

Abstract
Autosomal dominant polycystic kidney disease (ADPKD), primarily due to PKD1 or PKD2 mutations, causes progressive kidney cyst development and kidney failure. There is significant intrafamilial variability likely due to the genetic background and environmental/lifestyle factors; variability that can be modeled in PKD mice. Here, we characterized mice homozygous for the PKD1 hypomorphic allele, p.Arg3277Cys (Pkd1RC/RC), inbred into the BALB/cJ (BC) or the 129S6/SvEvTac (129) strains, plus F1 progeny bred with the previously characterized C57BL/6J (B6) model; F1(BC/B6) or F1(129/B6). By one-month cystic disease in both the BC and 129 Pkd1RC/RC mice was more severe than in B6 and continued with more rapid progression to six to nine months. Thereafter, the expansive disease stage plateaued/declined, coinciding with increased fibrosis and a clear decline in kidney function. Greater severity correlated with more inter-animal and inter-kidney disease variability, especially in the 129-line. Both F1 combinations had intermediate disease severity, more similar to B6 but progressive from one-month of age. Mild biliary dysgenesis, and an early switch from proximal tubule to collecting duct cysts, was seen in all backgrounds. Preclinical testing with a positive control, tolvaptan, employed the F1(129/B6)-Pkd1RC/RC line, which has moderately progressive disease and limited isogenic variability. Magnetic resonance imaging was utilized to randomize animals and provide total kidney volume endpoints; complementing more traditional data. Thus, we show how genetic background can tailor the Pkd1RC/RC model to address different aspects of pathogenesis and disease modification, and describe a possible standardized protocol for preclinical testing.
Keywords: ADPKD, animal models, disease modifiers, PKD1, preclinical testing
Autosomal dominant polycystic kidney disease (ADPKD) is a common monoallelic disease,1–3 characterized by progressive cyst development/expansion and accounting for 5% to 10% of end-stage kidney disease worldwide.4,5 Tolvaptan, a vasopressin receptor antagonist, is the only approved therapy6,7 that slows but does not prevent disease; hence, there is a need for complementary therapies. ADPKD is mainly caused by mutations to PKD1 or PKD2,8 encoding polycystin-1 (PC1) or polycystin-2. PKD1, especially due to truncating mutations, is the most severe ADPKD form,9 with male sex being a risk factor for earlier end-stage kidney disease. Severe polycystic liver disease is the most common extrarenal phenotype.10,11 PC1 and polycystin-2 form a complex on primary cilia interfacing between extra-cellular cues and cellular differentiation, possibly via calcium and/or cyclic adenosine monophosphate (cAMP) signaling.12,13 Reduction/loss of the functional polycystin (PC) complex impacts proliferation, fluid secretion, and injury/repair.14–16 PC1 cleavage at the G-protein coupled receptor proteolytic site and interaction with polycystin-2 is necessary for localization and function.17
ADPKD manifests with significant intrafamilial phenotypic variability, which is rarely associated with genic/allelic complexity,8,18,19 but is mainly driven by modifier genes, kidney injury, and environmental/lifestyle factors; body mass index and salt intake may influence the phenotype.20–22 ADPKD animal models potentially provide insights into genetic (and environmental/lifestyle) modifiers, with murine genetic background influencing kidney size, function, and toxicity.23–27 Wild-type mice in the C57BL/6J (B6) background are protected from glomerulosclerosis,28–31 kidney hypertrophy,32 and kidney injury33,34; the Nphp3Pcy/Pcy polycystic kidney disease (PKD) B6 model has less severe cystic burden than does the DBA background.35
Mice matching the human disease genotype, Pkd1+/— or Pkd2+/—, develop only few kidney (and liver) cysts even into old age, whereas mice biallelic for inactivating mutations are embryonically lethal.36,37 Conditional/inducible null mice manifest with a spectrum of PKD severity depending on the temporal/spatial onset of PC loss triggered by various Cre recombinases.13,38 These models are valuable for preclinical testing, mimic the proposed 2-hit mechanism in human ADPKD,39,40 and allow phenotypic evaluation of Pkd1/Pkd2 loss in other organs.17
Biallelic ADPKD models can be viable if at least 1 allele is hypomorphic. The Pkd2WS25 hypermutable allele results in somatic loss of wild-type polycystin-2 and slowly progressive PKD and polycystic liver disease in the Pkd2WS25/— model.40 The Pkd1nl and Pkd1V alleles are splicing or G-protein coupled receptor proteolytic site cleavage mutants, respectively, generating only a small fraction of functional PC141,42; homozygous models develop rapidly progressive cystic kidney expansion with early lethality (P14–P60). These models mimic a dosage model with reduced functional PC1 in all cells, similar to patients with PKD1, and enable the analysis of phenotypes beyond the kidney.16,43 Pkd1nl/nl mice in the F1(B6 × 129Ola/Hsd) background have longer survival (with rapid kidney size expansion followed by increased fibrosis and size reduction) compared to a mixed background, illustrating modifying effects also with these models.27,41,44
The PKD1 variant p.Arg3277Cys (RC) in humans is hypomorphic and associated with typical ADPKD in homozygosity, monoallelically with just a few cysts, and causes very early onset ADPKD in trans with a second pathogenic allele.45–47 A mouse mimicking this variant (Pkd1RC/RC) is viable to at least 1 year with slowly progressive PKD, whereas Pkd1RC/−mice have very early onset PKD with median death at ~P25.43 The combination of Pkd1RC with a floxed allele (Pkd1RC/fl) and Ksp-Cre results in an intermediate phenotype.48 PC1RC has a mild G-protein coupled receptor proteolytic site cleavage defect but also folds and traffics inefficiently, with only ~40% of mature and localized protein.43 Digenic models—Pkd1RC/RC with Pkd2WS25/− or with an autosomal recessive PKD model (Pkhd1−/−; with very mild PKD)—have synergistic increases in kidney disease severity.49,50 The Pkd1RC/RC model in the B6 background has a milder phenotype than does the model in the original mixed background, but it has been used extensively for preclinical testing.27,41,44,48,51–59
Here, we characterize the Pkd1RC/RC phenotype in 3 fully inbred backgrounds and 2 F1 combinations. These studies illustrate that the genetic background significantly modifies the kidney phenotype and describe an improved protocol for preclinical testing.
METHODS
Study approval
Experiments were conducted under the Institutional Animal Care and Use Committee protocols: A56913, A3539, and A24015.
Generation of inbred Pkd1RC/RC mice and F1 animals
B6 Pkd1RC/RC mice were inbred (>99%) into the 129S6/SvEvTac (129) or BALB/cJ (BC) backgrounds by using speed congenics, with the level of each background determined per generation by using microsatellite analysis (Idexx BioAnalytics). These mice were bred with inbred B6 Pkd1RC/RC mice to generate F1 progeny, F1(129/B6) or F1(BC/B6). Mouse housing and diet are described in Supplementary Methods.
Generation of the study cohort
Mice from the 5 backgrounds were killed and analyzed at 1, 3, 6, 9, and 12 months for the adult time points, or P1, P4, P6, P12, and P18 for the perinatal time points. We strove to obtain n = 5 of each sex per background for the adult time points and n = 3 for the perinatal time points. Where possible, the same animals were analyzed for the histological and biochemical endpoints; some substitutions were used where material was depleted or extra numbers were helpful (see Table 1). Differences in group sizes were not due to mortality. For the total kidney volume (TKV) analysis, different mice were used for each time point and were not the same as those used for the histological analysis.
Table 1 |.
Progression of cystic disease in Pkd1RC/RC mice across each genetic background
| Endpoint | B6 | BC | F1(BC/B6) | 129 | F1(129/B6) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| KW/BW, % | M | F | M | F | M | F | M | F | M | F | |||||
| 1 mo | 5 | 5 | 1.62 ± 0.13 | 5 | 5 | 2.58 ± 0.93d | 5 | 5 | 1.96 ± 0.33 | 5 | 5 | 2.45 ± 1.07 | 5 | 5 | 1.80 ± 0.32 |
| 3 mo | 5 | 5 | 1.85 ± 0.26 | 5 | 5 | 3.24 ± 0.86f | 5 | 5 | 1.92 ± 0.22 | 5 | 5 | 3.08 ± 0.84e | 6 | 4 | 2.64 ± 1.06 |
| 6 mo | 5 | 5 | 1.98 ± 0.28 | 5 | 5 | 3.73 ± 0.58a.f | 5 | 5 | 2.40 ± 0.37 | 5 | 5 | 4.93 ± 1.89c.f | 3 | 6 | 3.59 ± 1.01c.f |
| 9 mo | 5 | 5 | 2.24 ± 0.26a | 5 | 5 | 3.98 ± 0.96b.f | 5 | 5 | 3.60 ± 0.63c.e | 5 | 5 | 4.10 ± 0.88b.f | 5 | 5 | 2.90 ± 1.24a |
| 12 mo | 5 | 5 | 3.37 ± 1.06c | 5 | 5 | 4.10 ± 1.14b | 5 | 5 | 3.42 ± 0.49c | 5 | 5 | 4.27 ± 0.51b | 6 | 4 | 2.45 ± 0.71 |
| Cystic area, % | |||||||||||||||
| 1 mo | 5 | 5 | 3.13 ± 1.10 | 3 | 5 | 18.07 ± 5.59f | 4 | 5 | 5.10 ± 5.10 | 5 | 5 | 15.56 ± 6.72f | 5 | 5 | 7.02 ± 4.00 |
| 3 mo | 5 | 5 | 4.20 ± 2.15 | 5 | 5 | 18.15 ± 6.06f | 5 | 5 | 9.87 ± 5.49 | 5 | 4 | 26.48 ± 14.37a.f | 6 | 3 | 8.46 ± 3.73 |
| 6 mo | 5 | 5 | 4.83 ± 3.61 | 5 | 5 | 23.23 ± 4.71f | 4 | 5 | 9.33 ± 3.44 | 5 | 5 | 30.23 ± 10.20b.f | 2 | 6 | 17.31 ± 4.64b,f |
| 9 mo | 4 | 4 | 5.54 ± 3.55 | 4 | 5 | 21.02 ± 2.47f | 5 | 5 | 17.57 ± 3.99c,f | 4 | 5 | 25.14 ± 4.20f | 5 | 5 | 12.06 ± 7.28d |
| 12 mo | 5 | 5 | 14.43 ± 8.02c | 5 | 4 | 27.35 ± 10.75a,f | 5 | 5 | 16.02 ± 4.29c | 4 | 5 | 18.01 ± 5.64 | 6 | 4 | 16.95 ± 8.36c |
| Cyst number | |||||||||||||||
| 1 mo | 5 | 5 | 17 ± 8 | 3 | 5 | 94 ± 33d | 4 | 5 | 35 ± 22 | 3 | 5 | 62 ± 22 | 5 | 5 | 24 ± 9 |
| 3 mo | 3 | 4 | 19 ± 6 | 5 | 5 | 148 ± 55f | 4 | 5 | 62 ± 30 | 5 | 4 | 93 ± 27d | 4 | 5 | 54 ± 35 |
| 6 mo | 5 | 5 | 44 ± 38 | 5 | 5 | 205 ± 34c,f | 4 | 5 | 106 ± 57b,d | 5 | 5 | 166 ± 49c,f | 2 | 6 | 87 ± 26 |
| 9 mo | 2 | 4 | 75 ± 36c | 4 | 6 | 288 ± 81c.f | 4 | 5 | 216 ± 42c,f | 2 | 6 | 221 ± 46c,f | 5 | 5 | 116 ± 46b,e |
| 12 mo | 5 | 5 | 80 ± 30c | 5 | 5 | 197 ± 51b,f | 5 | 5 | 213 ± 44c,f | 4 | 5 | 201 ± 54c,f | 6 | 4 | 170 ± 67c,f |
| BUN, mg/dl | |||||||||||||||
| 1 mo | 5 | 5 | 17.7 ± 3.0 | 3 | 5 | 21.3 ± 4.0 | 5 | 5 | 16.4 ± 3.4 | 5 | 5 | 19.0 ± 4.2 | 3 | 3 | 15.5 ± 2.3 |
| 3 mo | 4 | 5 | 24.1 ± 1.6 | 5 | 5 | 20.5 ± 4.5 | 5 | 5 | 18.0 ± 3.3 | 5 | 5 | 23.0 ± 4.2 | 7 | 2 | 23.2 ± 2.6a |
| 6 mo | 5 | 5 | 24.0 ± 2.8 | 2 | 5 | 23.4 ± 3.3 | 5 | 5 | 18.6 ± 4.1 | 5 | 5 | 27.3 ± 4.9b | 6 | 2 | 30.4 ± 3.3b |
| 9 mo | 4 | 3 | 24.6 ± 5.2 | 5 | 5 | 32.1 ± 7.6 | 5 | 5 | 21.0 ± 4.5 | 3 | 5 | 30.5 ± 5.0c | 3 | 4 | 24.2 ± 3.5a |
| 12 mo | 5 | 5 | 30.5 ± 13.7c | 5 | 5 | 36.5 ± 16.7b | 5 | 5 | 23.0 ± 5.6b,d | 4 | 5 | 30.4 ± 8.1c | 7 | 2 | 35.0 ± 9.1c |
| Fibrotic area, (%) | |||||||||||||||
| 1 mo | 3 | 4 | 1.18 ± 0.78 | 3 | 3 | 1.10 ± 0.74 | 3 | 3 | 0.60 ± 0.44 | 3 | 5 | 0.65 ± 0.53 | 3 | 3 | 0.19 ± 0.15 |
| 3 mo | 3 | 3 | 1.16 ± 0.71 | 3 | 3 | 1.18 ± 1.57 | 3 | 4 | 0.58 ± 0.43 | 3 | 5 | 1.47 ± 0.52 | 3 | 2 | 1.34 ± 0.55 |
| 6 mo | 3 | 3 | 1.59 ± 0.63 | 3 | 3 | 2.37 ± 0.96 | 3 | 3 | 1.06 ± 0.71 | 5 | 5 | 3.45 ± 1.77 | 3 | 3 | 0.98 ± 1.06 |
| 9 mo | 3 | 3 | 4.23 ± 2.01b | 3 | 3 | 4.42 ± 4.05 | 3 | 3 | 5.02 ± 3.57a | 5 | 5 | 6.09 ± 2.50c | 3 | 4 | 3.44 ± 2.57 |
| 12 mo | 3 | 3 | 10.46 ± 2.91c | 3 | 4 | 8.64 ± 3.98c | 3 | 3 | 6.29 ± 3.96b,d | 5 | 5 | 14.17 ± 4.72c,d | 7 | 2 | 5.82 ± 4.17b,e |
| cAMP, pmol/mg protein | |||||||||||||||
| 1 mo | 5 | 5 | 6.96 ± 3.97 | 3 | 5 | 3.83 ± 1.70 | 5 | 4 | 9.57 ± 1.86 | 4 | 3 | 13.17 ± 5.22 | 5 | 5 | 7.34 ± 2.10 |
| 3 mo | 5 | 5 | 5.72 ± 2.10 | 5 | 5 | 3.81 ± 2.30 | 5 | 5 | 7.21 ± 1.29 | 3 | 3 | 14.26 ± 5.67d | 5 | 5 | 4.29 ± 0.94 |
| 6 mo | 5 | 4 | 7.90 ± 2.68 | 5 | 5 | 8.82 ± 5.06 | 4 | 5 | 9.71 ± 5.32 | 3 | 2 | 31.48 ± 10.71a,f | 4 | 5 | 10.35 ± 4.46 |
| 9 mo | 4 | 4 | 16.49 ± 6.38c | 5 | 5 | 11.56 ± 2.2a | 5 | 5 | 15.10 ± 5.81 | 4 | 3 | 45.31 ± 12.75c,f | 3 | 4 | 11.25 ± 4.79a |
| 12 mo | 2 | 5 | 18.40 ± 6.49c | 5 | 5 | 23.08 ± 10.13c | 5 | 5 | 19.02 ± 10.26b | 1 | 3 | 38.02 ± 12.75b,f | 7 | 2 | 7.76 ± 2.03 |
| TKV, mm3 | |||||||||||||||
| 1 mo | 3 | 7 | 442 ± 92 | 2 | 1 | 1022 ± 224e | 0 | 1 | 403 | 5 | 5 | 597 ± 201 | 5 | 5 | 448 ± 98 |
| 3 mo | 2 | 8 | 603 ± 145 | 1 | 2 | 1375 ± 247f | 3 | 4 | 735 ± 177 | 5 | 5 | 1175 ± 431c,f | 5 | 5 | 999 ± 267c,f |
| 6 mo | 3 | 7 | 745 ± 196a | 1 | 1 | 1225 ± 169 | 0 | 1 | 1021 | 2 | 4 | 1683 ± 165c,f | 5 | 5 | 1060 ± 410c,d |
| 9 mo | 5 | 4 | 750 ± 100a | 0 | 1 | 2122 | 0 | 0 | NA | 2 | 5 | 1876 ± 303c,f | 5 | 5 | 1479 ± 239c,f |
| 12 mo | 2 | 5 | 919 ± 163c | 0 | 0 | NA | 0 | 0 | NA | 2 | 1 | 1660 ± 273c,f | 6 | 4 | 1312 ± 269c,e |
129, 129S6/SvEvTac; ANOVA, analysis of variance; B6, C57BL/6J; BC, BALB/cJ; BUN, blood urea nitrogen; cAMP, cyclic adenosine monophosphate; KW/BW, kidney weight/body weight; NA, not available; Pkd1RC/RC mice, mice homozygous for the PKD1 hypomorphic allele p.Arg3277Cys; TKV, total kidney volume.
One-way ANOVA followed by Dunnett’s multiple comparison test compared to 1-month mice within each background:
P < 0.05,
P < 0.01,
P < 0.001. Two-way ANOVA followed by Dunnett’s multiple comparison test indicating significant difference from age-matched B6 mice:
P < 0.05,
P < 0.01,
P < 0.001. Numbers of male (M) and female (F) mice for each analysis are shown. Statistical analysis was performed only on groups that contained n ≥ 3.
Histological, immunofluorescence, biochemical, and magnetic resonance imaging analysis
These methods were performed as previously outlined43,50,60,61 and described in detail in Supplementary Methods.
Preclinical trial screening of F1(129/B6) mice
Twelve male and female mice per group were scanned by magnetic resonance imaging (MRI) at 3 weeks, treated with sham or tolvaptan for 10 weeks, and scanned by MRI before killing and histological analysis at 13 weeks. Mice were randomized into treatment groups on the basis of the initial TKV that was adjusted for animal length (LnTKV) and sex to ensure similar baseline cystic phenotypes across each treatment group. Siblings were also distributed as evenly as possible between the treatment groups. Tolvaptan (Carbosynth China Limited) was dissolved in dimethylsulfoxide at a final concentration of 37.5%. Hydroxypropyl methylcellulose (1%) was added to tolvaptan and animals were gavaged with 100 μl/10 g of body weight (BW) to obtain a final dose of 75 mg/kg every morning. Mice were also fed 45 g of food (Labdiet 5053; PMI LabDiet)/100 g of BW, with tolvaptan mixed at a concentration of 0.1% in the evening. Gavage as well as the drug added to food was designed to ensure tolvaptan was active in mice for a 24-hour period, as monitored by urine output.
Statistical analysis
Statistical measurements were obtained using PRISM 8 (GraphPad Software, Inc.). See figure legends for specific tests used for each experimental data set. Statistical analysis was performed only on groups with n ≥ 3.
RESULTS
Cystic kidney disease progression in inbred B6, BC, and 129 Pkd1RC/RC mice
B6 Pkd1RC/RC mice were fully inbred into the BC and 129 backgrounds and the phenotypes compared. Compared to the B6 background, by 1 month, BC mice had a greater percent cystic area (CA)/total kidney area, and cyst number (counted in histological sections), that was maintained up to 12 months, while percent kidney weight/BW (KW/BW) was greater up to 9 months (Figures 1 and 2a–c and Table 1). Similarly, 129 mice had more severe cystic disease than did B6 mice, with greater KW/BW (3–9 months), CA (1–9 months), and cyst number (3–12 months), although the expansive stage peaked by 6 to 9 months. Analysis within backgrounds also showed more rapid disease progression in the BC and 129 lines than in B6 (Figure 2a–c). Fibrotic area was greater at 9 or 12 months from 1 month in all backgrounds, but only the 129 strain differed from B6, at 12 months (~14%), a time corresponding to declining CA (Figure 2b and d and Table 1). Blood urea nitrogen, measuring kidney function, was elevated by 6 months in the 129 background but only by 12 months in the BC and B6 lines (Figure 2e and Table 1). The cAMP concentration in kidney tissue, which is often elevated in PKD,62 was increased in the 129 line by 6 months (highest level overall) and in the BC and B6 lines by 9 months (Table 1; Supplementary Figure S1A). Previously in the B6 background, the disease was determined to be significantly more severe in female mice than male mice by 9 months.63 Here, no significant differences between the sexes were observed in the structural (Figure 3a–d) and functional (Figure 3e) outcomes before 6 months (Supplementary Table S1), and the tendency for more severe disease in female mice was less dramatical or not present in the BC and 129 lines.
Figure 1|. Cystic kidney disease progression differs in Pkd1RC/RC mice in various genetic backgrounds.
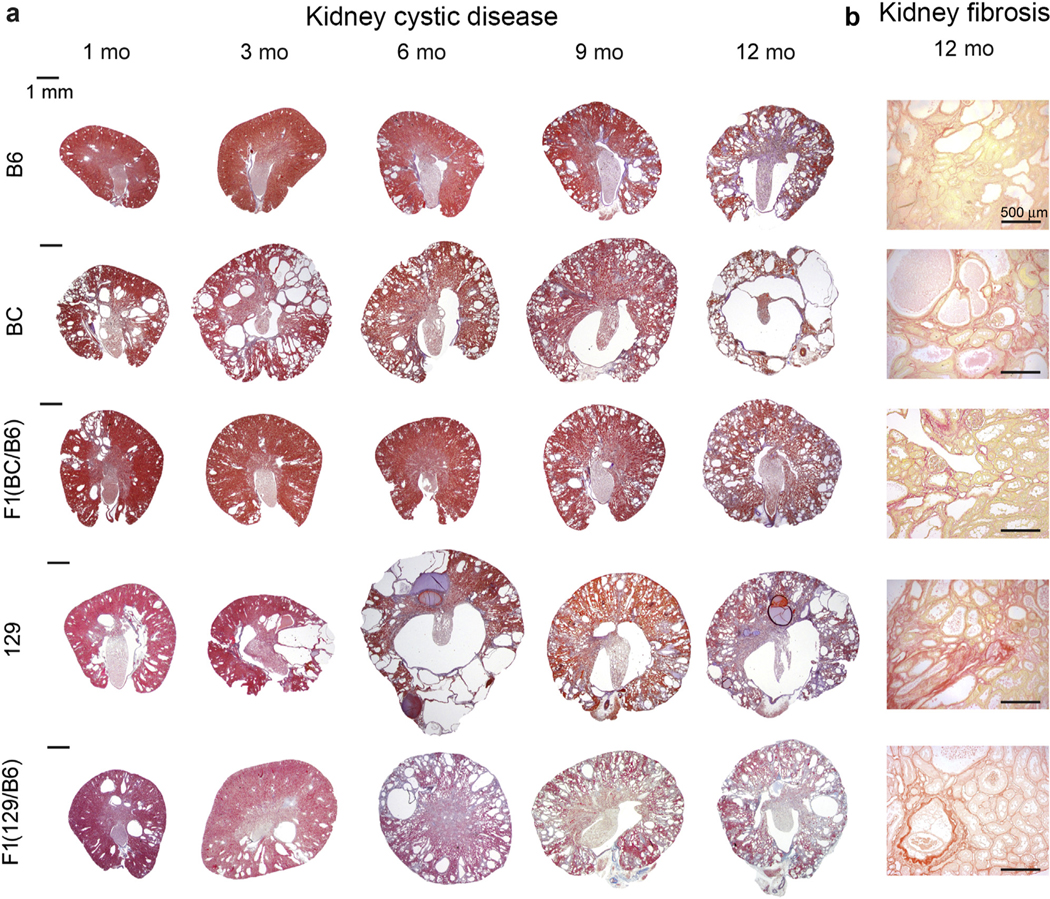
(a) Masson’s trichrome–stained kidney sections of mice at 1, 3, 6, 9, and 12 months of age showing the varying cystic disease progression between the lines. Bars = 1 mm. (b) Kidney sections of 12-month mice stained with picrosirius red illustrating fibrosis in the different lines. Bars = 500 μm. B6, C57BL/6J; BC, BALB/cJ; 129, 129S6/SvEvTac. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Figure 2 |. Characterization of the different rates of cystic kidney disease progression in Pkd1RC/RC mice in various genetic backgrounds.

(a) Percent kidney weight/body weight (KW/BW), (b) percent cystic area, (c) cyst number, (d) percent fibrotic area, and (e) blood urea nitrogen (BUN) were determined in mice from each background that were killed at 1, 3, 6, 9, and 12 months (see Table 1 for group sizes). The mean ± SD are shown on each scatter plot. Change in cystic disease within each background compared to the 1-month baseline was performed using a 1-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test within each genetic background: *P < 0.05, **P < 0.01, ***P < 0.001. In addition, each time point in each background was compared to corresponding B6 mice to determine differences in cystic disease by using a 2-way ANOVA followed by Dunnett’s multiple comparison test: †P < 0.05, ††P < 0.01, †††P < 0.001. Where possible, the same animals were measured for each endpoint but not all groups had equal numbers of male and female mice (Table 1; see Figure 3 and Supplementary Tables S1 and S2 for sex-specific data). B6, C57BL/6J; BC, BALB/cJ; 129, 129S6/SvEvTac.
Figure 3 |. Background contributes to sex effects on cystic kidney disease progression.

(a) Percent kidney weight/body weight (KW/BW), (b) percent cystic area, (c) cyst number, (d) percent fibrotic area, and (e) blood urea nitrogen (BUN) were determined for male and female mice from each background that were killed at 6, 9, and 12 months (see Table 1 for numbers of male and female mice for each endpoint). Two-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison test was performed between male and female groups for age-matched mice of each genetic background: *P < 0.05, **P < 0.01, ***P < 0.001. Each time point in each background was also compared to corresponding B6 mice to determine differences per sex in cystic disease by using a 2-way ANOVA followed by Dunnett’s multiple comparison test: †P < 0.05, ††P < 0.01, †††P < 0.001 (see Supplementary Table S1 for details of younger animals). Statistical analysis was performed only on groups that contained N ≥ 3, and groups not analyzed are marked with X. B6, C57BL/6J; BC, BALB/cJ; 129, 129S6/SvEvTac.
Disease progression in F1 Pkd1RC/RC models: BC × B6, F1(BC/B6) and 129 × B6, F1(129/B6)
We hypothesized that F1 mice combining the more rapidly progressing BC or 129 and milder B6 backgrounds would result in an intermediate phenotype with less variability due to heterozygosity at loci that differ between the parental backgrounds. For the structural endpoints, this was true for both F1 lines, but with a tendency for both to be more similar to B6; fibrotic area was smaller than that in either parental background (Figures 1 and 2a–d and Table 1). Interestingly, although the cAMP level remained low in the F1(129/B6) line, blood urea nitrogen was statistically elevated at all times compared with the 1-month time point (Figure 2e and Table 1; Supplementary Figure S1A). A significant sex difference, with female disease being more severe, was seen for both F1 lines evident by several endpoints and for the F1(129/B6) line sometimes even more marked than for the B6 line (Figure 3a–e; Supplementary Figure S1B and Supplementary Table S1). Because of the marked effect of sex on disease progression, a comparison of all outcomes by sex in each background was performed and significant differences from 1-month values in the same sex are shown (Supplementary Table S2).
Analysis of liver disease in different Pkd1RC/RC backgrounds
In previous publications, dilated and duplicated bile ducts and fibrosis were found earlier in the B6 line (3 months) than in the original mixed background (12 months).43,63 Analysis here showed biliary abnormalities in all 5 backgrounds by 6 months, with the first evident at 3 months (BC and 129), and a tendency to be more widespread in the 129 line (not statistically significant) (Figure 4a and b). The nature of the abnormality appeared to differ between the lines, with more evidence of dilation in the BC and F1(BC/B6) lines as compared with more bile duct proliferation in the 129 and F1(129/B6) lines (Figure 4a).
Figure 4 |. Characterization of the liver phenotypes in Pkd1RC/RC mice observed in different backgrounds.
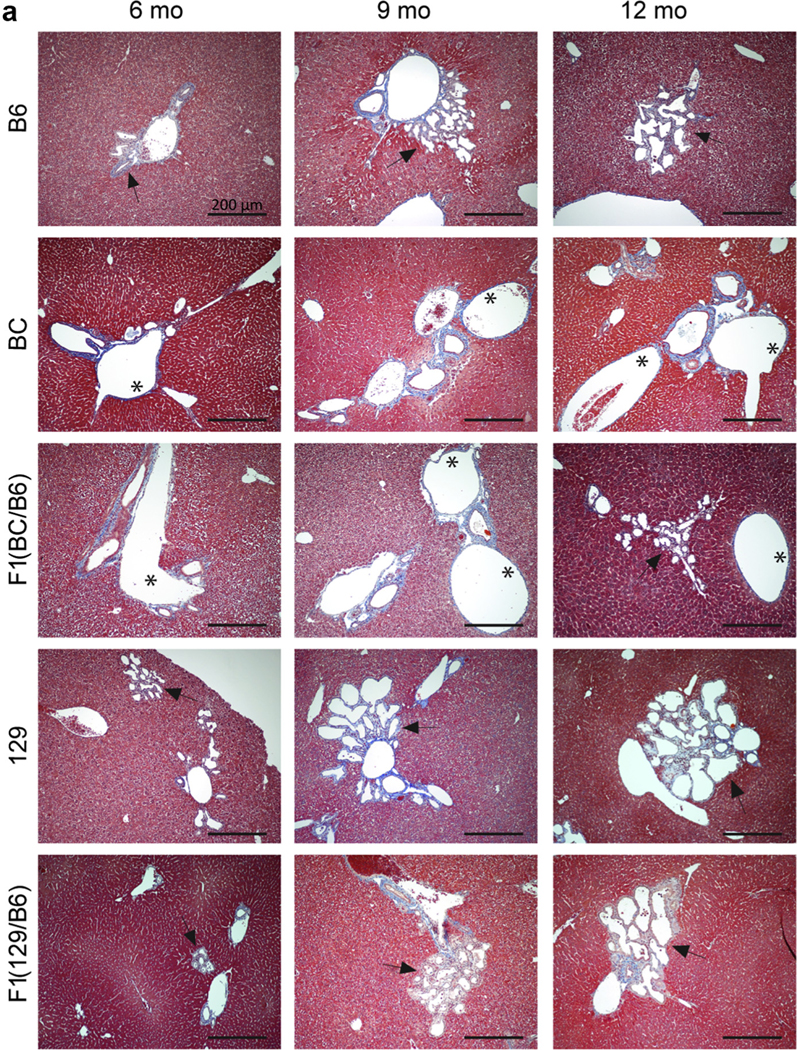

(a) Masson’s trichrome–stained liver sections of mice at 6, 9, and 12 months of age showing the biliary abnormalities in various lines. Asterisks indicate dilated bile ducts, and arrows indicate bile duct proliferation. Bar = 200 μm. (b) Incidence of biliary dysgenesis per animal in each background at 3, 6, 9, and 12 months. There was no evidence of biliary dysgenesis in any background at 1 month. Liver dysgenesis at each time point and background was compared to corresponding B6 mice by using a 2-way analysis of variance followed by Dunnett’s multiple comparison test, but no statistical significance was evident. B6, C57BL/6J; BC, BalbC/cJ; 129, 129S6/SvEvTac. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Disease progression in perinatal mice
We previously observed that the Pkd1RC/RC model exhibited an early burst of proximal tubule cysts (by P1) whereas collecting duct (CD) cysts, the predominant type in the adult, were more common by P12.43,63 Here, predominantly proximal tubule cysts contributed to greater CA at P1, which declined in all lines by P6 to P12 (Supplementary Figure S2A–D). CA in the BC line, and less pronounced in the 129 line, began to increase after P6, accompanied by an increase in CD cysts (Supplementary Figure S2A–D). The BC, 129, and F1(129/B6) lines had predominantly CD cysts by P18, whereas the others had an increase in both unstained (non–proximal tubule or non-CD) and CD cysts (Supplementary Figure S2C and D). Analysis of proliferating cells in cyst linings at 5 time points between P1 and P18 showed more proliferation at P1, when the CA tended to be greatest, than at P18 in all backgrounds, with overall similarities in the profile for both measures (Supplementary Figures S2B and S3A and B). Even at P18, the proliferation level in each background tended to be higher than the corresponding wild-type data, although this was not significant [F1(129/B6) not available] (Supplementary Figure S3). However, some exceptions between CA and proliferation rate were seen. For the 129 line at P1, only low proliferation was seen despite the large CA, and in the BC line, by P18 the expanding CA was not reflected in increased proliferation (Supplementary Figures S2B and S3B).
MRI to measure disease severity
During the course of the study, we introduced MRI to analyze the severity of disease throughout both kidneys in live animals.60 The foci of these analyses were the B6, 129, and F1(129/B6) lines (Figure 5a), because small litter sizes and problems with breeding homozygous BC mice made this model and the F1(BC/B6) line less attractive. However, examples of images of the BC (1–9 months) and F1(BC/B6) (1– 6 months) lines are provided (Supplementary Figure S4A). TKVs were calculated from MR images to quantify cystic disease,61 a key means to follow disease progression in patients with early-stage ADPKD (Figure 5a and b and Table 1; Supplementary Figure S4A and B and Supplementary Table S1).64 The more rapidly progressive nature of the cystic disease in the 129 and F1(129/B6) (and BC) lines than B6 is illustrated by a greater TKV at 3 months that is maintained to 12 months. The TKV increase is progressive up to 9 months in the 129 and F1(129/B6) lines and then declines, paralleling the histological data (Figures 1, 2a–d, and 5a and b, and Table 1).
Figure 5 |. Magnetic resonance (MR) imaging of the kidney from live Pkd1RC/RC mice in different backgrounds from 1 to 12 months.

(a) Representative kidney coronal sections in C57BL/6J (B6), 129S6/SvEvTac (129), and F1(129/B6) Pkd1RC/RC mice at 1, 3, 6, 9, and 12 months. Note that different animals were imaged at each time point. The arrow indicates urine retention, and total kidney volume (TKV, in cubic millimeters) is shown. Bars = 1 cm. (b) Quantification of TKVs from MR images. Significance of the difference within each background compared to 1 month baseline was determined using a 1-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test: *P < 0.05, **P < 0.01, ***P < 0.001. Significance of the TKV difference from age-matched B6 mice was determined using a 2-way ANOVA followed by Dunnett’s multiple comparison test: †P < 0.05, ††P < 0.01, †††P < 0.001. Statistical analysis was performed only on groups that contained N ≥ 3. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
MRI analysis of the 129 line revealed mild hydronephrosis (HN) from 3 months, with apparently mild HN sometimes seen in the BC background (Figure 5a; Supplementary Figure S4A), clearly contributing to TKV. This phenotype could also be seen in 129 (and BC) kidney sections as an expanded pelvis to ureter junction and as an apparent loss of medullary tissue (Figure 1). We also noticed significant inter-kidney variability in individual Pkd1RC/RC 129 mice, possibly HN related, and illustrated as extensive localized polar cystic disease (Supplementary Figure S5A). This variability resulted in a greater difference between the volume of right and left kidneys in the 129 than the F1(129/B6) model, although there was no clear preference for larger left or right kidneys (Supplementary Figure S5A–C). This inter-kidney variability may account for some of the extreme minimum and maximum of TKV, KW/BW, and CA seen in the 129 line (Figures 2a and b and 5b and Table 1).
Optimizing the Pkd1RC/RC model for preclinical testing
Because of slow disease progression, preclinical studies in the B6 Pkd1RC/RC model have usually lasted at least 5 months, and sometimes only started at 9 months, making them expensive and with possible sex differences.43,48,53–59,63 The more aggressive disease in the 129 and BC lines would potentially enable more rapid preclinical testing, but mild HN, breeding issues (BC), and inter-kidney and inter-mouse variability (129) are problems complicating obtaining efficacious outcomes. Therefore, we selected the F1(129/B6) strain because of the ease of breeding, lack of mild HN/extreme inter-animal/inter-mouse kidney variability, and relatively rapid disease progression from 1 month (Figures 1, 2a–c, and 5a and b; Supplementary Figure S5B and C).
We therefore designed a study to test tolvaptan against a vehicle control in the F1(129/B6) line. The study started at 3 weeks and extended for 10 weeks until 13 weeks. Group sizes were 12 male and 12 female mice, and tolvaptan was added to the food and also given daily by gavage (see Methods for details). To ensure that the treatment and control groups were matched, mice were analyzed by MRI before the study and the initial TKV was calculated and the cystic appearance of the kidneys observed. TKV was adjusted for animal length (LnTKV, similar to height-adjusted TKV used in human studies),11,65,66 which was found to correlate slightly better than TKV with the KW/BW and CA endpoints (Supplementary Figure S6A and B). As expected, KW and TKV and length-adjusted KW and LnTKV also correlated well (Supplementary Figure S6C and D). Mice that had very severe (largest ~3% of LnTKV) or very mild (smallest ~10% LnTKV) disease were excluded from the study to remove very slowly progressing animals and decrease variability within the treatment groups. Mice were randomized by initial LnTKV per sex (Figure 6a and b), appearance of the kidneys, and originating litter.
Figure 6 |. Preclinical testing of tolvaptan in F1(129/B6) Pkd1RC/RC mice.


(a) Magnetic resonance images at the start (3 weeks) and end (13 weeks) of the study in the sham and tolvaptan-treated groups (same animals at both time points). Bars = 1 cm. (b) Initial total kidney volume (TKV) adjusted for animal length (LnTKV) (3 weeks) in the sham and tolvaptan-treated groups. Corresponding final data (13 weeks) for the endpoints: (c) LnTKV, (d) percent change in LnTKV, (e) percent kidney weight/body weight (KW/BW), (f) cyst volume, (g) cyst number, (h) percent cystic area, (i) blood urea nitrogen (BUN), and (j) cyclic adenosine monophosphate (cAMP) for the sham and tolvaptan-treated groups (see also Supplementary Table S3). Significant difference between the sham and tolvaptan-treated groups was determined using an unpaired t test: *P < 0.05, **P < 0.01, ***P < 0.001. B6, C57BL/6J; 129, 129S6/SvEvTac. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
The endpoints that were significant included LnTKV, percent change in LnTKV (%ΔLnTKV), and KW/BW (Figure 6a and c–e; Supplementary Table S3). The final LnTKV was 25.5% smaller in tolvaptan-treated mice, the KW/BW differed by 20.3%, and the overall difference in % ΔLnTKV was 49.6%. The importance of randomization and of including mice with significantly severe disease was illustrated by the strong correlation of the initial LnTKV and final LnTKV (Supplementary Figure S7A) and a greater response to treatment of mice with a larger initial LnTKV (Supplementary Figure S7B). There was a trend for smaller CA in the treated group, but this was not significant (Figure 6h); however, a calculated cystic volume (TKV × CA) was significant (Figure 6f), as was cyst number (Figure 6g). Interestingly, even in these young animals, blood urea nitrogen and cAMP were also significantly lower in the treated group (Figure 6i and j).
Analysis of sex differences in these younger animals showed that although initial BW and LnTKV did not differ, final BW and %ΔLnTKV were greater in male mice in the sham and treatment groups and LnTKV and cystic volume in the tolvaptan group (Supplementary Figure S8A–H and Supplementary Table S4). KW/BW and cyst number did not differ by sex, but the treatment effect was significant only in female mice (Supplementary Figure S8G and I). The greater size increase in male mice detected here contrasts with the more severe female disease seen in older F1(129/B6) mice, but reflects (nonsignificant) TKV data seen in younger F1(129/B6) animals (Figures 3a–e and 5b; Supplementary Table S1). Overall, tolvaptan was more effective in female mice than male mice (Supplementary Figure S8D and F–I and Supplementary Table S4). This may be partly due to more variability in sham-treated male mice than female mice, although we note in humans that %ΔTKV/yr between the tolvaptan and placebo groups was greater in females than males (nonsignificant).6
DISCUSSION
The Pkd1RC/RC model reflects the progressive disease in human ADPKD. Mice are viable to >12 months, generally breed well as homozygotes (avoiding genotyping), and have a global reduction of functional PC1 in every cell.43 Genetic background makes a very significant difference to kidney disease severity, analogous to the intrafamilial variability seen in human ADPKD or patients with the same mutation (or mutation type).11,63 By using different backgrounds (or digenic combinations), many questions about disease pathogenesis, including factors controlling the expansive and fibrotic stages, identifying disease modifiers, and preclinical testing, can be addressed.43,48–50 The detailed description of the phenotype and the protocol for preclinical testing bring novelty to the study and illustrate the practical value of the model.
We found the B6 background to have the most slowly progressive disease, consistent with other cystic models.35 This may be related to resistance to kidney damage/injury,33 factors known to play a role in PKD progression.67 Because the disease severity in the Pkd1RC/RC model was clearly different by 1 month between B6 and 129 or BC, genetic variants that subtly change prenatal and early postnatal kidney development, such as proximal tubule differentiation, CD tubule elongation, and tubule segmentation, may be significant.68,69 In human ADPKD, in utero/infantile kidney growth is an important factor influencing disease severity.11 The BC and 129 models have more cysts than does B6 at all time points, including at early disease stages.19 One background-associated manifestation that may be contributing to disease severity, mild HN, was seen in the 129 and, to some extent, the BC line. It is unclear what genetic factors contribute to this phenotype, but mutations in a number of genes resulting in dysregulation of hedgehog signaling cause more severe HN,70–75 and ADPKD mouse models (and humans) also have decreased urine osmolality and higher urine output.76,77 However, it is difficult to know the mechanism(s) by which genetic background is influencing Pkd1RC/RC kidney disease severity, with many pathways and processes, cAMP, intracellular Ca2+, and factors controlling proliferation, secretion, immune response, metabolism, and ciliary function/structure, to name but a few, implicated in PKD pathogenesis.12,13 The background-related differences provide an opportunity via genome analyses of F2 animals to map and identify these modifying factors, the only way to determine which mechanisms are significant in this model—mechanisms that may also be important in human disease.78 The ease of next generation sequencing and available genomic sequences make this a less daunting task than was once the case.
Later disease progression also differed between the backgrounds. In the B6 line, cystic expansion occurred for 12 months but in the 129 line and, to some extent, BC and F1 lines, the expansive phase plateaued by 6 to 9 months, often declining thereafter. Human ADPKD is characterized by decades of exponential TKV growth with questions as to whether this slows as end-stage kidney disease approaches.11,64 The end of the expansive stage was accompanied by an increase in fibrosis, a change that also characterizes later stage human ADPKD.79 The length of the expansive stage and timing of the switch to more fibrosis is associated with disease severity. In models with a low level of functional PC1, such as Pkd1nl/nl and Pkd1RC/−, the expansive stage ends within the first month, with surviving mice having dramatic declines in kidney size and high levels of fibrosis, similar to progression in human autosomal recessive PKD.12,43,44 The mainly expansive disease in human ADPKD probably reflects the only 50% reduction of functional PC1 associated with a fully penetrant PKD1 mutation. It is unclear whether our observed background severity differences are directly associated with the level of functional PC1 or due to modifiers working in other ways.
Male mice have more severe disease in human ADPKD, but in our published studies, the B6 model had significantly more severe disease in older female mice.11,63 Here, although there was a similar tendency in older 129 and BC animals, this was often not significant. However in both F1 lines, the B6 sex difference was evident, suggesting “dominant” transmission of this trait. Interestingly, in younger F1(129/B6) mice used for the preclinical trial, the predominant sex influence was greater kidney growth in male mice. Inflammatory pathways and cAMP signaling, pathways affected in ADPKD, are known to affect sex differences,80–83 and in inducible Pkd1fl/fl lines with very early onset PKD, more severe male disease has been linked to differences in lipid metabolism.84 However, what hormonal, developmental, or other factors are specifically driving the background-related sex differences is presently unclear.
The significant phenotypic variability in Pkd1RC/RC mice was observed even in the same sex and background, and overall there was more variability between animals as they manifested more severe disease. Even within inbred lines, there is some genetic variability, including around the disease locus where single-nucleotide polymorphisms may persist even after multiple rounds of inbreeding. At other loci, homozygosity may be lethal, and there can be tissue selection for particular alleles.85–87 Continued inbreeding of homozygotes can lead to phenotypic drift, most often to a fitter, milder phenotype by selection within the residue variability in the founder animals.88 We have outbred lines every 5 to 10 generations to try to limit this drift. When a limited number of animals of the B6, BC, and 129 lines were shipped to establish new colonies, including a round of backcrossing onto wild-type mice of the same background, in each case the cystic kidney phenotype became more severe.54 This is presumably because of founder effects, although subtle differences in diet and housing, and perhaps epigenetic differences, may also play a role. Variability was also evident in the F1 lines, even though loci differing in the parental lines should be heterozygous in the F1 line. One explanation is comparative transcript expression, with studies in F1(B6/DBA) mice showing variable inter-tissue expression of transcripts from each parental background and allelic bias at 41% of loci in at least 1 tissue.86 Although there is a modulating effect of quite widespread heterozygosity, F1 lines may not simply have an intermediate of both parental mice, with dominance of the B6 alleles in both of our F1 lines. Stochastic factors and vulnerabilities in some backgrounds to kidney damage and even structural differences can contribute to variably within isogenic lines.33 This is most clear for the 129 line where superimposed mild HN seemed to significantly influence variability, including inter-kidney disease severity. Once localized cyst formation and expansion takes hold, positive feedback by secretion of proexpansion factors from existing cysts may also promote the growth of adjacent cysts.89
For decades-long progressive diseases such as ADPKD, the goal for preclinical testing is to model progressive disease that allows trial completion within a few months; hence, a balance between disease severity and variability is essential. The variability seen in the Pkd1RC/RC model, especially in some backgrounds, seems common in PKD models (and human ADPKD), with preclinical testing with conditional models sometimes showing even greater variability.90 Here we have illustrated preclinical testing with the F1(129/B6) line that balances disease severity and variability, with relatively rapid disease progression between 3 and 13 weeks, and sex differences limited to faster kidney growth in male mice. A key step in our protocol is MRI that has higher resolution than ultrasound imaging.60,91 Importantly, MRI and automated determination of TKV61 were used to remove outliers and randomize animals with similar disease severity in the treatment groups, the criterion standard in human studies.6 Excluding very mild animals likely increases the chance of study significance. Also, the initial LnTKV provides a baseline measurement from which to determine %ΔLnTKV, similar to human studies.65 Here, multiple endpoints were significant, although a significant sex difference was seen for some, illustrating the importance of sex-specific analyses. MRI-determined TKV fully allows the disease in both kidneys to be measured, whereas CA just analyzes parts of 1 kidney.
Although our study is comprehensive, we understand that there are limitations. Ideally, equal sized male and female groups and the same animals would have been used for each endpoint phenotypic analysis, but depletion of some groups did not make this always possible. Male and female group sizes were sometimes unbalanced that can alter the outcome when sex is an important disease modifier, as seen here. We have tried to overcome this by showing sex-specific analyses. Analyzing the same animals by imaging throughout the study, as we did for the preclinical trial, would have provided more information about progression in individual animals, but histological comparisons at younger time points would not have been possible. Our use of MRI is, we think, a strength of the study with precise analysis of live animals, but we understand that this methodology is expensive and not available everywhere, with ultrasound analysis being another viable option.43 Factors associated with kidney function (blood urea nitrogen) or cAMP were barely significant in these younger animals, and so later/longer treatment may be helpful for these outcomes and fibrotic area to be reliable endpoints.48–50,53 We have also not determined mechanistically why genetic background alters disease severity, but as indicated above, this will require further analysis.
Supplementary Material
Table S1. Details of cystic kidney disease in the different genetic backgrounds in Pkd1RC/RC mice by sex.
Table S2. Progression of disease by sex compared to 1m within each genetic background.
Table S3. Comparison of Sham vs. Tolvaptan-treated Mice.
Table S4. Sex-specific comparison of Sham vs.Tolvaptan-treated mice.
Figure S1. Analysis of kidney cAMP across different genetic backgrounds in Pkd1RC/RC mice. (A) Kidney cAMP was determined in mice from each background at 1, 3, 6, 9 and 12 months. Mean values ± SD are shown on the scatter plot. The significance of the difference in the cAMP level in each background compared to the 1-m baseline value was performed using a 1-way ANOVA followed by Dunnett’s multiple comparison test: *P < 0.05, **P < 0.01, ***P < 0.001. The significance of cAMP levels in each background were also determined compared to age matched B6 data using a 2-way ANOVA followed by Dunnett’s multiple comparison test: †P < 0.05, †††P < 0.001. (B) Mice at each age and background were analyzed for sex-specific differences in cAMP levels. Two-way ANOVA followed by Bonferroni’s multiple comparison test between males and females for age-matched mice of each genetic background: *P < 0.05. Each timepoint in each background was also compared to corresponding B6 mice to determine differences per sex in cystic disease using 2-way ANOVA followed by Dunnett’s multiple comparison test: †P < 0.05, †††P < 0.001. Statistical analysis was performed only on groups that contained N ≥ 3, and groups not analyzed are marked with X. Number of animals per group are shown in Table 1.
Figure S2. Characterization of cystic disease in early perinatal Pkd1RC/RC mice across the genetic backgrounds. (A) Masson Trichrome stained cross-sectional images of mice at P1, P4, P6, P12 and P18 in the different backgrounds. Bar = 250 μm. (B) CA measured from kidney cross sections for each background and timepoint shown as a scatter plot with the mean indicated. Change in cystic disease compared to the P1 baseline within each genetic background was analyzed with 1-way ANOVA followed by Dunnett’s multiple comparison test: *P < 0.05, **P < 0.01, ***P < 0.001. Each background was also compared to age-matched B6 mice to determine differences in cystic disease by 2-way ANOVA followed by Dunnett’s multiple comparison test compared to age-matched B6 mice: †P < 0.05, ††P < 0.01, †††P < 0.001. (C) Tubular segment origin of cysts for each of the backgrounds determined by LTA (proximal tubule [PT], white) or AQP2 (collecting duct [CD], green) staining or unstained/another segment (blue). Difference in %CD cysts compared to P1 within each genetic background was analyzed with 1-way ANOVA followed by Dunnett’s multiple comparison test: *P < 0.05, **P < 0.01, ***P < 0.001. Difference in %CD cysts from age-matched B6 mice was determined by 2-way ANOVA followed by Dunnett’s multiple comparison test: ††P < 0.01, †††P < 0.001. (D) Examples of immunofluorescence imaging of cyst linings to determine origin of cysts (LTA, PT [white]; AQP2, CD [green]; DAPI, nuclei [blue]). Bar = 50 μm. Statistical analysis was performed only on groups that contained N ≥ 3, and groups not analyzed are marked with X.
Figure S3. Proliferation in cyst linings in early postnatal mice. (A) Immunofluorescence imaging of cyst linings detected for PCNA (red), LTA (PT; white), AQP2 (CD; green), and DAPI (blue) to determine proliferating cystic epithelia (%PCE). PCNA-positive nuclei were counted as representative of a cell undergoing proliferation and only determined in cyst linings. Cysts were identified as tubules with a diameter >200 μm2. Bar = 50 μm. (B) Quantitative analysis of PCNA-positive cells in cyst linings showing mean values. Three wild-type animals were collected and analyzed at P18 for each background, except F1(129/B6). Mean P18 wild-type %PCNA-positive tubule epithelial cells: B6, 2.6; BC, 5.6; F1(BC/B6), 3.0; and 129, 1.3. The significance of differences in PCE compared to P1 baseline within each background was tested with a 1-way ANOVA followed by Dunnett’s multiple comparison test: *P < 0.05, **P < 0.01, ***P < 0.001. The significance of differences between each background compared to age-matched B6 data was determined with a 2-way ANOVA followed by Dunnett’s multiple comparison test: †P < 0.05, ††P < 0.01.
Figure S4. Comparison of live Pkd1RC/RC mice in the BC and F1(BC/B6) backgrounds over time by MR imaging. (A) Representative kidney coronal MR sections from BC and F1(BC/B6) mice. Bar = 1 cm. Note that different animals were imaged at each timepoint. (B) Quantification of TKV determined from MR images. Arrow indicates urine retention. The significance of differences in TKV within each background compared to 1-m baseline was determined with a 1-way ANOVA followed by Dunnett’s multiple comparison test: *P < 0.05. The significance of differences in TKV in each background compared to age-matched B6 mice was determined with a 2-way ANOVA followed by Dunnett’s multiple comparison test: ††P < 0.01, †††P < 0.001. Statistical analysis was only performed on groups that contained N ≥ 3, and groups not analyzed are marked with X.
Figure S5. Visualization of interkidney volume variability in individual 129 and F1(129/B6) Pkd1RC/RC mice. Coronal MR sections of kidneys from 129 (A) and F1(129/B6) (B) Pkd1RC/RC mice at 1 month and 3 months exhibiting interkidney cystic variability especially in the 129-line. The left kidney volumes (LKV) and right kidney volumes (RKV) are shown. Bar = 1 cm. (C) Chart showing the interkidney difference in TKV by percentage for each mouse. Significant difference in 129 versus F1(129/B6) at 1 month and 3 months was determined using an unpaired t test. ***P < 0.001.
Figure S6. Comparison of final TKV and LnTKV to KW/BW (A), cystic area (CA; B), KW (C), and LnKW (D). Combined data from the preclinical trial comparing these endpoints at 13 weeks. R2 was determined by linear regression analysis.
Figure S7. Change in LnTKV from 3 to 13 weeks. (A) Change in sham-treated Pkd1RC/RC F1(129/B6) males and females from start (3 weeks) to end (13 weeks) of preclinical study. (B) Change in LnTKV in both sham and tolvaptan-treated males and females during the 10-week study. A paired t test comparing animals at 3 weeks and 13 weeks resulted in P < 0.001 for all 4 groups.
Figure S8. Preclinical testing of tolvaptan in F1(129/B6) Pkd1RC/RC mice; sex-specific data. Initial BW (3 weeks) (A) and LnTKV (B) in the sham and tolvaptan-treated male and female groups. Corresponding final data (13 weeks) for the endpoints: BW (C), LnTKV (D), %ΔLnTKV (E), KW (F), KW/BW (G), CV (H), and CN (I). Significant differences between males versus females within treatment groups was determined using an unpaired t test. *P < 0.05, ***P < 0.001. Differences between sex-matched animals between treatment groups were also determined using an unpaired t test. †P < 0.05, ††P < 0.01, †††P < 0.001 (Supplementary Table S4).
Translational Statement.
Autosomal dominant polycystic kidney disease is a common inherited cause of end-stage kidney disease, with limited treatment options presently available. Orthologous animal models with appropriate rates of disease progression are required for preclinical testing, a vital step to developing new treatments. The C57BL/6J Pkd1RC/RC mouse is an established autosomal dominant polycystic kidney disease model, but in the present genetic background, it is slowly progressive. Here, by inbreeding into 2 other backgrounds and F1 derivatives of these backgrounds, models with more rapidly progressive disease were developed. A protocol for drug testing that mimics clinical trials is also described that will improve preclinical testing and thereby the pipeline for clinical trials and new treatments of autosomal dominant polycystic kidney disease.
ACKNOWLEDGMENTS
We thank the pathologists Roger K. Moreira and Mariam P. (Priya) Alexander for analysis of the histological data; Slobodan I. Macura, Prasanna K. Mishra, Ryan Meloche, and the NMR Core Facility for assistance in animal imaging; and Lisa Vaughan for statistical discussions. This study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases grant R01DK058816 (PCH) and Polycystic Kidney Disease Center grant P30 DK090728 (VET). The study was also supported by the Mayo Clinic Robert M. and Billie Kelley Pirnie Translational Polycystic Kidney Disease Center, Navitor Pharmaceuticals (PCH) and the Zell Family Foundation. JA and CJS were supported by the Mayo Nephrology T32 Training grant DK007013, and JA was also supported by F31 grant DK105778.
Footnotes
DISCLOSURE
PCH, KH, and Mayo Clinic have a financial interest in the licensed Pkd1RC/RC model. Not directly associated with the study, VET has grants from and has advised Otsuka Pharmaceuticals, Palladio Biosciences, Mironid, and Blueprint Medicines and has advised Sanofi and Reata and PCH has grants from and has advised Otsuka Pharmaceuticals and Acceleron and has advised Mitobridge, Regulus, and Vertex Pharmaceuticals. All the other authors declared no competing interests.
REFERENCES
- 1.Iglesias CG, Torres VE, Offord KP, et al. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota. Am J Kidney Dis. 1983;2:630–639. [DOI] [PubMed] [Google Scholar]
- 2.Dalgaard OZ. Bilateral polycystic disease of the kidneys: a follow-up of two hundred and eighty-four patients and their families. Acta Med Scand. 1957;328(suppl):1–255. [PubMed] [Google Scholar]
- 3.Suwabe T, Shukoor S, Chamberlain AM, et al. Epidemiology of autosomal dominant polycystic kidney disease in Olmsted County. Clin J Am Soc Nephrol. 2020;15:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reule S, Sexton DJ, Solid CA, et al. ESRD from autosomal dominant polycystic kidney disease in the United States, 2001–2010. Am J Kidney Dis. 2014;64:592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.United States Renal Data System. 2018 USRDS Annual Data Report: Epidemiology of kidney disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2018. Available at: https://adr.usrds.org/. Accessed September 1, 2020. [Google Scholar]
- 6.Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367: 2407–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N Engl J Med. 2017;377: 1930–1942. [DOI] [PubMed] [Google Scholar]
- 8.Cornec-Le Gall E, Torres VE, Harris PC. Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J Am Soc Nephrol. 2018;29:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cornec-Le Gall E, Audrezet MP, Chen JM, et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol. 2013;24:1006–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chebib FT, Jung Y, Heyer CM, et al. Effect of genotype on the severity and volume progression of polycystic liver disease in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2016;31: 952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lavu S, Vaughan LE, Senum SR, et al. The value of genotypic and imaging information to predict functional and structural outcomes in ADPKD. JCI Insight. 2020;5:e138724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018;4:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest. 2014;124: 2315–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torres VE, Harris PC. Mechanisms of disease: autosomal dominant and recessive polycystic kidney disease. Nat Clin Prac Nephrol. 2006;2:40–55. [DOI] [PubMed] [Google Scholar]
- 15.Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol. 2014;25:18–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ong AC, Harris PC. A polycystin-centric view of cyst formation and disease: the polycystins revisited. Kidney Int. 2015;88:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu J, Harris PC. Regulation of polycystin expression, maturation and trafficking. Cell Signal. 2020;72:109630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hopp K, Cornec-Le Gall E, Senum SR, et al. Detection and characterization of mosaicism in autosomal dominant polycystic kidney disease. Kidney Int. 2020;97:370–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris PC, Bae KT, Rossetti S, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2006;17:3013–3019. [DOI] [PubMed] [Google Scholar]
- 20.Nowak KL, You Z, Gitomer B, et al. Overweight and obesity are predictors of progression in early autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2018;29:571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Torres VE, Abebe KZ, Schrier RW, et al. Dietary salt restriction is beneficial to the management of autosomal dominant polycystic kidney disease. Kidney Int. 2017;91:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ilatovskaya DV, Levchenko V, Pavlov TS, et al. Salt-deficient diet exacerbates cystogenesis in ARPKD via epithelial sodium channel (ENaC). EBioMedicine. 2019;40:663–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hackbarth H, Hackbarth D. Genetic analysis of renal function in mice. 1. Glomerular filtration rate and its correlation with body and kidney weight. Lab Anim. 1981;15:267–272. [DOI] [PubMed] [Google Scholar]
- 24.Fontaine DA, Davis DB. Attention to background strain is essential for metabolic research: C57BL/6 and the International Knockout Mouse Consortium. Diabetes. 2016;65:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song HK, Hwang DY. Use of C57BL/6N mice on the variety of immunological researches. Lab Anim Res. 2017;33:119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arif E, Solanki AK, Nihalani D. Adriamycin susceptibility among C57BL/6 substrains. Kidney Int. 2016;89:721–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holditch SJ, Nemenoff RA, Hopp K. Polycystic Kidney Disease. Boca Raton, FL: CRC Press; 2020:193–243. [Google Scholar]
- 28.He C, Esposito C, Phillips C, et al. Dissociation of glomerular hypertrophy, cell proliferation, and glomerulosclerosis in mouse strains heterozygous for a mutation (Os) which induces a 50% reduction in nephron number. J Clin Invest. 1996;97:1242–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng F, Plati AR, Potier M, et al. Resistance to glomerulosclerosis in B6 mice disappears after menopause. Am J Pathol. 2003;162:1339–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng F, Striker GE, Esposito C, et al. Strain differences rather than hyperglycemia determine the severity of glomerulosclerosis in mice. Kidney Int. 1998;54:1999–2007. [DOI] [PubMed] [Google Scholar]
- 31.Esposito C, He CJ, Striker GE, et al. Nature and severity of the glomerular response to nephron reduction is strain-dependent in mice. Am J Pathol. 1999;154:891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hackbarth H, Harrison DE. Changes with age in renal function and morphology in C57BL/6, CBA/HT6, and B6CBAF1 mice. J Gerontol. 1982;37:540–547. [DOI] [PubMed] [Google Scholar]
- 33.Hartner A, Cordasic N, Klanke B, et al. Strain differences in the development of hypertension and glomerular lesions induced by deoxycorticosterone acetate salt in mice. Nephrol Dial Transplant. 2003;18:1999–2004. [DOI] [PubMed] [Google Scholar]
- 34.Garrett MR, Korstanje R. Using genetic and species diversity to tackle kidney disease. Trends Genet. 2020;36:499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi H, Calvet JP, Dittemore-Hoover D, et al. A hereditary model of slowly progressive polycystic kidney disease in the mouse. J Am Soc Nephrol. 1991;1:980–989. [DOI] [PubMed] [Google Scholar]
- 36.Lu W, Peissel B, Babakhanlou H, et al. Perinatal lethality with kidney and pancreas defects in mice with a targeted Pkd1 mutation. Nature Genet. 1997;17:179–181. [DOI] [PubMed] [Google Scholar]
- 37.Lu W, Fan X, Basora N, et al. Late onset of renal and hepatic cysts in Pkd1-targeted heterozygotes. Nat Genet. 1999;21:160–161. [DOI] [PubMed] [Google Scholar]
- 38.Piontek K, Menezes LF, Garcia-Gonzalez MA, et al. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med. 2007;13:1490–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87:979–987. [DOI] [PubMed] [Google Scholar]
- 40.Tan AY, Zhang T, Michaeel A, et al. Somatic mutations in renal cyst epithelium in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2018;29:2139–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet. 2004;13:3069–3077. [DOI] [PubMed] [Google Scholar]
- 42.Yu S, Hackmann K, Gao J, et al. Essential role of cleavage of polycystin-1 at G protein-coupled receptor proteolytic site for kidney tubular structure. Proc Natl Acad Sci U S A. 2007;104:18688–18693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hopp K, Ward CJ, Hommerding CJ, et al. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest. 2012;122:4257–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Happe H, van der Wal AM, Salvatori DC, et al. Cyst expansion and regression in a mouse model of polycystic kidney disease. Kidney Int. 2013;83:1099–1108. [DOI] [PubMed] [Google Scholar]
- 45.Rossetti S, Kubly VJ, Consugar MB, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009;75:848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vujic M, Heyer CM, Ars E, et al. Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol. 2010;21: 1097–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Durkie M, Chong J, Valluru MK, et al. Biallelic inheritance of hypomorphic PKD1 variants is highly prevalent in very early onset polycystic kidney disease. Genet Med. 2021;23:689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hajarnis S, Lakhia R, Yheskel M, et al. microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat Commun. 2017;8:14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gainullin VG, Hopp K, Ward CJ, et al. Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J Clin Invest. 2015;125:607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olson RJ, Hopp K, Wells H, et al. Synergistic genetic interactions between Pkhd1 and Pkd1 result in an ARPKD-like phenotype in murine models. J Am Soc Nephrol. 2019;30:2113–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hassane S, Leonhard WN, van der Wal A, et al. Elevated TGFbeta-Smad signalling in experimental Pkd1 models and human patients with polycystic kidney disease. J Pathol. 2010;222:21–31. [DOI] [PubMed] [Google Scholar]
- 52.Hopp K, Wang X, Ye H, et al. Effects of hydration in rats and mice with polycystic kidney disease. Am J Physiol Renal Physiol. 2015;308:F261–F266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Warner G, Hein KZ, Nin V, et al. Food restriction ameliorates the development of polycystic kidney disease. J Am Soc Nephrol. 2016;27: 1437–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kleczko EK, Marsh KH, Tyler LC, et al. CD8+ T cells modulate autosomal dominant polycystic kidney disease progression. Kidney Int. 2018;94: 1127–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kashyap S, Hein KZ, Chini CC, et al. Metalloproteinase PAPP-A regulation of IGF-1 contributes to polycystic kidney disease pathogenesis. JCI Insight. 2020;5:e135700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holditch SJ, Brown CN, Atwood DJ, et al. A study of sirolimus and mTOR kinase inhibitor in a hypomorphic Pkd1 mouse model of autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol. 2019;317: F187–F196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holditch SJ, Brown CN, Atwood DJ, et al. The consequences of increased 4E-BP1 in polycystic kidney disease. Hum Mol Genet. 2019;28: 4132–4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lakhia R, Yheskel M, Flaten A, et al. PPARa agonist fenofibrate enhances fatty acid β-oxidation and attenuates polycystic kidney and liver disease in mice. Am J Physiol Renal Physiol. 2018;314:F122–F131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lakhia R, Yheskel M, Flaten A, et al. Interstitial microRNA miR-214 attenuates inflammation and polycystic kidney disease progression. JCI Insight. 2020;5:e133785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Irazabal MV, Mishra PK, Torres VE, Macura SI. Use of ultra-high field MRI in small rodent models of polycystic kidney disease for in vivo phenotyping and drug monitoring. J Vis Exp. 2015:e52757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Edwards ME, Periyanan S, Anaam D, et al. Automated total kidney volume measurements in pre-clinical magnetic resonance imaging for resourcing imaging data, annotations, and source code. Kidney Int. 2021;99:763–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamaguchi T, Nagao S, Kasahara M, et al. Renal accumulation and excretion of cyclic adenosine monophosphate in a murine model of slowly progressive polycystic kidney disease. Am J Kidney Dis. 1997;30: 703–709. [DOI] [PubMed] [Google Scholar]
- 63.Hopp K, Hommerding CJ, Wang X, et al. Tolvaptan plus pasireotide shows enhanced efficacy in a PKD1 model. J Am Soc Nephrol. 2015;26: 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354:2122–2130. [DOI] [PubMed] [Google Scholar]
- 65.Chapman AB, Bost JE, Torres VE, et al. Kidney volume and functional outcomes in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2012;7:479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Irazabal MV, Rangel LJ, Bergstralh EJ, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol. 2015;26:160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takakura A, Contrino L, Zhou X, et al. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet. 2009;18:2523–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Costantini F, Kopan R. Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Dev Cell. 2010;18:698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lindstrom NO, Lawrence ML, Burn SF, et al. Integrated β-catenin, BMP, PTEN, and Notch signalling patterns the nephron. Elife. 2015;3:e04000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sheybani-Deloui S, Chi L, Staite MV, et al. Activated hedgehog-GLI signaling causes congenital ureteropelvic junction obstruction. J Am Soc Nephrol. 2018;29:532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yao X, Cheng F, Yu W, et al. Cathepsin S regulates renal fibrosis in mouse models of mild and severe hydronephrosis. Mol Med Rep. 2019;20:141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bose J, Grotewold L, Ruther U. Pallister-Hall syndrome phenotype in mice mutant for Gli3. Hum Mol Genet. 2002;11:1129–1135. [DOI] [PubMed] [Google Scholar]
- 73.Ye H, Wang X, Constans MM, et al. The regulatory 1a subunit of protein kinase A modulates renal cystogenesis. Am J Physiol Renal Physiol. 2017;313:F677–F686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.D’Cruz R, Stronks K, Rowan CJ, Rosenblum ND. Lineage-specific roles of hedgehog-GLI signaling during mammalian kidney development. Pediatr Nephrol. 2020;35:725–731. [DOI] [PubMed] [Google Scholar]
- 75.Nino F, Ilari M, Noviello C, et al. Genetics of vesicoureteral reflux. Curr Genomics. 2016;17:70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Verschuren EHJ, Mohammed SG, Leonhard WN, et al. Polycystin-1 dysfunction impairs electrolyte and water handling in a renal precystic mouse model for ADPKD. Am J Physiol Renal Physiol. 2018;315:F537–F546. [DOI] [PubMed] [Google Scholar]
- 77.Ahrabi AK, Terryn S, Valenti G, et al. PKD1 haploinsufficiency causes a syndrome of inappropriate antidiuresis in mice. J Am Soc Nephrol. 2007;18:1740–1753. [DOI] [PubMed] [Google Scholar]
- 78.Mrug M, Zhou J, Yang C, et al. Genetic and informatic analyses implicate Kif12 as a candidate gene within the Mpkd2 locus that modulates renal cystic disease severity in the Cys1cpk mouse. PLoS One. 2015;10: e0135678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grantham JJ, Mulamalla S, Swenson-Fields KI. Why kidneys fail in autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2011;7: 556–566. [DOI] [PubMed] [Google Scholar]
- 80.Juul KV, Bichet DG, Nielsen S, Norgaard JP. The physiological and pathophysiological functions of renal and extrarenal vasopressin V2 receptors. Am J Physiol Renal Physiol. 2014;306:F931–F940. [DOI] [PubMed] [Google Scholar]
- 81.Davis KE, Carstens EJ, Irani BG, et al. Sexually dimorphic role of G protein-coupled estrogen receptor (GPER) in modulating energy homeostasis. Horm Behav. 2014;66:196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Auger AP. Sex differences in the developing brain: crossroads in the phosphorylation of cAMP response element binding protein. J Neuroendocrinol. 2003;15:622–627. [DOI] [PubMed] [Google Scholar]
- 83.Liu J, Sharma N, Zheng W, et al. Sex differences in vasopressin V2 receptor expression and vasopressin-induced antidiuresis. Am J Physiol Renal Physiol. 2011;300:F433–F440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Menezes LF, Lin CC, Zhou F, Germino GG. Fatty acid oxidation is impaired in an orthologous mouse model of autosomal dominant polycystic kidney disease. EBioMedicine. 2016;5:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Keane TM, Goodstadt L, Danecek P, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477:289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carneiro AM, Airey DC, Thompson B, et al. Functional coding variation in recombinant inbred mouse lines reveals multiple serotonin transporter-associated phenotypes. Proc Natl Acad Sci U S A. 2009;106: 2047–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Doran AG, Wong K, Flint J, et al. Deep genome sequencing and variation analysis of 13 inbred mouse strains defines candidate phenotypic alleles, private variation and homozygous truncating mutations. Genome Biol. 2016;17:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sur C, Wafford KA, Reynolds DS, et al. Loss of the major GABA(A) receptor subtype in the brain is not lethal in mice. J Neurosci. 2001;21:3409–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Leonhard WN, Zandbergen M, Veraar K, et al. Scattered deletion of PKD1 in kidneys causes a cystic snowball effect and recapitulates polycystic kidney disease. J Am Soc Nephrol. 2015;26:1322–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Leonhard WN, Song X, Kanhai AA, et al. Salsalate, but not metformin or canagliflozin, slows kidney cyst growth in an adult-onset mouse model of polycystic kidney disease. EBioMedicine. 2019;47:436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kline TL, Irazabal MV, Ebrahimi B, et al. Utilizing magnetization transfer imaging to investigate tissue remodeling in a murine model of autosomal dominant polycystic kidney disease. Magn Reson Med. 2016;75:1466–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Details of cystic kidney disease in the different genetic backgrounds in Pkd1RC/RC mice by sex.
Table S2. Progression of disease by sex compared to 1m within each genetic background.
Table S3. Comparison of Sham vs. Tolvaptan-treated Mice.
Table S4. Sex-specific comparison of Sham vs.Tolvaptan-treated mice.
Figure S1. Analysis of kidney cAMP across different genetic backgrounds in Pkd1RC/RC mice. (A) Kidney cAMP was determined in mice from each background at 1, 3, 6, 9 and 12 months. Mean values ± SD are shown on the scatter plot. The significance of the difference in the cAMP level in each background compared to the 1-m baseline value was performed using a 1-way ANOVA followed by Dunnett’s multiple comparison test: *P < 0.05, **P < 0.01, ***P < 0.001. The significance of cAMP levels in each background were also determined compared to age matched B6 data using a 2-way ANOVA followed by Dunnett’s multiple comparison test: †P < 0.05, †††P < 0.001. (B) Mice at each age and background were analyzed for sex-specific differences in cAMP levels. Two-way ANOVA followed by Bonferroni’s multiple comparison test between males and females for age-matched mice of each genetic background: *P < 0.05. Each timepoint in each background was also compared to corresponding B6 mice to determine differences per sex in cystic disease using 2-way ANOVA followed by Dunnett’s multiple comparison test: †P < 0.05, †††P < 0.001. Statistical analysis was performed only on groups that contained N ≥ 3, and groups not analyzed are marked with X. Number of animals per group are shown in Table 1.
Figure S2. Characterization of cystic disease in early perinatal Pkd1RC/RC mice across the genetic backgrounds. (A) Masson Trichrome stained cross-sectional images of mice at P1, P4, P6, P12 and P18 in the different backgrounds. Bar = 250 μm. (B) CA measured from kidney cross sections for each background and timepoint shown as a scatter plot with the mean indicated. Change in cystic disease compared to the P1 baseline within each genetic background was analyzed with 1-way ANOVA followed by Dunnett’s multiple comparison test: *P < 0.05, **P < 0.01, ***P < 0.001. Each background was also compared to age-matched B6 mice to determine differences in cystic disease by 2-way ANOVA followed by Dunnett’s multiple comparison test compared to age-matched B6 mice: †P < 0.05, ††P < 0.01, †††P < 0.001. (C) Tubular segment origin of cysts for each of the backgrounds determined by LTA (proximal tubule [PT], white) or AQP2 (collecting duct [CD], green) staining or unstained/another segment (blue). Difference in %CD cysts compared to P1 within each genetic background was analyzed with 1-way ANOVA followed by Dunnett’s multiple comparison test: *P < 0.05, **P < 0.01, ***P < 0.001. Difference in %CD cysts from age-matched B6 mice was determined by 2-way ANOVA followed by Dunnett’s multiple comparison test: ††P < 0.01, †††P < 0.001. (D) Examples of immunofluorescence imaging of cyst linings to determine origin of cysts (LTA, PT [white]; AQP2, CD [green]; DAPI, nuclei [blue]). Bar = 50 μm. Statistical analysis was performed only on groups that contained N ≥ 3, and groups not analyzed are marked with X.
Figure S3. Proliferation in cyst linings in early postnatal mice. (A) Immunofluorescence imaging of cyst linings detected for PCNA (red), LTA (PT; white), AQP2 (CD; green), and DAPI (blue) to determine proliferating cystic epithelia (%PCE). PCNA-positive nuclei were counted as representative of a cell undergoing proliferation and only determined in cyst linings. Cysts were identified as tubules with a diameter >200 μm2. Bar = 50 μm. (B) Quantitative analysis of PCNA-positive cells in cyst linings showing mean values. Three wild-type animals were collected and analyzed at P18 for each background, except F1(129/B6). Mean P18 wild-type %PCNA-positive tubule epithelial cells: B6, 2.6; BC, 5.6; F1(BC/B6), 3.0; and 129, 1.3. The significance of differences in PCE compared to P1 baseline within each background was tested with a 1-way ANOVA followed by Dunnett’s multiple comparison test: *P < 0.05, **P < 0.01, ***P < 0.001. The significance of differences between each background compared to age-matched B6 data was determined with a 2-way ANOVA followed by Dunnett’s multiple comparison test: †P < 0.05, ††P < 0.01.
Figure S4. Comparison of live Pkd1RC/RC mice in the BC and F1(BC/B6) backgrounds over time by MR imaging. (A) Representative kidney coronal MR sections from BC and F1(BC/B6) mice. Bar = 1 cm. Note that different animals were imaged at each timepoint. (B) Quantification of TKV determined from MR images. Arrow indicates urine retention. The significance of differences in TKV within each background compared to 1-m baseline was determined with a 1-way ANOVA followed by Dunnett’s multiple comparison test: *P < 0.05. The significance of differences in TKV in each background compared to age-matched B6 mice was determined with a 2-way ANOVA followed by Dunnett’s multiple comparison test: ††P < 0.01, †††P < 0.001. Statistical analysis was only performed on groups that contained N ≥ 3, and groups not analyzed are marked with X.
Figure S5. Visualization of interkidney volume variability in individual 129 and F1(129/B6) Pkd1RC/RC mice. Coronal MR sections of kidneys from 129 (A) and F1(129/B6) (B) Pkd1RC/RC mice at 1 month and 3 months exhibiting interkidney cystic variability especially in the 129-line. The left kidney volumes (LKV) and right kidney volumes (RKV) are shown. Bar = 1 cm. (C) Chart showing the interkidney difference in TKV by percentage for each mouse. Significant difference in 129 versus F1(129/B6) at 1 month and 3 months was determined using an unpaired t test. ***P < 0.001.
Figure S6. Comparison of final TKV and LnTKV to KW/BW (A), cystic area (CA; B), KW (C), and LnKW (D). Combined data from the preclinical trial comparing these endpoints at 13 weeks. R2 was determined by linear regression analysis.
Figure S7. Change in LnTKV from 3 to 13 weeks. (A) Change in sham-treated Pkd1RC/RC F1(129/B6) males and females from start (3 weeks) to end (13 weeks) of preclinical study. (B) Change in LnTKV in both sham and tolvaptan-treated males and females during the 10-week study. A paired t test comparing animals at 3 weeks and 13 weeks resulted in P < 0.001 for all 4 groups.
Figure S8. Preclinical testing of tolvaptan in F1(129/B6) Pkd1RC/RC mice; sex-specific data. Initial BW (3 weeks) (A) and LnTKV (B) in the sham and tolvaptan-treated male and female groups. Corresponding final data (13 weeks) for the endpoints: BW (C), LnTKV (D), %ΔLnTKV (E), KW (F), KW/BW (G), CV (H), and CN (I). Significant differences between males versus females within treatment groups was determined using an unpaired t test. *P < 0.05, ***P < 0.001. Differences between sex-matched animals between treatment groups were also determined using an unpaired t test. †P < 0.05, ††P < 0.01, †††P < 0.001 (Supplementary Table S4).