

Abstract
Introduction:
Blood pressure (BP) lowering reduces the risk for cognitive impairment and the progression of cerebral white matter lesions. It is unclear whether hypertension control also influences plasma biomarkers related to Alzheimer’s disease and non-disease-specific neurodegeneration.
Methods:
We examined the effect of intensive (<120 mm Hg) vs standard (<140 mm Hg) BP control on longitudinal changes in plasma Aβ40 and Aβ42, total tau, and neurofilament light chain (NfL) in a subgroup of participants from the Systolic Blood Pressure Intervention Trial (N=517).
Results:
Over 3.8 years, there were no significant between-group differences for Aβ40, Aβ42, Aβ42 / Aβ40, or total tau. Intensive treatment was associated with larger increases in NfL compared to standard treatment. Adjusting for kidney function, but not BP, attenuated the association between intensive treatment and NfL.
Discussion:
Intensive BP treatment was associated with changes in NfL, which were correlated with changes in kidney function associated with intensive treatment.
TRIAL REGISTRATION:
clinicaltrials.gov Identifier: NCT01206062
1. INTRODUCTION
Meta-analyses of randomized trials have indicated that blood pressure (BP) lowering reduces the incidence of dementia [1,2]. Observational data [3,4], the Systolic Blood Pressure Intervention Trial (SPRINT) [5], and other randomized trials [6,7] have shown hypertension and its control are also associated with the presence and progression of white matter lesions. While cerebrovascular mechanisms are a likely explanation for the reduction in cognitive impairment observed in SPRINT [8], studies have also shown that vascular risk is associated the presence of brain amyloid deposition based on positron emission tomography [9] and that antihypertensive treatment is associated with a reduced risk of incident dementia and Alzheimer’s disease (AD) [10]. It is, however, unknown whether intensive BP control may impact biomarkers of AD pathology, which frequently co-occurs with vascular contributors to dementia. There is considerable observational evidence that vascular damage may contribute to the progression of AD neuropathology [11,12], but causal experimental evidence to this effect is lacking [13]. Intensive BP control in SPRINT has also been associated with greater decreases in total brain and hippocampal volumes [5,14], but not with changes in other magnetic resonance imaging (MRI) markers that are sensitive, but not specific for, AD-related neurodegeneration [14].
The characterization of AD has transitioned to a biological definition of the disease, relying upon quantitative measurements of beta-amyloid, tau, and neurodegeneration via imaging or cerebrospinal fluid [15]. Given the expense of imaging, patient burden related to imaging and cerebrospinal fluid sampling, and resulting potential selection biases, there is a major effort to use and calibrate blood based protein biomarkers [16–18] to reduce both the burden and cost of identifying pathologic AD. Using stored samples from a subgroup of SPRINT participants, here we examine the effect of intensive BP control on changes in plasma biomarkers related to AD and neurodegeneration more broadly (beta-amyloid, total tau, and neurofilament light chain). In addition, given the known effects of intensive treatment on kidney function [19,20], an important modulator of the composition of the blood proteome [21], we also explored the association of these biomarkers with changes in kidney function.
2. METHODS
2.1. Trial design
The trial design and methods have been published previously [22,23]. Briefly, we conducted a multicenter randomized clinical trial that compared two strategies for managing systolic BP (SBP) in older adults with hypertension who were at increased risk of cardiovascular disease. Participants were aged 50 years or older and had an SBP between 130 and 180 mm Hg at the screening visit, depending on the number of anti-hypertensive agents prescribed. Participants were considered to have an increased cardiovascular risk if they had clinical or subclinical cardiovascular disease, chronic kidney disease (defined by an estimated glomerular filtration rate of <60 mL/min/1.73 m2), or a Framingham Risk Score of 15% or greater or if they were aged 75 years or older. Individuals residing in a nursing home, persons with a diagnosis of dementia (based on medical record review), and those treated with medications primarily used for dementia therapy were excluded, as were persons with prevalent diabetes mellitus, history of stroke, proteinuria > 1 gram per day, or polycystic kidney disease. Individuals at 102 sites in the United States and Puerto Rico were randomized (1:1) to a SBP goal of less than 120 mm Hg (intensive treatment group, n = 4678) or a goal of less than 140 mm Hg (standard treatment group, n = 4683), using random permuted blocks with the randomization stratified by clinic site. The algorithms and formulary for the trial are listed in the published study protocol [8,23]. Trial enrollment began in November 2010 and ended in March 2013, with follow-up through July 1, 2016. The study was approved by the institutional review board at each participating site, and each participant provided written informed consent. The study is registered at ClinicalTrials.gov (NCT01206062).
2.2. Magnetic resonance imaging sub-study
A subset of participants (n = 2913) were recruited into a cognitive function sub-study to more extensively evaluate the effects of intensive SBP control on specific domains of cognitive function [24]. MRI scans were obtained in a further subset of these participants to evaluate brain structure [5]. All participants accessible to any one of 7 designated MRI sites (drawing from 27 clinic sites) were screened for the MRI sub-study, and eligible participants provided written informed consent. Exclusion criteria for the MRI sub-study included any implanted electrical medical device, such as a pacemaker, any MRI-incompatible or MRI compatibility unknown metallic foreign material, or claustrophobia. Structural MRI of the brain included 1-mm isotropic T1, T2, and fluid-attenuated inversion recovery imaging, and was processed using an automated pipeline [5].
2.3. Core laboratory measures
Participants were instructed to fast overnight for the randomization visit and for annual follow-up assessments. Blood was collected by venipuncture at the clinical sites into EDTA-plasma tubes, chilled for 20-30 minutes in a refrigerator, and then centrifuged for 10-15 minutes at 1800-1900 x g. Plasma was transferred into transport tubes, refrigerated, and shipped overnight on ice-cold gel packs the day of collection to the SPRINT Central Laboratory at the University of Minnesota. On receipt in the Laboratory, samples were aliquoted into 0.5 mL cryovials and stored at −70°C. Serum and urine creatinine were measured using a method traceable to isotope dilution mass spectrometry. Urine creatinine was measured with the Siemens ProSpec nephelometric analyzer. We calculated the estimated glomerular filtration rate (eGFR) with the Chronic Kidney Disease Epidemiology Collaboration equation [25]. Serum bicarbonate was measured at baseline using an enzymatic method with phosphoenolpyruvate carboxylase using Roche CO2-L reagent and Roche Cobas 6000 Chemistry Analyzers (Roche Diagnostics Corporation).
2.4. Plasma biomarkers
This work was intended as a pilot study. We included participants from the MRI sub-study that were 60 years or older at the time of randomization because biomarker changes would be most likely to be observed in older trial participants (Figure S1). We used stored plasma samples from the baseline visit, and then, if available, from a single follow-up visit for each participant (median follow-up of 3.8 years [interquartile range, 3.5 to 4.0 years]). For participants who completed the follow-up MRI assessment, we utilized the stored follow-up sample nearest to the date of the follow-up MRI. For participants who did not complete the follow-up MRI, we utilized their latest available stored follow-up sample (Table S1). Assays for plasma human beta-amyloid 40 (Aβ40), beta-amyloid 42 (Aβ42), total tau, and neurofilament light chain (NfL) were performed at the University of Kentucky on a single molecule-array (Simoa) HD-1 analyzer platform. NfL was measured using the Simoa Nf-light advantage kit. Aβ40, Aβ42, and total tau were assessed using the Simoa Human Neurology 3-Plex A assay. Frozen plasma samples from the SPRINT Central Laboratory were shipped on dry ice without thawing to the University of Kentucky where they were stored at −80°C. Samples were then thawed on ice and centrifuged at maximum speed for 10 minutes at 4°C. All samples were assayed in duplicate and were run with kits from the same lot for each analyte. Samples were randomly distributed across assay batches, with paired baseline and follow-up samples always performed within the same assay batch. While the median coefficients of variation by assay batch were generally <10% for Aβ40, Aβ42, and total tau, coefficients of variation were consistently higher for NfL (10-20%, Table S2).
2.5. Assessment of cognitive function
Methods for neuropsychological testing of cognitive function have been previously described [8,24]. All participants in the MRI sub-study were administered a comprehensive cognitive battery at baseline including the Montreal Cognitive Assessment, Logical Memory I and II, Digit Symbol Coding, Trail Making Test Parts A and B, Hopkins Verbal Learning Test-Revised, Modified Rey-Osterreith Complex Figure (Copy and Immediate Recall), Digit Span, Category Fluency – Animals, and the 15-item Boston Naming Test. Centrally trained and certified examiners administered the cognitive tests. They were administered in either English or Spanish, depending on the participant’s preferred language. Here we utilize composite cognitive domain scores at baseline representing memory, processing speed, executive function, language, and global cognitive function. Each composite domain score was calculated as the average of specific standardized test scores [24].
2.6. Statistical analyses
Because of skewed distributions for some of the biomarkers, Spearman rank correlations and partial correlation coefficients [26] adjusting for age (or age and eGFR) were used to describe correlations between measures at baseline. Robust linear mixed models, including random effects for participant and assay batch, were used to estimate the change in each plasma biomarker between the BP treatment groups, including time since randomization as a covariate [27]. Intuitively, robust mixed models address skewed outcome distributions by down-weighting observations with large residuals or random effects, reducing their influence on model estimates. We also conducted several sensitivity analyses. First, we fit models adjusting for baseline and follow-up SBP, diastolic BP, or eGFR measured at the same study visit as the biomarkers. Second, we examined treatment group differences as a function of the baseline level of each biomarker and white matter lesion volumes. All hypothesis tests were 2-sided, and P values less than .05 were considered statistically significant. No adjustments for multiple comparisons were made.
2.7. Role of the funding source
The National Institutes of Health and the US Department of Veterans Affairs had roles in the design and conduct of the trial; collection, management, analysis, and interpretation of the data; and in the preparation and review of the manuscript. Neither was involved in formal approval of the manuscript or the decision to submit the manuscript for publication. The Alzheimer’s Association had a role in the design and conduct of the biomarker study; but not in the collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or the decision to submit the manuscript for publication.
3. RESULTS
3.1. Baseline characteristics
Table 1 contains baseline information for participants 60 years or older in this biomarker sub-study, with 283 randomized to intensive treatment and 234 to standard treatment. At baseline, participants had a mean age of 69.9 ± 7.1 years, 42.9% were female, and 28.2% were Black. Participants had a mean SBP of 138.3 ± 16.8 mm Hg, and mean eGFR of 70.6 ± 18.6 ml/min/1.73 m2, with 29.8% having an eGFR <60 ml/min/1.73 m2. In comparison to non-included trial participants who were 60 years or older at the time of randomization (Table S3), participants in the biomarker sub-study were younger, more likely to be female, were less likely to have a history of cardiovascular disease, had higher mean eGFR levels, and higher baseline cognitive test scores. Table S4 includes comparative data from the Rotterdam Study (mean age 71.9 ± 7.5 years, 58.0% female), where plasma biomarkers were measured using the same Simoa kits [28]. In comparison to that population, participants in this sub-study had higher levels of Aβ42 and total tau, lower levels of Aβ40, with similar levels of NfL.
Table 1.
Baseline characteristics of participants in the blood biomarker sub-study by treatment group
| Characteristic | Intensive Treatment N = 283 |
Standard Treatment N = 234 |
|---|---|---|
|
| ||
| Age, years, mean (SD) | 69.8 (6.9) | 69.9 (7.4) |
| Age ≥ 75 years, No. (%) | 82 (29.0) | 65 (27.8) |
|
| ||
| Female sex, No. (%) | 126 (44.5) | 96 (41.0) |
|
| ||
| Race/Ethnicity, No. (%) | ||
| White | 192 (67.8) | 152 (65.0) |
| Black | 81 (28.6) | 65 (27.8) |
| Hispanic | 6 (2.1) | 14 (6.0) |
| Other | 4 (1.4) | 3 (1.3) |
|
| ||
| History of Cardiovascular Disease, No. (%) | 42 (14.8) | 37 (15.8) |
|
| ||
| Systolic Blood Pressure, mm Hg, mean (SD) | 138.0 (17.6) | 138.7 (15.9) |
|
| ||
| Diastolic Blood Pressure, mm Hg, mean (SD) | 75.7 (10.5) | 77.0 (12.3) |
|
| ||
| eGFR, ml/min/1.73 m2, mean (SD) | 71.1 (19.0) | 70.1 (18.0) |
| eGFR<60 ml/min/1.73 m2 | 77 (27.9) | 74 (32.0) |
|
| ||
| Urine Albumin to Creatinine Ratio, mg/g, median [IQR] | 9.4 [5.3 to 21.3] | 9.9 [6.2 to 20.4] |
|
| ||
| Use of Statin, No. (%) | 125 (44.3) | 109 (47.2) |
|
| ||
| Use of Aspirin, No. (%) | 156 (55.1) | 131 (56.0) |
|
| ||
| Montreal Cognitive Assessment, median [IQR]a | 24 [21 to 26] | 24 [21 to 27] |
|
| ||
| Logical Memory II, median [IQR]b | 9 [6.5 to 12] | 8 [6 to 11] |
|
| ||
| Digit Symbol Coding, median [IQR]c | 52 [43. to 60] | 52 [41 to 62] |
|
| ||
| Total Brain Volume, cm3, mean (SD) | 1124.7 (116.8) | 1133.4 (114.4) |
|
| ||
| WML Volume, cm3, median [IQR] | 3.6 [1.7 to 7.5] | 3.9 [2.0 to 6.8] |
|
| ||
| Aβ40, pg/ml, median [IQR] | 194.9 [133.9 to 277.5] | 186.0 [123.1 to 267.5] |
|
| ||
| Aβ42, pg/ml, median [IQR] | 21.0 [16.2 to 26.4] | 21.6 [16.5 to 28.8] |
|
| ||
| Aβ42 / Aβ40, median [IQR] | 0.10 [0.07 to 0.15] | 0.10 [0.08 to 0.22] |
|
| ||
| Total Tau, pg/ml, median [IQR] | 7.7 [6.3 to 9.3] | 8.1 [6.1 to 9.7] |
|
| ||
| Neurofilament Light Chain, pg/ml, median [IQR] | 13.7 [9.3 to 20.1] | 14.9 [8.6 to 23.4] |
eGFR denotes estimated glomerular filtration rate based on the CKD-EPI equation, IQR Interquartile Range, SD Standard Deviation, and WML White Matter Lesion.
Scores range from 0 to 30, with higher scores denoting better cognitive function.
Subtest of the Wechsler Memory Scale. Scores range from 0 to 14, with higher scores denoting better cognitive function.
Subtest of the Wechsler Adult Intelligence Scale. Scores range from 0 to 135, with higher scores denoting higher cognitive function.
3.2. Cross-sectional associations with plasma biomarkers
Figure 1 displays baseline cross-sectional correlations between the plasma biomarkers and age, vital signs, and laboratory measures. All of the biomarkers were positively correlated with age, with the strongest associations observed for NfL (Spearman’s rho = 0.43, p<0.001) and Aβ40 (rho = 0.27, p <0.001). Adjusting for age, NfL (partial Spearman’s correlation (PSC) = −0.30, p<0.001) and Aβ40 (PSC = −0.32, p<0.001) were also negatively correlated with eGFR. Table S5 displays baseline levels for each biomarker stratified by eGFR. With the exception of the ratio Aβ42 / Aβ40, all of the biomarkers exhibited increasing levels with lower eGFR. Baseline correlations between the biomarkers, neuropsychological test scores, and structural MRI measures were generally much weaker, with no significant partial correlations after accounting for age and eGFR (Figure 1).
Figure 1.
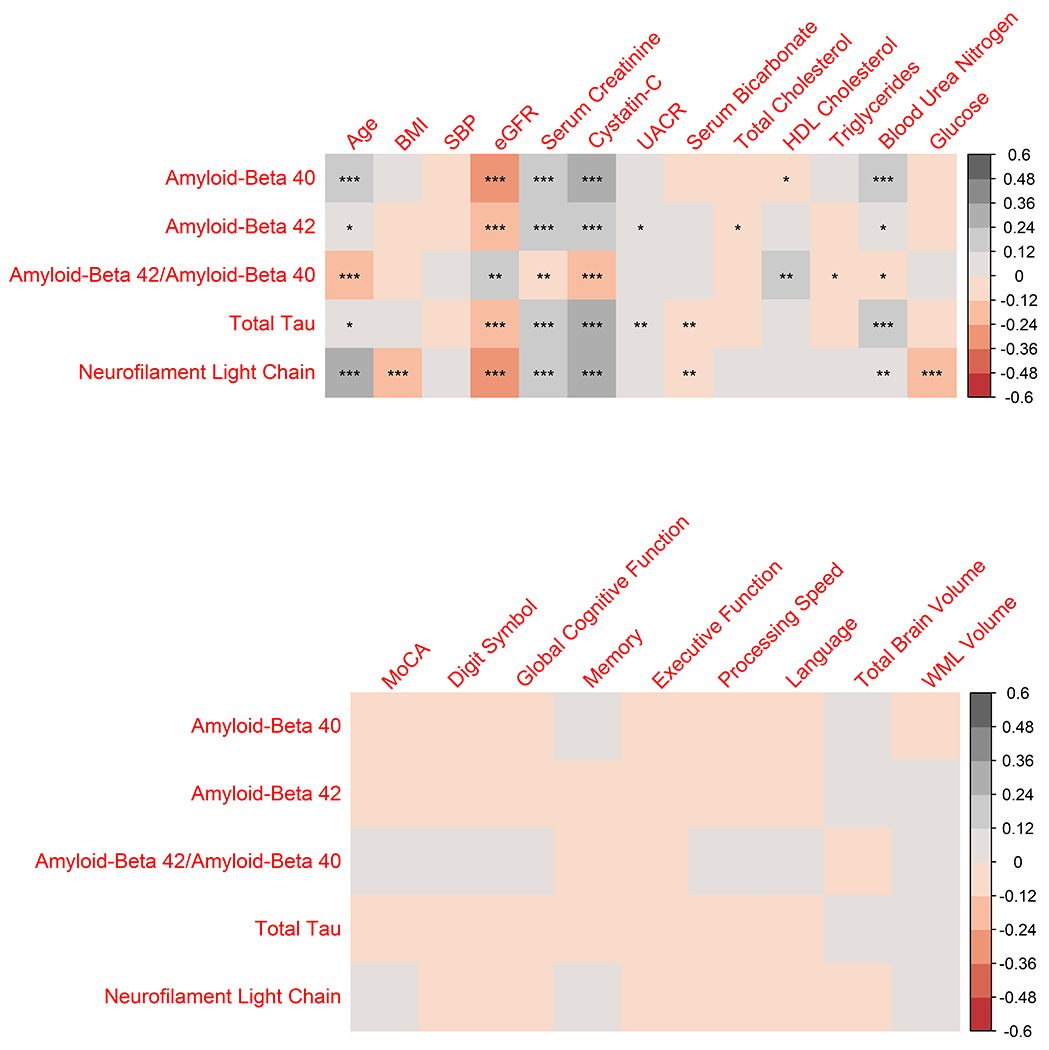
Baseline correlations between plasma biomarkers and age, vital signs, laboratory measures, cognitive test scores, and structural MRI measures
For age, values represent Spearman’s rank correlation for each biomarker. For all other variables, values represent a partial Spearman’s correlation adjusting for age (top panel), or adjusting for age and eGFR (bottom panel). BMI denotes body mass index, eGFR estimated glomerular filtration rate based on the CKD-EPI equation, HDL high density lipoprotein, UACR urine albumin to creatinine ratio, MoCA Montreal Cognitive Assessment, and WML white matter lesion. The memory composite domain score consisted of scores from the HVLT-R immediate and delayed recall, the ROCFm immediate recall, and Logical Memory I and II; Processing speed included the TMT-Parts A and B and Digit Symbol Coding; Executive function included the TMT – Part B minus Part A and Digit Span; Language included the Boston Naming and Category Fluency; and global cognitive function consisted of all tests included in the above domain scores. *** denotes P value<0.001, ** P value<0.01, and * P value<0.05.
3.3. Association of intensive treatment with changes in plasma biomarkers
Figure 2 displays the association between changes in each biomarker with BP treatment group. For each of the biomarkers, with the exception of Aβ42 / Aβ40 [29], increases would generally be associated with a more pathogenic state and increased risk for dementia. The mean change per year (MCPY) for Aβ40 was an increase of 11.8 pg/ml (95% CI: 9.2 to 14.4) with intensive treatment, as compared to 7.8 pg/ml (95% CI: 4.9 to 10.8) for standard treatment (p=0.05, Table S6). We observed larger mean increases in NfL with intensive treatment (between-group difference in MCPY = 0.8 pg/ml, 95% CI: 0.3 to 1.2, p=0.002). There were no significant differences for change in Aβ42, Aβ42 / Aβ40, or total tau between the treatment groups.
Figure 2.
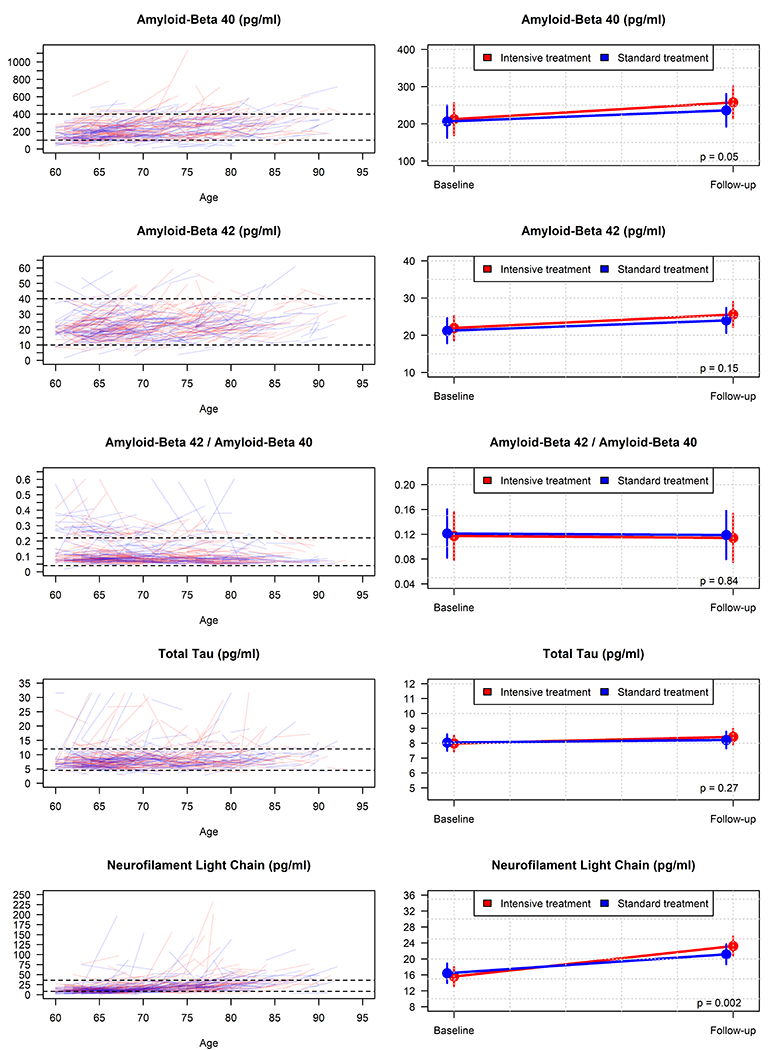
Change in plasma biomarkers comparing intensive treatment to standard treatment
Left panels display raw trajectories for each biomarker by participant as a function of baseline age. Dashed horizontal lines correspond to the y-axis limits for the right panel figures. Right panels display mean estimates from a robust linear mixed model with follow-up values computed at 3.81 years since randomization.
3.4. Sensitivity analyses
We also investigated how changes in eGFR might influence our results, given the early initial decline in eGFR associated with intensive BP treatment (Figure S2) [30]. During follow-up, 17 participants (6.0%) in the intensive treatment group experienced a ≥30% decline in eGFR on or before the collection of the follow-up blood sample used in this study, as compared to 2 (0.8%) in the standard treatment group. While based on a small number of participants, we found that the 19 participants that experienced a ≥30% decline in eGFR had significantly larger increases for all of the biomarkers with the exception of Aβ42 / Aβ40 (Table S7). For example, the MCPY for Aβ40 was 30.0 pg/ml (95% CI: 19.8 to 40.1) for participants who experienced a ≥30% decline in eGFR, as compared to a MCPY of 9.3 pg/ml (95% CI: 7.4 to 11.3) for participants that did not (between-group difference = 20.6 pg/ml, 95% CI: 10.3 to 30.9, p<0.001). Experiencing a 30% decline in eGFR was similarly associated with larger increases in NfL (between-group difference in MCPY = 2.2 pg/ml, 95% CI: 0.9 to 2.4, 0.001). When we adjusted treatment group comparisons for both baseline and follow-up eGFR, the between-group difference for NfL was attenuated (Table 2). In comparison, the effect on NfL was not attenuated when we analogously adjusted for either SBP or diastolic BP (Table 2).
Table 2.
Association of intensive treatment versus standard treatment on plasma biomarkers adjusted for kidney function and blood pressure
| Adjusted for eGFR | Intensive Treatment | Standard Treatment | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Biomarker | Baseline Mean (95% CI) |
Change / Year (95% CI) |
Baseline Mean (95% CI) |
Change / Year (95% CI) |
Difference (95% CI) |
P value |
|
| ||||||
| Aβ40 (pg/ml) | 216.7 (173.8, 259.5) |
7.6 (4.8, 10.3) |
208.7 (165.6, 251.7) |
6.6 (3.6, 9.7) |
1.0 (−3.1, 5.0) |
0.65 |
| Aβ42 (pg/ml) | 22.1 (18.6, 25.6) |
0.7 (0.5, 0.9) |
21.2 (17.7, 24.7) |
0.7 (0.4, 0.9) |
0.0 (−0.3, 0.3) |
0.89 |
| Aβ42 / Aβ40 | 0.118 (0.079, 0.156) |
0.000 (−0.001, 0.001) |
0.122 (0.083, 0.161) |
0.000 (−0.002, 0.001) |
0.001 (−0.001, 0.002) |
0.43 |
| Total Tau (pg/ml) | 8.1 (7.5, 8.6) |
0.0 (−0.1, 0.1) |
8.0 (7.5, 8.5) |
0.0 (−0.1, 0.1) |
0.0 (−0.1, 0.1) |
0.96 |
| NfL (pg/ml) | 16.8 (14.1, 19.6) |
1.1 (0.6, 1.7) |
16.8 (14, 19.5) |
1.1 (0.5, 1.7) |
0.0 (−0.8, 0.8) |
0.92 |
|
| ||||||
| Adjusted for SBP | Intensive Treatment | Standard Treatment | ||||
|
| ||||||
| Biomarker | Baseline Mean (95% CI) |
Change / Year (95% CI) |
Baseline Mean (95% CI) |
Change / Year (95% CI) |
Difference (95% CI) |
P value |
|
| ||||||
| Aβ40 (pg/ml) | 212.4 (168.8, 256.0) |
11.2 (8.3, 14.0) |
206.3 (162.4, 250.1) |
7.7 (4.7, 10.7) |
3.5 (−0.6, 7.5) |
0.09 |
| Aβ42 (pg/ml) | 21.9 (18.6, 25.3) |
0.9 (0.7, 1.1) |
21.2 (17.8, 24.6) |
0.7 (0.5, 0.9) |
0.2 (−0.1, 0.5) |
0.27 |
| Aβ42 / Aβ40 | 0.12 (0.08, 0.16) |
−0.001 (−0.002, 0.001) |
0.12 (0.08, 0.16) |
−0.001 (−0.002, 0.001) |
0.000 (−0.002, 0.002) |
0.97 |
| Total Tau (pg/ml) | 8.0 (7.5, 8.5) |
0.1 (0.0, 0.2) |
8.0 (7.5, 8.6) |
0.0 (0.0, 0.1) |
0.1 (−0.1, 0.2) |
0.23 |
| NfL (pg/ml) | 15.6 (13.2, 18.0) |
2.1 (1.7, 2.4) |
16.4 (13.9, 18.9) |
1.3 (0.9, 1.6) |
0.8 (0.3, 1.3) |
0.001 |
|
| ||||||
| Adjusted for DBP | Intensive Treatment | Standard Treatment | ||||
|
| ||||||
| Biomarker | Baseline Mean (95% CI) |
Change / Year (95% CI) |
Baseline Mean (95% CI) |
Change / Year (95% CI) |
Difference (95% CI) |
P value |
|
| ||||||
| Aβ40 (pg/ml) | 211.4 (168.4, 254.4) |
9.5 (6.7, 12.3) |
206.6 (163.4, 249.9) |
7.1 (4, 10.1) |
2.4 (−1.6, 6.5) |
0.24 |
| Aβ42 (pg/ml) | 21.9 (18.5, 25.3) |
0.8 (0.6, 1.0) |
21.3 (17.9, 24.6) |
0.7 (0.4, 0.9) |
0.1 (−0.2, 0.4) |
0.45 |
| Aβ42 / Aβ40 | 0.12 (0.08, 0.16) |
0.000 (−0.001, 0.001) |
0.12 (0.08, 0.16) |
0.000 (−0.002, 0.001) |
0.000 (−0.001, 0.002) |
0.68 |
| Total Tau (pg/ml) | 8.0 (7.4, 8.5) |
0.1 (0.0, 0.2) |
8.1 (7.5, 8.6) |
0.0 (−0.1, 0.1) |
0.0 (−0.1, 0.2) |
0.45 |
| NfL (pg/ml) | 15.5 (13.2, 17.9) |
1.7 (1.4, 2.1) |
16.6 (14.1, 19.0) |
1.1 (0.8, 1.5) |
0.6 (0.1, 1.1) |
0.03 |
CI denotes confidence interval, DBP diastolic blood pressure, eGFR denotes estimated glomerular filtration rate based on the CKD-EPI equation, NfL neurofilament light chain, and SBP systolic blood pressure. Estimates are from a robust linear mixed model with random effects for participant and batch, adjusting for time since randomization.
Figure 3 shows estimated mean differences between intensive and standard treatment on follow-up levels for each plasma biomarker as a function of baseline concentration. We did not observe evidence of statistical interactions with baseline levels for Aβ40, Aβ42, total tau, or NfL. While there was nominal evidence of heterogeneity with Aβ42 / Aβ40, estimates were consistent with a null effect of intensive treatment overall and were driven by a small number of participants with high values for Aβ42 / Aβ40. There were also no significant interactions in follow-up plasma concentrations as a function of baseline white matter lesion volume (Figure S3).
Figure 3.
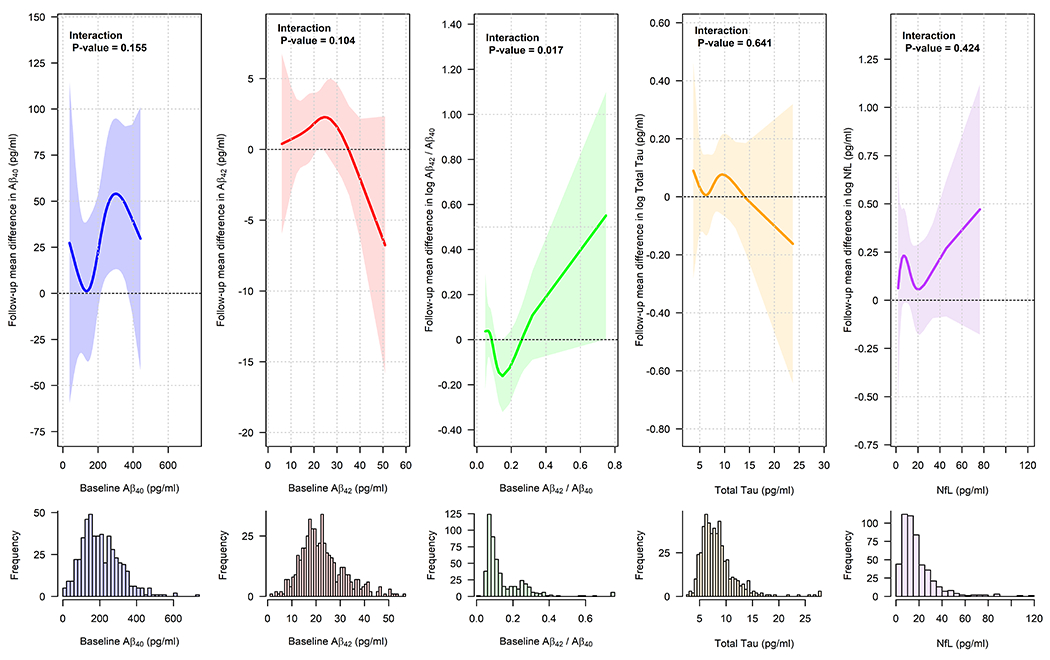
Association of intensive treatment versus standard treatment on follow-up plasma biomarkers as a function of baseline levels
Estimates based on linear model for follow-up plasma biomarker levels, adjusted for time since randomization, including an interaction between baseline levels and treatment group, with the effect of baseline levels modeled using cubic splines. Lines represent estimated treatment group difference at 3.81 years post-randomization (intensive treatment minus standard treatment) with associated 95% simultaneous confidence bands.
4. DISCUSSION
In this sub-study of a large randomized clinical trial, intensive SBP control resulted in greater increases in plasma NfL, which was attenuated by accounting for treatment-related changes in kidney function, as assessed by eGFR. There were not significant between-group differences for longitudinal change in Aβ40, Aβ42, Aβ42 / Aβ40, and total tau. SPRINT [5] and other randomized trials [6,7] have shown that intensive SBP control reduces the progression of cerebral white matter lesions measured via MRI. These results add to other observations that intensive BP treatment is associated with larger changes, albeit small, for several non-specific measures of atrophy and neurodegeneration, including TBV [5], hippocampal volume [14], and now plasma NfL. However, because all of these results emanate from the smaller and non-representative MRI sub-study in SPRINT, the implications of these biomarker results for the differences in adjudicated cognitive status observed in SPRINT remains unclear [8]. Several explanations are possible, though the most likely are selection biases due to sampling for the MRI sub-study and/or random variation. The lack of effect of intensive treatment on beta-amyloid and total tau is challenging to interpret with respect to effects on AD pathology. While plasma Aβ42 / Aβ40 is a sensitive marker for amyloid positivity measured by either positron emission tomography or cerebrospinal fluid, inference is limited by the somewhat lower accuracy of the Simoa platform relative to mass spectrometry [29] as well as the absence of a measure of phosphorylated tau, which is more specific for AD tauopathy [31–33].
A somewhat unexpected aspect of our results was the strength of the association between the plasma biomarkers and kidney function. Previous cross-sectional studies have noted associations between serum creatinine and plasma amyloid and NfL [34–39]. However, to our knowledge, this is the first study to demonstrate changes in these biomarkers within the context of an intervention known to affect kidney function [19,20] and also slow the development of cognitive impairment [8]. Declines in eGFR occurred more frequently with intensive treatment, though this effect is thought to reflect acute hemodynamic effects with BP lowering, as it did not lead to increases in urinary kidney injury markers or kidney failure [19,20,40]. The majority of studies to date investigating the potential diagnostic utility of plasma and serum AD biomarkers have largely ignored kidney function [16–18]. Chronic kidney disease has a high prevalence in older adults, estimated to affect one in five adults 65 to 79 years and half of those 80 years or older in the United States [41], and is a known risk factor for mild cognitive impairment and dementia [42]. With movement towards the amyloid, tau, and neurodegeneration (AT[N]) research framework as part of diagnostic screening and future clinical trials [15], these findings suggest a need to clarify the diagnostic interpretation of plasma and serum biomarkers and dementia more broadly within the context of impaired kidney function as well as populations with generally normal cognitive function. Similar to the use of brain natriuretic peptide in the diagnosis of heart failure, appropriate clinical thresholds for dementia biomarkers may differ in the context of chronic kidney disease [43].
Another non-intuitive result was that none of the plasma biomarkers, after accounting for age and eGFR, were associated with neuropsychological test scores or structural brain MRI measures at baseline. One explanation is that cognitive heterogeneity at baseline was limited given that dementia was a specific exclusion criterion. Other cohorts primarily comprised of individuals with normal cognition have generally shown rather weak cross-sectional correlations between cognitive test scores, white matter lesions, and amyloid biomarkers [44,45]. In addition, weak cross-sectional associations would certainly be expected viewing the plasma biomarkers as risk factors for subsequent cognitive decline. Unfortunately, very few participants included in this sub-study were adjudicated with cognitive impairment during the course of follow-up, precluding meaningful analyses correlating the plasma biomarkers with subsequent changes in cognitive function.
This study has several additional limitations that should be considered. First, there are several differences between participants included in this sub-study versus the much larger group of trial participants that were not. As such, our results should be considered preliminary, with a future need to study plasma biomarkers related to AD and vascular contributors to dementia in a larger, more representative group of participants both in SPRINT and in other populations. Second, the population in SPRINT was free of diagnosed dementia at baseline, follow-up was limited to a median of roughly 4 years, and the intensive blood pressure intervention was stopped early, all of which may have limited power to detect differential changes in these biomarkers. Third, the prevalence of AD pathology, indicated by amyloid positivity, in this cohort was likely low at baseline (<10-15%) on the basis of plasma Aβ42/Aβ40 levels [18].
In summary, within a subgroup of SPRINT participants, intensive treatment did not lead to significant changes in several plasma biomarkers of AD and neurodegeneration. Intensive treatment did, however, lead to larger increases in NfL, but this effect was explained by changes in kidney function. Future studies of blood-based dementia biomarkers should consider kidney function and distinguish between elevated biomarker levels due to increased production versus reduced clearance, especially within the context of chronic kidney disease.
Supplementary Material
RESEARCH IN CONTEXT.
Systematic review: The PubMed database was searched to review literature concerning the relationship between vascular mechanisms and Alzheimer’s disease pathology, and whether the treatment of hypertension influences biomarkers of Alzheimer’s disease and neurodegeneration.
Interpretation: Findings from a sub-study of a randomized trial indicate that intensive treatment of hypertension (target systolic blood pressure <120 mm Hg), as compared to less aggressive treatment (<140 mm Hg), is not associated with changes in plasma biomarkers of plasma beta-amyloid and total tau. Intensive treatment was associated with greater increases in plasma neurofilament light chain, though this difference was attenuated after accounting for changes in kidney function.
Future directions: Future studies addressing the interpretation of blood-based biomarkers should evaluate populations prior to the onset of symptomatic cognitive impairment, and examine the role that kidney function plays in circulating levels of these biomarkers.
Acknowledgements
We thank Maria C. Carrillo, PhD and Heather M. Snyder, PhD from the Alzheimer’s Association for scientific guidance in the conduct of this study.
Funding and Disclosure Information
Funding for this project was provided by the Alzheimer’s Association. The Systolic Blood Pressure Intervention Trial was funded by the National Institutes of Health (NIH), including the National Heart, Lung, and Blood Institute (NHLBI), the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), the National Institute on Aging (NIA), and the National Institute of Neurological Disorders and Stroke (NINDS) under contracts HHSN268200900040C, HHSN268200900046C, HHSN268200900047C, HHSN268200900048C, and HHSN268200900049C and interagency agreement A-HL-13-002-001. It was also supported in part with resources and use of facilities through the Department of Veterans Affairs (VA). Azilsartan and chlorthalidone (combined with azilsartan) were provided by Takeda Pharmaceuticals International Inc. Additional support was provided through the following National Center for Advancing Translational Sciences clinical and translational science awards: UL1TR000439 (awarded to Case Western Reserve University); UL1RR025755 (Ohio State University); UL1RR024134 and UL1TR000003 (University of Pennsylvania); UL1RR025771 (Boston University); UL1TR000093 (Stanford University); UL1RR025752, UL1TR000073, and UL1TR001064 (Tufts University); UL1TR000050 (University of Illinois); UL1TR000005 (University of Pittsburgh); 9U54TR000017-06 (University of Texas Southwestern Medical Center); UL1TR000105-05 (University of Utah); UL1 TR000445 (Vanderbilt University); UL1TR000075 (George Washington University); UL1 TR000002 (University of California, Davis); UL1 TR000064 (University of Florida); and UL1TR000433 (University of Michigan); and by National Institute of General Medical Sciences, Centers of Biomedical Research Excellence award NIGMS P30GM103337 (awarded to Tulane University).
N.M. Pajewski has received funding from the NIA, NHLBI, NIDDK, and the Duke Endowment; and is serving on Data and Safety Monitoring Boards (DSMBs) for the NIA and National Institute of Nursing Research (unpaid). F.M. Elahi is the vice chair of the vascular cognitive disorders Profession Interest Area of ISTAART. M. Kurella Tamura is supported by R01DK092241, serves on DSMBs for NIDDK and the VA (unpaid), and serves on advisory boards for the Clinician – Scientists Transdisciplinary Aging Research Coordinating Center (Clin-STAR) and Paul B. Beeson Emerging Leaders Career Development Award in Aging, for which she receives an honorarium from the American Federation of Aging Research. J.D. Hinman is supported by UH3NS100608; receives funding from the NIA, NINDS, American Heart Association, and American Neurological Association; has provided expert testimony for Bertoldo Baker Carter & Smith, Federal Public Defender - Central District of California, Popham Law Firm, WarrenAllen, LLP, Robert Miller Attorney at Law, Kjar McKenna & Stockalper, LLP, Matthew Millea Attorney at Law, Koskoff Koskoff & Bieder PC, and Goodwin Law; owns stock in Sage Cerebrovascular Diagnostics; and his institution has received drugs from Constant Therapeutics. I.M. Nasrallah receives funding from the NIH and the American Society of Neuroradiology; and has served as an educational speaker for Biogen; J.H. Ix receives funding from the NIDDK; has received consulting fees from AstraZeneca, Jnana Pharmaceuticals, and Ardelyx Pharmaceuticals; has received speaking honorarium from the National Kidney Foundation; serves on a DSMB for Sanifit International; serves on the Scientific Advisory Board for AlphaYoung; and has received donated study drug from Genentech Pharmaceticals unrelated to the present work. L.M. Miller was supported by T32DK104717. L.J. Launer has nothing to disclose. C.B. Wright has received royalties from UpToDate.com. M.A. Supiano is supported by R01AG055606; has received textbook royalties from McGraw-Hill; serves on a DSMB for the Depressed MIND study (unpaid); and serves on the board of the American Geriatrics Society. A. J. Lerner has received funding from Premier Applied Biosciences and the Elizabeth Prentiss Foundation; has received book royalty payments from Elsevier; and has received speaking honorarium from the Puerto Rican Academy of Neurology. T.L. Sudduth has nothing to disclose. A.A. Killeen has received funding from the NIDDK, Centers for Disease Control and Prevention, Diasorin, Inc., and ARKRAY Inc.; has received book royalty payments from Elsevier; has received consulting fees from the American Association for Clinical Chemistry; has held leadership roles for the American Association for Clinical Chemistry, College of American Pathologists, and the Academy of Clinical Laboratory Physicians and Scientists; and has received laboratory reagents from Roche Diagnostics for research studies unrelated to the present work. A.K. Cheung has received funding from the NIH; has received royalty payments from UpToDate.com; and has received consulting fees from Boehringer-Ingelheim and Calliditas. D.M. Reboussin is supported by R01AG055606; has received funding from the Alzheimer’s Association and the NIH; and serves on DSMBs for the NIH (unpaid). D.M. Wilcock is supported by UH3NS100606; receives funding from the NIA and NINDS; has received consulting fees from Alector and AC Immune and honoraria from the Neuroscience Education Institute, University of California Irvine, Louisiana State University-Shreveport; and has received travel support from the Alzheimer’s Association, Alzheimer’s and Parkinson’s Diseases Conference, and the Brightfocus Foundation. J.D Williamson is supported by R01AG055606; has received funding from the NIH; and serves on DSMBs for the China National Center for Cardiovascular Diseases and the NIH (unpaid). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Alzheimer’s Association, National Institutes of Health, the U.S. Department of Veterans Affairs, or the United States Government.
Footnotes
Presented at the Alzheimer’s Association International Conference (Virtual), July, 2020.
Data Sharing Statement
De-identified participant data will be available in the BioLinCC repository (https://biolincc.nhlbi.nih.gov/studies/sprint).
REFERENCES
- [1].Peters R, Warwick J, Anstey KJ, Anderson CS. Blood pressure and dementia: What the SPRINT-MIND trial adds and what we still need to know. Neurology 2019;92:1017–8. 10.1212/WNL.0000000000007543. [DOI] [PubMed] [Google Scholar]
- [2].Hughes D, Judge C, Murphy R, Loughlin E, Costello M, Whiteley W, et al. Association of Blood Pressure Lowering With Incident Dementia or Cognitive Impairment: A Systematic Review and Meta-analysis. JAMA 2020;323:1934–44. 10.1001/jama.2020.4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Liao D, Cooper L, Cai J, Toole JF, Bryan NR, Hutchinson RG, et al. Presence and severity of cerebral white matter lesions and hypertension, its treatment, and its control. The ARIC Study. Atherosclerosis Risk in Communities Study. Stroke 1996;27:2262–70. [DOI] [PubMed] [Google Scholar]
- [4].Basile AM, Pantoni L, Pracucci G, Asplund K, Chabriat H, Erkinjuntti T, et al. Age, hypertension, and lacunar stroke are the major determinants of the severity of age-related white matter changes. The LADIS (Leukoaraiosis and Disability in the Elderly) Study. Cerebrovasc Dis 2006;21:315–22. 10.1159/000091536. [DOI] [PubMed] [Google Scholar]
- [5].SPRINT MIND Investigators for the SPRINT Research Group, Nasrallah IM, Pajewski NM, Auchus AP, Chelune G, Cheung AK, et al. Association of Intensive vs Standard Blood Pressure Control With Cerebral White Matter Lesions. JAMA 2019;322:524–34. 10.1001/jama.2019.10551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Murray AM, Hsu F-C, Williamson JD, Bryan RN, Gerstein HC, Sullivan MD, et al. ACCORDION MIND: results of the observational extension of the ACCORD MIND randomised trial. Diabetologia 2017;60:69–80. 10.1007/s00125-016-4118-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].White WB, Wakefield DB, Moscufo N, Guttmann CRG, Kaplan RF, Bohannon RW, et al. Effects of Intensive Versus Standard Ambulatory Blood Pressure Control on Cerebrovascular Outcomes in Older People (INFINITY). Circulation 2019;140:1626–35. 10.1161/CIRCULATIONAHA.119.041603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].SPRINT MIND Investigators for the SPRINT Research Group, Williamson JD, Pajewski NM, Auchus AP, Bryan RN, Chelune G, et al. Effect of Intensive vs Standard Blood Pressure Control on Probable Dementia: A Randomized Clinical Trial. JAMA 2019;321:553–61. 10.1001/jama.2018.21442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gottesman RF, Schneider ALC, Zhou Y, Coresh J, Green E, Gupta N, et al. Association Between Midlife Vascular Risk Factors and Estimated Brain Amyloid Deposition. JAMA 2017;317:1443–50. 10.1001/jama.2017.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ding J, Davis-Plourde KL, Sedaghat S, Tully PJ, Wang W, Phillips C, et al. Antihypertensive medications and risk for incident dementia and Alzheimer’s disease: a meta-analysis of individual participant data from prospective cohort studies. Lancet Neurol 2020;19:61–70. 10.1016/S1474-4422(19)30393-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zlokovic BV Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 2011;12:723–38. 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science (80- ) 2019;365:eaav9518. 10.1126/science.aav9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Scheffer S, Hermkens DMA, van der Weerd L, de Vries HE, Daemen MJAP. Vascular Hypothesis of Alzheimer Disease: Topical Review of Mouse Models. Arterioscler Thromb Vasc Biol 2021;41:1265–83. 10.1161/ATVBAHA.120.311911. [DOI] [PubMed] [Google Scholar]
- [14].Nasrallah IM, Gaussoin SA, Pomponio R, Dolui S, Erus G, Wright CB, et al. Association of Intensive vs Standard Blood Pressure Control With Magnetic Resonance Imaging Biomarkers of Alzheimer Disease: Secondary Analysis of the SPRINT MIND Randomized Trial. JAMA Neurol 2021;78:568–77. 10.1001/jamaneurol.2021.0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018;14:535–62. 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mattsson N, Zetterberg H, Janelidze S, Insel PS, Andreasson U, Stomrud E, et al. Plasma tau in Alzheimer disease. Neurology 2016;87:1827–35. 10.1212/WNL.0000000000003246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Doré V, et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018;554:249–54. 10.1038/nature25456. [DOI] [PubMed] [Google Scholar]
- [18].Palmqvist S, Janelidze S, Stomrud E, Zetterberg H, Karl J, Zink K, et al. Performance of Fully Automated Plasma Assays as Screening Tests for Alzheimer Disease–Related β-Amyloid Status. JAMA Neurol 2019;76:1060. 10.1001/jamaneurol.2019.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Beddhu S, Rocco MV, Toto R, Craven TE, Greene T, Bhatt U, et al. Effects of Intensive Systolic Blood Pressure Control on Kidney and Cardiovascular Outcomes in Persons Without Kidney Disease: A Secondary Analysis of a Randomized Trial. Ann Intern Med 2017;167:375–83. 10.7326/M16-2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cheung AK, Rahman M, Reboussin DM, Craven TE, Greene T, Kimmel PL, et al. Effects of Intensive BP Control in CKD. J Am Soc Nephrol 2017;28:2812–23. 10.1681/ASN.2017020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yang J, Brody EN, Murthy AC, Mehler RE, Weiss SJ, DeLisle RK, et al. Impact of Kidney Function on the Blood Proteome and on Protein Cardiovascular Risk Biomarkers in Patients With Stable Coronary Heart Disease. J Am Heart Assoc 2020;9:e016463. 10.1161/JAHA.120.016463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ambrosius WT, Sink KM, Foy CG, Berlowitz DR, Cheung AK, Cushman WC, et al. The design and rationale of a multicenter clinical trial comparing two strategies for control of systolic blood pressure: the Systolic Blood Pressure Intervention Trial (SPRINT). Clin Trials 2014;11:532–46. 10.1177/1740774514537404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].SPRINT Research Group, Wright JT, Williamson JD, Whelton PK, Snyder JK, Sink KM, et al. A Randomized Trial of Intensive versus Standard Blood-Pressure Control. N Engl J Med 2015;373:2103–16. 10.1056/NEJMoa1511939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rapp SR, Gaussoin SA, Sachs BC, Chelune G, Supiano MA, Lerner AJ, et al. Effects of intensive versus standard blood pressure control on domain-specific cognitive function: a substudy of the SPRINT randomised controlled trial. Lancet Neurol 2020;19:899–907. 10.1016/S1474-4422(20)30319-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, Feldman HI, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009;150:604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu Q, Li C, Wanga V, Shepherd BE. Covariate-adjusted Spearman’s rank correlation with probability-scale residuals. Biometrics 2018;74:595–605. 10.1111/biom.12812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Koller M robustlmm: An R Package for Robust Estimation of Linear Mixed-Effects Models. J Stat Softw 2016;75:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].de Wolf F, Ghanbari M, Licher S, McRae-McKee K, Gras L, Weverling GJ, et al. Plasma tau, neurofilament light chain and amyloid-β levels and risk of dementia; a population-based cohort study. Brain 2020:1220–32. 10.1093/brain/awaa054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Keshavan A, Pannee J, Karikari TK, Rodriguez JL, Ashton NJ, Nicholas JM, et al. Population-based blood screening for preclinical Alzheimer’s disease in a British birth cohort at age 70. Brain 2021;144:434–49. 10.1093/brain/awaa403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Beddhu S, Shen J, Cheung AK, Kimmel PL, Chertow GM, Wei G, et al. Implications of Early Decline in eGFR due to Intensive BP Control for Cardiovascular Outcomes in SPRINT. J Am Soc Nephrol 2019;30:1523–33. 10.1681/ASN.2018121261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Karikari TK, Pascoal TA, Ashton NJ, Janelidze S, Benedet AL, Rodriguez JL, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol 2020;19:422–33. 10.1016/S1474-4422(20)30071-5. [DOI] [PubMed] [Google Scholar]
- [32].Janelidze S, Mattsson N, Palmqvist S, Smith R, Beach TG, Serrano GE, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med 2020;26:379–86. 10.1038/s41591-020-0755-1. [DOI] [PubMed] [Google Scholar]
- [33].Palmqvist S, Janelidze S, Quiroz YT, Zetterberg H, Lopera F, Stomrud E, et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA 2020;324:772. 10.1001/jama.2020.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Arvanitakis Z, Lucas JA, Younkin LH, Younkin SG, Graff-Radford NR. Serum Creatinine Levels Correlate With Plasma Amyloid β Protein. Alzheimer Dis Assoc Disord 2002;16:187–90. 10.1097/00002093-200207000-00009. [DOI] [PubMed] [Google Scholar]
- [35].Luchsinger JA, Tang M-X, Miller J, Green R, Mehta PD, Mayeux R. Relation of Plasma Homocysteine to Plasma Amyloid Beta Levels. Neurochem Res 2007;32:775–81. 10.1007/s11064-006-9207-7. [DOI] [PubMed] [Google Scholar]
- [36].Ruiz A, Pesini P, Espinosa A, Pérez-Grijalba V, Valero S, Sotolongo-Grau O, et al. Blood Amyloid Beta Levels in Healthy, Mild Cognitive Impairment and Alzheimer’s Disease Individuals: Replication of Diastolic Blood Pressure Correlations and Analysis of Critical Covariates. PLoS One 2013;8:e81334. 10.1371/journal.pone.0081334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lopez OL, Chang Y, Ives DG, Snitz BE, Fitzpatrick AL, Carlson MC, et al. Blood amyloid levels and risk of dementia in the Ginkgo Evaluation of Memory Study (GEMS): A longitudinal analysis. Alzheimer’s Dement 2019;15:1029–38. 10.1016/j.jalz.2019.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Korley FK, Goldstick J, Mastali M, Van Eyk JE, Barsan W, Meurer WJ, et al. Serum NfL (Neurofilament Light Chain) Levels and Incident Stroke in Adults With Diabetes Mellitus. Stroke 2019;50:1669–75. 10.1161/STROKEAHA.119.024941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chatterjee P, Cheong Y-J, Bhatnagar A, Goozee K, Wu Y, McKay M, et al. Plasma metabolites associated with biomarker evidence of neurodegeneration in cognitively normal older adults. J Neurochem 2020. 10.1111/jnc.15128. [DOI] [PubMed] [Google Scholar]
- [40].Zhang WR, Craven TE, Malhotra R, Cheung AK, Chonchol M, Drawz P, et al. Kidney Damage Biomarkers and Incident Chronic Kidney Disease During Blood Pressure Reduction: A Case-Control Study. Ann Intern Med 2018;169:610–8. 10.7326/M18-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Murphy D, McCulloch CE, Lin F, Banerjee T, Bragg-Gresham JL, Eberhardt MS, et al. Trends in Prevalence of Chronic Kidney Disease in the United States. Ann Intern Med 2016;165:473–81. 10.7326/M16-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Viggiano D, Wagner CA, Martino G, Nedergaard M, Zoccali C, Unwin R, et al. Mechanisms of cognitive dysfunction in CKD. Nat Rev Nephrol 2020;16:452–69. 10.1038/s41581-020-0266-9. [DOI] [PubMed] [Google Scholar]
- [43].Han X, Zhang S, Chen Z, Adhikari BK, Zhang Y, Zhang J, et al. Cardiac biomarkers of heart failure in chronic kidney disease. Clin Chim Acta 2020;510:298–310. 10.1016/j.cca.2020.07.040. [DOI] [PubMed] [Google Scholar]
- [44].Hilal S, Akoudad S, van Duijn CM, Niessen WJ, Verbeek MM, Vanderstichele H, et al. Plasma Amyloid-β Levels, Cerebral Small Vessel Disease, and Cognition: The Rotterdam Study. J Alzheimers Dis 2017;60:977–87. 10.3233/JAD-170458. [DOI] [PubMed] [Google Scholar]
- [45].Jansen WJ, Ossenkoppele R, Tijms BM, Fagan AM, Hansson O, Klunk WE, et al. Association of Cerebral Amyloid-β Aggregation With Cognitive Functioning in Persons Without Dementia. JAMA Psychiatry 2018;75:84–95. 10.1001/jamapsychiatry.2017.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.