

Abstract

The adenosine A2B receptor (A2BAR) belongs to the rhodopsin-like G protein-coupled receptor (GPCR) family. It is upregulated under hypoxic conditions, in inflammation and cancer. Previous studies indicated the coupling of the A2BAR to different G proteins, mainly Gs, but in some cases Gq/11 or Gi, depending on the cell type. We have now utilized novel technologies, (i) heterologous expression of individual members of the Gαq/11 protein family (Gαq, Gα11, Gα14, and Gα15) in Gαq/11 knockout cells, and (ii) the TRUPATH platform, allowing the direct observation of Gα protein activation for each of the Gα subunits by bioluminescence resonance energy transfer (BRET) measurements. Three structurally diverse A2BAR agonists were studied: the cognate agonist adenosine, its metabolically stable analog NECA, and the non-nucleosidic partial agonist BAY 60-6583. Adenosine and NECA activated most members of all four Gα protein families (Gαs, Gαq/11, Gαi, and Gα12/13). Significant differences in potencies and efficacies were observed; the highest efficacies were determined at the Gα15, Gαs, and Gα12 proteins, and for NECA additionally at the Gαi2 protein. In contrast, the partial agonist BAY 60-6583 only activated Gα15, Gαs, and Gα12 proteins. Adenosine deaminase, an allosteric modulator of ARs, selectively increased the potency and efficacy of NECA and BAY 60-6583 at the Gα15 protein, while it had no effect or decreased efficacy at the other Gα proteins. We conclude that the A2BAR is preferably coupled to the Gα15, Gαs, and Gα12 proteins. Upon upregulation of receptor or Gα protein expression, coupling to further Gα proteins likely occurs. Importantly, different agonists can display different activation profiles.
Keywords: adenosine, BAY 60-6583, G protein coupling, Gq/Gs/Gi/G12/13 proteins, HEK293 cells, NECA
The adenosine A2B receptor (A2BAR) belongs to the α-branch of rhodopsin-like G protein-coupled receptors (GPCRs). Four different subtypes exist, designated A1-, A2A-, A2B-, and A3ARs. The alkaloid caffeine is a non-selective AR antagonist employed to improve lung function of pre-term babies and for pain management in combination with analgesics.2,3 Moreover, the A2A-selective antagonist istradefylline is used for the treatment of Parkinson’s disease,4,5 while the A2A-selective agonist regadenoson and the cognate agonist adenosine are employed for cardiac imaging to induce vasodilation.6 Moreover, adenosine is therapeutically applied to treat arrhythmia in paroxysmal supraventricular tachycardia.1 All AR subtypes constitute promising drug targets, especially in the context of inflammation, immunity, and cancer.2,3,7−12 The A2BAR is mostly expressed in low density and only activated by relatively high, micromolar concentrations of adenosine, which are typically only present under pathological, e.g., inflammatory and hypoxic, conditions.13,14 There are exceptions: e.g., some epithelial cells, particularly in the gut, display rather high expression levels of the A2BAR.15 Moreover, A2BAR expression is upregulated in inflamed tissues and on many cancer cells.16−18 Therefore, A2BARs have significant potential as future drug targets for a range of diseases. Agonists have, e.g., been proposed for the treatment of stroke, obesity-induced diabetes, atherosclerosis, and wound healing, while antagonists have shown potential for treating inflammatory diseases (e.g., asthma, pulmonary and liver fibrosis, inflammatory bowel disease, multiple sclerosis), pain, cancer, and infections.3,12 On the other hand, anti-inflammatory effects induced by A2BAR activation have also been described.14
Agonists on GPCRs trigger intracellular signaling cascades by activation of heterotrimeric guanine nucleotide-binding proteins (G proteins), consisting of α-, β-, and γ-subunits. The Gα-subunits are crucial for the activation of various second messenger systems. They are subdivided into four families: Gαs, Gαi/o, Gαq/11, and Gα12/13.19,20 During G protein activation, the helical and the Ras-like domains of the Gα-subunit separate from each other, thereby allowing the dissociation of the bound guanosine diphosphate (GDP), which is replaced by guanosine triphosphate (GTP).21 The GTP-bound Gα-subunit then dissociates from the associated Gβγ-dimer and subsequently interacts with its effector proteins. Depending on the Gα protein subunit, specific effects are triggered in the cell, e.g., stimulation of adenylate cyclase (AC) by Gαs proteins, inhibition of AC by Gαi/o proteins, calcium mobilization by Gαq/11 proteins, or activation of Rho guanine exchange factors (Rho-GEFs) by Gα12/13 proteins.20,22
In the past, it was challenging to investigate the activation of specific Gα protein subunits by GPCR subtypes and their agonists. Recent technological advances, namely the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 technology,23 and especially the development of novel biosensors for measuring G protein activation, have provided tools to unambiguously investigate the activation of individual Gα protein subunits. Human embryonic kidney (HEK) 293 cells depleted of Gα proteins by CRISPR-Cas9 knockout followed by heterologous expression of individual Gα protein subunits have enabled specific studies.24−28 BRET sensors, e.g., the TRUPATH assay and similar previously described biosensors,29,30 allow the measurement of BRET ratios between renilla luciferase-8 (RLuc8)-fused Gα protein subunits and a green fluorescent protein 2 (GFP2)-tagged Gγ subunit. This ratio decreases upon G protein activation due to the dissociation of the α- and β,γ-subunits, resulting in an increased distance between BRET donor and acceptor.31 Further recently established methods for direct measurement of G protein activation include (i) split-luciferase assays (where Gα and, e.g., Gγ proteins are labeled by luciferase fragments that dissociate upon receptor activation, resulting in a decrease in luminescence),32−35 (ii) effector-membrane translocator assays (EMTAs, in which luciferase-tagged effectors interact exclusively with GTP-bound Gα subunits and are thereby brought into close proximity to a membrane-anchored BRET acceptor),36,37 and transforming growth factor-α (TGF-α) shedding assays (in which Gαq/11 protein activation results in cleavage and release of a soluble alkaline phosphatase (AP) fragment from a membrane-bound TGF-α-AP fusion protein by the metalloprotease ADAM17).34,38
The A2BAR was reported to couple to Gαs and Gαq proteins; however Gαq protein coupling has only been observed in some of the investigated cell types,39−41 and the reason for this has remained unclear. Few individual publications indicated additional coupling to Gαi/o proteins.40,42,43 Thus, the A2BAR appears to be endowed with an amazing, and at the same time confusing, promiscuity.40,42 In the present study, we utilized novel techniques, that only recently became accessible, to monitor the activation of individual Gα protein subunits by the A2BAR upon stimulation with structurally diverse agonists—namely the endogenous agonist adenosine, its metabolically stable analog NECA, and the A2B-selective partial agonist BAY 60-658339 (for structures see Figure S1A). Our first approach was to stably express each Gαq/11 protein family member (Gαq, Gα11, Gα14, and Gα15) in Gαq/11-deficient HEK293 cells (HEK293-ΔGαq/11) that natively express a low level of the human A2BAR40,44 and then directly measure intracellular calcium mobilization (see Figure S1B). Next, we employed the BRET-based biosensor platform TRUPATH31 to probe A2BAR-induced activation of virtually each of the Gα protein subunits, directly at the G protein level (see Figure S1C). We discovered that the A2BAR is preferably coupled to Gα15, Gαs, and Gα12 proteins. While the full A2BAR agonists activated almost all Gα protein subunits, their potencies and efficacies were significantly different. Efficacy in particular appears to play a decisive role for the resulting physiological and pharmacological effects.
Results
Calcium Mobilization Studies
Signaling via the Gq/11 protein family was initially studied by calcium mobilization assays in HEK293 cells, which natively express the A2BAR.40,44 In previously published experiments, Hinz et al.39 and Gao et al.40 demonstrated calcium mobilization upon A2BAR stimulation with the adenosine analog NECA, while the A2BAR-selective partial agonist BAY 60-6583 had not induced calcium mobilization in those studies. In order to investigate this discrepancy in more detail, we generated stable (polyclonal) cell lines that exclusively expressed a single Gα protein subunit of the Gαq protein family, either Gαq, Gα11, Gα14, or Gα15, by retroviral transfection of HEK293-ΔGαq/11 cells, resulting in similar expression levels for Gαq, Gα11, and Gα14 proteins (the expression level of Gα15 could not be quantified on a protein level).25 These cells were subsequently used to identify the Gα protein(s) involved in A2BAR-induced intracellular calcium release. Our aim was to examine (i) whether the A2BAR favors signaling via a specific Gαq subfamily member and/or (ii) if structurally diverse A2BAR agonists would result in specific or selective activation of certain Gα protein subunits and thus show biased signaling. Carbachol (CCh)-mediated activation of the muscarinic M3 receptor (M3R), which is endogenously expressed in HEK293 cells, was used as a control (for results see Figure 1A–E and Table S1).
Figure 1.

Concentration–response curves of NECA, adenosine (Ado), BAY 60-6583 (BAY), and carbachol (CCh) at HEK293 ΔGαq/11 cells recombinantly expressing (A) Gαq, (B) Gα11, (C) Gα14, or (D) Gα15 proteins. Data points are from at least three separate experiments, each performed in triplicates. (E) Heatmaps of A2BAR-mediated calcium signaling depicting potency (pEC50, left) and efficacy (% of CCh signal at the highest tested concentration, right).
In HEK293-ΔGαq/11 cells not expressing any Gαq/11 family protein, calcium mobilization was neither induced by adenosine nor by NECA (see below). CCh exhibited a preference for signaling via Gαq and Gα11 proteins, both of which were activated by submicromolar concentrations of the muscarinic receptor agonist. Higher CCh concentrations (20- to 30-fold) were required to activate calcium mobilization in HEK-Gα14 and HEK-Gα15 cells.
The physiological agonist adenosine and its closely related derivative NECA displayed similar activity showing a preference for Gα15-induced intracellular calcium release (pEC50 adenosine: 5.52; NECA, 5.79) (see Figure 1A,D and Table S1) followed by Gαq-mediated calcium mobilization (pEC50 adenosine: 4.84, NECA: 4.37). The efficacy also appeared to be higher for Gα15- as compared to Gαq-induced calcium release. Only minor or negligible activation of calcium mobilization via Gα11 or Gα14 proteins was observed (see Figure 1B,C). These results indicate that adenosine preferably activates Gα15 within the Gαq/11 protein family. Interestingly, the A2B-selective non-nucleosidic partial agonist BAY 60-6583 did not activate any of the Gαq/11 family proteins in these calcium mobilization experiments (fluorescence monitored at the highest tested concentration of BAY 60-6583 did not significantly differ from basal values).
In order to confirm that the observed effects were actually due to the activation of A2BARs endogenously expressed in the HEK293 cells, we preincubated the cells with AR subtype-selective antagonists. Calcium mobilization induced by NECA was almost completely blocked by the A2BAR-antagonist PSB-603 (Figure 2A), but not by selective A1-, A2A-, and A3AR antagonists. The A1AR-selective antagonist PSB-36 only inhibited calcium mobilization moderately at a concentration of 100 nM, but not at 10 nM, consistent with its affinity for the A2BAR (Ki A2B, 187 nM; Ki A1 0.7 nM).45 The A2AAR-selective agonist CGS-21680 did not induce calcium mobilization (Figure 2B), nor did adenosine or NECA in Gαq/11 knockout cells (Figure 2C).
Figure 2.

NECA-induced calcium mobilization in HEK293-Gα15 cells is inhibited by A2B-selective AR antagonists. (A) NECA-induced calcium mobilization (8.8 μM NECA corresponding to its EC80 value) in the presence of the AR antagonists PSB-36 (A1AR), MSX-2 (A2AAR), PSB-603 (A2BAR), PSB-10, and PSB-11 (both A3AR) in HEK293-Gα15 cells. Cells were preincubated with the indicated concentrations of AR antagonist for 30 min before measurement. Values were normalized to controls in the absence of antagonist (100%) and in the absence of agonist (0%). Three (PSB-36) or four (all other) independent experiments were performed in triplicates; bars represent means ± SEM. (B) The A2AAR-selective agonist CGS-21680 did not induce calcium mobilization in HEK293-Gα15 cells at concentrations of up to 30 μM. (C) Adenosine (Ado) and NECA did not induce calcium mobilization in HEK293-ΔGαq/11 cells.
Since the observed effects downstream of A2BAR-mediated Gα protein activation might be modulated by unknown factors, we next measured direct activation of Gα protein subunits.
Direct Measurement of G Protein Activation
G protein activation by A2BAR agonists was monitored using the TRUPATH assay platform, a BRET2-based method that allows to observe the dissociation of Gα from Gβγ protein subunits.31 Biosensors are available for all Gα protein subunits with the exception of transducins, Gα14, and Gαolf. For each biosensor, the RLuc8-Gα subunit was paired with an optimized Gβγ-GFP2 combination to yield maximum BRET2 ratio shifts upon GPCR activation.31 The biosensors were transiently expressed in native HEK293 cells together with the receptor of interest, and the BRET ratio shifts in response to agonist stimulation were measured. To validate the assay, we expressed the Gαi1-, and the Gαq biosensor, respectively, together with a GPCR that is known to couple to the respective Gα protein subunits (Figure S2), namely the thromboxane receptor (TP) for the Gαq biosensor, and the A3AR for the Gαi1 biosensor. Transfected cells were activated by a full agonist of the respective receptor. The U46619-activated TP receptor elicited a maximum ΔBRET shift of −0.2. Using the Gαi1 biosensor, we measured a maximum ΔBRET shift of −0.23 for the NECA-activated A3AR. The Emax values for the TP receptor and the A3AR were nearly identical to the Emax values reported for model receptors (isoprotenerol-activated β2 adrenoceptor for the Gαs family, DAMGO-activated μ-opioid receptor for the Gαi/o family, neurotensin-activated neurotensin 1 receptor for the Gαq/11 and Gα12/13 families) in the original publication.31 To assess the efficacy of receptor–G protein coupling we therefore decided to compare Emax values determined in the present study for the A2BAR upon coupling to various Gα protein subunits to Emax values for the same subunits observed for full agonists with standard GPCRs reported in the original publication using the same procedures and conditions.31
We first investigated direct activation of the Gαq/11 family members induced by A2BAR agonists to enable comparison with our previous results from calcium mobilization assays. Next, we studied the A2BAR’s canonical effector, Gαs (short isoform Gαs-s). Endogenous A2BAR expression in HEK293 cells was found to be not sufficient to trigger measurable activation of Gαq, Gα11, and Gαs-s biosensors in this system (Figure 3A-D; similarly, carbachol, an agonist at the endogenously expressed M3R, failed to activate Gαq, Gα11, and Gα15 biosensors in the TRUPATH BRET2 assays, data not shown). Only the Gα15 biosensor was activated in these cells by adenosine and NECA in a concentration-dependent manner. BAY 60-6583 showed a very small effect typical of a weak partial agonist (Figure 3C).
Figure 3.

Concentration–response curves of the agonists adenosine, NECA and BAY 60-6583 at HEK293 cells transiently transfected with TRUPATH biosensors for the indicated G protein subunits. A-D. ΔBRET values measured in HEK293 cells with native A2BAR expression levels and overexpression of the indicated biosensors A. Gαq, B. Gα11, C. Gα15, or D. Gαs-s. E-H. ΔBRET values measured in HEK293 cells with overexpression of the A2BAR and the indicated biosensors Gαq (e), Gα11 (f), Gα15 (g), or Gαs-s (h) biosensors. Data points are presented as means ± SEM of three or more independent experiments; pEC50 values and efficacy values are listed in Tables S2 and S3 of Supporting Information.
Next, the A2BAR was (moderately) overexpressed in HEK293 cells as recommended by the TRUPATH assay protocol31 (100 ng pCDNA3.1-human ADORA2B per 106 cells, transient expression) (see Figure 3E-H; and Tables S2 and S3 in Supporting Information). Now, adenosine and NECA activated all four investigated Gα proteins, Gαq, Gα11, Gα15 and Gαs-s, in a concentration-dependent manner, while BAY 60-6583 activated Gα15 and Gαs-s only. NECA appeared to be significantly more potent than adenosine in all cases, while BAY 60-6583 was equipotent to adenosine at Gα15 and Gαs-s proteins, where it showed activation. At the Gαq protein, NECA was more efficacious than adenosine while at all other investigated Gα subunits both nucleosidic agonists displayed similar efficacy. BAY 60-6583 was as efficacious as adenosine at the Gα15 and Gαs-s subunit.
Since we had not observed any calcium mobilization induced by BAY 60-6583 in Gα15-expressing HEK293 cells, we wondered whether this was due to the low A2BAR expression level in this cell line combined with the partial agonistic properties of BAY 60-6583. Thus, we studied calcium mobilization in HEK293 cells overexpressing A2BARs by BAY 60-6583 (Figure 4). To this end, we transiently overexpressed the A2BAR using two different cDNA concentrations, and recorded concentration-dependent intracellular calcium release induced by adenosine, NECA, and BAY 60-6583. Adenosine showed an increase in efficacy with increasing amounts of cDNA (compared to the maximal effect of CCh) and a moderate increase in potency (Emax adenosine: 100 ng DNA/well, 76%; 1000 ng DNA/well, 119%; pEC50 adenosine: 100 ng DNA/well, 5.38; 1000 ng/well, 5.72; Figure 4A). NECA displayed a minor increase in efficacy and a similar potency with increasing cDNA concentrations (Emax NECA: 100 ng DNA/well, 111%; 1000 ng DNA/well, 122%; pEC50 NECA: 100 ng DNA/well, 6.72; 1000 ng/well, 6.37; Figure 4B). In HEK-Gα15 cells with overexpression of the A2BAR, BAY 60-6583 was able to induce calcium mobilization; both potency and efficacy of BAY 60-6583 increased with increasing cDNA amounts used for transfection (Emax: BAY 60-6583 100 ng DNA/well = 27%, 1000 ng/DNA well = 71%; pEC50 BAY 60-6583 100 ng DNA/well = 6.73, 100 ng/well = 6.95, Figure 4C). Thus, the partial agonist BAY 60-6583 can induce calcium mobilization via Gα15-activation in HEK cells, but requires high A2BAR expression levels.
Figure 4.

Calcium mobilization induced by A2BAR agonists in HEK293-Gα15 cells transiently overexpressing A2BARs. The human A2BAR was transiently overexpressed with either a low amount of cDNA (100 ng/well) or a high amount of cDNA (1000 ng/well). A2BAR activation by A. adenosine (Emax adenosine: 100 ng DNA/well, 76 ± 10%; 1000 ng DNA/well, 119 ± 5%; pEC50 adenosine: 100 ng DNA/well, 5.38 ± 0.19; 1000 ng/well, 5.72 ± 0.21), B. NECA (Emax NECA: 100 ng DNA/well, 111 ± 3%; 1000 ng DNA/well, 122 ± 7%; pEC50 NECA: 100 ng DNA/well, 6.72 ± 0.36; 1000 ng/well, 6.37 ± 0.11), and C. BAY 60-6583 (Emax BAY 60-6583:100 ng DNA/well, 27 ± 1%; 1000 ng/DNA well, 71 ± 17%; pEC50 BAY 60-6583:100 ng DNA/well, 6.73 ± 0.11; 100 ng/well, 6.95 ± 0.15), normalized to the effect of 100 μM CCh. Data are means ± SEM of three individual experiments performed in duplicates.
Effect of adenosine deaminase
The signaling molecule adenosine is ubiquitous and may be released by the cells or formed from ATP by ectonucleotidases.46,47 To avoid an interfering effect of adenosine potentially present in the HEK293 cell culture, we additionally performed TRUPATH assays with the agonists NECA and BAY 60-6583 in the presence of adenosine deaminase (ADA) which converts adenosine to inosine, but has no effect on NECA or BAY 60-6583. NECA-induced Gαq and Gα11 biosensor activation was not significantly different in terms of potency and efficacy in the presence and absence of ADA (Figure 5; Supp. Tables 2,3). At the Gα15 protein, the potency of NECA and BAY 60-6583 was enhanced by ADA (∼10-fold for NECA, ∼ 120-fold for BAY), while the efficacy was concomitantly increased by about 2-fold for BAY as well as for NECA. Potencies at the Gαs-s biosensor observed in the presence of ADA were again similar to those without ADA, but ADA reduced the efficacy of NECA- and BAY 60-6583-induced Gαs-s activation by approximately 2-fold (see Table S3). In summary, ADA selectively increased potency and/or efficacy of NECA and BAY 60-6583 in Gα15 protein activation, while it had no effect or decreased efficacy at the other Gα proteins.
Figure 5.
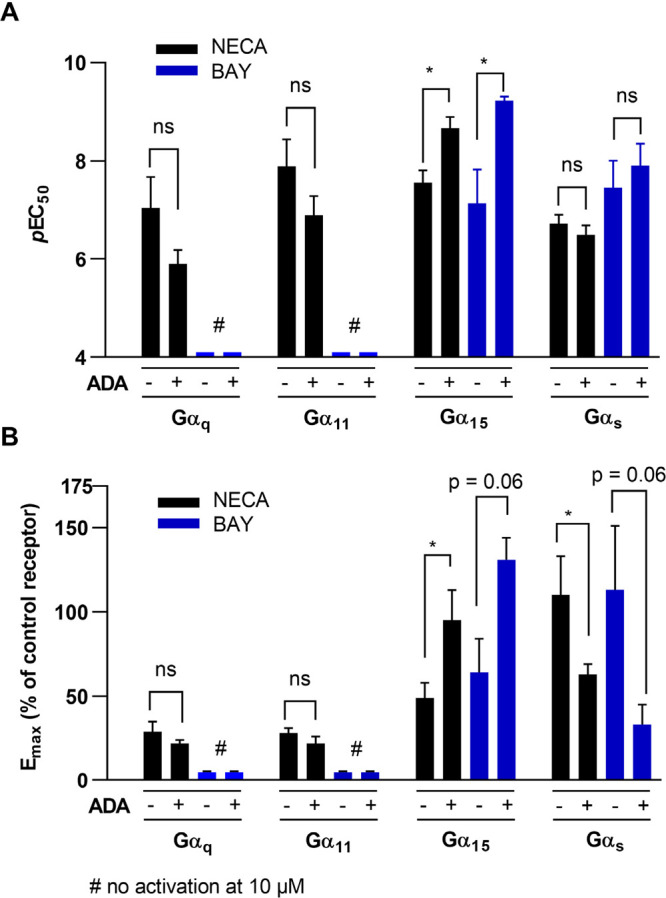
A. Potency and B. efficacy of NECA (black) or BAY 60-6583 (blue) for A2BAR stimulation determined in TRUPATH BRET2 assays using the Gαq, Gα11, Gα15, or the Gαs-s biosensor in the absence (−) or presence (+) of ADA. Data are presented as means ± SEM of three or more independent experiments; pEC50 and Emax values are summarized in Table S2 and S3. Statistical analysis was performed with multiple unpaired t-tests corrected for multiple comparisons by the Holm-Sidak-method (as implemented in GraphPad PRISM v. 8.0; ns – p > 0.05, * – p < 0.05, *** – p < 0.001).
The promiscuous A2BAR couples to all Gα protein families
Next, we investigated the potential coupling of the A2BAR to further, noncanonical Gα protein subunits, specifically to members of the Gα12/13 and the Gαi/o families (see Figure 6; pEC50 and Emax values are collected in Tables S2 and S3).
Figure 6.

ΔBRET curves of the A2BAR-agonists adenosine, NECA, and BAY 60-6583 at HEK293 cells transiently transfected with the A2BAR and the indicated TRUPATH biosensors: A. Gα12, B. Gα13, C. Gαi1, D. Gαi2, E. Gαi3, F. GαoA, G. GαoB, H. Gαz, I. Gαgust. Curves for NECA and BAY 60-6583 were determined in the presence of 2 U/ml ADA. Data points were obtained in 3–7 independent experiments, each performed with two technical replicates, and are represented as mean ± SEM. All pEC50 and Emax values are collected in Tables S2 and S3.
All three agonists were able to activate the Gα12 protein (Figure 6A). BAY 60-6583 acted as a partial agonist displaying ca. 36% efficacy relative to adenosine. Similar to the situation at Gα15 and Gαs-s proteins, BAY 60-6583 and NECA were much more potent than adenosine. At the Gα13 protein, adenosine and NECA displayed similar potency as at the Gα12 protein, but adenosine was less efficacious than NECA, and BAY 60-6583 was inactive under the employed conditions (Figure 6B).
In further experiments, the Gαi/o protein family was investigated. Again, both adenosine and NECA displayed concentration-dependent Gαi protein activation. NECA was about 2 orders of magnitude more potent than adenosine in activating the Gαi1–3, both agonists being similarly efficacious. In contrast, BAY 60-6583 did not activate any of the Gαi proteins (Figure 6C-E). Likewise, GαoA and GαoB biosensors were activated exclusively by adenosine and NECA, but not by BAY 60-6583; however, the potency of NECA was markedly lower at these Gα protein subunits than at Gαi1–3, while the potency of adenosine was similar in both cases (Figure 6F-G).
At the Gαz protein, a ubiquitously expressed, PTX-insensitive member of the Gαi/o-family, adenosine displayed a slightly higher potency (Figure 6H) than at other Gαi/o proteins while the gustatory G protein Gαgust was not activated by any of the investigated A2BAR agonists (Figure 6I).
In summary, all Gα protein subunits (with the exception of the Gαgust protein) could be activated by adenosine-stimulated A2BARs. The agonist NECA generally displayed higher potencies than adenosine, especially regarding the activation of Gαs, Gαi1–3, and Gα15 proteins. BAY 60-6583 exclusively activated Gαs, Gα15, and Gα12 protein subunits.
The extremely high promiscuity of the A2BAR, activating virtually all Gα protein subunits with pEC50 values for adenosine ranging from 5.0 to 6.5 as determined in the TRUPATH assay, raises the question: How does this actually translate into signal transduction? For example, Gαs proteins activate adenylate cyclase, while Gαi/o proteins inhibit the enzyme. Many of these G proteins, including Gαs and Gαi, are coexpressed by a large number of cells. Why does Gs signaling in most cells win over Gi signaling induced by A2BARs? Besides differences in expression levels, different efficacies in activation of distinct Gα proteins by the agonist-stimulated A2BAR might play a decisive role.
Efficacy of A2BAR-Induced G Protein Activation
As demonstrated using the TRUPATH assay, the A2BAR induces activation, measured by dissociation from the Gβγ-subunits, of nearly all Gα protein subunits. However, the A2BAR is generally considered to be primarily a Gαs-coupled and secondarily a Gαq/11-coupled receptor. Besides potency (pEC50), we additionally calculated efficacy (Emax) of agonist-induced A2BAR-Gα protein interactions. Thus, we compared the Emax values determined in the A2BAR-dependent assays with the Emax values measured for model receptors, the isoprotenerol-activated β2 adrenoceptor for the Gαs family, the DAMGO-activated μ-opioid receptor for the Gαi/o family, and the neurotensin-activated neurotensin 1 receptor for the Gαq/11 and the Gα12/13 families.31 The adenosine-activated A2BAR was observed to couple to Gαs proteins (efficacy: 120%) and also to the Gα15 protein (88%) with an efficacy close to that of the control receptors (set at 100%); the efficacy in activating the Gα12 protein was also still relatively high (64%) (see Figure 7 and Table S3; see Figure S3 for statistical analysis). A similar efficacy fingerprint was obtained for the A2BAR activated by NECA, but here we observed higher efficacy at the Gα15 protein than at Gαs and Gα12 proteins. NECA additionally activated the Gαi2 subunit with relatively high efficacy (76%). At all other Gα proteins, the adenosine- and NECA-activated A2BAR displayed an efficacy below 50% of controls. The partial agonist BAY 60-6583 exclusively activated subunits displaying a high coupling efficacy for adenosine and NECA. It partially activated Gαs (32% efficacy) and the Gα12 proteins (23%), and fully activated the Gα15 protein (in comparison to the endogenous agonist adenosine as well as the control receptors).
Figure 7.

Heatmaps depicting potency (pEC50 values) and efficacy (Emax values given as % of maximal efficacy (Emax) of control receptors: isoprotenerol-activated β2 adrenoceptor for the Gαs family, DAMGO-activated μ-opioid receptor for the Gαi/o family, neurotensin-activated neurotensin 1 receptor for the Gαq/11 and the Gα12/13 family), and a transduction coefficient expressed as log (Emax/EC50).31 Data for NECA and BAY 60-6583 (BAY) were determined in the presence of ADA. Units for log(Emax/EC50): Emax is in %, EC50 in mol/L.
The log(Emax/EC50) calculation is used to describe ligand bias toward a specific signaling pathway to provide a coefficient that includes both potency and efficacy.48 These data are presented in Figure 7 for comparison. When this term is applied, the EC50 values appear to predominate while the efficacy values are underappreciated. Thus, we conclude that it is important to consider potencies and efficacies separately, in agreement with other authors.31 Nevertheless, these data confirm the superior importance of the Gα15 signaling pathway for all three investigated A2BAR agonists.
Discussion
Previous studies on A2BAR signaling indicated that it could signal via multiple Gα proteins depending on the cell line studied.14,39−42 However, only recently, novel methods have become available to unambiguously study the direct activation of specific Gα protein subunits upon stimulation with agonists.31,35,37,38 In the present study, we determined agonist-dependent coupling of the human A2BAR to specific Gα protein subunits using two different approaches: (i) calcium mobilization assays utilizing CRISPR-Cas9-Gαq/11-KO cells with re-expression of specific Gαq protein subunits (Gαq, Gα11, Gα14, or Gα15),25 and (ii) TRUPATH BRET2 assays measuring dissociation of the G protein heterotrimer.31 We investigated the coupling behavior induced by three different A2BAR agonists, the cognate agonist adenosine, its metabolically stable analog NECA, and the non-nucleosidic A2B-selective partial agonist BAY 60-6583.39 Based on our data, we conclude and confirm that the A2BAR is a promiscuous receptor. Upon activation with the full agonists adenosine or NECA it potentially interacted with all Gα protein subunits, with the exception of the Gαgust and the Gα14 protein (the latter could only be tested in the calcium assays since a TRUPATH biosensor for Gα14 has not been available). Activation of the A2BAR with the non-nucleosidic (partial) agonist BAY 60-6583, however, resulted exclusively in Gα15, Gαs, and Gα12 coupling, with high efficacy at Gα15, but low efficacy at the other Gα proteins.
Interestingly, the treatment of HEK293 cells with the adenosine-metabolizing enzyme ADA increased the potency of the agonists NECA (10-fold) and BAY 60-6583 (120-fold) exclusively at the Gα15 subunit in TRUPATH BRET2 assays, while the efficacy of both agonists was approximately doubled (Figure 5). This may be explained by an allosteric modulation of the A2BAR by ADA as previously postulated.49−51 ADA is proposed to act as an allosteric modulator facilitating a receptor conformation that exhibits high affinity for the Gα15 protein. Another explanation could be that in cases where ADA appears to increase the potency of NECA, this could be due to the enzymatic activity of the enzyme.
It should be kept in mind that the employed test systems are highly artificial, the TRUPATH assays requiring overexpression of the A2BAR and control receptors. In calcium mobilization assays using HEK cells with low native A2BAR expression and recombinant expression of a single Gαq protein subunit, the full agonists adenosine and NECA sufficiently activated Gα15 and Gαq proteins to induce calcium mobilization, but not Gα11 and Gα14 (see Figure 1). The partial agonist BAY 60-6583 only induced a calcium signal in cells recombinantly expressing a higher A2BAR level via the Gα15 protein subunit (see Figure 4). These results point to a prominent role of the efficacy of a particular A2BAR agonist to activate a specific Gα protein subunit (see Figure 8). It appears that the efficacy fingerprint is an excellent indicator for the actual G protein coupling observed in native cells. Efficacies, however, can hardly be predicted at present, which may be the reason why computer programs have failed to correctly predict the G protein-coupling for the A2BAR (see precog.russellab.org).52 Focus on potency rather than efficacy, or log(Emax/EC50) as previously proposed,53 may not provide optimal results, at least for the A2BAR.
Figure 8.
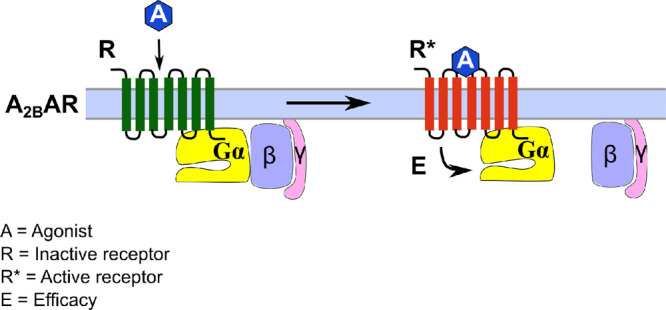
G protein dissociation induced by A2BAR activation. The agonist (A) binds to the receptor (R) with an agonist-specific affinity and induces an active conformation (R*). Different active conformations are conceivable depending on the employed agonist. The activated receptor binds to the heterotrimeric G protein (Gαβγ) with a receptor (R*)- and Gα-subunit-dependent affinity, and induces dissociation of the Gα- from the βγ-subunit with a specific efficacy (E) that depends on the nature and concentration of the agonist A, the receptor R*, and the Gα protein. The maximal efficacy (Emax) corresponds to the maximal effect observed for a specific agonist in a defined system.
The A2BAR couples most efficaciously to Gαs (Emax 120%), Gα15 (88%) and Gα12 proteins (64%) when activated by adenosine, and is therefore biased toward these signaling pathways. The (partial) agonist BAY 60-6583 only activated these three G proteins at all, with low efficacy in case of Gαs and Gα12 (see Figure 7). Notably, the adenosine analog NECA additionally activated the Gαi2 protein with relatively high efficacy (Emax 76%, compared to 49% for adenosine). Thus, the activation of specific G proteins is dependent on the employed agonist, and synthetic agonists do not necessarily imitate the activity of the physiological ligand.
The preferential potent and efficacious coupling of the A2BAR to Gα15 proteins is of great importance in the context of immunology and immunotherapy since Gα15 proteins are exclusively expressed on hematopoietic cells including immune cells.54
Studies on the Gα protein coupling of a large number of GPCRs have been recently published utilizing biochemical probes, such as the TRUPATH BRET2 assay,31 the enhanced bystander BRET or EMTA assays,37 and the TGFα shedding assay34,38). Inoue et al. employed the TGF-α shedding assay and found concentration-dependent coupling of the adenosine-activated A2BAR to all investigated Gα protein subunits (Gαi1, Gαi3, Gαo, Gαz, Gα12, Gα13, Gαs, Gαolf, Gαq, Gα14) with the exception of the Gα15 protein.34 In contrast, the Gα15 protein was potently and efficaciously activated by A2BAR stimulation, both in TRUPATH BRET2 and calcium mobilization assays in the present study. The previously published study relied on chimeric Gαq proteins, which only harbored the six C-terminal residues of the Gα15 protein to investigate its coupling. It appears likely that the interaction between the A2BAR and the Gα15 protein is based on other Gα15-specific protein–protein interactions apart from the C-terminal residues. Thus, the Gα15 protein is presumed to have a unique mode of engagement with and activation by the GPCR. In agreement with the present results, a study by Avet et al. using ebBRET/EMTA assays identified exclusively the Gαs and Gα15 proteins as coupling partners for the adenosine-activated A2BAR.37 These proteins were likewise identified as the most efficacious coupling partners for the A2BAR in the present study using TRUPATH BRET2 assays (Figure 7).
At increasing A2BAR or G protein density, e. g. in pathological scenarios such as inflammation and cancer,12,14,55 A2BAR activation by adenosine could result in efficacious activation of additional G proteins, such as Gαq, Gα11, Gαi/o or Gα13 proteins.
In summary, each investigated agonist displays a characteristic signaling fingerprint regarding potency and especially efficacy at individual Gα protein subunits. The efficacy of the agonist-activated A2BAR at certain Gα proteins appears to be decisive for biological significance of the respective signaling pathway. Since the cellular response to A2BAR activation is dependent on the investigated cell type and the employed agonist, results from biological studies have to be interpreted with great care. BAY 60-6583 being a partial agonist does not imitate adenosine action, but nevertheless activates Gα15, Gαs, and Gα12 proteins—presumably the most important downstream effectors of the A2BAR. A highly potent A2B-selective agonist that imitates adenosine action is currently not available, but would be most useful.
Materials and Methods
Materials and Reagents
HEK293 Gαq/11-KO cells were prepared by A. Inoue as previously described.56 Dulbecco’s modified Eagle medium (DMEM) and Hank’s balanced salt solution (HBSS) buffer, supplemented with CaCl2 and MgCl2, were purchased from Sigma-Aldrich (Merck, Darmstadt, Germany). Fetal calf serum (FCS), G418, and penicillin-streptomycin were ordered from PAN Biotech (Aidenbach, Germany). Recombinant HEK293 cell lines stably expressing exclusively Gαq/11 family subunits were created from CRISPR-Cas9-modified HEK293-ΔGαq/11 cells as previously described.25 Fluo-4-AM was purchased from Invitrogen (ThermoFisher, Waltham, MA, USA). Adenosine and ATP disodium salt were from Sigma (St. Louis, MO, USA); NECA was purchased from SantaCruz (Dallas, TX, USA), carbachol was purchased from AlfaAesar (Haverhill, MA, USA), BAY 60-6583 was purchased from Tocris (Bristol, UK). PSB-10,57 PSB-11,58 PSB-36,59 and PSB-60360 were synthesized as previously described. Lipofectamine 2000 was ordered from ThermoFisher Scientific (Waltham, MA, USA). U46619 was ordered from SantaCruz.61 Flat white-bottom 96-well plates were purchased from Greiner BioOne disposable materials such as pipet tips and cell culture flasks were purchased from Sarstedt (Nümbrecht, Germany).
Cultivation of Cells
Cells were cultured at 37 °C and 5% CO2 in DMEM supplemented with 10% FCS, 0.1 mg/mL streptomycin, and 100 U/ml penicillin (growth medium). If cells stably expressed recombinant genes, G418 (200 μg/mL) was added to the medium. At 70–80% confluency, cells were detached by trypsination, diluted with growth medium, and transferred into a fresh cell culture flask. Retroviral transfection of HEK293-ΔGαq/11 cells was performed as previously described.25
Calcium Mobilization Assay
Calcium mobilization was measured as previously described for the P2Y2 receptor.62 In short, cells were trypsinized from cell culture flasks and seeded into a black clear-bottom 96-well plate (Corning 3340, Corning, Amsterdam, NL) in a final volume of 200 μL per well (40,000 cells per well) 1 day before the assay. Cells were incubated overnight in 96-well plates. On the next day, the supernatant medium was discarded and the following solution (40 μL) was added: HBSS buffer containing 3 μM fluo-4-AM + 0.075% w/v Pluronic F-127 (“dye solution”). All steps of the assay were performed at room temperature. The cells were incubated with the dye solution for 60 min while gently shaking. Then, the dye solution was removed and a mixture of 178 μL HBSS + 2 μL DMSO (without or with inhibitor) was added. If no inhibitor was present, the measurement was performed after a 2 min equilibration period. All assays in the presence of inhibitory compounds were preincubated for 30 min with the inhibitor before the measurement started. Cells were transferred to a plate reader (NovoStar, BMG Labtech, Offenburg, Germany). The A2BAR was activated by the addition of 20 μL of agonist solution in HBSS (negative control: pure HBSS buffer). Fluorescence at 525 nm was measured for 40 s per well. Raw data were obtained in arbitrary fluorescence units (AU) and were corrected for the background signal (fluorescence intensity in the absence of agonist). If data were normalized, the normalization procedure was performed as indicated (see Figures). Each data point was recorded in duplicate or triplicate in three or more independent experiments. Dose–response curves were generated with Graph Pad Prism 8.0 (GraphPad, San Diego, CA, USA) using the “sigmoidal dose-response, variable slope” function. The assay principle is visualized in Figure S1B.
When the A2BAR was transiently overexpressed for calcium mobilization assays, cells were transfected for 12 h with 100 or 1000 ng of hADORA2B-pcDNA3.1 per well in a 6-well plate (denoted as 100 ng DNA/well and 1000 ng DNA/well, respectively), 4 h after seeding the cells, using Lipofectamine 2000 according to the manufacturer’s protocol. At the end of the 12 h period, the medium was exchanged. Transfected cells were harvested and transferred to 96-well plates 24 h after the end of the transfection, and from thereon the assay procedure was continued as described above.
BRET2 TRUPATH Assay
G protein heterotrimer dissociation was measured in HEK293 cells using the TRUPATH assay kit.31 TRUPATH plasmids were a gift from Bryan Roth (University of North Carolina on Chapel Hill, NC, USA) and shipped via Addgene (Addgene kit no. 1000000163). The measurements were performed as indicated in the original publication with minor modifications to the original protocol. On the first day, HEK293 cells cultivated in growth medium were detached from cell culture flasks by trypsination. Cells were seeded into 6-well plates at a density of approximately 500 000 cells per well in a volume of 2 mL, and incubated at 37 °C for 2 h before transfection. Transient transfection of HEK293 cells with the TRUPATH biosensors (100 ng of each pcDNA5/FRT/TO-Gα-RLuc8, pcDNA3.1-Gβ, and pcDNA3.1-Gγ-GFP2 per well) and the receptor of interest (100 ng receptor cDNA in pcDNA3.1 per well) was performed with Lipofectamine 2000 (2.5 μL per μg cDNA, ThermoFisher, Waltham, MA, USA). Lipofectamine was diluted in OptiMEM, incubated at room temperature for 10 min, and subsequently added to the DNA mixture in OptiMEM to a final volume of 500 μL per well. The mixture was added to the cells and the transfection was performed overnight. On the second day, media were removed, and cells were detached by pipetting and transferred to 96-well white bottom plates (Greiner BioOne, Frickenhausen, Germany) at a density of 30 000 cells per well in 60 μL of growth medium. On the third day, the medium was carefully aspirated, and cells were gently washed with assay buffer (HBSS + 20 mM HEPES pH 7.4). Assay buffer (60 μL per well; in experiments with adenosine deaminase (ADA), the buffer was supplemented with 5 μg ADA (Roche, Basel, Switzerland) per ml of buffer) and luciferase solution (50 μM coelenterazine 400a (Biomol, Hamburg, Germany) in assay buffer) were added to the cells. After a 5 min equilibration period, agonist solution (30 μL; 1% final DMSO concentration diluted in assay buffer) was added to the cells. After another 5 min equilibration period, luminescence and fluorescence were measured on a Mithras LB940 plate reader, using 395 and 510 nm emission filters for the RLuc8 and GFP2 signals, respectively. During assay optimization, the measurments were performed for several time points between 5 and 15 min after substrate addition.While BRET ratios remained stable, the absolute RLuc and GFP2 counts decreased over time probably due to degradation of the substrate. The ratio between the GFP2 and the RLuc8 signal intensity was computed and corrected for the baseline signal to obtain ΔBRET values, which were then fit to the “log (agonist) vs. response (three parameters)” equation in GraphPad PRISM 8 (GraphPad, San Diego, CA, USA) as suggested by Olsen et al.31 to determine pEC50 values and maximum efficacy (top-bottom of the sigmoidal concentration–response curve). The assay principle is visualized in Figure S1C.
Data Analysis
All data are expressed as mean ± standard error of the mean (SEM) of at least three independent experiments performed with at least two replicates per experiment. To test for significant differences between two groups, an unpaired t-test was employed. To assess significant differences among three or more groups, a one-way analysis of variance (ANOVA) was used. Significant thresholds are defined as follows: not significant (ns) p > 0.05, * p < 0.05, ** p < 0.01, *** p < 0.001. Data analysis was performed with GraphPad PRISM v8.0 (GraphPad, San Diego, CA, USA).
To assess differences in potency and efficacy for each individual agonist across all Gα protein subunits for all results obtained in the TRUPATH BRET2 assays, mean efficacies and potencies of each agonist (depicted in Figure 7) were compared to each other by a one-way ANOVA. The mean of each column was compared with the mean of each other column and corrected for multiple comparisons by Turkey’s test. All p-values, depicted in Figure S3, are multiplicity-adjusted.
Acknowledgments
We thank Dr. Bryan Roth, University of North Carolina School of Medicine, Chapel Hill, NC, USA, for sharing the TRUPATH Biosensor Platform.
Glossary
Abbreviations
- A1AR
adenosine A1 receptor
- A2AR
adenosine A2A receptor
- A2BAR
adenosine A2B receptor
- A3R
adenosine A3 receptor
- AC
adenylate cyclase
- ADA
adenosine deaminase
- Ado
adenosine
- AR
adenosine receptor
- ATP
adenosine triphospate
- BRET2
bioluminescence resonance energy transfer type 2
- CCh
carbachol
- CRISPR-Cas9
clustered regularly interspaced short palindromic repeats - CRISPR-associated protein 9
- EMTA
effector-membrane translocator assays
- GDP
guanosine diphosphate
- GFP2
green fluorescent protein-2
- GPCR
G protein-coupled receptor
- GTP
guanosine triphosphate
- HEK293
human embryonic kidney type 293
- M3R
muscarinic M3 receptor
- NECA
5′-N-ethylcarboxamidoadenosine
- Rluc8
Renilla luciferase
- TGF-α
transforming growth factor-α
- TP
thromboxane receptor
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.2c00020.
Tables S1–S3, listing all determined potencies and efficacies in calcium and BRET2 assays; Figures S1 and S2, displaying structures of the employed agonists, the assay principles, and reference curves obtained during establishment of TRUPATH BRET2 assays (PDF)
J.H.V., A.B.M., and C.E.M. were supported by the Deutsche Forschungsgemeinschaft (DFG, FOR2372, GRK1328). J.H.V., A.B.M., and C.E.M. are grateful for funding by the BMBF-funded Bonn International Graduate School Drug Sciences (BIGS DrugS). C.E.M. was supported by the EU COST Action ERNEST CA18133. A.B.M. is grateful for funding by the Ministry of Finance of Indonesia in the scheme of Indonesia Endowment Fund for Education (Lembaga Pengelola Dana Pendidikan (LPDP). A.I. was funded by the Japan Society for the Promotion of Science (JSPS) KAKENHI grants 21H04791 and 21H05113, FOREST JPMJFR215T from the Japan Science and Technology Agency (JST), LEAP JP20gm0010004 and BINDS JP20am0101095 from the Japan Agency for Medical Research and Development (AMED), the Takeda Science Foundation, the Daiichi Sankyo Foundation of Life Science, and The Uehara Memorial Foundation.
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Pharmacology & Translational Science virtual special issue “GPCR Signaling”.
Supplementary Material
References
- Müller C. E.; Baqi Y.; Namasivayam V. Agonists and antagonists for purinergic receptors. Methods Mol. Biol. 2020, 2041, 45–64. 10.1007/978-1-4939-9717-6_3. [DOI] [PubMed] [Google Scholar]
- Müller C. E.; Baqi Y.; Hinz S.; Namasivayam V.. Medicinal chemistry of A2B adenosine receptors. In The adenosine receptors; Borea P. A., Varani K., Gessi S., Merighi S., Vincenzi F., Eds.; The Receptors Vol. 34; Springer International Publishing, 2018; pp 137–168. 10.1007/978-3-319-90808-3_6 [DOI] [Google Scholar]
- Jenner P.; Mori A.; Aradi S. D.; Hauser R. A. Istradefylline - a first generation adenosine A2A antagonist for the treatment of Parkinson’s disease. Exp. Rev. Neurother. 2021, 21 (3), 317–333. 10.1080/14737175.2021.1880896. [DOI] [PubMed] [Google Scholar]
- Chen J.-F.; Cunha R. A. The belated US FDA approval of the adenosine A2A receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal. 2020, 16 (2), 167–174. 10.1007/s11302-020-09694-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghimire G.; Hage F. G.; Heo J.; Iskandrian A. E. Regadenoson: a focused update. J. Nucl. Cardiol. 2013, 20 (2), 284–288. 10.1007/s12350-012-9661-3. [DOI] [PubMed] [Google Scholar]
- Rankin A. C.; Brooks R.; Ruskin J. N.; McGovern B. A. Adenosine and the treatment of supraventricular tachycardia. Am. J. Med. 1992, 92, 655–664. 10.1016/0002-9343(92)90784-9. [DOI] [PubMed] [Google Scholar]
- Borea P. A.; Gessi S.; Merighi S.; Vincenzi F.; Varani K. Pharmacology of adenosine receptors: the state of the art. Physiol. Rev. 2018, 98 (3), 1591–1625. 10.1152/physrev.00049.2017. [DOI] [PubMed] [Google Scholar]
- Jamwal S.; Mittal A.; Kumar P.; Alhayani D. M.; Al-Aboudi A. Therapeutic potential of agonists and antagonists of A1, A2A, A2B and A3 adenosine receptors. Curr. Pharm. Des. 2019, 25 (26), 2892–2905. 10.2174/1381612825666190716112319. [DOI] [PubMed] [Google Scholar]
- Chandrasekaran B.; Samarneh S.; Jaber A. M. Y.; Kassab G.; Agrawal N. Therapeutic potentials of A2B adenosine receptor ligands: current status and perspectives. Curr. Pharm. Des. 2019, 25 (25), 2741–2771. 10.2174/1381612825666190717105834. [DOI] [PubMed] [Google Scholar]
- Cacciari B.; Pastorin G.; Bolcato C.; Spalluto G.; Bacilieri M.; Moro S. A2B adenosine receptor antagonists: recent developments. Mini Rev. Med. Chem. 2005, 5 (12), 1053–1060. 10.2174/138955705774933374. [DOI] [PubMed] [Google Scholar]
- Kotańska M.; Szafarz M.; Mika K.; Dziubina A.; Bednarski M.; Müller C. E.; Sapa J.; Kieć-Kononowicz K. PSB 603 - a known selective adenosine A2B receptor antagonist - has anti-inflammatory activity in mice. Biomed. Pharmacother. 2021, 135, 111164. 10.1016/j.biopha.2020.111164. [DOI] [PubMed] [Google Scholar]
- Gao Z.-G.; Jacobson K. A. A2B adenosine receptor and cancer. Int. J. Mol. Sci. 2019, 20 (20), 5139. 10.3390/ijms20205139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta A.; Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front. Immunol. 2014, 5, 304. 10.3389/fimmu.2014.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feoktistov I.; Biaggioni I. Role of adenosine A2B receptors in inflammation. Adv. Pharmacol. 2011, 61, 115–144. 10.1016/B978-0-12-385526-8.00005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick J.-S.; MacManus C. F.; Scully M.; Glover L. E.; Eltzschig H. K.; Colgan S. P. Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J. Immunol. 2009, 182, 4957–4964. 10.4049/jimmunol.0801324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xaus J.; Mirabet M.; Lloberas J.; Soler C.; Lluis C.; Franco R.; Celada A. IFN-gamma up-regulates the A2B adenosine receptor expression in macrophages: a mechanism of macrophage deactivation. J. Immunol. 1999, 162 (6), 3607–3614. [PubMed] [Google Scholar]
- Sepúlveda C.; Palomo I.; Fuentes E. Role of adenosine A2B receptor overexpression in tumor progression. Life Sci. 2016, 166, 92–99. 10.1016/j.lfs.2016.10.008. [DOI] [PubMed] [Google Scholar]
- Long J. S.; Crighton D.; O’Prey J.; Mackay G.; Zheng L.; Palmer T. M.; Gottlieb E.; Ryan K. M. Extracellular adenosine sensing-a metabolic cell death priming mechanism downstream of p53. Mol. Cell. 2013, 50 (3), 394–406. 10.1016/j.molcel.2013.03.016. [DOI] [PubMed] [Google Scholar]
- Simon M. I.; Strathmann M. P.; Gautam N. Diversity of G proteins in signal transduction. Science 1991, 252 (5007), 802–808. 10.1126/science.1902986. [DOI] [PubMed] [Google Scholar]
- Milligan G.; Kostenis E. Heterotrimeric G-proteins: A short history. Br. J. Pharmacol. 2006, 147 (Suppl 1), S46–S55. 10.1038/sj.bjp.0706405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror R. O.; Mildorf T. J.; Hilger D.; Manglik A.; Borhani D. W.; Arlow D. H.; Philippsen A.; Villanueva N.; Yang Z.; Lerch M. T.; Hubbell W. L.; Kobilka B. K.; Sunahara R. K.; Shaw D. E. Signal Transduction. Structural basis for nucleotide exchange in heterotrimeric G proteins. Science 2015, 348 (6241), 1361–1365. 10.1126/science.aaa5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N.; Hajicek N.; Kozasa T. Regulation and physiological functions of G12/13-mediated signaling pathways. Neurosignals 2009, 17 (1), 55–70. 10.1159/000186690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F. A.; Hsu P. D.; Wright J.; Agarwala V.; Scott D. A.; Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8 (11), 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annala S.; Feng X.; Shridhar N.; Eryilmaz F.; Patt J.; Yang J.; Pfeil E. M.; Cervantes-Villagrana R. D.; Inoue A.; Häberlein F.; Slodczyk T.; Reher R.; Kehraus S.; Monteleone S.; Schrage R.; Heycke N.; Rick U.; Engel S.; Pfeifer A.; Kolb P.; König G.; Bünemann M.; Tüting T.; Vázquez-Prado J.; Gutkind J. S.; Gaffal E.; Kostenis E. Direct targeting of Gαq and Gα11 oncoproteins in cancer cells. Sci. Signal. 2019, 12 (573), aau5948. 10.1126/scisignal.aau5948. [DOI] [PubMed] [Google Scholar]
- Kuschak M.; Namasivayam V.; Rafehi M.; Voss J. H.; Garg J.; Schlegel J. G.; Abdelrahman A.; Kehraus S.; Reher R.; Küppers J.; Sylvester K.; Hinz S.; Matthey M.; Wenzel D.; Fleischmann B. K.; Pfeifer A.; Inoue A.; Gütschow M.; König G. M.; Müller C. E. Cell-permeable high-affinity tracers for Gq proteins provide structural insights, reveal distinct binding kinetics, and identify small molecule inhibitors. Br. J. Pharmacol. 2020, 177 (8), 1898–1916. 10.1111/bph.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okashah N.; Wan Q.; Ghosh S.; Sandhu M.; Inoue A.; Vaidehi N.; Lambert N. A. Variable G protein determinants of GPCR coupling selectivity. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (24), 12054–12059. 10.1073/pnas.1905993116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patt J.; Alenfelder J.; Pfeil E. M.; Voss J. H.; Merten N.; Eryilmaz F.; Heycke N.; Rick U.; Inoue A.; Kehraus S.; Deupi X.; Müller C. E.; König G. M.; Crüsemann M.; Kostenis E. An experimental strategy to probe Gq contribution to signal transduction in living cells. J. Biol. Chem. 2021, 296, 100472. 10.1016/j.jbc.2021.100472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A. D.; Jean-Alphonse F. G.; Fang F.; Peña K. A.; Liu S.; König G. M.; Inoue A.; Aslanoglou D.; Gellman S. H.; Kostenis E.; Xiao K.; Vilardaga J.-P. Gq/11-dependent regulation of endosomal cAMP generation by parathyroid hormone class B GPCR. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (13), 7455–7460. 10.1073/pnas.1918158117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galés C.; van Durm J. J. J.; Schaak S.; Pontier S.; Percherancier Y.; Audet M.; Paris H.; Bouvier M. Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat. Struct. Mol. Biol. 2006, 13, 778–786. 10.1038/nsmb1134. [DOI] [PubMed] [Google Scholar]
- Galés C.; Rebois R. V.; Hogue M.; Trieu P.; Breit A.; Hébert T. E.; Bouvier M. Real-time monitoring of receptor and G-protein interactions in living cells. Nat. Methods 2005, 2, 177–184. 10.1038/nmeth743. [DOI] [PubMed] [Google Scholar]
- Olsen R. H. J.; DiBerto J. F.; English J. G.; Glaudin A. M.; Krumm B. E.; Slocum S. T.; Che T.; Gavin A. C.; McCorvy J. D.; Roth B. L.; Strachan R. T. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 2020, 16, 841. 10.1038/s41589-020-0535-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laschet C.; Dupuis N.; Hanson J. A dynamic and screening-compatible nanoluciferase-based complementation assay enables profiling of individual GPCR-G protein interactions. J. Biol. Chem. 2019, 294 (11), 4079–4090. 10.1074/jbc.RA118.006231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q.; Okashah N.; Inoue A.; Nehmé R.; Carpenter B.; Tate C. G.; Lambert N. A. Mini G protein probes for active G protein-coupled receptors (GPCRs) in live cells. J. Biol. Chem. 2018, 293 (19), 7466–7473. 10.1074/jbc.RA118.001975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A.; Raimondi F.; Kadji F. M. N.; Singh G.; Kishi T.; Uwamizu A.; Ono Y.; Shinjo Y.; Ishida S.; Arang N.; Kawakami K.; Gutkind J. S.; Aoki J.; Russell R. B. Illuminating G protein-coupling selectivity of GPCRs. Cell 2019, 177 (7), 1933–1947.e25. 10.1016/j.cell.2019.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon A. S.; Schwinn M. K.; Hall M. P.; Zimmerman K.; Otto P.; Lubben T. H.; Butler B. L.; Binkowski B. F.; Machleidt T.; Kirkland T. A.; Wood M. G.; Eggers C. T.; Encell L. P.; Wood K. V. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 2016, 11 (2), 400–408. 10.1021/acschembio.5b00753. [DOI] [PubMed] [Google Scholar]
- Wright S. C.; Lukasheva V.; Le Gouill C.; Kobayashi H.; Breton B.; Mailhot-Larouche S.; Blondel-Tepaz É.; Antunes Vieira N.; Costa-Neto C.; Héroux M.; Lambert N. A.; Parreiras-E-Silva L. T.; Bouvier M. BRET-based effector membrane translocation assay monitors GPCR-promoted and endocytosis-mediated Gq activation at early endosomes. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (20), e2025846118. 10.1073/pnas.2025846118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avet C.; Mancini A.; Breton B.; Le Gouill C.; Hauser A. S.; Normand C.; Kobayashi H.; Gross F.; Hogue M.; Lukasheva V.; St-Onge S.; Carrier M.; Héroux M.; Morissette S.; Fauman E. B.; Fortin J.-P.; Schann S.; Leroy X.; Gloriam D. E.; Bouvier M. Effector membrane translocation biosensors reveal G protein and β-arrestin coupling profiles of 100 therapeutically relevant GPCRs. eLife 2022, 11, 74101. 10.7554/eLife.74101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A.; Ishiguro J.; Kitamura H.; Arima N.; Okutani M.; Shuto A.; Higashiyama S.; Ohwada T.; Arai H.; Makide K.; Aoki J. TGFα shedding assay: an accurate and versatile method for detecting GPCR activation. Nat. Methods 2012, 9 (10), 1021–1029. 10.1038/nmeth.2172. [DOI] [PubMed] [Google Scholar]
- Hinz S.; Lacher S. K.; Seibt B. F.; Müller C. E. BAY60-6583 acts as a partial agonist at adenosine A2B receptors. J. Pharm. Exp. Ther. 2014, 349 (3), 427–436. 10.1124/jpet.113.210849. [DOI] [PubMed] [Google Scholar]
- Gao Z.-G.; Inoue A.; Jacobson K. A. On the G protein-coupling selectivity of the native A2B adenosine receptor. Biochem. Pharmacol. 2018, 151, 201–213. 10.1016/j.bcp.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden J.; Thai T.; Figler H.; Jin X.; Robeva A. S. Characterization of human A2B adenosine receptors: radioligand binding, western blotting, and coupling to Gq in human embryonic kidney 293 cells and HMC-1 mast cells. Mol. Pharmacol. 1999, 56 (4), 705–713. [PubMed] [Google Scholar]
- Cohen M. V.; Yang X.; Downey J. M. A2B adenosine receptors can change their spots. Br. J. Pharmacol. 2010, 159 (8), 1595–1597. 10.1111/j.1476-5381.2010.00668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Xin W.; Yang X.-M.; Kuno A.; Rich T. C.; Cohen M. V.; Downey J. M. A2B adenosine receptors inhibit superoxide production from mitochondrial complex I in rabbit cardiomyocytes via a mechanism sensitive to Pertussis toxin. Br. J. Pharmacol. 2011, 163 (5), 995–1006. 10.1111/j.1476-5381.2011.01288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood B. K.; Lopez J.; Wager-Miller J.; Mackie K.; Straiker A. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics 2011, 12, 14. 10.1186/1471-2164-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alnouri M. W.; Jepards S.; Casari A.; Schiedel A. C.; Hinz S.; Müller C. E. Selectivity is species-dependent: Characterization of standard agonists and antagonists at human, rat, and mouse adenosine receptors. Purinergic Signal. 2015, 11 (3), 389–407. 10.1007/s11302-015-9460-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiringhelli F.; Bruchard M.; Chalmin F.; Rébé C. Production of adenosine by ectonucleotidases: a key factor in tumor immunoescape. J. Biomed. Biotechnol. 2012, 2012, 473712. 10.1155/2012/473712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann H. History of ectonucleotidases and their role in purinergic signaling. Biochem. Pharmacol. 2021, 187, 114322. 10.1016/j.bcp.2020.114322. [DOI] [PubMed] [Google Scholar]
- Winpenny D.; Clark M.; Cawkill D. Biased ligand quantification in drug discovery: from theory to high throughput screening to identify new biased μ opioid receptor agonists. Br. J. Pharmacol. 2016, 173 (8), 1393–1403. 10.1111/bph.13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera C.; Casadó V.; Ciruela F.; Schofield P.; Mallol J.; Lluis C.; Franco R. Adenosine A2B receptors behave as an alternative anchoring protein for cell surface adenosine deaminase in lymphocytes and cultured cells. Mol. Pharmacol. 2001, 59 (1), 127–134. 10.1124/mol.59.1.127. [DOI] [PubMed] [Google Scholar]
- Gracia E.; Farré D.; Cortés A.; Ferrer-Costa C.; Orozco M.; Mallol J.; Lluís C.; Canela E. I.; McCormick P. J.; Franco R.; Fanelli F.; Casadó V. The catalytic site structural gate of adenosine deaminase allosterically modulates ligand binding to adenosine receptors. FASEB J. 2013, 27 (3), 1048–1061. 10.1096/fj.12-212621. [DOI] [PubMed] [Google Scholar]
- Moreno E.; Canet J.; Gracia E.; Lluís C.; Mallol J.; Canela E. I.; Cortés A.; Casadó V. Molecular evidence of adenosine deaminase linking adenosine A2A receptor and CD26 proteins. Front. Pharmacol. 2018, 9, 106. 10.3389/fphar.2018.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G.; Inoue A.; Gutkind J. S.; Russell R. B.; Raimondi F. PRECOG: PREdicting COupling probabilities of G-protein coupled receptors. Nucleic Acids Res. 2019, 47 (W1), W395–W401. 10.1093/nar/gkz392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser A. S.; Avet C.; Normand C.; Mancini A.; Inoue A.; Bouvier M.; Gloriam D. E. Common coupling map advances GPCR-G protein selectivity. eLife 2022, 11, 74107. 10.7554/eLife.74107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatruda T. T.; Steele D. A.; Slepak V. Z.; Simon M. I. G alpha 16, a G protein alpha subunit specifically expressed in hematopoietic cells. Proc. Natl. Acad. Sci. U. S. A. 1991, 88 (13), 5587–5591. 10.1073/pnas.88.13.5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onfroy L.; Galandrin S.; Pontier S. M.; Seguelas M.-H.; N’Guyen D.; Senard J.-M.; Gales C. G protein stoichiometry dictates biased agonism through distinct receptor-G protein partitioning. Sci. Rep. 2017, 7 (1), 7885. 10.1038/s41598-017-07392-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage R.; Schmitz A.-L.; Gaffal E.; Annala S.; Kehraus S.; Wenzel D.; Büllesbach K. M.; Bald T.; Inoue A.; Shinjo Y.; Galandrin S.; Shridhar N.; Hesse M.; Grundmann M.; Merten N.; Charpentier T. H.; Martz M.; Butcher A. J.; Slodczyk T.; Armando S.; Effern M.; Namkung Y.; Jenkins L.; Horn V.; Stößel A.; Dargatz H.; Tietze D.; Imhof D.; Galés C.; Drewke C.; Müller C. E.; Hölzel M.; Milligan G.; Tobin A. B.; Gomeza J.; Dohlman H. G.; Sondek J.; Harden T. K.; Bouvier M.; Laporte S. A.; Aoki J.; Fleischmann B. K.; Mohr K.; König G. M.; Tüting T.; Kostenis E. The experimental power of FR900359 to study Gq-regulated biological processes. Nat. Commun. 2015, 6, 10156. 10.1038/ncomms10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller C. E.; Burbiel J.; Thorand M. Improved, efficient synthesis for multigram-scale Production of PSB-10, a potent antagonist at human A3 adenosine receptors. Heterocycles 2003, 60 (6), 1425. 10.3987/COM-03-9753. [DOI] [Google Scholar]
- Müller C. E.; Diekmann M.; Thorand M.; Ozola V. [3H]8-Ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-5-one ([3H]PSB-11), a novel high-affinity antagonist radioligand for human A3 adenosine receptors. Bioorg. Med. Chem. Lett. 2002, 12 (3), 501–503. 10.1016/S0960-894X(01)00785-5. [DOI] [PubMed] [Google Scholar]
- Weyler S.; Fülle F.; Diekmann M.; Schumacher B.; Hinz S.; Klotz K.-N.; Müller C. E. Improving potency, selectivity, and water solubility of adenosine A1 receptor antagonists: xanthines modified at position 3 and related pyrimido[1,2,3-cd]purinediones. ChemMedChem. 2006, 1 (8), 891–902. 10.1002/cmdc.200600066. [DOI] [PubMed] [Google Scholar]
- Borrmann T.; Hinz S.; Bertarelli D. C. G.; Li W.; Florin N. C.; Scheiff A. B.; Müller C. E. 1-Alkyl-8-(piperazine-1-sulfonyl)phenylxanthines: development and characterization of adenosine A2B receptor antagonists and a new radioligand with subnanomolar affinity and subtype specificity. J. Med. Chem. 2009, 52 (13), 3994–4006. 10.1021/jm900413e. [DOI] [PubMed] [Google Scholar]
- Baqi Y.; Alshaibani S.; Ritter K.; Abdelrahman A.; Spinrath A.; Kostenis E.; Müller C. E. Improved synthesis of 4-/6-substituted 2-carboxy-1H-indole-3-propionic acid derivatives and structure–activity relationships as GPR17 agonists. Med. Chem. Commun. 2014, 5 (1), 86–92. 10.1039/C3MD00309D. [DOI] [Google Scholar]
- Rafehi M.; Neumann A.; Baqi Y.; Malik E. M.; Wiese M.; Namasivayam V.; Müller C. E. Molecular recognition of agonists and antagonists by the nucleotide-activated G protein-coupled P2Y2 receptor. J. Med. Chem. 2017, 60 (20), 8425–8440. 10.1021/acs.jmedchem.7b00854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.