

Abstract
This article reviews the discovery of PCSK9, its structure–function characteristics, and its presently known and proposed novel biological functions. The major critical function of PCSK9 deduced from human and mouse studies, as well as cellular and structural analyses, is its role in increasing the levels of circulating low-density lipoprotein (LDL)-cholesterol (LDLc), via its ability to enhance the sorting and escort of the cell surface LDL receptor (LDLR) to lysosomes. This implicates the binding of the catalytic domain of PCSK9 to the EGF-A domain of the LDLR. This also requires the presence of the C-terminal Cys/His-rich domain, its binding to the secreted cytosolic cyclase associated protein 1, and possibly another membrane-bound “protein X”. Curiously, in PCSK9-deficient mice, an alternative to the downregulation of the surface levels of the LDLR by PCSK9 is taking place in the liver of female mice in a 17β-estradiol-dependent manner by still an unknown mechanism. Recent studies have extended our understanding of the biological functions of PCSK9, namely its implication in septic shock, vascular inflammation, viral infections (Dengue; SARS-CoV-2) or immune checkpoint modulation in cancer via the regulation of the cell surface levels of the T-cell receptor and MHC-I, which govern the antitumoral activity of CD8+ T cells. Because PCSK9 inhibition may be advantageous in these processes, the availability of injectable safe PCSK9 inhibitors that reduces by 50% to 60% LDLc above the effect of statins is highly valuable. Indeed, injectable PCSK9 monoclonal antibody or small interfering RNA could be added to current immunotherapies in cancer/metastasis.
Keywords: β-cells, cancer/metastases, hypercholesterolemia, major histocompatibility complex I, sepsis
Graphical Abstract
Graphical Abstract.

By modulating the trafficking of key secretory proteins, PCSK9 is implicated in the regulation of major diseases. Secreted PCSK9 shortens the half-life of cell surface receptors, such as LDLR and MHC-I, by escorting them into the lysosomal pathway. The functional consequences of PCSK9 activity in different diseases is indicated.
Essential Points.
PCSK9 is the third gene implicated in familial hypercholesterolemia, after LDLR and APOB genes.
PCSK9 mAbs or siRNA over statin treatment reduce plasma LDL-cholesterol levels by a further 60%.
PCSK9 is highly expressed and secreted by hepatocytes and β-cells.
Circulating (liver) or locally secreted PCSK9 shortens the half-life of cell surface receptors, such as the LDLR, VLDLR, or MHC-I, by directing them to the lysosomal degradation pathway.
Thereby, PCSK9 modulates major functions, such as liver and peripheral lipoprotein uptake or antitumor immunity.
The first critical connection between cholesterol and heart disease was initially reported in 1913 in St. Petersburg by Nikolai Nikolajewitsch Anitschkow based on his seminal observation of the accelerated development of atherosclerosis in the arterial intima of rabbits fed a high cholesterol diet [reviewed in (1)]. Key epidemiological studies (2) and the similarity of atherosclerosis to the human pathology of heart disease catalyzed a 60-year-long search for cholesterol-lowering agents. In 1973, Akira Endo and his team screened 3800 fungal strains to discover a potent β-hydroxy β-methylglutaryl-CoA (HMG-CoA) reductase reversible inhibitor, mevastatin (also called compactin) (3). In 1987, a team at Merck Pharmaceutical Co. developed a chemically related compound, lovastatin (trademark Mevacor), which became the first statin to reach patients (4). This was followed by a successive improvement in the potency and specificity of statins: Zocor (simvastatin), Lipitor (atorvastatin), and Crestor (rosuvastatin), which led to 25% to 50% reduction of low-density lipoprotein cholesterol (LDLc) (5). Up to 2003, the reported major function of statins was to decrease LDLc levels by modulating cholesterol homeostasis in the liver. Mechanistically, by reducing the synthesis of cholesterol, treatment with statins results in the activation of the sterol regulatory element binding protein SREBP-2 in the endoplasmic reticulum (ER) (6), whereupon its N-terminal domain reaches the nucleus and enhances the synthesis of the LDL receptor (LDLR) following its association with sterol regulatory elements (SREs) in the LDLR promoter (7). This in turn activates cholesterol biosynthetic genes, leading to an increased LDLc uptake from circulation. This exquisite feedback mechanism balances cellular cholesterol levels and decreases the amount of circulating LDLc (for excellent reviews see (8, 9)).
The level of plasma cholesterol is influenced not only by its de novo biosynthesis, but also by the absorption of dietary cholesterol and the removal of cholesterol from the blood. Blocking the absorption of dietary cholesterol by the gut through inhibition of Niemann-Pick C1-Like 1 protein on small intestinal epithelial cells and hepatocytes is the basis of the mechanism of action of ezetimibe, a drug first described by Schering–Plough (10). When combined with a statin, ezetimibe further reduces LDLc by 15% to 20% (11) and improves cardiovascular outcomes (12).
In this review, we will introduce the family of proprotein convertases (PCs) and the implication of some members in cholesterol and fatty acid metabolism. We will then concentrate on PCSK9 implicated in the enhanced degradation of the LDLR, and hence clearance of LDLc from circulation. We will then present some of the multifaceted functions of this fascinating protein and highlight knowledge gaps that remain to be addressed. We do not aim to exhaustively cover all information regarding the various tissue-specific functions of PCSK9 in health and disease, but rather our focus has been placed on specific novel avenues, which we believe deserve more extensive investigations.
The Family of Proprotein Convertases
Secretory proteins undergo various post-translational modifications (PTMs) before reaching their intra/extracellular destination(s), thereby expanding and diversifying their functions (13). N- and O-glycosylation are major irreversible modifications that result in substantial structural rearrangements that affect many functions and fates of secretory proteins (14, 15). Reversible phosphorylation at specific Ser, Thr, and Tyr residues in secretory proteins is performed by a limited number of secretory kinases, FAM20C being the best studied one for Ser/Thr phosphorylation (16, 17). One of the major irreversible PTMs is the limited proteolysis of selected peptide bonds (18), which depends on the specificity of processing enzyme(s) and the accessibility of scissile peptide bonds to the attacking protease (19). Inactive precursor protein processing, often occurring in response to a stimulus, results in a prompt physiological response including but not limited to blood coagulation, fibrinolysis, active hormone production, fertilization, metamorphosis, and digestion. While some PTMs occur after the protein is secreted, precursor processing within the secretory pathway plays a prominent role in the activation of a wide variety of secretory proteins such as polypeptide hormones, growth factors, receptors, enzymes, and surface glycoproteins (19, 20).
Since the 1960s, it was realized that polypeptide hormones such as insulin (21, 22), melanotropins (α- and β-melanotropin stimulating hormone [MSH]) (23) and β-endorphin (24-26) are produced from longer, primarily inactive, precursor proteins via successive cleavages at paired basic amino acids (aa). Such a concept of limited proteolysis of prohormones by cellular proteases was later extended to many secretory proteins and even to infectious pathogens, and shown to occur at single and paired basic residues within the motif: (K/R)-Xn-(K/R)↓, or more commonly (K/R)-Xn-(R)↓, where Xn are spacer residues comprising 0, 2, 4, or 6 aa (27). It took more than 15 years to identify the cognate processing enzymes that recognize these motifs. They constitute a family of 7 basic aa-specific serine proteases, the PCs, related to subtilisin/kexin (SK), and their genes were mostly given the name PCSKs (for extensive reviews see (27, 28)). In essence, PC1 (gene PCSK1) and PC2 (gene PCSK2)/7B2 (7B2: chaperone of PC2) are the principal convertases responsible for the generation of regulated bioactive hormones stored in dense-core secretory granules, such as insulin; ACTH/α-MSH/β-endorphin, glucagon/GLP1,2; gastrin; enkephalins; TRH, etc. The other members are either ubiquitously expressed (type-I membrane-bound Furin (gene Fur) and PC7 (gene PCSK7)), widely expressed (PC5 (gene PCSK5) and PACE4 (gene PCSK6)), or synthesized only in gonadal tissues (PC4 (gene PCSK4)). All PCs are activated following 1 or 2 autocatalytic cleavages that remove their intramolecular N-terminal chaperone-like prodomain, starting in the ER (except for PC2) and continuing in the trans-Golgi network (Furin), immature secretory granules (PC1 and PC2), at the cell surface (PC5, PACE4, and possibly PC4) or in early endosomes (PC7). Thus, no basic aa-specific PC is active in the ER or early Golgi compartments, as they remain noncovalently associated with their respective inhibitory prodomain until they reach their destination(s), whereupon the cellular conditions (pH, calcium, etc.) are suitable for prodomain removal either by further autocatalytic cleavage (PC1, PC2, Furin, PC5, PACE4) or physical separation (PC4, PC7). Although most PC cleavages result in the activation of precursor proteins or generation of novel functions, some cleavages lead to substrate inactivation (29). For example, N-cadherin is activated by Furin, but inactivated by PC5 (29), and as discussed later PCSK9 is inactivated by Furin (30).
It was also realized that processing of precursor proteins can also occur by cleavage at nonbasic residues within the early secretory pathway. Typical examples are the sterol regulatory element binding proteins 1 and 2 (SREBP-1, 2) (31) and the surface glycoproteins of hemorrhagic fever viruses of Lassa virus (32) or Crimean Congo hemorrhagic fever virus (33) that are activated in the cis/medial Golgi. Using degenerate oligonucleotides hybridizing the active site encoding mRNA of convertases and amplification by polymerase chain reaction, we identified in 1999 the eighth member of the PCSK family and named it subtilisin-kexin isozyme 1 (SKI-1) (34). This enzyme cleaves pro-Brain Derived Neurotrophic Factor at R-G-LT↓S-L, where the bold and italic residues are critical for cleavage by SKI-1 (34). The latter turned out to be the same protease (called site 1 protease; S1P) identified in 1998 that activates SREBP-1, -2 (35) and later on the ER stress factor ATF6 (36). Indeed, in all subsequent substrates of SKI-1/S1P (gene MBPTS1) (37) the general consensus cleavage motif was found to be R-X-Aliphatic-Z↓, where X is any residue except Pro and Cys, and Z is any aa (best Leu) except Val, Pro, Cys, or Glu (34, 37-40). For an in-depth analysis of the multiple functions of SKI-1/S1P, including cholesterol and fatty acid regulation, neuronal development, lysosomal homeostasis, hypertension, activation of membrane-bound transcription factors and viral infections the reader is referred to recent reviews on this ever-expanding subject (28, 41-49).
The Discovery of PCSK9
The identification of SKI-1/S1P as a convertase that cleaves precursor proteins in the early cis/medial Golgi and regulate important physiological functions, such as cholesterol and fatty acid metabolism (35), ER stress (36), as well as the phosphorylation of mannose residues of proteins destined for lysosomal sorting (50), suggested to us that other homologous members of the PCSK family could also exist. Here again, we used polymerase chain reaction to amplify mRNAs that may encode a SKI-1/S1P homolog. This approach led us to clone the cDNA and analyze the biosynthesis of the 9th and last member of the PC family, a putative convertase originally called neural apoptosis-regulated convertase-1 (51). Upon screening databases, we noticed that Millennium Pharmaceuticals and Eli Lilly had released part of its cDNA, either in a patent database of a group of genes upregulated upon induction of apoptosis in primary cerebellar neurons by serum withdrawal (patent no. WO 01/57081 A2) or via the global cloning of secretory proteins (related LP251 sequence; patent no. WO 02/14358 A2). We completed the human, rat and mouse cDNA sequences and renamed the ortologous soluble protein as PCSK9 and its gene PCSK9 (52). PCSK9 belongs to the proteinase K family of subtilases, and its catalytic domain exhibits 25% of protein sequence identity to that of its closest family member SKI-1/S1P (51). The human PCSK9 mRNA (NM_174936.3) spans 3710 bp over 12 exons encoding a 692-aa protein (NP_777596.2). In situ hybridization and tissue/cell lines analyses by Northern blots revealed that liver and small intestine are the major sources of PCSK9 synthesis in the adult mouse, rat, and human. This report first appeared in February 2003 (51).
In a serendipitous twist of fate, the mapping of PCSK9 on the short arm of chromosome 1p32 (51) revealed that it was nearby to the 1p34.1p32 locus identified in large French families potentially encoding a third gene for autosomal dominant familial hypercholesterolemia (FH3), where the LDLR and apolipoprotein B (APOB) genes were excluded. This locus was associated with increased hepatic secretion of cholesterol associated with very low–density lipoprotein (VLDL), which after its secretion is converted into LDLc (53, 54). Armed with this information and the demonstration of the major PCSK9 expression in the liver (51), we contacted the French team led by Catherine Boileau, who was keenly interested in this region of chromosome 1. Intensive genetic analyses in 23 French families, with no LDLR or APOB variations, by a positional cloning approach using genetic mapping, physical mapping, and sequencing of several genes led to their identification of a missense variant, S127R, in the 22-kb PCSK9 gene in 2 large families and another variant, F216L, in a third family (52). This fruitful and joyful collaboration between a Canadian team working on PCs and a French one deciphering the genetics of hypercholesterolemia culminated in the first report that appeared in June 2003, with first author Marianne Abifadel revealing that human PCSK9 was the FH3 gene critical for LDLc regulation (52).
Analysis of PCSK9 biosynthesis in cells expressing human, rat or mouse orthologues revealed that, like other PCSKs, this enzyme also undergoes autocatalytic processing of its N-terminal prodomain in the ER (51) at the VFAQ152↓ site (55, 56), where the italic bold residues are critical for processing (57). However, PCSK9 is the only PCSK that always remains noncovalently associated with its prodomain (58), even when secreted (51). This was confirmed by 3D-analysis (59, 60) of circulating PCSK9 secreted from liver (61). Thus, PCSK9 acts as a protease only once, upon its autocatalytic processing in the ER, suggesting that secreted PCSK9 regulates LDLc levels by a nonenzymatic mechanism, and rationalizing the existence of gain-of-function (GOF) variants, unusual for an enzyme.
PCSK9 Promotes the Degradation of the LDLR and Other Family Members by a Nonenzymatic Mechanism
The first critical clues on the mechanism of action of PCSK9 came from 2 reports (early 2004 and late 2005) led by Maxwell and Breslow, who demonstrated that PCSK9 overexpression dramatically reduced the protein, but not mRNA, levels of the LDLR by inducing its degradation (62, 63) within the acidic endosomal/lysosomal pathway (56, 62, 64). The same authors had shown a few months earlier by microarray analysis that PCSK9 was strongly downregulated by dietary cholesterol and highly upregulated by SREBP-1a and SREBP-2, providing evidence that PCSK9 is a cholesterol-regulated gene (65). This conclusion was confirmed in a similar study (66) and by the ability of statins to enhance PCSK9 transcription (67). Thus, although PCSK9 and LDLR mRNA levels were both positively regulated by the lack of cholesterol and statin treatment, PCSK9 induced LDLR protein degradation (68). This clarified the mechanism behind the reported human mutations leading to hypercholesterolemia (52, 69). Thus, PCSK9 GOF result in exacerbated PCSK9-induced degradation of the LDLR.
A definite further support for PCSK9 function came with 2 elegant studies by Cohen et al., who sequenced PCSK9 in individuals exhibiting very low cholesterol levels, thereby unambiguously associating 2 prevalent heterozygote loss-of-function (LOF) variants Y142X and C679X in African Americans (2% combined frequency) with ~40% reductions in LDLc (70), and amazingly with an 88% lower incidence of coronary heart disease during a 15-year follow-up period (71). This was the first strong evidence that PCSK9 may be acting stoichiometrically on the LDLR, rather than as a protease, as enzymes usually require >90% loss activity to have a noticeable effect on their function (72). In 2005, Pcsk9 inactivation in mouse confirmed that the loss of PCSK9 expression was associated with ~3-fold higher LDLR levels in the liver and with a dramatic reduction of plasma LDLc (73). The viability of these knockout (KO) mice as well as the discovery of the first individuals completely lacking functional PCSK9 (74, 75) established this protein as an attractive therapeutic target for LDLc reduction.
In 2007, another key step was achieved by revealing that PCSK9 does not carry any protease activity in trans. The first reported crystal structures of PCSK9 (59, 60) showed that the C-terminus of the autocatalytically excised prodomain was solidly embedded in the substrate-binding groove, likely blocking access to any substrate. These structural data confirmed our original observation of the secretion of mature PCSK9 as a noncovalent complex with its inhibitory prodomain (51). In agreement, co-expression of the prodomain of PCSK9 with a catalytically dead mutant of mature PCSK9 in which the active site Ser386 was mutated to Ala (S386A) led to a reconstituted fully functional and secreted PCSK9 that can mediate LDLR degradation, similar to wild-type (WT) PCSK9 (76). This conclusion was later confirmed in another study using a similar approach with a PCSK9 mutant of the active site His226 (H226A) that also resulted in a similar degradation pathway of other family members VLDLR and ApoER2 (77). Thus, PCSK9 acts as a protease only once during its autocatalytic zymogen processing in the ER.
Insights From the Structure–Function of PCSK9, LDLR, and Their Complex
The 3D structures of PCSK9 revealed the existence of 3 distinct structural domains (Fig. 1): the prodomain (aa 31-152), the catalytic subunit (aa 152-421), and the C-terminal Cys/His-rich domain (CHRD; aa 453-692) (59, 60), each playing critical roles in the regulation of PCSK9 and its intracellular traffic.
Figure 1.

Schematic of PCSK9 and its potential partners. PCSK9 undergoes an autocatalytic cleavage between its catalytic and N-terminal prodomains, but the latter remains associated to the protein that has no other substrates. The C-terminal domain, CHRD, is composed of 3 tandem repeats (M1, M2, and M3) rich in Cys and His residues (Cys/His-rich domain). The LDLR binds the catalytic domain of PCSK9, while the MHC-I complex interacts with the M2 repeat of the CHRD. On the other hand, CAP1 binds the M1 and M3 domains of the CHRD and enhances PCSK9 activity.
The Prodomain (aa 31-152)
Following cleavage of the signal peptide, the precursor of PCSK9 (proPCSK9) is autocatalytically cleaved at VFAQ152↓SI very early in the ER (55, 56), but different from the other PCSKs, PCSK9 remains associated to its prodomain, even after its secretion (51). The prodomain and its cleavage are necessary for the exit of PCSK9 from the ER (51, 76, 77). Any variant that blocks ER exit, such as Q152H (57, 78), prevents its secretion and results in a LOF and hypocholesterolemia (79). The X-ray crystallography analyses of PCSK9 at both neutral and acidic pH (59, 60, 80, 81) revealed that the structure of the N-terminal Gln31–Thr60 segment is not resolved, suggesting a disordered, mobile region. Interestingly, PCSK9 lacking most of this segment composed of 25% of Glu and Asp residues (PCSK9-Δ33-53) enhanced by >7-fold the affinity of PCSK9 for the LDLR (81) and by ~3-fold the degradation of the LDLR in endosomes/lysosomes of hepatocytes (82, 83). This suggests that the acidic N-terminal segment of PCSK9 may bind an important ligand/partner (84). More than 40 PCSK9 GOF or LOF variations were reported in this sequence (84), including the common LOF R46L (71, 85) associated with protection against heart disease (71, 84), as well as Tyr38-sulphation (30) and Ser47-phosphorylation (86).
The Catalytic Domain (aa 153-421)
Degradation of the LDLR by PCSK9 was shown to depend on the internalization of the PCSK9-LDLR complex into clathrin-coated acidic endosomes (87-89). Through its catalytic subunit, secreted/plasma PCSK9 (90) binds the EGF-A domain of the LDLR (80, 81, 91). This stable complex is directed toward endosomes/lysosomes for degradation by a yet unknown mechanism(s), preventing LDLR recycling to the cell surface (56, 87, 88, 90). PCSK9 may also escort LDLR for degradation directly from the trans-Golgi network, although this intracellular pathway does not fully resemble the extracellular route (61, 92-94). However, for still obscure reasons, the pathway utilizing extracellular/circulating PCSK9 is the primary functional pathway in hepatocytes (90, 95, 96), small intestine (97), and pancreas (98, 99). In agreement, serum LDLc levels correlate directly with those of circulating PCSK9 levels (100-104), and statins regulate the levels of LDLc in part by upregulating those of circulating PCSK9 in mice and human (73, 104, 105). Within the catalytic domain, 3 reported PCSK9 LOF variants F216L (52), R218S (106) and R215H (107) led to the demonstration that Furin inactivates PCSK9 by cleavage at RFHR218↓ (30, 108). The latter site lays in an exposed mobile loop not resolved in any crystal structure (59, 60), in agreement with its accessibility to proteases such as Furin. Among the GOF variants, the most powerful is the Anglo-Saxon D374Y (69), which exhibits a 10- to 20-fold higher binding affinity for the LDLR (59, 109) and resistance to Furin cleavage (30). Reciprocally, an LDLR-H306Y variant within the EGF-A domain enhances PCSK9 binding to the LDLR and hence is associated with hypercholesterolemia (110). The crystal structure of the PCSK9-EGF-A(H306Y) complex revealed that indeed, Tyr306 forms a hydrogen bond with Asp374 in PCSK9 at neutral pH, which strengthens the extracellular complex (110).
The Hinge-CHRD Domain (aa 422-692)
The first crystal structures of PCSK9 (59, 60) and its alignment with other PCSKs revealed that the catalytic domain is followed by a relatively disordered hinge region (aa 422-452), and an ordered C-terminal 240 aa CHRD. The latter is composed of 3 tandem repeats tightly packed into structurally similar modules named M1 (aa 453-529), M2 (aa 530-603), and M3 (aa 604-692), whose frontiers are delimited by their β-sheet structures (Fig. 1). Each module exhibits a 6-bladed domain containing 6 Cys within the motif: (X)4C (X)17-19C (X)8-9C (X)18-25C (X)11-23C (X)2-13 forming 3 disulfide bridges connecting Cys 1 to Cys 6, Cys 2 to Cys 5, and Cys 3 to Cys 4 within each repeat (59, 60). Interestingly, 9 out the 14 His residues in the CHRD are present within the M2 module, suggesting that they may exert a pH-dependent role, especially at the acidic pH of endosomes. The M1–M2–M3 modules exhibit a structural homology to the homotrimer resistin (111), a small cytokine associated with obesity and diabetes, and whose plasma levels have been linked to inflammation, cancer, atherosclerosis, and cardiovascular disease (112). Most of the reported PCSK9 variations in the C-terminal domains are within the M1 and M3 modules. The hinge region and M2 module exhibit a lower rate of genetic variations, but a greater structural mobility (59, 60), suggesting that they are subject to a stronger selection pressure to interact with putative partners.
Complete LOF Variants of PCSK9 Exhibit a Healthy Profile
Since 2003, a large number of substitutions/deletions have been reported in each of the PCSK9 domains (for reviews see (84, 95, 113, 114)), some having major effects on PCSK9 activity through various underlying mechanisms (57, 114-117). Interestingly, 2 unrelated Canadian FH patients who are refractory to intensive statin therapy exhibit a whole gene duplication of PCSK9 (118). They presented severely elevated LDLc levels and premature cardiovascular events, and one of them had PCSK9 levels of ~5000 ng/mL, thus >20-fold higher than normal ones.
The few individuals lacking functional PCSK9, namely ΔR97/Y142X (74), C679X/C679X (75), and the monoallelic double variant R110C+V114A (116), seem healthy and exhibit circulating levels of LDLc of ~14 to 16 ng/mL, a value that is ~8-fold lower than the normal levels (~110-150 ng/mL) (71, 75, 116). In the latter case, a double mutation in the prodomain abolishes the autocatalytic processing of PCSK9, generating an unfolded proPCSK9 remaining in the ER that acts as a dominant negative by preventing the exit from the ER of the processed WT PCSK9 that is encoded by the intact allele (116). Similarly, French Canadian individuals carrying a Q152H substitution at the autocatalytic VFAQ152↓ cleavage site present an almost complete loss of circulating PCSK9, due to a dominant negative effect of proPCSK9-Q152H (57, 78), resulting in ~3-fold lower levels of LDLc (~40-50 ng/mL) (78, 79). Even though the Q152H substitution leads to the accumulation of proPCSK9 in the ER, the 30 heterozygote and 3 homozygote individuals identified seem to be protected against ER-stress (79). Indeed, the ER-retained proPCSK9-Q152H may act as a co-chaperone by increasing the levels of the ER-resident chaperones GRP94 (119) and GRP78, which play central roles in the regulation of protein folding and the unfolded-protein response (120). This results in a protection against ER stress-induced apoptosis, as also reported for other proteins that increase the stability of GRP78, such as Bag5 (121) and GALNT6 (122). These data suggested that inhibition of the autocatalytic cleavage of proPCSK9 in the ER may represent a safe and unique approach for the management of hyperlipidemia, with the added benefit of preventing liver dysfunction (79). The above human genetic studies suggested that humans lacking functional PCSK9 can live normal lives, and heterozygote complete LOF variants largely (88%) protect individuals from the development of cardiovascular complications and coronary heart disease over their lifetime (71). The unusually large contribution of some heterozygote LOF variants (C679X, R110C+V114A, Q152H) seems to be in part related to a dominant negative effect of the protein carrying the mutant allele on the trafficking of the WT PCSK9, preventing its exit from the ER (57, 78, 116).
The CHRD Is Critical for the PCSK9-Induced Degradation of the LDLR
PCSK9 and the LDLR primarily interact through their catalytic and EGF-A domains, respectively (80, 91). A minor interaction between Leu108 of the PCSK9 prodomain and Leu626 in the β-propeller domain of the LDLR was also reported (80, 123). Notably, the PCSK9 GOF L108R variant may reinforce the interaction of these 2 domains by transforming the initial hydrophobic bond into an electrostatic one between Arg108 in the PCSK9 variant and Glu605 of the β-propeller domain of the LDLR (80, 123).
Cellular data revealed that the CHRD of PCSK9 is required to trigger LDLR degradation (87, 124-127), but its absence in PCSK9-ΔCHRD still allows the binding of PCSK9 (or proPCSK9) to the LDLR (87, 126). On the other hand, the PCSK9-D374Y variant was able to degrade the LDLR in autosomal recessive hypercholesterolemia protein–independent manner (128), as well as the LDLR mutant K811X that lacks the cytosolic tail (129) or a chimeric LDLR carrying the C-terminal domain of the transferrin receptor (130). These data indicated that a third partner is required for the efficient LDLR degradation by PCSK9 (129, 131). It was hypothesized that this “protein X” binds the CHRD and allows the sorting of the PCSK9-LDLR complex to acidic degradation compartments (96). Intriguingly, it was reported that the CHRD could be substituted by the C-terminal domain of DsRed-Express fluorescent protein, an unrelated protein of comparable size and overall positive charge, and promote LDLR degradation (126). Because mutations of multiple histidines in the CHRD are needed to affect its activity, the authors suggested that the overall positive charge was more important than specific sequences within the CHRD. However, this may not be the whole story. Indeed, monoclonal antibodies (mAbs) (127, 132) and single domain antibodies (133, 134) that target the M1 and/or M3 domains of the CHRD inhibit the PCSK9-induced LDLR degradation without blocking the binding of PCSK9 to the LDLR. Although Sortilin and APLP2 can bind the CHRD, they were shown not to be credible “protein X” candidates (135). To date, none of the studies that investigated the trafficking of the PCSK9-LDLR complex (87, 131, 135-137), have identified all the critical partners needed to escort the complex into late endosomes/lysosomes for degradation.
A recent study proposed that the cytosolic adenyl cyclase-associated protein 1 (CAP1) is a candidate “protein X” (95, 135). The authors were inspired by its ability to bind resistin (138), a protein that bears a structural homology to the CHRD (111), as we mentioned above. CAP1 seems to bind the M1 and M3 domains and enhances degradation of the PCSK9-LDLR complex in lysosomes (138). They also showed that the PCSK9 E670G and S668R LOF variations in M3 considerably reduced the ability of PCSK9 to bind CAP1, likely due to the loss of phosphorylation at both Ser666 and Ser668 (86). These Ser phosphorylations are performed by FAM20C at Ser-X-Glu or Ser-X-phosphoserine sites (16). In that context, we previously reported that PCSK9 phosphorylation at these Ser residues located within the M3 domain enhanced LDLR degradation (86), suggesting that CAP1 may best bind phosphorylated PCSK9. CAP1 is a cytosolic protein, but “protein X” was predicted to be a secretory membrane–bound protein (96). The authors proposed that CAP1 can somehow flip from the inner-leaflet to the other side of the hepatocyte plasma membrane for exposure to the extracellular matrix (138), thereby binding extracellular PCSK9 to mediate caveolae-dependent endocytosis and lysosomal degradation of the PCSK9-LDLR complex.
However, because some cytosolic proteins can be secreted via an unconventional secretory pathway (139), we cannot exclude that this could also apply to CAP1, as for the cytosolic Annexin A2 that also binds the CHRD (140). Note that the PCSK9-Annexin A2 complex is internalized via clathrin-coated vesicles, but not caveolae (138). Nevertheless, CAP1 seems to be an important positive regulator of PCSK9 function on the LDLR, possibly by exposing a domain in the CHRD (eg, M2 domain) that may best bind the elusive membrane-bound “protein X” (141).
PCSK9 Mouse Models
We originally showed that statins upregulate the expression of both LDLR and PCSK9 mRNA and protein (67), revealing a conundrum in LDLc regulation since PCSK9-mediated LDLR degradation will blunt the contribution of higher levels of LDLR transcripts (68). This effect explains the higher sensitivity of apoB levels to statins in PCSK9 KO mice (73). Indeed, statins have a more prominent effect in KO than in WT mice, where hepatic LDLR protein levels accumulate in the absence of PCSK9. This has paved the way for the combined statin/PCSK9 mAb therapy. In agreement, LDLR labeling of primary mouse hepatocytes lacking PCSK9 (142), and of liver sections from mice lacking PCSK9 in the 2 first Pcsk9 KO models (73, 143), especially in male mice (61), revealed a strong accumulation of the LDLR protein at the hepatocyte cell surface.
Analysis of mouse and rat PCSK9 mRNA expression showed that it is restricted to discrete areas of the brain during development (51, 95, 144), and that its antisense morpholino-oligonucleotide knockdown in zebrafish leads to embryonic death with major neuronal deficits (145), although CRISPR/Cas9 deletion of its gene in zebrafish did not (146). Based on the previously observed embryonic lethality of Furin, PC5 and SKI-1/S1P KO in mice (28), we opted to develop a conditional mouse Pcsk9 KO strategy. This long and tedious approach led to both complete (143) and tissue-specific Pcsk9 KO mice, namely in hepatocytes (61, 143), enterocytes (97) and pancreatic β-cells (98). PCSK9-deficient mice are viable, with no apparent neuronal phenotype (73, 144).
Two other KO models were reported but not widely used thereafter. The first one was a whole-body KO model even though it derived from a conditional strategy. Comparison of the generated KO mice with transgenic mice overexpressing PCSK9 in liver demonstrated that overexpressed PCSK9 did not regulate LDLR, VLDLR, and apoER2 in the brain, likely because it does not cross the blood–brain barrier (147). The second KO model, homozygous for the Y119X mutation, was generated by N-ethyl N-nitrosourea mutagenesis and backcrossed into the C57BL/6J background using speed congenics (148). Thorough plasma, liver, and bile acid analyses in WT and KO mice treated or not with atorvastatin revealed that cholesterol metabolism and hepatic homeostasis were well maintained in the absence of PCSK9.
Mice lacking PCSK9 exhibit 40% to 50% lower circulating cholesterol, ~80% less LDLc, and ~3- to 4-fold higher levels of total liver LDLR protein (73, 143). Whole body and hepatocyte-specific PCSK9 KO mice revealed that, in the liver, PCSK9 is exclusively expressed in hepatocytes, which also represent the only source of circulating PCSK9 (61, 98, 143). Single LDLR or double LDLR/PCSK9 KO mice exhibit similar cholesterol profiles, indicating that PCSK9 regulates plasma cholesterol homeostasis essentially through the LDLR. Finally, PCSK9 KO regenerating livers exhibited necrotic lesions that were prevented by a high-cholesterol diet, suggesting that the absence of PCSK9 severely reduces tissue cholesterol levels and confers resistance to liver steatosis (143). Indeed, despite the 4-fold higher levels of LDLR at the hepatocyte surface of male mice, as measured by immunohistochemistry or liver fractionation, there is no accumulation of cholesterol in their liver (73, 143, 149). This is likely due to the 5-fold lower circulating LDLc levels, preventing any significant upsurge of cholesterol in hepatocytes. In the β-cells of the same KO mice, the low levels of circulating LDLc resulted in increased LDLR mRNA and protein levels, indicating that a cholesterol deficit was not fulfilled by the higher LDLR levels in these cells, as reflected by an upregulated the SREBP-2 pathway (98). Thus, even though both hepatocytes and β-cells of male PCSK9 KO mice express higher levels of LDLR, they do not accumulate cholesterol because of the low circulating levels of LDLc. Early data showed that like insulin, circulating PCSK9 is dramatically reduced (>95%) in fasted in mice (150). In addition, circulating PCSK9 strongly reduces the surface levels of the VLDLR protein in adipose tissue (61), and those of CD36 (151). Thus, PCSK9 may favor cholesterol uptake by peripheral tissues via hepatic LDLR degradation and reduced visceral adipogenesis.
The first indication that PCSK9 enhances the development of atherosclerosis was the demonstration that mice fed a high cholesterol diet for 15 weeks and expressing the GOF PCSK9-D374Y at physiological levels exhibited extensive atherosclerotic plaques compared with WT mice (152). This was further confirmed following a single injection of recombinant adeno-associated viral vectors encoding PCSK9-D374Y that rapidly induced atherosclerosis in mice and eliminated the need for germ-line genetic modifications (153). These data suggested that high levels or activity of PCSK9 may exacerbate the development of atherosclerosis and possibly enhance inflammation. To prove that the reverse is true in mice lacking PCSK9, we used mouse models of accelerated atherosclerosis consisting of either WT mice, PCSK9 KO or transgenic mice in ApoE or LDLR KO backgrounds. The data demonstrated a direct relationship between PCSK9 and atherosclerosis, whereby PCSK9 overexpression is proatherogenic and its absence is protective (154). A later report demonstrated that PCSK9 mRNA silencing suppressed atherosclerosis directly through inhibiting the TLR4/NF-κB signaling, which induces the expression of pro-inflammatory mediators, without affecting plasma cholesterol levels in high fat diet-fed ApoE KO mice (155). It was suggested that this may implicate the intermediary oxidized-LDL, a potent proinflammatory mediator of atherosclerosis (156), which induces PCSK9 expression in macrophages via activation of TLR4/NF-κB signaling. Indeed, PCSK9 seems to interact with the oxidized-LDL receptor-1 (LOX-1) in a mutually facilitative fashion (157). Finally, a PCSK9-specific vaccine reduced total cholesterol, vascular inflammation, and atherosclerosis in the atherogenic APOE*3Leiden.CETP mouse (158). In this context, in an inflammatory milieu, elevated levels of PCSK9 potently stimulate the expression of LOX-1 and oxidized-LDL uptake in macrophages, and thus contribute to the process of atherogenesis (159). Accordingly, there may be additional clinical benefits of PCSK9 inhibition on mechanisms unrelated to increased LDLR activity, but rather to a reduction of vascular inflammation.
PCSK9 is expressed in β-cell-derived cell lines (51), as well as in pancreatic islets (160). According to single-cell transcriptomics performed on dissociated cells from 20 murine tissues and compiled in the Tabula Muris database (161), β-cells constitute the cell type richest in PCSK9 expression, closely followed by hepatocytes. Two recent studies, including our data on β-cell-specific PCSK9 KO mice (98), show that endogenous PCSK9 in β-cells is well secreted and induces LDLR degradation, but does not affect glucose-stimulated insulin secretion (98, 99). Although β-cell PCSK9 cannot contribute significantly to the circulating pool of PCSK9 (98), its absence leads to an adaptative downregulation of the pancreatic SREBP-2 pathway. This reflects a higher rate of cholesterol internalization by unchanged LDLR protein levels and indicates that endogenous PCSK9 synthesized in the β-cell does indeed target the LDLR for degradation. However, in mice completely lacking PCSK9, pancreatic LDLR protein levels are increased by 2-fold, showing that the 5-fold lower circulating LDLc in these mice cannot satisfy the high requirement that β-cells have for cholesterol (98). Altogether, these data are reassuring in relation to the concern of a possible toxic effect of the absence of PCSK9 via excessive cholesterol internalization in β-cells (98, 99).
Even though the absence of PCSK9 results in 3- to 4-fold higher levels of the LDLR protein in the liver of both sexes, the cell surface levels of the receptor increased in male livers, as previously reported (73, 143), but not in female ones (149). This dimorphism was not restricted to the liver. Pancreatic islets also exhibited a stronger cell surface density of the LDLR in PCSK9 KO male mice vs female ones, while the opposite was observed for the intestinal epithelium, with PCSK9 KO female mice presenting the strongest surface LDLR labeling (149). Supplementation of ovariectomized mice with placebo or 17β-estradiol (E2) generated typical male and female labeling patterns, respectively. These observations were confirmed biochemically by Western blot analysis of liver fractions enriched in plasma membrane, in which LDLR protein levels were 3- to 4-fold higher in male than female fractions (149). Thus, in PCSK9-deficient female mice, E2 seems to take control over LDLR density at the hepatocyte surface. Whether E2 inhibits the targeting of the LDLR to the cell surface or favors its elimination from the cell surface remains to be determined. Intriguingly, despite the E2-mediated modulation of surface LDLR, male and female mice exhibit similar circulating levels of LDLc, indicating that the higher density of surface LDLR in male hepatocytes does not have a major effect on the LDLc clearance.
Major Cardiovascular Outcome Trials Using PCSK9 Inhibitors
Lipid-modifying agents are often prescribed to reduce atherogenic lipid levels and to prevent atherosclerotic cardiovascular disease (162). In view of the major effects of the lack of PCSK9 on the substantial reduction of circulating LDLc, and the overall safety of PCSK9 LOF or its complete absence in animal models (73, 143) and humans (71, 74, 75, 116), it was likely that potent PCSK9 inhibitors (PCSK9is) could represent a viable strategy to reduce LDLc. Various approaches have been proposed to reduce the circulating PCSK9 activity via mAbs or protein/peptide inhibitors, or through silencing its mRNA expression or translation in liver (163, 164). So far, major efforts by pharmaceutical companies led to the successful development of 2 distinct, robust, safe, and long-term PCSK9i (Fig. 2): (1) PCSK9 mAbs (evolocumab/Repatha and alirocumab/Praluent) that inhibit the interaction of circulating PCSK9 with the LDLR, thereby silencing its extracellular activity; (2) PCSK9 small interfering RNA (siRNA) in lipid nanoparticles (Inclisiran) specifically delivered to the liver that prevents PCSK9 translation (165), thereby abrogating both intracellular and extracellular functions of PCSK9 (92) in hepatocytes. Both approaches result in a ~50% to 60% reduction in LDLc above that obtained with statins alone. The reader is referred to very recent reviews on the history, efficacy, safety, and potential clinical uses and outcomes of these PCSK9i (163, 165-174). Below, we briefly describe salient features that emanated from the major clinical trials using either mAb or siRNA approaches, the targeted populations, and their outcomes.
Figure 2.
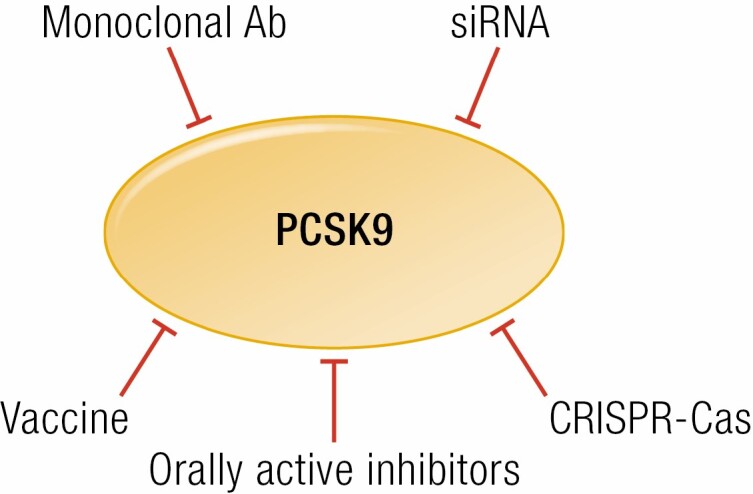
PCSK9 inhibition. Monoclonal Abs and siRNAs are safe and temporary inhibitors of PCSK9. Irreversible inhibition may be achieved through vaccination or PCSK9 gene modification via CRISPR-Cas. Finally, a more affordable inhibition based on orally active inhibitors is in development. See text for references.
The mAb Approach to Inhibit Circulating PCSK9 Activity on the LDLR
The liver is the primary source of circulating PCSK9, and the latter is a target of mAbs that recognize the catalytic subunit and sterically prevent the binding of PCSK9 to the EGF-A domain of the LDLR in hepatocytes and other tissues (175). A single subcutaneous injection of a neutralizing PCSK9 mAb to monkeys resulted in an ~80% reduction of LDLc after a single injection (176). Following this initial proof-of-principle and impressive efficacy, fully humanized mAb were developed. evolocumab (Repatha; Amgen) and alirocumab (Praluent; Sanofi) are presently the only 2 humanized mAb that successfully sailed through all the multiple clinical trials in hypercholesterolemic patients exhibiting or not FH mutations. Starting from 2015/2016, these mAb have been prescribed to patients in more than 30 countries as they generate a robust and sustained ~50% to 60% reduction in LDLc that has been consistently observed over the last 3 to 5 years. Their safety and tolerability profile are excellent, especially for hypercholesterolemic patients or those who do not optimally respond to or tolerate statin treatment. Only few patients exhibited mild injection site reactions. Below, we describe some outcome data that provide concrete evidence for the long-term clinical benefits of reducing circulating PCSK9 activity.
In patients with LDLc levels between 100 and 190 mg/dL, mAb monotherapy reduced LDLc levels by 50% to 55% after 12 to 24 weeks of treatment (177, 178), and similar LDLc reduction was observed in statin-intolerant patients (179). Thus, it became apparent that PCSK9 mAb treatment was efficacious in a broad range of patients with similar outcomes with respect to LDLc reductions (180). The combination of these mAbs with other lipid-lowering agents such as statins or the ezetimibe/fenofibrate resulted in a modest (≤15%) further decrease in LDLc (181, 182). Interestingly, all studies showed that treatment with PCSK9 mAb leads to significantly higher reductions in LDLc in the presence of a statin treatment, even though statins enhance the expression of PCSK9 via activation of the SREBP-2 pathway (67, 105). The increased levels of circulating PCSK9 in response to statins and/or ezetimibe may in part rationalize why PCSK9-antibodies are highly effective in reducing LDLc as an add-on therapy (183). Since PCSK9 primarily reduces LDLR levels in hepatocytes, the mAb approach works very well for normal and heterozygote FH (HeFH) patients (184, 185) and only partially for homozygous FH (HoFH) patients who exhibit residual LDLR activity due to incomplete silencing of its function (186).
In 2017 and 2018, 2 landmark outcome trials were reported on the use of evolocumab (187, 188) and alirocumab (189) as therapeutic mAb inhibitors of circulating PCSK9 activity in combination with statins.
• In the FOURIER trial a total of 27 564 patients with a mean age of 62.5 years (25% women) and receiving at least 20 mg of atorvastatin were randomized to subcutaneous doses of evolocumab (140 mg every 2 weeks or 420 mg monthly) or placebo. After 48 months, evolocumab treatment effectively lowered LDLc to a mean value of 30 mg/dL (maintained in follow up studies), and significantly decreased the risk of recurrent cardiovascular events in patients with established atherosclerotic cardiovascular disease (187) and/or type 2 diabetes (188). Evolocumab reduced the relative risk of the primary endpoint by 15% with a hazard ratio of 0.85 (95% CI 0.79-0.92) during a median follow-up of 2.2 years. The risk of the key secondary endpoint, a composite of cardiovascular death, myocardial infarction, or stroke, had a relative reduction of 20%. Effectively, this means that the number of patients to treat to have a highly significant prevention outcome is between 5 and 7 patients. Interestingly, LDLc lowering with evolocumab was also highly effective in reducing the risk of myocardial infarcts (190). In general, it was observed that patients with higher cardiovascular risk due to the presence of multiple cardiovascular risk factors in their genome or those suffering from more severe atherosclerotic disease had the largest clinical benefit as measured by the absolute risk reduction. Treatment with evolocumab for 2.2 years did not result in an increased risk of developing diabetes, or worsening glycemia (188), in agreement with recent studies on complete and β-cell specific Pcsk9 KO mice (98), and on β-cells isolated from human pancreas treated with alirocumab or siRNA silencing of PCSK9 expression (99). Patients with metabolic syndrome, having a higher event rate than patients without, exhibited similar reductions in LDLc and risk for primary and key secondary endpoints, without increasing new-onset diabetes, worsening glycemic control, or other major safety events (191). Overall, within the follow-up period of FOURIER, treatment with evolocumab resulted in very low LDLc and was not accompanied by any adverse medical events. It was also observed that treatment with evolocumab resulted in a median reduction of ~27% in Lp(a), an independent cardiovascular risk factor (192, 193). Whether this is due to the higher levels of LDLR in liver induced by evolocumab (194, 195) or to a lower rate of Lp(a) secretion (196, 197) is still under debate.
• The ODYSSEY outcomes study included 18 924 patients with a mean age of 59 years (25% women), and alirocumab treatment targeted patients with a history of an acute coronary syndrome within 1 to 12 months before randomization (189). Atorvastatin 40 mg or rosuvastatin 20 mg were included, and patients were randomized to alirocumab 75 mg every 2 weeks or placebo subcutaneously in a 1:1 ratio, with dose adjustment allowed during treatment. In contrast to the FOURIER trial in which no dose adjustments were allowed, at 48 months of alirocumab + statin treatments, LDLc was reduced to a mean of 66 mg/dL. The primary endpoint was a composite of coronary death, myocardial infarction, ischemic stroke, and unstable angina requiring hospitalization. After a median follow-up of 2.8 years, the relative risk for the primary endpoint was reduced by 15% with a hazard ratio of 0.85 (95% CI 0.78-0.93), although death from coronary artery disease or from cardiovascular causes was not. These values are comparable to those obtained in the FOURIER trial using evolocumab + statin. Notably, treatment with alirocumab resulted in an absolute 16.2% reduction in death risk in patients suffering from a composite coronary artery, peripheral artery, and cerebrovascular disease (198). Finally, extensions of this trial revealed that longer PCSK9 inhibition has the potential to reduce mortality after an acute coronary syndrome, especially in patients with elevated risk due to high LDLc.
The siRNA Approach to Silence Liver PCSK9 mRNA Expression
Since loss of PCSK9 expression in liver resulted in substantial reduction of LDLc, without apparent negative consequences in human (71, 74, 75, 116) or mice (73, 143), silencing of its hepatic expression was tested using an injectable lipid nanoparticle formulation carrying an antisense siRNA to PCSK9. Accordingly, an optimized siRNA version (Inclisiran) is composed of complementary 21 sense and 23 antisense oligonucleotide sequences that have been modified for durability and low immunogenicity. The sense strand is conjugated to triantennary N-acetylgalactosamine (GalNAc) to facilitate siRNA uptake by the liver via binding to the asialoglycoprotein receptor 1 (ASGR1) (199), which is highly expressed in hepatocytes (200). This approach enhances the level of the liver-specific delivery of the antisense strand and its efficacy to silence PCSK9 expression. After endosomal uptake, a small part of the siRNA is released into the cytoplasm, where the dissociated antisense strand binds the PCSK9 mRNA and recruits several proteins that assemble the RNA-induced silencing complex (RISC) resulting in the PCSK9 mRNA degradation over a prolonged period of time (171). Accordingly, Inclisiran subcutaneously injected twice a year substantially reduces by 50% to 60% the levels of LDLc (168, 170). Various ORION clinical trials were designed to test the efficacy and safety of Inclisiran in the presence of statins. Currently, the phase III cardiovascular outcome study ORION-4 is ongoing with completion expected in 2024 (https://clinicaltrials.gov/ct2/show/NCT03705234). Another study involves patients with atherosclerotic cardiovascular disease and elevated LDLc (≥70 mg/dL) despite receiving maximally tolerated statin therapy, with completion expected in 2023 (https://clinicaltrials.gov/ct2/show/NCT04929249?term=inclisiran&draw=2&rank=1). So far, it looks that Inclisiran administration in various subgroups of patients generates a consistent and sustained ≥50% LDLc reduction with no safety concern, except for some mild/moderate injection site effects that are not persistent. These overall positive data should encourage patient compliance with this siRNA treatment.
It should be underlined that the mAb approach requires repeated injections at 2- to 4-week intervals (13 or 26 injections/year), whereas the siRNA treatment is administered twice a year. Inclisiran received marketing authorization in the European Union in December 2020 for use in adults with primary hypercholesterolemia or mixed dyslipidemia, but its authorization in the United States still awaits the Food and Drug Administration approval. So far, both approaches seem safe following 2 to 5 years of clinical use, but we will have to await longer treatments for a more thorough assessment of the long-term effects of reducing liver vs circulating PCSK9.
From 2008 to 2018, the consumption of cholesterol-lowering agents defined as standard units consumed per 1000 inhabitants increased by ~4.1%/year, with statins representing the lion share of a market that presently targets >170 million people worldwide (201). However, PCSK9i are very useful when maximally tolerated statin therapy does not reduce LDLc sufficiently and in statin-intolerant patients. Compared to 2015, the year when PCSK9 mAbs were first approved by the Food and Drug Administration, prescriptions of PCSK9i increased by >5-fold during the years 2016-2018, especially in higher income countries (201). Over the last 6 years the beneficial effects of PCSK9i administration have been widely recognized, encouraging their prescription both in the primary and secondary prevention of cardiovascular incidents. However, the major stumbling block preventing their wider use has been their exorbitant cost (recently reduced to ~5800$/year in the United States). It is likely that prescriptions of PCSK9i would be much more widely adopted if the cost was substantially further reduced (202, 203).
Future Strategies Beyond mAb and siRNA
PCSK9-vaccination is a permanent approach proposed to silence PCSK9. Peptide-based vaccines have so far shown promising results in mice or macaques, including the use of bacteriophage virus-like particles displaying PCSK9-derived peptides (204, 205). Whether vaccination against PCSK9 using modified lipid nanoparticles (206) or effective capsid-like particles (207) expressing full-length PCSK9 will be approved to treat severe pathologies will have to be carefully evaluated for any harmful side effects of such an irreversible therapy (163).
In 2012, the advent of clustered regularly interspaced short palindromic repeats (CRISPR)-associated (Cas) system of gene deletion/editing (208) has revolutionized the field of molecular biology, leading to powerful approaches relying on Cas9/sgRNA ribonucleoprotein complexes to edit DNA. Multiple applications are thus made possible in various pathologies and gene replacement therapies (209-211). Very early, proof-of-concept Pcsk9 inactivation was reported using an adeno-associated viral vector-9 delivery of CRISPR-Cas9 to mouse liver (212), or in utero injections of an adenoviral vector expressing the safer SpCas9 base editor 3, which enables single-base conversions without generating double-stranded breaks (213). In both cases, cholesterol levels were significantly reduced. The small size of Cas12a also promoted its use in gene editing that led to a ~48% reduction in the expression of PCSK9 in mice (214). Finally, intravenous injection of nonviral lipid nanoparticles (LNPs) that deliver Cas9/sgRNA ribonucleoprotein complexes was reported to significantly decrease serum PCSK9 levels in mice, even though the latter are not specifically delivered to the liver (215). More recently, a single infusion of LNPs allowed the delivery of the more advanced base-editing CRISPR-ABE8.8 system that successfully modified the PCSK9 gene of cynomolgus monkeys and achieved ~90% and ~60% reductions of plasma PCSK9 and LDLc, respectively, for more than 8 months (216). These impressive reductions of LDLc should even be reinforced by using LNPs that specifically target the liver.
PCSK9 and Sepsis
Septic shock is often a fatal complication following sepsis, a multiorgan disease caused by complex arrays of bacterial and pathogenic molecules that trigger an uncontrolled systemic inflammatory response and subsequent organ failure (217). Beyond antibiotic therapy, there are still no effective treatments for septic shock (218). The first evidence that the loss of PCSK9 may be beneficial was provided by Walley et al. in 2014, both in humans and mice (219). Notably, PCSK9 KO mice were protected from septic shock induced by lipopolysaccharide (LPS) injections. Subsequently, PCSK9 LOF variants were reported to exhibit less frequent septic shocks and organ failures (220, 221), whereas the reverse is true in transgenic mice overexpressing PCSK9 (222). The LDLR clears, in an LDL-dependent mechanism, gram-positive lipoteichoic acid and gram-negative LPS, known to exacerbate sepsis pathophysiology (223). Because the protective effect of the lack of PCSK9 is abrogated in LDLR KO mice, it was proposed that the lack of PCSK9, or the use of PCSK9 inhibitors (224, 225), would enhance pathogen lipid clearance via the LDLR. Using a cecal ligation and puncture model of sepsis, PCSK9 KO mice were indeed reported to have lower bacterial concentrations in the blood, lungs, and peritoneal cavity fluid than WT animals, suggesting that loss of PCSK9 improves bacterial suppression/clearance (222). In support of this notion, clinical observations revealed that patients carrying a combination of 3 LOF variant alleles of PCSK9 (R46L, A53V, I474V), but not each individually (226), exhibited a >50% higher 1-year survival rate and protection from recurrent infections, likely due to enhanced resolution of infection and/or bacterial clearance after the initial infection (227). This likely reflects that strong LOF variants of PCSK9 may be beneficial but weak single LOF variants are not. PCSK9 is also known to sort VLDLR, ApoER2 (77) and CD36 (151) toward lysosomal degradation, some of which are highly expressed in adipose tissue. LPS can be sequestered in the latter tissue via the VLDLR. In that respect, a homozygote intronic GOF variant (SNP rs7852409; CC genotype) of VLDLR has been associated with improved sepsis survival, especially in patients with a body mass index below 25 (228).
However, there are translational challenges between mouse studies to human applications (229), and these should be overcome before the widespread prescription of PCSK9i to sepsis patients. Furthermore, the complex nature of sepsis is species specific, putting the widespread utility of the rodent models of sepsis to question (230). Indeed, mice and rats have a significantly higher resistance to sepsis than humans. However, recently humanized mouse models of sepsis were reported to mitigate somewhat this restriction (231). Nevertheless, although PCSK9 inhibitors may increase survival of adult patients, they may not be beneficial for young children with sepsis, as LOF of PCSK9 was associated with decreased survival in juvenile mice (14 day old) and young children (1-6 years old) (232). The rationale behind this observation is not clear, though it may be related to age-dependent pleiotropic effects of PCSK9 on other receptors or inflammation. Thus, until this paradoxical effect of PCSK9 LOF is clarified, it was suggested that children should be excluded from sepsis clinical trials involving PCSK9 inhibitors (232).
It should also be noted that patients with septic shock and lower PCSK9 levels on day 1 (within the first quartile) showed the highest 28- and 90-day mortality rate compared with other quartiles (233), suggesting that low levels of circulating PCSK9 1 day after the onset of sepsis are not correlated with a favorable disease outcome (234, 235). However, this does not necessarily exclude the preventive use of PCSK9 inhibitors, eg, before surgery, to neutralize circulating PCSK9, and therefore increase LDLR levels before the onset of sepsis. Moreover, PCSK9 does not significantly affect HDL levels (52), known to be critical, as patients with declining HDL during sepsis are at much greater risk of succumbing to organ failure and death (236). In conclusion, we have to wait for the data from 2 ongoing clinical trials (https://clinicaltrials.gov/ct2/show/NCT03869073and/NCT03634293) to know whether the quick removal of bacterial components through PCSK9 neutralization with a mAb is beneficial.
PCSK9 and Viral Infection
Proprotein convertases (PCs), especially Furin and SKI-1/S1P, are implicated in the processing of enveloped viruses resulting in enhanced viral entry into host cells and widespread infectivity (47). There has also been a limited number of cases where PCSK9 was linked to viral infectivity, these include infections by hepatitis C virus (HCV), Dengue virus (DENV), and possibly SARS-CoV-2, the etiological agent of COVID-19.
The LDLR has been proposed as one of the host factors implicated in HCV entry into hepatocytes (237), but its role remains controversial (238). Recently, induced pluripotent stem cells (iPSC) were isolated from an LDLR-null patient, allowing their induced-differentiation into hepatocytes. Although cells lacking functional LDLR can be well infected by HCV, viral production was significantly increased upon re-expression of functional LDLR, indicating a role of the receptor in HCV packaging, which is tightly linked to the lipid metabolism of the producing cell, rather than to virus entry (239).
Recently, direct-acting antivirals (DAAs; interferon-free) revolutionized HCV therapy by providing a highly sustained virological response rate (240). It was shown that plasma PCSK9 was significantly increased in patients responding well to an interferon-based therapy with added first generation DAAs, (241). The anticipated protective effect of circulating PCSK9 was confirmed by its ability to inhibit Huh-7.5.1 cell infection by HCV (241). In agreement, lower levels of circulating PCSK9 and LDLc were found in untreated patients chronically infected with HCV genotype 3 (HCV-G3), likely resulting from upregulated LDLR activity, than in patients infected with HCV-G1, which is more dependent on SR-B1 (scavenger receptor class B type I that mediates the uptake of HDL) for viral entry (242). In contrast, 4 weeks after the start of DAA therapy primarily in HCV-G1 patients (76%), circulating PCSK9 levels were reduced and yet LDLc was increased, suggesting that these changes are not functionally related (243). Whether circulating PCSK9 levels, usually measured by enzyme-linked immunosorbent assay, represent a good biomarker of HCV infection remains to be proven. Indeed, mass spectrometry assessment of the phosphorylation state and the ratio of active vs Furin-cleaved inactive PCSK9 would better evaluate circulating PCSK9 activity (86). Nevertheless, caution should be exerted before prescribing PCSK9i to patients infected by HCV (244, 245), as this would raise LDLR levels in hepatocytes, which may enhance viral packaging and infectivity (239).
Dengue virus (DENV) is a single positive-stranded RNA virus of the family Flaviviridae, transmitted to human by the urban-adapted Aedes mosquitoes, which yearly infects >400 million individuals worldwide, resulting in ~25 000 death/year, mostly children from southeast Asia (246). Recently, we showed that DENV infection induces expression of PCSK9 in hepatocytes (247), thereby reducing cell surface levels of LDLR and LDLc uptake, resulting in enhanced de novo cholesterol synthesis by the SREBP-2 pathway (248). DENV exploits this mechanism for viral packaging. Interestingly, following DENV infection, increased cholesterol levels in the ER causes a significant reduction in the antiviral type I interferon (IFN) response of the host liver hepatocytes due to the cholesterol-induced suppression of the phosphorylation and activation of STING. The latter is an ER-resident stimulator of the antiviral IFN-regulated genes. This cellular observation was supported by the detection of elevated plasma PCSK9 levels in patients infected with DENV resulting in high levels of viremia and increased severity of plasma leakage (247). This unexpected role of PCSK9 in Dengue pathogenesis led us to test the effect of blocking PCSK9 function by an inhibitory mAb alirocumab. Befittingly, this treatment resulted in higher LDLR levels and lower viremia. Thus, different from HCV, our data suggested that PCSK9 inhibitors could be beneficial for patients with DENV via increased levels of the antiviral IFN-response genes (247). Double-blind clinical studies are now needed to support this proposal.
In a separate study, it was reported that proPCSK9 (eg, proPCSK9-Q152H) in the ER can, in some way, reduce cellular IFN-β expression by inhibiting the functional activity of the cytosolic/nuclear activating transcription factor 2 (ATF2) (249). However, whether the cytosolic ATF2 communicates with the ER-localized proPCSK9 via another intermediate factor has not been addressed. Nevertheless, if confirmed by other studies, this anti-IFN-β effect of proPCSK9 via ATF2 inhibition, combined to the anti-IFN effect of mature PCSK9 via cholesterol upregulation in the ER and inhibition of STING (see above for DENV), would solidify PCSK9’s role as a key inhibitor of IFN expression, and thus a target of choice to inhibit for combating viral infection.
In the present COVID-19 pandemic, statins have been shown to increase the expression of the SARS-CoV-2 receptor angiotensin converting enzyme 2 (ACE2) in animal models (250, 251), but they were also reported to significantly improve the prognosis of COVID-19 patients >65 year-old (252-254). This likely reflects the ability of statins to improve endothelial function and to reduce blood coagulation and inflammation via cholesterol-dependent and cholesterol-independent mechanisms (255). Because PCSK9 mAb further reduces LDLc by ~60% and cardiovascular events in statin-treated patients, it was suggested to also treat selected COVID-19 patients with PCSK9i, especially those who would benefit most from a boost in their IFN levels (256). This is especially relevant for patients presenting LOF variants in the Toll-like receptor 3 (TLR3)- and interferon regulatory factor 7 (IRF7)-dependent IFN immunity (257).
Time will tell whether PCSK9 inhibition coupled with statins also alleviates the symptoms of patients carrying other viruses, such as the human cytomegalovirus (HCMV) that, like DENV, is more infectious when cellular cholesterol levels are upregulated in absence of LRP1 (258), another PCSK9 target (131).
PCSK9 and Cancer/Metastasis
The 2 leading causes of death in industrialized countries are cardiovascular diseases and cancer (https://www.cdc.gov/nchs/fastats/leading-causes-of-death.htm). LDLs not only promote atherosclerosis in a dose-dependent manner but also provide cholesterol to peripheral cells, including cancer cells, which have higher cholesterol requirements to sustain tumor growth (259). Cholesterol plays a key role in a plethora of cellular metabolic processes, particularly for highly demanding anabolic steps such as cell growth and division (260). In agreement, upregulation of the LDLR in tumors is associated with cancer progression (260-262). In the 1980s it was suggested that suppression of the SREBP pathway, would result in lower mevalonate synthesis (263). In turn this would lead to lower levels of isoprenylating intermediates that are needed for farnesylated-RAS insertion into the plasma membrane, resulting in reduced cell proliferation (264). Thus, it was proposed that specific farnesyl-protein transferase (FPTase) inhibitors may reduce the growth of certain Ras-dependent tumors (263). Indeed, it was shown that combined treatment of HeLa cells with cisplatin and an oral inhibitor of FPTase (L-744 832) significantly enhances the cytostatic potency of cisplatin (265). Other FPTase inhibitors have since been evaluated in clinical trials implicating various cancers (https://clinicaltrials.gov/ct2/results?term=farnesyl-protein+transferase+inhibitor+&Search=Search).
The roles of SREBP-2 activation and/or circulating vs tumor-derived PCSK9 in tumor LDLR regulation are not clear. Individuals with high tumoral PCSK9 mRNA expression exhibit worse overall survival than those with low expression in different patient cohorts (266). Interestingly, PCSK9 mAbs that raise LDLR levels in the liver and other tissues but reduce circulating LDLc were not associated with a significant change in cancer incidence, at least in the short term (162, 187). However, upon PCSK9 neutralization, extremely low circulating LDLc levels (originating from liver VLDL) will not result in excessive internalization by supernumerary LDLR (98), and may rather reduce tumor growth (267, 268). Indeed, it was reported that PCSK9 deficiency reduces melanoma metastasis in mouse liver (269) and that LOF and GOF variants are associated with lower and higher incidence of human breast cancer, respectively (270). Accordingly, PCSK9 expression could be a valuable biomarker for the clinical prognostic outcome of certain types of malignancies, including hepatic, gastric, kidney, pancreatic and breast cancers (271).
Early reports showed that PCSK9 is well expressed in the spleen and thymus (51, 95). Until very recently, its function(s) in these regulatory immune tissues remained obscure. A recent report shows that in CD8+ T cells the LDLR makes a complex with the T-cell receptor (TCR), which is activated upon binding antigenic peptides that are presented on tumor cells by the major histocompatibility complex (MHC). Because the LDLR association with TCR promotes its recycling to the cell surface and enhances CD8+ T cell antitumor activity (272), combining PCSK9 inhibition (163) with other immune modulating therapies, such as immune checkpoint inhibitors, may be quite promising (273, 274).
Independently from the LDLR, PCSK9 was also reported to bind MHC-I, but not MHC-II receptors and to target the former to endosomes/lysosomes for degradation (266). However, while the catalytic domain of PCSK9 is responsible for LDLR binding, it is the CHRD M2 domain that interacts with an exposed R-X-E70 motif in the N-terminal region of MHC-I α-chain (HLA-C; https://www.uniprot.org/uniprot/P10321). Alignment of α-chain sequences of several species refined the motif to R-(Y, M, E, G)-E. (266). In this context, it will be worth verifying whether the Furin-cleaved PCSK9 (aa 219-692) (30) detected in human (275) and mouse (108) plasma competes with intact PCSK9 for MHC-I receptor binding.
The hemochromatosis protein HFE (high Fe2+; https://www.uniprot.org/uniprot/Q30201), which is mutated in hereditary haemochromatosis, binds to the transferrin receptor 1 (hTfR1), and somehow reduces cell surface LDLR levels. HFE LOF variants, such as C282Y, are associated with higher levels of the LDLR (276). Since HFE shares a strong homology with some MHC-I α-chain (eg, HLA-C) and the presence of an exposed R-X-E69 motif, we recently hypothesized that it may interact with PCSK9 to modulate the levels of the LDLR. Ongoing modeling and site-directed mutagenesis should define more precisely the interaction between the M2 domain of PCSK9 and the R-X-E motif of MHC-I α-chain, and possibly HFE. Thus, whether the type I transmembrane MHC-I, HFE or family members, which are highly expressed in hepatocytes, are candidates for “protein X” remains to be defined.
Recently, Liu et al. (266) demonstrated that PCSK9-deficient tumor cells that were inoculated in syngeneic mice had reduced abilities to form tumors (breast 4T1 and colon CT26 cancer cells in Balb/c, or melanoma B16F10 and colon MC38 cancer cells in C57BL/6 mice). Moreover, injection of a programmed death 1 (PD-1) mAb, an immune checkpoint inhibitor widely used in immunotherapy (277), synergized with PCSK9 deficiency in all models to further suppress tumor growth, indicating that inhibition of tumor-derived PCSK9 overcomes tumor resistance to anti-PD-1 therapy. Amazingly, a similar synergistic effect of anti-PD-1 and PCSK9 mAbs was observed in WT mice, an astonishing result since PCSK9 mAbs target the catalytic domain (176, 278), whereas MHC-I binds the M2 domain of PCSK9 (266). Whether PCSK9 mAbs sterically hinder the M2 domain and prevent its ability to bind MHC-I or a putative “protein X” remains to be elucidated. In conclusion, inhibition of tumor-derived and/or circulating PCSK9, which contribute to tumor growth in mice, synergizes with that of immune checkpoint receptors. This unexpected role for PCSK9, aside from its canonical LDLR regulation, shows that the role of PCSK9 in immunity and/or in inflammation (279) remains to be better elucidated. In this respect, the generation of humanized mice with patient-derived xenografts for cancer immunotherapy studies provides an exciting new approach to achieve more robust mouse to human translational applications and preclinical evaluation of specific immunotherapies (280, 281).
Even though a strong body of evidence indicates that cholesterol plays a role in cancer development, the evaluation of the impact of extremely low cholesterol levels (LDLc < 25 mg/dL) on human tumorigenesis, cancer progression and/or metastasis should be addressed. Namely, the combination of cholesterol lowering drugs (PCSK9i, statins, ezetimibe) that drastically reduce LDLc (282) and enhance surface LDLR levels may reduce tumor growth and/or metastasis via cholesterol depletion and increased TCR and MHC-I activities. Pairing this approach with chemotherapy/immunotherapy has been proposed (268). In this vein, the drastic reduction of LDLc in patients with metastatic pancreatic adenocarcinoma combined with Folfirinox, a chemotherapy regimen for advanced pancreatic cancer (283), is presently being evaluated in a phase I clinical trial (https://clinicaltrials.gov/ct2/show/NCT04862260). Whether irreversible silencing of PCSK9, such as gene editing using CRISPR-Cas, or vaccination will have future applications in cancer/metastasis depends first on their careful evaluation for any potential harmful side effects (163).
Knowledge Gaps to Address Going Forward
Over the last 18 years, our understanding of the PCSK9 biology and its implications in various physiological and pathophysiological processes have tremendously advanced, in large part due to its robust upregulation of LDLc and the clinically proven safety of mAb and siRNA inhibitors of PCSK9 to reduce by >50% LDLc consistently and reliably in variety of patients. However, gaps do remain in our mechanistic understanding of the various PCSK9 functions during fetal development and in adults. Below we briefly touch on some of the questions remaining to have a more comprehensive view of the PCSK9 biology and its clinical applications:
What are the roles of PCSK9 in extrahepatic tissues such as gut, kidney, immune system, and brain?
What are the functions of PCSK9 (possibly independent of the LDLR) during development in liver and other tissues?
Is there any sex specificity in PCSK9-mediated effects?
What are the domains of PCSK9 that interact with CD36 and receptors other than the LDLR?
Is there any evidence of an intracellular degradation pathway of PCSK9 targets in vivo?
What are the long-term effects of PCSK9i and irreversible silencing of PCSK9?
Can the cost of PCSK9i be reduced (oral inhibitor) for worldwide affordability (Fig. 2)?
-
Can PCSK9i reduce
human cancer/metastasis, alone or in combination with other therapies?
some viral infections and/or the severity of human sepsis?
allergies and inflammation?
Concluding Remarks
The discovery of PCSK9 in 2003 led to an astonishingly fast progress in our deeper understanding of cholesterol homeostasis and the implication of PCSK9 in various pathologies. These include hypercholesterolemia, atherosclerosis, inflammation, sepsis, viral infections, cancer/metastasis, and likely many others. PCSK9 protein acts as a chaperone to escort selected surface receptors toward endosomes/lysosomes for degradation. While the catalytic domain of PCSK9 binds to certain receptors (eg, LDLR, VLDLR, ApoER2, LRP1), the CHRD is implicated in the binding of other ones (eg, MHC class I receptor) (51, 95, 96, 266). Since 2003, a barrage of biological, genetic, and epidemiological studies in mouse and/or human has resulted in the development of the first potent PCSK9i as mAbs in 2015. Such PCSK9 “revolution” starting from discovery to the safe clinical applications of inhibitors to a host target protein within a period of 12 years is exemplary (284), and may in part be related to the limited targets of PCSK9 and its major expression in hepatocytes.
The reason why the lack of PCSK9 activity has not yet been associated with defined negative outcomes is surprising and begs the question of why PCSK9 was kept during mammalian evolution, but lost in some species, and very rarely in human. PCSK9 LOF variations reducing circulating LDLc levels might have been problematic for our ancestors, such as the hunter-gatherer Denisovans (285), who probably had a cholesterol-poor diet, but can now be particularly advantageous for people on cholesterol-rich diets, such as those living in industrialized countries. In fact, it has been suggested that in some placental mammalian species PCSK9 underwent “pseudogenization” leading to the loss of active full length PCSK9 protein (286, 287). A recent study revealed a dramatic difference in the patterns of PCSK9 retention and loss between the 4 major clades of placental mammals (288). Namely, the Euarchontoglires clade that includes primates, rodents, and rabbits exhibit a strong pressure for gene preservation, whereas the Laurasiatheria clade is characterized by multiple independent events that led to the loss of PCSK9 in many species, such as pigs, bovine, camels, whales, bats, cats, dogs, bears, and horses (289). The loss of PCSK9 expression in pigs, suggests that studies of cholesterol metabolism therein would be advantageous for understanding cholesterol-related human diseases, as recently reported in D374Y-PCSK9 transgenics (152) resulting in hypercholesterolemic minipigs, a porcine model of advanced coronary atherosclerosis (290).
It is possible that in the future other hidden, unsuspected functions of PCSK9 will be uncovered, which may be inhibited by PCSK9i. This could open the door to novel applications of PCSK9i, and more advanced technologies that are still under development, for the treatments of pathologies other than hypercholesterolemia, such as cancer/metastasis, inflammation, and viral infections. Such translational applications deduced from animal models will surely need rigorous validations before they can be safely and successfully applied to human pathologies.
Acknowledgments
The authors would like to acknowledge the expert secretarial help of Habiba Oueslati in the preparation of this manuscript.
Financial Support: This work was funded thanks to grants to N.G.S. including a Canadian Institutes of Health Research Foundation Scheme grant (# 148363) and a Canada Chair in Precursor Proteolysis (# 950-231335).
Conflict of Interest Statement: The authors declare no conflict of interest.
Glossary
Abbreviations
- aa
amino acids
- CAP1
cyclase-associated protein 1
- CHRD
Cys/His-rich domain
- DAA
direct-acting antiviral
- DENV
Dengue virus
- E2
17β-estradiol
- ER
endoplasmic reticulum
- FH
familial hypercholesterolemia
- GOF
gain-of-function
- HCV
hepatitis C virus
- KO
knockout
- IFN
interferon
- LDLc
low-density lipoprotein cholesterol
- LDLR
LDL receptor
- LNP
lipid nanoparticle
- LOF
loss-of-function
- MHC
major histocompatibility complex
- PC
proprotein convertase
- PD
programmed death
- PTM
post-translational modification
- SK
subtilisin/kexin
- SRE
sterol regulatory element
- TCR
T-cell receptor
- WT
wild type
Contributor Information
Nabil G Seidah, Laboratory of Biochemical Neuroendocrinology, Montreal Clinical Research Institute (IRCM, affiliated to the University of Montreal), Montreal, QC, Canada.
Annik Prat, Laboratory of Biochemical Neuroendocrinology, Montreal Clinical Research Institute (IRCM, affiliated to the University of Montreal), Montreal, QC, Canada.
Additional Information
Disclosure Summary: Nothing to disclose.
References
- 1. Buja LM. Nikolai N. Anitschkow and the lipid hypothesis of atherosclerosis. Cardiovasc Pathol. 2014;23(3):183-184. [DOI] [PubMed] [Google Scholar]
- 2. Frantz ID Jr, Moore RB. The sterol hypothesis in atherogenesis. Am J Med. 1969;46(5):684-690. [DOI] [PubMed] [Google Scholar]
- 3. Endo A. A historical perspective on the discovery of statins. Proc Jpn Acad Ser B Phys Biol Sci 2010; 86(5):484-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li JJ. Triumph of the Heart: The Story of Statins. Oxford: University Press. [Google Scholar]
- 5. Jones P, Kafonek S, Laurora I, Hunninghake D. Comparative dose efficacy study of atorvastatin versus simvastatin, pravastatin, lovastatin, and fluvastatin in patients with hypercholesterolemia (the CURVES study). Am J Cardiol. 1998;81(5):582-587. [DOI] [PubMed] [Google Scholar]
- 6. Shimano H. Sterol regulatory element-binding proteins (SREBPs): transcriptional regulators of lipid synthetic genes. Prog Lipid Res. 2001;40(6):439-452. [DOI] [PubMed] [Google Scholar]
- 7. Amemiya-Kudo M, Shimano H, Hasty AH, et al. Transcriptional activities of nuclear SREBP-1a, -1c, and -2 to different target promoters of lipogenic and cholesterogenic genes. J Lipid Res. 2002;43(8):1220-1235. [PubMed] [Google Scholar]
- 8. Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29(4):431-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davignon J. The cardioprotective effects of statins. Curr Atheroscler Rep. 2004;6(1):27-35. [DOI] [PubMed] [Google Scholar]
- 10. Rosenblum SB, Huynh T, Afonso A, et al. Discovery of 1-(4-fluorophenyl)-(3R)-[3-(4-fluorophenyl)-(3S)-hydroxypropyl]-(4S)-(4 -hydroxyphenyl)-2-azetidinone (SCH 58235): a designed, potent, orally active inhibitor of cholesterol absorption. J Med Chem. 1998;41(6):973-980. [DOI] [PubMed] [Google Scholar]
- 11. Smith BA, Wright C, Davidson M. Role of ezetimibe in lipid-lowering and cardiovascular disease prevention. Curr Atheroscler Rep. 2015;17(12):72. [DOI] [PubMed] [Google Scholar]
- 12. Giugliano RP, Cannon CP, Blazing MA, et al. ; IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial) Investigators . Benefit of adding ezetimibe to statin therapy on cardiovascular outcomes and safety in patients with versus without diabetes mellitus: results from IMPROVE-IT (improved reduction of outcomes: vytorin efficacy international trial). Circulation. 2018;137(15):1571-1582. [DOI] [PubMed] [Google Scholar]
- 13. Schjoldager KT, Clausen H. Site-specific protein O-glycosylation modulates proprotein processing—deciphering specific functions of the large polypeptide GalNAc-transferase gene family. Biochim Biophys Acta 2012; 1820(12):2079-2094. [DOI] [PubMed] [Google Scholar]
- 14. Schjoldager KT, Narimatsu Y, Joshi HJ, Clausen H. Global view of human protein glycosylation pathways and functions. Nat Rev Mol Cell Biol. 2020;21(12):729-749. [DOI] [PubMed] [Google Scholar]
- 15. Aebersold R, Agar JN, Amster IJ, et al. How many human proteoforms are there? Nat Chem Biol. 2018;14(3):206-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tagliabracci VS, Wiley SE, Guo X, et al. A single kinase generates the majority of the secreted phosphoproteome. Cell. 2015;161(7):1619-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cui J, Xiao J, Tagliabracci VS, Wen J, Rahdar M, Dixon JE. A secretory kinase complex regulates extracellular protein phosphorylation. Elife. 2015;4:e06120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Neurath H. Proteolytic processing and physiological regulation. Trends Biochem Sci. 1989;14(7):268-271. [DOI] [PubMed] [Google Scholar]
- 19. Neurath H, Walsh KA. Role of proteolytic enzymes in biological regulation (a review). Proc Natl Acad Sci U S A. 1976;73(11):3825-3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lazure C, Seidah NG, Pélaprat D, Chrétien M. Proteases and posttranslational processing of prohormones: a review. Can J Biochem Cell Biol. 1983;61(7):501-515. [DOI] [PubMed] [Google Scholar]
- 21. Steiner DF, Cunningham D, Spigelman L, Aten B. Insulin biosynthesis: evidence for a precursor. Science. 1967;157(3789):697-700. [DOI] [PubMed] [Google Scholar]
- 22. Steiner DF. On the discovery of precursor processing. Methods Mol Biol. 2011;768:3-11. [DOI] [PubMed] [Google Scholar]
- 23. Chrétien M, Li CH. Isolation, purification, and characterization of gamma-lipotropic hormone from sheep pituitary glands. Can J Biochem. 1967;45(7):1163-1174. [DOI] [PubMed] [Google Scholar]
- 24. Chrétien M, Benjannet S, Dragon N, Seidah NG, Lis M. Isolation of peptides with opiate activity from sheep and human pituitaries: relationship to beta-lipotropin. Biochem Biophys Res Commun. 1976;72(2):472-478. [DOI] [PubMed] [Google Scholar]
- 25. Chrétien M. How the prohormone theory solved two important controversies in hormonal and neural Peptide biosynthesis. Front Endocrinol (Lausanne). 2013;4:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seidah NG, Lis M, Gianoulakis C, Schller P, Chrétien M. Letter: fragment of sheep beta-lipotropin with morphine-like activity. Lancet. 1976;1(7967):1017. [DOI] [PubMed] [Google Scholar]
- 27. Seidah NG, Chrétien M. Proprotein and prohormone convertases: a family of subtilases generating diverse bioactive polypeptides. Brain Res. 1999;848(1-2):45-62. [DOI] [PubMed] [Google Scholar]
- 28. Seidah NG, Prat A. The biology and therapeutic targeting of the proprotein convertases. Nat Rev Drug Discov. 2012;11(5):367-383. [DOI] [PubMed] [Google Scholar]
- 29. Seidah NG, Sadr MS, Chrétien M, Mbikay M. The multifaceted proprotein convertases: their unique, redundant, complementary, and opposite functions. J Biol Chem. 2013;288(30):21473-21481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Benjannet S, Rhainds D, Hamelin J, Nassoury N, Seidah NG. The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: functional consequences of natural mutations and post-translational modifications. J Biol Chem. 2006;281(41):30561-30572. [DOI] [PubMed] [Google Scholar]
- 31. Rawson RB, Cheng D, Brown MS, Goldstein JL. Isolation of cholesterol-requiring mutant Chinese hamster ovary cells with defects in cleavage of sterol regulatory element-binding proteins at site 1. J Biol Chem. 1998;273(43):28261-28269. [DOI] [PubMed] [Google Scholar]
- 32. Lenz O, ter Meulen J, Klenk HD, Seidah NG, Garten W. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc Natl Acad Sci U S A. 2001;98(22):12701-12705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vincent MJ, Sanchez AJ, Erickson BR, et al. Crimean-Congo hemorrhagic fever virus glycoprotein proteolytic processing by subtilase SKI-1. J Virol. 2003;77(16):8640-8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seidah NG, Mowla SJ, Hamelin J, et al. Mammalian subtilisin/kexin isozyme SKI-1: A widely expressed proprotein convertase with a unique cleavage specificity and cellular localization. Proc Natl Acad Sci U S A. 1999;96(4):1321-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sakai J, Rawson RB, Espenshade PJ, et al. Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol Cell. 1998;2(4):505-514. [DOI] [PubMed] [Google Scholar]
- 36. Ye J, Rawson RB, Komuro R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6(6):1355-1364. [DOI] [PubMed] [Google Scholar]
- 37. Seidah NG. Proprotein convertases SKI-1/S1P and PCSK9. In: Minamino N, Kastin A, eds. Handbook of the Biologically Active Peptides. Academic Press; 2012:1-8. [Google Scholar]
- 38. Espenshade PJ, Cheng D, Goldstein JL, Brown MS. Autocatalytic processing of site-1 protease removes propeptide and permits cleavage of sterol regulatory element-binding proteins. J Biol Chem. 1999;274(32):22795-22804. [DOI] [PubMed] [Google Scholar]
- 39. Pullikotil P, Vincent M, Nichol ST, Seidah NG. Development of protein-based inhibitors of the proprotein of convertase SKI-1/S1P: processing of SREBP-2, ATF6, and a viral glycoprotein. J Biol Chem. 2004;279(17):17338-17347. [DOI] [PubMed] [Google Scholar]
- 40. Pasquato A, Pullikotil P, Asselin MC, et al. The proprotein convertase SKI-1/S1P. In vitro analysis of Lassa virus glycoprotein-derived substrates and ex vivo validation of irreversible peptide inhibitors. J Biol Chem. 2006;281(33):23471-23481. [DOI] [PubMed] [Google Scholar]
- 41. Seidah NG, Prat A. Precursor convertases in the secretory pathway, cytosol and extracellular milieu. Essays Biochem. 2002;38:79-94. [DOI] [PubMed] [Google Scholar]
- 42. Seidah NG, Prat A. The proprotein convertases are potential targets in the treatment of dyslipidemia. J Mol Med (Berl). 2007;85(7):685-696. [DOI] [PubMed] [Google Scholar]
- 43. Pasquato A, Cendron L, Kunz S. Cleavage of the glycoprotein of arenaviruses. Activat Viruses Host Proteases 2018:47-70. [Google Scholar]
- 44. Velho RV, De Pace R, Klünder S, et al. Site-1 protease and lysosomal homeostasis. Biochim Biophys Acta Mol Cell Res. 2017;1864(11 Pt B):2162-2168. [DOI] [PubMed] [Google Scholar]
- 45. Tassew NG, Charish J, Seidah NG, Monnier PP. SKI-1 and Furin generate multiple RGMa fragments that regulate axonal growth. Dev Cell. 2012;22(2):391-402. [DOI] [PubMed] [Google Scholar]
- 46. Nakagawa T, Suzuki-Nakagawa C, Watanabe A, et al. Site-1 protease is required for the generation of soluble (pro)renin receptor. J Biochem. 2017;161(4):369-379. [DOI] [PubMed] [Google Scholar]
- 47. Seidah NG, Pasquato A, Andreo U. How do enveloped viruses exploit the secretory proprotein convertases to regulate infectivity and spread? Viruses 2021;13(7):1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kondo Y, Fu J, Wang H, et al. Site-1 protease deficiency causes human skeletal dysplasia due to defective inter-organelle protein trafficking. JCI Insight 2018;3(14):e121596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ye J. Transcription factors activated through RIP (regulated intramembrane proteolysis) and RAT (regulated alternative translocation). J Biol Chem. 2020;295(30):10271-10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marschner K, Kollmann K, Schweizer M, Braulke T, Pohl S. A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science. 2011;333(6038):87-90. [DOI] [PubMed] [Google Scholar]
- 51. Seidah NG, Benjannet S, Wickham L, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A. 2003;100(3):928-933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154-156. [DOI] [PubMed] [Google Scholar]
- 53. Varret M, Rabès JP, Saint-Jore B, et al. A third major locus for autosomal dominant hypercholesterolemia maps to 1p34.1-p32. Am J Hum Genet. 1999;64(5):1378-1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hunt SC, Hopkins PN, Bulka K, et al. Genetic localization to chromosome 1p32 of the third locus for familial hypercholesterolemia in a Utah kindred. Arterioscler Thromb Vasc Biol. 2000;20(4):1089-1093. [DOI] [PubMed] [Google Scholar]
- 55. Naureckiene S, Ma L, Sreekumar K, et al. Functional characterization of Narc 1, a novel proteinase related to proteinase K. Arch Biochem Biophys. 2003;420(1):55-67. [DOI] [PubMed] [Google Scholar]
- 56. Benjannet S, Rhainds D, Essalmani R, et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. 2004;279(47):48865-48875. [DOI] [PubMed] [Google Scholar]
- 57. Benjannet S, Hamelin J, Chrétien M, Seidah NG. Loss- and gain-of-function PCSK9 variants: cleavage specificity, dominant negative effects, and low density lipoprotein receptor (LDLR) degradation. J Biol Chem. 2012;287(40):33745-33755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Saavedra YG, Zhang J, Seidah NG. PCSK9 prosegment chimera as novel inhibitors of LDLR degradation. PLoS One. 2013;8(8):e72113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cunningham D, Danley DE, Geoghegan KF, et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat Struct Mol Biol. 2007;14(5):413-419. [DOI] [PubMed] [Google Scholar]
- 60. Piper DE, Jackson S, Liu Q, et al. The crystal structure of PCSK9: a regulator of plasma LDL-cholesterol. Structure. 2007;15(5):545-552. [DOI] [PubMed] [Google Scholar]
- 61. Roubtsova A, Munkonda MN, Awan Z, et al. Circulating proprotein convertase subtilisin/kexin 9 (PCSK9) regulates VLDLR protein and triglyceride accumulation in visceral adipose tissue. Arterioscler Thromb Vasc Biol. 2011;31(4):785-791. [DOI] [PubMed] [Google Scholar]
- 62. Maxwell KN, Fisher EA, Breslow JL. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci U S A 2005; 102(6):2069-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Maxwell KN, Breslow JL. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci U S A. 2004;101(18):7100-7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Park SW, Moon YA, Horton JD. Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J Biol Chem. 2004;279(48):50630-50638. [DOI] [PubMed] [Google Scholar]
- 65. Maxwell KN, Soccio RE, Duncan EM, Sehayek E, Breslow JL. Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J Lipid Res. 2003;44(11):2109-2119. [DOI] [PubMed] [Google Scholar]
- 66. Horton JD, Shah NA, Warrington JA, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100(21):12027-12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dubuc G, Chamberland A, Wassef H, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24(8):1454-1459. [DOI] [PubMed] [Google Scholar]
- 68. Attie AD, Seidah NG. Dual regulation of the LDL receptor–some clarity and new questions. Cell Metab. 2005;1(5):290-292. [DOI] [PubMed] [Google Scholar]
- 69. Timms KM, Wagner S, Samuels ME, et al. A mutation in PCSK9 causing autosomal-dominant hypercholesterolemia in a Utah pedigree. Hum Genet. 2004;114(4):349-353. [DOI] [PubMed] [Google Scholar]
- 70. Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37(2):161-165. [DOI] [PubMed] [Google Scholar]
- 71. Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264-1272. [DOI] [PubMed] [Google Scholar]
- 72. Yang J, Goldstein JL, Hammer RE, Moon YA, Brown MS, Horton JD. Decreased lipid synthesis in livers of mice with disrupted Site-1 protease gene. Proc Natl Acad Sci U S A. 2001;98(24):13607-13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rashid S, Curtis DE, Garuti R, et al. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci U S A. 2005;102(15):5374-5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79(3):514-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis. 2007;193(2):445-448. [DOI] [PubMed] [Google Scholar]
- 76. McNutt MC, Lagace TA, Horton JD. Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J Biol Chem. 2007;282(29):20799-20803. [DOI] [PubMed] [Google Scholar]
- 77. Poirier S, Mayer G, Benjannet S, et al. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem. 2008;283(4):2363-2372. [DOI] [PubMed] [Google Scholar]
- 78. Mayne J, Dewpura T, Raymond A, et al. Novel loss-of-function PCSK9 variant is associated with low plasma LDL cholesterol in a French-Canadian family and with impaired processing and secretion in cell culture. Clin Chem. 2011;57(10):1415-1423. [DOI] [PubMed] [Google Scholar]
- 79. Lebeau PF, Wassef H, Byun JH, et al. The loss-of-function PCSK9Q152H variant increases ER chaperones GRP78 and GRP94 and protects against liver injury. J Clin Invest. 2021; 131(2):e128650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lo Surdo P, Bottomley MJ, Calzetta A, et al. Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 2011;12(12):1300-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kwon HJ, Lagace TA, McNutt MC, Horton JD, Deisenhofer J. Molecular basis for LDL receptor recognition by PCSK9. Proc Natl Acad Sci U S A. 2008;105(6):1820-1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Holla ØL, Laerdahl JK, Strøm TB, et al. Removal of acidic residues of the prodomain of PCSK9 increases its activity towards the LDL receptor. Biochem Biophys Res Commun. 2011;406(2):234-238. [DOI] [PubMed] [Google Scholar]
- 83. Benjannet S, Saavedra YG, Hamelin J, et al. Effects of the prosegment and pH on the activity of PCSK9: evidence for additional processing events. J Biol Chem. 2010;285(52):40965-40978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Seidah NG. The elusive inhibitory function of the acidic N-terminal segment of the prodomain of PCSK9: the plot thickens. J Mol Biol. 2019;431(5):904-907. [DOI] [PubMed] [Google Scholar]
- 85. Berge KE, Ose L, Leren TP. Missense mutations in the PCSK9 gene are associated with hypocholesterolemia and possibly increased response to statin therapy. Arterioscler Thromb Vasc Biol. 2006;26(5):1094-1100. [DOI] [PubMed] [Google Scholar]
- 86. Ben Djoudi Ouadda A, Gauthier MS, Susan-Resiga D, et al. Ser-Phosphorylation of PCSK9 (Proprotein Convertase Subtilisin-Kexin 9) by Fam20C (family with sequence similarity 20, Member C) kinase enhances its ability to degrade the LDLR (low-density lipoprotein receptor). Arterioscler Thromb Vasc Biol. 2019;39(10):1996-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nassoury N, Blasiole DA, Tebon Oler A, et al. The cellular trafficking of the secretory proprotein convertase PCSK9 and its dependence on the LDLR. Traffic. 2007;8(6):718-732. [DOI] [PubMed] [Google Scholar]
- 88. Qian YW, Schmidt RJ, Zhang Y, et al. Secreted PCSK9 downregulates low density lipoprotein receptor through receptor-mediated endocytosis. J Lipid Res. 2007;48(7):1488-1498. [DOI] [PubMed] [Google Scholar]
- 89. Holla ØL, Cameron J, Berge KE, Ranheim T, Leren TP. Degradation of the LDL receptors by PCSK9 is not mediated by a secreted protein acted upon by PCSK9 extracellularly. BMC Cell Biol. 2007;8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lagace TA, Curtis DE, Garuti R, et al. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006;116(11):2995-3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhang DW, Lagace TA, Garuti R, et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007;282(25):18602-18612. [DOI] [PubMed] [Google Scholar]
- 92. Poirier S, Mayer G, Poupon V, et al. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J Biol Chem. 2009;284(42):28856-28864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Susan-Resiga D, Girard E, Kiss RS, et al. The proprotein convertase subtilisin/kexin type 9-resistant R410S low density lipoprotein receptor mutation: a novel mechanism causing familial hypercholesterolemia. J Biol Chem. 2017;292(5):1573-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mikaeeli S, Susan-Resiga D, Girard E, et al. Functional analysis of natural PCSK9 mutants in modern and archaic humans. FEBS J. 2020;287(3):515-528. [DOI] [PubMed] [Google Scholar]
- 95. Seidah NG, Abifadel M, Prost S, Boileau C, Prat A. The proprotein convertases in hypercholesterolemia and cardiovascular diseases: emphasis on proprotein convertase subtilisin/kexin 9. Pharmacol Rev. 2017;69(1):33-52. [DOI] [PubMed] [Google Scholar]
- 96. Seidah NG, Awan Z, Chrétien M, Mbikay M. PCSK9: a key modulator of cardiovascular health. Circ Res. 2014;114(6):1022-1036. [DOI] [PubMed] [Google Scholar]
- 97. Garçon D, Moreau F, Ayer A, et al. Circulating rather than intestinal PCSK9 (proprotein convertase subtilisin kexin type 9) regulates postprandial lipemia in mice. Arterioscler Thromb Vasc Biol. 2020;40(9):2084-2094. [DOI] [PubMed] [Google Scholar]
- 98. Peyot ML, Roubtsova A, Lussier R, et al. Substantial PCSK9 inactivation in β-cells does not modify glucose homeostasis or insulin secretion in mice. Biochim Biophys Acta Mol Cell Biol Lipids. 2021;1866(8):158968. [DOI] [PubMed] [Google Scholar]
- 99. Ramin-Mangata S, Thedrez A, Nativel B, et al. Effects of proprotein convertase subtilisin kexin type 9 modulation in human pancreatic beta cells function. Atherosclerosis. 2021;326:47-55. [DOI] [PubMed] [Google Scholar]
- 100. Alborn WE, Cao G, Careskey HE, et al. Serum proprotein convertase subtilisin kexin type 9 is correlated directly with serum LDL cholesterol. Clin Chem. 2007;53(10):1814-1819. [DOI] [PubMed] [Google Scholar]
- 101. Mayne J, Raymond A, Chaplin A, et al. Plasma PCSK9 levels correlate with cholesterol in men but not in women. Biochem Biophys Res Commun. 2007;361(2):451-456. [DOI] [PubMed] [Google Scholar]
- 102. Lambert G, Ancellin N, Charlton F, et al. Plasma PCSK9 concentrations correlate with LDL and total cholesterol in diabetic patients and are decreased by fenofibrate treatment. Clin Chem. 2008;54(6):1038-1045. [DOI] [PubMed] [Google Scholar]
- 103. Lakoski SG, Lagace TA, Cohen JC, Horton JD, Hobbs HH. Genetic and metabolic determinants of plasma PCSK9 levels. J Clin Endocrinol Metab. 2009;94(7):2537-2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Dubuc G, Tremblay M, Paré G, et al. A new method for measurement of total plasma PCSK9: clinical applications. J Lipid Res. 2010;51(1):140-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Awan Z, Seidah NG, MacFadyen JG, et al. Rosuvastatin, proprotein convertase subtilisin/kexin type 9 concentrations, and LDL cholesterol response: the JUPITER trial. Clin Chem. 2012;58(1):183-189. [DOI] [PubMed] [Google Scholar]
- 106. Allard D, Amsellem S, Abifadel M, et al. Novel mutations of the PCSK9 gene cause variable phenotype of autosomal dominant hypercholesterolemia. Hum Mutat. 2005;26(5):497. [DOI] [PubMed] [Google Scholar]
- 107. Cameron J, Holla ØL, Ranheim T, Kulseth MA, Berge KE, Leren TP. Effect of mutations in the PCSK9 gene on the cell surface LDL receptors. Hum Mol Genet. 2006;15(9):1551-1558. [DOI] [PubMed] [Google Scholar]
- 108. Essalmani R, Susan-Resiga D, Chamberland A, et al. In vivo evidence that furin from hepatocytes inactivates PCSK9. J Biol Chem. 2011;286(6):4257-4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bottomley MJ, Cirillo A, Orsatti L, et al. Structural and biochemical characterization of the wild type PCSK9-EGF(AB) complex and natural familial hypercholesterolemia mutants. J Biol Chem. 2009;284(2):1313-1323. [DOI] [PubMed] [Google Scholar]
- 110. McNutt MC, Kwon HJ, Chen C, Chen JR, Horton JD, Lagace TA. Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 cells. J Biol Chem. 2009;284(16):10561-10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hampton EN, Knuth MW, Li J, Harris JL, Lesley SA, Spraggon G. The self-inhibited structure of full-length PCSK9 at 1.9 A reveals structural homology with resistin within the C-terminal domain. Proc Natl Acad Sci U S A. 2007;104(37):14604-14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Filková M, Haluzík M, Gay S, Senolt L. The role of resistin as a regulator of inflammation: Implications for various human pathologies. Clin Immunol. 2009;133(2):157-170. [DOI] [PubMed] [Google Scholar]
- 113. Dron JS, Hegele RA. Complexity of mechanisms among human proprotein convertase subtilisin-kexin type 9 variants. Curr Opin Lipidol. 2017;28(2):161-169. [DOI] [PubMed] [Google Scholar]
- 114. Elbitar S, Susan-Resiga D, Ghaleb Y, et al. New sequencing technologies help revealing unexpected mutations in autosomal dominant hypercholesterolemia. Sci Rep. 2018;8(1):1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kotowski IK, Pertsemlidis A, Luke A, et al. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet. 2006;78(3):410-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Cariou B, Ouguerram K, Zaïr Y, et al. PCSK9 dominant negative mutant results in increased LDL catabolic rate and familial hypobetalipoproteinemia. Arterioscler Thromb Vasc Biol. 2009;29(12):2191-2197. [DOI] [PubMed] [Google Scholar]
- 117. Abifadel M, Rabès JP, Devillers M, et al. Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum Mutat. 2009;30(4):520-529. [DOI] [PubMed] [Google Scholar]
- 118. Iacocca MA, Wang J, Sarkar S, et al. Whole-gene duplication of PCSK9 as a novel genetic mechanism for severe familial hypercholesterolemia. Can J Cardiol. 2018;34(10):1316-1324. [DOI] [PubMed] [Google Scholar]
- 119. Poirier S, Mamarbachi M, Chen WT, Lee AS, Mayer G. GRP94 regulates circulating cholesterol levels through blockade of PCSK9-induced LDLR degradation. Cell Rep. 2015;13(10):2064-2071. [DOI] [PubMed] [Google Scholar]
- 120. Zhu G, Lee AS. Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J Cell Physiol. 2015;230(7):1413-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Gupta MK, Tahrir FG, Knezevic T, et al. GRP78 interacting partner Bag5 responds to ER stress and protects cardiomyocytes from ER stress-induced apoptosis. J Cell Biochem. 2016;117(8):1813-1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lin J, Chung S, Ueda K, Matsuda K, Nakamura Y, Park JH. GALNT6 stabilizes GRP78 protein by O-glycosylation and enhances its activity to suppress apoptosis under stress condition. Neoplasia. 2017;19(1):43-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Abifadel M, Guerin M, Benjannet S, et al. Identification and characterization of new gain-of-function mutations in the PCSK9 gene responsible for autosomal dominant hypercholesterolemia. Atherosclerosis. 2012;223(2):394-400. [DOI] [PubMed] [Google Scholar]
- 124. Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. 2009;50(Suppl):S172-S177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zhang DW, Garuti R, Tang WJ, Cohen JC, Hobbs HH. Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc Natl Acad Sci U S A. 2008;105(35):13045-13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Holla ØL, Cameron J, Tveten K, et al. Role of the C-terminal domain of PCSK9 in degradation of the LDL receptors. J Lipid Res. 2011;52(10):1787-1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Ni YG, Condra JH, Orsatti L, et al. A proprotein convertase subtilisin-like/kexin type 9 (PCSK9) C-terminal domain antibody antigen-binding fragment inhibits PCSK9 internalization and restores low density lipoprotein uptake. J Biol Chem. 2010;285(17):12882-12891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Fasano T, Sun XM, Patel DD, Soutar AK. Degradation of LDLR protein mediated by ‘gain of function’ PCSK9 mutants in normal and ARH cells. Atherosclerosis. 2009;203(1):166-171. [DOI] [PubMed] [Google Scholar]
- 129. Strøm TB, Holla ØL, Tveten K, Cameron J, Berge KE, Leren TP. Disrupted recycling of the low density lipoprotein receptor by PCSK9 is not mediated by residues of the cytoplasmic domain. Mol Genet Metab. 2010;101(1):76-80. [DOI] [PubMed] [Google Scholar]
- 130. Tveten K, Strøm TB, Cameron J, Holla ØL, Berge KE, Leren TP. Characterization of a naturally occurring degradation product of the LDL receptor. Mol Genet Metab. 2012;105(1):149-154. [DOI] [PubMed] [Google Scholar]
- 131. Canuel M, Sun X, Asselin MC, Paramithiotis E, Prat A, Seidah NG. Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One. 2013;8(5):e64145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Schiele F, Park J, Redemann N, Luippold G, Nar H. An antibody against the C-terminal domain of PCSK9 lowers LDL cholesterol levels in vivo. J Mol Biol. 2014;426(4):843-852. [DOI] [PubMed] [Google Scholar]
- 133. Weider E, Susan-Resiga D, Essalmani R, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9) single domain antibodies are potent inhibitors of low density lipoprotein receptor degradation. J Biol Chem. 2016;291(32):16659-16671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Essalmani R, Weider E, Marcinkiewicz J, et al. A single domain antibody against the Cys- and His-rich domain of PCSK9 and evolocumab exhibit different inhibition mechanisms in humanized PCSK9 mice. Biol Chem. 2018;399(12):1363-1374. [DOI] [PubMed] [Google Scholar]
- 135. Butkinaree C, Canuel M, Essalmani R, et al. Amyloid precursor-like protein 2 and sortilin do not regulate the PCSK9 convertase-mediated low density lipoprotein receptor degradation but interact with each other. J Biol Chem. 2015;290(30):18609-18620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Poirier S, Hamouda HA, Villeneuve L, Demers A, Mayer G. Trafficking dynamics of PCSK9-induced LDLR degradation: focus on human PCSK9 mutations and C-terminal domain. PLoS One. 2016;11(6):e0157230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Galvan AM, Chorba JS. Cell-associated heparin-like molecules modulate the ability of LDL to regulate PCSK9 uptake. J Lipid Res. 2019;60(1):71-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Jang HD, Lee SE, Yang J, et al. Cyclase-associated protein 1 is a binding partner of proprotein convertase subtilisin/kexin type-9 and is required for the degradation of low-density lipoprotein receptors by proprotein convertase subtilisin/kexin type-9. Eur Heart J. 2020;41(2):239-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Kim J, Gee HY, Lee MG. Unconventional protein secretion - new insights into the pathogenesis and therapeutic targets of human diseases. J Cell Sci. 2018;131(12):jcs213686. [DOI] [PubMed] [Google Scholar]
- 140. Mayer G, Poirier S, Seidah NG. Annexin A2 is a C-terminal PCSK9-binding protein that regulates endogenous low density lipoprotein receptor levels. J Biol Chem. 2008;283(46):31791-31801. [DOI] [PubMed] [Google Scholar]
- 141. Saavedra YG, Day R, Seidah NG. The M2 module of the Cys-His-rich domain (CHRD) of PCSK9 protein is needed for the extracellular low-density lipoprotein receptor (LDLR) degradation pathway. J Biol Chem. 2012;287(52):43492-43501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Seidah NG. Insights into a PCSK9 structural groove: a harbinger of new drugs to reduce LDL-cholesterol. Nat Struct Mol Biol. 2017;24(10):785-786. [DOI] [PubMed] [Google Scholar]
- 143. Zaid A, Roubtsova A, Essalmani R, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology. 2008;48(2):646-654. [DOI] [PubMed] [Google Scholar]
- 144. Rousselet E, Marcinkiewicz J, Kriz J, et al. PCSK9 reduces the protein levels of the LDL receptor in mouse brain during development and after ischemic stroke. J Lipid Res. 2011;52(7):1383-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Poirier S, Prat A, Marcinkiewicz E, et al. Implication of the proprotein convertase NARC-1/PCSK9 in the development of the nervous system. J Neurochem. 2006;98(3):838-850. [DOI] [PubMed] [Google Scholar]
- 146. Jacome Sanz D, Saralahti AK, Pekkarinen M, et al. Proprotein convertase subtilisin/kexin type 9 regulates the production of acute-phase reactants from the liver. Liver Int. 2021;41(10):2511-2522. [DOI] [PubMed] [Google Scholar]
- 147. Liu M, Wu G, Baysarowich J, et al. PCSK9 is not involved in the degradation of LDL receptors and BACE1 in the adult mouse brain. J Lipid Res. 2010;51(9):2611-2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Parker RA, Garcia R, Ryan CS, et al. Bile acid and sterol metabolism with combined HMG-CoA reductase and PCSK9 suppression. J Lipid Res. 2013;54(9):2400-2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Roubtsova A, Chamberland A, Marcinkiewicz J, et al. PCSK9 deficiency unmasks a sex- and tissue-specific subcellular distribution of the LDL and VLDL receptors in mice. J Lipid Res. 2015;56(11):2133-2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Costet P, Cariou B, Lambert G, et al. Hepatic PCSK9 expression is regulated by nutritional status via insulin and sterol regulatory element-binding protein 1c. J Biol Chem. 2006;281(10):6211-6218. [DOI] [PubMed] [Google Scholar]
- 151. Demers A, Samami S, Lauzier B, et al. PCSK9 induces CD36 degradation and affects long-chain fatty acid uptake and triglyceride metabolism in adipocytes and in mouse liver. Arterioscler Thromb Vasc Biol. 2015;35(12):2517-2525. [DOI] [PubMed] [Google Scholar]
- 152. Herbert B, Patel D, Waddington SN, et al. Increased secretion of lipoproteins in transgenic mice expressing human D374Y PCSK9 under physiological genetic control. Arterioscler Thromb Vasc Biol. 2010;30(7):1333-1339. [DOI] [PubMed] [Google Scholar]
- 153. Bjørklund MM, Hollensen AK, Hagensen MK, et al. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114(11):1684-1689. [DOI] [PubMed] [Google Scholar]
- 154. Denis M, Marcinkiewicz J, Zaid A, et al. Gene inactivation of proprotein convertase subtilisin/kexin type 9 reduces atherosclerosis in mice. Circulation. 2012;125(7):894-901. [DOI] [PubMed] [Google Scholar]
- 155. Tang ZH, Peng J, Ren Z, et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis. 2017;262:113-122. [DOI] [PubMed] [Google Scholar]
- 156. Mehta JL. Oxidized or native low-density lipoprotein cholesterol: which is more important in atherogenesis? J Am Coll Cardiol. 2006;48(5):980-982. [DOI] [PubMed] [Google Scholar]
- 157. Ding Z, Pothineni NVK, Goel A, Lüscher TF, Mehta JL. PCSK9 and inflammation: role of shear stress, pro-inflammatory cytokines, and LOX-1. Cardiovasc Res. 2020;116(5):908-915. [DOI] [PubMed] [Google Scholar]
- 158. Landlinger C, Pouwer MG, Juno C, et al. The AT04A vaccine against proprotein convertase subtilisin/kexin type 9 reduces total cholesterol, vascular inflammation, and atherosclerosis in APOE*3Leiden.CETP mice. Eur Heart J. 2017;38(32):2499-2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ding Z, Liu S, Wang X, et al. PCSK9 regulates expression of scavenger receptors and ox-LDL uptake in macrophages. Cardiovasc Res. 2018;114(8):1145-1153. [DOI] [PubMed] [Google Scholar]
- 160. Langhi C, Le May C, Gmyr V, et al. PCSK9 is expressed in pancreatic delta-cells and does not alter insulin secretion. Biochem Biophys Res Commun. 2009;390(4):1288-1293. [DOI] [PubMed] [Google Scholar]
- 161. The Tabula Muris Consortium. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018;562(7727):367-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111-188. [DOI] [PubMed] [Google Scholar]
- 163. Seidah NG, Prat A, Pirillo A, Catapano AL, Norata GD. Novel strategies to target proprotein convertase subtilisin kexin 9: beyond monoclonal antibodies. Cardiovasc Res. 2019;115(3):510-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Tombling BJ, Zhang Y, Huang YH, Craik DJ, Wang CK. The emerging landscape of peptide-based inhibitors of PCSK9. Atherosclerosis. 2021;330:52-60. [DOI] [PubMed] [Google Scholar]
- 165. Fitzgerald K, White S, Borodovsky A, et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N Engl J Med. 2017;376(1):41-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Catapano AL, Pirillo A, Norata GD. New pharmacological approaches to target PCSK9. Curr Atheroscler Rep. 2020;22(7):24. [DOI] [PubMed] [Google Scholar]
- 167. Xu Q, Deng Y, Xiao J, et al. Three musketeers for lowering cholesterol: statins, ezetimibe and evolocumab. Curr Med Chem. 2021;28(5):1025-1041. [DOI] [PubMed] [Google Scholar]
- 168. Nambi V, Agha A. Inclisiran: a game changer in a changing game? J Am Coll Cardiol. 2021;77(9):1194-1196. [DOI] [PubMed] [Google Scholar]
- 169. Voutyritsa E, Damaskos C, Farmaki P, et al. PCSK9 antibody-based treatment strategies for patients with statin intolerance. In Vivo. 2021;35(1):61-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Hardy J, Niman S, Pereira E, et al. A critical review of the efficacy and safety of inclisiran. Am J Cardiovasc Drugs 2021. doi: 10.1007/s40256-021-00477-7 [DOI] [PubMed] [Google Scholar]
- 171. Katzmann JL, Gouni-Berthold I, Laufs U. PCSK9 inhibition: insights from clinical trials and future prospects. Front Physiol. 2020;11:595819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Ruscica M, Ferri N, Santos RD, Sirtori CR, Corsini A. Lipid lowering drugs: present status and future developments. Curr Atheroscler Rep. 2021;23(5):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Guedeney P, Sorrentino S, Giustino G, et al. Indirect comparison of the efficacy and safety of alirocumab and evolocumab: a systematic review and network meta-analysis. Eur Heart J Cardiovasc Pharmacother. 2021;7(3):225-235. [DOI] [PubMed] [Google Scholar]
- 174. Kosmas CE, Muñoz Estrella A, Sourlas A, Pantou D. Inclisiran in dyslipidemia. Drugs Today (Barc). 2021;57(5):311-319. [DOI] [PubMed] [Google Scholar]
- 175. Stein EA, Raal F. Reduction of low-density lipoprotein cholesterol by monoclonal antibody inhibition of PCSK9. Annu Rev Med. 2014;65:417-431. [DOI] [PubMed] [Google Scholar]
- 176. Chan JC, Piper DE, Cao Q, et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci U S A. 2009;106(24):9820-9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Koren MJ, Lundqvist P, Bolognese M, et al. ; MENDEL-2 Investigators . Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol. 2014;63(23):2531-2540. [DOI] [PubMed] [Google Scholar]
- 178. Roth EM, Taskinen MR, Ginsberg HN, et al. Monotherapy with the PCSK9 inhibitor alirocumab versus ezetimibe in patients with hypercholesterolemia: results of a 24 week, double-blind, randomized Phase 3 trial. Int J Cardiol. 2014;176(1):55-61. [DOI] [PubMed] [Google Scholar]
- 179. Benhuri B, Ueyama H, Takagi H, Briasoulis A, Kuno T. PCSK9 Inhibitors and ezetimibe monotherapy in patients not receiving statins: a meta-analysis of randomized trials. Curr Vasc Pharmacol. 2021;19(4):390-397. [DOI] [PubMed] [Google Scholar]
- 180. Gouni-Berthold I, Descamps OS, Fraass U, et al. Systematic review of published Phase 3 data on anti-PCSK9 monoclonal antibodies in patients with hypercholesterolaemia. Br J Clin Pharmacol. 2016;82(6):1412-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Ito MK, Santos RD. PCSK9 inhibition with monoclonal antibodies-modern management of hypercholesterolemia. J Clin Pharmacol. 2017;57(1):7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Rey J, Poitiers F, Paehler T, et al. Relationship between low-density lipoprotein cholesterol, free proprotein convertase subtilisin/kexin type 9, and alirocumab levels after different lipid-lowering strategies. J Am Heart Assoc. 2016;5(6):e003323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Silbernagel G, Steiner LK, Hollstein T, et al. The interrelations between PCSK9 metabolism and cholesterol synthesis and absorption. J Lipid Res. 2019;60(1):161-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Ginsberg HN, Tuomilehto J, Hovingh GK, et al. Impact of age on the efficacy and safety of alirocumab in patients with heterozygous familial hypercholesterolemia. Cardiovasc Drugs Ther. 2019;33(1):69-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Kastelein JJ, Hovingh GK, Langslet G, et al. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 monoclonal antibody alirocumab vs placebo in patients with heterozygous familial hypercholesterolemia. J Clin Lipidol. 2017;11(1):195-203.e194. [DOI] [PubMed] [Google Scholar]
- 186. Thedrez A, Blom DJ, Ramin-Mangata S, et al. Homozygous familial hypercholesterolemia patients with identical mutations variably express the LDLR (low-density lipoprotein receptor): implications for the efficacy of evolocumab. Arterioscler Thromb Vasc Biol. 2018;38(3):592-598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Sabatine MS, Giugliano RP, Keech AC, et al. ; FOURIER Steering Committee and Investigators . Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713-1722. [DOI] [PubMed] [Google Scholar]
- 188. Sabatine MS, Leiter LA, Wiviott SD, et al. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: a prespecified analysis of the FOURIER randomised controlled trial. Lancet Diabetes Endocrinol. 2017;5(12):941-950. [DOI] [PubMed] [Google Scholar]
- 189. Schwartz GG, Steg PG, Szarek M, et al. ; ODYSSEY OUTCOMES Committees and Investigators . Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379(22):2097-2107. [DOI] [PubMed] [Google Scholar]
- 190. Wiviott SD, Giugliano RP, Morrow DA, et al. Effect of evolocumab on type and size of subsequent myocardial infarction: a prespecified analysis of the FOURIER randomized clinical trial. JAMA Cardiol. 2020;5(7):787-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Deedwania P, Murphy SA, Scheen A, et al. Efficacy and Safety of PCSK9 inhibition with evolocumab in reducing cardiovascular events in patients with metabolic syndrome receiving statin therapy: secondary analysis from the FOURIER randomized clinical trial. JAMA Cardiol. 2021;6(2):139-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Erqou S, Kaptoge S, Perry PL, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Boffa MB, Koschinsky ML. The journey towards understanding lipoprotein(a) and cardiovascular disease risk: are we there yet? Curr Opin Lipidol. 2018;29(3):259-267. [DOI] [PubMed] [Google Scholar]
- 194. Romagnuolo R, Scipione CA, Boffa MB, Marcovina SM, Seidah NG, Koschinsky ML. Lipoprotein(a) catabolism is regulated by proprotein convertase subtilisin/kexin type 9 through the low density lipoprotein receptor. J Biol Chem. 2015;290(18):11649-11662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Romagnuolo R, Scipione CA, Marcovina SM, et al. Roles of the low density lipoprotein receptor and related receptors in inhibition of lipoprotein(a) internalization by PCSK9. PLoS One. 2017;12(7):e0180869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Blanchard V, Chemello K, Hollstein T, et al. The size of apolipoprotein (a) is an independent determinant of the reduction in lipoprotein (a) induced by PCSK9 inhibitors. Cardiovasc Res. 2021: cvab247. doi: 10.1093/cvr/cvab247 [DOI] [PubMed] [Google Scholar]
- 197. Villard EF, Thedrez A, Blankenstein J, et al. PCSK9 modulates the secretion but not the cellular uptake of lipoprotein(a) ex vivo: an effect blunted by alirocumab. JACC Basic Transl Sci. 2016;1(6):419-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Jukema JW, Szarek M, Zijlstra LE, et al. ; ODYSSEY OUTCOMES Committees and Investigators . Alirocumab in patients with polyvascular disease and recent acute coronary syndrome: ODYSSEY OUTCOMES trial. J Am Coll Cardiol. 2019;74(9):1167-1176. [DOI] [PubMed] [Google Scholar]
- 199. Tanowitz M, Hettrick L, Revenko A, Kinberger GA, Prakash TP, Seth PP. Asialoglycoprotein receptor 1 mediates productive uptake of N-acetylgalactosamine-conjugated and unconjugated phosphorothioate antisense oligonucleotides into liver hepatocytes. Nucleic Acids Res. 2017;45(21):12388-12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Susan-Resiga D, Girard E, Essalmani R, et al. Asialoglycoprotein receptor 1 is a novel PCSK9-independent ligand of liver LDLR cleaved by furin. J Biol Chem. 2021;297(4):101177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Blais JE, Wei Y, Yap KKW, et al. Trends in lipid-modifying agent use in 83 countries. Atherosclerosis. 2021;328:44-51. [DOI] [PubMed] [Google Scholar]
- 202. Dayoub EJ, Eberly LA, Nathan AS, et al. Adoption of PCSK9 inhibitors among patients with atherosclerotic disease. J Am Heart Assoc. 2021;10(9):e019331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Grundy SM, Stone NJ. 2018 Cholesterol clinical practice guidelines: synopsis of the 2018 American Heart Association/American College of Cardiology/Multisociety cholesterol guideline. Ann Intern Med. 2019;170(11):779-783. [DOI] [PubMed] [Google Scholar]
- 204. Crossey E, Amar MJA, Sampson M, et al. A cholesterol-lowering VLP vaccine that targets PCSK9. Vaccine. 2015;33(43):5747-5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Pan Y, Zhou Y, Wu H, et al. A therapeutic peptide vaccine against PCSK9. Sci Rep. 2017;7(1):12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Toth S, Pella D, Fedacko J. Vaccines targeting PSCK9 for the treatment of hyperlipidemia. Cardiol Ther. 2020;9(2):323-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Fougeroux C, Goksøyr L, Idorn M, et al. Capsid-like particles decorated with the SARS-CoV-2 receptor-binding domain elicit strong virus neutralization activity. Nat Commun. 2021;12(1):324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816-821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Nidhi S, Anand U, Oleksak P, et al. Novel CRISPR-cas systems: an updated review of the current achievements, applications, and future research perspectives. Int J Mol Sci. 2021;22(7):3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Collins FS, Doudna JA, Lander ES, Rotimi CN. Human molecular genetics and genomics—important advances and exciting possibilities. N Engl J Med. 2021;384(1):1-4. [DOI] [PubMed] [Google Scholar]
- 211. Broeders M, Herrero-Hernandez P, Ernst MPT, van der Ploeg AT, Pijnappel WWMP. Sharpening the molecular scissors: advances in gene-editing technology. Iscience. 2020;23(1):100789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Ding Q, Strong A, Patel KM, et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res. 2014;115(5):488-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Rossidis AC, Stratigis JD, Chadwick AC, et al. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat Med. 2018;24(10):1513-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Sun W, Wang J, Hu Q, Zhou X, Khademhosseini A, Gu Z. CRISPR-Cas12a delivery by DNA-mediated bioresponsive editing for cholesterol regulation. Sci Adv. 2020;6(21):eaba2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Wei T, Cheng Q, Min YL, Olson EN, Siegwart DJ. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat Commun. 2020;11(1):3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Musunuru K, Chadwick AC, Mizoguchi T, et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. 2021;593(7859):429-434. [DOI] [PubMed] [Google Scholar]
- 217. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801-810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Russell JA. Management of sepsis. N Engl J Med. 2006;355(16):1699-1713. [DOI] [PubMed] [Google Scholar]
- 219. Walley KR, Thain KR, Russell JA, et al. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Sci Transl Med. 2014;6(258):258ra143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Boyd JH, Fjell CD, Russell JA, Sirounis D, Cirstea MS, Walley KR. Increased plasma PCSK9 levels are associated with reduced endotoxin clearance and the development of acute organ failures during sepsis. J Innate Immun. 2016;8(2):211-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Walley KR. Role of lipoproteins and proprotein convertase subtilisin/kexin type 9 in endotoxin clearance in sepsis. Curr Opin Crit Care. 2016;22(5):464-469. [DOI] [PubMed] [Google Scholar]
- 222. Dwivedi DJ, Grin PM, Khan M, et al. Differential expression of PCSK9 modulates infection, inflammation, and coagulation in a murine model of sepsis. Shock. 2016;46(6):672-680. [DOI] [PubMed] [Google Scholar]
- 223. Grin PM, Dwivedi DJ, Chathely KM, et al. Low-density lipoprotein (LDL)-dependent uptake of Gram-positive lipoteichoic acid and Gram-negative lipopolysaccharide occurs through LDL receptor. Sci Rep. 2018;8(1):10496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Yuan Y, Wu W, Sun S, Zhang Y, Chen Z. PCSK9: a potential therapeutic target for sepsis. J Immunol Res. 2020;2020:2687692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Leung AKK, Genga KR, Topchiy E, et al. Reduced Proprotein convertase subtilisin/kexin 9 (PCSK9) function increases lipoteichoic acid clearance and improves outcomes in Gram positive septic shock patients. Sci Rep. 2019;9(1):10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Feng Q, Wei WQ, Chaugai S, et al. A genetic approach to the association between PCSK9 and sepsis. JAMA Netw Open. 2019;2(9):e1911130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Genga KR, Lo C, Cirstea MS, et al. Impact of PCSK9 loss-of-function genotype on 1-year mortality and recurrent infection in sepsis survivors. Ebiomedicine. 2018;38:257-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Shimada T, Topchiy E, Leung AKK, et al. Very low density lipoprotein receptor sequesters lipopolysaccharide into adipose tissue during sepsis. Crit Care Med. 2020;48(1):41-48. [DOI] [PubMed] [Google Scholar]
- 229. Efron PA, Mohr AM, Moore FA, Moldawer LL. The future of murine sepsis and trauma research models. J Leukoc Biol. 2015;98(6):945-952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Shaler CR, Choi J, Rudak PT, et al. MAIT cells launch a rapid, robust and distinct hyperinflammatory response to bacterial superantigens and quickly acquire an anergic phenotype that impedes their cognate antimicrobial function: defining a novel mechanism of superantigen-induced immunopathology and immunosuppression. PLoS Biol. 2017;15(6):e2001930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Laudanski K. Humanized mice as a tool to study sepsis—more than meets the eye. Int J Mol Sci. 2021;22(5):2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Atreya MR, Whitacre BE, Cvijanovich NZ, et al. Proprotein convertase subtilisin/kexin type 9 loss-of-function is detrimental to the juvenile host with septic shock. Crit Care Med. 2020;48(10):1513-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Vecchié A, Bonaventura A, Meessen J, et al. ; ALBIOS Biomarkers Study Investigators . PCSK9 is associated with mortality in patients with septic shock: data from the ALBIOS study. J Intern Med. 2021;289(2):179-192. [DOI] [PubMed] [Google Scholar]
- 234. Innocenti F, Gori AM, Giusti B, et al. Plasma PCSK9 levels and sepsis severity: an early assessment in the emergency department. Clin Exp Med. 2021;21(1):101-107. [DOI] [PubMed] [Google Scholar]
- 235. Rannikko J, Jacome Sanz D, Ortutay Z, et al. Reduced plasma PCSK9 response in patients with bacteraemia is associated with mortality. J Intern Med. 2019;286(5):553-561. [DOI] [PubMed] [Google Scholar]
- 236. Trinder M, Boyd JH, Brunham LR. Molecular regulation of plasma lipid levels during systemic inflammation and sepsis. Curr Opin Lipidol. 2019;30(2):108-116. [DOI] [PubMed] [Google Scholar]
- 237. Molina S, Castet V, Fournier-Wirth C, et al. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J Hepatol. 2007;46(3):411-419. [DOI] [PubMed] [Google Scholar]
- 238. Felmlee DJ, Hafirassou ML, Lefevre M, Baumert TF, Schuster C. Hepatitis C virus, cholesterol and lipoproteins–impact for the viral life cycle and pathogenesis of liver disease. Viruses. 2013;5(5):1292-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Caron J, Pène V, Tolosa L, et al. Low-density lipoprotein receptor-deficient hepatocytes differentiated from induced pluripotent stem cells allow familial hypercholesterolemia modeling, CRISPR/Cas-mediated genetic correction, and productive hepatitis C virus infection. Stem Cell Res Ther. 2019;10(1):221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. D’Ambrosio R, Pasulo L, Puoti M, et al. ; NAVIGATORE-Lombardia Study Group . Real-world effectiveness and safety of glecaprevir/pibrentasvir in 723 patients with chronic hepatitis C. J Hepatol. 2019;70(3):379-387. [DOI] [PubMed] [Google Scholar]
- 241. Hyrina A, Olmstead AD, Steven P, Krajden M, Tam E, Jean F. Treatment-induced viral cure of hepatitis C virus-infected patients involves a dynamic interplay among three important molecular players in lipid homeostasis: circulating microRNA (miR)-24, miR-223, and proprotein convertase subtilisin/kexin type 9. Ebiomedicine. 2017;23:68-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Bridge SH, Sheridan DA, Felmlee DJ, et al. PCSK9, apolipoprotein E and lipoviral particles in chronic hepatitis C genotype 3: evidence for genotype-specific regulation of lipoprotein metabolism. J Hepatol. 2015;62(4):763-770. [DOI] [PubMed] [Google Scholar]
- 243. Grimm J, Peschel G, Müller M, et al. Rapid decline of serum proprotein convertase subtilisin/kexin 9 (PCSK9) in non-cirrhotic patients with chronic hepatitis C infection receiving direct-acting antiviral therapy. J Clin Med. 2021;10(8):1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Labonté P, Begley S, Guévin C, et al. PCSK9 impedes hepatitis C virus infection in vitro and modulates liver CD81 expression. Hepatology. 2009;50(1):17-24. [DOI] [PubMed] [Google Scholar]
- 245. Seidah NG. New developments in proprotein convertase subtilisin-kexin 9’s biology and clinical implications. Curr Opin Lipidol. 2016;27(3):274-281. [DOI] [PubMed] [Google Scholar]
- 246. Bhatt S, Gething PW, Brady OJ, et al. The global distribution and burden of dengue. Nature. 2013;496(7446):504-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Gan ES, Tan HC, Le DHT, et al. Dengue virus induces PCSK9 expression to alter antiviral responses and disease outcomes. J Clin Invest. 2020;130(10):5223-5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89(3):331-340. [DOI] [PubMed] [Google Scholar]
- 249. Li Z, Liu Q. Proprotein convertase subtilisin/kexin type 9 inhibits interferon β expression through interacting with ATF-2. FEBS Lett. 2018;592(13):2323-2333. [DOI] [PubMed] [Google Scholar]
- 250. Tikoo K, Patel G, Kumar S, et al. Tissue specific up regulation of ACE2 in rabbit model of atherosclerosis by atorvastatin: role of epigenetic histone modifications. Biochem Pharmacol. 2015;93(3):343-351. [DOI] [PubMed] [Google Scholar]
- 251. Shin YH, Min JJ, Lee JH, et al. The effect of fluvastatin on cardiac fibrosis and angiotensin-converting enzyme-2 expression in glucose-controlled diabetic rat hearts. Heart Vessels. 2017;32(5):618-627. [DOI] [PubMed] [Google Scholar]
- 252. Zhang XJ, Qin JJ, Cheng X, et al. In-hospital use of statins is associated with a reduced risk of mortality among individuals with COVID-19. Cell Metab. 2020;32(2):176-187.e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Tan WYT, Young BE, Lye DC, Chew DEK, Dalan R. Statin use is associated with lower disease severity in COVID-19 infection. Sci Rep. 2020;10(1):17458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Memel ZN, Lee JJ, Foulkes AS, Chung RT, Thaweethai T, Bloom PP. Statins are associated with improved 28-day mortality in patients hospitalized with SARS-CoV-2 Infection. medRxiv 2021. doi: 10.1101/2021.03.27.212.54.373 [DOI] [Google Scholar]
- 255. Vuorio A, Kovanen PT. Prevention of endothelial dysfunction and thrombotic events in COVID-19 patients with familial hypercholesterolemia. J Clin Lipidol. 2020;14(5):617-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Vuorio A, Kovanen PT. PCSK9 inhibitors for COVID-19: an opportunity to enhance the antiviral action of interferon in patients with hypercholesterolaemia. J Intern Med. 2021;289(5):749-751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Zhang Q, Bastard P, Liu Z, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370(6515):eabd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Gudleski-O’Regan N, Greco TM, Cristea IM, Shenk T. Increased expression of LDL receptor-related protein 1 during human cytomegalovirus infection reduces virion cholesterol and infectivity. Cell Host Microbe. 2012;12(1):86-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Mayengbam SS, Singh A, Pillai AD, Bhat MK. Influence of cholesterol on cancer progression and therapy. Transl Oncol. 2021;14(6):101043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Cruz PM, Mo H, McConathy WJ, Sabnis N, Lacko AG. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: a review of scientific findings, relevant to future cancer therapeutics. Front Pharmacol. 2013;4:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Vasseur S, Guillaumond F. LDL Receptor: An open route to feed pancreatic tumor cells. Mol Cell Oncol. 2016;3(1):e1033586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Guillaumond F, Bidaut G, Ouaissi M, et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2015;112(8):2473-2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343(6257):425-430. [DOI] [PubMed] [Google Scholar]
- 264. Schafer WR, Kim R, Sterne R, Thorner J, Kim SH, Rine J. Genetic and pharmacological suppression of oncogenic mutations in ras genes of yeast and humans. Science. 1989;245(4916):379-385. [DOI] [PubMed] [Google Scholar]
- 265. Wesierska-Gadek J, Kramer MP, Schmid G. A combined treatment of HeLa cells with the farnesyl protein transferase inhibitor L-744 832 and cisplatin significantly increases the therapeutic effect as compared to cisplatin monotherapy. J Cell Biochem. 2008;104(1):189-201. [DOI] [PubMed] [Google Scholar]
- 266. Liu X, Bao X, Hu M, et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature. 2020;588(7839):693-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Almeida CR, Ferreira BH, Duarte IF. Targeting PCSK9: a promising adjuvant strategy in cancer immunotherapy. Signal Transduct Target Ther. 2021;6(1):111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Gangloff A, Calon F, Seidah NG. Can iPCSK9-induced hypocholesterolemia starve cancer cells? J Clin Lipidol. 2017;11(3):600-601. [DOI] [PubMed] [Google Scholar]
- 269. Sun X, Essalmani R, Day R, Khatib AM, Seidah NG, Prat A. Proprotein convertase subtilisin/kexin type 9 deficiency reduces melanoma metastasis in liver. Neoplasia. 2012;14(12):1122-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Nowak C, Ärnlöv J. A Mendelian randomization study of the effects of blood lipids on breast cancer risk. Nat Commun. 2018;9(1):3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Bhattacharya A, Chowdhury A, Chaudhury K, Shukla PC. Proprotein convertase subtilisin/kexin type 9 (PCSK9): a potential multifaceted player in cancer. Biochim Biophys Acta Rev Cancer. 2021;1876(1):188581. [DOI] [PubMed] [Google Scholar]
- 272. Yuan J, Cai T, Zheng X, et al. Potentiating CD8+ T cell antitumor activity by inhibiting PCSK9 to promote LDLR-mediated TCR recycling and signaling. Protein Cell. 2021;12(4):240-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Voutsadakis IA. Immune blockade inhibition in breast cancer. Anticancer Res. 2016;36(11):5607-5622. [DOI] [PubMed] [Google Scholar]
- 274. Park J, Kwon M, Shin EC. Immune checkpoint inhibitors for cancer treatment. Arch Pharm Res. 2016;39(11):1577-1587. [DOI] [PubMed] [Google Scholar]
- 275. Han B, Eacho PI, Knierman MD, et al. Isolation and characterization of the circulating truncated form of PCSK9. J Lipid Res. 2014;55(7):1505-1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Demetz E, Tymoszuk P, Hilbe R, et al. The haemochromatosis gene Hfe and Kupffer cells control LDL cholesterol homeostasis and impact on atherosclerosis development. Eur Heart J. 2020;41(40):3949-3959. [DOI] [PubMed] [Google Scholar]
- 277. Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun. 2020;11(1):3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Liang H, Chaparro-Riggers J, Strop P, et al. Proprotein convertase substilisin/kexin type 9 antagonism reduces low-density lipoprotein cholesterol in statin-treated hypercholesterolemic nonhuman primates. J Pharmacol Exp Ther. 2012;340(2):228-236. [DOI] [PubMed] [Google Scholar]
- 279. Liu X, Suo R, Chan CZY, Liu T, Tse G, Li G. The immune functions of PCSK9: Local and systemic perspectives. J Cell Physiol. 2019;234(11):19180-19188. [DOI] [PubMed] [Google Scholar]
- 280. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172(5):2731-2738. [DOI] [PubMed] [Google Scholar]
- 281. Jin KT, Du WL, Lan HR, et al. Development of humanized mouse with patient-derived xenografts for cancer immunotherapy studies: a comprehensive review. Cancer Sci. 2021;112(7):2592-2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Korneva V, Kuznetsova T, Julius U. The state of the problem of achieving extremely low LDL levels. Curr Pharm Des. 2021. doi: 10.2174/1381612827999210111182030 [DOI] [PubMed] [Google Scholar]
- 283. Conroy T, Hammel P, Hebbar M, et al. ; Canadian Cancer Trials Group and the Unicancer-GI–PRODIGE Group . FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med. 2018;379(25):2395-2406. [DOI] [PubMed] [Google Scholar]
- 284. Seidah NG. The PCSK9 revolution and the potential of PCSK9-based therapies to reduce LDL-cholesterol. J Glob Cardiol Sci Pract. 2017;2017(1):e201702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Mikaeeli S, Resiga DS, Girard E, et al. Functional analysis of natural PCSK9 Mutants in modern and archaic humans. FEBS J. 2020;287(3):515-528. [DOI] [PubMed] [Google Scholar]
- 286. Ding K, McDonough SJ, Kullo IJ. Evidence for positive selection in the C-terminal domain of the cholesterol metabolism gene PCSK9 based on phylogenetic analysis in 14 primate species. PLoS One. 2007;2(10):e1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Cameron J, Holla ØL, Berge KE, et al. Investigations on the evolutionary conservation of PCSK9 reveal a functionally important protrusion. FEBS J. 2008;275(16):4121-4133. [DOI] [PubMed] [Google Scholar]
- 288. Murphy WJ, Eizirik E, Johnson WE, Zhang YP, Ryder OA, O’Brien SJ. Molecular phylogenetics and the origins of placental mammals. Nature. 2001;409(6820):614-618. [DOI] [PubMed] [Google Scholar]
- 289. van Asch B, Teixeira da Costa LF. Patterns and tempo of PCSK9 pseudogenizations suggest an ancient divergence in mammalian cholesterol homeostasis mechanisms. Genetica. 2021;149(1):1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Poulsen CB, Pedrigi RM, Pareek N, et al. Plaque burden influences accurate classification of fibrous cap atheroma by in vivo optical coherence tomography in a porcine model of advanced coronary atherosclerosis. Eurointervention. 2018;14(10):1129-1135. [DOI] [PubMed] [Google Scholar]