

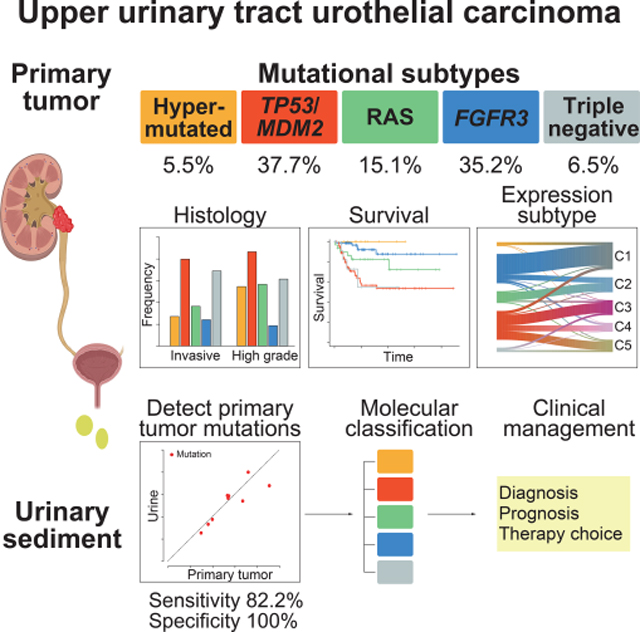
Summary
Upper urinary tract urothelial carcinoma (UTUC) is one of the common urothelial cancers. Its molecular pathogenesis, however, is poorly understood with no useful biomarkers available for accurate diagnosis and molecular classification. Through an integrated genetic study involving 199 UTUC samples, we delineate the landscape of genetic alterations in UTUC enabling genetic/molecular classification. According to the mutational status of TP53, MDM2, RAS, and FGFR3, UTUC is classified into five subtypes having discrete profiles of gene expression, tumor location/histology, and clinical outcome, which is largely recapitulated in an independent UTUC cohort. Sequencing of urine sediment-derived DNA has a high diagnostic value for UTUC with 82.2% sensitivity and 100% specificity. These results provide a solid basis for better diagnosis and management of UTUC.
Keywords: Upper urinary tract urothelial carcinoma, urothelial carcinoma, transcriptome, integrated molecular study, molecular classification, hypermutation, molecular diagnostic, FGFR3, RAS, TP53
eTOC blurb
Based on an integrated genomic analysis of 199 upper urinary tract urothelial carcinomas (UTUCs), Fujii et al. identify five genetic subtypes with discrete profile of gene mutation, expression, histology, and clinical outcome and also demonstrates a high diagnostic value of sequencing urinary sediment-derived DNA for non-invasive detection of UTUC.
Graphical Abstract
Introduction
Upper urinary tract urothelial carcinomas (UTUCs) account for 5–10% of all urothelial cancers. UTUC is more frequently involved with renal pelvis (56–63%) than ureter (37–44%) (Raman et al., 2011; Roupret et al., 2018). Although sharing many clinicopathological features with urothelial bladder carcinoma (UBC), UTUC has its distinct features. For example, UTUC is developed in mesoderm-derived epithelium, may be associated with the Lynch syndrome (Therkildsen et al., 2018) and could be induced by aristolochic acid (AA) (Chen et al., 2012); whereas UBC is rarely associated with the Lynch syndrome or AA exposure. These observations support that both cancers have distinct molecular pathogenesis. Previous genomic studies have revealed the genetic landscape of UBC (Guo et al., 2013; Hurst et al., 2017; Pietzak et al., 2017; Robertson et al., 2017). UBC has a relatively higher mutational burden compared to other common cancer types, which could be partly explained by the APOBEC-mediated mutagenesis. Frequently mutated genes in UBC include those involved in the RTK-Ras-PI3K pathway (such as FGFR3, HRAS, PIK3CA, and ERBB2), the p53-Rb pathway (such as TP53, RB1, CCND1, and CDKN2A), and chromatin modifiers (such as KMT2D, KMT2C, KDM6A, and CREBBP) (Guo et al., 2013; Hurst et al., 2017; Pietzak et al., 2017; Robertson et al., 2017).
By contrast, our knowledge of the molecular pathogenesis of UTUC is still limited, despite several genomic studies of UTUC have been published. One targeted-capture sequencing study of 196 UTUC cases has reported a mutation landscape of UTUC, clarifying a significant difference in mutation frequency in several genes between UTUC and UBC, such as HRAS, ERBB2, TP53, and RB1 (Audenet et al., 2019). However, this study has not provided unbiased approaches for the analysis of mutational burden, genomic signatures and gene expression profiles. In this regard, two studies (Moss et al., 2017; Robinson et al., 2019) have performed integrated analyses using whole exome sequencing (WES) and RNA sequencing. However, small numbers of samples analyzed (n=31 and 37, respectively) prevent an unbiased, comprehensive characterization of UTUC and preclude the detection of unique molecular subtypes with a clinical impact. This contrasts with the big number of large-scale integrated studies published for UBC, in which gene expression subtypes have significant impact on UBC biology and their clinical outcomes (Hedegaard et al., 2016; Kamoun et al., 2020; Robertson et al., 2017).
Molecular characterization would also aid the development of diagnostics, prognostication, and even therapeutics. In particular, UTUC is often invasive at initial diagnosis (60%) compared to UBC (15–25%) (Roupret et al., 2018), frequently associated with poor prognosis, which is at least partly due to difficulty in early detection. Urinary cytology is less sensitive for UTUC (41%) than for UBC (86%) and more invasive procedures are needed for definitive diagnosis (Messer et al., 2011; Tanaka et al., 2014). Thus, there is an urgent demand to establish non-invasive diagnostics reliably used for early diagnosis and prognostication. In this regard, detecting mutations in urine-derived DNA has diagnostic potential in UBC (Ou et al., 2020; Ward et al., 2019). However, only a few studies have evaluated the role of urine-derived DNA sequencing in UTUC (Hayashi et al., 2019; Springer et al., 2018), in which performance of detecting tumor-derived mutations and its potential in molecular diagnostics and prognostication has not been fully evaluated.
In this study, we conduct a comprehensive molecular study of UTUC using unbiased, multiplatform analyses, which we hypothesize should help fully understand the molecular pathogenesis of UTUC in terms of gene mutation, genomic copy number, DNA methylation, and gene expression. Through these analyses, we delineate distinct pathogenesis of UTUC and propose a molecular classification that provides personalized therapeutic options to UTUC patients. Built upon the molecular genetics findings of UTUC, we establish a sensitive platform based on urine-derived DNA sequencing, which enables non-invasive diagnosis with molecular classification and prognostication of UTUC.
Results
Multiplatform analysis of UTUC
We collected 199 fresh frozen tumor samples surgically removed from 198 patients, including one having bilateral tumors. None of the patients had history of presurgical treatments, and they were recruited from three institutions. These samples were subjected to WES (n=199) with a mean depth of 165x (101x-345x) (Figure S1A), single-nucleotide polymorphism (SNP) array karyotyping (n=199), TERT promoter sequencing (n=199), messenger RNA sequencing (n=158), and array-based DNA methylation analysis (n=86). In our cohort, non-papillary tumors tended to be underrepresented (15.1%) compared to previous reports (median of 27.5%; range, 15.7%-68.2%) (Raman et al., 2016; Shibing et al., 2016; Zhao et al., 2020). Typically presented as small flat tumors, non-papillary tumors are more likely to be missed in biopsy, which may partly explain their underrepresentation in our study, although the exact reason is unclear (Table S1).
Somatic mutations and CNAs in UTUC
In total, we detected 51,709 mutations, including 48,609 single nucleotide variants (SNVs), 128 dinucleotide variants (DNVs), and 2,972 insertion/deletions (indels), with a median of 2.3 (range, 0.3–182.3) mutations/Mb (hereafter, ‘mutations’ include SNVs, DNVs, and indels unless otherwise specified). Extremely large numbers of mutations were found in 11 (5.5%) samples (Figure 1A). Among these, 8 had biallelic defects in mismatch repair (MMR) genes and many (6/8) had a prior cancer history (Figure 1A; Table S2), which combined, are suggestive of Lynch syndrome. Except for these hypermutated cases, UTUC showed a smaller mutational burden, compared to UBC (5.8 mutations/Mb) (Figure S1B) (Robertson et al., 2017). Excluding those in hypermutated cases, SNVs in UTUC were dominated by C>T and C>G transitions. More frequent T>A substitutions were found in a single case (UTUC98T), in which exposure to AA was suspected (Figure S1C). Decomposition of SNVs using ‘pmsignature’ disclosed four single base substitution (SBS) signatures (Sigs. A-D) (Shiraishi et al., 2015), which explained 79.1% of all SNVs in non-hypermutated samples. Sigs. A-D corresponded to known COSMIC signatures: SBS2, SBS13, SBS1, and SBS16, respectively (Figure 1B). These are also signatures that have been implicated in APOBEC-a and APOBEC-b activities, aging, and transcription-coupled repair (TCR) (Alexandrov et al., 2020). Representing one of the most common mutational processes in human cancer, age-related Sig. C/SBS1 was also ubiquitously seen in UTUC samples, explaining 22% of all SNVs on average. It has not been highlighted in a previous UTUC study (Robinson et al., 2019). By contrast, frequency of APOBEC-related (SBS2, SBS13) and TCR-related (SBS16) SNVs was highly variable across samples (Figures S1D and S1E). Similar per-sample signature distributions were obtained using different inference algorithms, ‘deconstructSigs’ and ‘MutationalPatterns’, which showed high average cosine similarities to those obtained with ‘pmsignature’, 0.91 and 0.90, respectively (Figure S1F) (Blokzijl et al., 2018; Rosenthal et al., 2016). Sig. D/SBS16 has recently been implicated in alcohol consumption and aldehyde metabolism (Chang et al., 2017; Yokoyama et al., 2019) but was not reported in UBC samples from TCGA. Similar to other cancers enriched for SBS16 SNVs, Sig. D/SBS16 SNVs in our cohort was significantly and independently associated with the hypomorphic ALDH2 allele (A: rs671) and alcohol consumption (Figure 1C). When hypermutated cases were included, another signature (Sig. A’) was extracted, that has been correlated with defective MMR activity (Figure S1G). In fact, this signature was highly enriched in samples having defective MMR genes, while MMR-intact hypermutated cases were enriched for APOBEC signatures (Figure S1H). We found a total of 26 genes significantly mutated or positively selected in non-hypermutated UTUC (q < 0.1) (Table S3). In addition, frequent TERT promoter mutations were detected by PCR-based deep sequencing or Sanger sequencing (Table S4).
Figure 1. Multiplatform genetic analysis of UTUC.

(A) Mutational burden (blue) and their variability (red) in 199 UTUC samples (top) with mutation status of MMR genes, and history of cancer (bottom) for hypermutated samples. (B) Four predominant SBS signatures (left) and strand bias for each signature (right) in non-hypermutated UTUC samples. (C) Box plots of SNVs assigned to Sig. D, according to the allelic status of ALDH2 (rs 671) and history of alcohol consumption (left) and forest plots of odds ratios for indicated risk factors with 95% confidence intervals (CIs; right). P values calculated by two-sided Mann-Whitney U test with Bonferroni correction. The median, first and third quartiles, as well as outliners, are indicated with whiskers extending to the furthest value within 1.5 of the interquartile range in box plots. **P < 0.01, ***P < 0.001. (D and E) Comparisons of frequency of common genetic lesions (left), between invasive UBC (n = 375) and UTUC (n = 81) (D) and ureter (n = 54) and renal pelvis (n = 112) cancers (E). Log2-transformed odds ratios are also shown with 95% CIs (right). Significant enrichment is shown by asterisks (q < 0.1, two-sided Fisher’s exact test with Benjamini-Hochberg adjustment for multiple testing). TERT p., TERT promoter. (F) Summary of the molecular features in urothelial cancers according to tumor location. MSI, microsatellite instability.
See also Figures S1–S3 and Tables S2 and S3.
In the analysis of copy number alterations (CNAs), 37.2% of samples showed a highly complex karyotype with frequent focal CNAs, aneuploidy and chromothripsis (cluster 2), whereas the remaining samples showed rather a simple pattern with frequent arm-level aberrations involving chromosomes 1q, 3, 8, and 9 (Figure S2A). Overall, 23 chromosomal arms and 40 focal regions were recurrently affected (q < 0.1) (Figure S2B). We also identified 118 in-frame gene fusions in 68 samples in RNA sequencing. Most of these fusions were not recurrent, except for FGFR3-involving fusions found in nine samples. Seven of these had a well-known fusion partner, TACC3 (Guo et al., 2013), while the remaining two involved UBE2K and G3BP2, leading to in-frame FGFR3/UBE2K and FGFR3/G3BP2 fusions (Figure S2C). Tumors with FGFR3 fusions and SNVs showed significantly higher expression of FGFR3 than those without or normal urothelium samples (Figure S2D).
Combining these results, we obtained a comprehensive list of genetic alterations with different impacts on survival in UTUC (Figures S3A–C). Most frequently affected genes included the TERT promoter (49%), KMT2D (46%), CDKN2A (45%), FGFR3 (45%), and TP53 (35%). The driver genes significantly mutated in UTUC were largely overlapped to those identified in previous UBC studies (Guo et al., 2013; Robertson et al., 2017). As reported for UBC (Gui et al., 2011; Pietzak et al., 2017), frequencies of these genetic lesions differed between invasive and non-invasive tumors; TP53 and CCND1 were more commonly affected in invasive UTUC, whereas FGFR3, HRAS, and the TERT promoter mutations were more frequent in non-invasive tumors (Figure S3D). Despite high overall similarity in their frequency between UTUCs and UBCs, frequencies of several alterations substantially differed between both tumors; CDKN2A and KMT2D were preferentially affected in UTUC, while ERBB2 was more frequently mutated in UBC (Figures 1D and S3E). Frequencies of genetic lesions also differed depending on anatomical location of tumors; ureter cancers showed notably higher frequencies of KMT2D and TP53 mutations, whereas HRAS, KDM6A, and the TERT promoter were more commonly mutated in pelvic tumors (Figure 1E). Taken together, these results suggest that urothelial carcinomas showed a distinct pattern of genetic alterations depending on tumor location and progression status (Figure 1F).
Unique mutational subtypes of UTUC
Analysis of pair-wise relationships between mutations and CNAs across 188 non-hypermutated samples revealed a number of significantly co-occurring and mutual exclusive genetic lesions (Figures 2A and 2B), which allowed us to classify non-hypermutated UTUC into four distinct subtypes showing unique co-alteration/mutually exclusive patterns, based on a Bayesian model with minimum manual curations of a small number of overlapped cases (Papaemmanuil et al., 2016) (see STAR Methods; Figures S4A–E). These are characterized by the presence/absence of alterations in TP53 or MDM2, RAS (HRAS/KRAS/NRAS), and FGFR3 and combining hypermutated cases, a total of five subgroups were identified (Table 1; Figure 2C).
Figure 2. Unique Molecular subtypes of UTUC.

(A) Statistically significant (q < 0.1) positive (red) and negative (blue) correlations across common genetic lesions in non-hypermutated UTUC (Fisher’s exact test with Benjamini-Hochberg adjustment). (B) Statistically significant (q < 0.1, two-sided Fisher’s exact test with Benjamini-Hochberg adjustment) pairwise comparison of difference in MCFs between indicated gene pairs. (C) Landscape of somatic alterations in 199 UTUC cases grouped into five discrete subtypes. Significantly mutated (MutSigCV) and/or positively selected (dNdScv) driver genes are indicated by asterisks. At the bottom are SBS signatures. RTK, receptor tyrosine kinase; PI3K, phosphatidylinositol 3-kinase.
Table 1.
Characteristics of Mutational Subtypes in UTUC.
| Characteristic | Total | Hyper | TP53/MDM2 | RAS | FGFR3 | TN |
|---|---|---|---|---|---|---|
| Number | 199 | 11 | 75 | 30 | 70 | 13 |
| Gender – no. (%) | ||||||
| Male | 139 (69.8) | 6 (54.5) | 49 (65.3) | 23 (76.7) | 51 (72.9) | 10 (76.9) |
| Female | 60 (30.2) | 5 (45.5) | 26 (34.7) | 7 (23.3) | 19 (27.1) | 3 (23.1) |
| Age at surgery | ||||||
| Median | 71.9±9.5 | 72.2±5.8 | 71.4±9.4 | 65.0±10.1 | 74.4±9.7 | 71.2±7.3 |
| Range | 41–90 | 59–78 | 53–90 | 48–86 | 41–86 | 61–84 |
| T -category – no. (%) | ||||||
| Non–invasive | 98 (49.2) | 8 (72.7) | 15 (20.0) | 19 (63.3) | 53 (75.7) | 4 (30.8) |
| Invasive | 101 (50.8) | 3 (27.3) | 60 (80.0) | 11 (36.7) | 17 (24.3) | 9 (69.2) |
| Grade – no. (%) | ||||||
| Low | 90 (45.2) | 5 (45.5) | 10 (13.3) | 13 (43.3) | 57 (81.4) | 5 (38.5) |
| High | 109 (54.8) | 6 (54.5) | 65 (86.7) | 17 (56.7) | 13 (18.6) | 8 (61.5) |
| Morphology – no. (%) | ||||||
| Papillary | 169 (84.9) | 11 (100.0) | 52 (69.3) | 28 (93.3) | 70 (100.0) | 8 (61.5) |
| Non–papillary | 30 (15.1) | 0 (0.0) | 23 (30.7) | 2 (6.7) | 0 (0.0) | 5 (38.5) |
| UBC history – no. (%) | ||||||
| Previous | 18 (9.0) | 0 (0.0) | 10 (13.3) | 1 (3.3) | 4 (5.7) | 6 (46.2) |
| Simultaneous | 16 (8.0) | 1 (9.1) | 9 (12.0) | 0 (0.0) | 6 (8.6) | 0 (0.0) |
| None | 165 (83.0) | 10 (90.9) | 56 (74.7) | 29 (96.7) | 60 (85.7) | 7 (53.8) |
| Squamous differentiation – no. (%) | ||||||
| + | 23 (11.6) | 0 (0.0) | 13 (17.3) | 5 (16.7) | 2 (2.9) | 2 (15.4) |
| − | 176 (88.4) | 11 (100.0) | 62 (82.7) | 25 (83.3) | 68 (97.1) | 11 (84.6) |
| Metastasis – no. (%) | ||||||
| + | 52 (26.1) | 0 (0.0) | 33 (44.0) | 6 (20.0) | 7 (10.0) | 4 (30.8) |
| − | 147 (73.9) | 11 (100.0) | 42 (56.0) | 24 (80.0) | 63 (90.0) | 9 (69.2) |
| Tumor location – no. (%) | ||||||
| Pelvis | 123 (61.8) | 2 (18.2) | 29 (38.7) | 30 (100.0) | 53 (75.7) | 9 (69.2) |
| Ureter | 65 (32.7) | 8 (72.7) | 38 (50.6) | 0 (0.0) | 15 (21.4) | 4 (30.8) |
| Unknown | 11 (5.5) | 1 (9.1) | 8 (10.7) | 0 (0.0) | 2 (2.9) | 0 (0.0) |
| Tobacco history – no. (%) | ||||||
| + | 93 (46.7) | 3 (27.3) | 29 (38.7) | 18 (60.0) | 39 (55.7) | 5 (38.5) |
| − | 106 (53.3) | 8 (72.7) | 46 (61.3) | 12 (40.0) | 31 (44.3) | 8 (61.5) |
| Chemotherapy – no. (%) | ||||||
| AC | 56 (28.1) | 1 (9.1) | 33 (44.0) | 4 (13.3) | 10 (14.3) | 5 (38.5) |
Hyper, hypermutated; TP53/MDM2, TP53/MDM2-mutated; RAS, RAS-mutated; FGFR3, FGFR3-mutated, TN, triple-negative; UBC, urothelial bladder carcinoma; AC, adjuvant chemotherapy.
Enriched for invasive tumors, the largest (37.7%) UTUC subtype in our cohort was consisted of tumors carrying mutated TP53 or MDM2 amplification (TP53/MDM2-mutated subtype). Compared with other subtypes, the TP53/MDM2-mutated subtype was frequently accompanied by complex CNAs and CCND1 and KMT2D alterations while TERT promoter mutations were less common (Figures 2A, 3A, and S4F). In accordance with a previous report (Audenet et al., 2019), this subtype showed the most aggressive phenotype, with a high rate of metastasis (40.0%) and the shortest disease-specific survival (Figures 3A and 3B). No significant difference was observed across the four subtypes in terms of SBS signatures (Figure S4G).
Figure 3. Genetic and clinical features of molecular subtypes in UTUC.

(A) Frequencies of common genetic lesions and clinical/histological features across four non-hypermutated subtypes. Genes affected in more than two subtypes (CDKN2A, TERT promoter, and MDM2) are listed on the top. (B) Disease-specific survival for patients in each mutational subtype. Hazard ratios (HRs) and 95% CIs were calculated by Cox proportional-hazards model.
See also Figures S4 and S5.
The FGFR3-mutated subtype was also among the most common subtypes, accounting for 35.2% of the cohort, characterized by FGFR3 hotspot mutations at p.S249 and p.Y373 (Figure S3B) and significantly co-occurred with mutations in the TERT promoter, STAG2, PIK3CA, and KDM6A (Figures 2A and 3A). Significantly showing higher mutated cell fractions (MCFs) compared to other co-occurring mutations, FGFR3 mutations represented the founder mutations in most of the cases (Figure 2B). Arm-level CNAs were common in this subtype and most frequently involved chromosomes 1q, 7, 9, 11p, 17, and 20. By contrast, recurrent focal lesions were rare and when present, almost exclusively affected CDKN2A, MDM2, and/or IKZF2 (Figure S4F). Patients in this subtype tended to be at early stages and had a lower grade histology with morphologically papillary tumors and showed a favorable disease-specific survival (Figures 3A and 3B). Of interest, the majority (9/11) of hypermutated tumors had FGFR3 mutations, which has also been reported in a previous study, although the dominance of FGFR3 mutations was less conspicuous (11/17) (Donahue et al., 2018).
Another major subtype was characterized by hotspot mutations in RAS family genes (Figure S3B), accounting for 15.1% of the cohort. With the lowest mutational burden and age at diagnosis (Table 1; Figure S4H), the RAS-mutated subtype showed frequent DDX17 (26.7%) and TERT promoter (70.0%) mutations as well as CNAs involving chromosome 3, 8, 9, 19, and 20 and focal deletions targeting CDKN2A (Figures 2A, 3A, and S4F). All tumors in this subtype involved renal pelvis. With frequent high-grade histology and squamous differentiation, the RAS-mutated tumors showed a more aggressive phenotype than the FGFR3-mutated subtype and an intermediate prognosis, which was better than that in the TP53/MDM2-mutated and the triple-negative subtypes (Figures 3A and 3B).
The remaining, ‘triple-negative’ subtype, included 13 cases, none of which had alterations in subtype defining genes (i.e., TP53/MDM2, FGFR3, and RAS genes) or hypermutations. Patients in this subtype showed a poor prognosis comparable to that for the TP53/MDM2-mutated cases (Figure 3B). Of interest in this regard is a mutation at the splicing acceptor at the intron-exon boundary of TP53 exon 4 in a case (UTUC44T), potentially leading to mis-splicing and loss of TP53 function. Other three cases had complex chromosomal abnormalities characteristic of TP53/MDM2-mutated cases (Figure 2C), raising a possibility that some of these triple-negative cases might have had ‘masked’ TP53 lesions, such as exon skipping and other structural variations. Frequently mutated genes in this subtype included the TERT promoter and CDKN2A, as well as KDM6A and CCND1.
To validate the relevance of our genetic classification developed for UTUC, we analyzed an independent cohort of 123 UTUC patients from MSKCC, including 4 with hypermutated tumors (Audenet et al., 2019; Sfakianos et al., 2015). Frequencies of major driver alterations were largely similar between the Japanese and MSKCC cohorts, except for a lower frequency of alterations in several genes in the MSKCC cohort, including mutations in TP53, KMT2A, ELF3 and the TERT promoter, CCND1 amplification and CDKN2A deletions in invasive and/or non-invasive tumors (Figure S5A). Co-mutation patterns recapitulated those found in the Japanese cohort, with largely mutually exclusive alterations among TP53/MDM2, RAS, and FGFR3 genes (Figure S5B). As expected, non-hypermutated MSKCC cases were co-clustered with the Japanese cases into four distinct subtypes, and with similar impacts on survival (STAR Methods; Figures S5C–S5F). The analysis of MSKCC cases alone revealed only two clusters with and without FGFR3 mutations, likely due to a small number of cases (Figure S5D). MSKCC cases in each subtype showed genetic and clinical characteristics that were similar to those in the corresponding Japanese cases, although higher frequencies of pelvic/high-grade tumors and metastatic diseases were seen depending on subtype (Figure S5E). The mutation profile in each subtype was also largely similar between both cohorts, except for higher frequencies of CCND1 amplification, CDKN2A deletions, and TERT promoter mutations in the Japanese cohort (Figure S5E).
Although the five mutational subtypes were also identified in the TCGA UBC cohort, the clinical and histological differences were not as clear as those in our cohort (Figure S5G). Furthermore, cases included in the triple-negative subtype, which had no shared genetic lesions, accounted for 34.3% of the cohort, indicating that UBC is more genetically heterogeneous than UTUC and is difficult to be classified and characterized by simple rules of the presence/absence of gene alterations.
Gene expression and DNA methylation in UTUC
Next, we investigated gene expression profiles of UTUC. Through unbiased clustering analysis using RNA sequencing data from 158 UTUC samples, we identified five specific expression subtypes (C1-C5) (Figures 4 and S6A). We observed moderate to weak correlations with phi coefficients of 0.22–0.56 between expression and mutational subtypes (Figures 4A and 4B); the majority of the FGFR3-mutated and most of the hypermutated subtypes were classified in the C1 subtype, the TP53/MDM2-mutated and triple-negative subtypes largely separated into either of the C3-C5 subtypes, and most cases in the RAS-mutated subtype and a subset of the FGFR3-mutated cases belonged to the C2 subtype. This contrasted with the observations in the TCGA UBC cohort, in which, except for the FGFR3-mutated and the luminal-papillary subtypes, weak to no correlations between expression and mutational subtypes were observed (Figure S6B). As expected from significant prognostic impacts of mutational subtypes, gene expression subtypes also showed distinct prognostic profiles; C3–5 were associated with a worse prognosis, compared with C1 and to a lesser extent, C2 (Figure S6C).
Figure 4. Expression profiles of UTUC.

(A-B) Correlations (A) and phi coefficients (B) between mutational and expression subtypes in 158 UTUC cases in which both mutations and gene expression were analyzed. (C) Expression profiles of indicated gene pathways of biological relevance implicated in UBC. EMT, epithelial mesenchymal transition; ECM, extracellular matrix; APM, antigen presentation machinery.
To better understand the feature of each subtype, gene expression was compared between UTUC and UBC with respect to a set of functional pathways implicated in unique UBC subtypes, using the TCGA UBC dataset (Figures 4C and S6D; Table S5). In line with a previous report (Robinson et al., 2019), a substantial proportion of UTUC cases (C1, C2, and C5) were characterized by upregulated ‘luminal’ markers, which are hallmarks of three luminal subtypes in UBC. As expected from the enrichment for FGFR3 mutations, the C1 subtype showed the highest expression of FGFR3-associated markers. By contrast, the C3 and C4 subtypes were characterized by enhanced expression of ‘basal’ and ‘squamous’ markers, which is characteristic of the basal-squamous subtype in UBC. No significant association between female gender and basal tumors, which has been reported in previous UBC studies (Choi et al., 2014; Robertson et al., 2017), was observed in our UTUC cohort (Figure S6E). The expression level of PVRL4 and ERBB2, which encode target molecules of antibody-drug conjugates Nectin-4 and HER2, respectively, was lowest in C3, while comparable in the remaining four subtypes (Figure S6F). A subset of UTUC were also characterized by gene expression related to tumor microenvironment and immune responses. As opposed to a previous report, which reported a low frequency (12.5%) of immunologically active tumors in UTUC (Robinson et al., 2019), as many as 46.2% of UTUC comprising C3-C5 subtypes exhibited expression signatures suggestive of activated immune reactions, such as upregulation of genes related to CD8 T-cells and immune checkpoint, which was most prominent in C3 tumors. Among these subsets, C3 and C5 were also characterized by upregulation of genes related to extracellular matrix and smooth muscle signatures. Probably reflecting high involvement of immune cells and stromal components, samples belonging to C3 and C5 had lower tumor contents observed in these subgroups (Figure 5A). In agreement with these features of immune responses and stromal reactions, prominent cytolytic activities were noted in C3 and to a lesser extent, in C4 and C5 subtypes, while C3 and C5 showed enhanced expression of pan-fibroblast TGFβ response signature genes (F-TBRS), which is an indicator of TGFβ pathway activity in fibroblasts and implicated in resistance to immunotherapy by restricting T-cell infiltration (Figures 5B and 5C) (Mariathasan et al., 2018). We further classified C3 and C5 on the basis of expression of immune and stromal signature genes, aiming for more detailed analysis of heterogeneous tumor microenvironment in these subgroups (Figure S7A). C3 and C5 cases were further classified into C3a-C3d and C5a-C5c subclusters, respectively. C3a-C3d represent 60.0% of the C3 cases. C3a and C3c showed significantly higher cytolytic activities than other UTUC and UBC subgroups (Figure S7B). In addition, C3c tumors were characterized by high mutational burdens (Figure S7C). Thus, it is suggested that patients in these subtypes might potentially benefit from immune-checkpoint blockade (ICB) therapy. Of interest, C3a and C3b and C5b and C5c, which accounted for 46.7% of C3 and 87.0% of C5, respectively, were enriched for a stroma cell signature with a high F-TBRS (Figure S7D). These results suggest that a combination of immunotherapy and anti-TGFβ treatments might improve the clinical outcome of patients in C3a, C3b, C5b and C5c.
Figure 5. Association between mutation and gene expression/DNA methylation.

(A–C) Box plots showing tumor contents (A), cytotoxic score (B), and expression signature score for F-TBRS (C) in each expression subtype. The median, first and third quartiles, as well as outliers, are indicated with whiskers extending to the furthest value within 1.5 of the interquartile range. P values calculated by two-sided Mann-Whitney U test with Bonferroni correction. *P < 0.05, **P < 0.01, ***P < 0.001. (D) Number of UTUC samples assigned to consensus subtypes for UBC. (E) Disease-specific survival of patients assigned to the luminal papillary subtype according to UTUC expression subtypes (C1-C5). (F) Methylation profile in 86 UTUC and 8 normal urothelial tissue samples. Integrated view of DNA methylation clusters combined with mutation/expression subtypes (top) and consensus matrix (K = 3, bottom) are shown. (G) Hazard ratios (HRs) with 95% CIs for covariates significantly included in Cox proportional-hazards model of disease-specific survival. Factors that were significantly associated with disease-specific survival are shown in red. (H) Relative contribution of clinical and genetic factors to the hazard of disease-specific survival in Cox proportional-hazards model. Contribution of both combined and individual factors are depicted in horizontal and vertical axis, respectively.
See also Figures S6 and S7.
A recent report of muscle invasive bladder cancer provided a gene expression-based classification system to classify urothelial carcinoma into six consensus classes according to tumor transcriptome, which include luminal papillary (LumP), luminal unstable (LumU), stroma-rich, luminal non specified (LumNS), basal/squamous (Ba/Sq), and neuroendocrine-like (NE-like) (Kamoun et al., 2020). We investigated the similarity and difference between our UTUC expression classification with the one reported for muscle invasive bladder cancer in UTUC cases available to us. The majority (71.5%) of the UTUC samples were classified into the LumP subtype, which included most of the C1, C2 and C4 cases and to a lesser extent, C5 cases, whereas C3 cases were classified into the Ba/Sq subtype (Figure 5D). While overall the LumP subtype showed a superior prognosis compared to other consensus subtypes (Figure S7E), patients in LumP subtype had substantially variant prognosis depending on the C1-C5 classification subtype (Figure 5E). By contrast, other consensus subtypes showed a uniformly poor prognosis (Figure S7E). It is mostly due to C3 and C5 composition of the rest of the cases, many of which belong to the TP53/MDM2-mutated subtype (Figure S7F).
We also performed unsupervised clustering on the basis of the DNA methylation status of tumor-specific CpG islands and identified three clusters (Figures 5F, S7G, and S7H). Among these, two clusters showed extensive hypermethylation, which is a sign of CpG island methylator phenotypes (CIMPs) (67% of the total cases). Methylation patterns are associated with mutations in RAS genes and several epigenetic regulator genes (Figure S7G). Mutations in KMT2D, KMT2C, and EP300 were enriched in non-CIMP cases, while RAS mutations were associated with CIMP (Figure S7G). Using the Cluster of Cluster Assignments method (COCA) (Hoadley et al., 2014), we investigated the correlations between subtypes based on matched genetic lesions, gene expression, and methylation information which are available for total 82 samples (Figures S7I and S7J). The samples were first clustered into two major clusters belonging to the FGFR3-mutated and C1 subtypes (COCA1 and 2) and the TP53/MDM2-mutated categories (COCA3 and 4), each of which was further clustered into two, depending on the level of methylation. There was a large difference in prognosis between COCA1/2 and COCA3/4, while no difference of survival was observed between COCA1 vs. COCA2 or COCA3 vs. COCA4, which indicates there is no significant impact of methylation profile on survival (Figure S7H).
Multivariate analysis for survival
To evaluate the relative size of the impact of gene mutations on prognosis, we performed multivariate analysis of survival, using Cox proportional hazard modeling for the Japanese dataset (n = 197) and the model was validated in an independent dataset from MSKCC (n = 75). Mutational subtypes (the TP53/MDM2-mutated and RAS-mutated subtypes), age, and T-category (pT3 and pT4) were significantly associated with survival (Figure 5G). While the largest hazard was explained by clinical variables (~60% of the total hazard), the remaining hazard (~40%) was explained by mutational subtypes (Figure 5H). In fact, the size of impact is significantly increased if combining three unfavorable mutational subgroups as a single unfavorable factor (P = 0.004, the likelihood ratio test) (Figure S7K). The model was highly reproducible with a very high c-statistic value (0.85), when it was validated in the MSKCC cohort (Figure S7K). We also performed the same analysis on 157 Japanese cases where transcriptome data were available (Figure S7K). The finial model included T-category, tumor morphology, and two expression subtypes (C4 and C5) as significant prognostic factors in our cohort (Figure S7L). This model showed significant improvement compared to the clinical factor-alone model, and to the model including mutational subtypes (P = 0.01 and 0.02, the likelihood ratio test, respectively). However, the number of patients used in the modeling is relatively small compared to the number of variables and no validation cohort was available, thus the impact of the expression subtype needs further evaluation using a larger cohort.
Urinary sediment-derived DNA sequencing
Finally, we investigated the potential of targeted-capture sequencing of 30 genes commonly mutated in UTUC using urinary sediment-derived DNA for the diagnosis of UTUC with molecular classification (Figure S8A). We collected 41 preoperative (1–2 months before definitive surgery) and 25 postoperative (7 days after the definitive surgery) voided urine samples from 43 UTUC patients, together with 18 from those with non-urothelial cancer patients. Clinical and genetic backgrounds of these cohorts were comparable to the entire cohort (Table S6). In total, 67.0% (136/203) of mutations and 96.3% (26/27) of focal CNAs detected in primary tumors were also detected in preoperative samples, whereas these mutations/CNAs were detected in none of the postoperative or non-urothelial cancer urine samples (Tables S7 and S8). The sensitivity of cancer detection in sequencing of urinary sediment-derived DNA was significantly higher than that of urinary cytology (78.0% vs. 29.3%; P < 0.001) (Figure S8B). The results of these 43 cases were reproduced in newly recruited 35 surgically treated UTUC cases, where 83.1% (147/177) of mutations and 74.2% (23/31) of focal CNAs detected in primary tumors were confirmed in 87.5% (28/32) of preoperative urine samples. In general, mutations that were detectable in urine could all be confirmed in the primary tumors. However, in some cases, a subset of mutations were only detectable in urinary samples, and those mutations are the ones with low variant allele frequencies (< 0.10) (Figure S8C), suggesting the presence of minor clones. Overall, with 74.5% of mutation detection rate, sequencing of urinary sediment-derived DNA showed 82.2% sensitivity and 100% specificity (95% confidence interval [CI], 71.5–90.2% and 81.5%-100%, respectively) of cancer detection (Figure 6A; Table S8), which outperformed those of urinary cytology (32.9% [95% CI, 22.3–44.9%] and 88.9% [95% CI, 65.3–98.6%], respectively). The sensitivity was not affected by the genetic or histological background of the tumors or severity of pyuria and hematuria, this is a strength over urinary cytology, where a reduced sensitivity was observed for the diagnosis of low-grade tumors and tumors in the FGFR3-mutated and triple-negative subtypes (Figures 6B and S8D). Of interest, 8 out of the 13 samples showing a false-negative result were derived from patients with severe hydronephrosis, a condition that is considered to prevent a urinary flow and compromise mutation detection. When excluding these 8 samples, the sensitivity of detecting UTUC using urine sample was 92.3% (95% CI, 82.9–97.5%). In a case with severe hydronephrosis, when we analyzed the urine sample collected upstream from the obstruction using PCR-based amplicon sequencing, we successfully detected all mutations found in the primary tumors (n = 34), while only one mutation was detected in voided urine (Figure S8E). In line with this, the severity of hydronephrosis was significantly correlated with a reduced sensitivity and a decreased MCFs in urinary sediment samples (Figure 6C). In a case carrying bilateral tumors each of which induced severe and mild hydronephrosis, all mutations in the voided urine were originated from the tumor associated with mild hydronephrosis (Figure S8F).
Figure 6. Urinary sediment-derived DNA sequencing.

(A) Mutation profiles of paired samples of primary tumors and corresponding preoperative voided urinary sediments (n=73). The columns represent samples, and the rows represent genomic lesions in primary tumors (indicated by “T”) and preoperative urinary sediment (indicated by “U”). (B) Sensitivity of urinary sediment-derived DNA sequencing (yellow) and urinary cytology (brown) according to mutational subtypes and clinical/pathological features with 95% CI error bars. P values calculated by two-sided Fisher’s exact test. *P < 0.05, **P < 0.01, ***P < 0.001. (C) Bar plots showing the sensitivity of urinary sediment-derived DNA sequencing with 95% CI error bars (left) and box plots showing MCF ratios of preoperative urinary sediment samples and corresponding primary tumors (right) according to the severity of hydronephrosis. P values calculated by two-sided Fisher’s exact test and two-sided Mann-Whitney U test with Bonferroni correction, respectively. The median, first and third quartiles, as well as outliers, are indicated with whiskers extending to the furthest value within 1.5 of the interquartile range in box plots. *P < 0.05, **P < 0.01. (D) Summary of mutational subtypes prediction based on the results in urinary sediment-derived DNA sequencing. (E) Bar plots showing the accuracy (left) and positive prediction rate (right) in each subtype.
We also evaluated to what extent we could correctly predict the genetic subtype of tumors using urinary sediment-derived DNA, in which we applied our criteria for genetic classification of UTUC (See above section and Star Methods; Figures S8G and S8H). The subtype prediction using urinary sediment sequencing showed high accuracy (87.7–98.6%) for non-hypermutated subtypes, suggesting that urinary sediment sequencing has a potential value as a prognostic biomarker as well as a diagnostic tool. By contrast, we were not able to find a threshold for the number of mutations that efficiently discriminate hypermutated subtypes from non-hypermutated ones (Figures 6D and 6E).
Discussion
With a large number of samples analyzed by integrated multi-omics analysis, our study represents a comprehensive profiling of UTUC molecular alterations. Overall, UTUC and UBC show a similar set of affected driver genes. However, mutation frequency of several genes varies from UTUC to UBC, in addition, the mutation profiles of UTUC differ between tumors in ureter and renal pelvis. Thus, we suggest that while there seems to exist a common mechanism of positive selection in urothelial carcinogenesis, distinct mechanisms might play a role depending on tumor location in the urothelium. Among these differentially mutated genes, of particular interest is mutations in KMT2D. Two recent studies reported that this gene is the most frequent mutations found in normal urothelium and show different mutation frequencies between UTUC and UBC (Lawson et al., 2020; Li et al., 2020). KMT2D was more frequently mutated in normal ureter (33%) than bladder (9%) epithelium, which coincide with a difference of KMT2D mutation frequency variance between ureter and bladder cancers (85% vs. 25%). The frequency of KMT2D mutations in pelvic urothelium is unknown. These observations suggest that urothelial cancers are likely derived from positively selected clones in normal urothelium, in which distinct driver mutations play a role in the selection of clones in normal and cancer tissues, depending on the anatomical site.
We found four SBS signatures in UTUC, which are consistently found in UBC. In contrast to a relatively uniform contribution of age-related SNVs across samples, APOBEC- and TCR-related SNVs exhibited substantial interindividual variations. Interestingly, APOBEC-related SNVs were also reported in normal bladder urothelium and showed a distinct spatial variability (Lawson et al., 2020), which may explain the heterogeneous contribution of this signature across patients, although the underlying mechanism of this variability is unclear. Spatial and inter-individual difference in APOBEC mutagenesis in normal and cancer tissues were also seen in esophagus (Yokoyama et al., 2019). Heterogeneity in TCR-related mutagenesis could be explained by different habits of alcohol consumption and the state of the hypomorphic ALDH2 allele, which have been already demonstrated in other organs (Suzuki et al., 2020; Yokoyama et al., 2019). A higher prevalence of the hypomorphic ALDH2 allele, together with AA intake from herbal medicine, might contribute to the pathogenesis of UTUC in Asian patients.
Based on extensive co-alteration patterns across mutations and CNAs, UTUCs are classified into five distinctive subtypes. This molecular classification has a high clinical value, for its tight correlation with co-mutation pattern, gene expression profile, as well as clinicopathological feature of tumors, including histology and prognosis (Figure 7). As shown in multivariate analysis, the prediction power of UTUC survival is improved. Combined, genetic factors explained ~40% of total hazard, which was validated in an independent UTUC cohort, supporting the validity and relevance of our molecular classification. Recently, novel agents have been introduced to the metastatic urothelial carcinoma treatment and in this regard, the mutational subtypes we proposed might provide a potentially useful guidance for the choice of these therapeutic agents. For example, pan-FGFR inhibitors are promising therapeutic choices for patients in the hypermutated and the FGFR3-mutated subtypes. Hypermutated and a subset of TP53/MDM2-mutated patients might benefit from ICBs because of their high tumor mutational burden or high expression of immune checkpoint molecules, respectively. Recent report of a KRAS G12C inhibitor, AMG510, is also encouraging (Canon et al., 2019), given that KRAS-mutated patients in our cohort showed a significantly poor prognosis (Figure S3C).
Figure 7. Summary of UTUC genetic classification.

A flow chart showing classification of UTUC into five subgroups according to alteration status of TP53, MDM2, RAS, and FGFR3 and also to the presence/absence of hypermutation. Breakdown into gene expression subtypes C1-C5 is shown on the bottom.
Although diagnosis of UTUC has been substantially improved with minimally invasive procedures using advanced endoscopic devices, there still remain risks for adverse events such as ureter injury, infection, and intravesical tumor recurrences (De Coninck et al., 2020). In this regard, urinary sediment-derived DNA would be a plausible target of diagnostics, and only a few reports have investigated its potential for the diagnosis of UTUC (Hayashi et al., 2019; Springer et al., 2018). In this study, through the analysis of urine samples from 78 UTUC patients, we demonstrated that sequencing of urinary sediment-derived DNA had a high sensitivity and specificity for UTUC diagnosis, particularly in cases without hydronephrosis. Further studies involving a larger number of patients are warranted to evaluate the clinical utility of this non-invasive diagnostic approach. The use of urinary sediment-derived DNA for unbiased sequencing also enables the analysis of advanced, inoperable cases, which were not included in the current study due to the difficulty in obtaining tumor materials. A potential caveat is a possibility of false negative results in the case with hydronephrosis, in which a reduced urinary flow might prevent alterations from being detected. Even in such cases, an analysis of the catheterized urine upstream from the obstruction makes a molecular diagnosis possible and correct. Another problem is difficulty in diagnosing hypermutated tumors relying on sequencing of a small number of genes. However, given substantially reduced sequencing costs, whole exome/genome sequencing could be incorporated in this urinary sediment-derived DNA diagnostic approach and improve the sensitivity of detecting hypermutated tumors in the future.
In summary, we profiled molecular landscape of UTUC and identified five unique mutational subtypes of UTUC with distinct profiles of gene expression, and clinicopathological features. Sequencing of urinary sediment samples provides an alternative to conventional invasive diagnostic procedures for UTUC diagnosis. Our results contribute to the understanding of the pathophysiology of UTUC and improve its diagnostics for better stratification of UTUC patients.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Seishi Ogawa (sogawa-tky@umin.ac.jp).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All the whole exome sequencing (WES), RNA sequencing, single-nucleotide polymorphism (SNP) array, and methylation array data have been deposited in the European Genome Phenome Archive (http://www.ebi.ac.uk/ega/) under accession numbers EGAD0000100766, EGAD00001007667, EGAD00010002096, and EGAD00010002098.
We obtained public sequencing data of UBC and UTUC from previous genetic studies (Audenet et al., 2019; Cancer Genome Atlas Research, 2014; Pietzak et al., 2017; Robertson et al., 2017; Robinson et al., 2019; Sfakianos et al., 2015). Fastq files of whole exome and RNA sequencing data and CEL files of SNP array data were downloaded from the TCGA Data Portal (https://portal.gdc.cancer.gov/), all of which were analyzed with the same pipeline and methods as used in our UTUC data, as described in the following section. Results of targeted-capture sequencing of UBC and UTUC published from MSKCC were downloaded from cBioportal (http://www.cbioportal.org/) (Cerami et al., 2012; Gao et al., 2013). Samples with pTis and/or those which underwent presurgical treatment (NAC and BCG perfusion) were excluded from the following analysis. UTUC cases with preceding or simultaneous UBC occurrences were also excluded from the comparison analysis of alterations in driver genes.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects and materials
We included a total of 234 patients diagnosed with upper urinary tract urothelial carcinoma (UTUC) undergoing surgical resection at the University of Tokyo Hospital (n=225), the Fraternity Memorial Hospital (n=5), and Toranomon Hospital (n=4). We also included 26 non-urothelial cancer (UC) patients to obtain negative control samples. Out of 234 UTUC patients, 19 and 18 patients experienced preceding or simultaneous urothelial bladder cancer (UBC), respectively. Simultaneous bilateral occurrence of UTUCs was observed in two cases (UTUC229 and 260). There were eight and six patients who received platinum-based neoadjuvant chemotherapy (NAC) or Bacille Calmette-Guerin (BCG) perfusion treatment before surgery, respectively. Platinum-based adjuvant chemotherapy (AC) was recommended to all the patients fulfilling any of the following pathological features: ≥pT3, ≥pN1, ly+, and v+. AC was performed in 69 patients after obtaining agreement. Detailed information of therapy in each patient is summarized in Table S1. Patients with pTis were excluded from analysis due to the difficulty in proper sampling and the expected low tumor purity.
A total of 236 primary tumor samples (232 unilateral and 2 bilateral) and 234 matched germline samples were obtained from surgical resection biospecimens and the renal cortex and/or peripheral blood, respectively. As negative control samples for RNA sequencing and the DNA methylation array, normal urothelial epithelia samples were obtained from eight patients with kidney cancer undergoing nephrectomy at the University of Tokyo Hospital. In urinary sediment analysis, 20 ml of preoperative (1–2 months before definitive surgery) and/or postoperative (7 days after definitive surgery) voided urine samples were collected from 84 UTUC patients and 18 non-UC patients, of which half of the volume was subjected to cytology tests and the rest was immediately centrifuged for further urinary sediment analysis. In addition, a urine sample was also collected upstream from the tumor in one case (UTUC203). Cytology was based on the Papanicolaou’s classification (Papanicolaou, 1947), in which Class 1–5 indicate the absence of atypical cells (Class 1), reactive atypia (Class 2), atypical cells (Class 3), suspicious for malignancy (Class 4), and malignancy (Class 5) (Figure S8B). All the tumor/germline and urine samples were stored at −80 °C until sample processing. All the samples and clinical information were collected according to protocols approved by the ethics committee of the Graduate School of Medicine, the University of Tokyo, and other participating institutes. Written informed consents for this research was obtained from all patients. A full description of the samples including age at diagnosis and sex are provided in Table S1. No significant influence of sex was observed on the mutational classification result.
METHOD DETAILS
Sample selection
Of the 236 primary tumor samples, 199 were subjected to the integrated analysis, including WES (n=199), SNP array karyotyping (n=199), TERT promoter sequencing (n=199), messenger RNA sequencing (n=158), and array-based DNA methylation analysis (n=86). These 199 samples were obtained from 198 patients, including one having bilateral tumors, with no history of presurgical treatment (NAC and BCG perfusion) from 2000 to 2018. The remaining samples were subjected to WES and used for urinary sediment analysis regardless of the history of presurgical treatment.
Sample processing
Genomic DNA was extracted using the Gentra Puregene kit (QIAGEN) or QIAamp DNA Micro Kit (QIAGEN). RNA was extracted using the RNeasy Mini Kit (QIAGEN).
Whole exome sequencing
All the primary tumors and matched germline samples were subjected to WES. SureSelect Human All Exon v4/v5 kits (Agilent Technologies) and xGen Exome Research Panel v2 (IDT) were used for exome capture according to manufacturer’s instructions, followed by massively parallel sequencing by Hiseq 2500/NovaSeq 6000 (Illumina) or DNBSEQ-G400 (MGI) with a standard 100–150 bp pairedend mode, as previously described (Yoshida et al., 2011). The mean coverages were 162x (101x-244x), 177x (115x-345x), and 180x (92x-276x) in Hiseq 2500, NovaSeq 6000, and DNBSEQ-G400, respectively. Variant calling was performed using Genomon2 pipelines (https://genomon.readthedocs.io/ja/latest/), as previously reported (Kakiuchi et al., 2020; Yokoyama et al., 2019). Briefly, sequencing reads were aligned to the human genome reference (GRCh37) using Burrows-Wheeler Aligner (Li and Durbin, 2009) with default parameter settings. PCR duplicates were eliminated using biobambam2 (Tischler and Leonard, 2014). Somatic mutations were detected by eliminating polymorphisms and sequencing errors. To achieve this, Genomon2 first discards any of low-quality, unreliable reads and variants according to the following criteria: (i) mapping quality < 20, (ii) base call quality < 15. Variants were further filtered by the following criteria: (iii) both of tumor and normal depths ≥8, (iv) number of variant reads in tumor ≥ 4, (v) number of variant reads in normal ≤ 1 (Hiseq 2500, DNBSEQ-G400) / ≤ 2 (NovaSeq 6000, considering index hopping), (vi) variant allele frequencies (VAFs) in tumor ≥ 0.05, (vii) VAFs in germline control ≤ 0.02, (viii) Fisher’s exact test P < 10−1, and (ix) presenting in bidirectional reads. To select variants that were observed at significantly higher VAFs than expected for errors, we used the following criteria: (x) P ≤ 10−4, for which significance is evaluated by EBcall algorithm (Shiraishi et al., 2013) on the basis of an empirical distribution of VAFs as determined using WES data of non-paired germline samples (n=20). Candidate mutations were further filtered by removing mapping errors through visual inspection with the Integrative Genomics Viewer (IGV) (Thorvaldsdottir et al., 2013). Since slight tumor cell contamination was identified in germline samples from three patients (UTUC2T, 5T, and 7T), the following criteria was adopted in place of the criteria (v) and (vii): (xi) VAFs in germline control ≤ 0.05. Furthermore, in eight patients with moderate tumor cell contamination in their normal samples (UTUC4T, 37T, 58T, 77T, 127T, 187T, 447T, and 450T), non-paired filtering was performed with criteria (i) to (iv), (vi), (viii), (ix), and (x), as mentioned above. The candidate mutations were further filtered by the following criteria: (xii) not registered in the 1000 Genomes Project (May 2011 release), Exome Sequencing Project (ESP) 6500, or the Human Genome Variation Database (HGVD; October 2013 release) with frequencies > 0.001, (xiii) genes significantly mutated in UTUC as described in Significantly mutated genes in UTUC section, and (xiv) Estimated to be a pathogenic mutation as described in the Curation of oncogenic variants section. The results of these eight samples were only used for mutation landscape/mutation clustering analysis and excluded from mutational burden, significantly-mutated-gene identification, and single base substitution (SBS) signature analysis.
Detection of mismatch repair gene alterations
In the detection of alterations in mismatch repair (MMR) genes, in addition to the paired filtering of primary tumor samples, we also performed non-paired filtering of germline samples using the criteria (i)-(iv), (viii), (ix), (x), (xii), and (xiii) mentioned above. In two samples with MSH6 germline mutations but no somatic ones, we confirmed MSH6 inactivation by immunohistochemistry (UTUC260rt and UTUC260lt in a bilateral UTUC case). Results of MMR gene alterations in hypermutated samples are summarized in Table S2.
Validation of detected mutations
PCR-based deep sequencing was performed for validation of mutations detected in WES (Suzuki et al., 2015). In addition, one splice site mutation in TP53 in UTUC7T was also validated as the depth on this position in WES data did not fulfil criteria (iii) in the Whole exome sequencing section. In total, 207 single nucleotide variants (SNVs) and 9 insertion and deletion lesions (indels) were randomly selected and evaluated. Mutations were considered to be validated when (i) sequencing depths were ≥ 500 in both tumor and paired germline samples; (ii) VAFs in the tumor samples were 5 times higher than those in the corresponding germline samples; and (iii) VAFs in the tumor samples were ≥ 0.01 (Kakiuchi et al., 2020; Yokoyama et al., 2019). This allowed us to validate 98.6% (204/207) and 100% (9/9) of SNVs and indels, respectively.
Detection of TERT promoter sequencing
As the SureSelect v4/v5 baits do not cover the TERT promoter region, we performed Sanger sequencing and/or PCR-based deep sequencing. PCR-based deep sequencing was performed as previously reported (Suzuki et al., 2015). We adopted the following criteria: (i) sequencing depths ≥ 500 in both tumor and paired germline samples, (ii) VAFs in the tumor sample ≥ 0.05, (iii) VAFs in the germline sample ≤ 0.02, and (iv) mutations that have been previously reported to be pathogenic.
In six cases with contamination in their germline samples, we applied only criteria (i), (ii), and (iv). All the detected TERT promoter mutations by Sanger sequencing and/or PCR-based deep sequencing are summarized in Table S4.
Definition of hypermutation
We first ranked UTUC samples according to their mutational burden in descending order, and calculated the degree of variability, as previously reported (Nakamura et al., 2015). A huge change of mutational burden was observed at the dashed line in Figure 1A, which we chose as a cutoff to discriminate hypermutated and non-hypermutated samples. Using the same method for the discrimination of mutation status applied to TCGA WES data, two samples were regarded to be hypermutated. Although MSKCC datasets were analyzed by targeted-capture sequencing, which is difficult for estimating genome wide mutation burden, four samples in the UTUC dataset showed extremely large numbers of mutations, including MMR genes, thus we defined these samples as hypermutated.
Significantly mutated genes in UTUC
Genes in which mutations were significantly enriched or positively selected (q < 0.1) were confirmed by MutSigCV (Lawrence et al., 2013) and dNdScv (Martincorena et al., 2017) which identified 18 and 24 genes, respectively (Table S3). In this context, hypermutated cases were excluded from the analysis as the background mutation rate would be biased by their extremely large number of mutations.
Curation of oncogenic variants
Mutations in significantly mutated genes were further evaluated to determine if they were oncogenic or not. Basically, mutations were considered to be oncogenic if they were recurrently reported (≥10) in the Catalogue of Somatic Mutations in Cancer (COSMIC) databases v90. Mutations fulfilling the following criteria were also considered as oncogenic: (i) truncating variants (nonsense mutations, essential splicing mutations, or frameshift insertion/deletion) in genes that are oncogenic through loss of function, (ii) mutations within three amino acids from functional hotspots, and (iii) missense mutations that are computationally predicted as pathogenic by at least two of three algorithms such as FATHMM-MKL (score ≥ 0.7) (Shihab et al., 2015), GAVIN (Pathogenic) (van der Velde et al., 2017), and PredictSNP2 (deleterious) (Bendl et al., 2016).
SBS signature analysis
SBS signatures were extracted from all of 193 (11 hypermutated and 182 non-hypermutated) samples to assess overall signature profiles using the R package ‘pmsignature’ (Shiraishi et al., 2015). To avoid possible bias associated with hypermutations, 182 non-hypermutated samples were independently analyzed. The same analysis was also performed in TCGA UBC data (data not shown). Extracted signatures were compared with the COSMIC signatures using the R package ‘deconstructSigs’ (Rosenthal et al., 2016). We also used the same package to extract signatures from a case with frequent T>A/A>T transitions (UTUC98T). To evaluate the accuracy of extracted signature activities, per-sample signature contributions of Sig. A-D estimated by ‘pmsignature’ were compared with those of SBS1, 2, 13, and 16 estimated by the R packages ‘deconstructSigs’ and ‘MutationalPatterns’ (Blokzijl et al., 2018; Rosenthal et al., 2016). Cosine similarity was used to compare the contribution of signatures calculated by each program. In the analysis of association between Sig. D and alcohol metabolism/consumption, genotyping of SNPs in ALDH2 (rs671) was determined following the criteria previously reported (Yokoyama et al., 2019), and heavy alcohol consumption was defined as ≥20g alcohol/day on the basis of a recommendation from Ministry of Health, Labor and Welfare, Japan. Samples with < 40 SNVs were excluded from the analysis in Figures 1C, S1D and S1E.
Analysis of copy number alterations
Genome-wide copy number analysis was performed in primary tumor lesions of UTUC using GeneChip Human Mapping 250K Nspl (Affymetrix): SNP array karyotyping. To analyze and visualize the total and allele-specific copy number alterations (CNAs), we used the CNAG platform (Nannya et al., 2005; Yamamoto et al., 2007) for both the UTUC data in this study and the dataset available from TCGA. Ploidy estimation and chromothripsis calling were conducted on CEL files using ASCAT (Van Loo et al., 2010) and CTLPScanner (Yang et al., 2018) with default settings. Significant chromosomal and focal CNAs (q value < 0.1, brlen > 0.5) were detected using GISTIC2.0 (Mermel et al., 2011). Based on the GISTIC results, high level amplifications/deletions were further curated with visual inspection on each focal CNA. Previously reported putative responsible genes for each focal amplification and deletion are shown in Figure S2B. Hierarchical clustering of copy number data was performed by R function ‘hclust’ with Manhattan distance and Ward’s linkage algorithm. CNAs of urinary sediment and a subset of primary tumor samples (UTUC401T-450T) were evaluated on the basis of sequencing data using our in-house pipeline CNACs (https://github.com/papaemmelab/toil_cnacs) (Yoshizato et al., 2017).
Dirichlet process-model based clustering
To define robust groups of samples based on mutations and CNAs, we applied a Bayesian approach as previously reported (Papaemmanuil et al., 2016). The optimal number of clusters was learned by Markov Chain Monte Carlo (MCMC) methods using the Dirichlet process mixture model (https://github.com/nicolaroberts/hdp). We first created a binary matrix of genetic events in 188 non-hypermutated cases, which was comprised of 33 frequently observed (≥ 10 samples) and significantly mutated genes as well as significant focal CNAs. Cases with HRAS/KRAS/NRAS alterations were assembled into a single category as RAS. A case with no alterations in any of these factors was excluded from analysis (UTUC202). After 500 burn-in iterations and a subsequent 1,000 sample collection at intervals of 20 iterations, three subgroups with distinct genetic features were identified. Based on this result, we set class defining lesions as TP53, RAS, and FGFR3, which defined four categories; TP53-mutated, RAS-mutated, FGFR3-mutated, and triple-negative. Although cases with MDM2 amplification should be involved in the TP53-mutated subgroup considering its ability to inactivate TP53, there were also several cases of co-occurring FGFR3/RAS mutations. Interestingly, cases with MDM2 amplification showed totally different phenotypes and prognosis depending on the presence/absence of these co-occurring mutations, in which co-occurring cases showed quite favorable phenotypes and prognosis while cases with MDM2 amplification alone showed aggressive phenotype and poor prognosis similar to TP53-mutated cases (Figure S4E). Thus, we set additional criteria as follows: (i) samples with MDM2 amplification should be classified into TP53 subgroup unless they have mutations in FGFR3 or RAS.
Several cases still fulfilled more than two of the class criteria. Considering the clinical significance of each gene, we set several post processing criteria as follows: (ii) sample with FGFR3(+)/TP53(+) and RAS(+)/TP53(+) showed prognosis similar to FGFR3(−)/TP53(+) and RAS(−)/TP53(+)(Figures S4B and S4C), based on which FGFR3(+)/TP53(+) and RAS(+)/TP53(+) overlapping cases should be classified as the TP53 subgroup, (iii) although there were no significant differences in prognosis between RAS(+) and FGFR3(+) cases (Figure S4D), RAS(+) samples showed a more aggressive phenotype, based on which FGFR3 and RAS overlapping cases should be classified as the RAS subgroup. The results of the clustering analysis are summarized in Table S1. We performed a similar analysis of the combined data of non-hypermutated samples in our cohort and the MSKCC cohort. We found four categories which were defined by the presence/absence of alterations in TP53, RAS, and FGFR3. Criteria for overlapping cases were defined in the same manner as described above.
RNA sequencing
RNA was extracted from 199 tumor and eight normal urothelial epithelia samples, of which 158 and eight with enough RNA quality (RNA integrity number: RIN ≥ 7), respectively, were subjected to RNA sequencing following the recommendation of the RNA preparation kit (NEBNext Ultra RNA Library Prep kit for Illumina [New England BioLabs]). RIN was calculated by the Agilent 2200 TapeStation system. Libraries of messenger RNA were prepared and sequenced using the Hiseq 2500 platform with 125 bp paired-end reads. Sequenced reads were aligned to the human genome reference (GRCh37) using Bowtie (Langmead et al., 2009) and Blat (Kent, 2002). Following the method previously reported (Kataoka et al., 2015), fusion transcripts were detected using Genomon fusion and filtered by (i) supported reads ≥ 4; (ii) mapped to known exon/intron boundaries; (iii) mapped to non-repetitive regions. In addition, only in-frame transcripts were included.
Expression clustering
Raw read counts for each gene were counted with featureCounts from the Subread package (Liao et al., 2014), and were normalized to transcripts per million (TPM) and subjected to expression analysis. The TPM matrix was log2 transformed after adding 1 to each value, then further filtered to include genes with expression values > 2 in at least 10% of samples. 2,000 of the most variably expressed genes were selected based on the median absolute deviation (MAD) score to identify robust expression clusters. Consensus clustering was performed based on Ward and Euclidean algorithms with 1,000 iterations using R-package ‘ConsensusClusterPlus’ (Wilkerson and Hayes, 2010), in which the delta area plot suggested K = 3 as an optimal number of clusters. However, we found that with an increasing K value, one of the three clusters thus far obtained was further divided into three showing distinct biological features with K = 5 in terms of luminal, basal, stromal, p53 pathway, and immune signatures, suggesting that K = 5 provides a better clustering. In the detailed analysis of tumor microenvironment, previously reported genes that were upregulated in stroma and immune cells were adapted (Yoshihara et al., 2013). Hierarchical clustering was performed within each cluster by R function ‘hclust’ with Euclidean distance and Ward’s linkage algorithm. The results of clustering analysis are summarized in Table S1.
Expression signature analysis
To characterize each UTUC expression subgroup, we used expression signatures analyzed in the previous UBC studies (Robertson et al., 2017; Sjodahl et al., 2012). Several additional immune and stromal signatures were also adopted from other previous studies (Mariathasan et al., 2018; Yoshihara et al., 2013). To evaluate the expression levels of these signatures in UTUC, signature scores were calculated and compared with those of UBC (TCGA) and normal urothelial epithelia samples. For this purpose, we applied ComBat-Seq (Zhang et al., 2020) to raw read count data for removing batch effect, then processed data were converted to TPM. A mean of log2 (TPM+1) for marker genes in each signature was calculated for each sample, then the score was rank-normalized. Genes involved in each signature are listed in Table S5. Cytotoxic score was calculated as the geometric mean of TPM for GZMA and PRF1 as previously reported (Rooney et al., 2015). The R package ‘BLCAsubtyping’ was used to assign our UTUC samples to six consensus subtypes (Kamoun et al., 2020). The results of the classification are summarized in Table S1.
Estimation of mutated cell fraction
Mutated cell fraction (MCF) was calculated from copy number and observed VAF following the method previously reported (Yoshizato et al., 2017). Briefly, MCFs of mutations were calculated using VAFs, total copy number (TCN), and B allele frequency (BAF) of the region as follows:
MCF = 2 VAF for mutations with no copy number events.
MCF = TCN × VAF for mutations with deletions.
For mutations with uniparental disomy (UPD) regions, we applied the formula below depending on the order of mutations and UPD events;
MCF = 2 VAF + 2 BAF − 1 when mutations occurred earlier
MCF = 2VAF when mutations occurred later
For mutations with gains, we did not calculate MCFs as we cannot estimate the number of mutated alleles. Tumor content was estimated based on the maximum value of MCFs in each sample in Figure 5A.
Methylation array analysis
DNA methylation profiles of 86 tumors and eight normal urothelial epithelia samples were analyzed using the Infinium MethylationEPIC BeadChip Kit. After performing beta-mixture quantile normalization, we first eliminated probes with any missing values. Next, we selected probes that meet the following criteria: (i) annotated with “Promoter_Associated_Cell_type_specific” or “Promoter_Associated”, (ii) designed in “Island”, “N_Shore”, or “S_Shore”, (iii) not on the X or Y chromosomes. To capture tumor-specific hypermethylated events, we further applied exclusion criteria as follows: (iv) highly methylated in normal samples (mean β value > 0.2) and (v) methylated (β value > 0.3) in < 10% of tumor samples. Using top 4,000 in MAD-ranked probes, consensus clustering was performed based on Ward and Euclidean algorithms with 1,000 iterations using the R package ‘ConsensusClusterPlus’ (Wilkerson and Hayes, 2010). The result of clustering analysis is summarized in Table S1.
Urinary sediment-derived DNA sequencing
Since urinary sediment might contain many non-tumor cells derived from normal epithelia and blood, we performed PCR based deep sequencing (described above) and/or targeted capture sequencing for enough target depth. In PCR-based deep sequencing, all driver mutations and randomly selected passenger mutations detected in the corresponding primary tumors were examined. In addition to criteria (i) and (ii) in the validation analysis, the following VAF threshold criteria was adopted instead of (iii): (iv) VAFs in sample ≥ 0.05. In targeted deep sequencing, 1–100ng of genomic DNA from urinary sediments was enriched using custom bait libraries (xGen Predesigned Gene Capture Pools, xGen Custom Target Capture Probes, xGen CNV Backbone Panel; IDT). Predesigned and custom probes were designed to capture coding or promotor regions of 30 driver genes which are frequently mutated in UTUC and UBC (TP53, FGFR3, HRAS, KMT2D, KDM6A, STAG2, CDKN1A, ARID1A, ELF3, PIK3CA, CREBBP, KMT2C, RHOB, EP300, TSC1, KMT2A, ERCC2, RHOA, FBXW7, KRAS, NRAS, ATM, RB1, ERBB2, ERBB3, MDM2, CCND1, CDKN2A, TACC3, and TERT promoter) (Cancer Genome Atlas Research, 2014; Gui et al., 2011; Guo et al., 2013; Hurst et al., 2017; Pietzak et al., 2017; Robertson et al., 2017). We also utilized 16 blood samples from UTUC patients to exclude sequencing errors and single nucleotide polymorphisms. Library preparation was performed using the KAPA Hyper Prep Kit (Roche) or xGen Prism DNA Library Prep Kit (IDT), followed by sequencing with an average of 679x depth (103x- 2472x) using NovaSeq 6000 or DNBSEQ-G400. Sequencing reads were aligned to the human genome reference (GRCh37). Mutation calling were performed using Genomon2 pipelines. Mutations fulfilling criteria (i)–(iv), (viii)–(x), and (xii) described in the whole exome sequencing section were further curated by the following criteria: (xv) VAFs ≥ 0.03, and (xvi) exclude all missense SNVs with VAFs of 0.4–0.6 in copy-neutral regions, unless identical mutations were validated as somatic in the WES data of the corresponding primary tumor or recurrently reported (≥10) in COSMIC databases v90. The evaluation of hydronephrosis was based on preoperatively conducted computed tomography images as follows: patients with thin renal parenchyma or dilated calices were defined as “severe” and those with simple dilation of ureter or pelvis were defined as “mild.” To predict the hypermutated tumors on the basis of the total number of mutations in 30 panel genes, we first counted the number of mutations in these genes in primary tumor WES data. While 90.9% (10/11) of hypermutated samples had ≥ 10 mutations, 95.1% (176/185) of non-hypermutated samples had < 10 mutations. Considering the mutation detection rate (74.5%) in urinary samples, we set 8 as a threshold of hypermutated and non-hypermutated samples. All the patients’ urine information and detected mutations are summarized in Tables S1 and S7.
Survival analysis
In survival analysis, events of disease-specific survival were defined as disease-specific mortality. Hazard ratios and 95% confidence intervals were calculated using Cox proportional-hazards model. A patient who had bilateral tumors (UTUC229) was excluded from the analysis. The assumptions of Cox models were tested using cox.zph function in the R package ‘survival’. The proportional hazard assumptions were satisfied in Figures 3B, S3C, S5E, S5F, S6B, and S7H. P values were calculated by the algorithm Exact Log-rank Test (ExaLT) in Figure S3C (Vandin et al., 2015). In multivariate analysis, we first conducted univariate analyses for disease-specific survival. The factors we assessed included 10 clinical features (age, sex, history of alcohol and smoking, T-category, grade, tumor morphology, tumor location, squamous differentiation, and adjuvant chemotherapy) and three molecular subtypes (mutational, expression, and methylation subtypes), of which six (age, T-category, grade, tumor morphology, squamous differentiation, and adjuvant chemotherapy) and two subtypes (mutational and expression subtypes) were significantly associated with disease-specific survival. We used a stepwise backward selection method using Akaike’s Information Criterion (AIC) to control overfitting. We set pTa/pT1, the hypermutated/FGFR3-mutated subtypes, and the C1 subtype as references. C-statistic value was calculated using the R package ‘survcomp’ (Schroder et al., 2011). Relative hazard of each factor was calculated following the method previously reported (Yoshizato et al., 2017).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using R, version 3.5.0. All P values were calculated by two-sided analysis unless otherwise specified. Fisher’s exact test or Mann-Whitney U test was used for group comparisons. Pairwise Fisher’s exact test was used for co-occurring and mutual exclusive patterns among frequently mutated (≥ 5%) oncogenic alterations. In the comparison of driver gene alteration frequencies, frequently mutated (≥ 5%) genes in at least one of the cohorts were included. Multiple-hypothesis testing was corrected using the Bonferroni’s method for inter-group comparisons, with regard to ALDH2 genotypes (Figure 1C), mutational subtypes (the TP53/MDM2-, RAS-, and FGFR3-mutated and triple-negative subtypes (Figure 3C), gene expression subtypes (C1-C5) (Figure 5A–5C), tumor location (Figure S1B), and the type of FGFR3 alterations (Figure 2D). Otherwise, significance of the results in multiple testing was evaluated using the false discovery rate or q value (Figures 1D, 1E, 2A, 2B, S3C–E, S5A, S5B, and S5D). P values of less than 0.05 and q values of less than 0.1 were considered significant. Adjusted rand index was calculated using ARI function in the R package ‘aricode’ (Vinh et al., 2010).
Supplementary Material
Table S1, related to Table 1. Summary of clinical information and type of analysis in UTUC samples.
Table S7, related to Figure 6. Results of urinary sediment-derived DNA sequencing.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal anti-MSH6 | GeneTex | GTX62383; RRID:AB_10626440; clone name: EPR3945 |
| Biological Samples | ||
| Primary tumor and paired germline samples from UTUC patients | the University of Tokyo Hospital (Tokyo; Japan); the Fraternity Memorial Hospital(Tokyo; Japan); Toranomon Hospital (Tokyo; Japan) | See Table S1 |
| Normal urothelial epithelia samples from non-urothelial carcinoma patients | the University of Tokyo Hospital (Tokyo; Japan) | See Table S1 |
| Pre- and postoperatively collected urine sediment samples | the University of Tokyo Hospital (Tokyo; Japan) | See Table S1 |
| Critical Commercial Assays | ||
| Gentra Puregene Tissue Kit | Qiagen | 158667 |
| Gentra Puregene Blood Kit | Qiagen | 158445 |
| QIAamp DNA Micro Kit | Qiagen | 56304 |
| RNeasy Mini Kit | Qiagen | 74104 |
| SureSelectXT Human All Exon 50 Mb v4 Kit | Agilent | 5190-4632 |
| SureSelectXT Human All Exon 50 Mb v5 Kit | Agilent | 5190-6209 |
| GeneChip Human Mapping 250K Nsp | Affymetrix | 900768 |
| NEBNext Ultra RNA Library Prep Kit | Illumina | E7530 |
| MethylationEPIC Kit | Infinium | WG-317-1003 |
| xGen CNV Backbone Panel | IDT | 1080564 |
| xGen Exome Research Panel v2 | IDT | 10005153 |
| xGen Prism DNA Library Kit | IDT | 10006203 |
| KAPA Hyper Prep Kit | Roche | 07962363001 |
| Deposited Data | ||
| Whole exome sequencing data of UTUC | This paper | EGAD0000100766 |
| RNA sequencing data of UTUC | This paper | EGAD00001007667 |
| Methylation array data of UTUC | This paper | EGAD00010002096 |
| SNP array data of UTUC | This paper | EGAD00010002098 |
| Whole exome sequencing, RNA sequencing, and SNP-array data of UBC | Robertson et al., 2017 | https://portal.gdc.cancer.gov/ |
| Targeted–capture sequencing of UBC | Pietzak et al., 2017 | http://www.cbioportal.org/ |
| Targeted–capture sequencing of UTUC | Audenet et al., 2019, Sfakianos et al., 2015 | http://www.cbioportal.org/ |
| Oligonucleotides | ||
| Primer for TERT promoter sequencing | Suzuki et al., 2015 | N/A |
| Software and Algorithms | ||
| Burrows-Wheeler Aligner (v0.7.8) | Li and Durbin, 2009 | https://sourceforge.net/projects/bio-bwa/ |
| Biobambam2 (v2.0.85) | Tischler and Leonard, 2014 | https://github.com/gt1/biobambam |
| picard-tools (v1.39) | Broad Institute | http://picard.sourceforge.net/ |
| Genomon2 (v2) | Yokoyama et al., 2019 | https://genomon.readthedocs.io/ja/latest |
| EBCall (v2) | Shiraishi et al., 2013 | https://github.com/friend1ws/EBCall |
| Integrative Genomics Viewer (v2.3) | Thorvaldsdottir, 2013 | http://software.broadinstitute.org/software/igv/ |
| MutSigCV (v1.4) | Lawrence et al., 2013 | https://software.broadinstitute.org/cancer/cga/mutsig |
| dNdScv (v0.0.1) | Martincorena et al., 2017 | https://github.com/im3sanger/dndscv |
| FATHMM-MKL (v2.3) | Shihab et al., 2015 | http://fathmm.biocompute.org.uk/fathmmMKL.htm |
| GAVIN (r0.3) | van der Velde et al., 2017 | https://molgenis20.gcc.rug.nl/ |
| PredictSNP2 | Bendl et al., 2016 | https://loschmidt.chemi.muni.cz/predictsnp2/ |
| pmsignature (v0.3.0) | Shiraishi et al., 2015 | https://github.com/friend1ws/pmsignature |
| deconstructSigs (v1.8.0) | Rosenthal et al., 2016 | https://github.com/raerose01/deconstructSigs |
| MutationalPatterns (v3.12) | Blokzijl et al., 2018 | https://github.com/UMCUGenetics/MutationalPatterns |
| CNAG (v3.5.1) | Nannya et al., 2005 | http://www.genome.umin.jp/CNAG_DLpage/CNAG_top.html |
| CNACS | Yoshizato et al., 2017 | https://github.com/papaemmelab/toil_cnacs |
| ASCAT | Van Loo et al., 2010 | https://www.crick.ac.uk/research/labs/peter-van-loo/software |
| CTLPScanner | Yang et al., 2018 | http://47.88.3.162/CTLPScanner/about.php |
| GISTIC 2.0 | Mermel et al., 2011 | http://portals.broadinstitute.org/cgi-bin/cancer/publications/pub_paper.cgi?mode=view&paper_id=216&p=t |
| hdp | Papaemmanuil et al., 2016 | https://github.com/nicolaroberts/hdp |
| Bowtie (v1.2.3) | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| Blat (v3.2.1) | Kent, 2002 | http://www.kentinformatics.com/products.html |
| Genomon fusion | Kataoka et al., 2015 | https://github.com/Genomon/RNAseq_for_HGC |
| Subread (v1.5.3) | Liao et al., 2014 | http://subread.sourceforge.net/ |
| ConsensusClusterPlus (v1.53.0) | Wilkerson and Hayes, 2010 | https://bioconductor.org/packages/release/bioc/html/ConsensusClusterPlus.html |
| ComBat-Seq | Zhang et al., 2020 | https://github.com/zhangyuqing/ComBat-seq |
| BLCAsubtyping | Kamoun et al. 2019 | https://rdrr.io/github/cit-bioinfo/BLCAsubtyping/ |
| MASS | CRAN | https://cran.r-project.org/web/packages/MASS/index.html |
| survival | CRAN | https://cran.r-project.org/web/packages/survival/index.html |
| survcomp | Schroder et al., 2011 | https://www.bioconductor.org/packages/release/bioc/html/survcomp.html |
| ExaLT | Vandin et al., 2015 | https://github.com/fvandin/ExaLT |
| aricode | Vinh et al., 2010 | https://github.com/jchiquet/aricode |
Highlights.
A few genes differ on alteration frequencies in UTUC and UBC
UTUC comprises five molecular subtypes: hypermutated, TP53/MDM2, RAS, FGFR3, and TN
UTUC subtypes correlate with gene expression, histology, and clinical outcomes
Sequencing urinary sediment is a non-invasive diagnostic tools for UTUC
Acknowledgement
This work was supported by Grants-in-Aid from the Ministry of Health, Labor and Welfare of Japan (Health and Labor Sciences Research Expenses for Commission and Applied Research for Innovative Treatment of Cancer), the Project for Development of Innovative Research on Cancer Therapeutics from the Japan Agency for Medical Research and Development, AMED (JP15cm0106056h0005 and JP16cm0106501h0001) [Kyoto; S.O.], the Ministry of Education, Culture, Sports, Science and Technology of Japan, the High Performance Computing Infrastructure System Research Project (hp150232, hp160219; This research used computational resources of the K computer provided by the RIKEN Advanced Institute for Computational Science through the HPCI System Research project) [Kyoto; S.O., Tokyo; S. Miyano], Scientific Research on Innovative Areas (15H05909) [Kyoto; S.O., Tokyo; S. Miyano], “Stem Cell Aging and Disease” (14430052) [Nagoya; M.S.], Takeda Science Foundation [Kyoto; S.O., H.M., T.Y., Tokyo; Y. Sato], Uehara Memorial Foundation [Kyoto; H.S.], and The Japanese Urological Association (Young Researcher Promotion Grant) [Tokyo; Y. Sato], SGH Foundation [Tokyo; Y. Sato], The Yasuda Medical Foundation [Tokyo; Y. Sato]. S. O. is a recipient of JSPS Core-to-Core Program, A. Advanced Research Networks. None of other co-authors have any conflict of interests. The schematic illustration in the graphic abstract was created with BioRender.com. We thank Maki Nakamura and Takeshi Shirahari for technical assistance. We also thank Kenichi Hashimoto, Shoichi Nagamoto, Masaomi Ikeda, Toshikazu Okaneya, Yukimasa Matsuzawa, Kanade Hagiwara, Tomoyuki Kaneko, Hiroaki Nishimatsu, Yoshikazu Hirano, and all staffs at Department of Urology, The University of Tokyo Hospital for collecting samples. We thank iLAC, Co., Ltd. for sequencing support.
Footnotes
Conflict of interest
The authors declare no competing financial interests.
References
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. (2020). The repertoire of mutational signatures in human cancer. Nature 578, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audenet F, Isharwal S, Cha EK, Donoghue MTA, Drill EN, Ostrovnaya I, Pietzak EJ, Sfakianos JP, Bagrodia A, Murugan P, et al. (2019). Clonal Relatedness and Mutational Differences between Upper Tract and Bladder Urothelial Carcinoma. Clin Cancer Res 25, 967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendl J, Musil M, Stourac J, Zendulka J, Damborsky J, and Brezovsky J (2016). PredictSNP2: A Unified Platform for Accurately Evaluating SNP Effects by Exploiting the Different Characteristics of Variants in Distinct Genomic Regions. PLoS Comput Biol 12, e1004962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokzijl F, Janssen R, van Boxtel R, and Cuppen E (2018). MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med 10, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2014). Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al. (2019). The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Tan W, Ling Z, Xi R, Shao M, Chen M, Luo Y, Zhao Y, Liu Y, Huang X, et al. (2017). Genomic analysis of oesophageal squamous-cell carcinoma identifies alcohol drinking-related mutation signature and genomic alterations. Nat Commun 8, 15290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Dickman KG, Moriya M, Zavadil J, Sidorenko VS, Edwards KL, Gnatenko DV, Wu L, Turesky RJ, Wu XR, et al. (2012). Aristolochic acid-associated urothelial cancer in Taiwan. Proc Natl Acad Sci U S A 109, 8241–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, Roth B, Cheng T, Tran M, Lee IL, et al. (2014). Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 25, 152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coninck V, Keller EX, Somani B, Giusti G, Proietti S, Rodriguez-Socarras M, Rodriguez-Monsalve M, Doizi S, Ventimiglia E, and Traxer O (2020). Complications of ureteroscopy: a complete overview. World J Urol 38, 2147–2166. [DOI] [PubMed] [Google Scholar]
- Donahue TF, Bagrodia A, Audenet F, Donoghue MTA, Cha EK, Sfakianos JP, Sperling D, Al-Ahmadie H, Clendenning M, Rosty C, et al. (2018). Genomic Characterization of Upper-Tract Urothelial Carcinoma in Patients With Lynch Syndrome. JCO Precision Oncology, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, et al. (2011). Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet 43, 875–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Sun X, Chen C, Wu S, Huang P, Li Z, Dean M, Huang Y, Jia W, Zhou Q, et al. (2013). Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet 45, 1459–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Fujita K, Matsuzaki K, Matsushita M, Kawamura N, Koh Y, Nakano K, Wang C, Ishizuya Y, Yamamoto Y, et al. (2019). Diagnostic potential of TERT promoter and FGFR3 mutations in urinary cell-free DNA in upper tract urothelial carcinoma. Cancer Sci 110, 1771–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedegaard J, Lamy P, Nordentoft I, Algaba F, Hoyer S, Ulhoi BP, Vang S, Reinert T, Hermann GG, Mogensen K, et al. (2016). Comprehensive Transcriptional Analysis of Early-Stage Urothelial Carcinoma. Cancer Cell 30, 27–42. [DOI] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MDM, Niu B, McLellan MD, Uzunangelov V, et al. (2014). Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158, 929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst CD, Alder O, Platt FM, Droop A, Stead LF, Burns JE, Burghel GJ, Jain S, Klimczak LJ, Lindsay H, et al. (2017). Genomic Subtypes of Non-invasive Bladder Cancer with Distinct Metabolic Profile and Female Gender Bias in KDM6A Mutation Frequency. Cancer Cell 32, 701–715 e707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakiuchi N, Yoshida K, Uchino M, Kihara T, Akaki K, Inoue Y, Kawada K, Nagayama S, Yokoyama A, Yamamoto S, et al. (2020). Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature 577, 260–265. [DOI] [PubMed] [Google Scholar]
- Kamoun A, de Reynies A, Allory Y, Sjodahl G, Robertson AG, Seiler R, Hoadley KA, Groeneveld CS, Al-Ahmadie H, Choi W, et al. (2020). A Consensus Molecular Classification of Muscle-invasive Bladder Cancer. Eur Urol 77, 420–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, Totoki Y, Chiba K, Sato-Otsubo A, Nagae G, et al. (2015). Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet 47, 1304–1315. [DOI] [PubMed] [Google Scholar]
- Kent WJ (2002). BLAT--the BLAST-like alignment tool. Genome Res 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson ARJ, Abascal F, Coorens THH, Hooks Y, O’Neill L, Latimer C, Raine K, Sanders MA, Warren AY, Mahbubani KTA, et al. (2020). Extensive heterogeneity in somatic mutation and selection in the human bladder. Science 370, 75–82. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Du Y, Chen Z, Xu D, Lin T, Jin S, Wang G, Liu Z, Lu M, Chen X, et al. (2020). Macroscopic somatic clonal expansion in morphologically normal human urothelium. Science 370, 82–89. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, Davies H, Stratton MR, and Campbell PJ (2017). Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 171, 1029–1041 e1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome biology 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer J, Shariat SF, Brien JC, Herman MP, Ng CK, Scherr DS, Scoll B, Uzzo RG, Wille M, Eggener SE, et al. (2011). Urinary cytology has a poor performance for predicting invasive or high-grade upper-tract urothelial carcinoma. BJU Int 108, 701–705. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Arai Y, Totoki Y, Shirota T, Elzawahry A, Kato M, Hama N, Hosoda F, Urushidate T, Ohashi S, et al. (2015). Genomic spectra of biliary tract cancer. Nat Genet 47, 1003–1010. [DOI] [PubMed] [Google Scholar]
- Nannya Y, Sanada M, Nakazaki K, Hosoya N, Wang L, Hangaishi A, Kurokawa M, Chiba S, Bailey DK, Kennedy GC, and Ogawa S (2005). A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer research 65, 6071–6079. [DOI] [PubMed] [Google Scholar]
- Ou ZY, Li K, Yang T, Dai Y, Chandra M, Ning J, Wang YL, Xu R, Gao TJ, Xie Y, et al. (2020). Detection of bladder cancer using urinary cell-free DNA and cellular DNA. Clin Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al. (2016). Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med 374, 2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanicolaou GN (1947). Cytology of the urine sediment in neoplasms of the urinary tract. J Urol 57, 375–379. [DOI] [PubMed] [Google Scholar]
- Pietzak EJ, Bagrodia A, Cha EK, Drill EN, Iyer G, Isharwal S, Ostrovnaya I, Baez P, Li Q, Berger MF, et al. (2017). Next-generation Sequencing of Nonmuscle Invasive Bladder Cancer Reveals Potential Biomarkers and Rational Therapeutic Targets. Eur Urol 72, 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman JD, Messer J, Sielatycki JA, and Hollenbeak CS (2011). Incidence and survival of patients with carcinoma of the ureter and renal pelvis in the USA, 1973–2005. BJU Int 107, 1059–1064. [DOI] [PubMed] [Google Scholar]
- Raman JD, Warrick JI, Caruso C, Yang Z, Shuman L, Bruggeman RD, Shariat S, Karam JA, Wood C, Weizer AZ, et al. (2016). Altered Expression of the Transcription Factor Forkhead Box A1 (FOXA1) Is Associated With Poor Prognosis in Urothelial Carcinoma of the Upper Urinary Tract. Urology 94, 314 e311–317. [DOI] [PubMed] [Google Scholar]
- Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, Hinoue T, Laird PW, Hoadley KA, Akbani R, et al. (2017). Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 171, 540–556 e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson BD, Vlachostergios PJ, Bhinder B, Liu W, Li K, Moss TJ, Bareja R, Park K, Tavassoli P, Cyrta J, et al. (2019). Upper tract urothelial carcinoma has a luminal-papillary T-cell depleted contexture and activated FGFR3 signaling. Nat Commun 10, 2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney MS, Shukla SA, Wu CJ, Getz G, and Hacohen N (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, McGranahan N, Herrero J, Taylor BS, and Swanton C (2016). DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome biology 17, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roupret M, Babjuk M, Comperat E, Zigeuner R, Sylvester RJ, Burger M, Cowan NC, Gontero P, Van Rhijn BWG, Mostafid AH, et al. (2018). European Association of Urology Guidelines on Upper Urinary Tract Urothelial Carcinoma: 2017 Update. Eur Urol 73, 111–122. [DOI] [PubMed] [Google Scholar]
- Schroder MS, Culhane AC, Quackenbush J, and Haibe-Kains B (2011). survcomp: an R/Bioconductor package for performance assessment and comparison of survival models. Bioinformatics 27, 3206–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfakianos JP, Cha EK, Iyer G, Scott SN, Zabor EC, Shah RH, Ren Q, Bagrodia A, Kim PH, Hakimi AA, et al. (2015). Genomic Characterization of Upper Tract Urothelial Carcinoma. Eur Urol 68, 970–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibing Y, Liangren L, Qiang W, Hong L, Turun S, Junhao L, Lu Y, Zhengyong Y, Yonghao J, Guangqing F, et al. (2016). Impact of tumour size on prognosis of upper urinary tract urothelial carcinoma after radical nephroureterectomy: a multi-institutional analysis of 795 cases. BJU Int 118, 902–910. [DOI] [PubMed] [Google Scholar]
- Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day IN, Gaunt TR, and Campbell C (2015). An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31, 1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi Y, Sato Y, Chiba K, Okuno Y, Nagata Y, Yoshida K, Shiba N, Hayashi Y, Kume H, Homma Y, et al. (2013). An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res 41, e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi Y, Tremmel G, Miyano S, and Stephens M (2015). A Simple Model-Based Approach to Inferring and Visualizing Cancer Mutation Signatures. PLoS Genet 11, e1005657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjodahl G, Lauss M, Lovgren K, Chebil G, Gudjonsson S, Veerla S, Patschan O, Aine M, Ferno M, Ringner M, et al. (2012). A molecular taxonomy for urothelial carcinoma. Clin Cancer Res 18, 3377–3386. [DOI] [PubMed] [Google Scholar]
- Springer SU, Chen CH, Rodriguez Pena MDC, Li L, Douville C, Wang Y, Cohen JD, Taheri D, Silliman N, Schaefer J, et al. (2018). Non-invasive detection of urothelial cancer through the analysis of driver gene mutations and aneuploidy. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Katoh H, Komura D, Kakiuchi M, Tagashira A, Yamamoto S, Tatsuno K, Ueda H, Nagae G, Fukuda S, et al. (2020). Defined lifestyle and germline factors predispose Asian populations to gastric cancer. Sci Adv 6, eaav9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, Shimamura T, Niida A, Motomura K, Ohka F, et al. (2015). Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet 47, 458–468. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Kikuchi E, Kanao K, Matsumoto K, Shirotake S, Kobayashi H, Miyazaki Y, Ide H, Obata J, Hoshino K, et al. (2014). The predictive value of positive urine cytology for outcomes following radical nephroureterectomy in patients with primary upper tract urothelial carcinoma: a multi-institutional study. Urol Oncol 32, 48 e19–26. [DOI] [PubMed] [Google Scholar]
- Therkildsen C, Eriksson P, Hoglund M, Jonsson M, Sjodahl G, Nilbert M, and Liedberg F (2018). Molecular subtype classification of urothelial carcinoma in Lynch syndrome. Mol Oncol 12, 1286–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischler G, and Leonard S (2014). biobambam: tools for read pair collation based algorithms on BAM files. Source Code for Biology and Medicine 9, 13. [Google Scholar]
- van der Velde KJ, de Boer EN, van Diemen CC, Sikkema-Raddatz B, Abbott KM, Knopperts A, Franke L, Sijmons RH, de Koning TJ, Wijmenga C, et al. (2017). GAVIN: Gene-Aware Variant INterpretation for medical sequencing. Genome biology 18, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Loo P, Nordgard SH, Lingjaerde OC, Russnes HG, Rye IH, Sun W, Weigman VJ, Marynen P, Zetterberg A, Naume B, et al. (2010). Allele-specific copy number analysis of tumors. Proc Natl Acad Sci U S A 107, 16910–16915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandin F, Papoutsaki A, Raphael BJ, and Upfal E (2015). Accurate computation of survival statistics in genome-wide studies. PLoS Comput Biol 11, e1004071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinh NX, Epps J, and Bailey J (2010). Information Theoretic Measures for Clusterings Comparison: Variants, Properties, Normalization and Correction for Chance. J Mach Learn Res 11, 2837–2854. [Google Scholar]
- Ward DG, Gordon NS, Boucher RH, Pirrie SJ, Baxter L, Ott S, Silcock L, Whalley CM, Stockton JD, Beggs AD, et al. (2019). Targeted deep sequencing of urothelial bladder cancers and associated urinary DNA: a 23-gene panel with utility for non-invasive diagnosis and risk stratification. BJU Int 124, 532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson MD, and Hayes DN (2010). ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto G, Nannya Y, Kato M, Sanada M, Levine RL, Kawamata N, Hangaishi A, Kurokawa M, Chiba S, Gilliland DG, et al. (2007). Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of affymetrix single-nucleotide-polymorphism genotyping microarrays. Am J Hum Genet 81, 114–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu B, and Cai H (2018). Chromothripsis Detection and Characterization Using the CTLPScanner Web Server. Methods Mol Biol 1769, 265–278. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, Shiozawa Y, Sato Y, Aoki K, Kim SK, et al. (2019). Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 565, 312–317. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. (2011). Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69. [DOI] [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, Trevino V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizato T, Nannya Y, Atsuta Y, Shiozawa Y, Iijima-Yamashita Y, Yoshida K, Shiraishi Y, Suzuki H, Nagata Y, Sato Y, et al. (2017). Genetic abnormalities in myelodysplasia and secondary acute myeloid leukemia: impact on outcome of stem cell transplantation. Blood 129, 2347–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Parmigiani G, and Johnson WE (2020). ComBat-Seq: batch effect adjustment for RNA-Seq count data. bioRxiv, 2020.2001.2013.904730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Zhang L, Wu B, Zha Z, Yuan J, Jiang Y, and Feng Y (2020). The prognostic value of tumor architecture in patients with upper tract urothelial carcinoma treated with radical nephroureterectomy: A systematic review and meta-analysis. Medicine (Baltimore) 99, e22176. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, related to Table 1. Summary of clinical information and type of analysis in UTUC samples.
Table S7, related to Figure 6. Results of urinary sediment-derived DNA sequencing.
Data Availability Statement
All the whole exome sequencing (WES), RNA sequencing, single-nucleotide polymorphism (SNP) array, and methylation array data have been deposited in the European Genome Phenome Archive (http://www.ebi.ac.uk/ega/) under accession numbers EGAD0000100766, EGAD00001007667, EGAD00010002096, and EGAD00010002098.
We obtained public sequencing data of UBC and UTUC from previous genetic studies (Audenet et al., 2019; Cancer Genome Atlas Research, 2014; Pietzak et al., 2017; Robertson et al., 2017; Robinson et al., 2019; Sfakianos et al., 2015). Fastq files of whole exome and RNA sequencing data and CEL files of SNP array data were downloaded from the TCGA Data Portal (https://portal.gdc.cancer.gov/), all of which were analyzed with the same pipeline and methods as used in our UTUC data, as described in the following section. Results of targeted-capture sequencing of UBC and UTUC published from MSKCC were downloaded from cBioportal (http://www.cbioportal.org/) (Cerami et al., 2012; Gao et al., 2013). Samples with pTis and/or those which underwent presurgical treatment (NAC and BCG perfusion) were excluded from the following analysis. UTUC cases with preceding or simultaneous UBC occurrences were also excluded from the comparison analysis of alterations in driver genes.