation and NC-activation of the NLRP3 inflammasome in response to live E. coli were severely impaired in Nlrp3–/– and Pycard–/– macrophages, despite intact inflammasome priming (Fig. 4a,b), in contrast with the ability of virulent E. coli to elicit NLRP3- and ASC-independent caspase-11 cleavage in macrophages11. Although residual caspase-11 cleavage was detected in Nlrp3–/– and Pycard–/– macrophages at similar levels in response to either live or killed E. coli, it was not accompanied by the cleavage of GSDMD or release of LDH (as a readout of pyroptosis) observed in wild-type macrophages (Fig. 4a,b). A side-by-side comparison showed that, while both virulent and avirulent E. coli triggered inflammasome effector functions, such as caspase-1 cleavage and IL-1β cleavage and secretion, in an NLRP3- and ASC-dependent manner, only virulent E. coli elicited LDH release associated with caspase-11 and GSDMD cleavage in Nlrp3–/– and Pycard–/– macrophages (Fig. 4b).
Figure 4. Requirement of NLRP3 and ASC for caspase-11 activation in response to live avirulent Gram-negative bacteria.
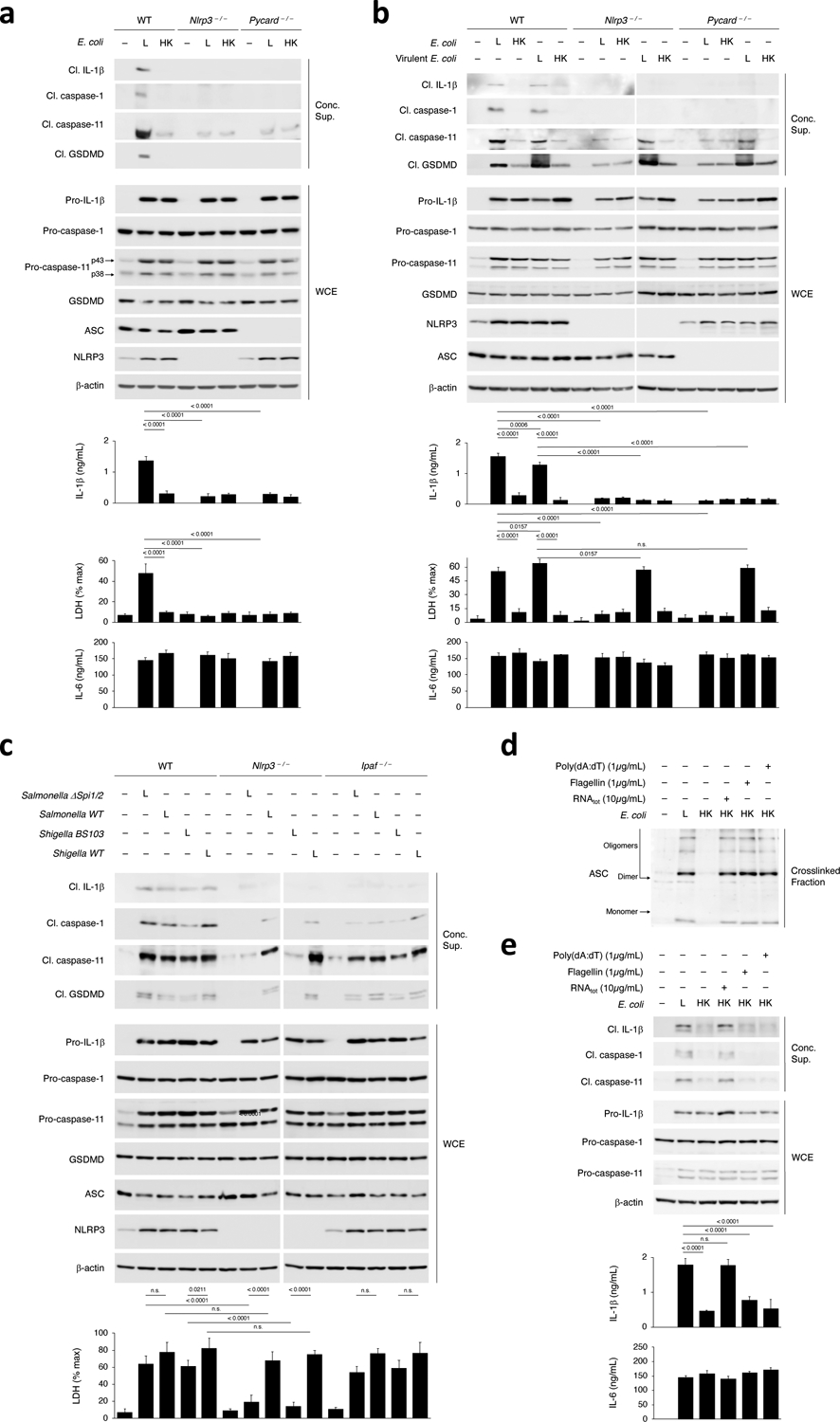
a-c, Immunoblots of macrophage concentrated supernatants (20hr) or WCE (6hr), and cytokine concentrations and LDH release in culture supernatants (20hr) post-stimulation of WT, Nlrp3–/– or Pycard–/– macrophages with L or HK E. coli (a), WT, Nlrp3–/– or Pycard–/– macrophages with L or HK avirulent or virulent E. coli (b), and WT, Nlrp3–/– or Ipaf–/– macrophages with Live Salmonella ΔSpi1/2 (avirulent, lacking the Salmonella pathogenicity islands 1 and 2 encoded type III secretion system) or WT (virulent) or Shigella BS103 (avirulent virulence plasmid cured) or WT (virulent) (note the doublet GSDMD bands here likely reflect the nature of the trigger) (c). d, Immunoblot of WT macrophage cross-linked fractions (16hr) post-stimulation with L, HK or HK E. coli supplemented with indicated doses of RNAtot, Flagellin or poly(dA:dT). e, Immunoblots of WT macrophage concentrated supernatants (20hr) or WCE (6hr), and cytokine concentrations in culture supernatants (20hr) post-stimulation as in d. LDH measured by cytotoxicity assay; IL-1β and IL-6 by ELISA. Error bars, mean ± s.e.m. One-way ANOVA followed by multiple comparisons Sidak tests and p values indicated in bar graphs in a, b, c and e (n=3). ns: non-significant. Bacteria:macrophage=20:1 for E. coli and virulent E. coli, 5:1 for all Salmonella and Shigella strains. Results represent at least 3 independent experiments.
To test whether the direct NLRP3- and ASC-independent pathway of caspase-11 activation depended on the bacterial expression of virulence factors, we used isogenic strains of clinically-relevant Salmonella typhimurium and Shigella flexneri Gram-negative bacteria that expressed (WT strains) or did not express (Salmonella typhimurium ΔSpi1/2 and Shigella flexneri BS103) virulence factors. Virulent and avirulent strains of Salmonella and Shigella elicited similar caspase-11 and GSDMD cleavage and pyroptosis in wild-type macrophages (Fig. 4c). However, only virulent Salmonella and Shigella elicited caspase-11 and GSDMD cleavage Nlrp3–/– macrophages (Fig. 4c). Avirulent Salmonella ΔSpi1/2 and Shigella BS103 induced considerably reduced amounts of cleaved caspase-11 in Nlrp3–/– macrophages, which was not associated with GSDMD cleavage or LDH release (Fig. 4c). This impairment was not detected in NLRC4-deficient (Ipaf–/–) macrophages (Fig. 4c). Virulent bacteria-triggered caspase-1 and pro-IL-β cleavage, which are canonical effector functions of the NLRP3 or NLRC4 inflammasomes, were strongly reduced in Nlrp3–/– and Ipaf–/– macrophages (Fig. 4c), in line with the ability of Salmonella and Shigella to activate the NLRP3 and NLRC4 inflammasomes30.
A larger percentage of macrophages showed rapid plasma membrane permeabilization (precedent to pyroptosis) and LDH release in response to virulent compared to avirulent bacteria, and beginning as early as 2 h post-stimulation (30% compared to 2.5%) (Extended Data Fig. 5a–d), a time point when the expression of inflammasome components, including NLRP3, had not yet peaked (Extended Data Fig. 5e), suggesting NLRP3 was not required for GSDMD cleavage and LDH release in response to virulent bacteria. Of note, cleaved caspase-1, caspase-11, GSDMD and IL-1β were detected at 6 h post-stimulation with virulent E. coli (Extended Data Fig. 5f). In contrast, a smaller percentage of macrophages (≤25%) were pyroptotic at any given time over the course of 72 hours post-stimulation with avirulent strains of E. coli, Salmonella and Shigella, and showed delayed kinetics of pyroptosis, which peaked 36 h post-stimulation (Extended Data Fig. 5b). Thus, caspase-11 activation, GSDMD cleavage, and pyroptosis elicited by avirulent bacteria were dependent on an assembled NLRP3 inflammasome, as they required both NLRP3 and ASC, while these events were independent of NLRP3 in response to virulent bacteria.
We next tested whether NLRP3 and ASC, and assembled inflammasomes, including inflammasomes other than NLRP3, provided a scaffold for caspase-11 activation. Stimulation of macrophages with a combination of killed E. coli and various canonical inflammasome inducers (flagellin, the NLRC4 inflammasome; poly(dA:dT), the AIM2 inflammasome) promoted ASC oligomerization similarly to killed E. coli plus RNAbac (Fig. 4d). However, only RNAbac plus killed E. coli induced caspase-11 cleavage and NLRP3 inflammasome effector functions (Fig. 4e), indicating a specific requirement of an assembled NLRP3 inflammasome for caspase-11 activation and that the NLRP3 inflammasome, but not the NLRC4 or AIM2 inflammasomes, underwent NC-activation by caspase-114. Macrophage stimulation with killed E. coli plus the bacterial toxin nigericin, a canonical NLRP3 inflammasome trigger, also coupled ASC oligomerization to caspase-11 cleavage and NLRP3 inflammasome effector functions (Extended Data Fig. 6a). Direct cytosolic delivery (in contrast to delivery with phagocytosed killed bacteria), of flagellin or poly(dA:dT) into LPS-primed macrophages, or their treatment with nigericin, elicited the cleavage of caspase-1, pro-IL-1β and GSDMD (Extended Data Fig. 6b). Altogether, these results indicated that a scaffold for procaspase-11 activation by LPS could only be provided by an assembled NLRP3 inflammasome and not by other assembled inflammasomes.
Caspase-11 is required for NLRP3 inflammasome assembly
We next investigated the requirement of caspase-11 for NC-activation of the NLRP3 inflammasome in response to Gram-negative bacteria. ASC oligomerization and all inflammasome effector functions were impaired in Casp11–/– macrophages following stimulation with live bacteria or killed bacteria plus RNA (compared to wild-type macrophages) (Fig. 5a–c), irrespective of whether ASC oligomerization was assessed early (8 h) or late (16 h) post-stimulation (Fig. 5b). Nascent ASC oligomers were detected in Casp1–/– macrophages 8 h post-stimulation with live E. coli, but not at 16 h post-stimulation with either live E. coli or killed E. coli plus RNAbac (Fig. 5a,b), indicating an inability to generate stable higher-order ASC clustering in the absence of caspase-1, consistent with prior observations27,31. ASC specks were also not detected in either Casp11–/– or Casp1–/– macrophages at 16 h post-stimulation with live bacteria (Fig. 5d). Pro-IL-1β expression was induced similarly by either live, killed or killed E. coli plus RNAbac in wild-type, Casp11–/– or Casp1–/– macrophages (Fig. 5a), although we noted a compensatory increase of procaspase-1 expression in Casp11–/– macrophages and procaspase-11 expression in Casp1–/– macrophages, while GSDMD and ASC expression were similar in all genotypes (Fig. 5a). Delivery of poly(dA:dT) with phagocytosed killed E. coli elicited ASC oligomerization in Casp11–/– macrophages (Fig. 5c), indicating Casp11–/– macrophages did not have a general impairment in ASC oligomerization. Yet, consistent with the specific requirement of an assembled NLRP3 inflammasome for caspase-11 activation, only ASC oligomerization triggered by killed E. coli with RNAbac, and not poly(dA:dT), was accompanied by cleavage of caspase-11, caspase-1, pro-IL-1β or GSDMD (Fig. 5c).
Figure 5. Procaspase-11 protein is required for assembly of the NLRP3 inflammasome in response to avirulent Gram-negative bacteria.
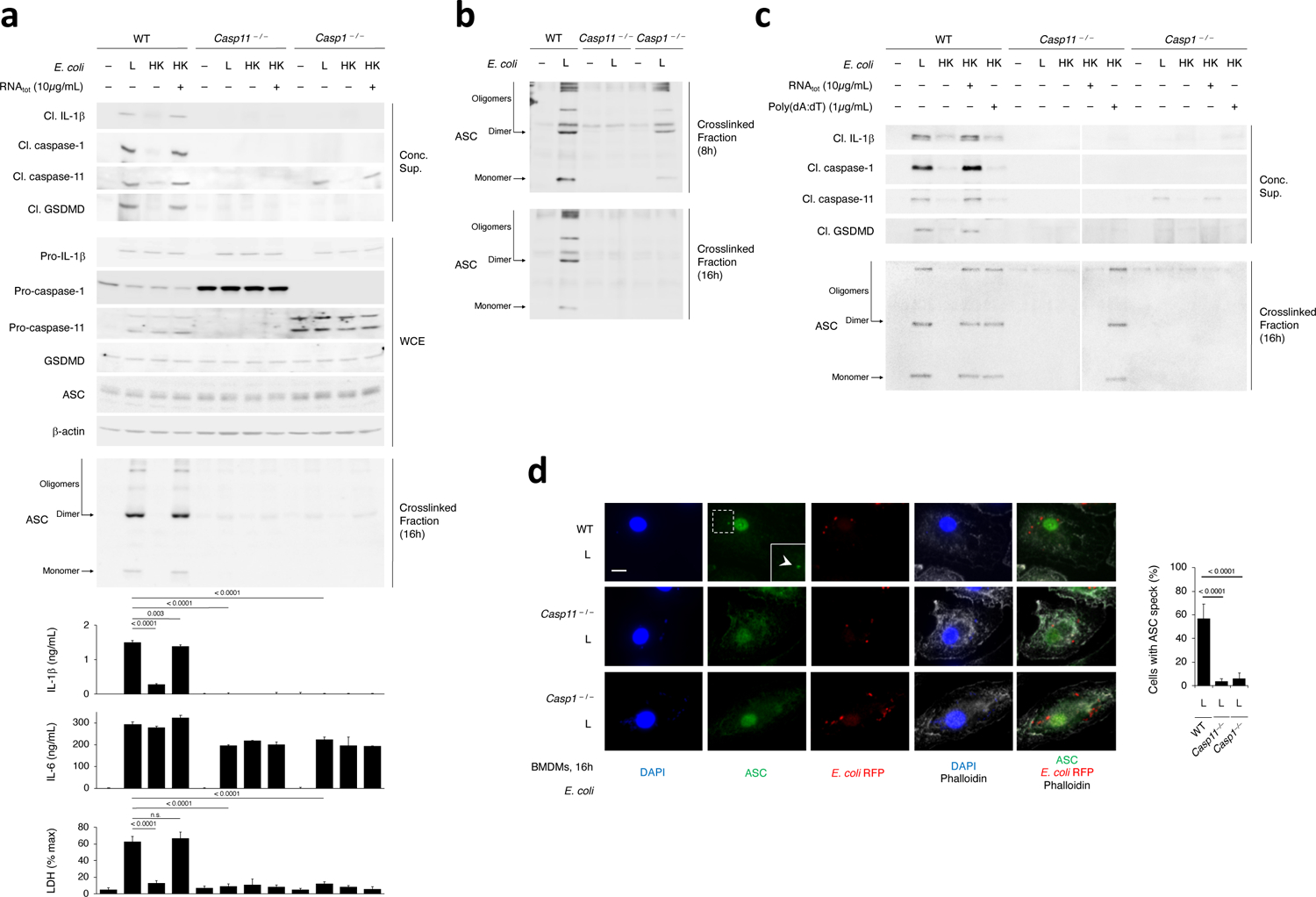
a, Immunoblots of WT, Casp11–/– or Casp1–/– macrophage concentrated supernatants (20hr) or WCE (6hr), and cytokine concentrations and LDH release in culture supernatants (20hr) post-stimulation with L, HK or HK E. coli supplemented with E. coli RNAtot. LDH release measured by cytotoxicity assay; IL-1β and IL-6 by ELISA. b, Immunoblot of WT, Casp11–/– or Casp1–/– macrophage cross-linked fractions (16hr) post-stimulation with L E. coli. c, Immunoblots of WT, Casp11–/– or Casp1–/– macrophage concentrated supernatants (20hr) (top panels) or cross-linked fractions (16hr) (bottom panels) post-stimulation with L, HK or HK E. coli supplemented with E. coli RNAtot or poly(dA:dT). d, Immunofluorescence confocal microscopy on WT, Casp11–/– or Casp1–/– macrophages stimulated for 16hr with L E. coli RFP. Bar graph, % cells exhibiting ASC speck. Inset shows magnification of indicated area. White arrowhead points to ASC speck. Phalloidin delineates the macrophage actin cytoskeleton. Scale bar=10μm. Error bars, mean ± s.e.m. One-way ANOVA followed by multiple comparisons Sidak tests and p values indicated in bar graphs in a (IL-1β, WT and Casp1–/– : n=3, Casp11–/– : n=4; LDH, n=3) and d (WT: n=6, Casp11–/– : n=7, Casp1–/– : n=8). ns: non-significant. Bacteria:macrophage=20:1. Results represent at least 3 independent experiments.
Cleaved caspase-11 was detected in concentrated supernatants from Casp1–/– macrophages in response to RNAbac and LPS (Fig. 5a,c), indicating that unlike the requirement for NLRP3 and ASC, caspase-1 was not required for caspase-11 activation. However, all inflammasome effector functions, including notably GSDMD cleavage and pyroptosis, were impaired in Casp1–/– macrophages compared to wild-type macrophages, despite intact caspase-11 activation (Fig. 5a,c), indicating that like IL-1β, GSDMD was cleaved by caspase-1 in response to RNAbac and LPS. Caspase-1, pro-IL-1β and GSDMD cleavage were not detected in Casp11–/– macrophages (Fig. 5a,c). Thus, IL-1β secretion and pyroptosis triggered by RNAbac and LPS were direct effector functions of caspase-11-mediated NC-activation of the NLRP3-ASC-procaspase-1 inflammasome complex.
RNAbac and LPS trigger a procaspase-11-NLRP3 interaction
We tested whether the functional reciprocal exclusivity of caspase-11 and NLRP3 was due to a physical interaction between the two. NLRP3 stimuli trigger disassembly of the trans-Golgi network (TGN) and recruitment of NLRP3 to the dispersed TGN (dTGN)32. Confocal microscopy showed vesicular subcellular colocalization of caspase-11 and NLRP3 in macrophages stimulated with bacteria, irrespective of bacterial viability or virulence, but not in resting bone marrow-derived macrophages (Fig. 6a and Extended Data Fig. 7a,b). We noted TGN dispersion and radiation away from the nucleus after stimulation with either live or dead, virulent or avirulent E. coli (Fig. 6a and Extended Data Fig. 7a), in wild-type, Nlrp3–/– or Casp11–/– macrophages (Extended Data Fig. 7b). NLRP3 was detected on dTGN in wild-type macrophages (Fig. 6a and Extended Data Fig. 7a), as reported in Pycard–/– macrophages in response to strong triggers such as nigericin, ATP or monosodium urate32. Caspase-11 colocalized with dTGN, and specifically to dTGN areas where NLRP3 was also present (Fig. 6a and Extended Data Fig. 7a). Caspase-11 did not localize to dTGN in Nlrp3–/– macrophages, while NLRP3 localized to dTGN in Casp11–/– macrophages (Extended Data Fig. 7b).
Figure 6. Coincident detection of bacterial RNA and LPS triggers a procaspase-11-NLRP3 interaction irrespective of LPS stimulatory activity.
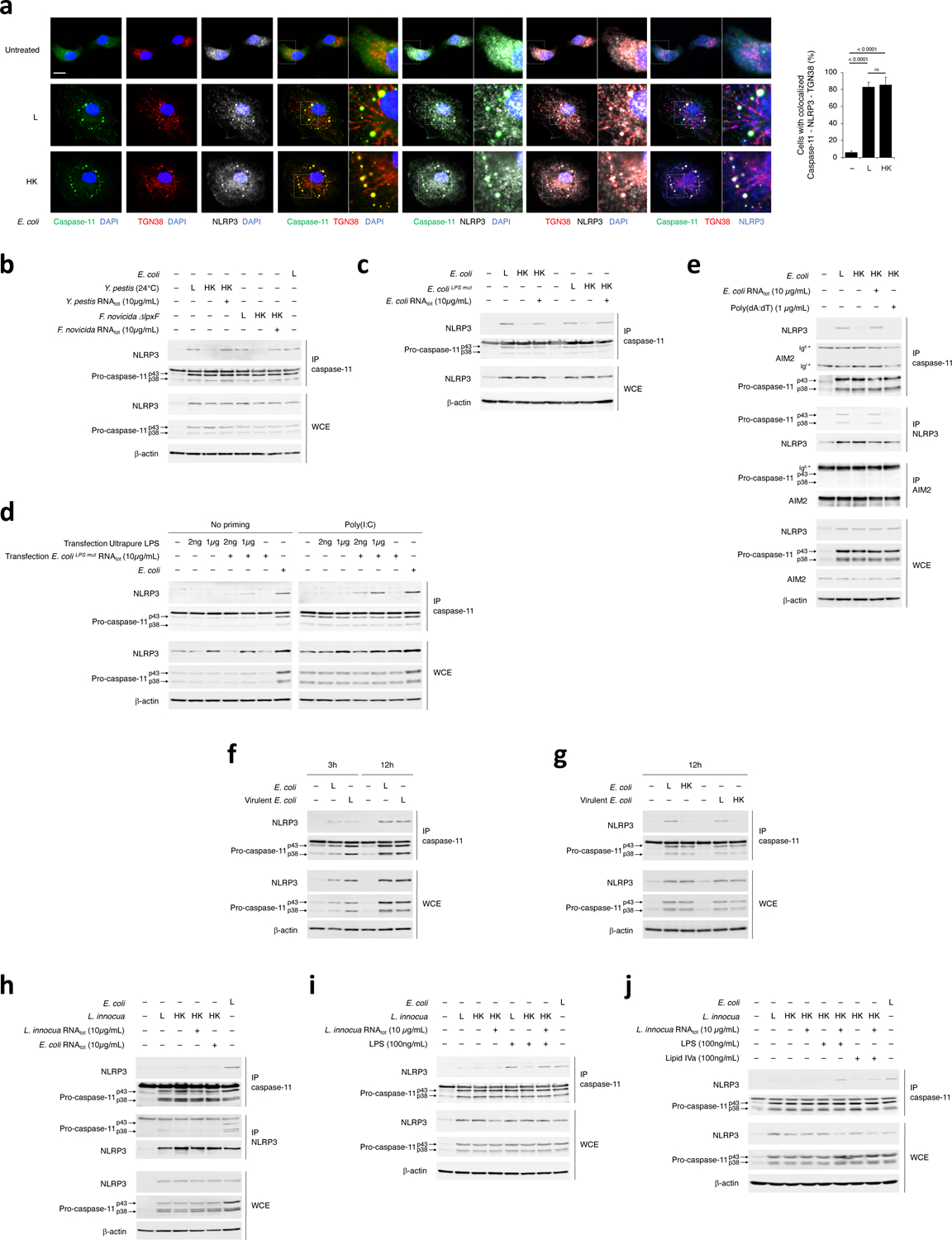
a, Immunofluorescence confocal microscopy on macrophages stimulated for 16hr with L or HK E. coli. Scale bar=10μm. For 3-color merges, magnification of indicated areas shown as insets to the right. Bar graphs, % cells with caspase-11-NLRP3-TGN38 colocalization. Error bars, mean ± s.e.m. One-way ANOVA followed by multiple comparisons Sidak tests were performed and P values are indicated in bar graphs (Unst.: n=10, L: n=9, HK: n=7). ns: non-significant. b-j, Immunoprecipitation (IP) of endogenous caspase-11, NLRP3 or AIM2 as indicated, and immunoblotting for coimmunoprecipitating proteins or WCE proteins from WT macrophages stimulated for 12hr with L, HK or HK Y. pestis (grown at 24°C) or F. novicida ΔlpxF supplemented with RNAtot from each bacterium (b), 12hr with L, HK or HK E. coli or E. coliLPSmut supplemented with E. coli RNAtot or E. coliLPSmut RNAtot, respectively (c), 4hr with L E. coli or after transfection with indicated doses of ultrapure LPS and E. coliLPSmut RNAtot +/− prior 12–16hr priming with 1μg/mL poly(I:C) (d), 12hr with L, HK or HK E. coli in combination with E. coli RNAtot or poly(dA:dT) (e), 3hr or 12hr with L or HK E. coli or virulent E. coli, (f,g), 12hr with L E. coli or L, HK or HK L. innocua supplemented with E. coli or L. innocua RNAtot (h-j), concomitant with LPS (h,i) or Lipid IVa (j) stimulation as indicated. All immunoblotted proteins are according to the labels to left of immunoblot panels. Bacteria:macrophage=20:1. Results represent at least 3 independent experiments.
We next tested whether caspase-11 interacted with NLRP3 in macrophages. NLRP3 coimmunoprecipitated with endogenous procaspase-11 efficiently following stimulation with live 24°C-grown Y. pestis, F. novicidaΔlpxF, and E. coli or killed forms of these bacteria supplemented with RNAbac, and not following stimulation with killed bacteria alone (Fig. 6b,c and Extended Data Fig. 7c). Conversely, the pro-forms of caspase-11 coimmunoprecipitated with endogenous NLRP3 in response to live E. coli or killed E. coli plus RNAbac, but not killed E. coli alone (Extended Data Fig. 7c). Transfection of either unprimed or poly(I:C)-primed macrophages with 2ng/mL ultrapure LPS and/or stimulatory-LPS-free RNAbac showed caspase-11 interacted with NLRP3 only when RNAbac and LPS were co-transfected (Fig. 6d). Killed E. coli with either RNAbac or nigericin, but not poly(dA:dT), promoted the procaspase-11-NLRP3 interaction (Fig. 6e and Extended Data Fig. 7d), indicating the interaction was specifically elicited by the combination of LPS and NLRP3 agonists. Procaspase-11 did not coimmunoprecipitate with AIM2 in response to either live or killed bacteria, or delivery of RNAbac or poly(dA:dT) with killed bacteria (Fig. 6e). Procaspase-11 and NLRP3 did not interact post-transfection of high dose (1μg/mL) of LPS (Fig. 6d), but did so post-co-transfection of RNAbac with 1μg/mL LPS (Fig. 6d). NLRP3 coimmunoprecipitated with procaspase-11 as early as 3 h post-stimulation with bacteria, and irrespective of bacterial virulence (Fig. 6f,g).
NLRP3 did not coimmunoprecipitate with procaspase-11, or the opposite, after macrophage stimulation with live Gram-positive L. innocua or killed L. innocua plus either Gram-negative or Gram-positive mRNAbac (Fig. 6h), suggesting LPS detection was required for procaspase-11-NLRP3 interaction. Addition of LPS, in conditions in which RNAbac was also present, to live L. innocua or killed L. innocua restored procaspase-11-NLRP3 interaction (Fig. 6i,j). Stimulation with either E. coliLPSmut (Fig. 6c) or the combination of Lipid IVa with killed L. innocua (Fig. 6j) induced procaspase-11-NLRP3 interaction as long as RNAbac was also present, suggesting that LPS binding to caspase-11, but not the stimulatory activity of LPS, was sufficient for mediating the procaspase-11-NLRP3 interaction. Thus, LPS binding to caspase-11 concurrent with RNAbac detection by NLRP3 triggers interaction of procaspase-11 with NLRP3.
Caspase-11 and NLRP3 interact prior to their activation
To investigate whether procaspase-11 and NLRP3 interacted prior to caspase-11 activation, we probed the procaspase-11-NLRP3 interaction in conditions where the activity of caspase-11 or its products were blocked. GSDMD-dependent cellular perturbations post-caspase-11 activation are a prerequisite for NLRP3 inflammasome activation4,5,14. In Gsdmd–/– macrophages, cleaved caspase-11 was detected in the whole cell extracts, but not in the concentrated supernatants, in response to both virulent and avirulent live E. coli or the combination of killed E. coli plus RNAbac (Fig. 7a and Extended Data Fig. 8a,b), while cleaved caspase-1 and IL-1β were not detected in the concentrated supernatants or the whole cell extracts (Fig. 7a and Extended Data Fig. 8c). Of note, procaspase-11 interacted with NLRP3 in Gsdmd–/– macrophages (Fig. 7b). The broad caspase inhibitor zVAD-FMK did not block the procaspase-11-NLRP3 interaction (Fig. 7c), although it abrogated inflammasome effector functions (Extended Data Fig. 8d). These observations indicated that the interaction of procaspase-11 with NLRP3 was upstream of caspase-11 activation.
Figure 7. Procaspase-11-NLRP3 interaction is upstream of NLRP3 inflammasome assembly and activation.
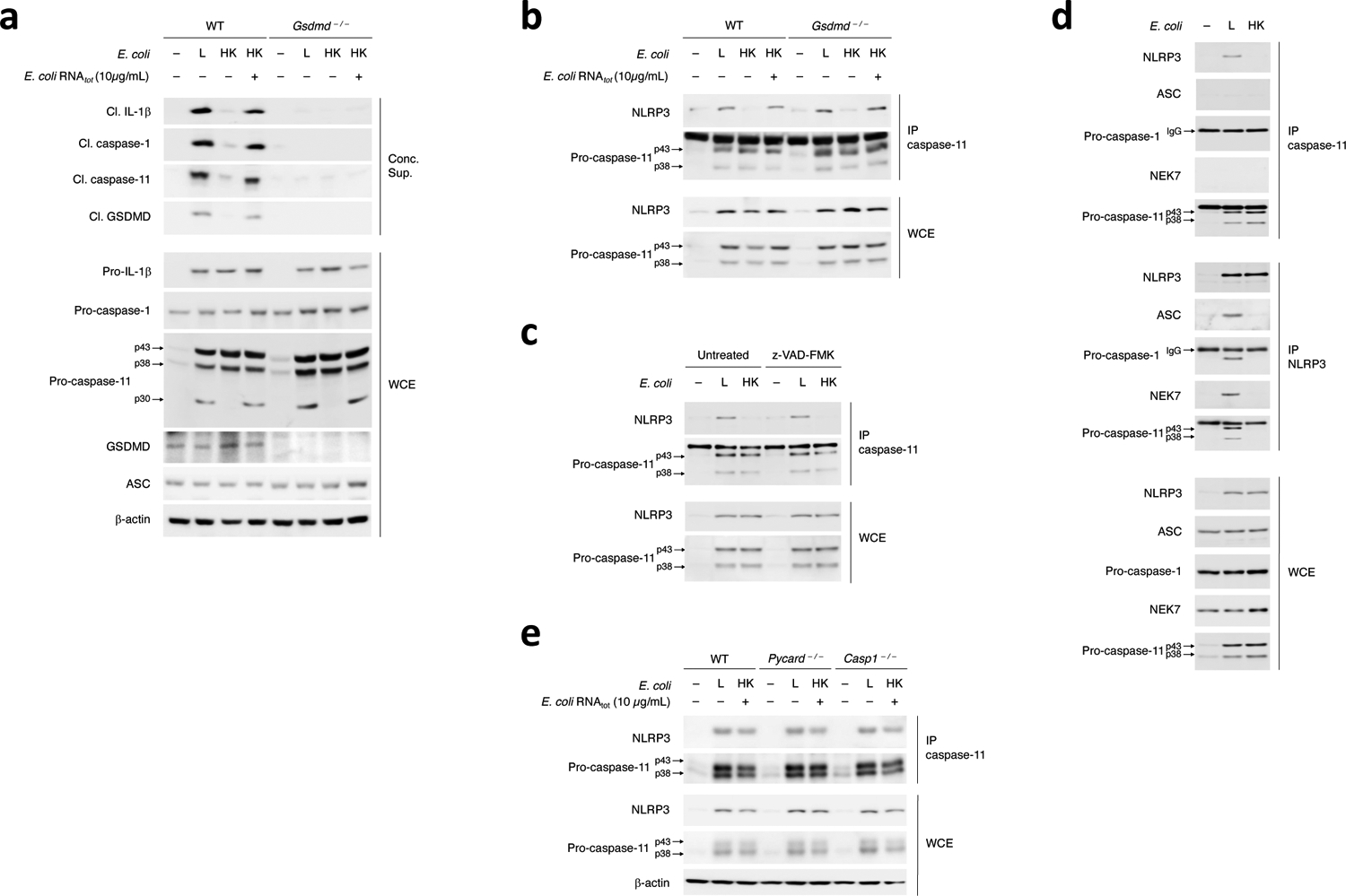
a, Immunoblots of macrophage concentrated supernatants (20hr) or WCE (6hr) as indicated post-stimulation of WT and Gsdmd–/– macrophages with L, HK or HK E. coli supplemented with E. coli RNAtot. b-e, Immunoprecipitation (IP) of endogenous caspase-11 or NLRP3 as indicated, and immunoblotting for coimmunoprecipitating and WCE proteins at 12hr post-stimulation of WT and Gsdmd–/– macrophages with L, HK or HK E. coli + E. coli RNAtot (b), WT macrophages with L or HK E. coli with or without caspases inhibition with 10μg/mL zVAD-FMK before stimulation with bacteria (c), WT macrophages with L or HK E. coli (d), and WT, Pycard–/– and Casp1–/– macrophages with L or HK E. coli supplemented with E. coli RNAtot (e). All immunoblotted proteins are according to the labels to left of immunoblot panels. Bacteria:macrophage=20:1. Results represent at least 3 independent experiments.
We tested whether an assembled NLRP3 inflammasome was required for the procaspase-11-NLRP3 interaction. Alongside procaspase-11, we detected ASC, procaspase-1, and NEK7, which binds NLRP3 and is required for its assembly with ASC and caspase-133,34, in the NLRP3 immunoprecipitates after macrophage stimulation with live, but not killed E. coli (Fig. 7d), suggesting that NLRP3 was ‘licensed’ for assembly41. On the other hand, NLRP3, but not ASC or NEK7, were detected in the procaspase-11 immunoprecipitates post-stimulation with live E. coli (Fig. 7d), indicating NLRP3, but not NEK7 or ASC, interacted with procaspase-11. Procaspase-11 and NLRP3 interacted in Pycard–/– or Casp1–/– macrophages following stimulation with live E. coli (Fig. 7e), indicating ASC and caspase-1 were not required and suggesting that the procaspase-11-NLRP3 interaction did not reflect caspase-11 recruitment to stable, caspase-1-containing NLRP3-ASC oligomers, following an event such as heterodimerization with caspase-112,13,35. These observations suggested that cytosolic co-detection of RNAbac and LPS from Gram-negative bacteria was necessary and sufficient to mediate the procaspase-11-NLRP3 interaction upstream of inflammasome assembly and caspase-11 activation (Extended Data Fig. 9).
Procaspase-11 SCAF interacts with NLRP3 LRR and PYD
To identify the domains of NLRP3 and procaspase-11 that are important for their interaction, we expressed in 293T cells full-length (NLRP3FL) and truncation mutant forms (NLRP3ΔPYD, NLRP3ΔLRR, NLRP3ΔNACHT ΔPYD, NLRP3ΔNACHT ΔLRR) of HA-tagged NLRP3, and full length (casp-11C254A FL) and truncation mutant forms (casp-11C254A ΔSCAF, casp-11C254A ΔCARD) of FLAG-tagged catalytically inactive caspase-11(C254A)3 (Extended Data Fig. 10a,b). HA-NLRP3FL did not efficiently coimmunoprecipitate with casp-11C254A ΔSCAF, even when this was loaded at a 4-fold higher concentration, compared to casp-11C254A FL and casp-11C254A ΔCARD (Fig. 8a and Extended Data Fig. 10c), indicating the CARD domain of caspase-11, which binds to LPS3, was not required for caspase-11 coimmunoprecipitation with NLRP3FL, while the other caspase domain, herein referred to as scaffold (SCAF) domain was required. Reverse immunoprecipitation of FLAG-casp-11C254A FL confirmed these results where NLRP3 FL could not be coimmunoprecipitated with casp-11C254A ΔSCAF in contrast to its coimmunoprecipitation with casp-11C254A FL (Fig. 8a).
Figure 8. Procaspase-11-NLRP3 interaction is mediated by the caspase-11 SCAF domain and NLRP3 LRR and PYD domains.
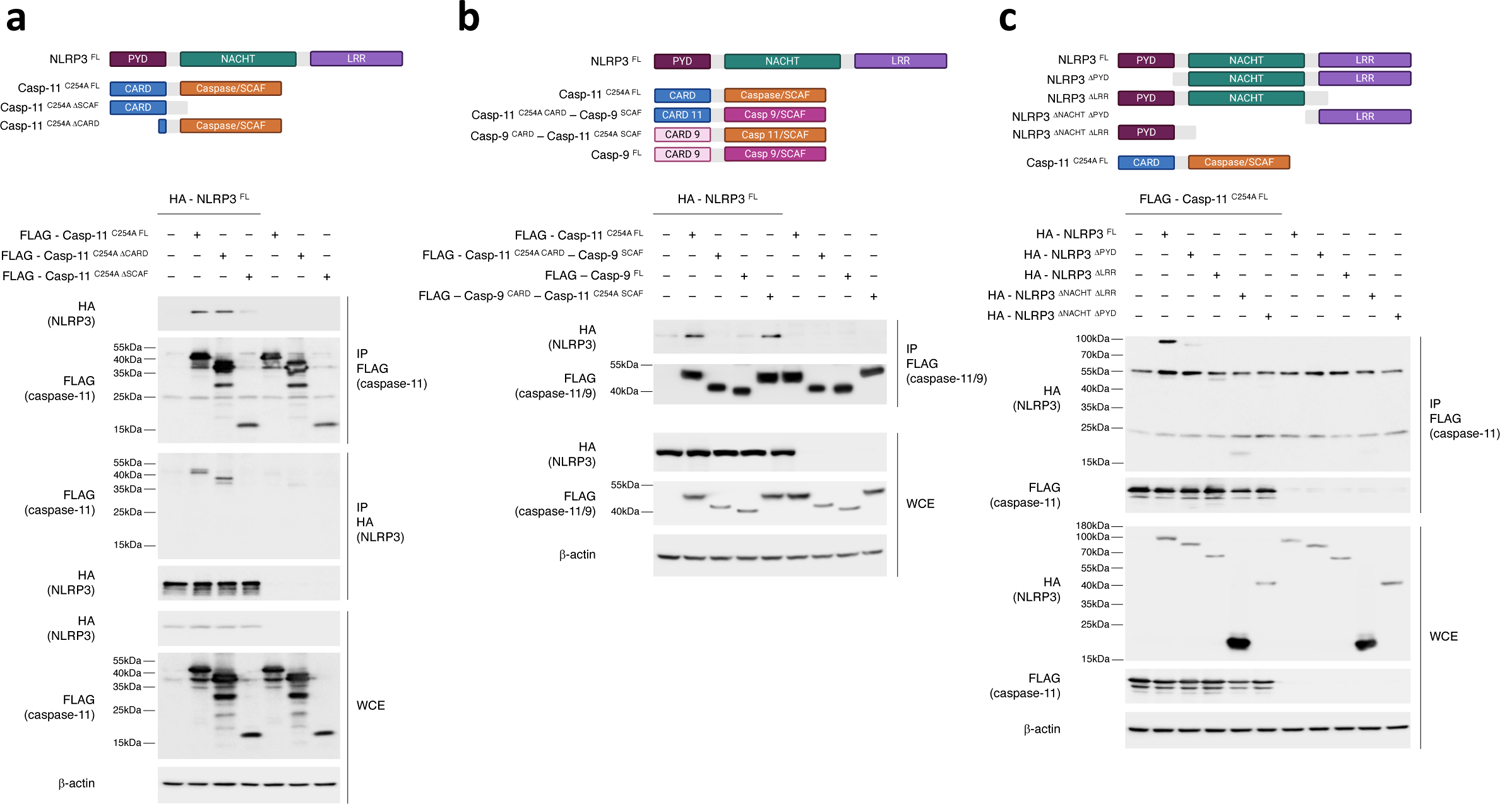
a, Immunoprecipitation and immunoblotting for coimmunoprecipitating and WCE proteins of overexpressed FLAG-casp-11C254A FL, FLAG-casp-11C254A ΔCARD or FLAG-casp-11C254A ΔSCAF with or without HA-NLRP3FL in 293T cells at 24hr post transfection. b, Immunoprecipitation and immunoblotting for coimmunoprecipitating and WCE proteins of overexpressed FLAG-caspase11–9 CARD-SCAF chimeras with or without HA-NLRP3FL in 293T cells as in a. c, Immunoprecipitation and immunoblotting for coimmunoprecipitating and WCE proteins of overexpressed HA-NLRP3 either as NLRP3FL, NLRP3ΔPYD, NLRP3ΔLRR, NLRP3ΔNACHT ΔLRR or NLRP3ΔNACHT ΔPYD with or without FLAG-casp-11C254A FL, in 293T cells as in a. All immunoblotted proteins and MW according to a protein standard are indicated to left of each immunoblot panel. Bacteria:macrophage=20:1. Results represent at least 3 independent experiments.
The SCAF domain is common to many caspases, including the inflammatory caspase-1 and caspase-11 and the apoptotic caspases, such as caspase-3, 7 and 9 (Extended Data Fig. 10d), and the SCAF domain of caspase-11 is highly homologous to the SCAF domains of the other caspases (Extended Data Fig. 10e,f). To determine if caspase-11-SCAF was unique in its interaction with NLRP3, we swapped the caspase-11-SCAF with the caspase-9-SCAF (Extended Data Fig. 10g). The FLAG-casp-11C254A CARD-casp-9SCAF chimeric molecule showed impaired coimmunoprecipitation of NLRP3FL compared to FLAG-casp-11C254A FL (Fig. 8b). Reversely, replacing caspase-9-SCAF domain with the caspase-11-SCAF (FLAG-casp-9CARD-casp-11C254A SCAF) allowed coimmunoprecipitation of NLRP3FL (Fig. 8b), indicating the SCAF domain of caspase-11 was required and sufficient for the interaction with NLRP3 in this ligand-independent expression system in 293T cells. On the other hand, NLRP3ΔPYD or NLRP3ΔLRR showed impaired coimmunoprecipitation with casp-11C254A FL compared to HA-NLRP3FL (Fig. 8c). These results indicated that the interaction between NLRP3 and procaspase-11 required the PYD and LRR domains of NLRP3 and the SCAF domain of procaspase-11.
Discussion
Here we show that coincident cytosolic detection of LPS with mRNAbac by macrophages simultaneously engaged NLRP3 and caspase-11, and enabled their interaction and interdependent reciprocal activation. Macrophage phagocytosis of live Gram-negative bacteria induced caspase-11 colocalization with NLRP3 at the dTGN, and a procaspase-11-NLRP3 interaction as early as 3 h. Procaspase-11 and NLRP3 may interact directly or indirectly through intermediary protein(s). mRNAbac and LPS together, and not each alone, at concentrations equivalent to those detected in the cytosol of macrophages after phagocytosis of bacteria, triggered procaspase-11-NLRP3 interaction upstream of the activation of either receptor and prior to NLRP3-ASC oligomerization. LPS binding to, but not stimulation of caspase-11, was necessary and sufficient as long as RNAbac was also present to engage NLRP3. Although ASC was not required for the procaspase-11-NLRP3 interaction, both NLRP3 and ASC (an assembled NLRP3 inflammasome) were required for procaspase-11 activation by LPS, indicating the upstream interaction with NLRP3 licensed LPS-bound procaspase-11 for activation. Reciprocally, caspase-11 protein and not its activity was required for mRNAbac-driven NLRP3 inflammasome assembly, which also required LPS, a requirement that may have been overlooked due to potential LPS contamination in RNAbac preparations17,28. Once assembled, the NLPR3 inflammasome, but not the AIM2 or NLRC4 inflammasomes, was associated with the activation of LPS-bound caspase-11. Procaspase-11 did not interact with AIM2 upon delivery of poly(dA:dT) with phagocytosed killed E. coli. It is possible that NLRP3 and procaspase-11 remain in a single complex upon NEK7 and ASC recruitment to NLRP3, which might explain the noted activation of caspase-11 upon assembly of the NLRP3, but not AIM2 inflammasome. Precisely how cytosolic co-detection of mRNAbac and LPS facilitates caspase-11 activation requires future investigation. Our findings suggest non-mutually exclusive modes of caspase-11 activation in macrophages dependent on the context of cytosolic LPS detection. A fast, NLRP3-independent mode was triggered by the concurrent expression of bacterial virulence factors during cell invasion by pathogenic Gram-negative bacteria2,11. A slower, NLRP3-dependent mode, was triggered by coincident detection of mRNAbac from phagocytosed live Gram-negative bacteria and independently of virulence factors.
We propose that the procaspase-11 interaction with NLRP3 nucleates a scaffold, the assembled NLRP3 inflammasome, in which NLRP3 and procaspase-11 reciprocally and exclusively activate one another. The importance of a platform for caspase signaling emerged from studies showing that caspase-4, 5 or 11 prefer large LPS micelles or LPS aggregates formed upon GBP1 polymerization36,37,38,39. LPS micelles occur at higher μM range concentrations compared to the LPS monomers at low concentrations40,41,42,43, which could explain how microgram levels of cytosolic LPS provide an activation surface for caspase-4, 5 and 11 without the requirement of mRNAbac. Under conditions when LPS cytosolic concentrations are lower than the μM range, such as following phagocytosis of Gram-negative bacteria, the procaspase-11-NLPR3 interaction upon co-detection of LPS and RNAbac from live bacteria, would provide a platform to mediate the activation of procaspase-11 and reciprocally, NLRP3.
The observation that NLRP3-ASC oligomerization was not sufficient for inflammasome activation is in contrast to the existing view that NLRP3-ASC oligomers are automatically activated, leading to the cleavage of caspase-1 and IL-1β. Our findings do not necessarily imply that the NLRP3-ASC oligomers elicited by LPS plus mRNAbac are different from the oligomers formed through the canonical pathway of inflammasome activation, either qualitatively or quantitatively. Future studies are necessary to decipher whether this is the case. Rather, the use of non-stimulatory LPS with mRNAbac ‘freeze-framed’ each step of the process, and revealed an additional regulatory layer to allow activation after NLRP3-ASC oligomerization. Our data indicated that the NLRP3-ASC oligomers did not activate caspase-1 for the subsequent cleavage of GSDMD unless caspase-11 was activated beforehand. Therefore, while the LPS-bound pro-form of caspase-11 was important for mRNAbac-driven NLRP3 inflammasome assembly, the active form of caspase-11 allowed assembled NLRP3-ASC oligomers to proceed to full activation manifesting in cleavage of caspase-1 and leading to IL-1β secretion and pyroptosis. The requirement for GSDMD in the process may reflect a positive feedback role to maintain NLRP3 inflammasome assembly and activity.
mRNAbac and LPS synergistically coordinated NC-activation of the NLRP3 inflammasome. mRNAbac was the NLRP3 trigger, similar to nigericin, and its requirement alongside LPS was not a function of inflammasome ‘priming’, which can be mediated by various PAMPs during macrophage phagocytosis of bacteria. However, LPS binding to procaspase-11 and the interaction of LPS-bound procaspase-11 with NLRP3 in the presence of RNAbac, ultimately set LPS apart from other PAMPs. Co-detection of LPS with eukaryotic mRNA did not elicit NC-activation of the NLRP3 inflammasome, likely due to differences between eukaryotic versus bacterial mRNAs at the 3’ ends44, which may be important for NLRP3 inflammasome activation17,45,46. Future investigations are necessary to elucidate how mRNAbac stimulate NLRP3.
Detection of mRNAbac in the context of a prototype vaccine comprised of live or dead Gram-negative bacteria elicited follicular CD4+ T helper (TFH) cell and amplified antibody responses18,20,47. Although Casp11–/– Casp1–/– mice were used in those studies18, the present work indicated that these triggers did not activate caspase-1 in the absence of caspase-11, pointing to a role for NC-activation of the NLRP3 inflammasome in mediating the TFH cell and antibody responses in vivo. Our findings provide a rationale for creating combination adjuvants based on LPS and mRNAbac to synergistically enhance immune responses that benefit from inflammasome activation18,20,47,48,49. While more work is necessary to extend our findings to human caspases-4 and 5, the establishment of mRNAbac and LPS as a combinatorial trigger for caspase-11 activation should inform much needed therapies for Gram-negative sepsis50.
Online Methods
Mice
C57BL/6J mice were from The Jackson Laboratory and bred in-house. Nlrp3–/– and Pycard–/– mice were from R. Flavell, Casp1–/– and Casp11–/– mice from T.D. Kanneganti (MTA with Genentech), and femurs from Gsdmd–/– mice from K. Fitzgerald. 6–12 week-old male and female mice were randomly used. Animal procedures were approved by the Icahn School of Medicine at Mount Sinai (ISMMS) and Weill Cornell Medicine (WCM) IACUC in accordance with the ‘Guide for the Care and Use of Laboratory Animals’ (NIH publication 86–23, revised 1985).
Bone marrow-derived macrophages
Bone marrow progenitors harvested from femurs of mice were differentiated into macrophages over 7–10 days in RPMI 1640 with M-CSF (conditioned medium from L929 cell line) and 10% fetal bovine serum, plus 100U/mL penicillin, 100μg/mL streptomycin, 2mM L-glutamine, 10mM HEPES, 1mM sodium pyruvate, 1% MEM non-essential amino acids, and 55μM β-Mercaptoethanol (Sigma).
Bacterial strains
Escherichia coli K12, DH5α (Invitrogen). Naturally occurring thymidine auxotrophs (thyA–) were selected and E. coli RFP thyA– were generated as described17. E. coli (Migula) Castellani and Chalmers strain (pathogenic E. coli serovar O1:K1:H7), ATCC® 11775. E. coli CMR30051 (E. coliLPSmut) provided by V. Dixit. All E. coli strains grown in Luria-Bertani (LB), supplemented with 500μg/mL Thymidine and 50μg/mL Trimethoprim (Sigma) when thyA–. Yersinia pestis KIM derivative 19 (D19), BEI Resources (NR-4681) and grown in Tryptic Soy Broth at 24 or 37°C. Francisella tularensis subsp. novicida (F. novicida WT) was from R. Ernst, and Francisella tularensis subsp. novicida strain Utah 112 (F. novicidaΔlpxF), BEI resources (NR-13); both grown in Brain Heart Infusion (BHI) Broth (Gibco). Yersinia pestis KIM D19 and F. novicida WT and F. novicidaΔlpxF were imported to the Blander laboratory with permits from the U.S. Department of Agriculture. J.M. was tested for circulating iron levels prior to work with Yersinia pestis KIM D19. Listeria innocua Seeliger (ATCC® 33091™) (Ref.52), and grown in BHI Broth (Gibco). Staphylococcus aureusΔAgrΔSar, lacking global regulators Sar (Staphylococcal accessory regulator) and Agr (Accessory gene regulator)53, were from L. Stuart and grown in BHI Broth (Gibco). Salmonella enterica serovar typhimurium SL1344 WT and Salmonella enterica serovar typhimurium ΔSpi1/2, lacking T3SS, ATCC, and grown in LB broth. Shigella flexneri WT and BS103 (virulence plasmid-cured strain) were from M. Goldberg and grown in Terrific Broth (TB). Work with all strains of bacteria was approved by the institutional Biosafety and EHS of ISMMS and WCM.
E. coli cultured in 2μg/mL Rifampicin (Sigma) were washed 3X in PBS, serial dilutions plated on LB agar containing 500μg/mL Thymidine and 50μg/mL Trimethoprim (Sigma), and colony counts determined. For heat-killing, bacteria were grown to mid-log phase, washed, resuspended in PBS and incubated at 60°C for 2 hr. E. coli RFP (E. coli mCherry) were killed by treatment with 50μg/mL Gentamicin sulfate at 37°C shaking for 12–16 hr. Killing confirmed by plating on LB agar.
Stimulation of macrophages
12–16hr before stimulation with bacteria, bone marrow derived macrophages (hereafter ‘macrophages’) were re-plated as follows (Refs.19,54): Cytokines or LDH measurements in culture supernatants, 2.5×105 macrophages/condition (1 well of 24-well plate, 2 cm2); Immunoblot of whole cell extracts (WCE), depending on number of immunoblots, at least 2.5×106 macrophages/condition (2 wells of 6-well plate, 20 cm2); cleavage of inflammasome components in macrophages supernatants after protein concentration (concentrated supernatants) by immunoblot, 3.75×106 macrophages/condition (3 wells of 6-well plate, 30 cm2); ASC oligomerization assay, 7.5×106 macrophages/condition (full 6-well plate, 60 cm2); immunoprecipitation of endogenous caspase-11, NLRP3 or AIM2, at least 2.25×107 macrophages/condition (3 full 6-well plates, 180 cm2). On day of stimulation, cell medium was replaced by medium without antibiotics, and cells were at a concentration of 250,000 cells/200μL/cm2 for all conditions or readouts.
Prior to macrophage stimulation, bacteria were grown to mid-log phase (optical density at 600nm of 0.6) in appropriate media, pelleted at 4,000×g and washed 3X in PBS, and counted. Successive centrifugations and washing of bacterial pellets excluded outer membrane vesicles (OMVs), which remain in bacterial growth medium after centrifugation at 4,000×g and can only be pelleted after ultracentrifugation. Heat-killed and Gentamicin-treated bacteria washed 3X in PBS and counted. All bacteria added at 20:1macrophage, except F. novicida WT or ΔlpxF (50:1), S. enterica WT or ΔSpi1/2 (5:1), S. flexneri WT or BS103 (5:1). After adding bacteria, macrophages were briefly centrifuged (325×g, 2 min). 1hr post-stimulation with bacteria, antibiotics (cocktail of Penicillin, Streptomycin and Gentamicin) were added to the medium and kept until end of experiments.
E. coli in mid log phase (doubling time 24 min) contain 211fg total RNA/cell55, see also: https://bionumbers.hms.harvard.edu/search.aspx?trm=Mass+RNA+per+cell. On day of stimulation, 250,000 macrophages/200μL/cm2 were plated for all conditions. At a bacteria:macrophage ratio=20:1, 250,000 macrophages received 5×106 bacteria i.e. 1μg RNA content (considering 211fg RNA/cell). When adding bacterial RNA (RNAbac) at 10μg/mL, 250,000 macrophages received 2μg RNAbac in 200μL, a 2-fold excess of exogenously added RNAbac compared to RNA from live bacteria, considering that added RNAbac may be subject to degradation in the medium (less likely with live bacteria which contain the RNA until its release in phagolysosomes), and the likelihood that not all RNAbac would be internalized with killed bacteria. ~100-fold lower concentrations of purified bacterial mRNA (mRNAbac) (100ng/mL) yielded comparable functional readouts to 10μg/mL RNAbac17. mRNAbac constitutes 1% of RNAbac, which translates into nanogram quantities. Based on these calculations, 5×106 bacteria would contain ~10ng mRNA. When adding mRNAbac at 100ng/mL, 250,000 macrophages received 20ng mRNAbac, ~2-fold higher than from live bacteria, which after accounting for degradation and fractions internalized, was estimated to be within physiological range.
For priming prior to stimulation or transfection, macrophages were treated with 100ng/mL LPS or 1μg/mL poly(I:C) for 12–16hr. Macrophage treatment with 10 μg/mL zVAD-FMK was 1hr before stimulation with bacteria.
Transfection of macrophages and 293T cells
Macrophages were transfected using Lipofectamine 2000 (ThermoFisher). For a well of 24-well plate, 1.5μl Lipofectamine/well was diluted in 50μl opti-MEM medium (Gibco) to transfect indicated concentrations of LPS (E. coli 055:B5, Sigma), ultrapure LPS (E. coli O111:B4, Invivogen) or RNAbac. For indication, when transfected with 2ng/mL LPS, one well of 24-well plate containing 500,000 macrophages plated at 250,000 macrophages/200μL/cm2 received 0.8ng LPS, while one well of 6-well plate received 4ng LPS. When transfected with 1μg/mL LPS, the same number of cells received respectively 400ng and 2μg LPS.
293T cells were transfected using JetPrime (Polyplus Transfection). Transfections were according to manufacturer’s instructions. For 10cm2 plate, 10μg of DNA was diluted in 500μL JetPrime buffer before adding 20μL JetPrime Reagent.
Preparation of bacterial RNA (RNAbac)
RNAbac was isolated from mid-log phase growing bacteria using RNeasy prep kits (Qiagen) following manufacturer’s instructions (Ref.54). To enrich mRNAbac, ribosomal 16S and 23S RNA (rRNA) removed by magnetic bead-based capture hybridization using MICROBExpress kit (Ambion/Applied Biosystems). Remaining mRNAbac purified using RNeasy mini prep (Qiagen) following the RNA cleanup protocol. RNA concentration and purity determined by nanodrop measuring 260/280nm and 260/230 nm absorbance ratios, and RNA integrity by 1% agarose gel electrophoresis.
Immunoblots
Macrophages were washed in cold PBS, then lysed in 50mM Tris-HCl (pH 7,9), 300mM NaCl, 1% Triton X-100, supplemented with protease and phosphatase inhibitor cocktails (respectively Complete Protease and Phostop, Roche). WCE were centrifuged at 3,640×g, 10 min at 4°C. Protein concentrations were determined using the Bradford method. Samples were denatured in Laemmli sample buffer for SDS-PAGE resolution. Proteins were transferred onto a PVDF membrane (Millipore). Membranes were blocked with 7% evaporated milk in PBS 0.2% Tween and incubated with primary antibodies and peroxidase-conjugated secondary antibodies (all diluted in PBS 7% milk 0.2% Tween). Bound antibodies were visualized using the Amersham™ ECL or Pierce® ECL2 detection reagents, and imaged using developing machine or Amersham™ Imager 600 (GE Healthcare). All immunoblots represent at least 3 independent experiments. All blots were probed for β-Actin as a loading control.
Concentration of supernatants using TCA precipitation
Proteins of macrophage supernatants (prepared in serum-free Opti-MEM) were concentrated using Trichloroacetic acid (TCA) precipitation and washed in cold acetone (Ref.54). Precipitated proteins were resuspended and denatured in 50μL 2X Laemmli buffer for SDS-PAGE resolution on 12% polyacrylamide gels.
ASC oligomerization
Following a protocol described in Ref.56, Macrophages were resuspended in hypotonic buffer (20mM HEPES, 10mM KCl, 1.5mM MgCl2, 1mM EDTA, with protease inhibitor cocktail) and mechanically lysed by passing 20X through a 27G gauge needle. Intact cells, debris and nuclei were removed by centrifugation at 340×g for 8 min. Supernatant containing soluble and insoluble proteins was collected and mixed V1/1 with CHAPS buffer (20mM HEPES, 5mM MgCl2, 0.5mM EGTA 0.1% CHAPS, protease inhibitors). ASC oligomers were pelleted by centrifugation at 2,650×g for 8 min and crosslinked in CHAPS buffer containing 25mM DSS (ThermoScientific) for 30 min at room temperature before centrifugation at 2,650×g for 8 min. Pellets were resuspended in 2X Laemmli buffer and denatured 5 min at 95°C. Cross-linked fractions were resolved by SDS-PAGE on 12% polyacrylamide gels. Detection of ASC oligomers was initially performed using the anti-ASC N-15 antibody (sc-22514-R, Santa Cruz technologies), which was discontinued. Later experiments used anti-ASC AL177 (AG-25B-0006-C100, Adipogen). ASC oligomers were 1) detected at the expected molecular weight: monomer (~22kDa), dimer (~45kDa) and polymers (above 45kDa), and 2) impaired in ASC-deficient macrophages validating assay specificity.
Immunoprecipitation
For immunoprecipitation of endogenous NLRP3 and AIM2, macrophages were harvested, washed in cold PBS, lysed in 50mM Tris-HCl (pH 7,9), 150mM NaCl, 0.25% Triton X-100 supplemented with protease and phosphatase inhibitor cocktails (respectively Complete Protease and Phostop, Roche), and immunoprecipitation performed in the same buffer. Alternatively, for immunoprecipitation of endogenous caspase-11, lysis and immunoprecipitation were performed in 50mM Tris-HCl (pH 7,9), 300mM NaCl and 0.5% Triton X-100 supplemented with protease inhibitor and phosphatase inhibitor cocktails.
For immunoprecipitation of overexpressed NLRP3 and Caspase-11, 24 hr-transfected 293T cells were lysed in 50mM Tris-HCl (pH 7,9), 300mM NaCl, 1% Triton X-100 supplemented with protease and phosphatase inhibitor cocktails. For all IP conditions, portions of cleared WCE were pre-adsorbed on Protein G-agarose beads for 30 min (Sigma) before incubation with anti-caspase-11, anti-NLRP3, anti-AIM2, anti-HA or anti-FLAG M2 antibodies for 2 hr at 4°C in the respective lysis/IP buffer, and final binding on Protein G-agarose beads for 30 min. After 3 washes in the corresponding lysis/IP buffer, beads pellets were resuspended and denatured in 40μL 2X Laemmli buffer, before SDS-PAGE of both immunoprecipitates and WCE. Alternatively, for anti-FLAG M2 immunoprecipitates, elution was performed using 100μg/mL 3X FLAG peptide (Sigma #F4799) in lysis/IP buffer for 30 min before denaturation in Laemmli buffer.
Pulldown of active caspases
5μM biotinylated zVAD-FMK (#1135688-15-1, Cayman Chemical) was applied to macrophages at 2 hr before bacterial stimulation or transfection. For zVAD-FMK pull-down of entire macrophage output (WCE plus supernatant), cells were lysed directly by adding 1% IGEPAL CA630 in the cell medium. Mixed lysates were incubated on ice for 5 min in the presence of protease and phosphatase inhibitors. Pulldown was performed using Neutravidin agarose beads (#29200, ThermoScientifc). Beads were washed 5X in 50 mM Tris-HCl (pH 7.9), 300mM NaCl, 1% IGEPAL CA630 buffer before resuspension in Laemmli 2X and denaturation 10 min at 95°C.
In vitro caspase activity assay
Immunoprecipitation of endogenous caspase-11 was performed as described in Immunoprecipitation methods. Hydrolysis of fluorogenic substrate zVAD-AMC by immunoprecipitates was performed by mixing 50μL of immunoprecipitates with 50μL assay buffer (50mM HEPES pH 7.5, 150mM NaCl, 3mM EDTA, 0.005% (v/v) Tween-20 and 10mM DTT). zVAD-AMC was added into the reaction at a final concentration of 75μM and samples were incubated at 37°C for 30 min. Substrate cleavage was monitored by measuring fluorescence intensity of free AMC hydrolyzed from zVAD-AMC (emission 450nm, excitation 365nm) on SpectraMax iD3 fluorimeter.
Measurement of cytosolic LPS
Macrophages were washed several times in cold PBS 2 hr after transfection with LPS or stimulation with E. coli to remove extracellular LPS or bacteria. WCE were prepared and LPS levels were determined using Limulus Amebocyte Lysate (LAL) Pyrochrome detection kit (Asscociates of Cape Cod Incorporated), using Glucashield reconstitution buffer to overcome interference from (1–3)-β-Glucan.
Preparation of cytosolic and residual fractions
Subcellular fractionation of macrophages was conducted by a digitonin-based method as described with modifications57. 2 hr after stimulation or transfection, macrophages were washed 5X 5 min in cold PBS with gentle shaking at 4°C to remove extracellular bacteria and LPS. Plated cells were treated with 0.05% digitonin buffer for 10 min and supernatant containing cytosol was collected. The residual cell fraction (containing cell membrane, organelles and nucleus) was collected in 0.1% CHAPS buffer. Cytosol and residual fractions were used for LPS quantification via LAL assay or for RT-qPCR quantification of RNAbac. Fractions were additionally immunoblotted for organelle and cytosolic markers to validate purity of cytosolic fractions.
Detection of RNAbac using Pepper RNA – tDeg system
We used Pepper RNA – tDeg for imaging RNAbac within cells58,59. Pepper RNA binds to and prevents proteasomal degradation of cytosolic tDeg-tagged mNeonGreen, resulting in increased fluorescence when Pepper RNA is detected. Plasmids encoded Pepper RNA tag (CMV-mCherry-(F30–2xPepper)10, Addgene #129401) and tDeg-tagged mNeonGreen (miniCMV-(mNeonGreen)4-tDeg, Addgene # 29402). We subcloned Pepper RNA tag into a bacterial expression vector, and tDeg-tagged mNeonGreen together with its miniCMV promotor into a lentiviral vector (pCDH-CMV-MCS-EF1α-Puro, System Biosciences). J2-immortalized macrophages and THP-1 cells expressing cytosolic (mNeonGreen)4-tDeg were generated by lentiviral transduction. Recombinant E. coli expressing Pepper RNA were generated by electroporation. J2-immortalized macrophages and THP-1 cells were stimulated with E. coli expressing Pepper RNA for 2 hr before WCE or cytosolic/residual extract preparation. mNeonGreen Fluorescence in extracts was measured using SpectraMax iD3 fluorimeter (excitation 495nm, emission 525nm).
Detection of RNAbac using ViewRNA ISH
Macrophages plated on coverslips were stimulated with live or Gentamicin-killed mCherry E. coli. At 6 hr, cells were fixed and stained using ViewRNA® Cell Plus Assay kit (Invitrogen), using sequence-specific probes for RNAs encoding for ectopic mCherry or endogenous GroES, both labeled with Alexa Fluor 647. Additional staining for LAMP1 protein or LPS was performed before the probe hybridization according to the kit manual. Fluorescence was analyzed by confocal microscopy.
Quantification of RNAbac by RT-qPCR
2hr post macrophage stimulation, cytosolic fractions were prepared and RNAs extracted using RNeasy mini kit. RNAs were treated with TURBO™ DNase (ThermoFisher), and ‘no RT-qPCR’ controls were run together to control the genomic DNA contamination. Reverse transcription was performed using SuperScript™ III Reverse Transcriptase (ThermoFisher) and qPCR using SYBR™ Green PCR Master Mix (ThermoFisher) for bacterial genes: groES (forward (F)primer: ACTAAATCTGCTGGCGGCAT; reverse (R)primer: CAACTTTCACATCCAGCGGC), groEL (Fprimer: CGTGGTCAGAACGAAGACCA; Rprimer: CTTCGCCGCAGTTCAATACG), era (Fprimer: AAGCGACTCATCACTTCCCG; Rprimer: ATCTCCACGGTCACGGAGTA) and dnaE (Fprimer: CGCTCTGGGACAATTCCAGT; reverse primer: ACCTGCTCCGAACCAATGAG). qPCR for gapdh (Fprimer: AGGTCGGTGTGAACGGATTTG; Rprimer: TGTAGACCATGTAGTTGAGGTCA) was used as Eukaryotic control and amplification of bacterial genes was reported to gapdh.
Cytokine ELISA
Supernatants from cultured macrophages were collected 20 hr post-stimulation or at indicated times. Enzyme-linked immunosorbent assay (ELISA) capture/detection antibodies used were: IL-6, MP5-20F3/MP5-32C11 (BDPharmingen); IL-1β, B12/rabbit polyclonal antibody (eBioscience); TNFα 14-7423-85 (eBioscience)/13-7341-85 (Invitrogen). All antibodies were at 2μg/mL capture and 0.5 μg/mL detection, except IL-6 capture, which was at 1μg/ml. Detection antibodies were biotinylated and labeled by streptavidin-conjugated horseradish peroxidase (HRP), and visualized by adding 3,3′, 5,5′-tetramethylbenzidine (TMB, KPL). Color development was stopped with TMB-Stop Solution (KPL). Recombinant cytokines served as standards (Peprotech). Absorbances at 450nm were measured on a microplate reader (Molecular Devices). Cytokine supernatant concentrations were calculated by extrapolating absorbance values from standard curves where known concentrations were plotted against absorbance using SoftMax Pro 5 software.
Measurement of inflammatory cell death
Cell death of macrophages was measured using the Cytotox96 cytotoxicity assay (Promega) following manufacturer’s instructions. The assay measures the release of lactate dehydrogenase (LDH) into the supernatant calculated as the percentage of maximum LDH content, measured from total cellular lysates (100%).
Measurement of cell death by SYTOX™ incorporation
Macrophages were plated in 96-well plates and stimulated with bacteria as indicated in triplicates. 1hr post-stimulation of macrophage, SYTOX™ red dead cell stain (Invitrogen) was added to the cell medium at a final concentration of 60nM. Kinetics of SYTOX™ incorporation into macrophages was determined using Incucyte (Sartorius) for 74 hr. Percentages of SYTOX™ + macrophages at each time point were calculated by reporting the amount of SYTOX™ + macrophages to the number of plated macrophages. Representative images were selected at different time points.
Confocal microscopy
Macrophages were plated on glass coverslips. After bacterial stimulation, coverslips were washed with PBS, fixed in 2% PFA, and quenched in 50mM NH4Cl. Cells were then permeabilized with 0.2% Triton X-100 in PBS before blocking with 10% FBS PBS and incubation with appropriate antibodies prepared in 10% FBS PBS. When indicated, Phalloidin Alexa 647 was used together with secondary fluorescent antibodies to stain Actin and delineate the cell area. Lastly, cells were stained with DAPI. Coverslips mounted with ProLong Diamond Antifade Mountant (Invitrogen). Images acquired on a Leica SP5 DM microscope with a 63x/1.4 NA oil immersion objective. The partial nuclear staining pattern noted in Extended Data Figure 4 has been reported60 and is ASC-specific given its absence in Pycard–/– macrophages.
Plasmids and constructs
The plasmid encoding Casp11 (NM_007609.3) was provided by Feng Shao; Nlrp3 cDNA clone (NM_145827.3) was purchased from OriGene. Full length and truncated Casp11 or Nlrp3 were polymerase chain reaction (PCR) amplified from the corresponding plasmids and subcloned into vector pCDH-CMV-MCS-EF1-Puro between XbaI and BamHI sites (for Casp11), or BstBI and NotI sites (for Nlrp3). 3xFLAG Tag and HA tag were introduced into the N-terminus of Casp11 and Nlrp3 respectively via PCR. Casp11 (C254A) mutation was generated using Agilent mutagenesis kit (Catalog #200521).
Antibodies
For Immunoblot: Cell Signaling Technology: anti-β-Actin (3700), anti-ERP72 (5033), anti-S6K (2708), anti-α-Tubulin (3873), anti-FLAG (14793). eBiosciences: anti-Caspase-1 p20 (14-9832-82), anti-AIM2 (14-6008-93). Santa Cruz Technologies: anti-ASC (sc-22514-R), anti-TGN38 (sc-166594), anti-HistoneH1 (sc393358). Sigma: anti-Caspase-11 (C1354), anti-GasderminD (G7422), anti-FlagM2 (F3165). Adipogen: anti-NLRP3 (AG-20B-0014-C100), anti-ASC (AG-25B-0006-C100). Millipore: anti-Phospho-p62 S403 (MABC186). Research & Development: anti-IL-1β (AF-401-NA). Roche: anti-HA (11815016001). Abcam: anti-GasderminD (ab219800), anti-NEK7 (ab133514), anti-Calreticulin (ab92516), anti-GM130 (ab52649), anti-CathepsinS (ab232740), anti-LPS (ab35654). Biolegend: anti-LAMP1 (121602). For immunoprecipitation of HA-NLRP3: Cell Signaling: anti-HA (3724).
Statistical analysis
For statistical analyses of 3+ group experiments, one-way ANOVA was performed followed by multiple comparisons Sidak tests to allow 2-by-2 comparisons. For experiments with 2 groups (Extended Data Fig. 1b and 4b), Student’s t-test was performed. Error bars, mean ± s.e.m. ns: non-significant, p values and group sizes are indicated in the corresponding figures and legends. Analyses were conducted on biological replicates.
Extended Data
Extended Data Fig. 1. Bacterial mRNA and stimulatory LPS are both required for caspase-11 and noncanonical inflammasome activation.
a, Immunofluorescence confocal microscopy on bone marrow derived macrophages (hereafter ‘macrophages’) 2hr post-stimulation with L or HK E. coli or untreated. b, Bar graph, % cells with LAMP1-LPS colocalization. c, Immunoblots of macrophage concentrated supernatants (20hr) or WCE (6hr), and cytokine concentrations and LDH release in culture supernatants (20hr) post-stimulation with L, HK or HK E. coli supplemented with 100ng/mL mRNA isolated from E. coli or eukaryotic cells. LDH measured by cytotoxicity assay; IL-1β, TNF and IL-6 by ELISA. Error bars, mean ± s.e.m. t-test was performed in b (n=3). ns: non-significant. Bacteria:macrophage=20:1. Results represent at least 3 independent experiments.
Extended Data Fig. 2. Cytosolic levels of LPS and bacterial mRNA after macrophage stimulation with live or heat-killed bacteria.
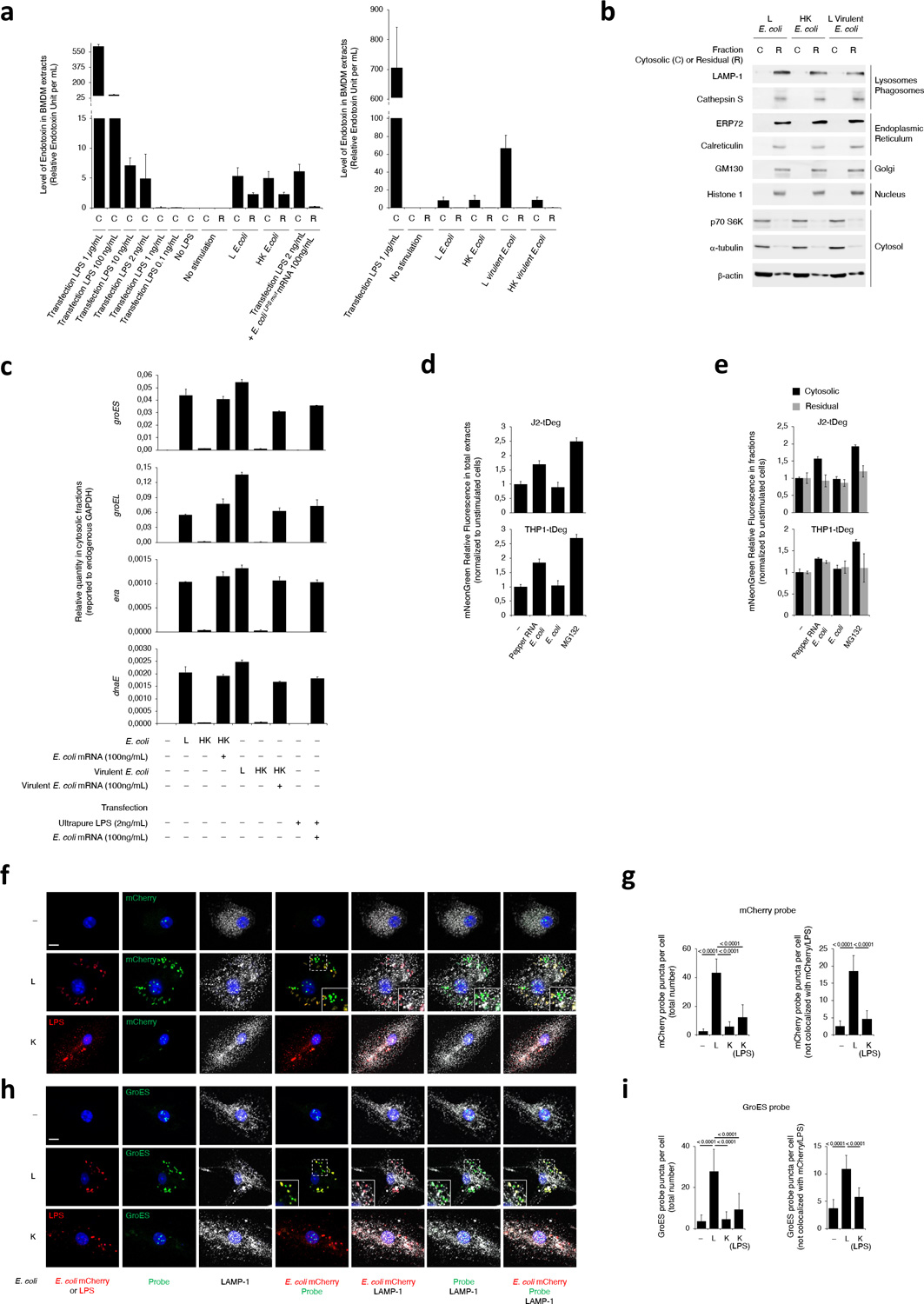
a, Measurements of LPS in the cytosolic (C) or residual (R) fractions prepared from macrophages 2hr post-transfection of indicated doses of LPS alone, co-transfection of 2ng/mL LPS with 100ng/mL E. coliLPSmut mRNA, or stimulation with L or HK E. coli or virulent E. coli as indicated. LPS was measured via Limulus Amebocyte Lysate method. b, Immunoblots of cytosolic (C) and residual (R) fractions from macrophages 2hr post-stimulation with L E. coli, HK E. coli or L virulent E. coli, and probed with marker antibodies for indicated subcellular compartments. c, RT-qPCR for the bacterial gene groES, groEL, era and dnaE on cytosolic fractions prepared 2hr post macrophage transfection with indicated doses of ultrapure LPS and mRNA prepared from E. coli, or stimulation with L, HK or HK E. coli or virulent E. coli supplemented with mRNA prepared from each bacterium. d,e, mNeonGreen fluorescence measurements on total extracts (d) or cytosolic versus residual fractions (e) of macrophages stably expressing tDeg-tagged mNeonGreen 2hr post-stimulation with E. coli expressing or not Pepper RNA, or after treatment with proteasome inhibitor MG132 as indicated. By expressing a Pepper RNA-regulated fluorogenic protein (mNeonGreen-tDeg) in the cytosol of macrophages, we noted 1.5–2-fold increase in mNeonGreen fluorescence following phagocytosis of recombinant Pepper RNA-expressing E. coli compared to the 2–2.5-fold increase with the proteasomal inhibitor MG132, indicating cytosolic access of Pepper RNA derived from phagocytosed E. coli bound to and stabilized the fluorogenic protein in the cytosol of macrophages. f,h, Confocal microscopy of direct fluorescence RNA in situ hybridization (ViewRNA ISH) to detect two RNAbac transcripts encoding for either mCherry (f) or endogenous GroES (h) from recombinant mCherry E. coli showed significantly more probe signal in macrophages at 6hr post-phagocytosis of live (L) compared to killed (K) bacteria. Killed bacteria were visualized by anti-LPS staining due to loss of mCherry fluorescence. RNA probe signal localized with live bacteria in lysosomes labeled with LAMP-1 as expected, but almost half of this signal did not colocalize to these bacteria suggesting cytosolic access. Inserts show magnification of indicated area. g,i, Bar graphs show quantification of probe signal in (f) and (h), respectively. Bacteria:macrophage=20:1. Error bars, mean ± s.e.m. One-way ANOVA followed by multiple comparisons Sidak tests and p values are indicated in bar graphs in g (Total number, Unst.: n=13, L: n=19, K: n=15, K(LPS): n=17 – Not colocalized, Unst.: n=11, L, K: n=14) and i (Total number, Unst.: n=11, L: n=14, K, K(LPS): n=10 – Not colocalized, n=10). Results represent at least 3 independent experiments.
Extended Data Fig. 3. Bacterial mRNA and LPS are both required for IL-1β secretion and generation of the active form of caspase-11.
a, Cytokine concentrations and LDH release in culture supernatants (20hr) post-transfection of WT or Nlrp3–/– macrophages as indicated with 2ng/mL ultrapure LPS and/or 10, 30 and 100 ng/mL of mRNA prepared from E. coli LPSmut, L. innocua, or in vitro transcribed (IVT). b, Immunoblots of macrophage concentrated supernatants, WCE, mixed concentrated supernatants and WCE, or pulldown of caspases with biotinylated zVAD-FMK from WT, Nlrp3–/– or Casp11–/– macrophages 20hr post-stimulation with L, HK or HK E. coli supplemented with E. coli total RNA (RNAtot), or transfection with indicated doses of ultrapure LPS and E. coli RNAtot. c, In vitro zVAD-AMC fluorescence post-incubation with immunoprecipitates of endogenous caspase-11 from macrophages stimulated for 6hr or 12hr as indicated with L, HK, or HK E. coli supplemented with E. coli RNAtot (10μg/mL). No IgG served as a control for Protein G-bound proteins alone. Bacteria:macrophage=20:1. Error bars, mean ± s.e.m. Results represent at least 3 independent experiments.
Extended Data Fig. 4. LPS is required alongside bacterial mRNA for assembly of the NLRP3 inflammasome and regardless of LPS stimulatory activity.
a-c, Immunofluorescence confocal microscopy on macrophages 16hr post-stimulation of indicated macrophage genotypes with L, gentamicin-killed (K) or K red fluorescent protein (RFP) expressing recombinant E. coli supplemented with E. coli RNAtot. Note, the partial nuclear staining pattern has previously been observed (see methods) and appears to be a specific signal given its absence in Pycard–/– macrophages. In a,b, insets show magnification of indicated areas. White arrowheads point to ASC specks. Phalloidin delineates the macrophage actin cytoskeleton. Scale bar=10μm. Bar graphs, % cells exhibiting ASC specks. Error bars, mean ± s.e.m. t-test was performed in b (n=5). One-way ANOVA followed by multiple comparisons Sidak tests were performed in a (n=5) and c (n=6). P values are indicated in bar graphs. Bacteria:macrophage=20:1. Results represent at least 3 independent experiments.
Extended Data Fig. 5. Kinetics of noncanonical activation of the NLPR3 inflammasome in macrophages in response to virulent and avirulent bacteria.
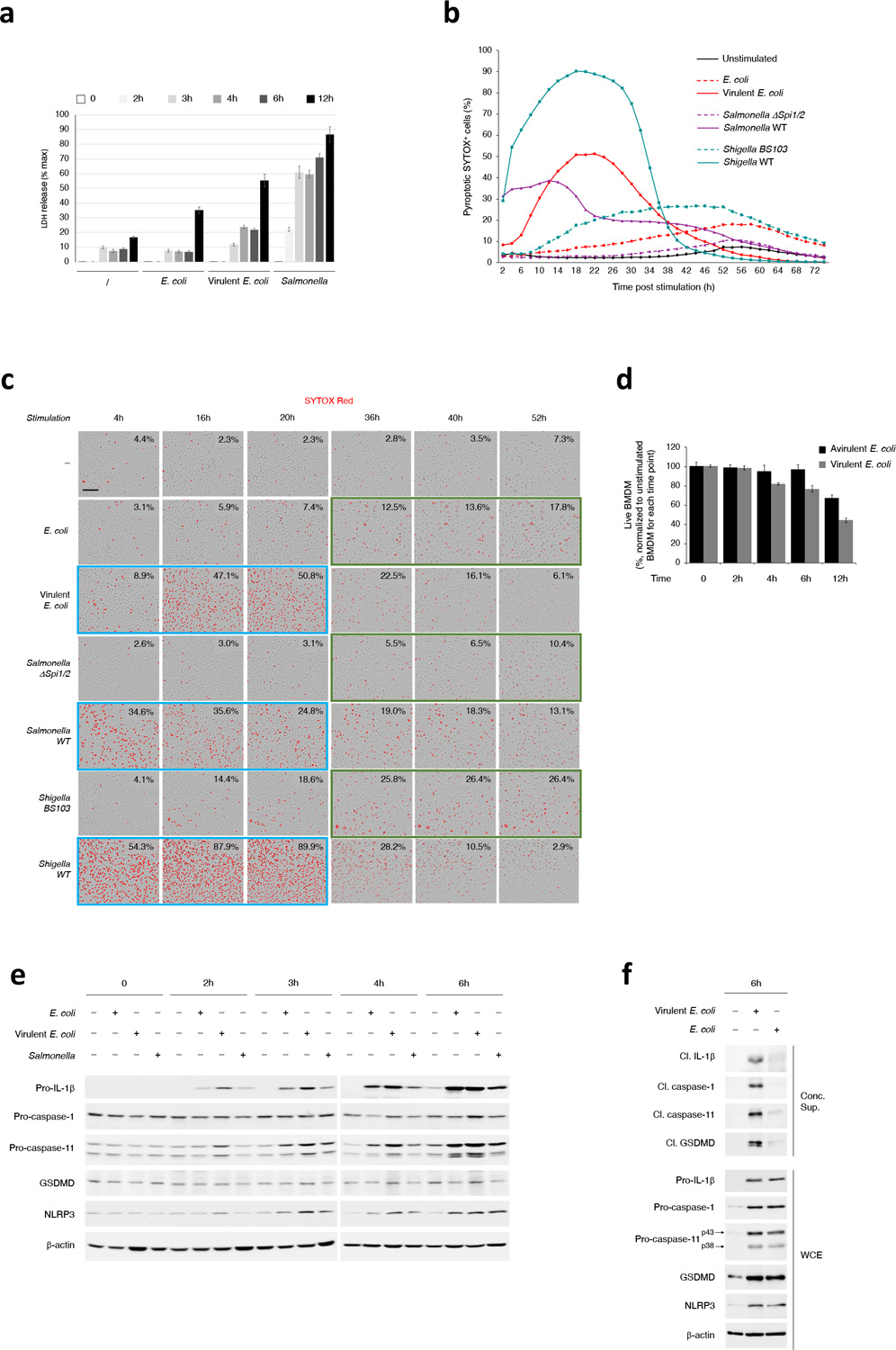
a, LDH released in culture supernatants of macrophages at indicated time points post-stimulation with Live E. coli (avirulent), virulent E. coli or virulent Salmonella. LDH measured by cytotoxicity assay. b,c, Kinetics of SYTOX Red incorporation over 72hr in macrophages stimulated with Live E. coli or virulent E. coli, Salmonella ΔSpi1/2 (avirulent) or WT (virulent), or Shigella BS103 (avirulent) or WT (virulent) (b), and representative images of SYTOX Red incorporation at selected time points (c). Peaks of SYTOX incorporation occurred faster (blue boxes) in response to virulent bacteria, before decreasing likely due to destruction of cell structure, while macrophages stimulated with avirulent bacteria were still incorporating SYTOX and peaked later (orange boxes). Scale bar=150μm. d, Counting of live macrophages at the indicated time points post-stimulation with Live E. coli or virulent E. coli. Counts were normalized to the unstimulated macrophages for each time point. e, Immunoblots of macrophage WCE at the indicated time points post-stimulation with Live E. coli, virulent E. coli or virulent Salmonella. f, Immunoblots of macrophage concentrated supernatants 6hr post-stimulation with Live E. coli or virulent E. coli. Error bars, mean ± s.e.m. Bacteria:macrophage =20:1 for E. coli and virulent E. coli, 5:1 for Salmonella and Shigella strains. Results represent at least 3 independent experiments.
Extended Data Fig. 6. NLRP3 stimuli specifically couple with LPS in mediating noncanonical activation of the NLRP3 inflammasome.
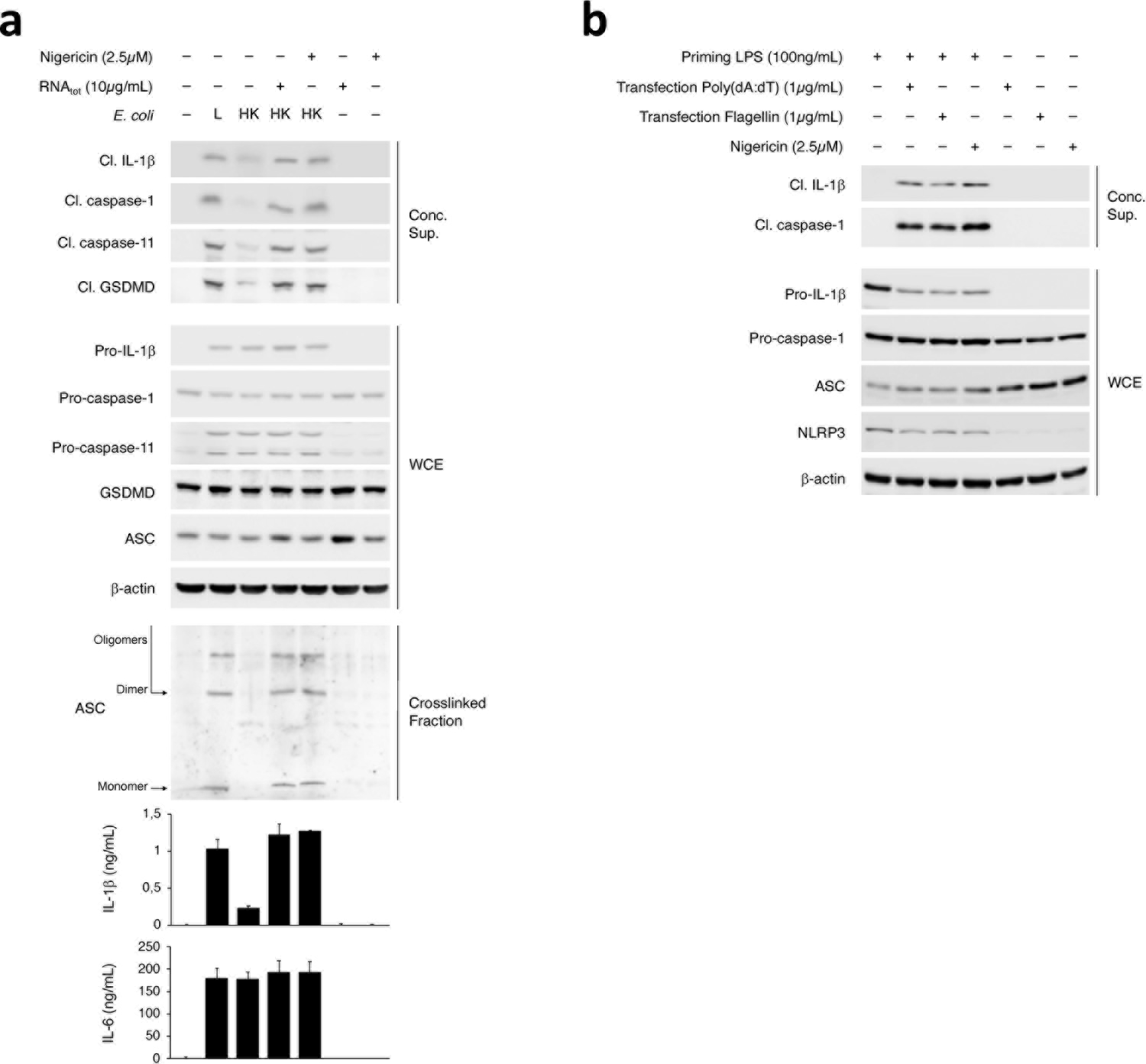
a,b, Immunoblots of macrophage concentrated supernatants (20hr), WCE (6hr), or cross-linked fractions (16hr), and cytokine concentrations in culture supernatants (20hr) as indicated post-stimulation with L, HK or HK E. coli supplemented with indicated doses of RNAtot or Nigericin (a), and post-treatment with Nigericin or transfection of indicated doses of poly(dA:dT) or Flagellin with or without prior priming with 100ng/mL LPS for 12–16hr (b). IL-1β and IL-6 by ELISA. Error bars, mean ± s.e.m.
Extended Data Fig. 7. dTGN colocalization and biochemical procaspase-11-NLRP3 interaction.
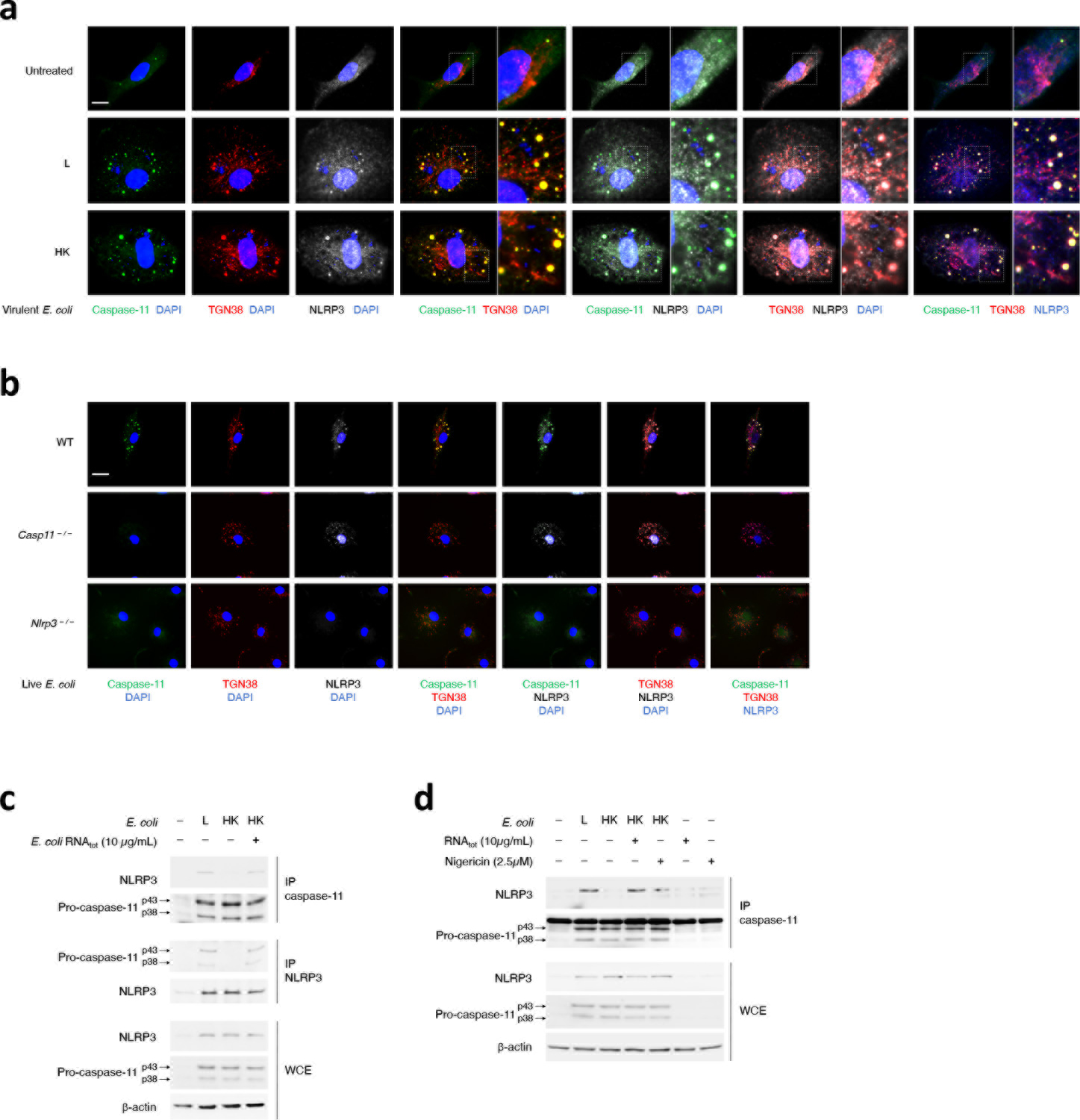
a,b, Immunofluorescence confocal microscopy 16hr post-stimulation of WT macrophages with L or HK virulent E. coli (a), and WT, Casp11–/– and Nlrp3–/– macrophages as indicated with L E coli (b). Scale bar=10μm. In (a), side micrograph insets in the triple merges show magnification of the indicated areas. c,d, Immunoprecipitation (IP) of endogenous caspase-11 or NLRP3 as indicated, and immunoblotting for co-immunoprecipitating proteins and WCE proteins (labels to left of immunoblot panels) from macrophages stimulated 12hr with L, HK or HK E. coli supplemented with indicated dose of E. coli RNAtot. (c) and L, HK or HK E. coli supplemented with indicated dose of E. coli RNAtot or Nigericin, E. coli RNAtot alone or Nigericin alone (d). Bacteria:macrophage=20:1. Results represent at least 3 independent experiments.
Extended Data Fig. 8. GSDMD is important for noncanonical activation of the NLRP3 inflammasome in response to Gram-negative bacteria.
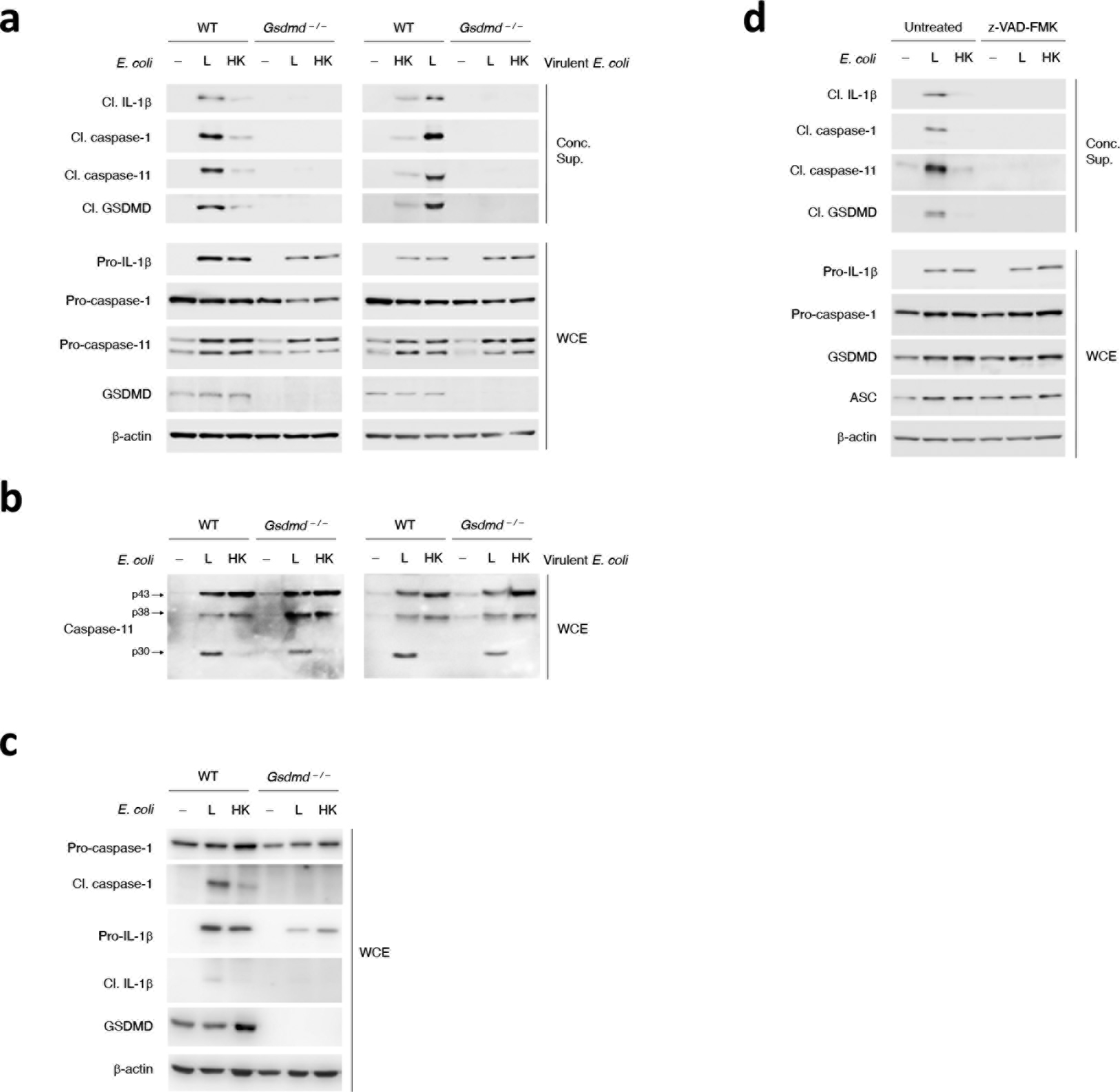
a-c, Immunoblots of macrophage concentrated supernatants (20hr) or WCE at 6hr (a,b) and 20hr (c) post-stimulation of WT and Gsdmd–/– macrophages with L or HK E. coli (avirulent) or virulent E. coli as indicated. (d) Immunoblots of macrophage concentrated supernatants (20hr) or WCE (6hr) post-stimulation of macrophages with L or HK E. coli with or without zVAD-FMK treatment.
Extended Data Fig. 9. Model for noncanonical activation of the NLRP3 inflammasome by live Gram-negative bacteria.
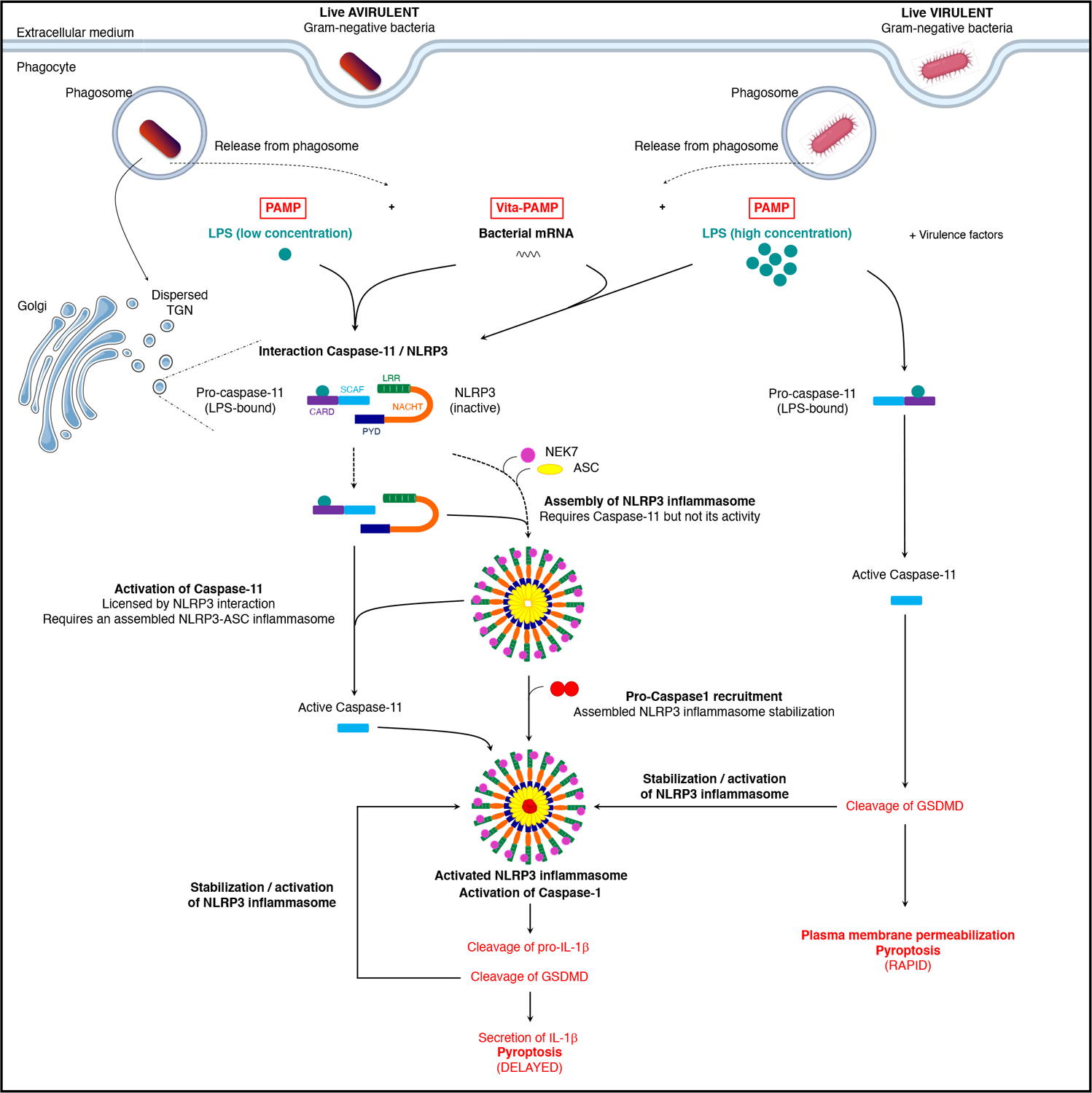
Following phagocytosis of live Gram-negative bacteria, two classes of PAMPs are exposed to cytoplasmic pattern recognition receptors: the classical PAMP LPS, shared by live and killed bacteria, and the vita-PAMP bacterial mRNA (mRNAbac), present only in live bacteria. Coincident detection of mRNAbac and LPS from virulent or avirulent Gram-negative bacteria alike promotes a physical and mutually exclusive interaction between NLRP3 and the intracellular LPS receptor procaspase-11. This interaction localizes to the dispersed Trans-Golgi Network (TGN) and is mediated through the procaspase-11 SCAF domain and the LRR and PYD domains of NLRP3. The interaction of NLRP3 and procaspase-11 is upstream of their activation: It does not require the ability of LPS to activate caspase-11 and can still occur in the absence of GSDMD. It also does not require ASC and caspase-1 which are important for NLRP3 activation. Besides their interaction, NLRP3 and procaspase-11 are reciprocally required for their function: LPS binding to but not activation of procaspase-11, is necessary for mRNAbac-mediated NLRP3 inflammasome assembly. Reciprocally, NLRP3 and ASC but not caspase-1 are required for procaspase-11 activation, indicating the necessity for ‘nascent’ NLRP3 inflammasome assembly upon sensing the viability of Gram-negative bacteria (detection of mRNAbac) and irrespective of bacterial virulence factor expression. Although NLRP3-ASC oligomers can form in the absence of procaspase-1, these oligomers are unstable indicating stabilization of the ‘nascent’ assembled NLRP3 inflammasome upon procaspase-1 recruitment. Furthermore, higher concentrations of intracellular LPS, likely due to virulence factor activity during infection with virulent cell-invasive Gram-negative bacteria, trigger faster kinetics of plasma membrane permeabilization/pyroptosis compared to avirulent bacteria, and independently of NLRP3 and ASC2,9. Collectively, the model that emerges demonstrates two modes of procaspase-11 activation by LPS, a fast NLRP3-independent mode triggered by the concurrent expression of bacterial virulence factors, and a slower NLRP3-dependent mode triggered by coincident detection of the vita-PAMP mRNAbac that signifies bacterial viability. (SCAF, scaffold; PYD, Pyrin; LRR, leucine rich repeat).
Extended Data Fig. 10. Characterization and mapping of the procaspase-11 and NLRP3 interaction.
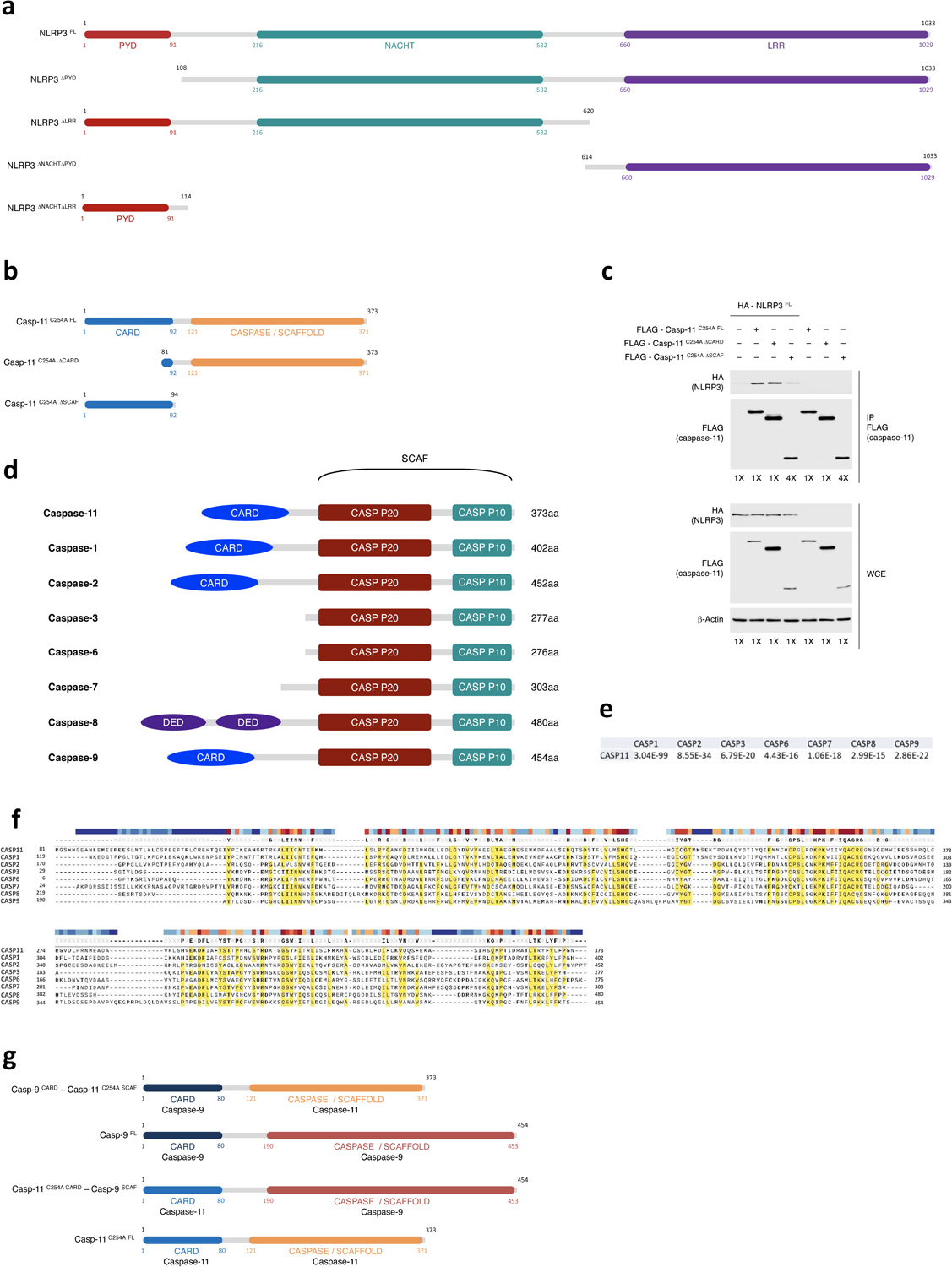
a, Schematic indicating the different truncated mutants of NLRP3 used for co-immunoprecipitation experiments in 293T cells. All forms were fused to 3xHA tag in N-Terminus. b, Schematic indicating the different truncated mutants of casp-11C254A used for co-immunoprecipitation experiments in 293T cells. All forms were fused to 3xFLAG tag in N-Terminus. c, Immunoprecipitation and immunoblotting for coimmunoprecipitating and WCE proteins of overexpressed FLAG-caspase-11 either casp-11C254A FL, Casp-11 C254A ΔCARD or Casp-11 C254A ΔSCAF with or without HA-NLRP3FL in 293T cells 24 hr post-transfection. To equilibrate the levels of FLAG-caspase-11 mutants in the immunoprecipitates, 4 times more protein extracts were submitted to anti-FLAG immunoprecipitation when FLAG-casp-11C254A ΔSCAF was expressed (labelled 4X) compared to when either FLAG-casp-11C254A FL or FLAG-casp-11C254A ΔCARD were expressed (labelled 1X). Therefore, note that all proteins from FLAG-casp-11C254A ΔSCAF samples (including HA-NLRP3FL) were 4 times more abundant during anti-FLAG immunoprecipitation, yet HA-NLRP3FL was still much less co-immunoprecipitated with FLAG-casp-11C254A ΔSCAF compared to FLAG-casp-11C254A FL. Immunoblotted proteins are indicated to left of each immunoblot panel. d, Schematic representation of CARD, DED, CASPASE p20 and CASPASE p10 domains of murine inflammatory and apoptotic caspases. SCAF domain is composed of CASPASE p20 and CASPASE p10 domains. All caspases, with the exception of the short forms of mouse and human caspase-12, have SCAF domains. The alignment E values were calculated using the NCBI alignment tool. e, Similarity coefficients between caspase-11 and other murine caspases. f, Protein sequence alignment for murine caspases. The alignment diagram was generated using the alignment module of SnapGene software. g, Schematic indicating the different caspase-11C254A and caspase-9 chimeras used for co-immunoprecipitation experiments in 293T cells. All forms were fused to 3xFLAG tag in N-terminus.
Supplementary Material
Acknowledgements
We are grateful to D. Amsen, N. Vabret, T. Kanneganti, K. Fitzgerald, V. Dixit, F. Shao, M. Goldberg, R. Ernst, J. Chipuk, J. Gelles-Hurwitz, and R. Flavell for reagents and discussions. We thank D. Filipescu, C. Brou, P. Chastagner, A. Israël, A. Zanin-Zhorov, S. Waksal, M.A. Blander, and S.J. Blander for their support.
Funding:
This work was supported by National Institutes of Health (NIH) grants AI127658 to JMB, AI139425 to J.C., R35NS111631 to S.R.J., and Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease Awards to J.M.B and to J.C. The Blander laboratory was supported by NIH grants DK072201, DK111862, AI095245 and AI123284 to J.M.B. J.M.B. was supported by the Leukemia & Lymphoma Society.
Footnotes
Competing interests: Authors declare no competing interest.
Data and materials availability:
Datasets generated or analyzed during this study are available on reasonable request.
References:
- 1.Hagar JA, Powell DA, Aachoui Y, Ernst RK & Miao EA Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kayagaki N et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Shi J et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Broz P & Dixit VM Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16, 407–420 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Broz P, Pelegrin P & Shao F The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol 20, 143–157 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Lieberman J, Wu H & Kagan JC Gasdermin D activity in inflammation and host defense. Sci Immunol 4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aachoui Y et al. Caspase-11 protects against bacteria that escape the vacuole. Science 339, 975–978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broz P et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–291 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Case CL et al. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc Natl Acad Sci U S A 110, 1851–1856 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gurung P et al. Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-beta (TRIF)-mediated caspase-11 protease production integrates Toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem 287, 34474–34483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kayagaki N et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Rathinam VA et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–619 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S et al. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92, 501–509 (1998). [DOI] [PubMed] [Google Scholar]
- 14.Kayagaki N et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Blander JM & Sander LE Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat Rev Immunol 12, 215–225 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Finethy R et al. Inflammasome Activation by Bacterial Outer Membrane Vesicles Requires Guanylate Binding Proteins. mBio 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sander LE et al. Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature 474, 385–389 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barbet G et al. Sensing Microbial Viability through Bacterial RNA Augments T Follicular Helper Cell and Antibody Responses. Immunity 48, 584–598 e585 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moretti J et al. STING Senses Microbial Viability to Orchestrate Stress-Mediated Autophagy of the Endoplasmic Reticulum. Cell 171, 809–823 e813 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blander JM & Barbet G Exploiting vita-PAMPs in vaccines. Curr Opin Pharmacol 41, 128–136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayward JA, Mathur A, Ngo C & Man SM Cytosolic Recognition of Microbes and Pathogens: Inflammasomes in Action. Microbiol Mol Biol Rev 82 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kailasan Vanaja S et al. Bacterial RNA:DNA hybrids are activators of the NLRP3 inflammasome. Proc Natl Acad Sci U S A 111, 7765–7770 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gros M & Amigorena S Regulation of Antigen Export to the Cytosol During Cross-Presentation. Front Immunol 10, 41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herskovits AA, Auerbuch V & Portnoy DA Bacterial ligands generated in a phagosome are targets of the cytosolic innate immune system. PLoS Pathog 3, e51 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakamura N et al. Endosomes are specialized platforms for bacterial sensing and NOD2 signalling. Nature 509, 240–244 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Dick MS, Sborgi L, Ruhl S, Hiller S & Broz P ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun 7, 11929 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu A et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanneganti TD et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Stutz A, Horvath GL, Monks BG & Latz E ASC speck formation as a readout for inflammasome activation. Methods Mol Biol 1040, 91–101 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Sanchez-Garrido J, Slater SL, Clements A, Shenoy AR & Frankel G Vying for the control of inflammasomes: The cytosolic frontier of enteric bacterial pathogen-host interactions. Cell Microbiol 22, e13184 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu A et al. Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat Struct Mol Biol 23, 416–425 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen J & Chen ZJ PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564, 71–76 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He Y, Zeng MY, Yang D, Motro B & Nunez G NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharif H et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 570, 338–343 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanders MG et al. Single-cell imaging of inflammatory caspase dimerization reveals differential recruitment to inflammasomes. Cell Death Dis 6, e1813 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santos JC et al. Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat Commun 11, 3276 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wandel MP et al. Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat Immunol 21, 880–891 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kutsch M et al. Direct binding of polymeric GBP1 to LPS disrupts bacterial cell envelope functions. EMBO J 39, e104926 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.An J et al. Caspase-4 disaggregates lipopolysaccharide micelles via LPS-CARD interaction. Sci Rep 9, 826 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aurell CA & Wistrom AO Critical aggregation concentrations of gram-negative bacterial lipopolysaccharides (LPS). Biochem Biophys Res Commun 253, 119–123 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Sasaki H & White SH Aggregation behavior of an ultra-pure lipopolysaccharide that stimulates TLR-4 receptors. Biophys J 95, 986–993 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bergstrand A, Svanberg C, Langton M & Nyden M Aggregation behavior and size of lipopolysaccharide from Escherichia coli O55:B5. Colloids Surf B Biointerfaces 53, 9–14 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Santos NC, Silva AC, Castanho MA, Martins-Silva J & Saldanha C Evaluation of lipopolysaccharide aggregation by light scattering spectroscopy. Chembiochem 4, 96–100 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Belasco JG All things must pass: contrasts and commonalities in eukaryotic and bacterial mRNA decay. Nat Rev Mol Cell Biol 11, 467–478 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eigenbrod T et al. Bacterial RNA mediates activation of caspase-1 and IL-1beta release independently of TLRs 3, 7, 9 and TRIF but is dependent on UNC93B. J Immunol 189, 328–336 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sha W et al. Human NLRP3 inflammasome senses multiple types of bacterial RNAs. Proc Natl Acad Sci U S A 111, 16059–16064 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ugolini M et al. Recognition of microbial viability via TLR8 drives TFH cell differentiation and vaccine responses. Nat Immunol 19, 386–396 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Deets KA & Vance RE Inflammasomes and adaptive immune responses. Nat Immunol 22, 412–422 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Munoz-Wolf N & Lavelle EC A Guide to IL-1 family cytokines in adjuvanticity. FEBS J 285, 2377–2401 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Cavaillon JM, Singer M & Skirecki T Sepsis therapies: learning from 30 years of failure of translational research to propose new leads. EMBO Mol Med 12, e10128 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reynolds CM & Raetz CR Replacement of lipopolysaccharide with free lipid A molecules in Escherichia coli mutants lacking all core sugars. Biochemistry 48, 9627–9640 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buchrieser C et al. Comparison of the genome sequences of Listeria monocytogenes and Listeria innocua: clues for evolution and pathogenicity. FEMS Immunol Med Microbiol 35, 207–213 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Cheung AL, Bayer AS, Zhang G, Gresham H & Xiong YQ Regulation of virulence determinants in vitro and in vivo in Staphylococcus aureus. FEMS Immunol Med Microbiol 40, 1–9 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Moretti J, Vabret N & Blander JM Measuring Innate Immune Responses to Bacterial Viability. Methods Mol Biol 1714, 167–190 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Nierlich DP Regulation of ribonucleic acid synthesis in growing bacterial cells. II. Control over the composition of the newly made RNA. J Mol Biol 72, 765–777 (1972). [DOI] [PubMed] [Google Scholar]
- 56.Lugrin J & Martinon F Detection of ASC Oligomerization by Western Blotting. Bio Protoc 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramsby M & Makowski G Differential detergent fractionation of eukaryotic cells. Cold Spring Harb Protoc 2011, prot5592 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Strack R A peck of Peppers. Nat Methods 16, 1075 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Wu J et al. Live imaging of mRNA using RNA-stabilized fluorogenic proteins. Nat Methods 16, 862–865 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bryan NB, Dorfleutner A, Rojanasakul Y & Stehlik C Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol 182, 3173–3182 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets generated or analyzed during this study are available on reasonable request.