

Abstract
The presence of microbes in the colon impacts host physiology. Therefore, microbes are being evaluated as potential treatments for colorectal diseases. Humanized model systems that enable robust culture of primary human intestinal cells with bacteria facilitate evaluation of potential treatments. Here, we describe a protocol that can be used to co-culture a primary human colon monolayer with aerotolerant bacteria. Primary human colon cells maintained as organoids are dispersed into single-cell suspensions and then seeded on collagen-coated Transwell inserts, where they attach and proliferate to form confluent monolayers within days of seeding. The confluent monolayers are differentiated for an additional four days and then co-cultured with bacteria. As an example application we describe how to co-culture differentiated colon cells for 8 h with four strains of B. thetaiotaomicron, each engineered to detect different colonic microenvironments via genetically-embedded logic circuits incorporating deoxycholic acid (DCA) and anhydrotetracycline (aTc) sensors. Characterization of this co-culture system reveals that barrier function remains intact in the presence of engineered B. thetaiotaomicron. The bacteria stay close to the mucus layer and respond in a microenvironment-specific manner to the inducers (DCA and aTc) of the genetic circuits. This protocol thus provides a useful mucosal barrier system to assess the effects of bacterial cells that respond to the colonic microenvironment, and may also be useful in other contexts to model human intestinal barrier properties and microbiota-host interactions.
EDITORIAL SUMMARY
Co-culture of a primary human colon monolayer derived from cells growing in colon organoids with aerotolerant bacteria, such as strains of B. thetaiotaomicron genetically engineered to respond to different stimuli in colonic microenviroments.
Introduction
Genetically engineered and unmodified microbes have great potential to treat chronic diseases by impacting host physiology. Comprehensive evaluation of the performance of these potentially therapeutic microbes is critical and requires both in vitro and in vivo models. The US Food and Drug Administration (FDA) supports the use of gut simulators to provide evidence for a claimed beneficial bacterial effect (e.g., Simulator of the Human Intestinal Microbial Ecosystem [SHIME]).1–4 These luminal microbial culture systems provide reproducible experiments, however, lack critical components of host-microbe interactions involving bacterial adhesion to, or entrainment in, epithelial-associated mucus; direct attachment to epithelial cells; and reaction to epithelial-secreted signaling molecules. To study the communication of bacteria with host mucosal barrier cells and evaluate attachment to cells and/or cell-produced mucus, various in vitro gut models have been developed. These gut models use either immortalized human cancer cell lines or primary cells expanded as organoids, in static cultures or continuous microfluidic systems,5–8 and have proved useful to study drug transport and drug delivery. 6–9
In this protocol, we describe how to generate primary human colon monolayers in a standard cell culture membrane insert (Transwell) using colon proliferative cells previously maintained as organoids (Figure 1). We provide detailed instructions for the passaging and expansion of the colon organoids, as well as detailed instructions for cell seeding and differentiation. We have used this protocol to co-culture colon monolayers with genetically engineered Bacteroides thetaiotaomicron strains.10
Figure 1. Schematic workflow to generate differentiated colon epithelial monolayers and co-culture with engineered B. thetaiotaomicron.
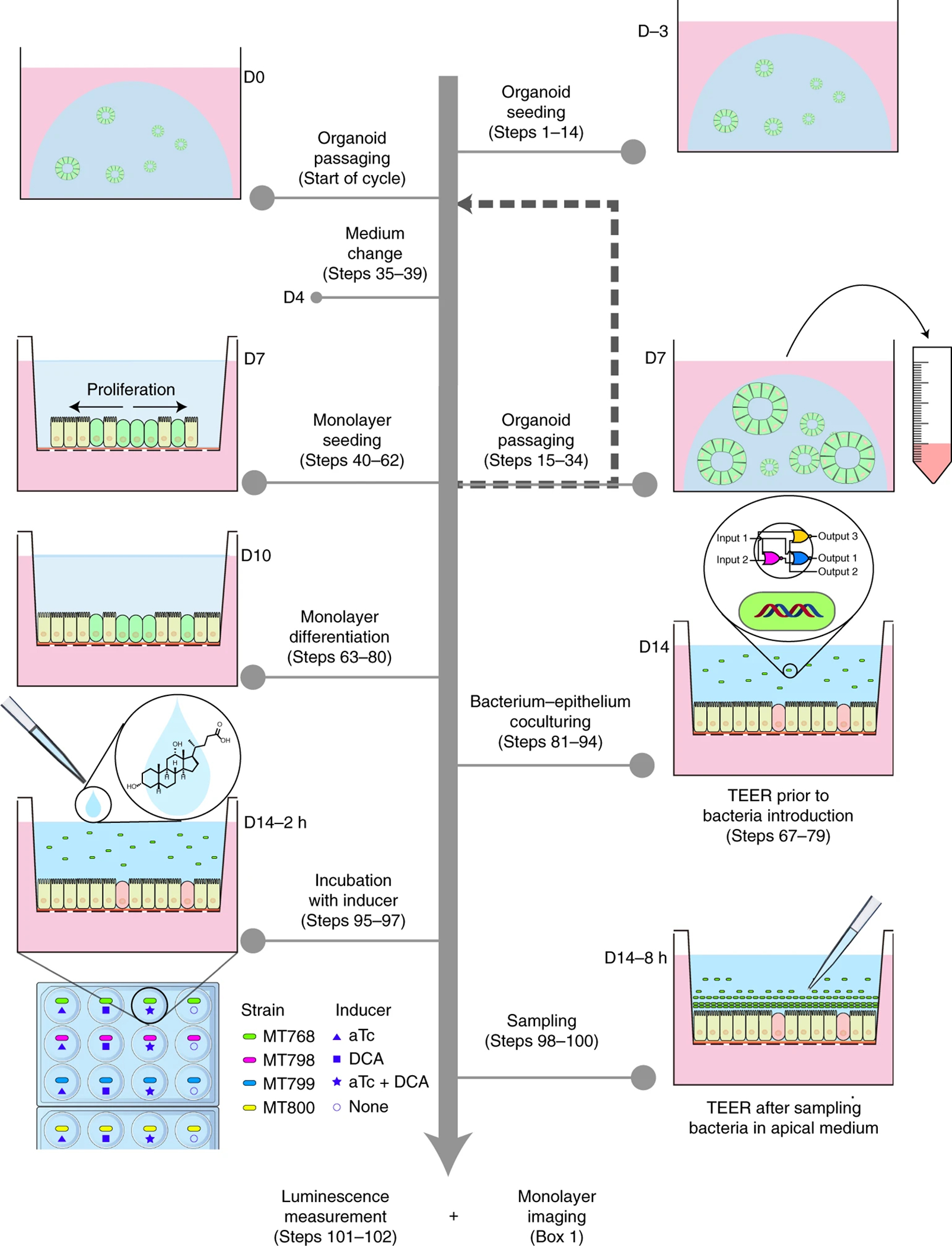
The workflow includes initial organoid seeding (steps 1–14), organoid passaging (steps 15–34), organoid medium change (steps 35–39), organoid processing for monolayer seeding (steps 40–62), monolayer differentiation (steps 63–80), bacteria-monolayer co-culture (steps 81–94), introduction of inducers (steps 95–97), and sampling for downstream analysis (steps 98102). Sampling refers to the collection of apical media containing bacteria as described in detail in steps 98–100 and in the original publication.10 In the co-culture experiments we previously conducted, 10 engineered B. thetaiotaomicron MT798, MT799, and MT800 were used. Each strain carried a genetic circuit with designed response to inducers anhydrotetracycline (aTc, an antibiotic analog) and/or deoxycholic acid (DCA, a secondary bile acid in human colon). As a control, B. thetaiotaomicron (MT768) engineered with constant expression of luminescence was used. TEER measurement is optional but we recommend it be performed at key timepoints (e.g. D7, D10, and D14), particularly prior to coculture with bacteria at D14 and after media collection at D14. The procedure for TEER measurement is described in steps 67–79.
Comparison to other models that evaluate mammalian colon cell and bacterial cell interactions
Engineered bacteria that harbor genetic circuits designed for specific therapeutic applications have been traditionally tested in simple in vitro bacterial culture models, such as the three-stage continuous culture systems; SHIME3 and PloyFermS,4 followed by evaluation in animal models, but without an evaluation in a mammalian cell culture model.11–13 For example, in a previous publication, we fed mice with Bacteroides engineered with an inducible gene circuit, then measured the bacterial inducible response (luciferase activity) in the feces.14 However, using this approach, it was difficult to characterize the detailed performance of the bacterial genetic circuit within the mice, in part due to difficulties visualizing bacterial adherence to the small intestinal/colon epithelium.15
Animal experiments are also low-throughput and expensive, and the method of bacterial administration (for example by gavage) adds complexity in the procedure. Further, animals and humans differ in their physiology, diet, mucus composition, mucosal immunology, molecular transport, and native microbiota composition, all of which can radically impact bacterial biology.16,17 Such differences can make it difficult to extrapolate the performance of an engineered bacterium from an animal model to humans (https://investor.synlogictx.com/news-releases/news-release-details/synlogic-discontinues-development-synb1020-treat-hyperammonemia).12 Evaluating bacterial performance in humans, however, is even slower and convoluted due to patient-to-patient variability and issues involving regulatory compliance. Thus, the ability to evaluate the performance of engineered bacteria in an in vitro model that mimics critical features of the human colonic environment relevant for the bacterial circuits in question is desirable. In contrast to animal models, in vitro gut models enable fast turnover (i.e., 2–4 weeks for monolayer preparation in Transwells) and intermediate throughput (16 Transwells or more per week).
In vitro gut models can vary on cellular, geometry, and operation capabilities to simulate human gut biology. For instance, an in vitro gut model can include an epithelial monolayer in combination with neuronal and immune cells using either commercial cell lines or tissue-derived primary cells with static or dynamic mechanical stimulation, which appears to promote mucus and villus-like structures.6–9 Native gut microbiota, such as Bacteroides spp., can be introduced into such devices for hours to weeks, depending on the culture configuration and luminal media exchange rates, to enable mechanistic studies to better understand the effects of the microbiota.18–26 Two dimensional culture of epithelial monolayers or bilayers with supporting cells in standardized transwell inserts are often used for evaluating drug interactions with human colon due to the easy access to both luminal and basal compartments,27–32 however these have not been widely deployed to test engineered bacteria safety and performance. These intestinal models were traditionally developed using commercially-available cell lines,33 and recently expanded to use primary rodent and human small intestinal cells as a starting material.27–32
More complex in vitro gut models that require highly specialized apparatus or 3D architecture19–21,24,34–37 have also been developed but often tailored for narrow and specific applications. For example, Jalili-Firoozinezhad et al. designed a customized anaerobic chamber that enable the culture of obligate anaerobes in two-channel microfluidic gut-chip.24 In another customized microfluidics-based gut model (HuMiX), Shah et al. created two microchambers separated by a nanoporous membrane to prevent intestinal cells from direct contact with bacterial cells during co-culture experiments.21 Unfortunately, the complexity of the gut devices makes them impractical for routine use in all but a few dedicated laboratories and are not always generalizable to desired readouts.
Simplified gut models using Transwell-type inserts have been used for a variety of applications including drug screening,27 drug toxicity,28 drug transport,30 cytokine secretion,38 host-parasite interactions,29 host-pathogen interactions,39,40 and host-norovirus interactions.41,42 Because the luminal side of the epithelial monolayer in Transwell culture is completely accessible, it is easy to introduce bacteria, add any stimuli, and collect samples over multiple time points. Therefore, monolayer cultures can be scaled up to screen multiple conditions and bacterial strains with significantly higher throughput than animal models. A drawback of static monolayer models is inability to maintain a super-strict anaerobic luminal environment or a constant supply of nutrients, making it difficult to establish long-term stable co-cultures due to short-life span of host cells18 and bacterial overgrowth.20 While the oxygen concentration at the surface of an epithelial monolayer in a static Transwell in the presence of bacteria can approach micro-aerobic or even moderately anaerobic states,43 static monolayers are therefore limited in application to aerobes and aerotolerant anaerobes. This limitation can be overcome by combining the transwell monolayer model with a microphysiological systems technology.20,26,43
Gut organoid models have emerged as a new tool to study host-microbe interactions.44 The culture of organoids from primary human and animal intestinal tissues enables the routine maintenance of intestinal organoids from different donors.27,44–46 In addition to tissue-derived organoids, intestinal organoids can also be derived from human pluripotent stem cells, although the differentiation requires up to 4 weeks and does not recapitulate features of adult tissue.47,48
Intestinal organoids comprise a single layer of epithelial cells surrounding a hollow central lumen, thus microbes are introduced into the lumen (apical side) using microinjection techniques49 that are labor intensive and require extensive technical expertise, hence is relatively low throughput. Further, depending on the types of readouts required (e.g., fluorescence, transcriptomic, proteomics, metabolomic. etc), multiple organoids may be required to create enough sample for testing. In addition, in organoid cultures, antibiotics must be added into the culture medium to keep the basolateral side free from bacteria49, resulting in alterations to intestinal function that are independent of antimicrobial activity.50
To utilize the strengths of the organoid and monolayer systems, whilst also overcoming some of the limitations discussed above, we and other researchers developed methods to generate primary human colon monolayers in a standard cell culture membrane insert (Transwell) from colon proliferative cells previously maintained as organoids.10,27,29,51,52 This protocol generates primary human colon epithelial monolayers that contain differentiated and proliferative cells. The monolayers exhibit physical and biological barrier functions as indicated by an increase in transepithelial electrical resistance (TEER) values over time. In contrast to other systems,19–21,24,34,35 the open configuration of the Transwell allows access to both the apical and basolateral sides of the epithelium for the incorporation of bacterial cells engineered with genetic circuits and their corresponding inducers at different time points during a given experiment.
Applications of the protocol
Human gut models can provide a physiological environment for testing safety and efficacy of biologics before progressing to animal or human trials.53 For instance, engineered bacteria with inducible gene circuits can be validated in standard bacterial culture conditions and then tested for efficacy in an in vitro gut model. We recently employed this strategy to test engineered bacteria with gene circuits that respond to different colon microenvironments, using a primary human colon mucosal barrier in a static culture configuration.10 Further, gut models can be combined with physiomimetic/microfluidic systems technology to recapitulate complex multi-cellular crosstalk. For example, we recently used a physiomimetic platform to study the crosstalk between primary human colon epithelial cells with oxygen-intolerant commensal Faecalibacterium prausnitzii. 26 In addition, we also built a physiomimetic platform to study multicellular crosstalk among gut-liver-immune cells in the context of ulcerative colitis.52
The methods described here have been used to successfully expand organoids from donors of different sex, age, colon regions, and disease state (mainly ulcerative colitis) (Table 1), thus, we anticipate that this method could be extended to other human intestinal tissues (e.g. small intestine) and other understudied mucosal barriers, such as oral cavity, endocervix, and endometrium. Indeed, the organoid and monolayer technique has been successfully applied to stem cell–derived small intestinal enteroids54 to study host-virus interactions in different microenvironments.55 In addition, we anticipate that this protocol can be adopted to study a variety of other important topics in mucosal epithelial biology including host-pathogen interactions, drug absorption and toxicity in the context of personalized medicine.
Table 1.
Application of the protocol to different donors that varied in age, sex, and colon biopsy region.
| Donor Number | Experiment Number | Patient Age | Patient Sex | Patient Pathology | Tissue Collected | Culture |
|---|---|---|---|---|---|---|
|
| ||||||
| 1 | HC2978 | 30 yr | Male | diverticulosis and diverticulitis | Rectosigmoid (Normal appearing region) | O, M |
| 2 | HC176 | 6 m | Female | Non-IBD | Ascending colon | O, M |
| 3 | HC465 | 18 yr | Male | UC | Ascending colon | O, M |
| 4 | HC100 | N/A | N/A | UC | Ascending colon | O, M |
| 5 | HC511 | 12 yr | N/A | UC | Ascending colon | O |
| 6 | HC457 | 19 yr | Female | Non-IBD | Sigmoid colon | O |
| 7 | HC443 | 15 yr | N/A | UC | Ascending colon | O |
O: organoid culture protocol applied to the indicated donor; M: monolayer culture protocol applied to the indicated donor. Donor 5, 6, and 7 were on the stage of generating organoids, thus were not tried to generate monolayers. UC: ulcerative colitis. IBD: inflammatory bowel diseases. N/A: not applicable (not identified)
Experimental design
The procedure comprises several stages (Figure 1) that start with established organoids in 3D culture. We first describe how to maintain, passage and expand organoids, then we describe how organoids are dissociated to generate single cells for seeding into Transwells inserts and generate functional colon monolayers. Finally, we outline the media regime to differentiate epithelial monolayers prior to the addition of bacteria. To evaluate the integrity of the epithelial monolayers we suggest monitoring the TEER values at various time points. We also suggest to only use intact monolayers that show an increase in TEER values over time. Alternatively, the epithelial barrier integrity can also be evaluated using other assays, such as the leakage of lucifer yellow from the apical to the basolateral side. The proliferative state of the monolayers can be evaluated using commercially available proliferative kits, such as the incorporation of nucleotide analogous during DNA synthesis (EdU staining) described in Box 1.
Box 1. EDU Staining to determine the proliferative state of the monolayer ●Timing set up 1 h, incubation 1 day, imaging 0.5 h.
After Step 75, the EdU staining can be performed to determine the proportion of proliferative cells. We do this using the Click-iT EdU Cell Proliferation Kit following the instructions given below.
Materials specific to EDU staining
Procedure
1) Make a 10 μM (1x) solution of EdU in both base medium and colon differentiation medium by diluting the 10 mM (1000x) stock solution of EdU (component A of the kit).
2) Add 500 μl of the 1x EdU in base medium solution onto the apical side of the monolayer, and 1.5 ml of the 1x EdU in colon differentiation medium onto the basolateral side of the monolayer
3) Return monolayers to the incubator (5% CO2, 37°C) and incubate overnight (16 h). ▲CRITICAL The incubation time can be increased or decreased depending on the proliferative state of the donor/cells being used but it needs to be kept consistent across all time points and samples in any given experiment.
4) Once incubation period is complete, remove the media from both sides of the monolayer, and replace it with fixative solution (4% paraformaldehyde in DPBS−/−) with 500 μl on both the apical and basolateral sides of the monolayer. Incubate at room temperature for 15 min
5) Aspirate the fixative solution and wash the monolayers at least twice with 3% BSA in DPBS−/− (1 ml on the apical side, 2 ml on the basolateral side of the monolayer).
6) Add 500 μl of the permeabilization solution (0.2% Triton X-100 in DPBS) to both apical and basolateral sides of the monolayer. Incubate at room temperature for 20 min.
7) Prepare the Click-iT reaction cocktail containing 1X Click-iT reaction buffer, CuSO4, Alexa Fluor azide, and reaction buffer additive, according to the manufacturer’s procedure.
8) Aspirate permeabilization solution and wash the monolayers at least twice with 3% BSA in PBS. Add 500 μl of the Click-iT reaction cocktail to the apical side of the monolayer. Incubate at room temperature for 30 min. ▲CRITICAL Incubation step must be performed protected from light.
9) Aspirate the Click-iT reaction cocktail, wash at least twice with 3% BSA in DPBS.
121. If additional staining is desired, follow steps 106–112 of the main procedure described in the kit. After carrying out the additional staining, immerse monolayers in DPBS (applying 500 μl to the apical and 1500 μl basolateral sides) and proceed with imaging and analysis. PAUSEPOINT If you wish to delay imaging, store monolayers at 4°C. To prevent fading, store monolayers protected from light and image within 1 week. If you wish to store monolayers for longer than a week before imaging, mount on glass microscope slides as described in steps 114–120.
END BOX
Co-culture:
As an example, we describe how to co-culture epithelial monolayers with engineered B. thetaiotaomicron bacteria. The bacterium B. thetaiotaomicron was engineered with genetic circuits that can turn on different programs in response to different external environments, where the output of the circuit is reported via expression and activity of luciferase. A logic circuit that could report out the presence of either secondary bile acid deoxycholic acid (DCA) or anhydrotetracycline (aTc ), or the absence of either, was engineered into B. thetaiotaomicron.10 The circuit was tested using luciferase as the sole reporter; hence 3 strains were required to measure the individual outputs as described previously.10 An additional strain of B. thetaiotaomicron (MT768) was constructed to constantly express luciferase and used as a control for standardization of the luciferase activity. The multi-input, multi-output, logic system was built using a combination of three NOR gates built with CRISPRi-dCas9, integrated into a DCA and aTc sensors as inputs and three different output promoters. This resulted in three bacterial strains (MT798, MT799 and MT800) that have the same circuit but report only one of the three output promoters.10 For the experimental set up, four separate bacterial co-cultures (M768, M798, M799, and MT800) were started once the monolayers show signs of differentiation (e.g. increase in TEER values over time), which generally takes 7 days. After 2 h of bacterial-epithelial co-culture, the bacterial genetic circuits were induced by the addition of inducers. Each strain was exposed to the four combinations of inducers (i.e., 62.5 μM DCA, 100 ng μl−1 aTc, DCA+aTc, and none (as a control)).
Evaluation of co-culture:
Eight hours after the start of co-culture (i.e. six hours after introduction of the inducers) we evaluated the effects of the different culture conditions on bacterial growth, gene circuit activation, and epithelial monolayer integrity. The bacteria cell density was measured using optical density at 600 nm. The gene circuit activation by the inducers was determined by measuring the luciferase activity in each stain and condition using the NanoLuc luciferase assay. The resulting bacteria cell density and the luminescence in the test (MT798, MT799 and MT800) and control bacterial strain (MT768) were used to calculate the output of the gene circuit, which was designated as relative promoter units (RPULs). In our experimental setting, the RPUL for each strain was greatest when exposed to the inducer corresponding to its designed genetic circuit compared to other inducers; e.g., strain MT798 exhibited greater output in response to DCA compared to aTC along, aTC plus DCA, or no inducer. In addition, the integrity of the epithelial monolayer was monitored via TEER measurement and then characterized by immunofluorescent staining to identify the location of bacteria in the epithelium-bacteria co-culture system (Figure 1). Although not assessed in this study, additional characterization of the epithelial monolayer permeability using molecular tracers could be employed.30 More detailed phenotypic insights could also be obtained by analyzing the mucin production and harvesting the epithelial monolayer to conduct expression analysis via RNAseq or qPCR.26
Limitations
A current limitation of the Transwell system is that it does not provide dynamic control of nutrient renewal, which can result in bacterial overgrowth within 10–12 h of culture. A short (< 8 hr) timescale is sufficient for testing the responses of engineered bacteria to specific inducers, but may limit other applications requiring longer periods of culture. This limitation can be overcome by integrating the transwell monolayer into a microphysiological system which controls apical flow to provide continuous nutrient replenishment for the bacterial compartment.26 Another limitation of the transwell monolayer is that the apical compartment is not anaerobic, thus can only be used for culturing aerotolerant bacteria such as B. thetaiotaomicron, B. fragilis, Bifidobacteria, and Lactobacillus. This challenge has been overcome by incorporating transwell monolayers into a specialized microphysiological systems.26 For the very short term (a few hours), a system without apical flow allows acute responses to be assessed.18,20,43,56 For the longer term (hours – days) a device called gut-microbe (GuMI) with anaerobic apical flow and aerobic basal flow maintains an oxygen gradient across the epithelial barrier similar to a healthy colon in vivo, enabling successful days-long co-culture of colon epithelial cells with oxygen-sensitive bacteria, such as F. prausnitzii and Eubacterium rectale.26 Collectively, the studies described above exemplify the potentially wide application of the epithelial monolayer method described in this protocol.
Donor-to-donor variability can also affect the success of establishing functional epithelial monolayers. Although we did not exhaustively test many donors for monolayer formation, we observed a variability in the successful rate of monolayer generation from three different donors tested (Figure 2 and Source Data Figure 2). For example, the successful rate for generating usable monolayers from one donor was only 33% compared to another donor in which all monolayers were usable (100%). The variability we have seen is not due to reagents or organoid culture conditions, since donors that have lower monolayer formation efficiency could be maintained as organoids in Matrigel. We speculate that intrinsic donor properties as well as the removal of Matrigel that supply unknown factors during organoid culture can account for the variability in monolayer formation. Therefore, we recommend that the users perform pilot experiments with organoids from multiple donors to test monolayer formation efficiency and monolayer stability prior to establish co-culture experiments. In our experience, monolayers from donors with low monolayer formation efficiency also showed areas in the transwells that are not covered by epithelial cells. Usually, these areas are located at the edge of the monolayers and can be easily identified using an inverted microscope and light microscopy (see details in the Troubleshooting).
Figure 2. The success rate of generating usable monolayers and the cell viability of single cells from dissociated organoids used for monolayer seeding.
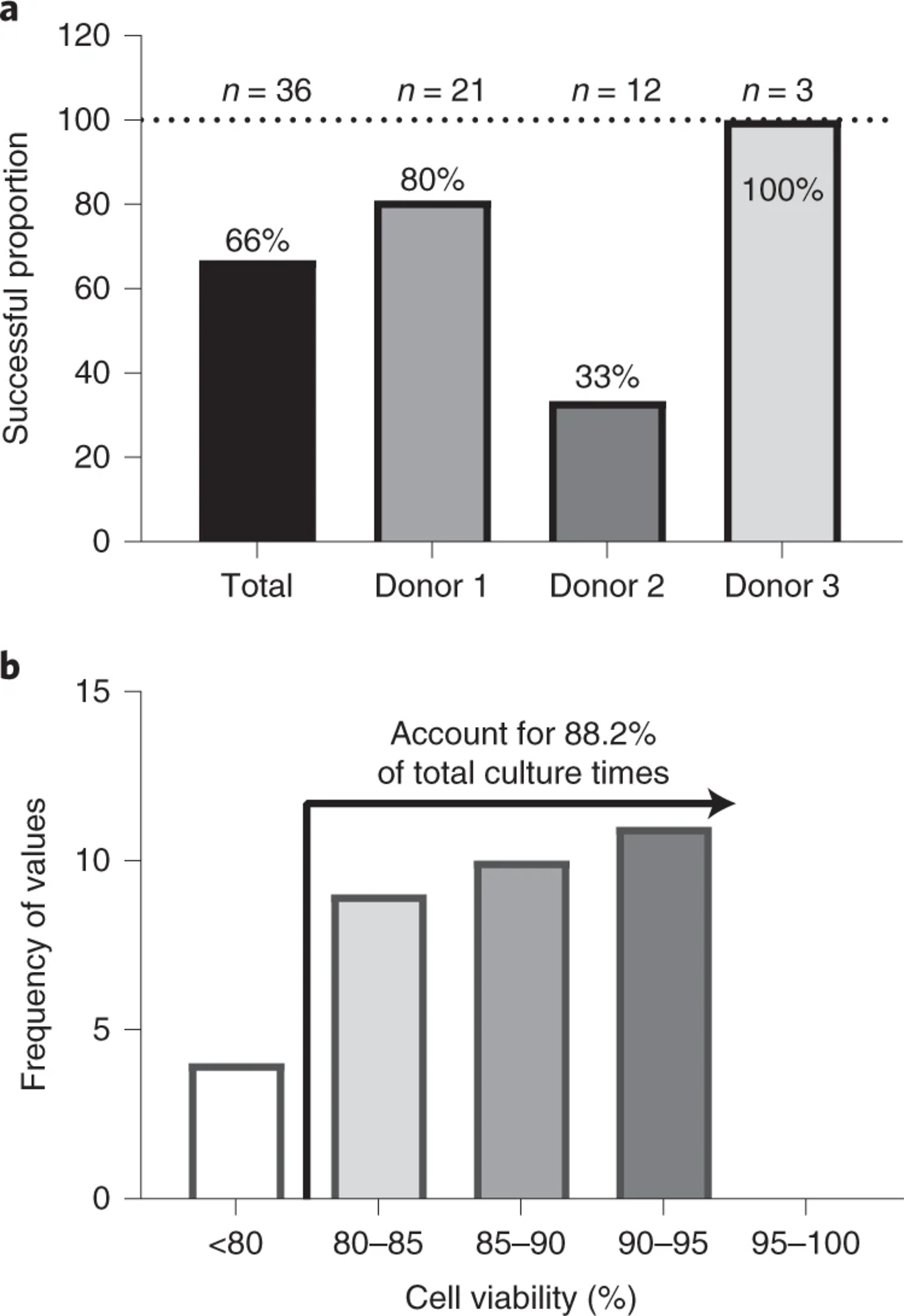
(a) The success rate in generating usable monolayers varied among three donors. n represents the number of attempts (weeks) to generate monolayer in the year 2019. (b) The viability of single cells obtained after processing organoids. In most cases the cell viability reached above 80%. Source Data Figure 2.
Materials
Biological materials
Colon organoids. Colon organoids used in this study were established by the Yilmaz lab at the Koch Institute/Massachusetts Institute of Technology and by the Breault lab at Boston Children’s Hospital, with details given in Table 1. To establish organoid cultures, endoscopic tissue biopsies were collected from the ascending colon of de-identified individuals at either Massachusetts General Hospital or Boston Children’s Hospital upon the donors informed consent. Methods were carried out in accordance to the Institutional Review Board of Boston Children’s Hospital (protocol number IRB-P00000529) and the Koch Institute Institutional Review Board Committee as well as the Massachusetts Institute of Technology Committee on the use of humans as experimental subjects. Once organoids are established, they can be used for monolayer formation after 2 passages from thawing.
!CAUTION The use of human tissues must follow all relevant ethical guidelines and informed consent should be obtained from the tissue donors and the protocol approved by the research institution. Authenticity of the cells should be regularly checked (we undertake a morphology check by microscope) and cultures should be regularly tested for mycoplasma contamination. All work on primary cells should be performed in Biosafety Level 2 labs or higher with proper precautions and personal protection equipment. All cell-containing samples need to be de-contaminated upon disposal as biohazard materials.
Bacteria of interest. We obtained glycerol stocks of B. thetaiotaomicron engineered to contain specific genetic circuits (MT768, MT798, MT799, and MT800) from the Voigt lab at the Massachusetts Institute of Technology.10
Reagents
Advanced DMEM/F12 (Gibco, cat. no. 12634–010, 500 ml)
DMEM/F12 (Sigma-Aldrich, cat. no. D6421–500ML)
HEPES Buffer (Gibco, cat. no. 15630–080, 100x)
Penicillin/Streptomycin (Gibco, cat. no. 15140–148, 100x)
Glutamax (Gibco, cat. no. 35050–061, 100x)
Fetal Bovine Serum, Certified, Heat Inactivated, US Origin (Gibco, cat. no. 10082147)
Dimethyl Sulfoxide (Sigma, cat. no. D2650–100ML)
Wnt, R-spondin, Noggin (WRN) Conditioned Medium. Produced by cell line (ATCC Cat. no. CRL-3276, RRID:CVCL_DA06) following established protocols.57 Alternatively, this media is commercially available, for example, via the Harvard Digestive Diseases Center (HDDC) Organoid Core.
R-Spondin 1 Conditioned Media. Produced by cell line (Sigma-Aldrich, cat. no. SCC111). Alternatively, this conditioned medium is commercially available, for example via the HDDC Organoid Core.
B27 Supplement (Gibco, cat. no. 17504–001, 50x)
N2 Supplement (Gibco, cat. no. 17502–001, 100x)
Nicotinamide (Sigma-Aldrich, cat. no. N0636)
N-acetyl L-cysteine (Sigma-Aldrich, cat. no. A9165)
Y-27632 dihydrochloride (Biogems, cat. no. 1293823)
SB202190 (Biogems; Tocris, cat. no. 1523072; 1264)
A83–01 (Biogems, cat. no. 9094360)
Murine EGF (Peprotech, cat. no. AF-315–09)
Human [leu15]-Gastrin I (Sigma-Aldrich, cat. no. G9145)
Prostaglandin E2 (PGE2, Biogems, cat. no. 3632462)
Thiazovivin (Sigma-Aldrich, cat. no. SML1045)
Human Noggin (Peprotech, cat. no. 120–10C)
DPBS without calcium and magnesium, pH 7.4 (DPBS−/−, Gibco, cat. no. 10010023)
DPBS with calcium and magnesium, (DPBS+/+, Gibco, cat. no. 14040133)
Collagen I protein from rat tail (Gibco, cat. no. A10483–01)
10X Trypsin Solution (Sigma, cat. no. T4549)
TypLE Express (Gibco, cat. no. 1260413)
UltraPure 0.5M EDTA, pH 8.0 (Invitrogen, cat. no. 15575020)
PREemt disinfectant solution (VWR, cat. no. 10822–516)
Growth Factor-Reduced, Phenol Red Free Matrigel (Corning, cat. no. 356231)
▲ CRITICAL For consistent organoid culture the total protein concentration in Matrigel should be above 8.5 mg/mL.
Cell Recovery Solution (Corning, cat. no. 354253)
Trypan Blue (Invitrogen, cat. no. T10282)
Phalloidin-iFluor 488 Reagent (ab176753–300TEST)
DAPI Solution (1 mg/ml) (Invitrogen, cat. no. 62248)
BlockAid Blocking Solution (Invitrogen, cat. no. B10710)
4% formaldehyde (Sigma-Aldrich, cat. no. 100496) !CAUTION Formaldehyde is a Group 1 carcinogen classified by International Agency for Research on Cancer. It should be used in a fume hood and disposed with precaution. Following the recommended manufacturer guidelines.
Equipment
Cell culture
Refrigerated benchtop centrifuge with rotor accommodating 15-mL tubes
Biosafety cabinet (Thermo Fisher Scientific, 1300 Series A2, cat. no. 1305)
CO2 incubator (Thermo Scientific Forma Steri-Cycle Model 370)
Olympus 24-well, flat bottomed, tissue culture treated plates (Olympus, cat. no. 25–107)
Pipettes and pipette tips (1000 μl, 200 μl, 20 μl, 10 μl)
Serological pipettes (Falcon, 5 ml, cat. no. 357543; 10 ml, cat. no. 357551)
15 ml conical tubes (Falcon, cat. no. 352097)
Aluminum cooling block for 1.5/2.0ml Tubes, 15-Well (Thomas Scientific, cat. no. 63615–01)
Ice bucket
Glass dish (150 mm × 75 mm, VWR, cat. no. 10754–778)
2.5 mg/ml Trypsin (Sigma, cat. no. T4549) diluted with 0.45 mM EDTA (Invitrogen, cat. no. 15575020) in DPBS−/−
Trypan Blue (Invitrogen, cat. no. T10282)
Countess Automated Cell Counter and chamber slides (Invitrogen, cat. no. AMQAX1000)
12-well transwell inserts, 0.4 μm pore size, polyester (Corning, cat. no. 3460)
Vacuum line
Colon monolayers on Transwell inserts
EndOhm Cup Chambers (12 mm; WPI, cat. no. ENDOHM-12G)
Epithelial Volt/Ohm Meter (WPI, cat. no. EVOM2) with EVOM2 cable (WPI, cat. no. 53330–01)
70% ethanol. !CAUTION Ethanol is highly flammable and causes serious eye irritation. Use with precaution and appropriate personal protection equipment.
Bacterial culture
Anaerobic workstation (Coy, Vinyl Anaerobic Chamber Type A Glove Box, no. 7000000)
Glycerol stock of B. thetaiotaomicron
Differentiated colon monolayers on transwell inserts
24-well tissue culture plate (Corning, cat. no. 3526)
YCFA media (Anaerobe System, cat. no. AS-680)
YCFA agar plates (Anaerobe System, cat. no. AS-675)
EndOhm chamber and the EVOM2 Epithelial Volt/Ohmmeter
Anaerobic workstation (Vinyl Type A, Coy Laboratory Products Inc, cat. no. 7150000) equipped with pipettes and pipette tips (1000-μl, 200-μl, 20-μl, 10-μl), multi-channel pipette (200-μl, 10-μl), disposable inoculating loop (Fisher Scientific, cat. no. 22–363-597), and 37 °C incubator (Model 2000, Coy Laboratory Products Inc, cat. no. 6100000)
Biosafety cabinet (Thermo Fisher Scientific, 1300 Series A2, cat. no. 1305). To avoid cross contamination we recommend dedicating a biosafety cabinet solely for work with bacteria.
Luminescence measurement
Synergy H1 Hybrid Reader (BioTek, cat. no. 8041000)
Transparent flat-bottom 96-well plate (Corning, cat. no. 351172)
Black walls with clear flat bottom 96-well microtiter plate (Nunc, cat. no. 165305)
Promega Nano-Glo Luciferase System (Promega, cat no. N1120)
Imaging
EVOS M5000 cell imaging system
Zeiss LSM 880 confocal microscope
ProLong Gold antifade reagent (Thermo Fisher, cat. no. P10144).
Microscope glass slide (VWR, cat. no. 16004–368)
Microscope glass coverslip (VWR, cat. no. 48382–042)
Scalpel (Exel International Inc. cat. no. 29550)
Tweezer (Excelta, KST-5)
CoverGrip coverslip sealant (Biotium, cat. no. 23005)
Pipettes and pipette tips (1000 μl, 200 μl, 20 μl, 10 μl)
Software
Zen 2.3 SP1 FP3, Version 14.0.20.201
Graphpad prism v8.0
Microsoft Office 365 ProPlus
Reagent setup
Base Medium. Advanced DMEM/F12, 2mM Glutamax, 10 mM HEPES, 1x Penicillin/Streptomycin. See Table 2Error! Reference source not found..
Antibiotic-free Base Medium. Advanced DMEM/F12, 2mM Glutamax, 10 mM HEPES
Organoid Freezing Medium. 70% base medium, 20% heat-inactivated FBS, 10% DMSO
Organoid Growth Medium. 65% L-WRN conditioned medium, 32% base medium, 1x B-27 Supplement, 1x N-2 Supplement, 10 mM Nicotinamide, 500 μM N-acetyl L-cysteine, 10 μM Y-27632 dihydrochloride, 10 μM SB202190, 500 nM A83–01, 50 ng/ml murine EGF, 10 nM human [Leu15]-Gastrin I, 5 nM PGE2. See Table 3Error! Reference source not found..
Washing Medium. DMEM/F-12, 10% heat-inactivated FBS, 2mM Glutamax, 1x Penicillin/Streptomycin.
Colon Seeding Medium. 65% L-WRN conditioned medium, 32% base medium, 1x B-27 Supplement, 1x N-2 Supplement, 500 μM N-acetyl L-cysteine, 10 μM SB202190, 500 nM A83–01, 2.5 μM thiazovivin, 50 ng/ml murine EGF, 10 nM human [Leu15]-Gastrin I, 5 nM PGE2. See Table 4.
Colon Differentiation Medium. 20% R-spondin1 conditioned medium, 80% antibiotic-free base medium, 1x B-27 Supplement, 1x N-2 Supplement, 500 μM N-acetyl-L-cysteine, 500 nM A83–01, 100 ng/ml noggin, 50 ng/ml murine EGF, 10 nM human [Leu15]-Gastrin I. See Table 5.
Table 2.
Base medium composition
| Component | Stock concentration | Volume added | Final concentration | Recommended storage condition |
|---|---|---|---|---|
|
| ||||
| Advanced DMEM/F12 | NA | 500 ml | NA | 4 °C fridge |
| HEPES buffer | 1 M | 5 ml | 10 mM | 4 °C fridge |
| Glutamax | 200 mM | 5 ml | 2 mM | 4 °C fridge |
| Penicillin/Streptomycin* | 10,000 units/ml | 5 ml | 100 units/ml | −20 °C freezer |
| Final volume | 515 ml | |||
This component is excluded for antibiotic-free culture medium. NA: not applicable. HEPES: 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (CAS no. 7365-45-9). DMEM/F12: Dulbecco’s Modified Essential Medium (DMEM) and Ham’s F-12 Medium.
Table 3.
Colon organoid growth medium composition
| Component | Stock concentration | Volume added | Final concentration | Recommended storage condition |
|---|---|---|---|---|
|
| ||||
| WRN-conditioned media | 100 × | 6.5 ml | 65 × | −20 °C freezer |
| Base media | 100 × | 3.2 ml* | 32 × | 4 °C fridge |
| B27 | 50 × | 200 μl | 1 × | −20 °C freezer |
| Nicotinamide | 1 M | 100 μl | 10 mM | −20 °C freezer |
| N2 | 100 × | 100 μl | 1 × | −20 °C freezer |
| N-acetyl cysteine | 500 mM | 10 μl | 500 μM | −20 °C freezer |
| A 83-01 | 500 μM | 10 μl | 500 nM | −20 °C freezer |
| Y-27632 dihydrochloride | 10 mM | 10 μl | 10 μM | −20 °C freezer |
| SB 202190 | 30 mM | 3.33 μl | 10 μM | −20 °C freezer |
| EGF | 250 μg/ml | 2 μl | 50 ng/ml | −20 °C freezer |
| human [Leu15]-Gastrin | 100 μM | 1 μl | 10 nM | −20 °C freezer |
| Prostaglandin E2 | 10 mM | 2 μl** | 5 nM | −80 °C freezer |
| Final volume | 10.14 ml | |||
Base media consists of Advanced DMEM/F12, 2 mM Glutamax, 10 mM HEPES, 100 units/ml penicillin/streptomycin, described in Table 2. WRN: Wnt, R-spondin 1, Noggin. EGF: epidermal growth factor.
Prostaglandin E2 should be diluted before adding. Make a dilution of 1 μl PGE stock to 349 μl BM. Add 2 μl PGE dilution for 10 ml media.
Table 4.
Details of colon seeding medium composition and preparation
| Component | Stock concentration | Volume added | Final concentration | Recommended storage condition |
|---|---|---|---|---|
|
| ||||
| WRN-conditioned media | 100× | 6.5 ml | 65× | −20 °C freezer |
| Base media* | 100× | 3.2 ml* | 32× | 4 °C fridge |
| B27 | 50× | 200 μl | 1× | −20 °C freezer |
| N2 | 100× | 100 μl | 1× | −20 °C freezer |
| N-acetyl cysteine | 500 mM | 10 μl | 500 μM | −20 °C freezer |
| A83-01 | 500 μM | 10 μl | 500 nM | −20 °C freezer |
| Thiazovivin** | 6.25 mM | 4 μl | 2.5 μM | −80 °C freezer |
| SB202190 | 30 mM | 3.33 μl | 10 μM | −20 °C freezer |
| EGF | 250 μg/ml | 2 μl | 50 ng/ml | −20 °C freezer |
| human [Leu15]-Gastrin | 100 μM | 1 μl | 10 nM | −20 °C freezer |
| Prostaglandin E2 | 10 mM | 2 μl*** | 5 nM | −80 °C freezer |
| Final volume | 10.03 ml | |||
Base media consists of Advanced DMEM/F12, 2mM Glutamax, 10 mM HEPES, 100 units/ml penicillin/streptomycin as described in Table 1.
Nicotinamide is removed and thiazovivin is used instead of Y-27632 dihydrochloride in seeding medium to allow differentiation into secretory cell types in monolayers as reported by Kozuka et al.27
Prostaglandin E2 should be diluted before use. Make a dilution of 1 μl PGE stock to 349 pl base media. Add 2 μl PGE dilution for 10 ml media
Table 5.
Colon differentiation medium composition
| Component | Stock concentration | Volume added | Final concentration | Recommended storage condition |
|---|---|---|---|---|
|
| ||||
| R-spondin 1 conditioned media | 100 × | 2 ml | 20 × | −20 °C freezer |
| Base media* | 100 × | 8 ml* | 80 × | 4 °C fridge |
| B27 | 50 × | 200 μl | 1 × | −20 °C freezer |
| N2 | 100 × | 100 μl | 1 × | −20 °C freezer |
| N-acetyl cysteine | 500 mM | 10 μl | 500 μM | −20 °C freezer |
| A83-01 | 500 μM | 10 μl | 500 nM | −20 °C freezer |
| Noggin | 100 μg/ml | 10 μl | 100 ng/ml | −80 °C freezer |
| EGF | 250 μg/ml | 2 μl | 50 ng/ml | −20 °C freezer |
| human [Leu15]-Gastrin | 100 μM | 1 μl | 10 nM | −20 °C freezer |
| Final volume | 10.03 ml | |||
Base media consists of Advanced DMEM/F12 (Gibco, 12634-01), 2mM Glutamax, 10 mM HEPES, 100 units/ml. Penicillin/Streptomycin. For antibiotic-free culture, penicillin/streptomycin is excluded, as described in Table 1.
▲ CRITICAL We recommend preparing the organoid growth medium, colon seeding medium, and colon differentiation medium fresh every time. We never used medium that was older than one week. Base medium, organoid freezing medium, and washing medium can be prepared ahead of time and stored at 4 °C for up to three months.
Procedure
Colon Organoid Seeding
-
Timing 3 days (organoid growth); 2–3 h (organoid seeding)
-
1
Set up the centrifuge to accommodate 15 ml conical tubes and cool down to 4 °C. ▲ CRITICAL Centrifuge needs to be at 4 °C for step 9.
-
2
Sterilize biosafety cabinet using PREempt disinfectant solution and 70% ethanol
-
3
Obtain a bucket of ice.
-
4
Thaw an appropriate number of Matrigel aliquots in a 4 °C fridge (1.2 ml Matrigel is required for a full 24-well plate of organoids droplets). ▲CRITICAL Matrigel needs to be thawed before step 11.
-
5
Prepare base medium, colon organoid medium and washing medium as described in Table 2, Table 3, and Materials. 15 ml of colon organoid medium is required for a full 24-well plate with organoid droplets.
-
6
Obtain a cryovial of organoids from the liquid nitrogen tank and place it into a 37°C water bath. Hold in the water bath until thawed (approximately 1 min)
-
7
Bring the cryovial into the biosafety cabinet after spaying the outer surface with 70% ethanol.
-
8
Add 1 mL of washing medium to the cryovial, resuspend the organoids gently and then transfer the content of the cryotube to the 15 mL conical tube that already contains 10 mL of washing medium. Fill the tube with additional washing medium up to 15 mL.
-
9
Centrifuge at 500 g for 5 min at 4°C. During centrifugation, prepare the material for the subsequent steps: Sterilize a glass dish with PREempt disinfectant solution and 70% ethanol. Add ice to 50–70% capacity of the glass dish, spray with 70% ethanol again, and place the dish into the biosafety cabinet. Put Matrigel aliquots into the aluminum cooling block. Spray thoroughly with 70% ethanol and place gently into the glass dish containing ice. ▲CRITICAL Ice is not sterile, use with extra caution in the biosafety cabinet.
-
10
After centrifugation, aspirate the supernatant (washing medium) with vacuum using a sterile Pasteur pipette.
-
11
Resuspend the organoid pellet with 1.2 mL of Matrigel and seed 50 μL droplets into each well of an Olympus 24-well, tissue culture treated plate. ▲CRITICAL It is important to use Olympus 24-well tissue culture treated plates as they maintain the Matrigel droplets spherical which is critical for 3D organoid growth.
-
12
Carefully transfer the plate(s) containing Matrigel droplets to the CO2 incubator. Incubate for 15–20 min to allow Matrigel gelation. After 15–20 min, check the Matrigel gelation by tilting the plates. If the Matrigel holds firm, they should be ready.
-
13
Place the plate(s) inside the biosafety cabinet and add 600 μl of colon organoid media to each well. Carefully dispense the medium into the side of the well to avoid disturbing the Matrigel droplet. At this point, organoids would be small. In some cases is possible to see fragmented organoids with an inverted bright field microscope.
-
14
After 3 days of culture, observe under an inverted microscope to ensure organoids increased in size (>100 microns in diameter) and proceed with the organoid passaging protocol (i.e. if thawed on Friday, passage on Monday).
-
1
Colon Organoid Passaging
-
Timing 7 days (organoid growth); 4–5 h (organoid passaging)
▲ CRITICAL Organoids should be passaged at a 7-day cycle (e.g. passage organoids every Monday). Avoid culturing organoids for more than 15 consecutive passages after thawing.
-
15
Set up the centrifuge to accommodate 15 ml conical tubes and cool down to 4 °C. ▲CRITICAL Centrifuge needs to be at 4 °C for steps 21 and 25.
-
16
repeat steps 2 and 3.
-
17
Check the organoid droplets under an inverted microscope to evaluate if the size of most organoids is above 100 μm in diameter or larger. Estimate the split ratio to use for passaging organoids to calculate the amount of Matrigel to thaw. ?TROUBLESHOOTING.
-
18
Thaw an appropriate number of Matrigel aliquots in a 4 °C fridge (1.2 ml Matrigel is required for a full 24-well plate of organoids droplets). Thaw 1.2 ml additional Matrigel in case higher split ratio will be used. ▲CRITICAL Matrigel needs to be thawed before step 27. Thaw extra Matrigel ahead in case higher splitting ratio is needed in step 27.
-
19
Prepare colon organoid medium (15 ml for every 24-well plate) by first making up base medium, as described in Table 2, and then using this as a component of colon organoid medium, made as described in Table 3. Alternatively, this media can be prepared during cell recovery solution (CRS) incubation (step 24).
-
20
Collect organoid droplets using the medium they are currently growing. Using a 1000-μl pipette, set the pipette to 650 μl (there should be a maximum volume in each well of 50 μl Matrigel and 600 μl medium). Scrape the bottom of the well with the pipette tip to dislodge the Matrigel droplet before drawing up the medium. Place medium and organoids into a 15 ml conical tube. One 24-well plate of organoids should fit into one 15 ml conical tube. ▲CRITICAL Allow space beneath the pipette tip when drawing up the Matrigel suspension (do not press against bottom of the well). If an obvious amount of Matrigel droplet remains adhered to the plate after drawing up the medium, return the medium to the well and repeat scraping to dislodge the remaining Matrigel. Small fragments of the Matrigel droplet will likely remain stuck to the plate even after repeating the procedure. As long as ≥ 90% of the droplet is collected that is acceptable, as negligible number of organoids will be left behind in these small fragments.
-
21
Centrifuge the collected Matrigel droplets at 1000 g for 5 min at 4°C. After centrifugation, there should be approximately 1 ml Matrigel pellet per a 24-well plate of organoid droplets collected.
-
22
Aspirate the supernatant (spent organoid medium) with vacuum using a sterile Pasteur pipette.
-
23
Resuspend the Matrigel pellet in CRS using a 1:10 dilution. For a full 24-well plate of organoids use 12 ml of CRS. Resuspend the Matrigel droplet by gently inverting the 15-ml conical tube. ▲CRITICAL Do not resuspend the droplet by repeated pipetting as organoid will adhere to the serological pipette and there might be some organoid loss.
-
24
Incubate the resuspended Matrigel in the ice bucket for 45–60 min, or until Matrigel is dissolved. Mix the Matrigel-CRS solution via gentle inversion every 10–15 min. Organoids will settle at the bottom of the tube when Matrigel is fully dissolved. ▲CRITICAL Do not exceed 60 min incubation as this can decrease cell viability. Do not vortex the organoid suspension.
-
25
Centrifuge at 1000 g for 5 min at 4 °C. After centrifugation, there should be a noticeable organoid pellet, approximately 200 μl volume of organoid, depending on the density of the original culture. During centrifugation, prepare the material for the subsequent steps: Sterilize a glass dish with PREempt disinfectant solution and 70% ethanol. Add ice to 50–70% capacity of the glass dish, spray with 70% ethanol again, and place the dish into the biosafety cabinet. Put Matrigel aliquots into the aluminum cooling block. Spray thoroughly with 70% ethanol and place it into the glass dish containing the ice. ▲CRITICAL Ice is not sterile, use with extra caution in the biosafety cabinet.
-
26
Aspirate CRS using vacuum and a sterile Pasteur pipette. After aspiration, place the conical tube into the glass dish containing ice. ▲CRITICAL Remove the CRS as much as possible, as remaining CRS could decrease the cell viability and compromise the integrity of the Matrigel matrix.
-
27
Calculate the volume needed for the desired splitting ratio allowing 50 μL of Matrigel per well (i.e., 1.2 ml Matrigel per 24-well plate). A typical splitting ratio is 1:3 to 1:5, but this varies and depends on the density of the original culture, length of culture since original thaw, and donor intrinsic growth behavior.
-
28
Keep the tube on ice and resuspend the organoid pellet in a maximum of 1 ml of Matrigel. If the total volume required is less than 1 ml, add the required volume, for example, if 900 μl is needed to achieve the desired splitting ratio, add 900 μl. ▲CRITICAL 1 ml is initially added since a larger volume makes it harder to dissociate the organoids and leads to a more diverse organoid fragment size.
-
29
Bend a 1000-μl pipette tip by pressing down the pipette tip on the inside of the cap for the conical tube. Using this bend tip to pipette the Matrigel suspension 25 times if organoids are in the first passage after thawing. Otherwise pipette the Matrigel suspension for 50 times. After resuspension check the level of organoid fragmentation under an inverted microscope. If further dissociation is needed, return conical tube to ice in the biosafety cabinet and continue pipetting with bent pipette tip for 25 more times and re-examine under the microscope. Repeat until mostly fragments remain. ▲CRITICAL Organoids are more fragile after the first thawing and will be dissociated into fragments much more easily. Do not observe organoid fragmentation under the microscope for longer than 10 s to avoid Matrigel polymerization. If more than 10 s is needed to evaluate the level of organoid fragmentation, look in 10 s intervals, returning the tube to the ice bucket between intervals. An alternative approach is to take a 5–10 μL aliquot of the organoid/Matrigel mixture using a wide orifice pipette tip, place the droplet on a microscope slide and assess organoid fragmentation. A Matrigel suspension with mostly organoid fragments is desired.
-
30
Once organoids have been fragmented, add the remaining Matrigel needed (calculated in step 14) to achieve the desired passaging ratio. Mix the Matrigel-organoid fragment suspension well before proceeding to the next step.
-
31
Seed 50 μl droplets of the fragment suspension into each well of an Olympus 24-well, tissue culture treated plate. ▲CRITICAL It is important to use Olympus 24-well tissue culture treated plates as they maintain the Matrigel droplets spherical which is critical for 3D organoid growth. Using Falcon tissue culture treated plates results in the spreading and flattening of the Matrigel droplet. Alternatively, Falcon non-tissue culture treated plates may be used, but as these do not allow adhesion of the droplets, there is a high risk of the Matrigel droplets dislodging from the well and being aspirated when changing media.
-
32
Carefully transfer the plate(s) containing Matrigel droplets to the CO2 incubator. incubate the plates for 15–20 min to allow Matrigel gelation. After 15–20 min, check the Matrigel gelation by tilting the plates. If the Matrigel holds firm, they should be ready.
-
33
Place the plate(s) in the biosafety cabinet. Add 600 μl of colon organoid media to each well by carefully loading into the side of the well to avoid disturbing the Matrigel droplet.
-
34
Incubate tissue culture plates in cell culture incubator at 37 °C, 95% air, and 5% CO2 for 4 days (i.e., if passaged on Monday, move to next step to change media on Friday).
-
15
Colon organoid media change
-
Timing 0.5–1.5 h
-
35
Sterilize biosafety cabinet with PREempt disinfectant solution and 70% ethanol
-
36
Prepare organoid medium (15 ml for every plate) according to Table 3Error! Reference source not found.. Media should be prepared fresh each time, as the small molecules and proteins can become unstable if they remain in the media for a long time.
-
37
Carefully aspirate the media from each of the wells using vacuum aspirator and a sterile Pasteur pipette. When aspirating, tilt the plate towards yourself and press the pipette tip against the bottom of the well to avoid disrupting the Matrigel droplet adhered in the center of the well.
-
38
Add 600 μl of media per well by carefully loading into the side of the well to avoid disturbing the Matrigel droplet.
-
39
Place plate back into incubator. Generally, organoids are ready for both passaging and monolayer seeding 3 days later (7 days after seeding).
-
35
Colon Monolayer Seeding (Single Cells) ● Timing 4–6 h; 2.5 h (transwell coating); 2–4 h (monolayer seeding)
Transwell coating
-
40
Obtain a bucket of ice and keep the collagen I on ice
-
41
Sterilize biosafety cabinet using PREempt disinfectant solution and 70% ethanol.
-
42
Prepare a coating solution containing 50 μg/ml collagen I in PBS−/−. For 12-well Transwells add 300 μl of coating solution on the apical side and 1200 μl on the basolateral side.
-
43
Incubate the Transwells at 37 °C for at least 2 h to allow collagen coating to occur.
Organoid collection, single cell dissociation and monolayer seeding
-
44
Prepare colon monolayer seeding medium as described in Table 4. At least 2 ml should be made for each Transwell plus an additional 1–2 ml for cell counting. Alternatively, media can be prepared during CRS incubation (step 46).
-
45
Collect organoids as described in steps 20–23.
-
46
After resuspending in CRS, incubate on ice for 45–60 min, or until Matrigel is dissolved. Resuspend the Matrigel via gentle inversion every 10–15 min and proceed to the next step 15 min before the end of incubation. ▲CRITICAL Do not exceed 60 min incubation time in CRS as this can decrease cell viability. The trypsin/EDTA used in following steps will degrade most of any remaining matrix.
-
47
With 15 min remaining for the CRS incubation, place a 1x Trypsin/EDTA aliquot into a 37 °C water bath.
-
48
Centrifuge organoids from step 46 at 1000 g for 5 min at 4 °C. After centrifugation, there should be a noticeable organoid pellet, approximately 50–200 μl in size depending on the density of the original culture.
-
49
During centrifugation, place pre-warmed trypsin/EDTA and washing medium inside the biosafety cabinet. Make aliquots of 15 mL of washing medium. One 15 mL aliquot is needed per plate of organoid culture.
-
50
After centrifugation, place the 15-ml conical tube containing the organoids inside the biosafety cabinet and aspirate the CRS using vacuum and a sterile Pasteur pipette. ▲CRITICAL While aspirating the CRS slowly tip the 15 mL tube and avoid placing the Pasteur pipette too close to the organoid pellet.
-
51
Resuspend the organoid pellet in 1 ml pre-warmed trypsin/EDTA via short and quick repeat pipetting. Incubate the organoid pellet in a 37 °C water bath for 5 min. ▲CRITICAL Avoid completely drawing up the organoid suspension/pellet when adding trypsin/EDTA. Organoids are very “sticky” after CRS incubation, so drawing the whole suspension into the pipette tip will result in organoid loss.
-
52
After incubation in trypsin/EDTA take the 15 mL conical tube inside a biosafety cabinet to initiate single cell preparation. To break up the organoids, first pre-wet a 1000 μl pipette tip in washing medium, then bend the pipette tip by pressing the tip on the inside of the 15-ml conical tube cap. Finally, to dissociate the organoids and generate single cells pipette the organoids 50 times and then inspect under the microscope for single cells. If a significant number of organoids and/or fragments remains, pipette with a bent tip for an additional 50 times before evaluating again. Repeat cycles of pipetting 50 times until the suspension is primarily single cells. ▲CRITICAL. Pipette tip needs to be wet before bending. This will decrease the organoid adherence to the pipette tip. Do not exceed 10 min total in trypsin/EDTA, because this will decrease cell viability. Even if the cells have not reached primarily single cells after 10 min of mechanical dissociation, proceed to next step. It is important to maintain the cell viability. Foaming might be present during single cell preparation, but it should not affect cell viability. With a new (non-bent) pipette tip, add washing medium to the cell suspension to neutralize the trypsin/EDTA. If cell dissociation resulted in a single-cell suspension in the previous step use a serological pipette to add a sufficient volume of washing medium to the cell suspension to reach 15 ml. If cell dissociation did not reach single-cell suspension, add 1 ml of washing medium to the cell suspension, and continue pipetting with a bent pipette tip until a single-cell suspension is reached. Then use a serological pipette to add a sufficient volume of washing medium to the cell suspension to reach 15 ml. ▲CRITICAL Do not mix with a serological pipette, as this results in cell loss.
-
53
Centrifuge single-cell suspension at 400 g for 5 min at 4°C. ▲CRITICAL Make sure the centrifuge is at no higher than 800 g to preserve high cell viability.
-
54
Aspirate and discard the supernatant using vacuum and a sterile Pasteur pipette. ▲CRITICAL Leave ~50 μl of liquid at the bottom of the tube to avoid cell loss that occurs from placing the Pasteur pipette too close to the cell pellet during aspiration.
-
55
Resuspend the cell pellet in 1 ml of colon monolayer seeding medium. Take a 15 μl aliquot of the cell suspension to determine cell density. ▲CRITICAL Ensure the suspension is well mixed before taking the aliquot. Take 15 μl from the middle of the cell suspension to make it as representative as possible of the entire suspension.
-
56
Add 15 μl of trypan blue to the 15 μl aliquot cell suspension and mix well via repeated pipetting. Count cells and assess viability by adding 10 μl of the suspension to each side of a disposable Countess chamber slide and inserting into countess machine. Determine cell density from both sides of the slide and record live cell density and % viability. ?TROUBLESHOOTING !CAUTION If there is a large discrepancy between the two cell counts, repeat cell counting with a new aliquot. An alternative protocol for cell counting is to use a hemocytometer following the manufacturer’s protocol for cell density determination.
-
57
Calculate the total number of cells needed from the cell suspension to seed the desired number of monolayers. For 12-well Transwells inserts seed 3.0×105 cells per Transwell. Alternatively, in case of low density during single cell preparation, seed 2.5×105 cells per Transwell insert.
-
58
From the original single cell preparation, transfer the volume of cells needed for seeding to a new 15 ml conical tube. Add additional colon monolayer seeding medium to reach necessary density for seeding transwells. For 12-well transwells, 0.5 ml per well is needed, which is equivalent to a density of 6×105 cells/ml.
-
59
Bring the coated transwells from step 43 from the incubator into the biosafety cabinet. Completely aspirate the coating solution from both sides of the transwells. Rinse each transwell at least once with PBS−/−. For 12-well transwells, rinse with 1 ml on the apical side and 2 ml on the basolateral side.
-
60
Mix the single cell suspension well via repeated pipetting to get it ready for seeding into transwells. Remove the PBS−/− from the apical side of each transwell insert prior to the addition of the cell suspension. For 12-well transwells, add 0.5 ml of the cell suspension into the apical side.
-
61
Add colon monolayer seeding medium to the basolateral side of each transwell. For 12-well Transwells, add 1.5 ml of medium per transwell.
-
62
Place the seeded Transwells in a 37 °C incubator at 5% CO2. Inspect the transwells every day under an inverted bright field microscope for confluency. ▲CRITICAL Colon cells are flat and hard to see when attached to the Transwell membrane, as time progresses the tight junctions will be more visible under phase contrast. Proceed to next step when the TEER values reaches approximately 200 Ω cm2 or higher. Typically, the first media change (to start differentiation) and TEER measurement occur on day three after seeding (i.e., if seeded on Mondays, media change and first TEER measurement take place on Thursdays).
Monolayer differentiation and transepithelial electrical resistance (TEER) monitoring ● Timing 1–2 h
▲CRITICAL This section describes how to monitor TEER using EndOhm Cup Chambers and Epithelial Volt/Ohm Meter, whilst we have not tested alternative systems it is likely they could be substituted, and the steps described here can be adapted for use with other systems. Independent of the instrument used to monitor TEER, first sterilize the biosafety cabinet with PREempt disinfectant solution and 70% ethanol.
-
63
Make aliquots of base media and PBS−/−. Typically, 45 ml of base media and 40 ml of PBS−/− should be sufficient for one 12-well plate of monolayers. Pre-warm the aliquots in a 37 °C water bath for about 10 min.
-
64
To disinfect the tweezers needed for transwell manipulation during TEER measurement, prepare two, 45 ml aliquots of 70% ethanol in 50 mL conical tubes.
-
65
Place a pair of long tweezers in one of the 70% ethanol aliquots for sterilization.
-
66
Prepare differentiation medium according to Reagent Setup and Table 5. ▲CRITICAL Media should be freshly prepared whenever possible.
-
67
If the cover to the EndOhm cup chamber is held shut with tape or parafilm, remove the cover. Spray the outer surface of the chamber and connecting wires thoroughly with 70% ethanol. Avoid getting excessive amounts of ethanol on the ends of the wire, as this can cause inaccurate measurements at later steps. Open the chamber and spray 70% ethanol on both the bottom and inside of the cap.
-
68
Bring the EndOhm chamber into the biosafety cabinet, leave the end of the wire that connects to the Ohmmeter outside the cabinet.
-
69
Place the Ohmmeter next to the biosafety cabinet (on a small, portable table) and connect the wire to the Ohmmeter. Keep the Ohmmeter turned off.
-
70
Rinse the EndOhm chamber with 6 ml of 70% ethanol and then fill with 6 ml of fresh 70% ethanol and let it sit for at least 5 min. !CAUTION Do not exceed 30 min in 70% ethanol. This could damage the wire contacts in the EndOhm chamber and lead to inaccurate measurements. For 12-well Transwells, 6 ml is sufficient to fill the chamber.
-
71
Aspirate the 70% ethanol completely from the EndOhm chamber. Wash and rinse the chamber completely at least three times as follows: After removing the 70% ethanol, wash the EndOhm chamber once with 6 ml of pre-warmed PBS−/−, then fill the chamber with 6 ml of pre-warm PBS−/−. Let it sit for 2 min. Aspirate the PBS−/−, then wash again with 6 ml of PBS−/−. After this wash, add 3 ml of PBS−/− to the EndOhm chamber, place the top cover on top of the chamber and set aside.
-
72
Turn on the Ohmmeter and record the background measurement when in PBS−/−. The background measurement will vary due to a wide range of factors and readings from −5 Ω to +5 Ω are acceptable for the 12-well chamber. ▲CRITICAL If the background measurement is abnormal, leave the system to sit and equilibrate in PBS−/− for longer.
-
73
After recording the ohms background, aspirate the PBS−/− from the chamber. Rinse completely with 6 ml of pre-warmed base media at least three times. ▲CRITICAL On the second rinse, let sit for 2 min to allow any remaining PBS−/− to leach out of the wire contacts. On the final rinse, add 2.5 ml of base medium needed in the chamber for TEER measurements.
-
74
Remove tweezers/forceps from 70% ethanol. Allow the alcohol to evaporate from the forceps inside the biosafety cabinet, making sure the tweezers/forceps tips are away from any surface.
-
75
Using a vacuum aspirator and Pasteur pipette, aspirate the media from both apical and the basolateral side of the Transwells. Tilt the plate towards yourself and aspirate with the pipette tip pressed against the bottom of the well in the basolateral side. Completely aspirate the basolateral side of every transwell first, then go back and aspirate the apical side of each transwell. This order of aspiration is recommended by the transwell manufacturers. ▲CRITICAL Be very careful when aspirating the apical side of the transwell. Do not completely aspirate the apical side (leave ~50 μl), to avoid damaging the monolayer. ▲CRITICAL If you wish to determine the proliferative state of the monolayer using EdU staining, follow the instructions given in Box 1.
-
76
Add base media to the apical side of each transwell. For 12-well transwells, add 500 μl to the apical side. ▲CRITICAL Dead/detached cells are expected to be floating on top of the monolayer at the first media change (i.e., 3 days after seeding), hence it is critical rinse the apical side with 500 μl of base media once before adding fresh 500 μl of base media. This extra rinse step will help remove dead cells and allow more stable TEER measurements.
-
77
Add base media to the basolateral side of the transwells. For 12-well transwells, add 1.5 ml to the basolateral side.
-
78
Using the alcohol-sterilized and dried forceps, carefully pick up a transwell and place it into the EndOhm chamber. Record the TEER measurement, then carefully place the transwell back into the 12-well plate. Repeat for all transwells. ▲CRITICAL Make sure the 70% alcohol is completely evaporated from the tweezers/forceps.
-
79
At the end of all TEER measurements, aspirate the media from the basolateral side and add differentiation media. For 12-well transwells, 1.5 ml differentiation media is required per well.
-
80
Return the monolayers to the incubator and check the monolayers under inverted microscope every day to inspect the confluency. Proceed to the next step to prepare bacteria for co-culture at day 7 after monolayer seeding. ?TROUBLESHOOTING
Co-culture of Monolayers with B. thetaiotaomicron and downstream analysis● Timing set up 2–3 h, whole process 2 d
Bacterial culture preparation
-
81
Place the bacterial glycerol stocks in a cold metal block in an anaerobic workstation. If you wish to replicate our study,10 use B. thetaiotaomicron strains MT768, MT798, MT799, and MT800.
-
82
Use a disposable inoculating loop to scrape the top of the glycerol stock and inoculate the scraped ice onto the YCFA agar plate, invert the agar plate, seal it into a plastic bag and place it into the incubator (37 °C) inside the anaerobic workstation for 24–36 h.
-
83
After incubation for colony formation, use a disposable inoculating loop to pick one colony and inoculate it into liquid YCFA media (~7 ml) and place it into the incubator (37 °C) inside the anaerobic workstation for 24 h. ?TROUBLESHOOTING
-
84
The next day, subculture the bacteria using a 1:1000 dilution and let it grow overnight. Once it has been incubated overnight, the bacterial culture is ready to use.
-
85
To determine the bacterial CFU bring a 96-well plate into the anaerobic workstation and add 90 μl of pre-reduced YCFA media or DPBS buffer into each well.
-
86
Use a 10-μl pipette to transfer 10 μl of bacterial culture into each well of the first row (row A), mix thoroughly, and make a serial dilution with rows A to H to cover dilution of 10−1 to 10−8 (Supplementary Figure 1).
-
87
Use a multi-channel pipette to transfer 2 μl of the diluted bacterial culture from all eight wells (rows A-H) onto a pre-reduced YCFA agar plate (Supplementary Figure 1). Culture the plate anaerobically for 24 h at 37 °C.
-
88
The next day, inspect the agar plate and count the bacterial colonies from wells in which the colonies can be easily counted with the naked eye. Record the number of colonies as N. ?TROUBLESHOOTING
-
89
Calculate the bacterial CFU per ml of original culture using the following formula: CFU/ml = N × 10(dilution) × 500.
-
90
To determine bacterial growth over time, repeat step 86–89 during bacterial growth of each strain in step 84 every 2–3 h. Record the OD600 value and calculate the CFU of 200-μl culture from the same bacterial culture at each time point.
-
91
Generate a standard curve by plotting the OD600 values and the bacterial concentration (CFU/ml) (Supplementary Table 1).
Co-culture of monolayer with bacteria
-
92
Carefully aspirate the apical media from each of the wells of the plates from step 80 using a vacuum aspirator and Pasteur pipette tip. ▲CRITICAL Tilt the plate towards yourself and aspirate with the pipette tip pressed against the wall of the transwell insert (avoid placing the Pasteur pipette tip too close to the monolayer). ▲CRITICAL We recommend additional TEER measurements be taken immediately prior to co-culture with bacteria. To do this, follow steps 63–80, note that it is not necessary to carry out steps 76–77 as medium is not being changed.
-
93
Sterilize the outer surface of the overnight-grown bacterial culture tubes from step 86 and the pre-reduced YCFA media with 70% ethanol and bring them into the biosafety cabinet.
-
94
Make a 1:100 dilution for the overnight-grown bacterial culture in step 93 with pre-reduced YCFA media. Add 500 μl of the diluted bacteria culture onto the apical side of the monolayer and immediately put the covered monolayer back into the incubator (5% CO2, 37 °C) for 2 h. ▲CRITICAL It is critical to use media that is not toxic to the monolayers. Experimentally we foud that two commonly used bacterial media; tryptone-yeast extract-glucose (TYG) and brain-heart-infusion (BHI) are toxic to the epithelial monolayers. Therefore, a pretest for the toxic effect of the bacterial media on the monolayers is recommended. ?TROUBLESHOOTING
-
95
After 2 h, remove the plate containing co-culture of monolayer and bacteria from the incubator. Use a 1000-μl pipette to transfer the apical media from each well into a new 24-well tissue culture plate and set aside. Also set aside the transwell for further use. ▲CRITICAL Label the 24-well plate containing the apical media with the corresponding transwell, as the media should be transferred back to the original transwell in step 95.
-
96
Add an appropriate inducer into the apical media collected in the 24-well tissue culture plate and mix it thoroughly but gently.
-
97
Use a 1000-μl pipette to transfer the inducer-containing apical media back onto the corresponding monolayer. Continue the co-culture in the incubator (5% CO2, 37 °C) for another 6 h.
-
98
After 6 more h, use a 1000-μl pipette to withdraw 450 μl of the apical media from the top of each well and transfer to a new 24-well plate. ▲CRITICAL Again, record which monolayer the media was removed from. Transfer 200 μl into a transparent 96-well plate for OD600 measurement. ▲CRITICAL Keep the pipette tips only on the surface of the apical medium when drawing the liquid to avoid disturbing the dense bacterial layer at the epithelial surface.
-
99
Use a 200-μl pipette to transfer the residual media (~ 50 μL) containing the bacterial cells settling on the surface of the epithelial cells into another new 24-well tissue culture plate.
-
100
Use a 20-μl pipette to take 20 μl of residual media from step 99 and dilute it into a transparent 96-well plate, make 1:10 dilutions with pre-reduced YCFA ready for OD600 measurement.
-
101
Use a 20-μl pipette to take 15 μl residual media from step 99 and mix it with 15 μl of Nanoluc reaction buffer (Promega Nano-Glo Luciferase System N1120).
-
102
Measure the luminescence of the plate prepared in steps 101 and measure the OD600 of the plate prepared in step 100. For luminescence we used a Synergy H1 Hybrid Reader. ▲CRITICAL For luminescence recording, 1 s at a gain setting of 100 was used.
Luminescence data analysis
-
103
Calculate the CFU for each sample from steps 98 and 102 by comparing the OD600 readings obtained with the standard curve generated in step 91 (Supplementary Table 2).
-
104
Calculate the relative luminescence unit (RLU) by dividing the raw luminescence values obtained from the samples prepared in step 102 by the CFU obtained from step 102 and standard curve from step 91.
-
105
Normalize the RLU value to relative promoter units (RPUL) by dividing the RLU values of target strain by that of an RPUL standard strain. For example, we divide the RLU values B. thetaiotaomicron strain MT798, MT799, and MT800 by that of the B. thetaiotaomicron strain MT768 (Supplementary Table 2).
Sample preparation for immunofluorescent imaging.
-
106
Aspirate the residual apical and basolateral media and replace them with 150 and 500 μl of fixative solution (4% paraformaldehyde in DPBS−/−) into the apical and basolateral sides, respectively. Incubate at room temperature for 10 min. !CAUTION Formaldehyde is Group 1 carcinogen classified by International Agency for Research on Cancer. It should be used in a fume hood and disposed of with precaution.
-
107
Manually aspirate the fixative solution using a 1000-μl pipette. ▲CRITICAL Gently remove the formaldehyde solution from the apical side to avoid bacterial cell loss.
-
108
Add 150 and 500 μl of 0.2% Triton-X into the apical and basolateral sides, respectively, and incubate at room temperature for 10 min. ▲CRITICAL It is not recommended to wash the monolayer with DPBS−/− as this would cause bacterial cell loss from the apical side.
-
109
Aspirate the Triton-X solution using a 1000-μl pipette. ▲CRITICAL Gently remove the Triton-X solution from the apical side to avoid bacterial cell loss. It is not recommended to wash the monolayer with DPBS−/− as this would cause bacterial cell loss from the apical side.
-
110
Prepare a staining solution by diluting Phalloidin-iFluor 488 and DAPI (both 1:1000 dilution) in Blockaid solution.
-
111
Add 200 μl and 1 ml staining solution into the apical and basolateral sides, respectively. Cover the plate with aluminum foil and incubate overnight at 4 °C.
-
112
Aspirate the staining solution using a 1000-μl pipette. ▲CRITICAL Gently remove the staining solution from the apical side to avoid bacterial cell loss.
-
113
Gently add 300 μl DPBS−/− to the apical side and 1.5 ml to the basolateral side and incubate at room temperature for 5 min.
-
114
Aspirate the DPBS−/− using a 1000-μl pipette. ▲CRITICAL Gently remove the staining solution from the apical side to avoid bacterial cell loss.
-
115
Flip the transwell insert and place it firmly on a flat surface.
-
116
Use the Scalpel cut the whole membrane containing the stained epithelial and bacterial cells.
-
117
Transfer the membrane to a glass slide with a tweezer. ▲CRITICAL Make sure the cell layer is facing up.
-
118
Add one drop of ProLong Gold antifade reagent onto the membrane.
-
119
Mount a cover slip on top of the membrane. ▲CRITICAL Place the cover slip to one side of the membrane and then slowly cover the whole membrane to avoid getting air bubbles. Tiny bubbles that cover <5% of the membrane are acceptable.
-
120
Seal the glass-membrane-cover slip sandwich by placing coverslip sealant on the edge of the cover slip. Wait for ~15 min to allow the sealant dry prior to imaging. PAUSE POINT After the sealant is dried, the slide can be stored at room temperature for <48 h.
Troubleshooting
For troubleshooting guidance, refer to Table 6.
Table 6:
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 17 | Organoid morphology is not spherical or cyst-like | The organoids are too differentiated, i.e., the shape is not cyst-like, but has thick and folded edges (Figure 3). | Break down the fragments more when passaging the organoids. Reduce cell density by increasing the split ratio |
| 17 | Organoid yield low | Organoids were seeded too sparsely or are not healthy (Figure 4). We observed that this occurs occasionally after thawing a new vial of organoids | Decrease the splitting ratio, (e.g., to 1:2 split ratio) Check the quality of the WRN-Conditioned medium by assessing Wnt activity.57 |
| 17 | Organoids stop growing after a few passages | Unknown – suspect genetics are deficient for growth, we observed that this is donor specific and organoids derived from diseased donors are more prone to stopping growing. We encountered organoids from one donor that stopped growing a few passages following thawing. Mycoplasma contamination |
Thaw a new vial of organoids, or switch to using organoids from another donor. Check the WRN-Conditioned medium has high Wnt activity.57 Check for mycoplasma contamination regularly. |
| 56 | Low cell viability after single cell dissociation (empirical threshold: 75%) | Dissociation too harsh, organoids not healthy. | Avoid incubating for too long in CRS or trypsin. Avoid too much pipetting during organoid dissociation. |
| 80 | Monolayer detaching from the edge of the Transwell membrane (Figure 5a) | High passage number in continuous organoid culture or monolayers are not mature enough at the start of differentiation. | Avoid culturing organoids for more than 15 consecutive passages after thawing. Let the monolayer grow in seeding medium for 1–2 more days before starting the differentiation. |
| 83, 88 | Bacteria do not form a colony when culture is started or during CFU plating assays | High oxygen levels, agar too dry | Exchange the instrument catalyzer in the anaerobic chamber regularly (at least once every two weeks), use freshly prepared and pre-reduced agar and preserve the agar in sealed bags. |
| 94 | Cells in monolayer die after introducing bacterial media | Bacterial media is toxic (Figure 5b) | Evaluate the toxicity of the bacterial media. |
Timing
Steps 1–14, organoid seeding: 2–3 h
Steps 15–34, organoid passaging: 4–5 h
Steps 35–38, organoid media change: 0.5–1.5 h
Steps 40–62, colon monolayer seeding (single cells): 4–6 h
Steps 63–80, monolayer differentiation and transepithelial electrical resistance (TEER) monitoring: 1–2 h,
Step 80, check the monolayer in differentiation media: 4 d
Steps 81–91 bacterial culture preparation: set up 1–1.5 h, bacterial growth 2 d; bacterial plating: 0.5 h, bacterial growth 1 d. whole process: 2 d
Steps 92–102 co-culture of monolayer with bacteria: set up 1 h, bacterial growth 8 h.
Steps 103–105: luminescence data analysis 0.5 h.
Steps 106–120: sample preparation for immunofluorescent imaging: set up 1 h, staining incubation 1 d.
Box 1: (optional) EdU staining for determining the proliferative states of monolayer: set up 1 h, incubation 1 day, imaging 0.5 h
Anticipated results
Integrity and differentiation of the colon monolayers prior to co-culture with bacteria
In this protocol, we describe how to maintain colon organoids and prepare monolayers from these cells. Representative images of organoids during organoid culture (i.e., during steps 15–34) are shown in Figure 6a. Organoid size and number should increase over time, with the round cyst-like morphology maintained throughout. Representative images of the morphology of monolayers over time (i.e., during steps 62 to 80) are depicted in Figure 6b. Images are shown for monolayers at three, five, and seven days after seeding with the increase on the corresponding TEER values over time in Figure 6c and Source Data Figure 6, suggesting that the monolayers are undergoing differentiation. In Figure 6d we show typical images of proliferative cells (EdU-positive) in a monolayer over time. The increased differentiation is accompanied by a decrease in proliferative cells. At day seven, (i.e., four days after switching to differentiation media), the proportion of proliferative cells is reduced compared to monolayer at day 3 (Figure 6e and Source Data Figure 6).
Figure 6. Representative images of organoids, monolayers and proliferative cells in those monolayers.
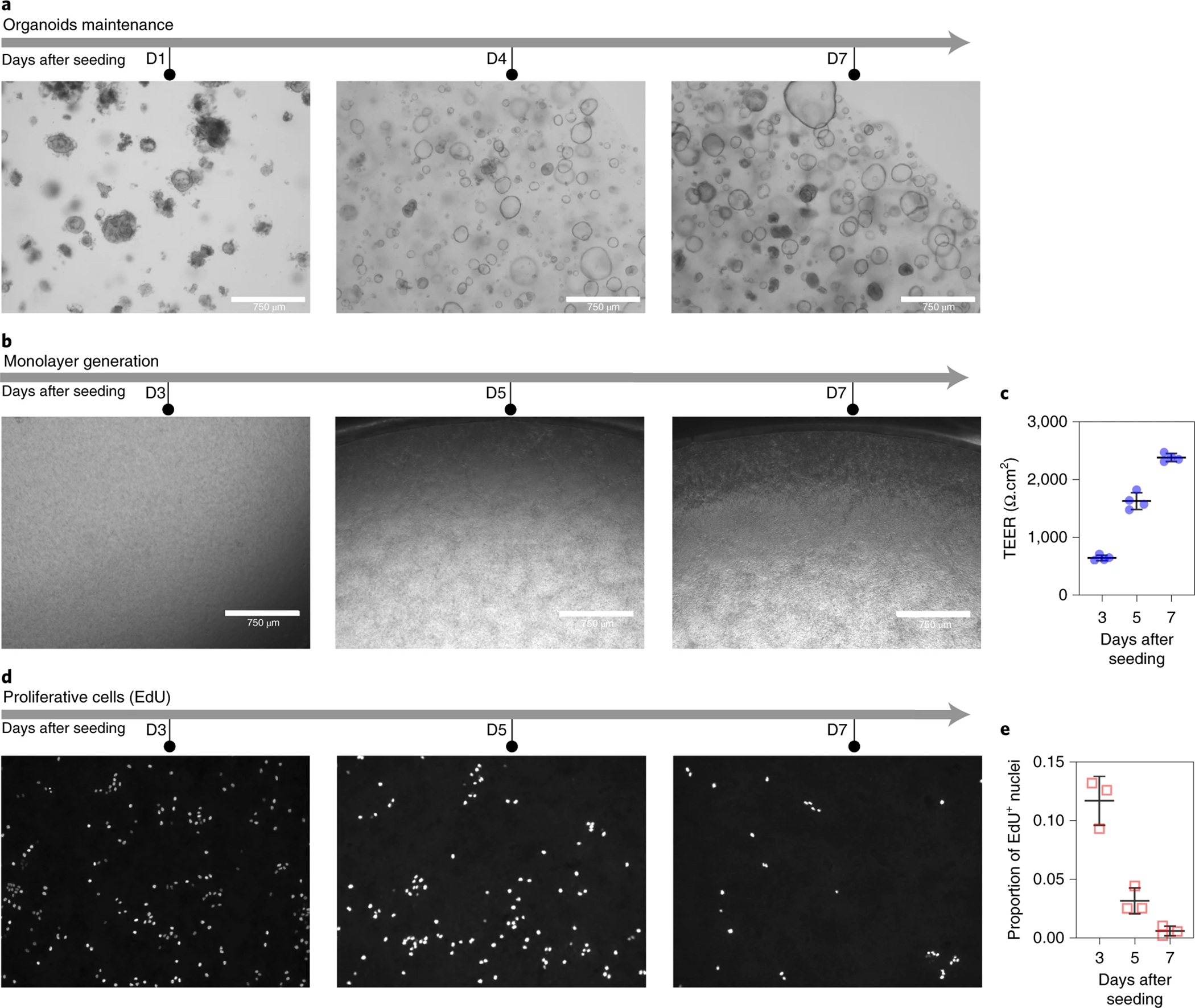
(a) Time course organoid growth at days one (D1), four (D4), and seven (D7) in Matrigel. (b) Bright field images of colon monolayers after three (D3), five (D5), and seven (D7) days post seeding. (c) Transepithelial electrical resistance (TEER) of monolayers at three (D3), five (D5), and seven (D7) days post seeding (n=4). Source Data Figure 6. (d) EdU staining of the cells in monolayer at three (D3), five (D5), and seven (D7) days post seeding. (e) The decrease in the proportion of EdU-positive cells 1316 upon differentiation, suggest a decrease of proliferative cells (n = 3). Source Data Figure 6. Data in (c) 1317 and (e) are presented as mean and standard deviation.
We observed donor-to-donor variability in monolayer formation. Therefore, we recommend testing a few donors for their ability to grow as monolayer in transwell and remain intact after induction of differentiation. Although we did not perform exhaustive testing, we observed that organoids derived from diseased donors are more difficult to expand as organoids in Matrigel and to generate stable monolayers in transwells. In Table 7, we provide guidelines to reduce donor-to-donor variability. First, it is important to maintain the organoids in an undifferentiated state (i.e., organoids displaying empty lumens and thin layer of cells) to increase cell viability during single cell disociation (above 85%) and monolayer seeding. Second, it is critical to monitor organoid growth in Matrigel to assess organoid growth based on shape, size and overall organoid morphology. If organoids are growing faster (or slower) that originally expected, the passaging schedule should be adjusted accordingly. Third, it is recommended to prepare more monolayers than the minimum required to complete your proposed study to allow for some monolayer failures. We typically prepared 30–50% additional transwells to guarantee sufficient usable monolayers.
Table 7.
Guidelines to increase the success rate in usable monolayers
| Steps | Good practices | Threshold |
|---|---|---|
| Organoid maintenance | High yield of organoids | Figure 4 |
| Appropriate size of organoids (>100 μm) | Figure 3, Figure 4 | |
| Short incubation in CRS | ≤60 min | |
| High cell viability diameter | ≥80% | |
| Use of low centrifugal force for pelleting organoids | ≤1000 g | |
| Monolayer generation | Short incubation in CRS | ≤60 min |
| High cell viability | ≥80% | |
| Sufficient cells for seeding | 300,000 | |
| Seeding of single cells | N/A | |
| Monolayer differentiation | Confluent monolayer prior to differentiation | Figure 6b |
| Moderate TEER values (donor dependent) prior to differentiation | Donor dependent (>200 Ω cm2) |
Integrity of monolayers and response of B. thetaiotaomicron to inducers after co-culture
We co-cultured colon monolayers with B. thetaiotaomicron MT768, MT798, MT799, and MT800.10 After 2-hour of co-culture, bacteria were exposed to four induction conditions: no inducer, anhydrotetracycline (aTc), deoxycholic acid (DCA), and aTc+DCA. Six hours post-induction, the apical media containing B. thetaiotaomicron was harvested to determine the effect of inducers on the bacteria present in the medium. Most bacterial cells were located close to the colon epithelium, as shown by the higher optical density and luminescence signal for the media recovered at the bottom of the apical compartment (Supplementary Table 3). In parallel, the monolayer’s integrity was monitored via TEER measurements. Representative brightfield images of the monolayers after 8 h of co-culture, are shown in Figure 7a. The monolayers appear intact with well-defined cell borders, which was confirmed by the relatively high TEER values (Figure 7b and Source Data Figure 7), compared to those seen in a bioprinted bilayer co-culture of primary human intestinal myofibroblasts and the colorectal cancer cell line (Caco-2) monolayers.30,58 Further evaluation of the monolayers using immunofluorescent staining demonstrated well-defined polygonal cell borders between epithelial cells (Figure 7c) and bacterial cells in co-culture with epithelial cells (Figure 7d).
Figure 7. Evaluation on the integrity of monolayers and response of B. thetaiotaomicron to inducers 1320 after co-culture.
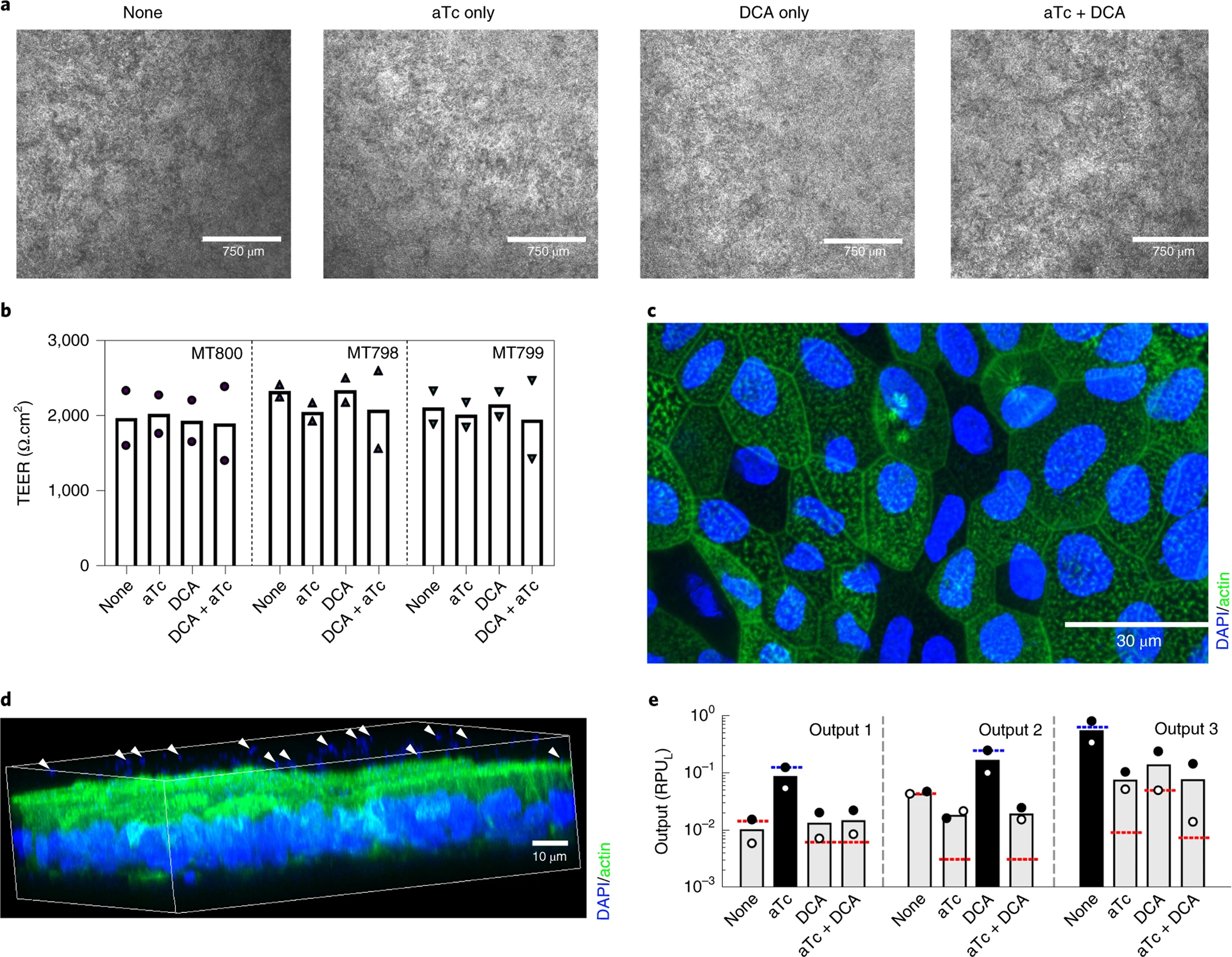
(a) representative brightfield images of monolayers after 8 h co-culture with B. thetaiotaomicron MT798 with inclusion of indicated inducers for the last 6 h of culture. Scale bar = 750μm. (b) TEER values of the monolayers co-cultured with B. thetaiotaomicron MT800 (circle), MT798 (upward triangle), and MT799 (downward triangle) at four different conditions (n=2), corresponding to the outputs in (e). Two independent experiments are depicted in the plot. Raw data can be found in Source Data Figure 7. (c) Epithelial cells in the monolayer with nuclear (blue) and beta-actin (green) staining. Scale bar =30 μm. (d) Three-dimensional rendering of the monolayer stained with DAPI (blue) and beta-actin (green), bacteria is present above the green actin layer of epithelial cells (see white arrows). Note that the scale bar in 3D rendered image is an approximation of the precise scale. (e) Luminescent output of B. thetaiotaomicron MT800 (Output 1), MT798 (Output 2), and MT799 (Output 3) in response to four different conditions (n=2). Dots indicate the values from two independent experiments. Figure 7b, 7d, and 7e were published previously.10 Raw data and the calculation of RPUL can be found in Supplementary Table S3.
Quantification of the relative bacterial promoter units (RPUL) in the co-culture of the three bacterial strains (B. thetaiotaomicron MT798, MT799, and MT800) demonstrated that each strain responded to the corresponding inducers (e.g., strain MT798 responded preferentially to the DCA inducer, whereas MT800 responded preferentially to aTc). These results are similar to the bacterial induction previously observed during the validation assay of the circuits (Figure 7e and Supplementary Table 3). This suggests that this gut in vitro model system can be used to assess the effect of engineered bacterial strains on human colon cells.
Supplementary Material
Figure 3. Morphology of primary human colon organoids.

(a) representative images of a panel of organoids showing different morphologies when undergoing differentiation (thick/folded edges [a1-a4], dark lumen [a2-a3], and non-cyst-like shape [a3-a4]). (b) a representative image of un-differentiated, cystlike colon organoid. (c) a side-by-side comparison of differentiated and un-differentiated organoids within the same Matrigel droplet. Note the relatively large and un-differentiated organoids (highlighted with a yellow asterisk) that show an empty lumen, and compare them to differentiated organoids that show thick-edges, budding, and a dark lumen (highlighted with a red rectangle)
Figure 4. Example of organoid expansion after each passaging.
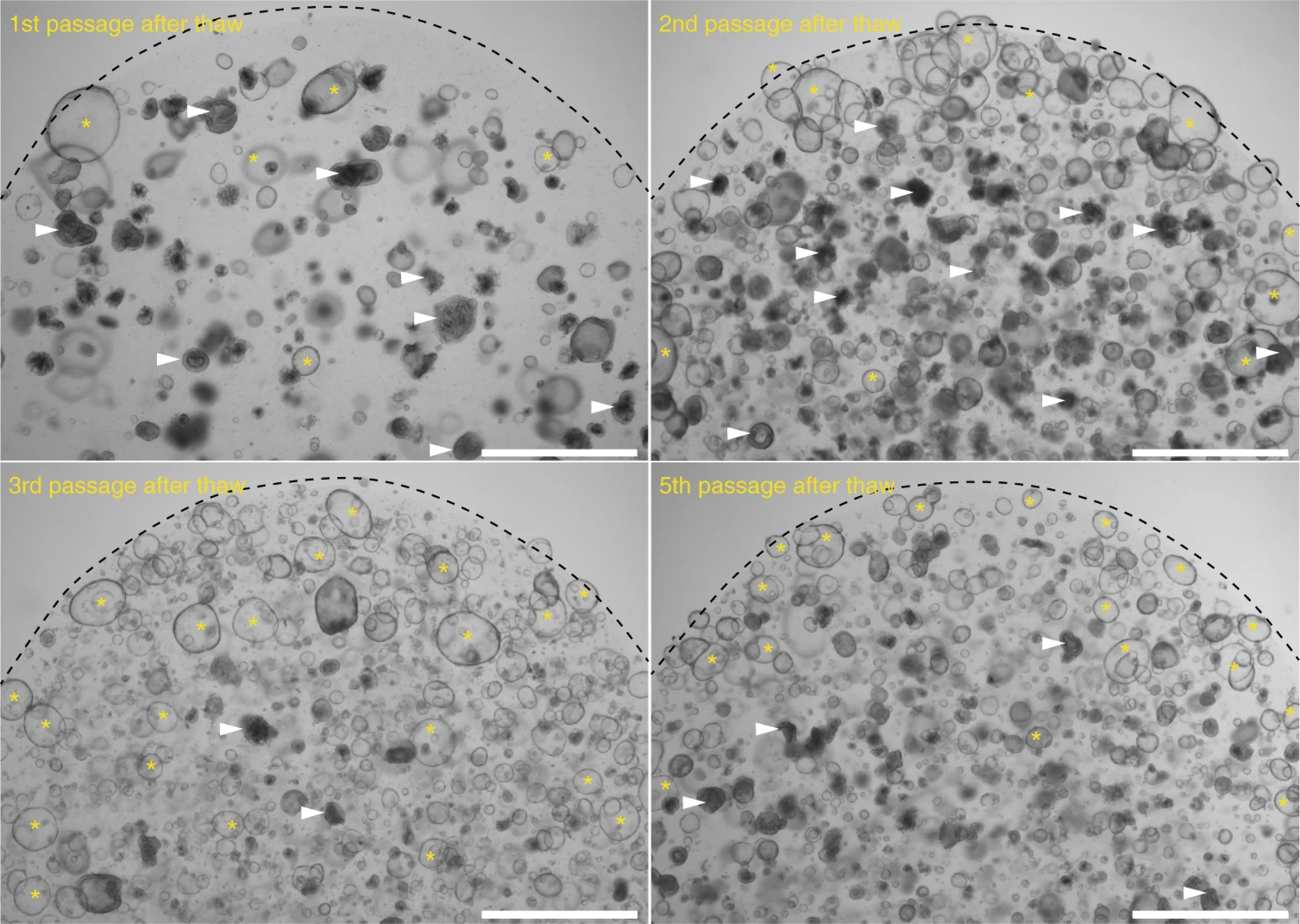
At the first passage after thawing, there is a low number of organoids. Some organoids display a thick layer of cells with dark lumens (highlighted with white triangles). Following subsequent passages, the number of organoids with a thin layer of cells and well-defined and clear lumen increases (highlighted by yellow asterisks). After the third passage, the number of undifferentiated organoids with thin layer of cells and clear lumen increases (yellow asterisk) and the number of differentiated organoids showing a thick layer and darker lumen decreases (white arrow). Dashed line indicates the approximate boundary of the Matrigel droplet edge.
Figure 5. Representative images of failed monolayers that show regions with holes or the presence of excessive dead cells after exposure to bacterial medium.
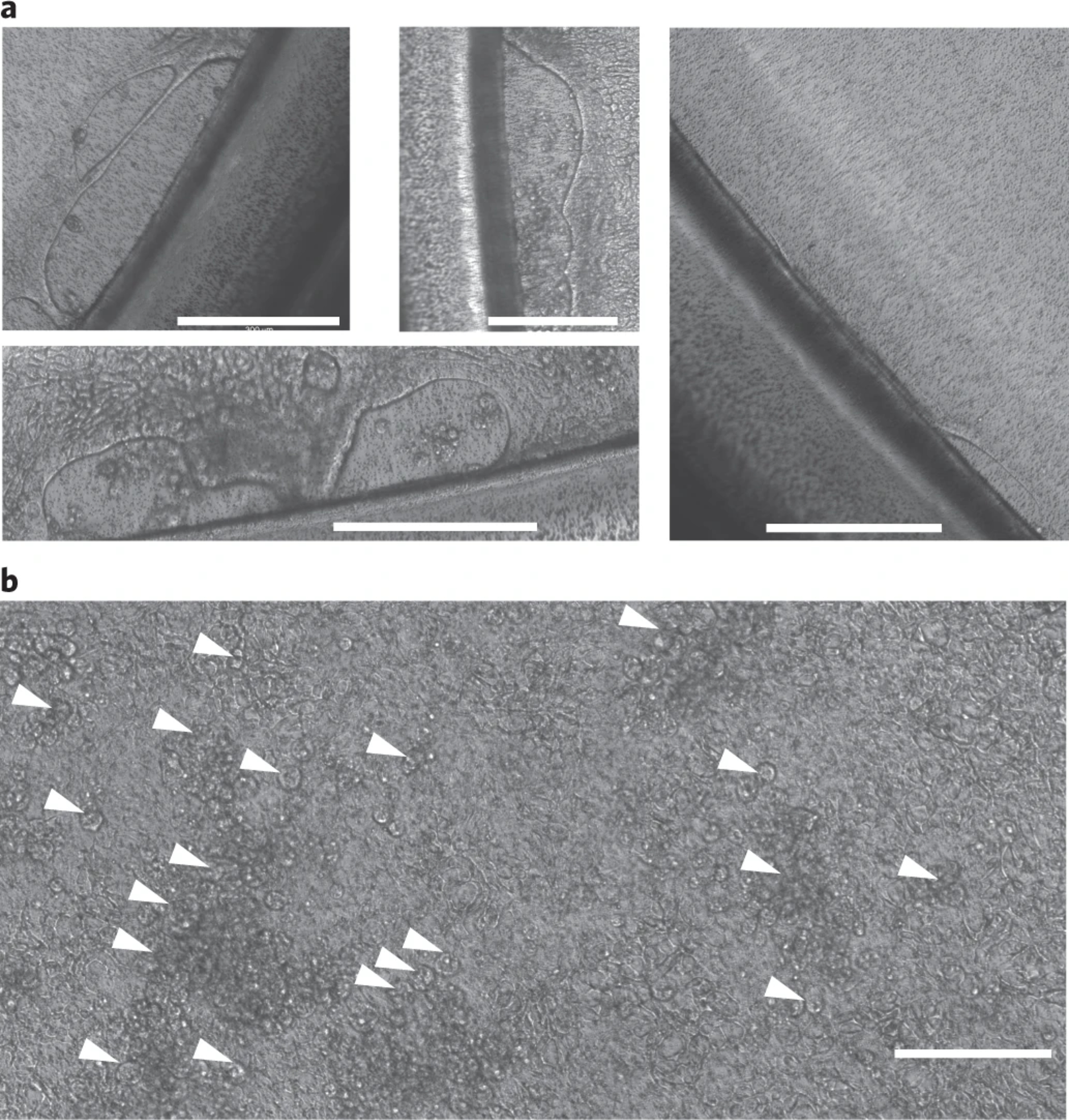
(a) Example of monolayers showing holes at the edge of the transwells after induction of differentiation. Note that monolayers with holes have relatively high TEER values (depending on the donor, TEER > 500 Ω cm2), therefore, we recommend that every monolayer be inspected under an inverted microscope prior to the addition of bacteria. (b) A monolayer that was exposed to bacterial medium (tryptone yeast extract glucose [TYG]) for 6 h. The arrows point to regions in the monolayer where epithelial cells lifted from the transwell membrane, indicating they were dying after exposure to the TYG medium alone. Scale bar = 300 μm.
Acknowledgement
This study was supported by the National Institute of Health R01EB021908, the Boehringer Ingelheim SHINE Program, the NIH P50 grant (P50-GM098792), Office of Naval Research Multidisciplinary University Research Initiatives Program (N00014–13-1–0074), Defense Agency Research Projects Agency Synergistic Discovery and Design (SD2; FA8750–17-C-0229) and National Science Foundation Semiconductor Synthetic Biology for Information Processing and Storage Technologies (SemiSynBio; CCF-1807575) program. We thank Dr. O. Yilmaz for providing the colon organoid of donor HC2978. We are grateful for the lab management support from H. Lee (MIT).
Footnotes
Competing interests
C.A.V. and M.T. have filed a provisional patent based on this work. All other authors have no competing interests.
Related links
Key references using this protocol
Taketani, M., Zhang, J., Zhang S., Triassi, A.J., Huang, Y-J., Griffith L.G., Voigt C.A. Nature Biotechnology. 38 (8), 962–969, 2020. https://doi.org/10.1038/s41587–020-0468–5 Zhang, J., Huang, Y-J., Yoon, J.Y., Kemmitt, J., Wright, C., Schneider, K., Sphabmixay, P., Hernandez-Gordillo, V., Holcomb, S.J., Bhushan, B., Rohatgi, G., Benton, K., Carpenter, D., Kester, J.C., Eng, G., Breault, D.T., Yilmaz, O., Taketani, M., Voigt, C.A., Carrier, R.L., Trumper, D.L., Griffith, L.G. Primary human colonic mucosal barrier crosstalk with super oxygen-sensitive Faecalibacterium prausnitzii in continuous culture. Med. 2(1), 74–98, 2021. doi:https://doi.org/10.1016/j.medj.2020.07.001.
Data availability
Additional imaging data that supports the findings of this study is in Supplementary Tables and Source Data or available from the corresponding author upon reasonable request.
References
- 1.Molly K, Vande Woestyne M & Verstraete W Development of a 5-step multi-chamber reactor as a simulation of the human intestinal microbial ecosystem. Applied Microbiology and Biotechnology 39, 254–258, doi: 10.1007/BF00228615 (1993). [DOI] [PubMed] [Google Scholar]
- 2.Venema K & van den Abbeele P Experimental models of the gut microbiome. Best Pract Res Cl Ga 27, 115–126, doi: 10.1016/j.bpg.2013.03.002 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Molly K, Woestyne MV, Smet ID & Verstraete W Validation of the Simulator of the Human Intestinal Microbial Ecosystem (SHIME) Reactor Using Microorganism-associated Activities. Microbial Ecology in Health and Disease 7, 191–200, doi: 10.3109/08910609409141354 (1994). [DOI] [Google Scholar]
- 4.Cinquin C, Le Blay G, Fliss I & Lacroix C New three-stage in vitro model for infant colonic fermentation with immobilized fecal microbiota. FEMS Microbiology Ecology 57, 324–336, doi: 10.1111/j.1574-6941.2006.00117.x (2006). [DOI] [PubMed] [Google Scholar]
- 5.Trapecar M, Goropevsek A, Gorenjak M, Gradisnik L & Slak Rupnik M A co-culture model of the developing small intestine offers new insight in the early immunomodulation of enterocytes and macrophages by Lactobacillus spp. through STAT1 and NF-kB p65 translocation. PLoS One 9, e86297, doi: 10.1371/journal.pone.0086297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim HJ, Li H, Collins JJ & Ingber DE Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proceedings of the National Academy of Sciences 113, E7–E15, doi: 10.1073/pnas.1522193112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim HJ & Ingber DE Gut-on-a-Chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr Biol (Camb) 5, 1130–1140, doi: 10.1039/c3ib40126j (2013). [DOI] [PubMed] [Google Scholar]
- 8.Zhou WD et al. Multifunctional Bioreactor System for Human Intestine Tissues. Acs Biomaterials Science & Engineering 4, 231–239, doi: 10.1021/acsbiomaterials.7b00794 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park GS et al. Emulating Host-Microbiome Ecosystem of Human Gastrointestinal Tract in Vitro. Stem Cell Rev Rep 13, 321–334, doi: 10.1007/s12015-017-9739-z (2017). [DOI] [PubMed] [Google Scholar]
- 10.Taketani M et al. Genetic circuit design automation for Bacteroides, applied to integrate signals from bile acid and antibiotic sensors. Nature Biotechnology 38, 962–969, doi: 10.1038/s41587-020-0468-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riglar DT et al. Engineered bacteria can function in the mammalian gut long-term as live diagnostics of inflammation. Nature Biotechnology 35, 653–+, doi: 10.1038/nbt.3879 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurtz CB et al. An engineered E coli Nissle improves hyperammonemia and survival in mice and shows dose-dependent exposure in healthy humans. Science Translational Medicine 11, doi:ARTN eaau7975 10.1126/scitranslmed.aau7975 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Isabella VM et al. Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nature Biotechnology 36, 857–+, doi: 10.1038/nbt.4222 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Mimee M, Tucker AC, Voigt CA & Lu TK Programming a Human Commensal Bacterium, Bacteroides thetaiotaomicron, to Sense and Respond to Stimuli in the Murine Gut Microbiota (vo 1, pg 62, 2015). Cell Systems 2, 214–214, doi: 10.1016/j.cels.2016.03.007 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Tropini C, Earle KA, Huang KC & Sonnenburg JL The Gut Microbiome: Connecting Spatial Organization to Function. Cell Host & Microbe 21, 433–442, doi: 10.1016/j.chom.2017.03.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rawls JF, Mahowald MA, Ley RE & Gordon JI Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 127, 423–433, doi: 10.1016/j.cell.2006.08.043 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arrieta MC, Walter J & Finlay BB Human Microbiota-Associated Mice: A Model with Challenges. Cell Host Microbe 19, 575–578, doi: 10.1016/j.chom.2016.04.014 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Ulluwishewa D et al. Live Faecalibacterium prausnitzii in an apical anaerobic model of the intestinal epithelial barrier. Cell Microbiol 17, 226–240, doi: 10.1111/cmi.12360 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Shin W et al. A Robust Longitudinal Co-culture of Obligate Anaerobic Gut Microbiome With Human Intestinal Epithelium in an Anoxic-Oxic Interface-on-a-Chip. Front Bioeng Biotechnol 7, 13, doi: 10.3389/fbioe.2019.00013 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fofanova TY et al. A novel human enteroid-anaerobe co-culture system to study microbial-host interaction under physiological hypoxia. bioRxiv, 555755, doi: 10.1101/555755 (2019). [DOI] [Google Scholar]
- 21.Shah P et al. A microfluidics-based in vitro model of the gastrointestinal human–microbe interface. Nature Communications 7, 11535–11535, doi: 10.1038/ncomms11535 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y et al. Robust bioengineered 3D functional human intestinal epithelium. Scientific Reports 5, doi:ARTN 13708 10.1038/srep13708 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Zhou WD, Roh T, Estes MK & Kaplan DL In vitro enteroid-derived three-dimensional tissue model of human small intestinal epithelium with innate immune responses. Plos One 12, doi:ARTN e0187880 10.1371/journal.pone.0187880 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jalili-Firoozinezhad S et al. Complex human gut microbiome cultured in anaerobic human intestine chips. Nature Biomedical Engineering, 421404, doi: 10.1101/421404 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadabad MS et al. A simple coculture system shows mutualism between anaerobic faecalibacteria and epithelial Caco-2 cells. Scientific Reports 5, doi:ARTN 17906 10.1038/srep17906 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J et al. Primary human colonic mucosal barrier crosstalk with super oxygen-sensitive Faecalibacterium prausnitzii in continuous culture. Med 2, 74–98, doi: 10.1016/j.medj.2020.07.001 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kozuka K et al. Development and Characterization of a Human and Mouse Intestinal Epithelial Cell Monolayer Platform. Stem Cell Reports 9, 1976–1990, doi: 10.1016/j.stemcr.2017.10.013 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhatt AP et al. Nonsteroidal Anti-Inflammatory Drug-Induced Leaky Gut Modeled Using Polarized Monolayers of Primary Human Intestinal Epithelial Cells. ACS Infect Dis 4, 46–52, doi: 10.1021/acsinfecdis.7b00139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilke G et al. in In Vitro Culture of Cryptosporidium parvum Using Stem Cell-Derived Intestinal Epithelial Monolayers (eds Mead Jan R. & Arrowood Michael J.) Ch. 20, 351–372 (Springer, 2020). [DOI] [PubMed] [Google Scholar]
- 30.Madden LR et al. Bioprinted 3D Primary Human Intestinal Tissues Model Aspects of Native Physiology and ADME/Tox Functions. iScience 2, 156–167, doi: 10.1016/j.isci.2018.03.015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freire R et al. Human gut derived-organoids provide model to study gluten response and effects of microbiota-derived molecules in celiac disease. Sci Rep 9, 7029, doi: 10.1038/s41598-019-43426-w (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y et al. Self-renewing Monolayer of Primary Colonic or Rectal Epithelial Cells. Cell Mol Gastroenterol Hepatol 4, 165–182 e167, doi: 10.1016/j.jcmgh.2017.02.011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hubatsch I, Ragnarsson EG & Artursson P Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat Protoc 2, 2111–2119, doi: 10.1038/nprot.2007.303 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Kasendra M et al. Development of a primary human Small Intestine-on-a-Chip using biopsy-derived organoids. Sci Rep 8, 2871, doi: 10.1038/s41598-018-21201-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kasendra M et al. Duodenum Intestine-Chip for preclinical drug assessment in a human relevant model. Elife 9, doi: 10.7554/eLife.50135 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y et al. In Vitro Generation of Mouse Colon Crypts. ACS Biomater Sci Eng 3, 2502–2513, doi: 10.1021/acsbiomaterials.7b00368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hinman SS, Wang Y, Kim R & Allbritton NL In vitro generation of self-renewing human intestinal epithelia over planar and shaped collagen hydrogels. Nat Protoc 16, 352–382, doi: 10.1038/s41596-020-00419-8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y et al. Analysis of Interleukin 8 Secretion by a Stem-Cell-Derived Human-Intestinal-Epithelial-Monolayer Platform. Anal Chem 90, 11523–11530, doi: 10.1021/acs.analchem.8b02835 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zamora CY et al. Application of a gut-immune co-culture system for the study of N-glycan-dependent host–pathogen interactions of Campylobacter jejuni. Glycobiology 30, 374–381, doi: 10.1093/glycob/cwz105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noel G et al. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci Rep 7, 45270, doi: 10.1038/srep45270 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ettayebi K et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 353, 1387–1393, doi: 10.1126/science.aaf5211 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costantini V et al. Human Norovirus Replication in Human Intestinal Enteroids as Model to Evaluate Virus Inactivation. Emerg Infect Dis 24, 1453–1464, doi: 10.3201/eid2408.180126 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim R et al. An in vitro intestinal platform with a self-sustaining oxygen gradient to study the human gut/microbiome interface. Biofabrication 12, 015006, doi: 10.1088/1758-5090/ab446e (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dutta D, Heo I & Clevers H Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol Med 23, 393–410, doi: 10.1016/j.molmed.2017.02.007 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Hernandez-Gordillo V et al. Fully synthetic matrices for in vitro culture of primary human intestinal enteroids and endometrial organoids. Biomaterials, 120125, doi: 10.1016/j.biomaterials.2020.120125 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sato T et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265, doi: 10.1038/nature07935 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Spence JR et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470, 105–109, doi: 10.1038/nature09691 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCracken KW, Howell JC, Wells JM & Spence JR Generating human intestinal tissue from pluripotent stem cells in vitro. Nat Protoc 6, 1920–1928, doi: 10.1038/nprot.2011.410 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williamson IA et al. A High-Throughput Organoid Microinjection Platform to Study Gastrointestinal Microbiota and Luminal Physiology. Cell Mol Gastroenterol Hepatol 6, 301–319, doi: 10.1016/j.jcmgh.2018.05.004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kester JC et al. Clostridioides difficile-Associated Antibiotics Alter Human Mucosal Barrier Functions by Microbiome-Independent Mechanisms. Antimicrob Agents Chemother 64, doi: 10.1128/AAC.01404-19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.In J et al. Enterohemorrhagic Escherichia coli reduce mucus and intermicrovillar bridges in human stem cell-derived colonoids. Cell Mol Gastroenterol Hepatol 2, 48–62 e43, doi: 10.1016/j.jcmgh.2015.10.001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trapecar M et al. Gut-Liver physiomimetics reveal paradoxical modulation of IBD-related inflammation by short-chain fatty acids. Cell Systems 10, 223–239, doi: 10.1016/j.cels.2020.02.008 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ozdemir T, Fedorec AJH, Danino T & Barnes CP Synthetic Biology and Engineered Live Biotherapeutics: Toward Increasing System Complexity. Cell Syst 7, 5–16, doi: 10.1016/j.cels.2018.06.008 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Ettayebi K et al. Replication of human noroviruses in stem cell–derived human enteroids. Science 353, 1387, doi: 10.1126/science.aaf5211 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murakami K et al. Bile acids and ceramide overcome the entry restriction for GII.3 human norovirus replication in human intestinal enteroids. Proceedings of the National Academy of Sciences 117, 1700, doi: 10.1073/pnas.1910138117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maier E, Anderson RC, Altermann E & Roy NC Live Faecalibacterium prausnitzii induces greater TLR2 and TLR2/6 activation than the dead bacterium in an apical anaerobic co-culture system. Cellular Microbiology 20, doi:ARTN e12805 10.1111/cmi.12805 (2018). [DOI] [PubMed] [Google Scholar]
- 57.VanDussen KL, Sonnek NM & Stappenbeck TS L-WRN conditioned medium for gastrointestinal epithelial stem cell culture shows replicable batch-to-batch activity levels across multiple research teams. Stem Cell Res 37, 101430, doi: 10.1016/j.scr.2019.101430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Srinivasan B et al. TEER Measurement Techniques for In Vitro Barrier Model Systems. Journal of Laboratory Automation 20, 107–126, doi: 10.1177/2211068214561025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Additional imaging data that supports the findings of this study is in Supplementary Tables and Source Data or available from the corresponding author upon reasonable request.