

Abstract
Background:
Familial hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac disease and is typically caused by mutations in genes encoding sarcomeric proteins that regulate cardiac contractility. HCM manifestations include left ventricular hypertrophy and heart failure, arrythmias, and sudden cardiac death. How dysregulated sarcomeric force production is sensed and leads to pathological remodeling remains poorly understood in HCM, thereby inhibiting the efficient development of new therapeutics.
Methods:
Our discovery was based on insights from a severe phenotype of an individual with HCM and a second genetic alteration in a sarcomeric mechanosensing protein. We derived cardiomyocytes from patient-specific induced pluripotent stem cells (iPSC-CMs) and developed robust engineered heart tissues (EHTs) by seeding iPSC-CMs into a laser-cut scaffold possessing native cardiac fiber alignment in order to study human cardiac mechanobiology at both cellular and tissue levels. Coupled with computational modeling for muscle contraction and rescue of disease phenotype via gene editing and pharmacological interventions, we have identified a new mechanotransduction pathway in HCM, shown to be essential in modulating the phenotypic expression of HCM in five families bearing distinct sarcomeric mutations.
Results:
Enhanced actomyosin crossbridge formation caused by sarcomeric mutations in cardiac myosin heavy chain (MYH7) led to increased force generation, which when coupled with slower twitch relaxation, destabilized the muscle LIM protein (MLP) stretch-sensing complex at the Z-disc. Subsequent reduction in the sarcomeric MLP level caused disinhibition of calcineurin–nuclear factor of activated T-cells (NFAT) signaling, which promoted cardiac hypertrophy. We demonstrate that the common MLP-W4R variant is an important modifier, exacerbating the phenotypic expression of HCM, but alone may not be a disease-causing mutation. By mitigating enhanced actomyosin crossbridge formation through either genetic or pharmacological means, we alleviated stress at the Z-disc, preventing the development of hypertrophy associated with sarcomeric mutations.
Conclusions:
Our studies have uncovered a novel biomechanical mechanism through which dysregulated sarcomeric force production is sensed and leads to pathological signaling, remodeling, and hypertrophic responses. Together, these establish the foundation for developing innovative mechanism-based treatments for HCM that stabilize the Z-disc MLP mechanosensory complex.
Keywords: Hypertrophic cardiomyopathy, hypertrophy, induced pluripotent stem cells, cardiomyocytes, engineered heart tissues, computational modelling, sarcomeric, biomechanical, mechanobiology, mechanosensing, mechanotransduction, crossbridge, Z-disc, modifier, muscle LIM protein, myosin heavy chain 7, myosin binding protein C3, heart failure
Introduction
Familial hypertrophic cardiomyopathy (HCM) is characterized by hypertrophic left ventricular (LV) thickening of the heart muscle, diminished outflow tract obstruction, and impaired diastolic relaxation1, 2. HCM affects 1 in 500 people and is the leading cause of sudden death in young adults3, 4. Mutations in sarcomeric genes, including MYH7 (encoding β-MHC), account for ~90% of genotype-positive familial HCM cases in humans5, 6. Enhanced and prolonged myofilament force generation has been implicated as a major physiological cause underlying HCM7–9. However, the mechanosignaling cascades through which the heightened sarcomeric force generation is sensed at the sarcomere and leads to abnormal nuclear hypertrophic signaling, have yet to be determined. This lack of mechanistic understanding has inhibited the development of precise and effective therapies. β-adrenergic and calcium blockers are standard current therapies for HCM10, but fail to halt or reverse the progression of disease. Mavacamten (a.k.a. MYK-461), a small molecule inhibitor of sarcomere contractility11, has recently been shown to reduce LV outflow obstruction, ameliorate symptoms, and decrease the expression of biomarkers of cardiac stress in HCM patients12, 13. However, the mechanisms of action and long-term safety and efficacy to prevent and/or reverse hypertrophy in humans requires further investigation. There is some concern with the initial observation that reductions of LV ejection fractions (LVEFs) below 45% or 50% (normal LVEF: ≥55%) were more common in mavacamten-treated patients compared with those in the placebo group12, 13. Thus, further understanding sarcomeric mechanosensing signaling may establish the foundation for the development of additional, complementary HCM therapeutics.
Muscle LIM protein (MLP), a component of the sarcomeric Z-disc, has been proposed to play an important role in mechanical stretch sensing and remodeling of cardiac myofilaments14, 15. A common MLP variant, MLP-W4R, is present in up to 1% amongst European Caucasians16, and may lead to defective mechanosensing at the Z-disc. However, the pathogenic role of MLP-W4R in human cardiomyopathy remains the subject of long-standing clinical debate14, 17, 18, as to whether it constitutes a disease-causing mutation or a benign polymorphism. Mouse models that express MLP-W4R and sarcomeric HCM mutations have suggested an association of MLP-W4R with HCM and cardiac dysfunction due to altered mechanical properties in the mutant sarcomere11, 16, 19. However, the isoform expression profiles of several proteins (β-MHC in particular) differ greatly between rodents and humans20, so that human cardiomyocyte-based systems for studying the mechanobiology of HCM are essential to achieve insight into human disease. Our ability to study human HCM mutations has improved significantly with the emergence of induced pluripotent stem cells (iPSCs), which can be derived from patient somatic cells by the introduction of stem cell factors21 and then differentiated into functional cardiomyocytes. Our group has incorporated patient-specific iPSC-derived cardiomyocytes (iPSC-CMs) into engineered heart tissues (EHTs) for precise mechanical stretching and disease modeling22–24. Here we used patient-specific iPSC-CMs, 3D functional EHTs, and computational modeling to uncover a new mechanotransduction pathway whereby force dysregulation arising from sarcomeric mutations is sensed at the sarcomere, leading to pathological signaling, remodeling, and severe hypertrophic cardiomyopathy. Our results indicate that the mechanosensing complex at the Z-disc could be an effective target for innovative pharmacologic approaches to treat HCM.
Methods
The authors declare that the data, analytic methods, and research agents generated or utilized in this study are available from the corresponding author, Dr. Yibing Qyang (yibing.qyang@yale.edu), upon reasonable request.
Animal use for mouse embryonic fibroblast (MEF) isolation
Pregnant CF1 mice on embryonic day (ED) 12.5–13.5 were purchased from Charles River Laboratories for the derivation of mouse embryonic fibroblasts (MEFs) for maintenance of induced pluripotent stem cells (iPSCs) under the approval of the Institutional Animal Care and Use Committee (IACUC) of the Yale School of Medicine. Detailed information can be found in the Expanded Methods in the SUPPLEMENTAL MATERIAL.
Patient biopsy and healthy heart tissues
We have abided by all relevant ethical regulations. The proband and a healthy donor heart tissues were obtained with Institutional Review Board approval from Yale University, Columbia University, and the New England Organ Bank. The proband (1.3-year-old male patient) showed severe heart failure, with thickened interventricular septum and obstruction of blood flow in the left ventricular outflow tract. Tissues derived from the proband septal myectomy were fixed for staining and histology. Healthy tissues of left ventricular free wall were taken from a healthy young adult (18-year-old) died in a car crash, and used as a control in the study. See the Expanded Methods in the SUPPLEMENTAL MATERIAL for details.
Human induced pluripotent stem cells
MLP-W4R;MYH7-R723C (proband), MLP-W4R (mother), and MYH7-R723C (father) iPSC lines were generated by infecting peripheral blood mononuclear cells (PBMCs) with Sendai viral particles that encode human OCT4, KLF4, SOX2, and c-MYC, under the approval of Yale Institutional Review Board. A control iPSC line used in this study was reported previously25. All iPSC lines were cultured and expanded on the MEF feeder. Detailed iPSC generation can be found in the Expanded Methods in the SUPPLEMENTAL MATERIAL.
Cardiac differentiation
Cardiac differentiation was performed as previously described26, and the details can be found in the Expanded Methods in the SUPPLEMENTAL MATERIAL.
Calcium imaging
See the Expanded Methods in the SUPPLEMENTAL MATERIAL for details.
Engineered heart tissue (EHT) generation and cell seeding
EHTs were created by seeding decellularized laser-cut porcine myocardium with iPSC-CMs as described previously22. iPSC-CMs on day 14 of cardiac differentiation were detached and resuspended in MEF medium containing 5 μM ROCK inhibitor. A cell suspension of 10-million/mL was made and 100 μL of cell suspension (1-million) was seeded into an assembled cassette for EHT generation (details in the Expanded Methods in the SUPPLEMENTAL MATERIAL).
Mechanical and functional testing of EHTs
EHT contractile mechanics were measured in a custom muscle mechanical setup at day 30 after cardiac differentiation as previously described22. Measurements were performed at 36°C with 1Hz field stimulus. Twenty twitches were recorded, averaged, and employed to quantify the peak force, time to peak force (TTP), and time from peak force to 50% relaxation (RT50). Details can be found in the Expanded Methods in the SUPPLEMENTAL MATERIAL.
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-mediated gene editing for MYH7-R723C correction
See the Expanded Methods in the SUPPLEMENTAL MATERIAL for details.
Transcription activator-like effector nuclease (TALEN)-mediated gene editing for MLP-W4R correction
See the Expanded Methods in the SUPPLEMENTAL MATERIAL for details.
Computational modeling
Muscle mechanics simulations were performed using a computational model of cooperative myofilament calcium-dependent activation with a three-state crossbridge cycle as previously described27, 28. In general, Ca2+ transients from the wild-type myosin (MYH7-WT) and mutant myosin (MYH7-R723C) were used to simulate force transients (i.e. twitches) with model parameters for normal and mutant myosin. See the Expanded Methods in the SUPPLEMENTAL MATERIAL for details.
Immunofluorescence staining, gene expression analysis, western blot analysis and immunoprecipitation, luciferase assay for measuring NFAT transcriptional activity, cloning and site-directed mutagenesis, lentivirus production, tissue histological analysis, and drug treatment.
See the Expanded Methods in the SUPPLEMENTAL MATERIAL for details.
Quantification and statistical analyses
Data quantification and statistical analyses were performed using GraphPad Prism 8. One-way ANOVA with Tukey’s post-hoc tests or Kruskal-Wallis test with Dunn’s post-hoc tests were applied to evaluate statistical differences amongst three or more experimental groups when appropriate. Two-way ANOVA followed by Tukey’s post-hoc tests was employed to evaluate the effect of two independent variables, in combination, on a dependent variable amongst experimental groups. A two-tailed Student’s t test or nonparametric Mann-Whitney U test was used to compare differences between two experimental groups as appropriate. For paired analysis with repeated measurements on the same samples, a paired Mann-Whitney U test was used. A p value lower than 0.05 was considered significant. Data were presented as mean ± standard error of the mean (S.E.M.). Statistical details for each experiment can also be found in the corresponding figure legends and related results.
Results
Patient Clinical Phenotypes and Recapitulation of HCM Defects with Patient-Specific iPSC-Derived Cardiomyocytes and Engineered Heart Tissues
To explore how sarcomeric mutations lead to pathological hypertrophy, we investigated a unique family in which the 1.3-year-old HCM proband presented with severe heart failure, markedly thickened interventricular septum (IVS; 2.3 cm), and significant obstruction of blood flow in the left ventricular outflow tract (LVOT gradient 88 mm Hg) (Figure 1A; Video S1). He underwent septal myectomy, reducing his LVOT gradient to <30 mm Hg. While his mother (35-year-old; II-4 in Figure 1B) was clinically healthy (Figure 1A; Video S2), his father (36-year-old; II-3 in Figure 1B) showed modest septum thickening (Figure 1A; Video S3). Several second-degree relatives demonstrated modest HCM, including the proband’s paternal grandmother (I-2) and uncle (II-2). There were no major cardiac abnormalities detected clinically in the proband’s maternal relatives (Figure 1B; Table S1).
Figure 1. Patient Clinical Phenotypes and HCM Modeling with Patient-Specific iPSC-Derived Cardiomyocytes and Engineered Heart Tissues.
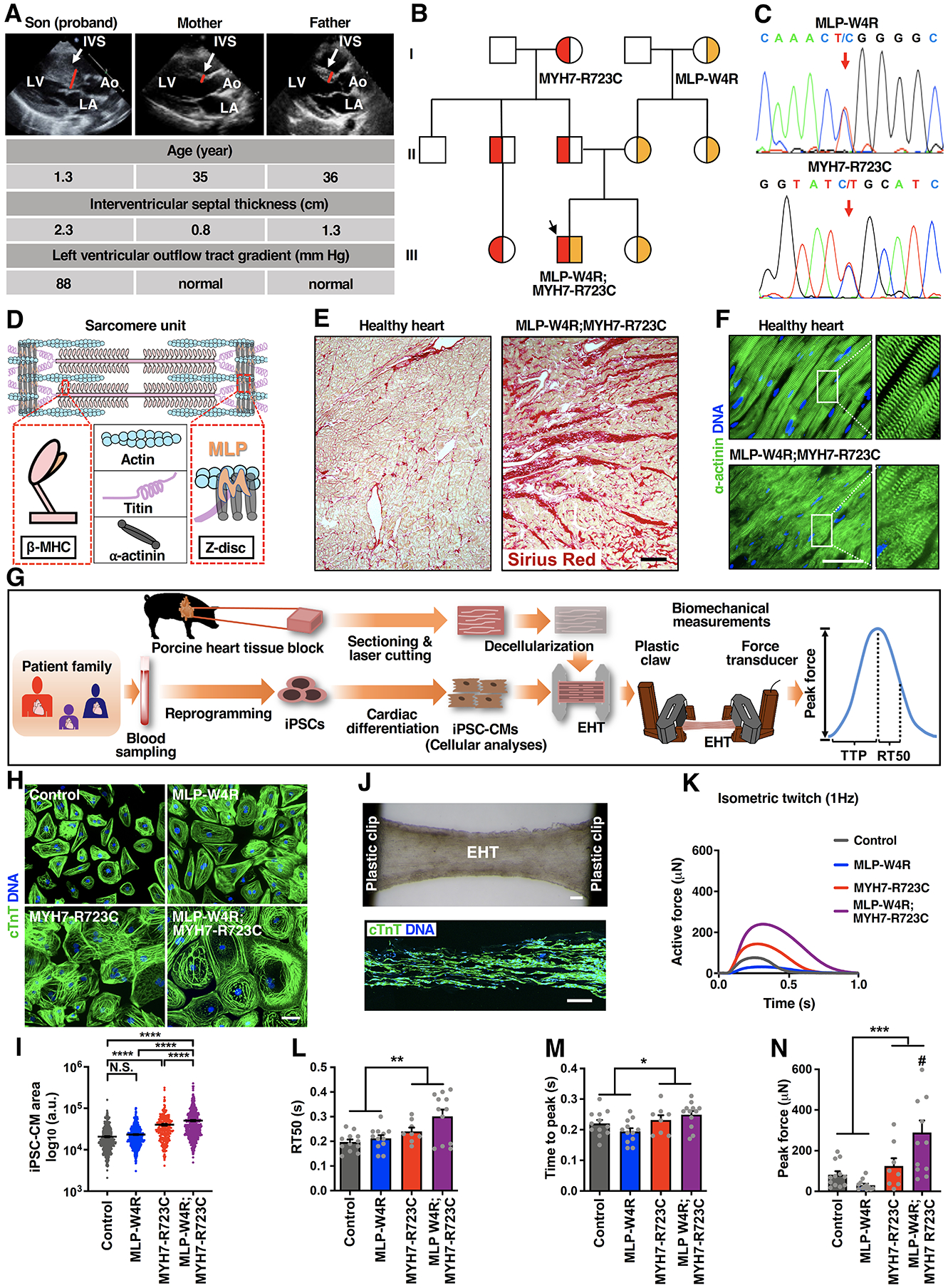
A, Echocardiographic images (parasternal long axis) showing severe interventricular septum (IVS) hypertrophy in the neonatal proband with no overt structural changes in the proband’s parents. LV: left ventricle; LA: left atrium; Ao: aorta. B, Schematic pedigree of the proband exhibiting co-inheritance of the MLP-W4R and MYH7-R723C heterozygous missense mutations (III-2, black arrow). Squares represent male family members and circles represent females. Red indicates the MYH7-R723C heterozygous mutation, and yellow indicates the MLP-W4R heterozygous mutation. See Table S1 for additional clinical characteristics of the proband and family members. C, Confirmation of the heterozygous MLP-W4R and MYH7-R723 mutations in the respective MLP (exon 2) and MYH7 (exon 20) genes via PCR and Sanger sequencing in the proband. Red arrows indicate MLP and MYH7 mutations. See Figure S1D for additional sequencing information. D, Schematic of a sarcomere unit that includes thick filament components β myosin heavy chain (β-MHC; encoded by the MYH7 gene) and titin, thin filament component actin, and Z-disc components α-actinin and MLP. E, Sirius red staining of patient myectomy tissues versus healthy human controls. Scale bar, 200 μm. F, α-actinin immunostaining of patient myectomy tissue versus healthy human control. DNA was counterstained by DAPI. Scale bar, 50 μm. G, Schematic illustrating patient iPSC generation, iPSC-CM derivation, cellular analyses, EHT production, and EHT biomechanical measurements including peak force, time to peak force (TTP), and time from peak force to 50% relaxation (RT50). H, cTnT immunostaining of day 35 control, MLP-W4R, MYH7-R723C, and MLP-W4R;MYH7-R723C iPSC-CMs. Scale bar, 100 μm. I, Quantification of iPSC-CM areas in panel H with ImageJ from more than four independent cardiomyocyte differentiation batches (≥50 cells per batch for each cell type). Two-way ANOVA with Tukey’s multiple comparisons test revealed that the MYH7-R723C and MLP-W4R mutations synergistically regulated cell size (F(1,1985)=7.213, p=0.0073; each mutation considered as an independent factor). Note that the F value indicates the ratio of explained variance between groups to unexplained variance due to experimental variations within groups and the degrees of freedom (df) represent the df for factor (genotype) interaction and the sum of the individual df for each genotype group, respectively. J, Top panel: A representative image of an EHT constructed by seeding day 14 iPSC-CMs into decellularized thin sections of native porcine myocardium followed by an additional 14-day culture. Scale bar, 200 μm. Bottom panel: cTnT immunostaining of a representative EHT section. Scale bar, 200 μm. K, Representative isometric twitches of control, MLP-W4R, MYH7-R723C, and MLP-W4R;MYH7-R723C EHTs under 1 Hz pacing. L-N, Quantification of EHT biomechanical properties (n≥8 per group from three or more independent cardiomyocyte differentiation batches). Two-way ANOVA with Tukey’s multiple comparisons test revealed that there were no statistically significant interactions between the MYH7-R723C and MLP-W4R mutations in regulating RT50 (F(1,40)=1.46, p=0.234) and TTP (F(1, 40)=2.991, p=0.092). The presence of the MYH7-R723C mutation significantly prolonged RT50 (L, F(1,40)=12.200, p=0.001) and TTP (M, F(1,40)=6.857, p=0.012). In addition, these two mutations synergistically increased peak force (N) in the proband EHTs (F(1,40)=8.080, p=0.007). # denotes that proband MLP-W4R;MYH7-R723C EHTs generated significantly higher peak force than MLP-W4R (p<0.0001), MYH7-R723C (p=0.029), or control (p=0.001) EHTs. Note that the degrees of freedom (df) represent the df for each factor (genotype) or factor interaction and the sum of the individual df for each genotype group, respectively (L-N). Each mutation is considered as an independent factor. Each data point represents a single iPSC-CM (I) or EHT (L-N) derived from at least three independent cardiomyocyte differentiation batches. All data are presented as mean ± S.E.M; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; N.S.: not significant.
We next performed whole exome-sequencing of a panel of 20 genes associated with HCM including ACTC1, ACTN2, JPH2, LDB3, MLP, MYBPC, MYH6, MYH7, MYL2, MYL3, MYLK2, MYOZ2, PLN, TCAP, TNNC1, TNNI3, TNNT2, TPM1, TTN, and VCL. Results revealed that the proband carried two heterozygous point mutations MYH7-R723C and MLP-W4R (Figure 1C and 1D; Table S1; Figure S1D). Further screening of family members revealed the proband inherited the MYH7-R723C mutation from his father and the MLP-W4R mutation from his mother (Figure 1B; Table S1). Although the MYH7-R723C mutation is implicated in HCM29, 30, as previously noted, the pathogenic role of the MLP-W4R variant in cardiomyopathy remains unresolved14, 17, 18. The lack of significant clinical cardiac abnormalities in the proband’s mother’s family members carrying the MLP-W4R mutations (Figure 1A; Table S1), in this and other studies17, 18 raised the intriguing possibility that this variant might be a modifier of HCM disease severity. Sirius red staining of tissue biopsies revealed marked fibrosis in the proband compared with a healthy young adult donor heart (Figure 1E). Moreover, α-actinin immunostaining showed noticeable sarcomeric disarray and disintegration (Figure 1F), consistent with pathologies reported in HCM patients1,10. Coinheritance of the MYH7-R723C and MLP-W4R mutations in the proband resulted in an aggressive disease presentation, supporting a unique mechanistic interaction between these two mutations.
To investigate potential molecular pathological interactions of MLP-W4R and MYH7-R723C, we generated iPSCs from the father, mother, and proband, based on non-integrating Sendai viral vectors encoding SOX2, OCT4, KLF4 and c-MYC in peripheral blood mononuclear cells31, 32 (Figure 1G). We used a previously reported iPSC line from a healthy individual as a control25. iPSC lines with normal chromosomal integrity and pluripotency (Figure S1A–1H) were expanded, differentiated26, and enriched via metabolic selection33 to achieve high purity cardiomyocytes (Figure 1H; Figure S1I). Since the proband exhibited severe asymmetric septal hypertrophy at a very young age (1.3-year-old), iPSC-CM disease modeling appeared particularly suitable for studying early onset (fetal/neonatal) disease, and elucidating the mechanisms responsible for alterations of MYH7 and MLP-mediated biochemical signaling. The iPSC-CMs were evaluated for cellular abnormalities including cell area, sarcomere disarray, and the expression levels of HCM-associated markers such as brain natriuretic peptide (BNP). The cell areas of the double heterozygote proband MLP-W4R;MYH7-R723C iPSC-CMs were significantly larger than those of control or single heterozygote iPSC-CMs (Figure 1H and 1I). Furthermore, the proband iPSC-CMs showed increased sarcomere disarray compared to either control or single heterozygote iPSC-CMs (Figure S1J and S1K). Additionally, BNP mRNA transcript expression appeared to be higher in the double mutant proband iPSC-CMs (Figure S1L). Thus, the double mutant proband iPSC-CMs demonstrated pathological abnormalities, characteristic of HCM, that were markedly more pronounced compared to those in the single mutant parental CMs, recapitulating clinical disease manifestations in the family.
To determine whether the observed cellular defects in the proband CMs are sufficient to initiate tissue level mechanical abnormalities, we generated EHTs (Figure 1G) made utilizing thin sections of decellularized porcine myocardium22–24. These unique engineered scaffolds possess native cardiac fiber alignment that is critical to realistic force generation and mechanical assessment. Day 14 differentiated iPSC-CMs were seeded into the scaffold, and the EHTs were cultured for 2 weeks. We quantified the EHT contractile mechanics by measuring the time to peak force (TTP), time from peak force to 50% relaxation (RT50), and peak force (Figure 1J–1N). Two-way ANOVA revealed no interaction between the MYH7-R723C and MLP-W4R mutations in regulating RT50 (interaction p=0.234) and TTP (interaction p=0.092), thus only permitting statistical analysis via data grouping of each individual mutation. Results revealed that while the MYH7-R723C mutation resulted in a significantly prolonged RT50 (p=0.001) and TTP (p=0.012), the MLP-W4R mutation exhibited a trend toward prolonged RT50 (p=0.052), with no effect on TTP (p=0.756) (Figure 1L and 1M). Importantly, the MYH7-R723C and MLP-W4R mutations synergistically upregulated the force production (interaction p=0.007) in the double mutant proband CMs (Figure 1N). These results suggest that while the MYH7-R723C mutation might drive the mechanical alterations in twitch kinetics, the MLP-W4R mutation could act as a modifier to alter phenotypic expression of hypertrophy by drastically augmenting the force development in the double mutant CMs (Figure 1K).
Genetic Correction and Computational Modeling of Muscle Contraction Reveal the Role of the Myosin Mutation in Initiating the HCM Phenotype
To further determine the role of MYH7-R723C and MLP-W4R mutations in HCM pathogenesis, we corrected either R723C (MYH7-corrected) or W4R (MLP-corrected) in the proband iPSCs using gene editing techniques (Figure 2A; Figures S2 and S3). After differentiating corrected iPSCs into CMs, cellular and mechanical defects were investigated in an isogenic setting. Correction of the MYH7-R723C or MLP-W4R mutation resulted in rescue of the HCM phenotype including reducing cell size and the expression of BNP (Figure 2B–2G). Further, EHTs with isogenic correction of the MYH7-R723C mutation revealed a significant attenuation of the mechanical defects including RT50, TTP, and the peak force (Figure 2H–2K). Notably, isogenically corrected MLP EHTs primarily rescued peak force abnormalities but not RT50 (Figure 2L–2O). Together, these data suggest that the MYH7-R723C mutation may be required for initiating the HCM phenotype and the MLP-W4R mutation may enhance these defects in the double mutant, leading to exacerbated hypertrophy and cardiac remodeling in the proband.
Figure 2. Mutant Cardiac Myosin Initiates HCM Phenotype.
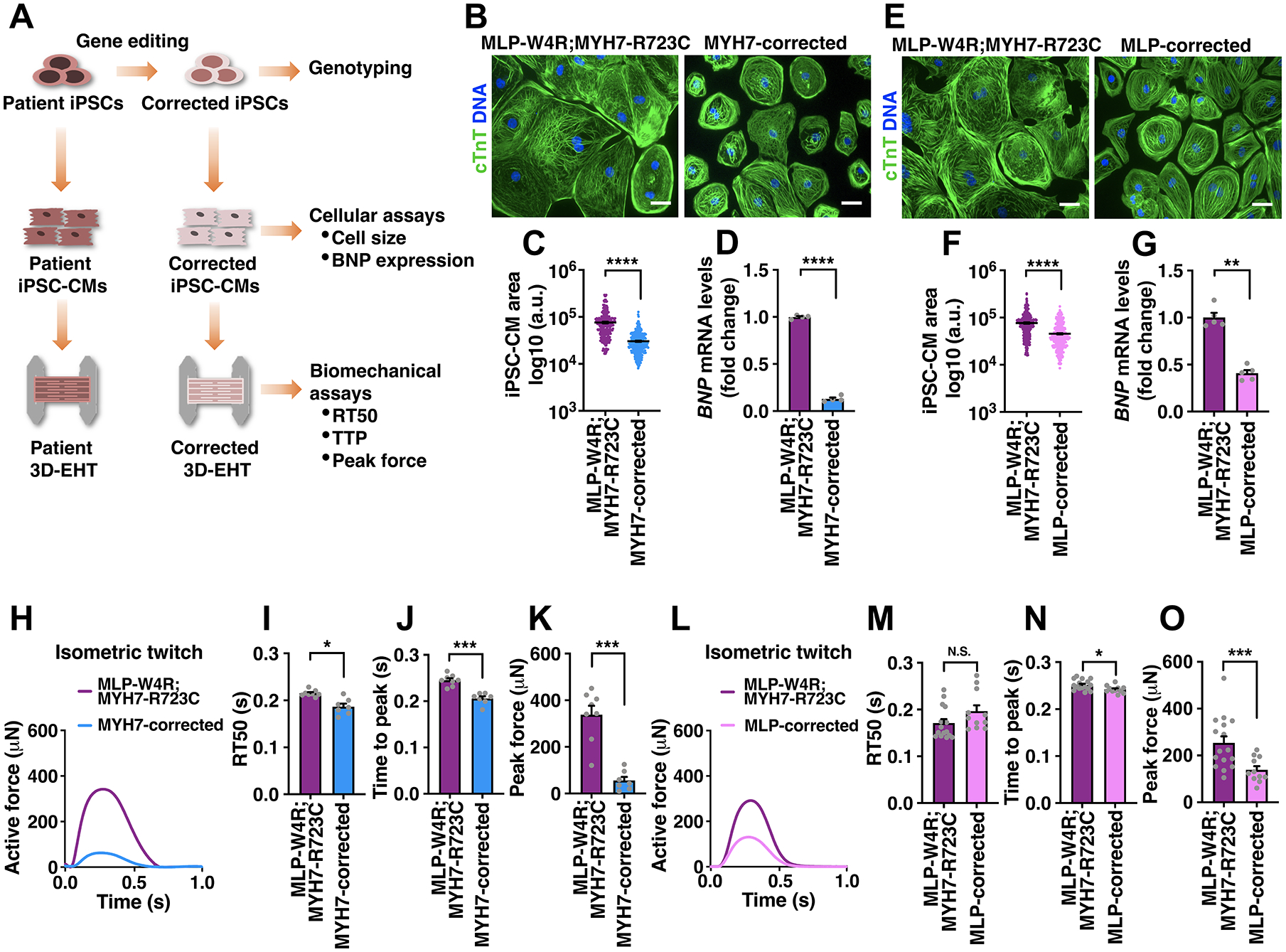
A, Schematic of correcting HCM-associated mutations in iPSCs via gene editing and phenotypic analyses of corrected iPSC-CMs and EHTs. See Figures S2 and S3 for CRISPR-Cas9- and TALEN-mediated correction of the myosin and MLP mutations. B-D, cTnT immunostaining in MLP-W4R;MYH7-R723C and MYH7-corrected (containing MLP-W4R) isogenic iPSC-CMs (B), followed by quantification of cell area based on cTnT staining (C) and analysis of hypertrophic molecular marker BNP via qRT-PCR (D). E-G, cTnT immunostaining in MLP-W4R;MYH7-R723C and MLP-corrected (containing MYH-R723C) isogenic iPSC-CMs (E), followed by quantification of cell area based on cTnT staining (F) and analysis of hypertrophic molecular marker BNP via qRT-PCR (G). Cell areas in panels B and E were quantified with ImageJ from three independent cardiomyocyte differentiation batches (≥100 cells per batch). mRNA expression was normalized to GAPDH and fold change relative to MLP-W4R;MYH7-R723C iPSC-CMs was presented (D: n=4 independent cardiomyocyte differentiation batches per group; G: n=5 independent cardiomyocyte differentiation batches per group). Scale bar (B, E), 100 μm. A two-tailed unpaired Student’s t test was used for cell area comparison (C, F) and a two-tailed unpaired Mann-Whitney U test for gene expression comparison (D, G). H, Representative twitches of EHTs constructed from MLP-W4R;MYH7-R723C and MYH7-corrected isogenic iPSC-CMs under 1 Hz pacing. I-K, Quantification of EHT biomechanical properties including RT50 (I), TTP (J), and peak force (K) (n≥7 per group from three independent cardiomyocyte differentiation batches). L, Representative twitches of EHTs constructed from MLP-W4R;MYH7-R723C and MLP-corrected isogenic iPSC-CMs under 1 Hz pacing. M-O, Quantification of EHT biomechanical properties including RT50 (M), TTP (N), and peak force (O) (n≥11 per group from at least three independent cardiomyocyte differentiation batches). A two-tailed unpaired Mann-Whitney U test was used for analysis between two groups (I-K, M-O). Each data point represents a single iPSC-CM (C and F), EHT (I-K and M-O), or sample generated from a batch of iPSC-CMs (D and G) from at least three independent cardiomyocyte differentiation batches. All data are presented as mean ± S.E.M; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; N.S: not significant.
The impaired relaxation and prolonged force generation observed in the proband EHTs prompted us to investigate whether alterations of intracellular Ca2+ transients, such as slower reuptake in the sarcoplasmic reticulum (SR), could underlie defective twitch kinetics. To test this, we measured Ca2+ transients in iPSC-CMs. Two-way ANOVA showed the MYH7-R723C mutation alone resulted in significantly higher diastolic Ca2+ but lower systolic Ca2+, independent of the MLP-W4R mutation (Figure S4A–4C). Surprisingly, the rate of Ca2+ decay (decay time constant from 80% peak Ca2+ [Tau80], Figure S4D and S4E) was faster in the MYH7-R723C mutant CMs (MYH7-R723C or MLP-W4R;MYH7-R723C), compared with the MYH7-wild-type (WT) CMs (control or MLP-W4R), indicating increased SR reuptake. This was associated with a concomitant upregulation of SERCA2a and the ratio of phosphorylated phospholamban (P-PLB) to total PLB (Figure S4F–4I), potentially underlying the enhanced Ca2+ SR reuptake in MYH7-R723C mutant CMs. These results suggest that the slower calcium SR reuptake was not likely to be the cause for the impaired relaxation and prolonged contraction in the MYH7-R723C mutant CMs, but most likely a compensatory mechanism.
We next investigated whether altered crossbridge cycling may account for the prolonged contraction in the MYH7-R723C mutant CMs. MYH7-R723C is a mutation located in the myosin converter domain (Figure 3A), which connects the α-helical light chain binding domain to the myosin catalytic domain and is implicated in modulating actomyosin binding during muscle contraction34–36. We extended our previously developed computational model of cooperative myofilament activation27, 28, 37 to predict the effect of MYH7-R723C on myofilament protein interactions using twitches and calcium transients collected experimentally (Figure 3A–3G). Representative experimental Ca2+ transients and twitches in the WT and MYH7-R723C mutant CMs were used as model inputs and outputs (Figure 3B and 3C). First, baseline model parameters were determined by fitting the WT twitch data driven by the WT Ca2+ transient (Figure 3B and 3D; Table S2). Twitches simulated using baseline model parameters driven by the MYH7-R723C mutant Ca2+ transients were shorter compared to the WT twitches (Figure 3B and 3E). In order to fit our model driven by the mutant MYH7 Ca2+ transient to the mutant MYH7 experimental twitches, a single parameter increase in the myosin duty cycle (δ; Table S2) by 30% was required (Figure 3B and 3F). This increased myosin duty cycle suggested that the MYH7-R723C mutation results in a higher ratio of strong actin-binding myosin heads relative to total crossbridge cycling compared to WT myosin. We then applied the mutant myosin model using WT Ca2+ transients, resulting in even more prolonged twitches compared with the MYH7-R723C mutant (Figure 3G). Altogether, these data suggest that the MYH7-R723C mutation alters contractile kinetics by increasing the actomyosin duty cycle during cardiac contraction and that the observed increase in the Ca2+ reuptake is the manifestation of a compensatory response to normalize the abnormal contractility and prolonged kinetics in the mutant CMs.
Figure 3. An Increase in Actomyosin Duty Cycle by Mutant Cardiac Myosin Leads to Prolonged Muscle Contraction in HCM.
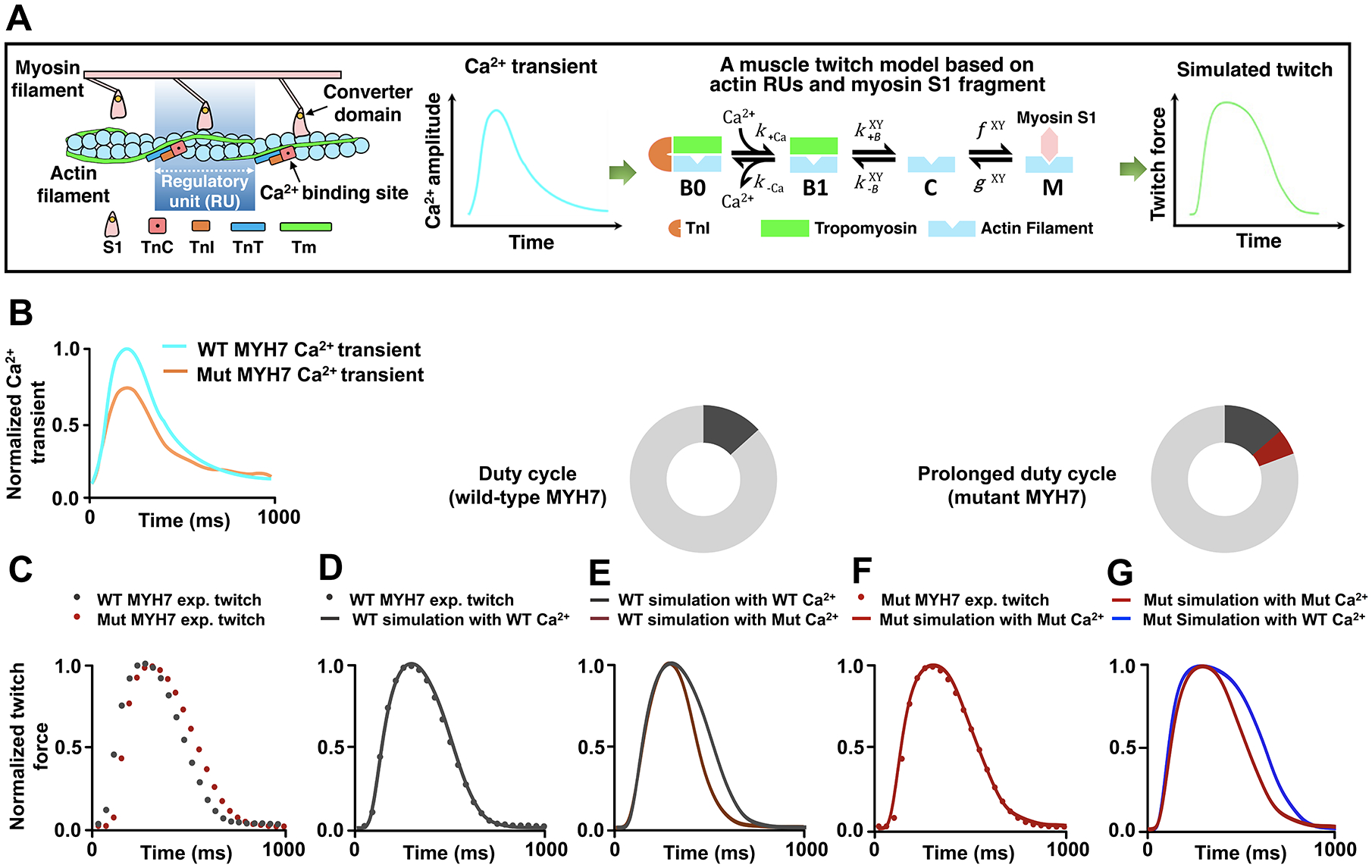
A, Schematic illustration of a computational model for muscle contraction based on cooperative myofilament activation27, 28, 37. Left panel: The model depicts the function of individual thin filament regulatory units (RUs), consisting of 7 actin monomers, TnC, TnI, and tropomyosin (Tm), in conjunction with the S1 fragment of myosin head. Right panel: The model takes a calcium transient as input and outputs activation of the myofilament dependent on the model parameters which describe myofilament protein interactions. Each RU exists in one of four states illustrating Ca2+ binding (from Ca2+-free B0 to Ca2+-bound B1 blocked states), Tm shifting (from B1 blocked to the closed [C] states), and myosin attachment (from the closed [C] to open [M] states). Note that the transition between closed and open (M) states is determined by the simplified crossbridge attachment (f) and detachment (g) rates27, 28, 37. The B↔C and C↔M transition rates are functions of Tm states of the two nearest neighboring RUs (X and Y). Also see additional information in the Expanded Methods in the SUPPLEMENTAL MATERIAL. B, Experimental Ca2+ transients collected from both the wild-type (WT) and MYH7 mutant (Mut) iPSC-CMs. Ca2+ transients (Figure S4A–4C) were normalized and scaled to reflect relative average properties from the summary data, with the WT Ca2+ transient normalized at a diastolic value of 0.1 μM and a maximum value of 1.0 μM. C, Experimental twitches collected from both the WT and the MYH7 Mut EHTs and normalized to their individual maximum force for model analysis. D, The WT twitch was simulated by inputting the realistic WT Ca2+ transient into the model and optimizing the parameter set through minimization of the root mean square error using the particle swarm stochastic optimization algorithm. See details in the Expanded Methods. E, The MYH7 Mut Ca2+ transient was input into the model while keeping the parameters that fit the WT twitch. F, MYH7 Mut twitch was simulated using the MYH7 Mut Ca2+ transient and increasing the myosin duty cycle (δ) by 30% from the WT fit parameters (δWT 0.20; δMut 0.26). No other parameters were changed from the WT fit except for the myosin attachment rate f. See Table S2 for myofilament model parameter sets. G, The WT Ca2+ transient was input into the model with the mutant parameter set, representing only an increase in the myosin attachment rate.
The MLP-W4R Variant Exacerbates the Phenotypic Expression of HCM
As excessive force development in the double mutant proband (Figure 1K and 1N) might affect the integrity of sarcomeres and MLP stability at the Z-disc, we measured MLP protein levels in day 35 iPSC-CMs. While the expression of MLP was comparable in the control, MYH7-R723C, and MLP-W4R CMs, there was a significant reduction in the double mutant proband CMs (Figure 4A and 4B). Additionally, cycloheximide treatment to determine MLP protein stability revealed that MLP had a markedly higher decay rate in the double mutant CMs than that in the control CMs (Figure 4C and 4D). These results raised the intriguing possibility that a reduction of MLP expression due to decreased protein stability may contribute to HCM pathology. Consistent with this notion, lentiviral-mediated ectopic expression of HA-tagged wild-type MLP in the double mutant iPSC-CMs significantly rescued the HCM phenotype including the enlarged CM area and the elevated expression of BNP (Figure 4E–4G; Figure S5A–5C). Importantly, genetic correction of MLP-W4R resulted in restoration of MLP levels in the double mutant CMs (Figure 4H and 4I) and a significant rescue of the HCM defects (Figure 2E–2G, 2L, 2N, and 2O). Additionally, there were markedly reduced levels of MLP at the Z-disc in the double mutant proband heart tissue compared with that in the healthy control tissue (Figure 4J). Together, these results suggest that normal MLP expression and function are required for maintenance of effective cardiac function.
Figure 4. MLP-W4R Exacerbates the Phenotypic Expression of HCM.
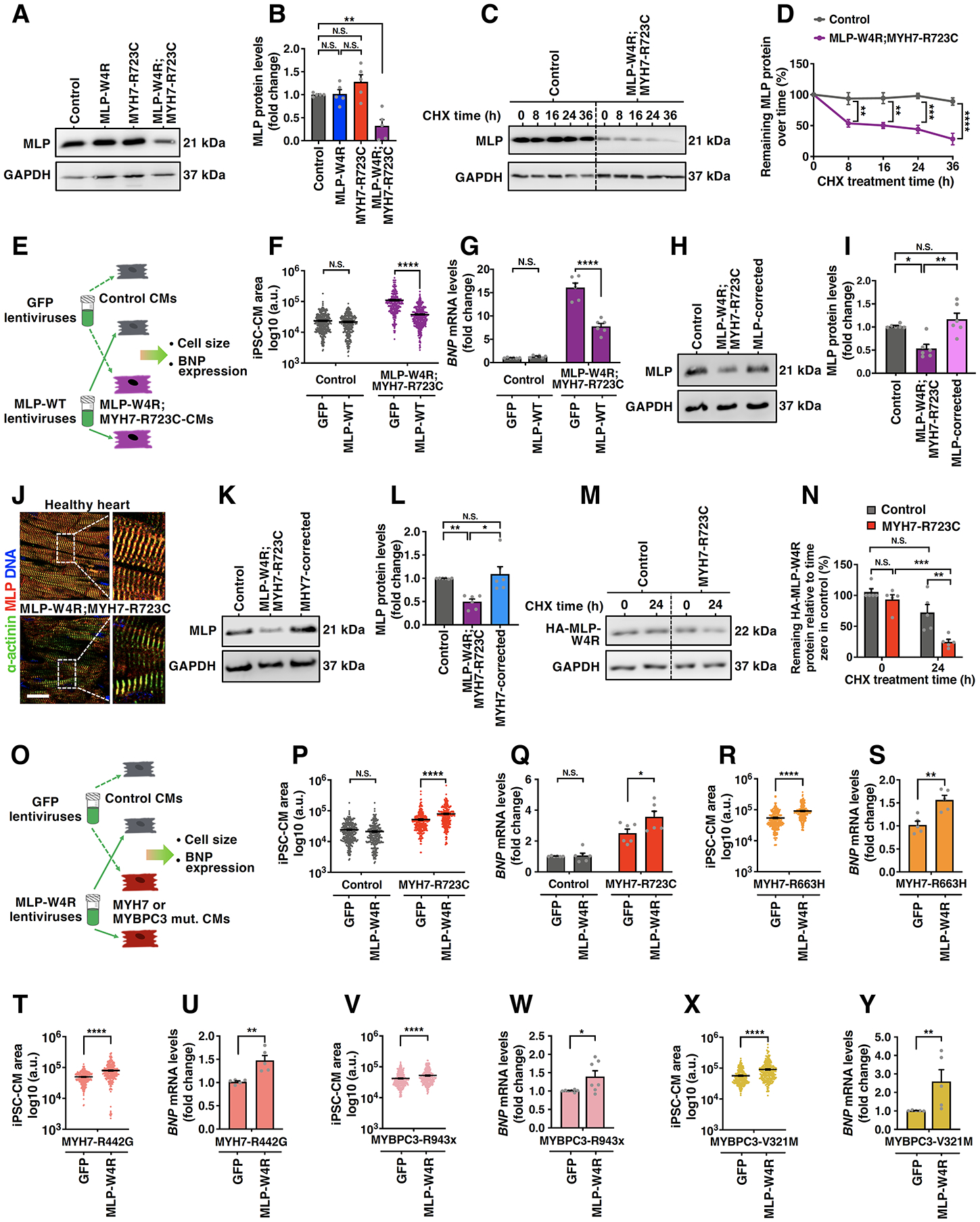
A, A representative western blot image showing MLP protein levels in day 35 control, MLP-W4R, MYH7-R723C, and MLP-W4R;MYH7-R723C iPSC-CMs. GAPDH was used as a loading control. B, Quantification of MLP protein in panel A (n=5 independent cardiomyocyte differentiation batches per group). Two-way ANOVA with Tukey’s multiple comparisons test revealed that there was a statistically significant interaction between the MLP-W4R and MYH7-R723C mutations in regulating MLP protein levels (F(1, 16)=17.940, p=0.0006; each mutation considered as an independent factor). C, Evaluation of MLP protein stability in control and MLP-W4R;MYH7-R723C iPSC-CMs via western blotting following cycloheximide (CHX, 50 μg/mL) treatments for 0, 8, 16, 24 and 36 hours. D, Quantification of the percentage of remaining MLP protein at different time points of CHX treatment normalized to time zero protein levels in panel C (n=3 independent cardiomyocyte differentiation batches per group). Two-way ANOVA with Tukey’s multiple comparisons test was used to evaluate the MLP levels between control and MLP-W4R;MYH7-R723C iPSC-CMs at each time point and revealed a significantly higher decay rate of MLP in the double mutant CMs than that in the control CMs 8, 16, 24, and 36 hours after CHX treatment (F(4,20)=6.811, p=0.001; genotype and treatment time considered as two independent factors). E, Schematic showing ectopic expression of GFP and HA-tagged wild-type MLP (MLP-WT) in control and MLP-W4R;MYH7-R723C day 35 iPSC-CMs and phenotypic analysis. F, Quantification of iPSC-CM cell area in control and MLP-W4R;MYH7-R723C iPSC-CMs from three independent cardiomyocyte differentiation batches (≥100 cells per batch). See Figure S5A–5C for cell staining images. G, qRT-PCR analysis of BNP mRNA expression levels in control and MLP-W4R;MYH7-R723C iPSC-CMs (n=6 independent cardiomyocyte differentiation batches per group). Two-way ANOVA with Tukey’s multiple comparisons test revealed a preferential normalization of cell area (F(1,1404)=180.200, p<0.0001) and BNP expression (F(1,20)=53.410, p<0.0001) by MLP-WT in MLP-W4R;MYH7-R723C iPSC-CMs (F, G). Note that genotype and type of ectopically expressed protein were considered as two independent factors. H, A representative western blot image showing MLP protein levels in day 35 control, MLP-W4R;MYH7-R723C, and MLP-W4R-corrected (containing MYH7-R723C) iPSC-CMs. I, Quantification of MLP protein in panel H (n=6 independent cardiomyocyte differentiation batches per group) by Kruskal–Wallis with Dunn’s multiple comparisons test (H(2)=11.000, p=0.0007). J, Immunostaining of MLP (red) and α-actinin (green) in young adult healthy heart tissue and MLP-W4R;MYH7-R723C proband heart tissue. Scale bar, 50 μm. K, A representative western blot image showing MLP levels in day 35 control, MLP-W4R;MYH7-R723C, and MYH7-corrected (containing MLP-W4R) iPSC-CMs. L, Quantification of MLP protein in panel K (n=6 independent cardiomyocyte differentiation batches per group) by Kruskal–Wallis with Dunn’s multiple comparisons test (H(2)=11.910, p=0.0002). M, A representative western blot image showing the expression of HA-tagged MLP-W4R protein in control or MYH7-R723C iPSC-CMs after CHX treatment. HA-MLP-W4R was ectopically expressed in day 21–22 iPSC-CMs via lentiviral infection. Infected iPSC-CMs were cultured for an additional 12–13 days followed by a 24-hour treatment of CHX (50 μg/mL) to examine the stability of HA-MLP-W4R via detection of HA signals. N, Quantification of the remaining HA-MLP-W4R protein in control and MYH7-R723C iPSC-CMs after CHX treatment in panel N (n=5 independent cardiomyocyte differentiation batches per group). Two-way ANOVA with Tukey’s multiple comparisons test revealed a significantly higher decay rate of HA-MLP-W4R protein in MYH7-R723C iPSC-CMs compared with that in control iPSC-CMs (F(1,16)=4.560, p=0.0485; genotype and type of ectopically expressed protein considered as two independent factors). O, Schematic showing ectopic expression of the GFP or HA-tagged MLP-W4R protein in control and MYH7-R723C iPSC-CMs via lentiviral infection and phenotypic analysis. P-Q, Quantification of cell area (P) and BNP mRNA levels (Q) in control and MHY7-R723C day 35 iPSC-CMs. See Figure S5D–5F for cell staining images. Two-way ANOVA with Tukey’s multiple comparisons test revealed a specific effect of HA-MLP-W4R in worsening HCM defects, including cell area (F(1,1289)=42.39, p<0.0001) and BNP expression (F(1,20)=4.457, p=0.0475), in MYH7-R723C iPSC-CMs but not in control iPSC-CMs (genotype and type of ectopically expressed protein considered as two independent factors). Note that cell area measurements were from three independent cardiomyocyte differentiation batches (≥100 cells/batch) and BNP expression analysis from six independent cardiomyocyte differentiation batches. R-Y, Quantification of cell area (R, T, V, and X) and BNP mRNA levels (S, U, W, and Y) in the corresponding day 35 MYH7-R663H, MYH7-R442G, MYBPC3-R943x, and MYBPC3-V321M iPSC-CMs transduced with GFP or MLP-W4R lentiviruses on day 21–22. See Figure S5G–5R for cell staining images. A two-tailed unpaired Student’s t test was used for cell area comparison (R, T, V, and X) and a two-tailed unpaired Mann-Whitney U test for BNP expression analysis (S, U, W, and Y). Cell area measurements in R and T were from three independent cardiomyocyte differentiation batches (≥100 cells/batch), and those in V and X from four independent batches (≥50 cells/batch). BNP expression analyses were from five or more (S, U, W, and Y) independent cardiomyocyte differentiation batches. Note that F values indicate the ratio of explained variance between groups to unexplained variance due to experimental variations within groups and the degrees of freedom (df) represent the df for factor interaction and the sum of the individual df for each experimental group, respectively (B, D, F, G, N, P, and Q). Additionally, H values indicate Kruskal-Wallis H Test statistics and df represent df for experimental groups (I and L). Each data point represents a single sample generated from a batch of iPSC-CMs (B, D, G, I, L, N, Q, S, U, W, and Y) or iPSC-CM (F, P, R, T, V, and X) derived from at least three independent cardiomyocyte differentiation batches. All data are presented as mean ± S.E.M; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; N.S: not significant.
We next examined whether the MYH7-R723C mutation is required for elevated MLP degradation in the double mutant CMs. Importantly, genetic correction of MYH7-R723C in the double mutant CMs resulted in a marked restoration of the MLP levels (Figure 4K and 4L) and an effective rescue of the HCM defects (Figure 2B–2D and 2H–2K). As MLP self-associates in a protein complex38, 39, this prompted us to hypothesize that the MLP-W4R-containing complex is more sensitized for degradation in the MYH7-R723C genetic background (i.e. double mutant proband) than in the MYH7-WT type background (MLP-W4R single mutant). To test this, HA-tagged MLP-W4R was ectopically expressed in the MYH7-R723C and WT iPSC-CMs, and HA-MLP-W4R protein stability was analyzed by cycloheximide treatment. Interestingly, HA-MLP-W4R exhibited a significantly higher decay rate in the MYH7-R723C mutant CMs than in the WT control CMs after cycloheximide treatment (Figure 4M and 4N). Taken together, these results suggest that MLP-W4R is more prone to degradation in the MYH7-R723C genetic background, providing molecular evidence for the aggressive hypertrophic pathology in the double mutant proband CMs, which have both a source of enhanced Z-disc mechanical stimulus (the myosin mutation) and a hypersensitive detector (MLP-W4R) of that stimulus.
Based on the prolonged, hypercontractile twitches caused by the MYH7-R723C mutation, the increased MLP-W4R decay in the MYH7-R723C background, and the more severe HCM defects in the double mutant proband CMs, we postulated that the myosin mutation might trigger the initial hypertrophic stimulus while the MLP-W4R mutation could be a modifier of the phenotypic expression of HCM. To test this, HA-tagged MLP-W4R was ectopically expressed in the MYH7-R723C and WT control iPSC-CMs. While MLP-W4R significantly exacerbated the enlarged cell area and elevated BNP expression in the MYH7-R723C mutant CMs, it did not affect the control CMs (Figure 4O–4Q; Figure S5D–5F). Additionally, MLP-W4R ectopic expression in iPSC-CMs of four other HCM patients (MYH7-R663H, MYH7-R442G, myosin binding protein C3 [MYBPC3-R943x; with a premature stop codon], and MYBPC3-V321M)7, 40–42 exacerbated the HCM defects (Figure 4R–4Y; Figure S5G–5R). These results suggest that MLP-W4R, likely via interfering with the formation of functional MLP-protein complexes38, 39, may have a broad role in modifying phenotypic expression of HCM associated with MYH7 and MYBPC3 mutations.
Inhibiting Calcineurin/NFAT or Mitigating Enhanced Actomyosin Crossbridge Formation in Proband iPSC-CMs Prevents Development of the HCM Phenotype
We next investigated the potential mechanism by which reduced expression of MLP in the double mutant proband CMs leads to severe hypertrophic defects. A previous murine study reported the colocalization of MLP and calcineurin, a pro-hypertrophic phosphatase that dephosphorylates NFAT and promotes its nuclear translocation, at the Z-disc43. MLP also appeared to have a role in anchoring calcineurin to the Z-disc in the mouse heart. We performed co-immunostaining of MLP and calcineurin in LV tissue sections from a healthy human donor heart, and observed colocalization of MLP and calcineurin at the Z-disc (Figure 5A). Additionally, MLP and calcineurin could be coimmunoprecipitated from lysates of healthy control iPSC-CMs, suggesting an interaction (direct or indirect) of MLP with calcineurin (Figure 5B). Moreover, co-immunostaining of calcineurin and α-actinin in sections from a healthy donor and the double mutant proband heart showed that while calcineurin primarily colocalized with α-actinin at the Z-discs in the healthy heart, there were readily appreciable numbers of disintegrated sarcomeres with calcineurin dissociated from the Z-discs (Figure 5C) in the proband heart, suggesting contribution of abnormal calcineurin signaling to proband HCM pathogenesis. To test this, we measured nuclear translocation of NFATc4 in control and double mutant proband iPSC-CMs. Nuclear translocation of NFATc4 in the double mutant CMs was markedly higher than that in the control CMs (Figure 5D and 5E). Further, treatment of double mutant CMs with calcineurin inhibitor FK506 resulted in reduced nuclear localization of NFATc4 (Figure 5F and 5G) and a significant rescue of hypertrophic defects including normalizing cell area and expression of BNP (Figure 5H and 5I). Additionally, consistent with enhanced nuclear localization of NFATc4 in proband CMs, a higher NFAT transcriptional activity was observed in proband CMs compared with controls based on NFAT-luciferase activity (Figure S5S and 5T).
Figure 5. Inhibiting Calcineurin/NFAT or Cardiac Myosin Mitigates Development of the HCM Phenotype in Proband iPSC-Derived Cardiomyocytes.
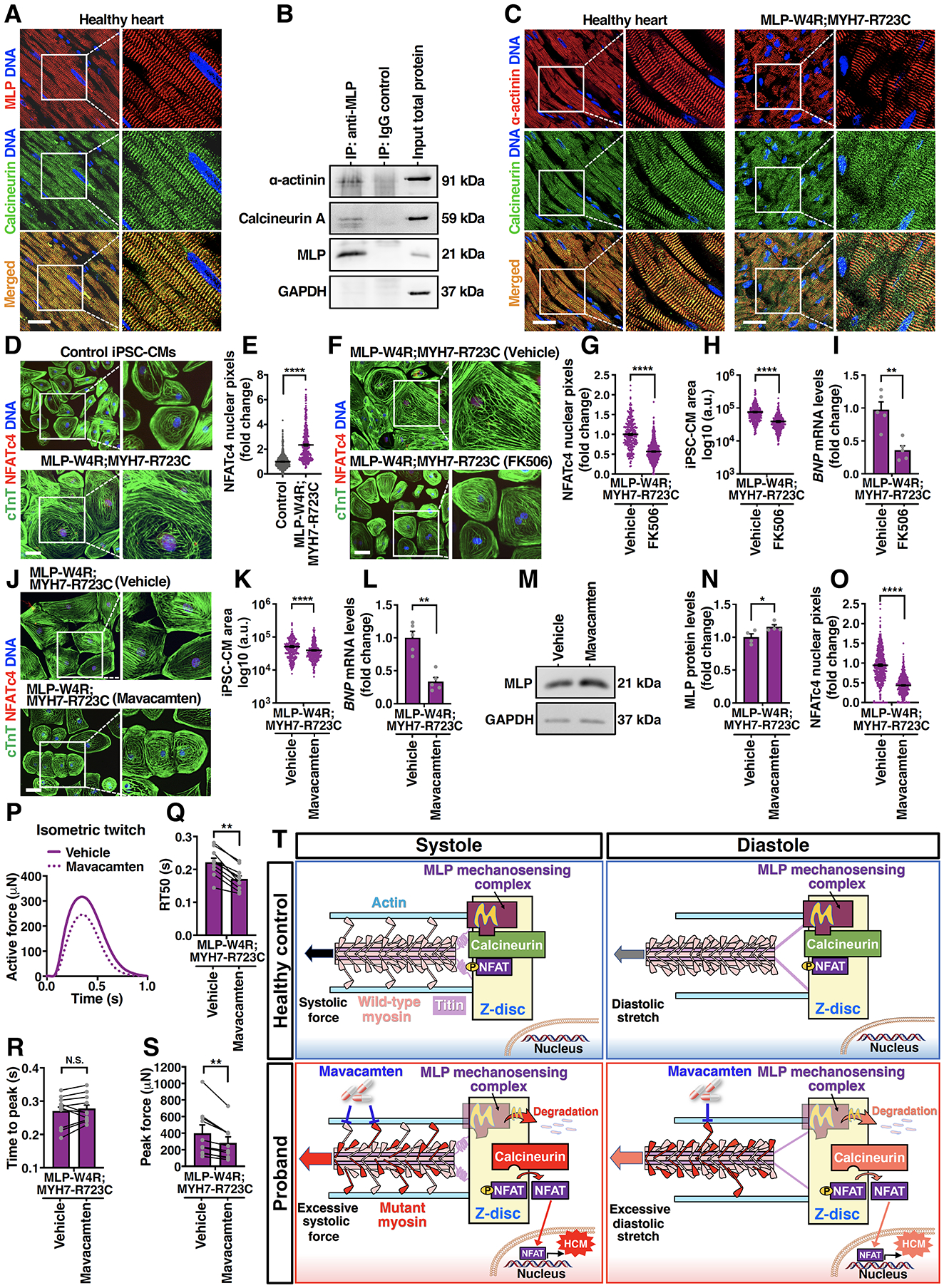
A, Immunostaining of calcineurin (green) and MLP (red) in the young adult healthy heart tissue. DNA was counterstained by DAPI. Scale bar, 50 μm. B, A representative immunoblot showing co-immunoprecipitation of MLP, calcineurin, and α-actinin in day 35 control iPSC-CMs. C, Immunostaining of calcineurin (green) and α-actinin (red) in the young adult healthy heart tissue and MLP-W4R;MYH7-R723C proband heart tissue. Scale bar, 50 μm. D, Immunostaining of NFATc4 (red) and cTnT (green) in day 35 control and MLP-W4R;MYH7-R723C iPSC-CMs. DNA was counterstained by DAPI. Scale bar, 100 μm. E, Quantification of NFATc4 nuclear signals in panel D (two-tailed unpaired Student’s t test). Nuclear NFATc4 pixels (gray value) were quantified by ImageJ from three independent cardiomyocyte differentiation batches (≥100 cells/batch). F, Immunostaining of NFATc4 (red) and cTnT (green) in MLP-W4R;MYH7-R723C iPSC-CMs treated with DMSO (vehicle) or 0.5 μg/mL calcineurin inhibitor FK506. Treatment was started on day 25 of cardiac differentiation for 4 days (G and H) and 24 hours (I). Scale bar, 100 μm. G-I, Quantification of NFATc4 nuclear signals (G), cell area (H), and BNP gene expression (I) in DMSO- or FK506-treated MLP-W4R;MYH7-R723C iPSC-CMs. A two-tailed unpaired Student’s t test was performed for nuclear NFATc4 and cell area analyses from three independent cardiomyocyte differentiation batches (≥100 cells/batch). A two-tailed unpaired Mann-Whitney U test was used for BNP gene analysis (n=5 independent cardiomyocyte differentiation batches per group; normalized to GAPDH). J, Immunostaining of NFATc4 (red) and cTnT (green) in MLP-W4R;MYH7-R723C iPSC-CMs treated with DMSO (vehicle) or 0.5 μM cardiac myosin ATPase inhibitor mavacamten. Treatment was started on day 25 of cardiac differentiation for four days (J, K, M, N, and O) and 24 hours (L). Scale bar, 100 μm. K-L, Quantification of cell area (K) and BNP gene expression (L) in DMSO- or mavacamten-treated MLP-W4R;MYH7-R723C iPSC-CMs. A two-tailed unpaired Student’s t test was performed for cell area analysis from three independent cardiomyocyte differentiation batches (≥100 cells/batch). A two-tailed unpaired Mann-Whitney U test was used for BNP gene analysis (n=5 independent cardiomyocyte differentiation batches per group; normalized to GAPDH). M, A representative western blot of MLP expression in vehicle- and mavacamten-treated MLP-W4R;MYH7-R723C iPSC-CMs. N, Quantification of MLP protein levels in panel M. A Mann-Whitney U test was used for MLP protein analysis (n=4 independent cardiomyocyte differentiation batches per group; normalized to GAPDH). O, Quantification of NFATc4 nuclear signals in vehicle and mavacamten-treated MLP-W4R;MYH7-R723C iPSC-CMs in panel J. A two-tailed unpaired Student’s t test was performed based on three independent cardiomyocyte differentiation batches (≥100 cells/batch). P, Representative paired measurement of proband MLP-W4R;MYH7-R723C iPSC-CM-derived EHTs treated with DMSO (vehicle) or 0.5 μM mavacamten to steady state (30 minutes). Q-S, Quantification of biomechanical properties including RT50 (Q), time to peak (R), and peak force (S) (ten EHTs generated from three independent proband cardiomyocyte differentiation batches). A two-tailed paired Mann-Whitney U test was used for analysis between two groups. T, Schematic of the proposed working model. Mechanical force is transmitted into the Z-disc primarily through pulling actin by myosin heads via actomyosin crossbridges during systolic contraction. Titin is the main mechanical element transferring load into the Z-disc during diastolic stretch, with very little mechanical input through actin due to the detachment of myosins from actin. MLP mechanosensing complex may keep calcineurin/NFAT signaling in check under normal systolic and diastolic conditions. In contrast, the MYH7-R723C mutation in the proband resulted in abnormally higher systolic force transmitted into the Z-disc due to more crossbridge formation. Additionally, residual mutant actomyosin crossbridges due to delayed relaxation in the proband could lead to elevated diastolic Z-disc stretching. Consequently, MLP stretch-sensing machinery would likely be sensitized, leading to MLP degradation and subsequent calcineurin-NFAT hypertrophic signaling. Importantly, treatment of proband CMs with mavacamten, a cardiac myosin ATPase inhibitor, could potentially normalize systolic force generation and diastolic stretching, resulting in a restored MLP level and rescue from HCM defects. See the main text for details. Each data point represents a single iPSC-CM (E, G, H, K, and O), sample generated from a batch of iPSC-CMs (I, L, and N), or EHT (Q-S) derived from at least three independent cardiomyocyte differentiation batches. All data are presented as mean ± S.E.M; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; N.S: not significant.
Since we showed that the MYH7 mutation caused increased myosin duty cycle and could prolong contraction and putatively drive pathological hypertrophy, we next investigated whether mavacamten (MYK-461), a small molecule that modulates myosin duty cycles by reducing myosin ATPase activity11, could attenuate the hypertrophic response in proband CMs. Indeed, there appeared to be a significant rescue of HCM defects in proband iPSC-CMs by mavacamten, including the enlarged cell area as well as elevated expression of BNP and other HCM markers (Figure 5J–5L; Figure S5U–5Z). We then explored whether mavacamten treatment might affect the expression of MLP, implicated in preventing hyperactivity of calcineurin-NFAT and pathological hypertrophy in the proband CMs (Figure 4A–4L and Figure 5C–5I). Notably, administration of mavacamten resulted in a reproducible upregulation of MLP (Figure 5M and 5N) as well as downregulation of nuclear NFATc4 (Figure 5J and 5O), its target gene MCIP1, and NFAT-luciferase activities (Figure S5Y’ and 5Z’) in the proband CMs. Moreover, double mutant proband EHTs treated with mavacamten exhibited reduced peak force and RT50 (Figure 5P–5S). These results indicate that reducing cardiomyocyte contractility can attenuate the severe hypertrophic signaling in the double mutant proband CMs, by indirectly blocking the degradation of MLP and preventing NFAT translocation. Increased myosin duty cycle appears to be necessary for driving hypertrophic signaling and can be attenuated with pharmacological interventions.
Discussion
Here we describe a unique clinical case that provides insights into a new biomechanical mechanism by which enhanced myofilament contractile force generation due to sarcomeric mutations destabilizes the MLP/Z-disc mechanosensory complex, leading to disinhibition of calcineurin-NFAT signaling and consequent hypertrophic responses (Figure 5T). Further, our studies report for the first time, evidence to address the long-standing knowledge gap and clinical debate over the role of the common MLP-W4R variant that appears in Caucasians of up to 1%16. Previous studies reported that while the human MLP-W4R variant associates with dilated cardiomyopathy (DCM)14, heterozygous knock-in of MLP-W4R in mice causes moderate HCM16. In contrast, CMs derived from MLP-W4R iPSCs did not show appreciable cardiac abnormalities (Figure 1H–1N; Figures S1J–1L and S4), consistent with the lack of overt cardiomyopathic defects in healthy MLP-W4R human carriers in our (Figure 1A; Table S1) and other research settings17, 18. Further, while ectopic expression of MLP-W4R in healthy control iPSC-CMs did not cause any abnormalities, it significantly worsened the HCM defects in iPSC-CMs derived from five HCM patients bearing different MYH7 and MYBPC3 mutations (Figure 4O–4Y). Our studies thus provide strong evidence that the MLP-W4R common variant is an important disease modifier, and in the presence of other cardiac risk factors, can promote cardiac pathology. However, it remains possible that the MLP-W4R variant associated with DCM in the original report14 may be in linkage disequilibrium with an unknown mutation that initiates DCM worsened by MLP-W4R. Additionally, these results highlight the possibility that significant physiological and genetic differences between mice and humans may contribute to the different cardiac phenotypes observed in the presence of the heterozygous MLP-W4R variant.
Our biophysical analysis, computer modeling, genetic-correction, and pharmacological normalization experiments, revealed a prolonged myosin duty cycle leading to elevated sarcomeric force generation in the MYH7-R723C mutation (Figures 2, 3 and 5). This is consistent with a recent report suggesting that mutations in the MYH7 converter domain including arginine at position 723 may result in an increased availability of myosin heads participating in force production44. Enhanced force generation by mutant myosin is expected to be transmitted into the Z-disc and trigger an abnormally high mechanical stimulus during systole (Figure 5T, Proband vs. Healthy control). Since the MYH7-R723C mutation also led to a prolonged twitch event with slower relaxation (Figures 1K, 1L and 2H, 2I), it is likely that some mutant crossbridges remained attached to the thin filament, even as Ca2+ fell to diastolic levels. These crossbridges would pull on actin as the sarcomere lengthens, add to the titin-based force already being transmitted to the Z-disc, and provide a higher-than-normal mechanical stimulus during diastole (Figure 5T, Proband vs. Healthy control). Notably, slower relaxation of the myosin mutant CMs was not due to delayed reuptake of intracellular Ca2+ to the SR, since Ca2+ decay was significantly faster in the MYH7-R723C mutant CMs (Figure S4D–4I), which likely represents a compensatory response to blunt the heightened contractility and prolonged kinetics in the mutant CMs. Abnormally high force transmission into the Z-disc in either systole or diastole would likely sensitize the MLP/Z-disc mechanosensory machinery, leading to MLP degradation and subsequent calcineurin-NFAT hypertrophic signaling (Figure 5T). Consistent with this model, restoration of MLP levels via genetic correction of either myosin or MLP mutation (Figure 4H, 4I, 4K, and 4L) or ectopic expression of wild-type MLP (Figure 4E) markedly rescued hypertrophic defects in the proband CMs (Figure 2A–2O and Figure 4F, 4G). Additionally, inhibiting calcineurin-NFAT led to significant rescue of hypertrophic defects in the proband CMs (Figure 5F–5I). These findings highlight the potentially important interaction of MLP and calcineurin in a protein complex localized to the Z-disc in healthy CMs (Figure 5A and 5B).
These results raise the intriguing possibility that MLP may prevent calcineurin-NFAT signaling from being activated by forming an MLP/calcineurin/NFAT protein complex at the Z-disc in healthy hearts (Figure 5T). Notably, MLP is implicated in anchoring calcineurin at the Z-disc, and compartmentalization of calcineurin and NFAT at the Z-disc appears to be essential for coordinated activation of NFAT via dephosphorylation in murine studies43, 45. It is possible that MLP has both inhibitory and facilitative effects on calcineurin activity. By localizing calcineurin/NFAT complex at the Z-disc, under normal physiological conditions, the MLP mechanosensing complex might keep calcineurin/NFAT in check to prevent hypertrophy in healthy hearts. In contrast, if a higher-than-normal mechanical stimulus is received by the MLP/Z-disc complex, MLP proteolytic degradation might allow for a rapid activation of a pre-assembled calcineurin/NFAT complex at the Z-disc and subsequent hypertrophic signaling (Figure 5T, Proband). Consistent with this model, normalization of hypercontractile force and prolonged relaxation in proband CMs with the actomyosin crossbridge inhibitor mavacamten resulted in an increase in the MLP level, a decrease in the calcineurin/NFAT activity, and a rescue of the hypertrophic defects (Figure 5J–5S; Figure S5X–5Z’). Future studies are warranted to further elucidate the molecular mechanisms that mediate the repression of calcineurin/NFAT by MLP, MLP protein degradation by stretch-sensing, and potentially additional targets mediating MLP mechanosignaling. Future work is also required to obtain further mechanistic insights into the marked enhancement of measured twitch force in the proband EHTs regarding potential contributions from increased cell mass, production of new isoforms of sarcomeric proteins, and post-translational modifications to sarcomeric and/or calcium handling proteins associated with the activation of calcineurin-NFAT signaling in the proband. Additionally, future endeavors will be made to optimize the EHT technology with further electromechanical conditioning, reproducible cardiac scaffolds, coupled with isogenic iPSC lines based on gene-editing, for robust disease modeling.
It is anticipated that the sarcomeric contraction/MLP/calcineurin mechanotransduction pathway described here would have relevance to many sarcomeric HCM mutations, not solely the three β-cardiac myosin and two myosin-binding protein C3 mutations that we studied. In addition, reduced MLP levels at the Z-disc and consequent calcineurin/NFAT complex activation associated with abnormally high mechanical stress, could lead to pathological remodeling in many types of heart failure, including other familial cardiomyopathies or acquired heart failure that develops in the context of hypertension, diabetes, or aging. To the extent that mavacamten therapy predisposes to reduced cardiac function in HCM patients12, 13, its use would be restricted in advanced heart failure patients with depressed LVEF. In this context, an alternative therapeutic strategy that enhances the Z-disc MLP mechanosensing complex might prove advantageous. Such an approach warrants further investigation and may hasten the development of effective, rationally designed therapeutics for a wide spectrum of heart failure patients.
Supplementary Material
Clinical Perspective.
What is new?
This study presents a novel biomechanical mechanism by which enhanced myofilament contractile force generation due to sarcomeric mutations destabilizes the MLP/Z-disc mechanosensory complex, leading to disinhibition of calcineurin-NFAT signaling and consequent hypertrophy.
Normalization of hypercontractile force in proband cardiomyocytes either with gene editing approaches or with the actomyosin crossbridge inhibitor mavacamten resulted in an increase in MLP levels, a decrease in the calcineurin/NFAT activity, and a rescue from HCM defects.
We also provide evidence that the common MLP-W4R variant is an important modifier that worsens the disease severity of HCM but alone does not appear sufficient to cause disease.
What are the clinical implications?
The sarcomeric contraction/MLP/calcineurin mechanotransduction pathway discovered from this study may play important roles in pathological hypertrophic remodeling caused by many sarcomeric HCM mutations.
Reduction in the sarcomeric MLP level associated with elevated Z-disc mechanical stress and consequent disinhibition of the calcineurin/NFAT signaling could contribute to the development of many types of heart failure, including other familial cardiomyopathies or acquired heart failure in the context of hypertension, diabetes, or aging.
New therapeutics that stabilize the sarcomeric MLP mechanosensory complex warrant further investigation and may prove fruitful in treating HCM and other heart failure patients.
Acknowledgements
Y.Q., S.G.C., D.L.J, M.R., J.P., L.R.S., Y.R., J.S., P.T.P, I.P.C., M.Y., L.Y., and C.C.M. conceived the study; M.R., J.P., L.R.S., Y.R., J.S., P.T.P., S.D., Y.H., and J.L. performed research; M.R., J.P., L.R.S., Y.R., J.S., P.T.P., S.D., Y.H., J.L., L.S., I.P.C., M.Y., L.Y., D.L.J, S.G.C., and Y.Q. analyzed data; M.R., J.P., L.R.S., and Y.Q. wrote the manuscript; M.R., J.P., L.R.S., M.W.E., G.T., J.H., L.Y., C.C.M., D.L.J., S.G.C., and Y.Q. edited the manuscript and provided critical feedback. We thank Christopher Anderson, Luke Batty, and Colleen Lopez from Yale Cardiovascular Research Center for their support on research and manuscript revision. We would also like to acknowledge and thank the patients and their family members for their participation in the study.
Source of Funding
This work was supported by an NIH K02HL101990, an R01HL131940, a Department of Defense (DOD) 12712285, and a DOD 11959515 (to YQ). Work was also supported by an R21HL126025 and an R01HL136590 (to S.G.C.), an R01DE025664 (to I.P.C.), a DOD W81XWH-20-1-0036 (to J.P.), an American Heart Association Predoctoral Fellowship, a P.D. Soros Fellowship for New Americans, and a National Institute of General Medical Sciences Medical Scientist Training Program Grant T32GM007205 (to L.R.S.), a Connecticut Regenerative Medicine Research Program 12-SCA-Yale-15 (to Y.R.), and an NIH F31-HL143924 and a T32-GM0007324 (to M.W.E.).
Non-standard Abbreviations and Acronyms
- ANOVA
analysis of variance
- BNP
brain natriuretic peptide
- CHX
cycloheximide
- CRISPR
clustered regularly interspaced short palindromic repeats
- DCM
dilated cardiomyopathy
- EHT
engineered heart tissue
- HCM
hypertrophic cardiomyopathy
- iPSC-CM
induced pluripotent stem cell–derived cardiomyocytes
- IVS
interventricular septum
- LV
left ventricular
- LVEF
left ventricular ejection fraction
- LVOT
left ventricular outflow tract
- MEF
mouse embryonic fibroblast
- MLP
muscle LIM protein
- MYBPC3
myosin binding protein C3
- MYH7
myosin heavy chain 7
- NFAT
nuclear factor of activated T-cells
- N.S.
not significant
- RT50
time from peak force to 50% relaxation
- S.E.M.
standard error of the mean
- TALEN
transcription activator-like effector nuclease
- TTP
time to peak force
Footnotes
Conflict of Interest Disclosures
S.G.C. has equity ownership in Propria LLC, which has licensed technology employed in the studies reported in this manuscript. Propria LLC did not fund these studies, and Propria LLC did not influence the conduct, description, or interpretation of the findings in this report. None of the other authors declares any competing financial interest.
References
- 1.Sen-Chowdhry S, Jacoby D, Moon JC and McKenna WJ. Update on hypertrophic cardiomyopathy and a guide to the guidelines. Nat Rev Cardiol. 2016;13:651–675. [DOI] [PubMed] [Google Scholar]
- 2.Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, Caleshu CA, Helms AS, Colan SD, Girolami F, Cecchi F, Seidman CE, Sajeev G, Signorovitch J, Green EM and Olivotto I. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018;138:1387–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–20. [DOI] [PubMed] [Google Scholar]
- 4.Ashrafian H and Watkins H. Reviews of translational medicine and genomics in cardiovascular disease: new disease taxonomy and therapeutic implications cardiomyopathies: therapeutics based on molecular phenotype. J Am Coll Cardiol. 2007;49:1251–64. [DOI] [PubMed] [Google Scholar]
- 5.Tardiff JC. Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev. 2005;10:237–48. [DOI] [PubMed] [Google Scholar]
- 6.Ho CY, Charron P, Richard P, Girolami F, Van Spaendonck-Zwarts KY and Pinto Y. Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc Res. 2015;105:397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, Han L, Yen M, Wang Y, Sun N, Abilez OJ, Hu S, Ebert AD, Navarrete EG, Simmons CS, Wheeler M, Pruitt B, Lewis R, Yamaguchi Y, Ashley EA, Bers DM, Robbins RC, Longaker MT and Wu JC. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis J, Davis LC, Correll RN, Makarewich CA, Schwanekamp JA, Moussavi-Harami F, Wang D, York AJ, Wu H, Houser SR, Seidman CE, Seidman JG, Regnier M, Metzger JM, Wu JC and Molkentin JD. A Tension-Based Model Distinguishes Hypertrophic versus Dilated Cardiomyopathy. Cell. 2016;165:1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohn R, Thakar K, Lowe A, Ladha FA, Pettinato AM, Romano R, Meredith E, Chen YS, Atamanuk K, Huey BD and Hinson JT. A Contraction Stress Model of Hypertrophic Cardiomyopathy due to Sarcomere Mutations. Stem Cell Reports. 2019;12:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spoladore R, Maron MS, D’Amato R, Camici PG and Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J. 2012;33:1724–33. [DOI] [PubMed] [Google Scholar]
- 11.Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, Henze M, Kawas R, Oslob JD, Rodriguez HM, Song Y, Wan W, Leinwand LA, Spudich JA, McDowell RS, Seidman JG and Seidman CE. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351:617–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho CY, Mealiffe ME, Bach RG, Bhattacharya M, Choudhury L, Edelberg JM, Hegde SM, Jacoby D, Lakdawala NK, Lester SJ, Ma Y, Marian AJ, Nagueh SF, Owens A, Rader F, Saberi S, Sehnert AJ, Sherrid MV, Solomon SD, Wang A, Wever-Pinzon O, Wong TC and Heitner SB. Evaluation of Mavacamten in Symptomatic Patients With Nonobstructive Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2020;75:2649–2660. [DOI] [PubMed] [Google Scholar]
- 13.Olivotto I, Oreziak A, Barriales-Villa R, Abraham TP, Masri A, Garcia-Pavia P, Saberi S, Lakdawala NK, Wheeler MT, Owens A, Kubanek M, Wojakowski W, Jensen MK, Gimeno-Blanes J, Afshar K, Myers J, Hegde SM, Solomon SD, Sehnert AJ, Zhang D, Li W, Bhattacharya M, Edelberg JM, Waldman CB, Lester SJ, Wang A, Ho CY, Jacoby D and investigators E-Hs. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;396:759–769. [DOI] [PubMed] [Google Scholar]
- 14.Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, McKenna W, Yokoyama M, Schork NJ, Omens JH, McCulloch AD, Kimura A, Gregorio CC, Poller W, Schaper J, Schultheiss HP and Chien KR. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111:943–55. [DOI] [PubMed] [Google Scholar]
- 15.Boateng SY, Senyo SE, Qi L, Goldspink PH and Russell B. Myocyte remodeling in response to hypertrophic stimuli requires nucleocytoplasmic shuttling of muscle LIM protein. J Mol Cell Cardiol. 2009;47:426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knoll R, Kostin S, Klede S, Savvatis K, Klinge L, Stehle I, Gunkel S, Kotter S, Babicz K, Sohns M, Miocic S, Didie M, Knoll G, Zimmermann WH, Thelen P, Bickeboller H, Maier LS, Schaper W, Schaper J, Kraft T, Tschope C, Linke WA and Chien KR. A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circ Res. 2010;106:695–704. [DOI] [PubMed] [Google Scholar]
- 17.Newman B, Cescon D, Woo A, Rakowski H, Erikkson MJ, Sole M, Wigle ED and Siminovitch KA. W4R variant in CSRP3 encoding muscle LIM protein in a patient with hypertrophic cardiomyopathy. Mol Genet Metab. 2005;84:374–5. [DOI] [PubMed] [Google Scholar]
- 18.Geier C, Gehmlich K, Ehler E, Hassfeld S, Perrot A, Hayess K, Cardim N, Wenzel K, Erdmann B, Krackhardt F, Posch MG, Osterziel KJ, Bublak A, Nagele H, Scheffold T, Dietz R, Chien KR, Spuler S, Furst DO, Nurnberg P and Ozcelik C. Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum Mol Genet. 2008;17:2753–65. [DOI] [PubMed] [Google Scholar]
- 19.Geisterfer-Lowrance AA, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE and Seidman JG. A mouse model of familial hypertrophic cardiomyopathy. Science. 1996;272:731–4. [DOI] [PubMed] [Google Scholar]
- 20.Lowey S, Lesko LM, Rovner AS, Hodges AR, White SL, Low RB, Rincon M, Gulick J and Robbins J. Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an alpha- or beta-myosin heavy chain backbone. J Biol Chem. 2008;283:20579–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K and Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. [DOI] [PubMed] [Google Scholar]
- 22.Schwan J, Kwaczala AT, Ryan TJ, Bartulos O, Ren Y, Sewanan LR, Morris AH, Jacoby DL, Qyang Y and Campbell SG. Anisotropic engineered heart tissue made from laser-cut decellularized myocardium. Sci Rep. 2016;6:32068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sewanan LR and Campbell SG. Modelling sarcomeric cardiomyopathies with human cardiomyocytes derived from induced pluripotent stem cells. J Physiol. 2020;598:2909–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng R, Sewanan LR, Stankey P, Li X, Qyang Y and Campbell S. Shortening Velocity Causes Myosin Isoform Shift in Human Engineered Heart Tissues. Circ Res. 2021;128:281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen IP, Fukuda K, Fusaki N, Iida A, Hasegawa M, Lichtler A and Reichenberger EJ. Induced pluripotent stem cell reprogramming by integration-free Sendai virus vectors from peripheral blood of patients with craniometaphyseal dysplasia. Cell Reprogram. 2013;15:503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park J, Anderson CW, Sewanan LR, Kural MH, Huang Y, Luo J, Gui L, Riaz M, Lopez CA, Ng R, Das SK, Wang J, Niklason L, Campbell SG and Qyang Y. Modular design of a tissue engineered pulsatile conduit using human induced pluripotent stem cell-derived cardiomyocytes. Acta Biomater. 2020;102:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campbell SG, Lionetti FV, Campbell KS and McCulloch AD. Coupling of adjacent tropomyosins enhances cross-bridge-mediated cooperative activation in a markov model of the cardiac thin filament. Biophys J. 2010;98:2254–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheikh F, Ouyang K, Campbell SG, Lyon RC, Chuang J, Fitzsimons D, Tangney J, Hidalgo CG, Chung CS, Cheng H, Dalton ND, Gu Y, Kasahara H, Ghassemian M, Omens JH, Peterson KL, Granzier HL, Moss RL, McCulloch AD and Chen J. Mouse and computational models link Mlc2v dephosphorylation to altered myosin kinetics in early cardiac disease. J Clin Invest. 2012;122:1209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watkins H, Thierfelder L, Hwang DS, McKenna W, Seidman JG and Seidman CE. Sporadic hypertrophic cardiomyopathy due to de novo myosin mutations. J Clin Invest. 1992;90:1666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Girolami F, Ho CY, Semsarian C, Baldi M, Will ML, Baldini K, Torricelli F, Yeates L, Cecchi F, Ackerman MJ and Olivotto I. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol. 2010;55:1444–53. [DOI] [PubMed] [Google Scholar]
- 31.Fusaki N, Ban H, Nishiyama A, Saeki K and Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seki T, Yuasa S, Oda M, Egashira T, Yae K, Kusumoto D, Nakata H, Tohyama S, Hashimoto H, Kodaira M, Okada Y, Seimiya H, Fusaki N, Hasegawa M and Fukuda K. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell. 2010;7:11–4. [DOI] [PubMed] [Google Scholar]
- 33.Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, Egashira T, Seki T, Muraoka N, Yamakawa H, Ohgino Y, Tanaka T, Yoichi M, Yuasa S, Murata M, Suematsu M and Fukuda K. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. 2013;12:127–37. [DOI] [PubMed] [Google Scholar]
- 34.Kohler J, Winkler G, Schulte I, Scholz T, McKenna W, Brenner B and Kraft T. Mutation of the myosin converter domain alters cross-bridge elasticity. Proc Natl Acad Sci U S A. 2002;99:3557–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swank DM, Knowles AF, Suggs JA, Sarsoza F, Lee A, Maughan DW and Bernstein SI. The myosin converter domain modulates muscle performance. Nat Cell Biol. 2002;4:312–6. [DOI] [PubMed] [Google Scholar]
- 36.Gunther LK, Rohde JA, Tang W, Walton SD, Unrath WC, Trivedi DV, Muretta JM, Thomas DD and Yengo CM. Converter domain mutations in myosin alter structural kinetics and motor function. J Biol Chem. 2019;294:1554–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sewanan LR, Moore JR, Lehman W and Campbell SG. Predicting Effects of Tropomyosin Mutations on Cardiac Muscle Contraction through Myofilament Modeling. Front Physiol. 2016;7:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boateng SY, Belin RJ, Geenen DL, Margulies KB, Martin JL, Hoshijima M, de Tombe PP and Russell B. Cardiac dysfunction and heart failure are associated with abnormalities in the subcellular distribution and amounts of oligomeric muscle LIM protein. Am J Physiol Heart Circ Physiol. 2007;292:H259–69. [DOI] [PubMed] [Google Scholar]
- 39.Hoffmann C, Moreau F, Moes M, Luthold C, Dieterle M, Goretti E, Neumann K, Steinmetz A and Thomas C. Human muscle LIM protein dimerizes along the actin cytoskeleton and cross-links actin filaments. Mol Cell Biol. 2014;34:3053–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han L, Li Y, Tchao J, Kaplan AD, Lin B, Li Y, Mich-Basso J, Lis A, Hassan N, London B, Bett GC, Tobita K, Rasmusson RL and Yang L. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc Res. 2014;104:258–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seeger T, Shrestha R, Lam CK, Chen C, McKeithan WL, Lau E, Wnorowski A, McMullen G, Greenhaw M, Lee J, Oikonomopoulos A, Lee S, Yang H, Mercola M, Wheeler M, Ashley EA, Yang F, Karakikes I and Wu JC. A Premature Termination Codon Mutation in MYBPC3 Causes Hypertrophic Cardiomyopathy via Chronic Activation of Nonsense-Mediated Decay. Circulation. 2019;139:799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu H, Yang H, Rhee JW, Zhang JZ, Lam CK, Sallam K, Chang ACY, Ma N, Lee J, Zhang H, Blau HM, Bers DM and Wu JC. Modelling diastolic dysfunction in induced pluripotent stem cell-derived cardiomyocytes from hypertrophic cardiomyopathy patients. Eur Heart J. 2019;40:3685–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heineke J, Ruetten H, Willenbockel C, Gross SC, Naguib M, Schaefer A, Kempf T, Hilfiker-Kleiner D, Caroni P, Kraft T, Kaiser RA, Molkentin JD, Drexler H and Wollert KC. Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proc Natl Acad Sci U S A. 2005;102:1655–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawana M, Sarkar SS, Sutton S, Ruppel KM and Spudich JA. Biophysical properties of human beta-cardiac myosin with converter mutations that cause hypertrophic cardiomyopathy. Sci Adv. 2017;3:e1601959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, Cseresnyes Z, Randall WR and Schneider MF. Activity-dependent nuclear translocation and intranuclear distribution of NFATc in adult skeletal muscle fibers. J Cell Biol. 2001;155:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seki T, Yuasa S and Fukuda K. Generation of induced pluripotent stem cells from a small amount of human peripheral blood using a combination of activated T cells and Sendai virus. Nat Protoc. 2012;7:718–28. [DOI] [PubMed] [Google Scholar]
- 47.Livak KJ and Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 48.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA and Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR and Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–8. [DOI] [PubMed] [Google Scholar]
- 50.Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, Howard TP, Takeda DY, Ly SH, Kim E, Gannon HS, Hurhula B, Sharpe T, Goodale A, Fritchman B, Steelman S, Vazquez F, Tsherniak A, Aguirre AJ, Doench JG, Piccioni F, Roberts CWM, Meyerson M, Getz G, Johannessen CM, Root DE and Hahn WC. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet. 2018;50:1381–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.