

Abstract
Background:
Vascular mechanisms may contribute to the accumulation of AD pathology.
Objective:
We examined whether the burden of vascular risk factors proximate to death is associated with amyloid-β and tau levels or modified their known association.
Methods:
We examined the brains of 1, 585 participants from two longitudinal community-based studies of older adults. Amyloid-β and tau were quantified by postmortem examination. The burden of vascular risk factors was summarized by calculating the Framingham general cardiovascular risk score (FRS) proximate to death. Using linear regressions, we examined the association of the FRS with the amyloid-β and tau levels and examined if the FRS modified the association of the amyloid-β with tau.
Results:
On average, participants were nearly 90 years old and two-thirds were women. The FRS was not associated with amyloid-β (Spearman r = −0.00, p = 0.918) or tau (r = 0.01, p = 0.701). However, the FRS as a whole (estimate = −0.022, SE = 0.008, p = 0.009), and specifically the systolic blood pressure (SBP) component (estimate = −0.033, SE = 0.012, p = 0.009), modified the association of the amyloid-β with tau. Further analysis showed that the association between amyloid-β and tau was stronger at lower levels of SBP.
Conclusion:
Late-life vascular risk scores were not related to postmortem levels of amyloid-β or tau. However, lower levels of vascular risk scores and SBP were associated with a stronger association between amyloid-β and tau. These data suggest that vascular risk factors may modify the relation of AD pathology markers to one another.
Keywords: Alzheimer’s disease, amyloid, autopsy, blood pressure, diabetes mellitus, risk factors, smoking, tau proteins
INTRODUCTION
Mid-life vascular risk factors are recognized as risk factors for late-life cognitive decline and dementia [1–3]. However, controversy exists regarding the association of late-life vascular risk factors with dementia. While higher levels of systolic blood pressure (SBP) at mid-life has been consistently reported to be associated with increased risk of late-life dementia [4, 5], longitudinal studies of late-life SBP have been mixed, with some showing increased risk for dementia [5], others showing protection against dementia [6], and yet others no association [4].
As Alzheimer’s disease (AD) is the most common single cause of late-life dementia, a major question is the association between vascular risk factors and pathological markers including amyloid-β (Aβ) and tau. Previous studies using in vivo assessments of Aβ and tau, by positron emission tomography (PET) or cerebrospinal fluid markers, showed inconsistent results. Some studies showed higher levels of Aβ [7–9] and tau [8, 10] in participants with a higher burden of vascular risk factors, while others did not [11]. In addition, a recent PET study found an interaction between Aβ and vascular risk factors on tau in a way that the association of tau and vascular risk factors was stronger in participants with higher levels of Aβ [12]. However, few clinical-pathologic studies have examined the association of vascular risk factors with neuropathologically-defined AD markers [13]. We previously showed that a higher level of late-life SBP was associated with an increasing number of brain infarcts [14]. In this study, we now examine the association of vascular risk factors with Aβ and tau levels using clinical and autopsy data from two ongoing community-based studies of aging, the Religious Orders Study (ROS) and the Rush Memory and Aging Project (MAP).
METHODS
Participants
The study participants came from one of two community-based, prospective clinical-pathologic studies of aging conducted at the Rush Alzheimer’s disease center, the ROS and the MAP [15]. ROS began enrolling Catholic nuns, priests, and brothers from across the United States in 1994. MAP participants enrolled from retirement centers and subsidized housings, from across the greater Chicago metropolitan area, beginning in 1997. Eligible participants in both cohorts were older adults who did not have known dementia at baseline. Both studies followed similar methods, including participants’ consenting to annual medical, cognitive, and other clinical evaluations, and brain donation at the time of death [15]. All participants signed an informed consent and Anatomic Gift Act. An Institutional Review Board of Rush University Medical Center approved both studies.
From 1994 until February 2020 at the time of analyses for this study, 1,468 participants were recruited in ROS and completed the baseline evaluation. From those recruited, 69 participants later withdrew (follow-up rate = 95%), and over time 867 died and 800 underwent autopsy (autopsy rate = 92%). For this study, 788 with neuropathological data were eligible for the current analyses; however, Aβ or tau measure was missing in 30 (Supplementary Figure 1), leaving 758 for these analyses. In the MAP, of 2, 174 participants with complete baseline evaluation 205 subsequently withdrew from the study (follow-up rate = 91%). During the follow-up, 1, 071 participants died, of which 888 had an autopsy (autopsy rate = 83%). Of 856 decedents who underwent autopsy, indices of Aβ or tau was missing in 29 leaving 827 included in these analyses (Supplementary Figure 2). Therefore, the total analytic sample consisted of 1, 585 participants with clinical and autopsy data. The analytic sample was younger in age at study entry and had fewer women compared with the participants excluded from this study (Supplementary Table 1).
Framingham risk score calculation
In the ROS and MAP annual visits, participants’ medical conditions and risk factors, including a history of hypertension, diabetes mellitus (DM), and smoking, are evaluated and summarized in a summary statistic of the count of hypertension, DM, and smoking present in a participant (1 point for each, with a total score range 0–3) [16, 17]. The annual clinical visits also include measurement of participants’ height and weight, and visual inspection of the participants’ medications. Medications’ names and dosages were recorded and were coded subsequently using Medi-Span Drug Data Base System [18]. SBP was measured by trained nurses using automated sphygmomanometers in standing and sitting positions, and an average was calculated as described previously [14]. Based on previously published methods, we calculated the cardiovascular Framingham risk score (FRS), separately for women and men, using 5 components: age, body mass index (BMI: calculated as weight in kilograms divided by height in meters squared), SBP, smoking, and DM [19]. The SBP component point was differentially calculated for individuals who were using versus not using medications to treat hypertension, as previously published [19]. In this study, DM and smoking components of FRS were scored as positive if a participant had at least one annual visit with positive relevant history. The SBP and BMI components for each participant were calculated by averaging all available measurements across the study. Lastly, the age component was calculated using the age of the participants at the last clinical visit that occurred on average 10.1 months (SD = 13.8 months, range = 23.7 hours – 13.2 years) from the time of death.
Assessment of postmortem pathological changes
The mean postmortem interval was 9.3 hours (SD = 8.1). Details of the autopsy procedures, which were done by staff blinded to clinical data, are described elsewhere [16]. One hemisphere was fixed and cut into 1 cm slabs. Tissue blocks were prepared from the slabs of predetermined brain regions and were cut into thin sections.
AD pathology indices
Quantification of the amount of Aβ and tau was done through immunostaining of tissue sections from eight cortical regions: anterior cingulate cortex, superior frontal cortex, mid frontal cortex, inferior temporal cortex, hippocampus, entorhinal cortex, angular gyrus/supramarginal cortex, and calcarine cortex. Then, by using computer-assisted sampling, image analyses (Aβ), and stereology (tau tangles), levels of Aβ and tau were quantified in each of the eight regions and summary measures for Aβ and tau were made by averaging relevant regional measures, as described in detail elsewhere [20]. Also, a modified Bielschowsky silver stain was used to visualize neuritic plaques, diffuse plaques, and neurofibrillary tangles [21], and a board-certified neuropathologist determined the pathologic diagnosis of AD based on published criteria [22].
Infarcts
Because vascular risk factors are stroke risk factors [23], we examined the association of FRS with infarcts and examined whether the association of FRS with AD pathology indices changed after adjustment for infarcts. As we have done in prior postmortem studies, we included only chronic infarcts that would be more reflective of the cumulative effects of the vascular risk burden [16, 24]. Slabs were inspected with the naked eye for the presence of gross infarcts, and suspected lesions were confirmed microscopically [25]. Microinfarcts are not visible to the naked eye and were identified under microscopy. For these analyses, we summarized infarcts as any infarct (gross infarcts or microinfarcts), gross infarcts, and microinfarcts, each as a dichotomous variable (presence versus absence).
Assessment of clinical stroke
In the annual clinical evaluations, participants answered questions about occurrence of a clinical stroke and its associated symptoms. Then, a neurologist validated the histories through reviewing the histories, taking history, and/or performing physical examination. Presence of a clinical stroke in the annual evaluations were summarized as a binary variable indicating whether a participant had experienced any clinical stroke before death.
Statistical analyses
We used a square root transformation of the Aβ and tau levels, due to the positively skewed distributions of these data. To calculate bivariate associations of FRS with AD pathological markers, t-test, chi-square, and Spearman correlation coefficient were used. Then, we employed two separate linear regressions with FRS as the predictor, and Aβ and tau as the separate outcomes, to examine the association of the FRS with Aβ and tau. Next, we used another linear regression with tau as the outcome, Aβ, FRS, and their interaction as the predictors, to test if FRS modified the association of Aβ with tau. After examining FRS, we replaced FRS with its components in subsequent models to find the component that drove the association or modification effect of FRS with Aβ and tau. Moreover, we did sensitivity analyses. In one sensitivity analysis, we excluded participants whose last visit was done more than 1 year before death, and in the other sensitivity analysis we recalculated FRS by using the last measurements of BMI and SBP instead of their averages across years of assessments. As the SBP component was the component with a modification effect on the association of Aβ with tau, we next examined the association of Aβ with tau at different levels of SBP for further exploration of the modification effect. Finally, we replaced SBP by diastolic blood pressure (DBP) to examine if the modification of the association between Aβ and tau by SBP was also seen by DBP. Analyses controlled for age at death, sex, and education. We used SAS version 9.4 and considered an alpha of 0.05, and a Bonferroni adjusted alpha for multiple comparison.
RESULTS
The demographic and clinical characteristics of the participants are shown in Table 1. On average, participants were nearly 90 years old at the time of death, two-thirds were women, and two-thirds had a pathological diagnosis of AD. The FRS ranged from 11 to 30 points (out of a possible range of −3 to 34), had a mean of 19.3 (SD = 3.2), and a median of 19. To contextualize the FRS, we calculated frequency of a clinical stroke at different quartiles of FRS. Clinical stroke was reported in 17% of participants with FRS score of 17 or less, 20% with FRS score between 17 and 19, 23% with FRS score between 19 and 21, and 25% with FRS score more than 21.
Table 1.
Demographic and clinical characteristics with postmortem AD indices of the participants
| Characteristic | All (n = 1,585) |
|---|---|
| Demographic | |
| Age at death, mean (SD) y | 89.3 (6.7) |
| Age at the last clinical visit, mean (SD) y | 88.4 (6.7) |
| Women, n (%) | 1,060 (67%) |
| Non-Hispanic white, n (%) | 1,498 (95%) |
| Education, mean (SD) years | 16.3 (3.7) |
| Clinical & genetic | |
| Any apolipoprotein ε4 | 403 (26%) |
| Hypertension, n (%) | 1,054 (67%) |
| Diabetes mellitus, n (%) | 342 (22%) |
| History of smoking, n (%) | 490 (31%) |
| Number of vascular risk factors,* median (Q1–Q3) | 1.0 (0–2) |
| Framingham risk score (FRS), mean (SD) | 19.3 (3.2) |
| FRS-age component score, median (Q1–Q3) | 15.0 (15–15) |
| FRS-Body mass index score, median (Q1–Q3) | 1.0 (0–0) |
| FRS-Systolic blood pressure score, median (Q1–Q3) | 3.0 (2–4) |
| FRS-Smoking score, median (Q1–Q3) | 0.0 (0–0) |
| FRS-Diabetes mellitus score, median (Q1–Q3) | 0.0 (0–0) |
| AD pathological indices | |
| NIA-Reagan AD Pathological diagnosis, n (%) | 1,020 (64%) |
| Square root of Aβ levels, mean (SD) | 1.5 (1.1) |
| Square root of tau levels, mean (SD) | 1.6 (1.3) |
Number of vascular risk factors is a summary statistic of the count of hypertension, diabetes mellitus, and smoking present in a participant (1 point for each, with a total score range 0– 3).
Association of the Framingham risk score with Aβ and tau
The FRS was not associated with a pathological diagnosis of AD (t(1583) = 0.30, p = 0.766), nor with the markers of Aβ (Spearman r = −0.00, p = 0.918) or tau (r = 0.01, p = 0.701). In linear regression analyses, FRS was not associated with Aβ (estimate = −0.005, SE = 0.009, p = 0.560) or tau levels (estimate = −0.017, SE = 0.010, p = 0.105) (Table 2). Then, we examined whether FRS modified the association of Aβ with tau. In a linear regression model including Aβ, FRS, and their interaction as the predictors, and tau as the outcome, the interaction term was significant and negative (estimate = −0.022, SE = 0.008, p = 0.009), suggesting that FRS modified the association of Aβ with tau in a way that the association was stronger with a lower FRS. These data suggest that in persons with fewer vascular risk factors the association of Aβ with tau was stronger.
Table 2.
Association of FRS with postmortem levels of Aβ and tau controlled for age at death, sex, and education
| Variable | Outcome Estimate (SE), p | |
|---|---|---|
| Aβ | Tau | |
| Age at death | 0.026 (0.004), <0.001 | 0.028 (0.005), <0.001 |
| Sex (women versus men) | 0.125 (0.060), 0.038 | 0.338 (0.072), <0.001 |
| Education | −0.018 (0.008), 0.019 | −0.019 (0.009), 0.040 |
| FRS | −0.005 (0.009), 0.560 | −0.017 (0.010), 0.105 |
From 2 separate linear regressions with Aβ and tau as the outcomes. The results indicate that FRS was not associated with levels of Aβ and tau. SE, standard error. Aβ and tau square root transformed.
To uncover if a particular FRS component(s) was (were) responsible for the interaction between FRS and Aβ in relation to tau pathology, we separately examined each of the five components of FRS. In each of the five linear regressions with tau levels as the outcome, one of the FRS components was examined together with Aβ and their interaction as the predictors. The SBP was the only FRS component showing an interaction (Table 3). This interaction was negative, indicating that the association of Aβ with tau was stronger at lower levels of SBP. To contextualize the effect size of the interaction, we compared its estimate (estimate = −0.022, SE = 0.008, p = 0.009) with the estimate of age at death (estimate = 0.014, SE = 0.005, p = 0.002) in their association with tau (Table 3, Model 1). The effect size associated with one score higher FRS-SBP was equivalent to the effect size associated with being 1.5 years younger in age.
Table 3.
Interaction of Framingham Risk Score (FRS) components with Aβ in separate linear regressions with tau levels as outcome
| Model | FRS term | Terms Estimate (SE), p | ||
|---|---|---|---|---|
| FRS component | Aβ | Interaction of FRS component with Aβ | ||
| 1 | FRS-Age | 0.358 (0.150), 0.017 | 1.413 (1.606), 0.379 | −0.056 (0.107), 0.602 |
| 2 | FRS-BMI | −0.018 (0.064), 0.785 | 0.607 (0.038), <0.001* | −0.057 (0.034), 0.096 |
| 3 | FRS-SBP | 0.030 (0.023), 0.187 | 0.654 (0.042), <0.001* | −0.033 (0.012), 0.009* |
| 4 | FRS-smoking | 0.068 (0.077), 0.376 | 0.576 (0.027), <0.001* | −0.050 (0.040), 0.211 |
| 5 | FRS-DM | 0.000 (0.029), 0.998 | 0.575 (0.029), <0.001* | −0.003 (0.015), 0.867 |
Using 5 separate linear regressions with terms for FRS components, Aβ, and their interactions as predictors and tau as the outcome controlled for age at death, sex, and education (except model 1 controlled for sex and education only). In each of the 5 models, one of the FRS components has been examined: age, body mass index (BMI), systolic blood pressure (SBP), smoking, and diabetes mellitus (DM). Cells’ parameters are estimates (SE), p-values. SE, standard error. Aβ and tau square root transformed.
Statistically significant at a Bonferroni corrected alpha of 0.01.
Because vascular risk factors increase stroke risk [14, 23], we examined whether FRS was associated with the odds of infarct in our sample. In a logistic regression model controlled for age at death, sex, and education, a 1-point higher FRS was associated with 7% higher odds of infarct of any size or location (OR = 1.07, 95%CI=1.04 1.10, p < 0.001). We next examined the association of FRS with gross infarcts versus microinfarcts. Higher FRS scores were associated with higher odds of gross infarcts (OR = 1.08, 95%CI=1.05 1.12, p < 0.001), but the association of FRS and microinfarcts was borderline (OR = 1.03, 95%CI=1.00 1.07, p = 0.054). In further analyses, we adjusted for infarcts of any size, gross infarcts, or microinfarcts in three separate groups of models that examined the association of FRS and its components, Aβ, and their interaction with tau (Supplementary Tables 2–4). We found that controlling for infarcts variables did not change the study findings: a) FRS and its components were not associated with tau; b) FRS and FRS-SBP component negatively modified the association of Aβ with tau.
Because a recent study using PET imaging found a positive interaction between FRS and Aβ on tau deposition in the inferior temporal cortex but not in the entorhinal cortex [12], we next examined for the interaction of FRS and Aβ in the inferior temporal cortex. Results showed no interaction of FRS with Aβ in the inferior temporal cortex, with tau levels in the inferior temporal cortex as the outcome (estimate = −0.021, SE = 0.013, p = 0.107).
Sensitivity analyses
We did two sensitivity analyses. First, we excluded participants (n = 280) whose last evaluation was done more than 1 year before death to shorten the time interval from the FRS data to autopsy. Exclusion of these participants did not change our findings that FRS was not related to the levels of Aβ and tau while FRS and its SBP components negatively modified the association of Aβ with tau (Supplementary Table 5). Second, we replaced the average of SBP and BMI measurements assessed across the study, which was presumed to be a closer surrogate of the cumulative burden of these risk factors, with the last visit measurements of the SBP and BMI, respectively, in the calculation of FRS. The new FRS was correlated with the original FRS (Spearman r = 0.66, p < 0.001). Like the original FRS, the new FRS was not associated with the Aβ and tau levels (Supplementary Table 6). However, in contrast to the results with the original FRS calculation, the new FRS and its SBP component did not modify the association of Aβ with tau (Supplementary Table 6). These findings suggest that a low level of vascular risk factors over years, rather than the level at the last visit prior to death, was associated with modifying the association of Aβ with tau.
Systolic blood pressure and the association of Aβ with tau levels
SBP was the only FRS component interacting with Aβ on tau. However, the FRS-SBP component is a function of SBP, the use of blood pressure medication, and sex. Therefore, in secondary analyses we examined the association of Aβ with tau at different levels of SBP. Following published guidelines on blood pressure classification [26], we classified the sample into four subsamples by their SBP levels: “<120 mmHg” (n = 253), “120–129 mmHg” (n = 395), “130–139 mmHg” (n = 437), and “≥140 mmHg” (n = 484). Then, we employed separate linear regressions in the four subsamples with tau levels as the outcome and Aβ levels as the predictor. In graphical inspection, the estimates of association between Aβ and tau were monotonically decreasing with increase of SBP except for the SBP group “over 140” (Fig. 1). For participants with low SBP (“<120”), the estimated association between Aβ and tau was 0.694 (Supplementary Table 7), equivalent to being 58 years older for every additional unit of Aβ. By contrast, for participants with SBP 130–139 the estimated association between Aβ and tau was 0.500 (Supplementary Table 7), equivalent to being 18 years older for every additional unit of Aβ.
Fig. 1.
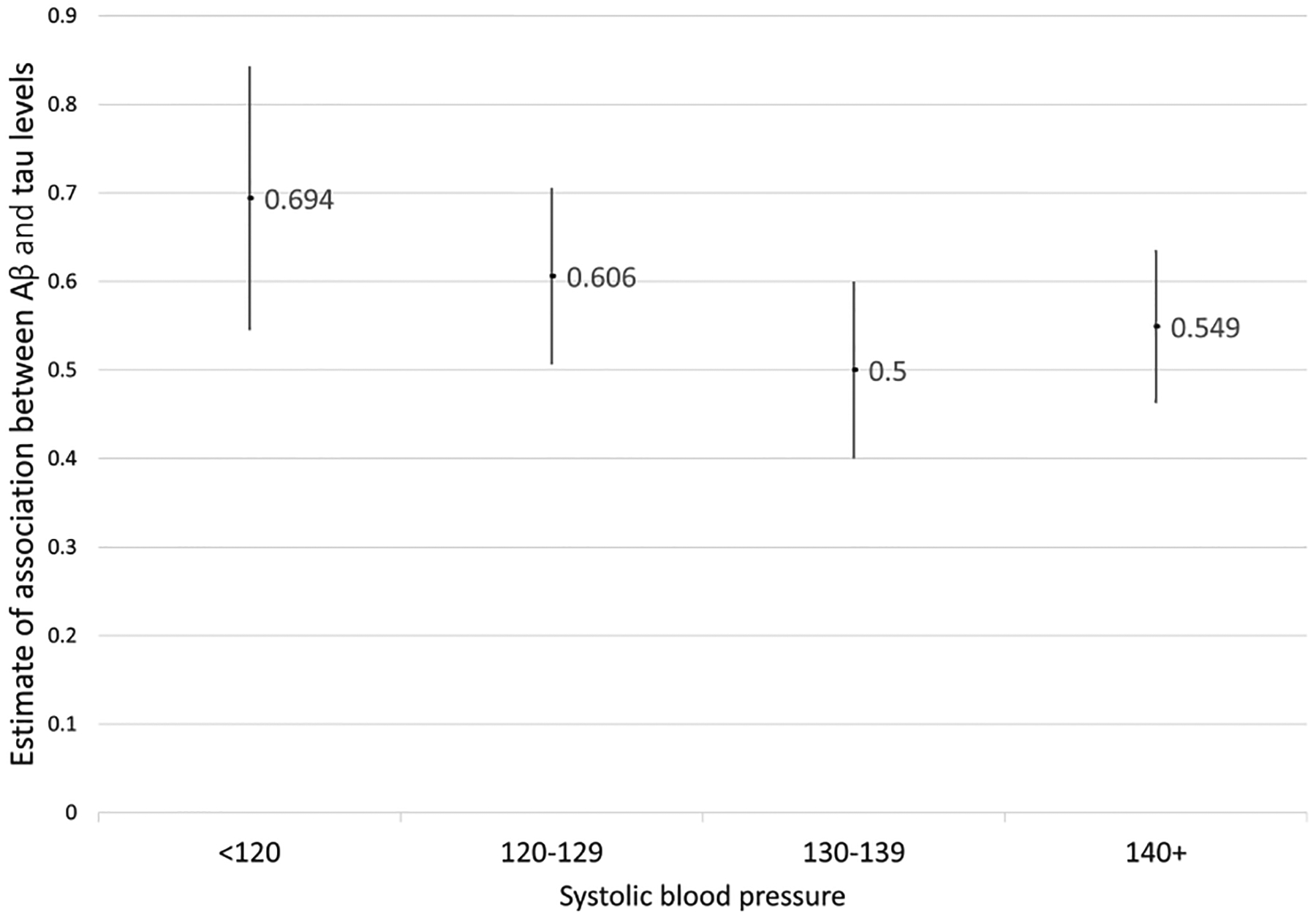
Association of Aβ with tau deposition using different systolic blood pressure categories. The estimates are derived from four linear regressions, including participants at four different systolic blood pressure categories. In each linear regression, tau was the outcome and Aβ was the predictor, and the model controlled for age at death, sex, and education. Graphical inspection indicates that the estimates of association between Aβ and tau are monotonically decreasing with increase of SBP except for the SBP group “over 140”. Aβ and tau square root transformed.
Diastolic blood pressure and the association of Aβ with tau levels
We examined the association of Aβ with tau at different levels of DBP rather than SBP. Following the guideline classification [26], we stratified the sample into 3 subsamples by their DBP levels: : “<80 mmHg” (n = 1, 358), “80–89 mmHg” (n = 193), and “≥90 mmHg” (n = 25). Because 86% of the participants were classified in one group with the 3 categories classification, we further stratified the data using 5 categories: “<60 mmHg” (n = 95), “60–69 mmHg” (n = 552), “70–79 mmHg” (n = 711), “80–89 mmHg” (n = 193), and “≥90 mmHg” (n = 25). In graphical inspection, the estimates of association between Aβ and tau were not changing monotonically across the 5 DBP categories (Supplementary Figure 1), in contrast to the monotonic decrease in the estimates seen across the SBP categories (Fig. 1).
DISCUSSION
Leveraging antemortem and postmortem data from nearly 1,600 deceased persons, we found that vascular risk factors, summarized as FRS, were not related to Aβ or tau levels. However, FRS, and more specifically its SBP component, showed a negative interaction with Aβ on tau levels, indicating that low levels of SBP in late-life were associated with higher Aβ related tau levels.
Mid-life vascular risk factors have consistently been found to be associated with late-life dementia. However, ambiguity exists regarding the association of late-life vascular risk factors with dementia. To clarify this ambiguity, examining the association of vascular risk factors with AD pathology is helpful. But, few autopsy studies have examined this association [13, 14, 27]. We extended the prior researches in three ways. First, we used FRS as a well-known composite vascular risk score to study the association with AD, rather than examining individual vascular risk factors like DM or hypertension. This strategy increased our power to simultaneously study different vascular risk factors in their association with AD pathology. Second, we examined not only the association of FRS with the AD markers but also the interaction of FRS with Aβ specifically, expanding on the factors that drive the association of Aβ with tau. Third, we leveraged data of approximately 1,600 deceased persons. This large sample size minimized the possibility for inadequate power, in the event of a finding that FRS was not associated with Aβ or tau, as was the case in this study.
Although FRS was found to modify the relationship of Aβ to tau in our study, we did not find an association between FRS and level of either Aβ or tau themselves, a finding in line with several previously published studies [13, 14, 27]. Vascular risk scores like FRS [13], and vascular risk factors like diabetes [27] and hypertension [14] have been found associated with vascular pathologies, including brain infarcts and cerebral vessel pathologies such as atherosclerosis and arteriolosclerosis. Our results which did not show an association between vascular risk factors and AD is in line with prior studies that did not find a synergism between vascular pathologies and AD pathology [24].
Studies using in vivo assessment of Aβ and tau have examined the association of vascular risk factors with AD biomarker levels [1, 8, 11, 28, 29]. The studies’ findings suggest that vascular risk factors were associated with higher levels of AD biomarkers when vascular risk factor assessment was done in midlife or late-midlife, ages younger than 65–70 years old. By contrast, no association was found between vascular risk factors and AD biomarkers when vascular risk factor assessment was done in old age, similar to this study’s age distribution. The age difference can also explain the differences between our study and a PET study [12] about the interaction of FRS and Aβ in their association with tau. We found a negative interaction while the PET study found a positive one indicating more tau per unit of Aβ at higher levels of FRS in the PET study [12]. Comparison of the participants’ ages of the two studies shows that the average age of our participants at the time of FRS calculation was 88.3 (SD = 6.7) years, while the PET study’s one was 73.5 (SD = 6.1), showing that our study participants were on average 15 years older. Another possibility for explaining the difference between the two studies is the fact that the amyloid and tau measured by PET may not provide the similar granularity that is obtained with IHC methods in autopsies, with the latter being the gold standard for estimating the sensitivity and specificity of the PET findings [30,31]. In a longitudinal study, the correlation coefficient between Aβ measured by PET one year before death and by immunohistochemistry in autopsies was 0.79 [31], indicating that imaging and autopsy findings on measures of AD indices and their related covariates may be different.
The finding that no association existed between levels of vascular risk score and AD pathology indices is in line with the complex relation of vascular risk factors and AD clinical phenotype (impaired cognition or dementia) throughout the life cycle, including in late-life [32–35]. Using the Rush Alzheimer’s Disease Center cohorts’ data, investigators showed that baseline late-life levels of SBP and DBP were not associated with the risk of AD dementia or with the rate of cognitive decline [32]. DM was associated with lower levels of semantic memory, not with levels of other cognitive systems or global cognition [33], and while not related to global cognitive decline, was related to decline in perceptual speed [34]. Lower, not higher, levels of BMI were associated with faster cognitive decline [35]. These data suggest that the high burden of vascular risk factors in late-life may not be a risk factor for cognitive decline. However, it is plausible that high vascular risk burden, even higher levels of BP in late life, is associated with cognitive impairment in persons with cerebrovascular pathologies, including atherosclerosis and arteriolosclerosis, and that this association is independent of AD pathological changes [36, 37].
We found that FRS modified the association of Aβ with tau. Further evaluation showed that the SBP component of FRS was the only component showing an interaction with Aβ in the association with tau. The negative interaction was in line with monotonic decrease in the estimates of association between Aβ and tau seen with increase of SBP until SBP ≥ 140 mmhg. We interpret these findings as suggesting that per one unit higher Aβ level more tau was seen at low SBP levels. Among several possible underlying mechanisms, we hypothesize that hypoperfusion may play a role in how lower SBP levels may modify the association between Aβ and tau. Indeed, animal models have shown increased levels of Aβ and phosphorylated tau following hypoperfusion [38, 39]. Furthermore, increased levels of peroxidation products [40], impaired mitochondrial functions [40], and changes in the microRNA transcription [41] are possible pathways via which hypoperfusion may result in higher levels of AD pathology indices. As tau is the AD pathological index that drives cognitive decline in late-life [42], we also hypothesize more dementia cases seen at low SBP levels. In fact, a prior cross-sectional study on Chicagoans older than 65 years found that odds of AD dementia was more than twice in individuals with “SBP<130 mmhg” compared with the reference group of “130–139 mmhg” [43]. However, further studies are needed to disentangle the complex relationship between SBP and cognition and cognitive decline in late-life.
In a sensitivity analysis, we replaced the average of SBP and BMI across years of measurements with the last measurement of SBP and BMI in the calculation of FRS. The new FRS and its SBP component did not modify the association of Aβ with tau. Therefore, a low level of vascular risk factors over years, rather than the level at the last visit prior to death, was associated with a higher level of tau per one unit higher Aβ. We hypothesize that the last measurements of SBP and BMI were influenced by a terminal decline phenomenon that is distinct from late-life pathological changes of brain. In prior studies, our group showed that terminal decline in cognition started on average 3.7 years before death [44] and was seen in one-third of decedents after controlling for dementia-related pathological changes [45]. Although terminal decline has also been reported for other metrics, including well-being [46], we did not find any reported studies examining terminal decline in vascular risk factors. As longitudinal changes of vascular risk factors including BMI does not follow a linear model [47], we need more sophisticated analyses with large sample sizes to test our hypothesis regarding terminal decline in vascular risk factors, and the differential association of AD pathology indices with FRS in the terminal decline period compared with the FRS in years beforehand.
The study has strengths. It leveraged data of a large number of deceased persons who were prospectively evaluated during life, and came to an autopsy which yielded neuropathological data derived from uniform protocols. Trained staff who collected clinical or pathological data were blinded to other data, reducing the potential for bias. However, the study has limitations too, including limitations to the generalizability of the results. Although only less than 10% of the ROS and the MAP participants were lost to follow up, the current study’s analytic sample enrolled in the ROS or MAP studies at a younger age and were more likely to be women, compared with participants not included in the current study. Results of this study will need to be replicated before interpretation about generalization of the findings can be made. The average age of participants at the time of death was 89 years old, making the results not generalizable to younger people. We had only 41 participants with average SBP more than 160 mmhg, and the study results cannot adequately inform about higher levels of SBP. Finally, the study participants were mainly Caucasians with high levels of education.
Supplementary Material
ACKNOWLEDGMENTS
We sincerely thank all the participants of the Religious Orders Study and the Rush Memory and Aging Project. Also, we appreciate the work of Traci Colvin, MPH, Tracey Nowakowski, MA, and Karen Skish, MS, for study coordination; Alysha Hodges, MS, for statistical programming; and the numerous other staff of the Rush Alzheimer’s Disease Center.
The study was supported by National Institute of Health grants P30AG10161, R01AG15819, R01 AG17917, R01NS84965, and RF1AG59621.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0412r2).
Footnotes
SUPPLEMENTARY MATERIAL
The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-200412.
REFERENCES
- [1].Gottesman RF, Albert MS, Alonso A, Coker LH, Coresh J, Davis SM, Deal JA, McKhann GM, Mosley TH, Sharrett AR, Schneider ALC, Windham BG, Wruck LM, Knopman DS (2017) Associations between midlife vascular risk factors and 25-year incident dementia in the Atherosclerosis Risk in Communities (ARIC) Cohort. JAMA Neurol 74, 1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Harrison SL, Ding J, Tang EYH, Siervo M, Robinson L, Jagger C, Stephan BCM (2014) Cardiovascular disease risk models and longitudinal changes in cognition: A systematic review. PLoS One 9, e114431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kivipelto M, Ngandu T, Fratiglioni L, Viitanen M, Kåreholt I, Winblad B, Helkala E-L, Tuomilehto J, Soininen H, Nissinen A (2005) Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch Neurol 62, 1556–1560. [DOI] [PubMed] [Google Scholar]
- [4].Lane CA, Barnes J, Nicholas JM, Sudre CH, Cash DM, Parker TD, Malone IB, Lu K, James S-N, Keshavan A, Murray-Smith H, Wong A, Buchanan SM, Keuss SE, Gordon E, Coath W, Barnes A, Dickson J, Modat M, Thomas D, Crutch SJ, Hardy R, Richards M, Fox NC, Schott JM (2019) Associations between blood pressure across adulthood and late-life brain structure and pathology in the neuroscience substudy of the 1946 British birth cohort (Insight 46): An epidemiological study. Lancet Neurol 18, 942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].McGrath ER, Beiser AS, DeCarli C, Plourde KL, Vasan RS, Greenberg SM, Seshadri S (2017) Blood pressure from mid- to late life and risk of incident dementia. Neurology 89, 2447–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Corrada MM, Hayden KM, Paganini-Hill A, Bullain SS, DeMoss J, Aguirre C, Brookmeyer R, Kawas CH (2017) Age of onset of hypertension and risk of dementia in the oldest-old: The 90+ Study. Alzheimers Dement 13, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gottesman RF, Schneider ALC, Zhou Y, Coresh J, Green E, Gupta N, Knopman DS, Mintz A, Rahmim A, Sharrett AR, Wagenknecht LE, Wong DF, Mosley TH (2017) Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA 317, 1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nation DA, Edland SD, Bondi MW, Salmon DP, Delano-Wood L, Peskind ER, Quinn JF, Galasko DR (2013) Pulse pressure is associated with Alzheimer biomarkers in cognitively normal older adults. Neurology 81, 2024–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Reed BR, Marchant NL, Jagust WJ, DeCarli CC, Mack W, Chui HC (2012) Coronary risk correlates with cerebral amyloid deposition. Neurobiol Aging 33, 1979–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Lowe VJ, Graff-Radford J, Roberts RO, Mielke MM, Machulda MM, Petersen RC, Jack CR (2017) Age, vascular health, and Alzheimer disease biomarkers in an elderly sample: Vascular Health and AD. Ann Neurol 82, 706–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rabin JS, Schultz AP, Hedden T, Viswanathan A, Marshall GA, Kilpatrick E, Klein H, Buckley RF, Yang H-S, Properzi M, Rao V, Kirn DR, Papp KV, Rentz DM, Johnson KA, Sperling RA, Chhatwal JP (2018) Interactive associations of vascular risk and β-amyloid burden with cognitive decline in clinically normal elderly individuals: Findings from the Harvard Aging Brain Study. JAMA Neurol 75, 1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rabin JS, Yang H-S, Schultz AP, Hanseeuw BJ, Hedden T, Viswanathan A, Gatchel JR, Marshall GA, Kilpatrick E, Klein H, Rao V, Buckley RF, Yau W-YW, Kirn DR, Rentz DM, Johnson KA, Sperling RA, Chhatwal JP (2019) Vascular risk and β-amyloid are synergistically associated with cortical tau: Vascular risk, Aβ, and tau. Ann Neurol 85, 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Conner SC, Pase MP, Carneiro H, Raman MR, McKee AC, Alvarez VE, Walker JM, Satizabal CL, Himali JJ, Stein TD, Beiser A, Seshadri S (2019) Mid-life and late-life vascular risk factor burden and neuropathology in old age. Ann Clin Transl Neurol 6, 2403–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Arvanitakis Z, Capuano AW, Lamar M, Shah RC, Barnes LL, Bennett DA, Schneider JA (2018) Late-life blood pressure association with cerebrovascular and Alzheimer disease pathology. Neurology 91, e517–e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA (2018) Religious Orders Study and Rush Memory and Aging Project. J Alzheimers Dis 64, S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Oveisgharan S, Arvanitakis Z, Yu L, Farfel J, Schneider JA, Bennett DA (2018) Sex differences in Alzheimer’s disease and common neuropathologies of aging. Acta Neuropathol (Berl) 136, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Oveisgharan S, Yu L, Dawe RJ, Bennett DA, Buchman AS (2020) Total daily physical activity and the risk of parkinsonism in community-dwelling older adults. J Gerontol Biol Sci Med Sci. 75, 702–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Medi-Span: Master drug data base documentation manual.
- [19].D’Agostino RB, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM, Kannel WB (2008) General cardiovascular risk profile for use in primary care: The Framingham Heart Study. Circulation 117, 743–753. [DOI] [PubMed] [Google Scholar]
- [20].Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE (2004) Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol 61, 378–384. [DOI] [PubMed] [Google Scholar]
- [21].Bennett DA, Wilson RS, Schneider JA, Evans DA, Aggarwal NT, Arnold SE, Cochran EJ, Berry-Kravis E, Bienias JL (2003) Apolipoprotein E epsilon4 allele, AD pathology, and the clinical expression of Alzheimer’s disease. Neurology 60, 246–252. [DOI] [PubMed] [Google Scholar]
- [22].Hyman BT, Trojanowski JQ (1997) Editorial on Consensus Recommendations for the Postmortem Diagnosis of Alzheimer Disease from the National Institute on Aging and the Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer Disease: J Neuropathol Exp Neurol 56, 1095–1097. [DOI] [PubMed] [Google Scholar]
- [23].O’Donnell MJ, Chin SL, Rangarajan S, Xavier D, Liu L, Zhang H, Rao-Melacini P, Zhang X, Pais P, Agapay S, Lopez-Jaramillo P, Damasceno A, Langhorne P, McQueen MJ, Rosengren A, Dehghan M, Hankey GJ, Dans AL, Elsayed A, Avezum A, Mondo C, Diener HC, Ryglewicz D, Czlonkowska A, Pogosova N, Weimar C, Iqbal R, Diaz R, Yusoff K, Yusufali A, Oguz A, Wang X, Penaherrera E, Lanas F, Ogah OS, Ogunniyi A, Iversen HK, Malaga G, Rumboldt Z, Oveisgharan S, Al Hussain F, Magazi D, Nilanont Y, Ferguson J, Pare G, Yusuf S (2016) Global and regional effects of potentially modifiable risk factors associated with acute stroke in 32 countries (INTERSTROKE): A case-control study. Lancet 388, 761–75. [DOI] [PubMed] [Google Scholar]
- [24].Schneider JA, Wilson RS, Bienias JL, Evans DA, Bennett DA (2004) Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology 62, 1148–1155. [DOI] [PubMed] [Google Scholar]
- [25].Schneider JA, Wilson RS, Cochran EJ, Bienias JL, Arnold SE, Evans DA, Bennett DA (2003) Relation of cerebral infarctions to dementia and cognitive function in older persons. Neurology 60, 1082–1088. [DOI] [PubMed] [Google Scholar]
- [26].Whelton PK, Carey RM, Aronow WS, Casey DE, Collins KJ, Dennison Himmelfarb C, DePalma SM, Gidding S, Jamerson KA, Jones DW, MacLaughlin EJ, Muntner P, Ovbiagele B, Smith SC, Spencer CC, Stafford RS, Taler SJ, Thomas RJ, Williams KA, Williamson JD, Wright JT (2018) 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 71, e13–e115. [DOI] [PubMed] [Google Scholar]
- [27].Abner EL, Nelson PT, Kryscio RJ, Schmitt FA, Fardo DW, Woltjer RL, Cairns NJ, Yu L, Dodge HH, Xiong C, Masaki K, Tyas SL, Bennett DA, Schneider JA, Arvanitakis Z (2016) Diabetes is associated with cerebrovascular but not Alzheimer’s disease neuropathology. Alzheimers Dement 12, 882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Köbe T, Gonneaud J, Pichet Binette A, Meyer P-F, McSweeney M, Rosa-Neto P, Breitner JCS, Poirier J, Villeneuve S, for the Presymptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease (PREVENT-AD) Research Group (2020) Association of vascular risk factors with β-amyloid peptide and tau burdens in cognitively unimpaired individuals and its interaction with vascular medication use. JAMA Netw Open 3, e1920780. [DOI] [PubMed] [Google Scholar]
- [29].Luchsinger JA, Palta P, Rippon B, Sherwood G, Soto L, Ceballos F, Laing K, Igwe K, He H, Razlighi Q, Teresi J, Moreno H, Brickman AM (2020) Pre-diabetes, but not type 2 diabetes, is related to brain amyloid in late middle-age. J Alzheimers Dis 75, 1241–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Clark CM (2011) Use of Florbetapir-PET for imaging β-amyloid pathology. JAMA 305, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Clark CM, Pontecorvo MJ, Beach TG, Bedell BJ, Coleman RE, Doraiswamy PM, Fleisher AS, Reiman EM, Sabbagh MN, Sadowsky CH, Schneider JA, Arora A, Carpenter AP, Flitter ML, Joshi AD, Krautkramer MJ, Lu M, Mintun MA, Skovronsky DM (2012) Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: A prospective cohort study. Lancet Neurol 11, 669–678. [DOI] [PubMed] [Google Scholar]
- [32].Shah RC, Wilson RS, Bienias JL, Arvanitakis Z, Evans DA, Bennett DA (2006) Relation of blood pressure to risk of incident Alzheimer’s disease and change in global cognitive function in older persons. Neuroepidemiology 26, 30–36. [DOI] [PubMed] [Google Scholar]
- [33].Arvanitakis Z, Bennett DA, Wilson RS, Barnes LL (2010) Diabetes and cognitive systems in older black and white persons. Alzheimer Dis Assoc Disord 24, 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA (2004) Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol 61, 661. [DOI] [PubMed] [Google Scholar]
- [35].Arvanitakis Z, Capuano AW, Bennett DA, Barnes LL (2018) Body mass index and decline in cognitive function in older black and white persons. J Gerontol Ser A 73, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA (2016) Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: A cross-sectional study. Lancet Neurol 15, 934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Oveisgharan S, Hachinski V (2010) Hypertension, executive dysfunction, and progression to dementia: The Canadian Study of Health and Aging. Arch Neurol 67, 187–192. [DOI] [PubMed] [Google Scholar]
- [38].Qiu L, Ng G, Tan EK, Liao P, Kandiah N, Zeng L (2016) Chronic cerebral hypoperfusion enhances Tau hyperphosphorylation and reduces autophagy in Alzheimer’s disease mice. Sci Rep 6, 23964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Park J-H, Hong J-H, Lee S-W, Ji HD, Jung J-A, Yoon K-W, Lee J-I, Won KS, Song B-I, Kim HW (2019) The effect of chronic cerebral hypoperfusion on the pathology of Alzheimer’s disease: A positron emission tomography study in rats. Sci Rep 9, 14102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Feng T, Yamashita T, Zhai Y, Shang J, Nakano Y, Morihara R, Fukui Y, Hishikawa N, Ohta Y, Abe K (2018) Chronic cerebral hypoperfusion accelerates Alzheimer’s disease pathology with the change of mitochondrial fission and fusion proteins expression in a novel mouse model. Brain Res 1696, 63–70. [DOI] [PubMed] [Google Scholar]
- [41].Sun L-H, Ban T, Liu C-D, Chen Q-X, Wang X, Yan M-L, Hu X-L, Su X-L, Bao Y-N, Sun L-L, Zhao L-J, Pei S-C, Jiang X-M, Zong D-K, Ai J (2015) Activation of Cdk5/p25 and tau phosphorylation following chronic brain hypoperfusion in rats involves microRNA-195 down-regulation. J Neurochem 134, 1139–1151. [DOI] [PubMed] [Google Scholar]
- [42].Yu L, Boyle PA, Leurgans S, Schneider JA, Bennett DA (2014) Disentangling the effects of age and APOE on neuropathology and late life cognitive decline. Neurobiol Aging 35, 819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Morris MC, Scherr PA, Hebert LE, Bennett DA, Wilson RS, Glynn RJ, Evans DA (2002) Association between blood pressure and cognitive function in a biracial community population of older persons. Neuroepidemiology 21, 123–130. [DOI] [PubMed] [Google Scholar]
- [44].Wilson RS, Yu L, Leurgans SE, Bennett DA, Boyle PA (2020) Proportion of cognitive loss attributable to terminal decline. Neurology 94, e42–e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wilson RS, Wang T, Yu L, Bennett DA, Boyle PA (2020) Normative cognitive decline in old age. Ann Neurol 87, 816–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gerstorf D, Hoppmann CA, Löckenhoff CE, Infurna FJ, Schupp J, Wagner GG, Ram N (2016) Terminal decline in well-being: The role of social orientation. Psychol Aging 31, 149–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Capuano AW, Wilson RS, Leurgans SE, Dawson JD, Bennett DA, Hedeker D (2018) Sigmoidal mixed models for longitudinal data. Stat Methods Med Res 27, 863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.