

Abstract
Medulloblastoma, a malignant childhood brain tumour, has been the recent focus of intensive molecular profiling efforts, profoundly advancing our understanding of biologically and clinically heterogeneous disease subgroups. Genomic, epigenomic, transcriptomic, and proteomic landscapes have now been mapped for an unprecedented number of bulk medulloblastoma patient samples, and more recently single medulloblastoma cells. These efforts have provided pivotal new insights into the diverse molecular mechanisms presumed to drive tumour initiation, maintenance, and recurrence across individual subgroups and subtypes. Translational opportunities stemming from this knowledge are continuing to evolve, providing a framework for improved diagnostic and therapeutic intervention. In this Review, we summarize recent advances illuminated through continued molecular characterization of medulloblastoma and contextualize this progress towards the deployment of more effective, molecularly informed treatments for affected children.
Introduction
Medulloblastoma (MB), a pediatric cerebellar tumour, is one of the most common malignant central nervous system (CNS) tumours in children (~500 new diagnoses/year in the United States) and a leading cause of cancer-related death in this age group1. During the past two decades, large-scale genomic efforts have helped disentangle the molecular basis of MB, especially biologically and clinically relevant intertumoural heterogeneity. Consensus molecular subgroups2, WNT, SHH, Group 3, and Group 4, each characterized by distinct ‘omic and clinical features, are now widely recognized. As a direct consequence, mechanistic, developmental, and preclinical studies are currently undertaken in a manner that is cognizant of molecular subgroup status. Moreover, clinical protocols have adopted molecular subgrouping strategies into routine diagnosis, treatment stratification, and patient selection for molecularly targeted therapies.
In 2012, following the formal recognition of consensus molecular subgroups, the initial wave of next-generation sequencing (NGS) studies conducted on primary MB samples were reported3–7. Since then, additional molecular characterization has ensued on increasingly large cohorts, resulting in the generation of a wealth of multidimensional ‘omics data. As a result, molecular classification of MB has evolved beyond the four consensus subgroups and new methods for robust and accurate assignment of patients into relevant subtypes have become mainstream. More recently, spatial and single-cell genomic approaches have been applied, delving into intratumoural heterogeneity, cellular composition, and developmental origins at single-cell resolution. We recently reviewed the epidemiologic, biologic, and therapeutic characteristics of MB in a report that was intended to be all encompassing of the human disease1. In this Review, we emphasize what has been learned from the ‘omic analysis of MB patients since the first NGS studies3, highlighting insights into the molecular and biological mechanisms underlying MB heterogeneity and translational implications emerging from these efforts.
Molecular Classification
WHO Consensus subgroups & subtypes within subgroups
First gene expression array studies confirmed that MB was molecularly distinct from other embryonal brain tumours, such as primitive neuroectodermal tumour (PNET) and atypical teratoid rhabdoid tumour (AT/RT)8. In this seminal report, gene expression comparison of classic versus desmoplastic histology MB identified specific up-regulation of genes involved in hedgehog signaling in desmoplastic tumours, including PTCH1, GLI1, and MYCN8. A parallel report comparing expression profiles of a series of genetically engineered mouse (GEM) models to normal cerebellar controls identified similar activation of hedgehog pathway-associated gene sets in MB GEM models9. Together, these early reports of gene expression signatures defining MBs in humans and mice would pave the way for the molecular era that would follow. Multiple independent gene expression array profiling studies conducted on patient cohorts described distinct molecular subgroups within MB that differ in their demographics, genetic alterations, and clinical outcomes10–13, culminating in the definition of consensus subgroups2. The WNT and SHH subgroups, which represent approximately 10% and 30% of all MB patients, respectively, are associated with activation of the WNT (Wingless) and SHH (Sonic hedgehog) signaling pathways. WNT and SHH subgroups are discrete across studies and technologies, providing a basis for their incorporation into the 2016 update of the WHO Classification of Central Nervous System Tumors14. Further subdivision within the SHH subgroup according to TP53 mutation status (SHH-TP53 wild type and SHH-TP53 mutant) is also recognized by the WHO. Group 3 and Group 4 subgroups, which represent the remaining 25% and 35% of all MB patients, respectively, exhibit some molecular and biological similarities (detailed below). Consequently, these subgroups are formally recognized by the WHO as non-WNT/non-SHH-MB, listing Group 3 and Group 4 designations as provisional entities.
Differential expression analysis between MB subgroups has led to the identification of select biomarkers that enable molecular classification based on immunohistochemistry12,15,16, NanoString17,18, and other panel-based gene expression assays19. While these assays are widely accessible in clinical practice, DNA methylation arrays, which can measure hundreds of thousands of methylation sites across the genome, have emerged as the platform of choice for MB classification. Owing to the relative stability of DNA, methylation arrays and derivative targeted DNA methylation-based assays, allow for routine analysis of clinical samples in a diagnostic setting as well as profiling of archival tumour material, with limited technical variation between institutions20–24.
In 2017, three independent studies25–27 used DNA methylation analysis to investigate additional substructure within subgroups, leading to a varying definition of molecular subtypes in each study (Figure 1; detailed below).
Figure 1 |. Comparison of MB DNA methylation-derived subtypes described across recent studies.

a | Correspondence between four molecular subtypes of the SHH-MB subgroup described by Cavalli et al. and the subtypes described in three additional studies. DNA methylation profiles from all samples of each additional study were used to classify patients into the four molecular subtypes using a machine learning approach. The height of each row corresponds to the fraction of samples per subtype in the Cavalli et al. study. Percentages indicate overlap of predicted subtypes with original subtype annotations in each additional study. No samples from the study by Robinson et al. predicted as SHH-δ, because the study only included patients under the age of six years.
b | Similar comparison between the eight molecular subtypes of Group 3/4-MB described by Northcott et al. and Sharma et al. and the subtypes described in two additional studies. Line width between consensus subgroups and DNA methylation subtypes indicate the fraction of samples per subtype originally classified as Group 3- or Group 4-MB.
WNT medulloblastoma.
WNT-MB primarily occurs in children after 4 years of age to early adulthood (median age ~11 years) and exhibits a balanced male:female ratio (Figure 2). WNT-MB tumours are usually of classic histology and infrequently metastatic at diagnosis. Outcomes for WNT-MB patients are favorable, experiencing 5-year survival rates of 95% or better. Somatic mutations in CTNNB1 are the hallmark genetic event defining this subgroup (~85% of patients)13,28, with most remaining patients typified by pathogenic APC variants in the germline29. Tumour genomes are mostly devoid of somatic copy-number alterations (SCNAs), except for loss of chromosome 6 (i.e. monosomy 6) in most patients.
Figure 2 |. Summary of demographic, clinical, and molecular features of novel MB subtypes.

Values for age and gender distribution, frequency of metastasis, and 5-year overall survival for the WNT and SHH subgroups are derived from the Cavalli et al. study. Driver events were additionally derived from the Kool et al. and Robinson et al. studies. Similarly, values for the Group 3 and Group 4 subgroups were derived from the Sharma et al. study. OS, overall survival; LCA, large cell anaplastic; MBEN, medulloblastoma with extensive nodularity; i17q, isochromosome 17q; BCOR, BCL6 Corepressor; CTDNEP1, CTD Nuclear Envelope Phosphatase 1; CTNNB1, Catenin Beta 1; DDX3X, DEAD-Box Helicase 3 X-Linked; GFI1, Growth Factor Independent 1 Transcriptional Repressor; GFI1B, Growth Factor Independent 1B Transcriptional Repressor; GLI2, GLI Family Zinc Finger 2; KBTBD4, Kelch Repeat and BTB Domain Containing 4; KDM6A, Lysine Demethylase 6A (UTX); KMT2C, Lysine Methyltransferase 2C (MLL3); KMT2D, Lysine Methyltransferase 2D (MLL2); MYC, MYC Proto-Oncogene BHLH Transcription Factor; MYCN, MYCN Proto-Oncogene BHLH Transcription Factor; OTX2, Orthodenticle Homeobox 2; PRDM6, PR/SET Domain 6; PTCH1, Patched 1; PTEN, Phosphatase and Tensin Homolog; SMARCA4, SWI/SNF Related Matrix Associated Actin Dependent Regulator of Chromatin Subfamily A Member 4; SMO, Smoothened Frizzled Class Receptor; SUFU, SUFU Negative Regulator of Hedgehog Signaling; TERT, Telomerase Reverse Transcriptase; TP53, Tumour Protein P53; ZMYM3, Zinc Finger MYM-Type Containing 3.
WNT-MB has been described to be largely homogenous between patients in regard to genome-wide expression and methylation profiles. However, two molecular subtypes, WNT α and WNT β, that differ in age at diagnosis (median age of 10 vs. 20 years, respectively) and frequency of monosomy 6 have been suggested25. Survival outcomes of adults with WNT-MB (i.e. WNT β25) have been inconsistent in the literature, with some reports describing outcomes to be comparably favorable to those of pediatric WNT-MB patients25,30, and others reporting reduced overall survival31.
SHH medulloblastoma.
SHH-MB displays an intriguing bimodal age distribution, representing the most common molecular subgroup in infants (<3 years of age) and adults (>17 years of age), with fewer cases diagnosed during childhood and adolescence (Figure 2). Demographically, SHH-MB is more common in males than females (approximately 2:1, male:female). Classic and desmoplastic/nodular (including medulloblastoma with extensive nodularity, MBEN) histology occur at similar frequencies (each accounting for ~40% of patients), with large-cell/anaplastic (LCA) histology making up the remainder. Mutations and focal SCNAs targeting genes in the SHH signaling pathway represent the most common genetic events, including inactivating germline or somatic mutations and deletions in PTCH1 and SUFU, activating mutations in SMO, and amplifications of GLI232. Frequent chromosomal alterations include loss of chromosomes 9q, 10q, 14q, and 17p, and gains of chromosomes 2 and 9p.
Age-associated molecular differences discriminating infant and adult SHH-MB have been observed by both gene expression and DNA methylation array profiling33,27,32. Compared to pediatric counterparts, adult SHH-MBs are characterized by a higher overall mutational burden, a higher prevalence of SHH-pathway associated mutations (including a higher incidence of PTCH1 and SMO alterations), a more expansive list of chromatin modifier mutations (i.e. BRPF1, CREBBP), and virtually all harbor TERT promoter mutations32,34–36. Intriguingly, transcriptional comparison of available SHH-MB mouse models to patient tumours suggested that, despite being primarily driven by inactivation of Ptch1 or activation of Smo – genetic events that occur in both pediatric and adult tumours, current mouse models are more molecularly similar to adult SHH-MB37.
More recently, four molecular subtypes of SHH-MB have been reported25 (Figure 1 & 2): SHH-β and SHH-γ correspond to infant subtypes (median age of 1.9 and 1.3 years, respectively), whereas SHH-α and SHH-δ respectively correspond to childhood/adolescent and adult subtypes (median age of 8 and 26 years, respectively). Subtype SHH-α is enriched for patients harboring TP53 mutations (~1/3 of SHH-α patients) and associated with an inferior outcome compared to SHH-δ patients. Infant subtype SHH-β showed lower 5-year survival than SHH-γ. Similar observations were made in an independent study reporting on a clinical trial in infants and young children (under 6 years of age) which identified two molecular subtypes, iSHH-I (equivalent to SHH-β) and iSHH-II (equivalent to SHH-γ and SHH-α)38 (Figure 1 & 2).
Group 3/4 medulloblastoma.
Group 3-MB occurs during infancy and childhood and is rarely seen in patients older than 18 years of age, whereas Group 4-MB occurs across all age groups (Figure 2). Male:female ratios are 2:1 or higher for both subgroups. LCA histology is more prevalent in Group 3 than Group 4, and 30–40% of patients are metastatic at diagnosis in both subgroups. MYC amplification is a common genetic feature of Group 3-MB and is associated with a particularly poor clinical outcome10,12. MYCN and CDK6 amplifications are notable genetic alterations seen in Group 4-MB6. Isochromosome 17q is a hallmark cytogenetic event in both subgroups, found in >50% of patients from either subgroup.
The definition and substructure of Group 3/4-MB has been a topic of debate since their initial discovery10–13. Indeed, early nomenclature did not always define Group 3 and Group 4 into distinct subgroups and in some cases described them as a single ‘mixed’ subgroup of patients designated as ‘non-WNT/non-SHH’ MB16. However, the recognition of Group 3 and Group 4 MB has continued to evolve in recent years, with the majority of the neuro-oncology community, and the WHO14, acknowledging their definition as mostly discrete molecular entities and supporting the utility of their distinction, as supported in the literature (see1,39–41 for recent reviews).
More recently, three independent studies identified a varying number of subtypes within Group 3 and Group 4 (Figure 1). Schwalbe and colleagues identified four molecular subtypes in Group 3- and Group 4-MB that split each subgroup into high- and low-risk subtypes27. Cavalli and colleagues identified three molecular subtypes in each subgroup: Group 3α, β, and γ, and Group 4α, β, and γ25. In a combined analysis of Group 3 and Group 4 cases, Northcott and colleagues identified eight molecular subtypes, designated I to VIII26. In order to harmonize subtype definitions, a joint analysis of 1,501 Group 3/4 MBs from all three studies was recently undertaken42. This analysis showed that separation into eight subtypes best unified the substructure observed in each of the aforementioned studies (Figure 1).
Subtype I represents the least common subtype and comprises a mix of Group 3 and Group 4 tumours (Figure 2). Subtype I tumour genomes are generally balanced and enriched for amplification of the OTX2 oncogene and activation of GFI1/GFI1B (described in detail below). Subtypes II, III and IV are bona fide Group 3 subtypes. Subtypes II and III are characterized by amplification of the MYC oncogene and are associated with poor outcomes. In comparison to other subtypes, Subtype IV is enriched for younger patients (median age of 3 years) and is associated with a favorable outcome; low progression free survival observed in infant Subtype IV patients suggests that survival rates are dependent on treatment with craniospinal axis radiation38. Subtypes V, VI and VII consist mostly of Group 4 MBs, but also include some Group 3 tumours. Subtype V genomes are characterized by amplification of both MYC and MYCN and are associated with modest outcomes. Subtype VIII is the most common and only pure Group 4 subtype, mostly occurring in older children (median age of 10 years). Subtype VIII tumours display a balanced genome, except for presence of isochromosome 17 (i17q) in most cases. Subtype VIII is associated with favorable 5-year survival; however, many patients are affected by late relapse and death, a feature that is unique to this subtype42.
Genome
Gene mutations.
MB NGS studies primarily detailed somatic non-synonymous mutations affecting protein-coding genes in relatively modest patient cohorts4,5,7,43. New recurrently mutated genes emerged from these analyses, including DDX3X, BCOR, CTDNEP1, and TBR1, among others. In addition, chromatin-modifying genes such as KMT2D (MLL2), KMT2C (MLL3), SMARCA4, and KDM6A, previously identified by large-scale exome-resequencing44, were also confirmed and their mutational frequencies and distribution contextualized by subgroup. Despite these advances, initial standalone studies were underpowered to adequately detail the broader scope of low frequency driver gene alterations contributing to MB, especially in Groups 3 and 4 which were heterogeneous and devoid of highly recurrent gene-centric mutations3.
Recently, an international collaborative effort aimed at comprehensive characterization of the MB genomic landscape summarized putative driver gene alterations across a series of 491 primary MB samples26. As expected, WNT- and SHH-MB subgroups were largely characterized by mutations and SCNAs affecting known genes (Figure 3). Of interest, functional annotation of recurrently altered genes identified somatic deregulation of SWI/SNF family chromatin remodeling genes in one third of WNT-MBs (namely SMARCA4, ARID1A, and ARID2) and recurrent targeting of histone acetyltransferase genes in nearly 20% of SHH-MBs (namely CREBBP, KANSL1, BRPF1, and others). Detailed mechanistic studies will be required to determine how somatic targeting of these chromatin-associated complexes potentiates MB pathogenesis in the affected subgroups.
Figure 3 |. Recurrently altered genes and pathways across MB subgroups.
Grouped barplot of recurrently altered genes identified in WNT- (blue), SHH- (red), Group 3- (yellow) and Group 4-MB (green). Altered genes are grouped by functional categories. The percentage of affected samples is indicated (y-axis). Mutations of CTNNB1 are identified in 85.7% of WNT-MB.
In Group 3/4-MB, the multidimensional molecular analysis performed on this broader set of patients (n=324 total) confirmed that recurrent gene-level mutations remained relatively rare (Figure 3). SMARCA4 mutations were seen in 9% of Group 3 and only 2% of Group 4 patients. KDM6A (7%), ZMYM3 (6%), and KMT2C (6%) represented the most commonly mutated genes in Group 4. Previously unknown somatic in-frame insertions affecting KBTBD4 were evenly distributed between Group 3- and Group 4-MB (6% of patients from either subgroup). Although poorly characterized to date, KBTBD4 is a member of the BTB-BACK-Kelch protein family and predicted to recruit protein substrates to cullin-RING ligases for targeted ubiquitination and protein degradation45. MB-associated KBTBD4 insertions were confined to the Kelch domain and deemed unlikely to disrupt the overall domain structure but instead converged on the known substrate-binding interface described for other BTB-BACK-Kelch protein family members. Recently, identical somatic in-frame insertions to those seen in MB have been reported in pineal parenchymal tumours of intermediate differentiation (PPTID)46, suggesting that common oncogenic mechanism(s) may be shared between affected MB and PPTID patients.
Structural alterations and enhancer hijacking.
Group 3/4-MB genomes are characterized by a preponderance of SCNAs and structural variants (SVs)6, suggesting that these alterations play an integral role in disease pathogenesis. Analyzing the genomes of 137 Group 3/4-MB samples identified a series of atypical SVs (i.e. deletions, duplications, inversions, and more complex genomic alterations) mapping to chromosome 9q34 that were specific to these subgroups47. Integration with sample-matched gene expression data uncovered pronounced, SV-associated up-regulation of GFI1B in affected samples. GFI1B is a transcriptional repressor that is primarily known for its role in T-cell and B-cell development, as well as in hematopoietic malignancies where it functions as an oncogene48. The related family member GFI1, was also determined to be aberrantly expressed in a mutually exclusive set of Group 3/4-MBs harboring SV breakpoints proximal to the GFI1 locus. Integration with histone chromatin immunoprecipitation sequencing (ChIP-seq) data for H3K27ac marking active enhancers, in addition to other supporting ‘omics data, suggested that activation of GFI1B and GFI1 expression in MB was accomplished via SV-dependent misappropriation of distal, highly active enhancers/super-enhancers to their normally repressed gene promoters. Similar to classical translocations leading to over-expression of established oncogenes, such as IgG-MYC in Burkitt lymphoma49,50, this mechanism of SV-dependent gene activation was designated ‘enhancer hijacking’ and has since been explored and documented in numerous follow-up studies in other cancer types51–55. Using an orthotopic transplantation approach, GFI1B and GFI1 were validated as novel MB oncogenes capable of cooperating with MYC to promote highly aggressive Group 3-like MB in mice47. Overall, enhancer hijacking-associated GFI1 and GFI1B activation is estimated to account for ~12–15% of Group 3/4-MB patients, with clear enrichment of these events in Subtype I and to a lesser extent Subtype II26.
Motivated by the discovery of enhancer hijacking above, a novel computational pipeline termed CESAM (Cis-expression structural alteration mapping)54 was developed to systematically identify additional enhancer hijacking events through integration of SV breakpoints and gene expression data. Applying CESAM to sample-matched genomic datasets derived from 164 MB patients discovered PRDM6 as a novel target of enhancer hijacking in 17% of Group 4 patients26. PRDM6 maps to chromosome 5q23, approximately 600kb downstream of SNCAIP; a locus known to be targeted by highly recurrent, stereotypical tandem duplications exclusively in Group 4-MB6. Through multi-omic data integration for a series of Group 4-MBs, a putative model of enhancer hijacking mediated activation of PRDM6 was proposed. PRDM6 is described as a transcriptional repressor, mediating gene silencing through intrinsic H4K20 methyltransferase activity in concert with known chromatin-associated repressive complexes56,57. To date, PRDM6 represents the most frequent somatically altered gene in Group 4-MB, and together with mutations targeting other chromatin-modifying genes, further implicates deregulation of physiological transcriptional control as an essential mechanism underlying Group 4-MB pathogenesis.
Transcriptome & Epigenome
Compared to childhood leukemias58 and other pediatric brain cancers (i.e. supratentorial ependymoma59, pilocytic astrocytoma60), recurrent gene fusions are rare in MB. Early transcriptome sequencing discovered recurrent PVT1 gene fusions in Group 3-MB that were linked to chromothripsis and MYC amplification on chromosome 8q246. Additional PVT1-associated fusion events have since been reported in Group 3, including PVT1-NDRG1, PVT1-LINC00964, PVT1-ZCH3, and others26. PVT1 encodes a long intergenic noncoding RNA (lincRNA) harboring a cluster of six annotated microRNAs (namely, miR-1204, miR-1205, miR-1206, miR-1207–5p, miR-1207–3p, and miR-1208). Several reports link PVT1 over-expression and activity with pro-tumourigenic phenotypes61–67. In contrast, a recent study suggested that the PVT1 gene promoter is a tumour suppressor DNA element that inhibits MYC expression through enhancer-promoter competition in cis68. Future mechanistic and phenotypic studies in relevant model systems will be necessary to decipher the role of PVT1 fusions, among others, seen in MB.
Transcriptome analysis of MB patient samples has also led to the identification of alternate promoters and transcriptional start sites (TSS), indicating that transcriptional regulation of specific genes might reside outside previously annotated promoter regions. Analysis of high-coverage RNA-seq data in 43 MB samples revealed 262 novel first exons that were spliced to internal exons in excess of 15kb upstream of the previously annotated TSS, with some being located more than 500kb away69. Many of these alternate transcripts were expressed in a subgroup-specific manner, which often coincided with patterns of differential DNA methylation in the region of the novel TSS. One notable example is the pluripotency factor LIN28B, which has been described to regulate multiple oncogenic processes including downregulation of the tumour-suppressive let-7 miRNA family70. In MB, LIN28B is expressed specifically in Group 3/4. In about half of Group 3 cases and nearly all Group 4 cases the annotated promoter region is fully hypermethylated, and transcription is initiated at a novel first exon that is spliced to the second annotated exon.
Systematic miRNA profiling of patient samples resulted in the identification of a number of differentially expressed miRNAs between MB subgroups and relative to normal controls10,71,72. Among the best studied is the oncogenic miR-17–92 cluster, which was described to be genetically amplified and over-expressed specifically in SHH-MB71,73 and required for the formation of SHH-MB in the Ptch1+/− mouse MB model74. Furthermore, LNA-mediated silencing prolonged survival in intracranial SHH-MB allografts75. Another well studied example is the miR-183–96–182 cluster that is highly expressed in WNT- and Group 3/4-MB, targeting the AKT/PI3K/mTOR pathway and regulating cell proliferation and migration in vitro and in vivo76–78. Other miRNAs are downregulated in MB and have been described as tumour suppressors, including miR-125b, miR-326 and miR-324–5p, which have been described to regulate the SHH signaling pathway79, and miR-124a and miR-9, regulating the REST complex and cell proliferation80–83. An extensive review on the role of miRNAs in MB has recently been published84.
Genome-wide analysis of differential DNA methylation between MB subgroups and control tissues showed that the classical notion of gene silencing through promoter hypermethylation was not a prominent feature in MB69. The most abundant pattern of differential methylation was identified in regions extending several kilobases downstream of the promoter into the gene-body, in which hypomethylation correlated with elevated gene expression (promoter downstream correlated regions, pdCRs). About 20% of genes exhibiting MB subgroup-specific expression contained a pdCR, which suggests that this pattern plays an important regulatory role in distinct tumour-specific transcriptomes. Large, megabase-scale blocks of reduced DNA methylation (partially methylated domains, PMDs) represented another pattern of differential methylation in MB69. PMDs are a prominent feature in many cancer types and coincide with nuclear lamina-associated domains and other heterochromatic regions85–87. In MB, PMDs are primarily detected in the WNT and Group 3 subgroups and can cover up to one third of the genome, often in a subgroup-specific manner.
Integration of MB RNA-seq and enhancer ChIP-seq (namely H3K27ac) datasets has further enlightened mechanisms of gene regulation88. Enhancer ChIP-seq data generated for 28 primary MBs and 3 MB cell lines enabled annotation of the active cis-regulatory landscape across MB subgroups. In total, nearly 80,000 enhancers were inferred, 25% of which had not been previously annotated by ENCODE or the Roadmap Epigenomics Consortium. Computationally linking highly active, subgroup-specific enhancers, or super-enhancers (SEs), to putative target genes revealed new insights into the gene regulatory networks underlying MB subgroup biology and identity. Known cancer-associated genes were identified as prominent SE-targets, including ALK in WNT-MB, GLI2, SMO, and NTRK3 in SHH-MB, LMO1, LMO2, and MYC in Group 3-MB, and ETV4 and PAX5 in Group 4-MB, among others. Differential enhancer/gene target analysis identified aberrant TGF β signaling activity that was specific to Group 3, substantiating prior genomic-based evidence implicating oncogenic TGF β signaling in a subset of tumours6.
Proteome
Proteins are the major functional product of the genome and dictate most cellular functions. Abundance, structure, stability, and post-translational modifications (PTMs) of proteins collectively increase the level of complexity and activity of the proteome. During the past decade, tremendous technical progress has been made towards deep detection and accurate quantification of the proteome89 and recent studies have begun to explore the proteomic landscape of MB. Quantitative mass-spectrometry (MS)-based proteomics of primary human MBs predominantly confirmed the classification of MB into consensus subgroups in three independent cohorts90–92. The proteome also revealed notable substructure within both SHH (SHH-a and SHH-b) and Group 3 (Group 3a and 3b) subgroups; although these observations have been limited to modest sample cohorts and were not immediately supported by companion DNA methylation or transcriptomic datasets.
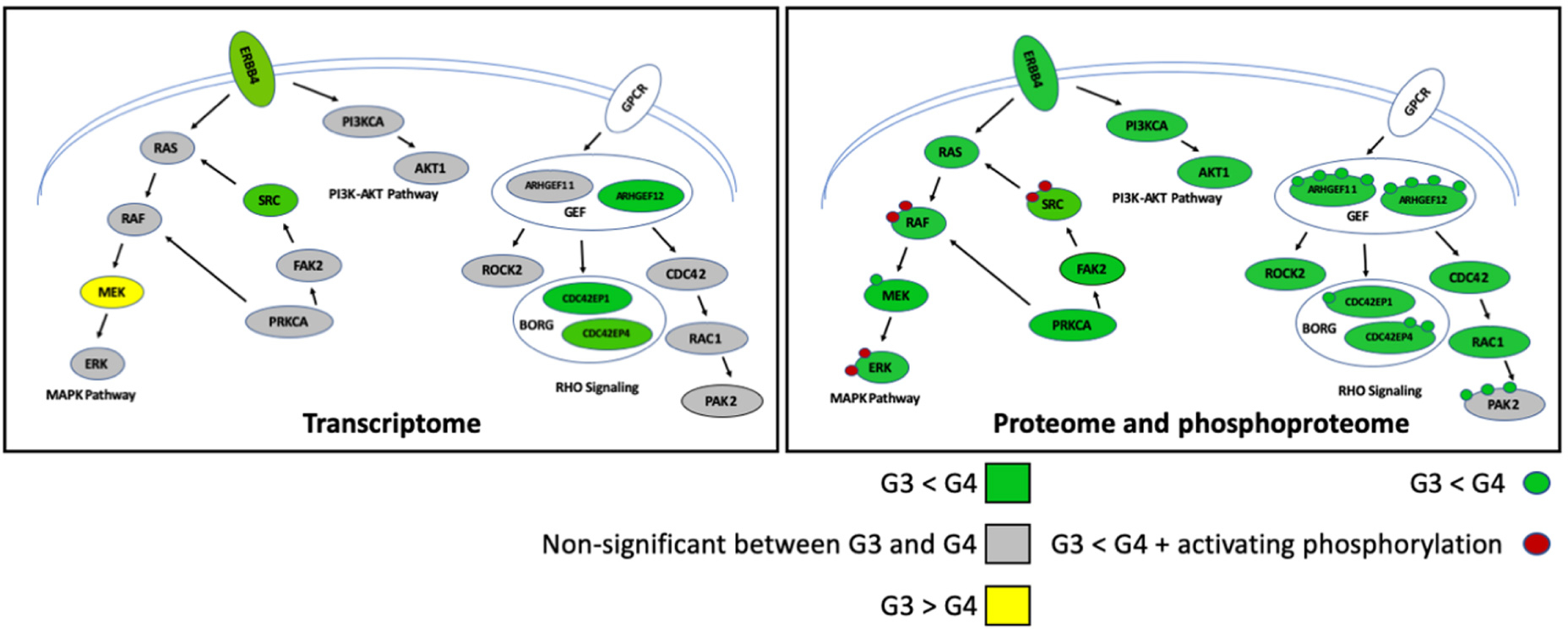
Concordance between mRNA transcript and protein abundance is known to vary between species and cell types, with only ~30–40% of protein variance explained by variance in mRNA abundance93. Group 3/4-MB exhibit the lowest correlation between mRNA and protein expression, emphasizing the potential role of post-transcriptional mechanisms in their underlying biology91,92. At the proteomic level, Group 3-MB exhibited elevated expression of eukaryotic initiation factor (eIF) subunits (EIF2s, EIF3s, EIF4Gs, and EIF4As)49, a complex implicated in initiation of protein synthesis in eukaryotes. Supporting this finding, pharmacological inhibition of the formation of the eIE4F complex reduced MB cell viability in vitro92. Interestingly, discrepancies between mRNA and protein expression revealed activation of receptor tyrosine kinase (RTK) signaling in Group 4, especially aberrant expression of ERBB4 and phosphorylated SRC (Figure 4). In utero electroporation-mediated over-expression of activated SRC in combination with dominant-negative Tp53 induced Group 4-like MB in vivo91, functionally substantiating observations gleamed through proteomics. While the mechanism(s) responsible for the mRNA/protein discrepancies seen in Group 3/4-MB are likely due to a multitude of factors, translational effects mediated through MYC or MYCN are suspected to play a role91,94,95.
Figure 4 |. Aberrant signaling pathways implicated in Group 3 and Group 4 MB by proteomics.
Colors indicate the relative abundance in mRNA (left part), protein and phosphoprotein (right part) between Group 4 and Group 3. Phosphorylation is indicated as a small circle. The half circle indicates the plasma membrane.
Phosphoproteomics can inform protein kinase activity and potential opportunities for therapeutic intervention through administration of pharmacological inhibitors. Bioinformatic analyses of MB phosphoproteomic data predicted activation of several prominent kinases, including GSK3 β (Group 4 and SHHb), PRKDC (Group 3 and WNT), CDK5 (Group 4) and CLK/CK2 (Group 3). Also, kinome analyses using high-throughput peptide phosphorylation profiling revealed two distinct protein-signaling signatures in MB: MYC-like protein-signaling observed in the majority of SHH and Group 3, and Protein-signaling profile-2, characterized by DNA-damage response, apoptotic, and neuronal signaling in the majority of Group 495. Hence, these findings reinforce the idea that protein activity might reveal unifying and/or distinct tumour biologies amongst MB subgroups. Also, several PTMs of key players in MB have been identified. For instance, in SHH-MB, deacetylation of Gli1 and Gli2 proteins induces their transcriptional activity96, while phosphorylation of Atoh1 by Jak2 controls tumour growth97. Besides phosphorylation and acetylation, a myriad of other PTMs, including, ubiquitinylation, methylation and others have yet to be investigated in MB. The field of proteomics is rapidly advancing and along with further technological and bioinformatic breakthroughs, a deeper characterization of MB subgroup biology will undoubtedly follow.
Intratumoural heterogeneity
Phenotypic heterogeneity of individual cells within tumours has been a longstanding interest in MB research. In 2003, paralleled by similar discoveries in other solid tumours and leukemia98,99, Peter Dirks and colleagues first reported the discovery of a stem-like tumour cell population in MB patient samples characterized by the neural stem cell surface marker CD133 (encoded by PROM1, positive for 6 to 21% of cells)100,101. These cells show marked capacity for proliferation, self-renewal, and neuronal differentiation both in vitro by neurosphere culture and in vivo by mouse xenotransplantation. As few as 1,000 CD133(+) cells were sufficient to initiate tumours in mice that phenotypically resemble the original tumour, whereas 50,000 CD133(−) cells failed to establish tumours. CD133 is most highly expressed in Group 3-MB102, but does not mark tumour-propagating cells in SHH-MB GEM models103,104. Instead, the neural stem cell surface antigen CD15 was found to enrich for tumour propagating cells in this subgroup. Further analysis of the Ptch1+/− SHH-MB GEM model identified a rare, quiescent Sox2(+) cell population that gave rise to rapidly cycling progenitors105.
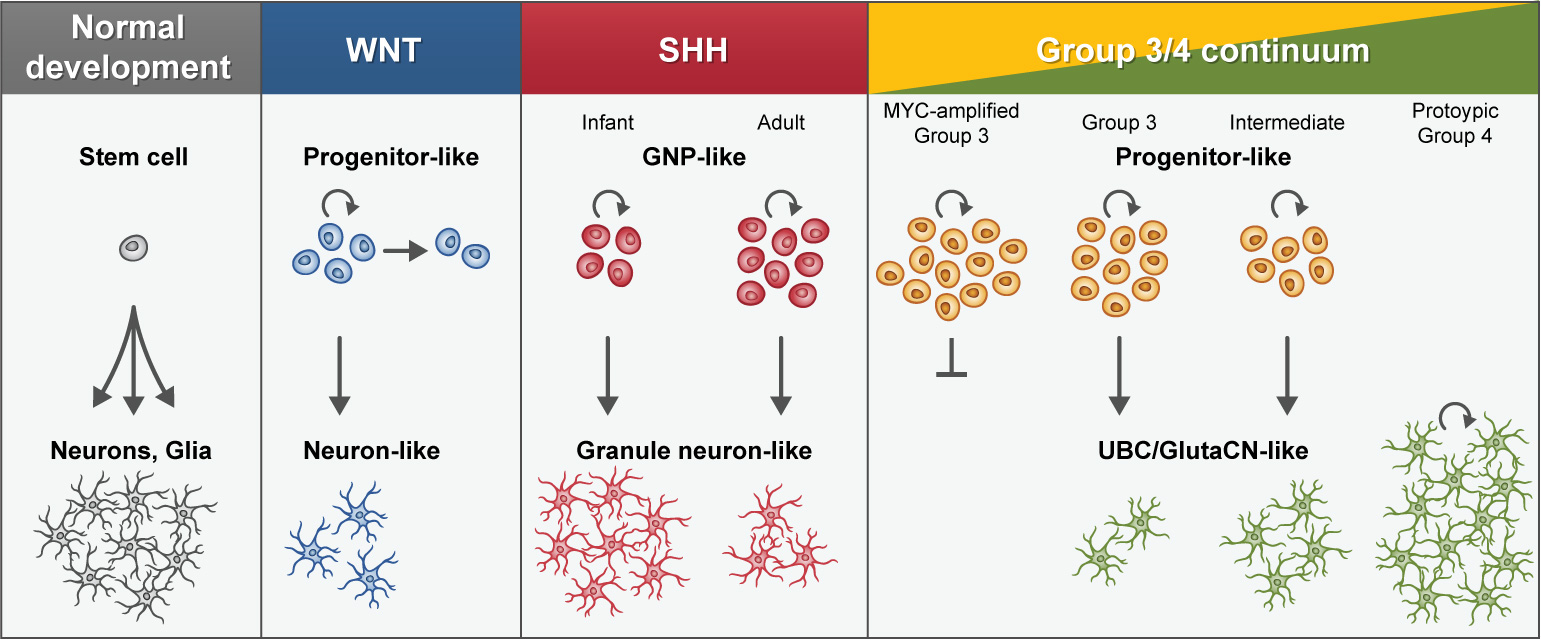
In recent years, single-cell transcriptome sequencing (i.e. scRNA-seq) has emerged as a powerful method to decipher cellular states in healthy and diseased tissues in an unbiased way, as exemplified in adult and pediatric gliomas106–110. Recently, two independent studies applied scRNA-seq to cohorts of primary MBs, showing that MB displays subgroup-specific transcriptional heterogeneity at the single-cell level111,112. Analyzing eight patients from the SHH-, Group 3-, and Group 4-MB subgroups, Vladoiu and colleagues demonstrated mixed populations of cells with divergent differentiation along cerebellar neuronal lineages111. Analysing 25 patients across all molecular subgroups, Hovestadt and colleagues identified subgroup-specific undifferentiated and differentiated neuronal-like malignant populations112 (Box 1). Both studies confirmed resemblance of SHH tumours to the cerebellar granule neuron lineage, in agreement with earlier experimental evidence9,113 (Figure 5; see Box 1). Interestingly, adult SHH-MB tumours showed a higher fraction of undifferentiated granule neuron progenitors (GNPs) compared to infant tumours, possibly linking to their divergent biology (Box 1). Combined analysis of Group 3/4-MB tumours revealed a related developmental trajectory from primitive progenitor-like to more mature neuronal-like cells, whose relative proportions distinguished these subgroups (Box 1). MYC-amplified Group 3-MBs only comprised undifferentiated progenitor-like cells and did not show any capacity to differentiate. Most other Group 3-MBs showed a small degree (<10%) of neuronal differentiation, indicating that tumour cells maintained the capacity to differentiate. Interestingly, a subset of tumours characterized as ‘intermediate’ cases by DNA methylation-based classification exhibited both undifferentiated and differentiated populations at varying proportions, providing an explanation for the challenges to confidently assign some Group 3/4-MB cases to either subgroup by bulk molecular profiling (Box 1). Prototypic Group 4-MBs were comprised almost exclusively of more differentiated neuronal-like cells resembling unipolar brush cells (UBCs) and glutamatergic cerebellar nuclei (GluCN) (Figure 5; see Box 1).
Box 1 | Mapping cellular origins of MB subgroups.
Graphical depiction of an embryonic mouse cerebellum (estimating development at ~13.5) highlighting cellular populations proposed to be developmentally linked to specific MB subgroups. GluCN, glutamatergic cerebellar nuclei; UBC, unipolar brush cell; GNP, granule neuron progenitor; LRL, lower rhombic lip; CB, cerebellar; URL, upper rhombic lip; EGL, external granule layer; VZ, ventricular zone; NTZ, nuclear transitory zone.

By performing parallel gene expression profiling and exome sequencing of 47 multiregional biopsies from eight patients, it was recently concluded that MB is characterized by spatially homogeneous transcriptomes, in which biopsies from the same patient were more similar to each other than to those of other patients114. This was contrary to findings in glioblastoma, in which different biopsies from the same patient classified as different transcriptional subtypes114. However, the same study reports high levels of genetic heterogeneity in MB at the level of SCNAs and somatic mutations. As affected genes included therapeutic targets, these observations put the representativity of single biopsies and the efficacy of monotherapies to treat MB into question.
Molecular insights into MB relapse and metastasis
Since MB relapse and metastases remain the most significant morbidity factors influencing patient outcomes115, an improved understanding of the molecular events driving treatment resistance and recurrence represents a major priority. To date, comparative studies of primary versus relapse disease have been exceedingly limited and restricted to relatively modest cohorts. Application of a NanoString-based molecular classification approach confirmed that MB subgroup status is conserved between primary and relapse disease, reinforcing the concept that individual MB subgroups constitute distinct diseases116. This observation was later independently confirmed using a complementary subgrouping approach117. Specific emergence of MYC/MYCN amplifications and TP53 pathway defects (namely TP53 mutation, CDKN2A deletion) at relapse was also reported117. By way of an elegant Sleeping Beauty transposon-based murine model of SHH-MB (Ptch1+/−/Math1-SB11/T2Onc or T2Onc2), compelling evidence for genetic divergence of primary versus recurrent tumours was substantiated by a paucity of shared genetic events between treatment-naïve and recurrent mouse MBs118. In the same study, a cohort of 36 primary/relapse MB patient tumours were subjected to genomic characterization. Recurrent tumours harbored a profound increase in mutational burden and SVs, the overwhelming majority of which were restricted to either primary or relapse disease, with minimal overlap observed between both disease compartments. More extensive efforts that are sufficiently powered to build on these studies and provide further understanding of the molecular basis of MB treatment failure and relapse are ongoing.
MB metastasis almost always occurs via leptomeningeal dissemination (LMD) that remains confined to the CNS and spinal cord115. Approximately one third of all MB patients are metastatic at diagnosis, with patterns and frequencies that vary considerably according to molecular subgroup119. Patients that fail conventional therapy and/or relapse metastatically share a universally dismal prognosis, with virtually all patients succumbing to their refractory disease. As such, there is a critical and urgent need to decipher the molecular and cellular mechanisms governing LMD in this unacceptably high fraction of affected MB patients. Through genomic analysis of simultaneously collected patient-matched primary MB, leptomeningeal metastatic MB, and peripheral blood, Garzia and colleagues inferred the presence of circulating tumour cells (CTCs) based on careful analysis of specific mutations that were clonal in the metastasis, clonal or subclonal in the primary tumour, and present at very low fractions in the peripheral blood120. In 3/6 of the evaluated peripheral blood samples, exceedingly rare CD56(+)/CD45(−) (both of which were used to mark malignant cells) morphologically abnormal cells were identified, further supporting the presence of rare CTCs in the blood of some MB patients. These molecular and cellular observations derived from patient samples were strengthened by a series of innovative mouse modeling experiments, including implementation of a parabiosis model that established hematogenous spread of implanted MB cells between surgically connected donor (implanted mouse) and recipient (sibling mouse) animals. Comparison of gene expression profiles for a limited set of patient-matched primary and metastatic MB samples in the same study identified over-expression of the chemokine CCL2 in the metastatic compartment, that was further substantiated through additional molecular and functional analyses, suggesting that aberrant CCL2 signaling may be an important mediator of LMD in MB.
Emerging clinical implications & translational opportunities
Risk stratification.
Clinically relevant insights uncovered during the molecular era have enabled cautious, yet steady transition of discoveries made in the research arena into the clinical setting. Indeed, clinical protocols for MB patients have begun to implement molecular subgroup-informed strategies for treatment stratification. SJMB12 (NCT01878617), an active trial for newly diagnosed MB patients includes separate treatment arms for WNT, SHH, and non-WNT/non-SHH patients (i.e. Group 3 and Group 4). On this protocol, clinically standard-risk WNT-MB patients (non-metastatic with near total surgical tumour resection) receive reduced craniospinal radiation (CSI; 15Gy as opposed to the standard dose of 23.4Gy administered to standard-risk MB patients), owing to the highly favorable outcomes that have been consistently reported for WNT-MB since 200528,121. Skeletally mature (i.e. females with a bone age ≥15 years or males with a bone age ≥17 years) SHH-MB patients enrolled on SJMB12 receive the SMO inhibitor, vismodegib, on top of standard of care chemotherapy and CSI. In contrast, non-WNT/non-SHH patients are further stratified into standard- and high-risk treatment arms based on a combination of clinical- and molecular-risk factors, including extent of resection, metastatic status, and MYC amplification status. A trial for newly diagnosed patients ongoing in Europe, International Society of Paediatric Oncology (SIOP) PNET 5 (NCT02066220), is likewise stratifying WNT-MB patients according to a low-risk treatment arm and administering reduced dose CSI (18Gy) compared to non-WNT-MB patients that are treated according to standard of care; an effort that is also being mirrored for WNT-MB patients by Children’s Oncology Group (ACNS1422; NCT02724579) in North America. Despite these examples of molecularly-informed risk-stratification in current MB trials, the clinical arena still lags behind the pace at which retrospective research studies enlighten new concepts. Indeed, recent molecular studies conducted on both large retrospective and trial cohorts have provided increased rationale for further molecularly-driven stratification in future protocols, especially within specific MB subgroups25,27,31,38,122.
Genetic predisposition.
Hereditary genetic predisposition to MB remains an underappreciated clinical challenge. Recent data collected on large retrospective MB cohorts suggests that especially in the case of SHH-MB, the proportion of patients with underlying cancer predisposition might approach ~25% or even higher29. In WNT-MB patients, the proportion appears to be in the range of 5–10%. At this stage, these estimates only account for known cancer predisposition genes and efforts to systematically analyze yet unknown pathogenic events in the germline of MB patients are currently ongoing. Based on published AACR guidelines123 that suggest genetic testing and counseling for specific tumour types that frequently occur in the context of genetic predisposition (threshold of ≥10%), the prevalence of clearly pathogenic germline events in known cancer predisposition genes certainly qualifies this disease (especially SHH-MB, but also WNT-MB) for a general recommendation to offer genetic testing and counselling prior to adjuvant therapy. This infrastructure, however, is currently not in place in the majority of treatment centers across the world. The largest MB predisposition study to date29 identified APC germline mutations in CTNNB1 mutation-negative WNT-MB patients, as well as significant enrichment of TP53, SUFU, PTCH1, BRCA2 and PALB2 pathogenic germline variants in SHH-MB. These genes can be tested at once by clinical grade whole-exome or panel sequencing, but also successively as single gene tests based on their age associations and, in some cases, family history. AACR surveillance guidelines are available for APC, TP53, SUFU, and PTCH1, whereas it remains to be determined how to appropriately manage MB patients with damaging heterozygous germline variants in BRCA2 and PALB2. Since this considerable proportion of MB patients with hereditary disease and their families require special clinical attention including potential treatment modifications, family testing, and surveillance, this challenge has to be tackled systematically, but expeditiously.
Therapeutic targets.
Arguably it was the discovery of small molecule hedgehog pathway antagonists (i.e. SMO inhibitors) and their preclinical potency against Ptch1+/− GEM models124 that sparked the need to molecularly identify MB patients that would benefit from these inhibitors. However, what emerged was molecularly far more complicated than first envisioned. SHH pathway gene alterations in SHH-MB differ according to patient age at diagnosis and subtype25,26,32,38, collectively accounting for the variable responses to SMO inhibitors. PTCH1 mutations (both germline and somatic) are the most common but present in less than half of all SHH-MB patients and mostly within infants and adults. Germline and somatic SUFU mutations are largely restricted to infant SHH-MBs, whereas activating somatic SMO mutations are enriched in adult SHH-MB patients. Similarly, germline and somatic TP53 mutations, predominantly coincident with GLI2 and MYCN amplifications, are exclusively found in children between the ages of 8–17 years old. These observations are of direct clinical relevance when considering treatment of SHH-MB patients with SMO inhibitors. Treatment of the Ptch1+/− GEM model124 and SHH-MB patient derived xenograft (PDX) model32 harboring PTCH1 mutations were determined to be sensitive to the SMO inhibitors (Hhantag, vismodegib, sonidegib), whereas a GEM model lacking Sufu125 or PDX harboring TP53 mutation and MYCN amplification exhibited primary resistance32. These preclinical findings were corroborated clinically in an early phase clinical trial on patients with relapsed SHH-MB when treated with the SMO inhibitor, vismodegib126. Once again, as molecularly predicted, responders to SMO inhibition were more likely to have tumours that harbored a PTCH1 mutation whereas no beneficial clinical responses were observed in patients harboring TP53 or SUFU mutations and concurrent MYCN and GLI2 amplifications. Moreover, another significant concern that has restricted the clinical use of SMO inhibitors is the emergence of permanent growth plate fusions in the long bones of small children127, a morbidity that was predicted in early preclinical studies on young mice128. Collectively, these findings demonstrate the importance of performing thorough preclinical and molecular testing to identify the appropriate patient populations as candidates for molecularly targeted therapy.
Mechanistically, GFI1 was recently shown to mitigate its oncogenicity in MB through direct interaction with KDM1A/LSD1129, a histone lysine demethylase associated with transcriptional repression. Treatment of MYC/GFI1-driven MBs with LSD1 inhibitors attenuated the malignant phenotype in vitro and in vivo using a flank PDX model. No treatment effect was observed when treating the same tumours in an orthotopic setting, demonstrating that this model might serve as a preclinical testing vehicle for blood-brain-/blood-tumour-barrier penetration. Taken together, these results provide preclinical support for treating affected GFI1/GFI1B-driven MB patients with LSD1 inhibitors, given that a brain penetrant LSD1 inhibitor will eventually become available.
With the identification of aberrant TGF β signaling in Group 3-MB and RTK signaling in Group 4-MB, new opportunities for preclinical testing of inhibitors to these pathways have likewise emerged91,130. Additional studies evaluating the efficacy of such agents in treating accurate preclinical MB models will be necessary to further substantiate their future implementation in the clinic.
Conclusion and future outlook
Through continued multi-omic analyses conducted on unprecedented patient cohorts, MB now undoubtedly represents one of the most extensively characterized cancer entities. Deep understanding of molecular substructure and assignment of known and novel driver gene alterations to specific disease subtypes has created a more refined understanding of tumour biology. This knowledge will enable the development of better models that more accurately recapitulate what is seen in patients. Moreover, these advances pave the way for critical functional studies required to determine the mechanistic contribution of newly discovered genes and molecular complexes to MB pathogenesis, in the appropriate cellular context. A continued transition from bulk tumour profiling to more detailed analyses of intratumoural heterogeneity in single-cells is to be expected, especially as methods for analyzing genetic alterations, the epigenome, transcriptome, and proteome at single-cell resolution continue to evolve and become attainable on archival clinical samples. These studies will prove essential for further resolving recently described molecular subtypes within subgroups, elucidating the molecular and cellular basis of MB recurrence and metastasis, and more.
Clinically, the separation of MB along molecular lines into subgroups and now into subtypes within subgroups generates concern among many treating physicians that the number of divisions far outpaces our ability to give subtype-specific care. However, while this argument against a complicated classification system is understandable given that the majority of subtype-specific findings are not immediately targetable by individual medicines, it ignores the clinical benefit of improved risk stratification. Despite reasonably high cure rates in the range of 70–75% for all, the cost of current therapy that combines surgery, CSI, and chemotherapy remains unacceptably high. Molecular subgrouping has already demonstrated pronounced differences in survival, and this has afforded the opportunity to trial judicious reductions in therapy to the favorable WNT subgroup while maintaining and optimizing intensive therapy for the unfavorable Group 3 patients. Moreover, the more precise and more well defined the subtypes become the better the opportunity to hone this therapeutic approach. For example, the new subtyping of very young children with MB is anticipated to fairly rapidly lead to new trials since the toxicities of CSI and chemotherapy remain so unacceptably high in this most vulnerable population. Furthermore, if promising targeted therapies such as SMO inhibitors, LSD1 inhibitors, and RTK inhibitors are to be successful, then it is imperative that these agents are given to the appropriately targeted populations. Even though the end of further evolution of MB molecular sub-classification may be in sight, the impact of these molecularly informed advances on therapy is only just beginning.
MB subgroup origins (Box 1)
As MB subgroups are enriched for specific genetic alterations and exhibit discriminatory epigenetic and transcriptional profiles, it has long been suspected that they arise from distinct cellular populations or developmental lineages131. Mouse models of the WNT and SHH subgroups have respectively substantiated lower rhombic lip progenitors (Blbp+) and GNP populations (Atoh1+) as probable cells-of-origin for these subgroups132. Multiple orthotopic and somatic gene transfer models have demonstrated that a variety of progenitor cell populations can be effectively transformed to replicate molecular and phenotypic features of Myc-driven Group 3-MB133–135. Mapping the MB enhancer landscape enabled inference of master transcription factors (TFs) governing subgroup-specific tumour biology. LMX1A, EOMES, and LHX2 were predicted to function as master regulators of Group 4-MB. Developmentally, these master TFs regulate lineage specification for restricted glutamatergic progenitor cell populations born out of the cerebellar upper rhombic lip during cerebellar morphogenesis, including glutamatergic cerebellar nuclei (GluCN; also known as deep cerebellar nuclei; DCN) and unipolar brush cells (UBCs). Since LMX1A, EOMES, and LHX2 were inferred to be highly specific SE-regulated candidate master TFs in Group 4-MB, the aforementioned glutamatergic populations expressing these markers were proposed as the putative lineage(s)-of-origin for this MB subgroup. More recently, comparative cross-species transcriptomic analyses have complemented these observations, utilizing single-cell transcriptional profiles of the developing mouse cerebellum as a reference for mapping MB subgroup origins111,112. Granule neuron lineage populations were shown to be highly correlated with SHH-MBs, reinforcing the expansive literature implicating GNPs as their developmental origin. Interestingly, GluCN and UBCs were both shown to be highly transcriptionally similar to Group 4-MBs, suggesting that these populations could represent bona fide cells-of-origin for this subgroup of patients. Early embryonic Nestin+ progenitor cells were suggested as being highly correlated with Group 3-MB111; although, these results were not supported in the companion study112. Deeper molecular analyses on these novel candidate populations and functional studies that mimic relevant candidate driver alterations in the correct lineage at the correct developmental stage are ongoing and will be required to further substantiate these initial findings.
Acknowledgements
P.A.N. is a Pew-Stewart Scholar for Cancer Research (Margaret and Alexander Stewart Trust) and recipient of The Sontag Foundation Distinguished Scientist Award. P.A.N. was also supported by the National Cancer Institute (R01CA232143–01), American Association for Cancer Research (NextGen Grant for Transformative Cancer Research), The Brain Tumour Charity (Quest for Cures), the American Lebanese Syrian Associated Charities (ALSAC), and St. Jude. V.H. is supported by a Human Frontier Science Program long-term fellowship (LT000596/2016-L). We acknowledge K. Smith, L. Bihannic, and T. Sharma for assistance with data organization and figure design. We thank Brandon Stelter for assistance with artwork.
Glossary
- Next-generation sequencing
Technologies enabling massively parallelized reading of amplified short nucleotide sequences (typically yielding hundreds of million reads 100–500 bp in length). Broad availability of these technologies has transformed genomic research in the last decade. In contrast, emerging third-generation sequencing technologies read sequences without prior amplification, yielding much longer reads, albeit with reduced accuracy and throughput.
- Molecular classification
Classification of patient tumour samples based on molecular markers, opposed to classification based on histomorphological appearance. Commonly used molecular markers in MB include genetic mutations (e.g. mutations in CTNNB1 in WNT-MB), copy-number alterations (e.g. MYC amplification in Group 3-MB), and genome-wide gene expression and DNA methylation patterns for classification into molecular subgroups and subtypes.
- Intratumoural heterogeneity
Observation that tumours comprise of distinct malignant and non-malignant (cells of the micro-environment) cell types. Heterogeneity of malignant cells encompasses genetic heterogeneity (e.g. different genetic subclones) and transcriptional heterogeneity (e.g. malignant cell states resembling normal development).
- DNA methylation
A central epigenetic mark in which a methyl group is added to cytosine bases in genomic DNA. Cytosine methylation is inherited through cell division and plays important roles in gene regulation during normal human development and diseases such as cancer. Genome-wide methylation patterns constitute very stable and informative molecular markers for tumour subgroup and subtype classification.
- Isochromosome
An abnormal chromosome in which both chromosome arms are identical. Isochromosome 17q is one of the most frequent copy-number alterations in Group 3/4-MB, in which a second q-arm is fused to the p-arm proximal to the centromere in most cases.
- Non-synonymous mutation
A genetic alteration that alters the amino acid sequence of an affected protein, possibly altering protein function. Most described recurrent mutations in MB are non-synonymous mutations. Other types of mutations include synonymous mutations (silent substitutions), which do not alter the protein sequence because of the degeneracy of the genetic code, and non-coding mutations, which affect regions that are not translated into proteins. Both synonymous mutations and non-coding mutations can have important gene regulatory functions. The most common non-coding mutation in MB are found in a hotspot in the promoter of the TERT gene.
- Chromatin
Describes the complex of genomic DNA and proteins. Its main functions are packaging of DNA and regulation of gene expression. The main type of proteins in chromatin are histones. Histone tails are frequently marked by post-translational modifications that mediate changes in chromatin structure and accessibility through interaction with histone modifiers and/or nucleosome remodeling complexes. Genome-wide patterns of histone modifications can be analyzed by chromatin immunoprecipitation-coupled sequencing (ChIP-seq).
- Germline mutation
Describes genetic variation within the germ cells of a carrier that can be passed on to offspring. If inherited, this variation is then present in all somatic and germline cells of the offspring (constitutional mutation). Germline mutations can predispose to cancer. Common examples of germline mutations associated with MB include mutations of PTCH1 (Gorlin syndrome) and TP53 (Li-Fraumeni syndrome).
- Patient derived xenograft
Models of cancer in which tumour cells from a patient are implanted and maintained in a non-human carrier, most commonly immunodeficient or humanized laboratory mice. PDX models are thought to be more closely resembling patient tumours than cell cultures. Orthotopic models describe models in which cells are implanted in the corresponding anatomical position, compared to heterotopic models in which cells are implanted in an area unrelated to the original tumour site (e.g. subcutaneously).
- Structural variation
Describes genomic variations such as deletions, duplications, inversions, and translocations that affect the structure the chromosome at the microscopic and submicroscopic level. Structural variants can result in fusion genes and enhancer hijacking. An extreme form of structural variation observed in MB is a phenomenon termed chromothripsis, in which a large number of rearrangements affect a single chromosome.
- Blood-brain barrier
Is a semipermeable border formed by endothelial cells lining the cerebral microvasculature that separates the brain from the circulating blood. The blood-brain barrier thereby protects the brain from fluctuations in plasma composition and circulating agents such as neurotransmitters and pathogens. The blood-brain barrier also presents a challenge for drug delivery when treating brain tumours.
- microRNA
An abundant class of small non-coding RNA molecules that are approx. 21–22 nucleotides in length. In complex with the RNA-induced silencing complex these molecules exert important gene regulatory functions by post-transcriptional gene silencing through base complementarity to target genes.
- CRISPR gene editing
A novel method that allows for efficient genetic manipulation to study the effect of coding and non-coding variants in model systems. Delivery of the CRISPR-Cas9 nuclease complex is guided by base complementarity of synthetic guide RNAs to genomic regions.
- Glutamatergic neurons
A class of neurons that release the excitatory neurotransmitter glutamate. The second major class of neurons are GABAergic neurons that release the inhibitory neurotransmitter GABA (gamma-aminobutyric acid).
- Mass-spectrometry
An analytical technique that allows accurate mass determination of large biomolecules such as proteins. Important applications of mass-spectrometry are the quantification of protein expression levels and the analysis of post-translational protein modifications (PTMs).
- Single-cell RNA-seq
Emerging technology that enables unsupervised characterization of transcriptional profiles in individual cells of healthy and diseased tissues. Throughput of technologies has steadily increased over recent years, now enabling profiling of tens-of-thousands of individual cells in a single experiment.
Footnotes
Competing interests
None.
References
- 1.Northcott PA et al. Medulloblastoma. Nat Rev Dis Primers 5, 11, doi: 10.1038/s41572-019-0063-6 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Taylor MD et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123, 465–472, doi: 10.1007/s00401-011-0922-z (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Northcott PA et al. Medulloblastomics: the end of the beginning. Nat Rev Cancer 12, 818–834, doi: 10.1038/nrc3410 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rausch T et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148, 59–71, doi: 10.1016/j.cell.2011.12.013 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pugh TJ et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488, 106–110, doi: 10.1038/nature11329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Northcott PA et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 488, 49–56, doi: 10.1038/nature11327 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones DT et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 488, 100–105, doi: 10.1038/nature11284 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pomeroy SL et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 415, 436–442, doi: 10.1038/415436a (2002). [DOI] [PubMed] [Google Scholar]
- 9.Lee Y et al. A molecular fingerprint for medulloblastoma. Cancer Res 63, 5428–5437 (2003). [PubMed] [Google Scholar]
- 10.Cho YJ et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29, 1424–1430, doi: 10.1200/JCO.2010.28.5148 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kool M et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One 3, e3088, doi: 10.1371/journal.pone.0003088 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Northcott PA et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29, 1408–1414, doi: 10.1200/JCO.2009.27.4324 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson MC et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24, 1924–1931, doi: 10.1200/JCO.2005.04.4974 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Louis DN et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131, 803–820, doi: 10.1007/s00401-016-1545-1 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Min HS, Lee JY, Kim SK & Park SH Genetic grouping of medulloblastomas by representative markers in pathologic diagnosis. Transl Oncol 6, 265–272, doi: 10.1593/tlo.12382 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellison DW et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol 121, 381–396, doi: 10.1007/s00401-011-0800-8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Northcott PA et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol 123, 615–626, doi: 10.1007/s00401-011-0899-7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leal LF et al. Reproducibility of the NanoString 22-gene molecular subgroup assay for improved prognostic prediction of medulloblastoma. Neuropathology 38, 475–483, doi: 10.1111/neup.12508 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Schwalbe EC et al. Rapid diagnosis of medulloblastoma molecular subgroups. Clin Cancer Res 17, 1883–1894, doi: 10.1158/1078-0432.CCR-10-2210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Capper D et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474, doi: 10.1038/nature26000 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hovestadt V et al. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol 125, 913–916, doi: 10.1007/s00401-013-1126-5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwalbe EC et al. DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin-fixed biopsies. Acta Neuropathol 125, 359–371, doi: 10.1007/s00401-012-1077-2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwalbe EC et al. Minimal methylation classifier (MIMIC): A novel method for derivation and rapid diagnostic detection of disease-associated DNA methylation signatures. Sci Rep 7, 13421, doi: 10.1038/s41598-017-13644-1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korshunov A et al. DNA-methylation profiling discloses significant advantages over NanoString method for molecular classification of medulloblastoma. Acta Neuropathol 134, 965–967, doi: 10.1007/s00401-017-1776-9 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Cavalli FMG et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 31, 737–754 e736, doi: 10.1016/j.ccell.2017.05.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Northcott PA et al. The whole-genome landscape of medulloblastoma subtypes. Nature 547, 311–317, doi: 10.1038/nature22973 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwalbe EC et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol 18, 958–971, doi: 10.1016/S1470-2045(17)30243-7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ellison DW et al. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol 23, 7951–7957, doi: 10.1200/JCO.2005.01.5479 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Waszak SM et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol 19, 785–798, doi: 10.1016/S1470-2045(18)30242-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Remke M et al. Adult medulloblastoma comprises three major molecular variants. J Clin Oncol 29, 2717–2723, doi: 10.1200/JCO.2011.34.9373 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Shih DJ et al. Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol 32, 886–896, doi: 10.1200/JCO.2013.50.9539 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kool M et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 25, 393–405, doi: 10.1016/j.ccr.2014.02.004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Northcott PA et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol 122, 231–240, doi: 10.1007/s00401-011-0846-7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Remke M et al. TERT promoter mutations are highly recurrent in SHH subgroup medulloblastoma. Acta Neuropathol 126, 917–929, doi: 10.1007/s00401-013-1198-2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koelsche C et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol 126, 907–915, doi: 10.1007/s00401-013-1195-5 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Lindsey JC et al. TERT promoter mutation and aberrant hypermethylation are associated with elevated expression in medulloblastoma and characterise the majority of non-infant SHH subgroup tumours. Acta Neuropathol 127, 307–309, doi: 10.1007/s00401-013-1225-3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poschl J et al. Genomic and transcriptomic analyses match medulloblastoma mouse models to their human counterparts. Acta Neuropathol 128, 123–136, doi: 10.1007/s00401-014-1297-8 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Robinson GW et al. Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol, doi: 10.1016/S1470-2045(18)30204-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Garancher A, Ramaswamy V & Wechsler-Reya RJ Medulloblastoma: From Molecular Subgroups to Molecular Targeted Therapies. Annu Rev Neurosci 41, 207–232, doi: 10.1146/annurev-neuro-070815-013838 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Ramaswamy V & Taylor MD Medulloblastoma: From Myth to Molecular. J Clin Oncol 35, 2355–2363, doi: 10.1200/JCO.2017.72.7842 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Ramaswamy V et al. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol 131, 821–831, doi: 10.1007/s00401-016-1569-6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharma T et al. Second-generation molecular subgrouping of medulloblastoma: an international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol, doi: 10.1007/s00401-019-02020-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson G et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 488, 43–48, doi: 10.1038/nature11213 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parsons DW et al. The genetic landscape of the childhood cancer medulloblastoma. Science 331, 435–439, doi: 10.1126/science.1198056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Canning P et al. Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases. J Biol Chem 288, 7803–7814, doi: 10.1074/jbc.M112.437996 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee JC et al. Recurrent KBTBD4 small in-frame insertions and absence of DROSHA deletion or DICER1 mutation differentiate pineal parenchymal tumor of intermediate differentiation (PPTID) from pineoblastoma. Acta Neuropathol 137, 851–854, doi: 10.1007/s00401-019-01990-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Northcott PA et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511, 428–434, doi: 10.1038/nature13379 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moroy T, Vassen L, Wilkes B & Khandanpour C From cytopenia to leukemia: the role of Gfi1 and Gfi1b in blood formation. Blood 126, 2561–2569, doi: 10.1182/blood-2015-06-655043 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Erikson J et al. Translocation of an immunoglobulin kappa locus to a region 3’ of an unrearranged c-myc oncogene enhances c-myc transcription. Proc Natl Acad Sci U S A 80, 7581–7585, doi: 10.1073/pnas.80.24.7581 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Croce CM et al. Transcriptional activation of an unrearranged and untranslocated c-myc oncogene by translocation of a C lambda locus in Burkitt. Proc Natl Acad Sci U S A 80, 6922–6926, doi: 10.1073/pnas.80.22.6922 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haller F et al. Enhancer hijacking activates oncogenic transcription factor NR4A3 in acinic cell carcinomas of the salivary glands. Nat Commun 10, 368, doi: 10.1038/s41467-018-08069-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin-Garcia D et al. CCND2 and CCND3 hijack immunoglobulin light-chain enhancers in cyclin D1(−) mantle cell lymphoma. Blood 133, 940–951, doi: 10.1182/blood-2018-07-862151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zimmerman MW et al. MYC Drives a Subset of High-Risk Pediatric Neuroblastomas and Is Activated through Mechanisms Including Enhancer Hijacking and Focal Enhancer Amplification. Cancer Discov 8, 320–335, doi: 10.1158/2159-8290.CD-17-0993 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weischenfeldt J et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat Genet 49, 65–74, doi: 10.1038/ng.3722 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ryan RJ et al. Detection of Enhancer-Associated Rearrangements Reveals Mechanisms of Oncogene Dysregulation in B-cell Lymphoma. Cancer Discov 5, 1058–1071, doi: 10.1158/2159-8290.CD-15-0370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu Y et al. PRDM6 is enriched in vascular precursors during development and inhibits endothelial cell proliferation, survival, and differentiation. J Mol Cell Cardiol 44, 47–58, doi: 10.1016/j.yjmcc.2007.06.008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davis CA et al. PRISM/PRDM6, a transcriptional repressor that promotes the proliferative gene program in smooth muscle cells. Mol Cell Biol 26, 2626–2636, doi: 10.1128/MCB.26.7.2626-2636.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hunger SP & Mullighan CG Acute Lymphoblastic Leukemia in Children. N Engl J Med 373, 1541–1552, doi: 10.1056/NEJMra1400972 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Parker M et al. C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature 506, 451–455, doi: 10.1038/nature13109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jones DT et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68, 8673–8677, doi: 10.1158/0008-5472.CAN-08-2097 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He F et al. Long noncoding RNA PVT1–214 promotes proliferation and invasion of colorectal cancer by stabilizing Lin28 and interacting with miR-128. Oncogene 38, 164–179, doi: 10.1038/s41388-018-0432-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu Z & Zhang H LncRNA plasmacytoma variant translocation 1 is an oncogene in bladder urothelial carcinoma. Oncotarget 8, 64273–64282, doi: 10.18632/oncotarget.19604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tian Z et al. LncRNA PVT1 regulates growth, migration, and invasion of bladder cancer by miR-31/ CDK1. J Cell Physiol 234, 4799–4811, doi: 10.1002/jcp.27279 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Tseng YY et al. PVT1 dependence in cancer with MYC copy-number increase. Nature 512, 82–86, doi: 10.1038/nature13311 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Z, Su M, Xiang B, Zhao K & Qin B Circular RNA PVT1 promotes metastasis via miR-145 sponging in CRC. Biochem Biophys Res Commun 512, 716–722, doi: 10.1016/j.bbrc.2019.03.121 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Yang T et al. lncRNA PVT1 and its splicing variant function as competing endogenous RNA to regulate clear cell renal cell carcinoma progression. Oncotarget 8, 85353–85367, doi: 10.18632/oncotarget.19743 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao J et al. LncRNA PVT1 promotes angiogenesis via activating the STAT3/VEGFA axis in gastric cancer. Oncogene 37, 4094–4109, doi: 10.1038/s41388-018-0250-z (2018). [DOI] [PubMed] [Google Scholar]
- 68.Cho SW et al. Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell 173, 1398–1412 e1322, doi: 10.1016/j.cell.2018.03.068 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hovestadt V et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 510, 537–541, doi: 10.1038/nature13268 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Balzeau J, Menezes MR, Cao S & Hagan JP The LIN28/let-7 Pathway in Cancer. Front Genet 8, 31, doi: 10.3389/fgene.2017.00031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Northcott PA et al. The miR-17/92 polycistron is up-regulated in sonic hedgehog-driven medulloblastomas and induced by N-myc in sonic hedgehog-treated cerebellar neural precursors. Cancer Res 69, 3249–3255, doi: 10.1158/0008-5472.CAN-08-4710 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferretti E et al. MicroRNA profiling in human medulloblastoma. Int J Cancer 124, 568–577, doi: 10.1002/ijc.23948 (2009). [DOI] [PubMed] [Google Scholar]
- 73.Uziel T et al. The miR-17~92 cluster collaborates with the Sonic Hedgehog pathway in medulloblastoma. Proc Natl Acad Sci U S A 106, 2812–2817, doi: 10.1073/pnas.0809579106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zindy F et al. Role of the miR-17 approximately 92 cluster family in cerebellar and medulloblastoma development. Biol Open 3, 597–605, doi: 10.1242/bio.20146734 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Murphy BL et al. Silencing of the miR-17~92 cluster family inhibits medulloblastoma progression. Cancer Res 73, 7068–7078, doi: 10.1158/0008-5472.CAN-13-0927 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bai AH et al. MicroRNA-182 promotes leptomeningeal spread of non-sonic hedgehog-medulloblastoma. Acta Neuropathol 123, 529–538, doi: 10.1007/s00401-011-0924-x (2012). [DOI] [PubMed] [Google Scholar]
- 77.Weeraratne SD et al. Pleiotropic effects of miR-183~96~182 converge to regulate cell survival, proliferation and migration in medulloblastoma. Acta Neuropathol 123, 539–552, doi: 10.1007/s00401-012-0969-5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang Z, Li S & Cheng SY The miR-183 approximately 96 approximately 182 cluster promotes tumorigenesis in a mouse model of medulloblastoma. J Biomed Res 27, 486–494, doi: 10.7555/JBR.27.20130010 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ferretti E et al. Concerted microRNA control of Hedgehog signalling in cerebellar neuronal progenitor and tumour cells. EMBO J 27, 2616–2627, doi: 10.1038/emboj.2008.172 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Su X et al. Abnormal expression of REST/NRSF and Myc in neural stem/progenitor cells causes cerebellar tumors by blocking neuronal differentiation. Mol Cell Biol 26, 1666–1678, doi: 10.1128/MCB.26.5.1666-1678.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wu J & Xie X Comparative sequence analysis reveals an intricate network among REST, CREB and miRNA in mediating neuronal gene expression. Genome Biol 7, R85, doi: 10.1186/gb-2006-7-9-r85 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pierson J, Hostager B, Fan R & Vibhakar R Regulation of cyclin dependent kinase 6 by microRNA 124 in medulloblastoma. J Neurooncol 90, 1–7, doi: 10.1007/s11060-008-9624-3 (2008). [DOI] [PubMed] [Google Scholar]
- 83.Silber J et al. Expression of miR-124 inhibits growth of medulloblastoma cells. Neuro Oncol 15, 83–90, doi: 10.1093/neuonc/nos281 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang X et al. miR miR on the wall, who’s the most malignant medulloblastoma miR of them all? Neuro Oncol 20, 313–323, doi: 10.1093/neuonc/nox106 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hansen KD et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet 43, 768–775, doi: 10.1038/ng.865 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Berman BP et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet 44, 40–46, doi: 10.1038/ng.969 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hon GC et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res 22, 246–258, doi: 10.1101/gr.125872.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin CY et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 530, 57–62, doi: 10.1038/nature16546 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aebersold R & Mann M Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355, doi: 10.1038/nature19949 (2016). [DOI] [PubMed] [Google Scholar]
- 90.Archer TC et al. Proteomics, Post-translational Modifications, and Integrative Analyses Reveal Molecular Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 34, 396–410 e398, doi: 10.1016/j.ccell.2018.08.004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Forget A et al. Aberrant ERBB4-SRC Signaling as a Hallmark of Group 4 Medulloblastoma Revealed by Integrative Phosphoproteomic Profiling. Cancer Cell 34, 379–395 e377, doi: 10.1016/j.ccell.2018.08.002 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Rivero-Hinojosa S et al. Proteomic analysis of Medulloblastoma reveals functional biology with translational potential. Acta Neuropathol Commun 6, 48, doi: 10.1186/s40478-018-0548-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vogel C & Marcotte EM Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 13, 227–232, doi: 10.1038/nrg3185 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Staal JA et al. Proteomic profiling of high risk medulloblastoma reveals functional biology. Oncotarget 6, 14584–14595, doi: 10.18632/oncotarget.3927 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zomerman WW et al. Identification of Two Protein-Signaling States Delineating Transcriptionally Heterogeneous Human Medulloblastoma. Cell Rep 22, 3206–3216, doi: 10.1016/j.celrep.2018.02.089 (2018). [DOI] [PubMed] [Google Scholar]
- 96.Canettieri G et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol 12, 132–142, doi: 10.1038/ncb2013 (2010). [DOI] [PubMed] [Google Scholar]
- 97.Klisch TJ, Vainshtein A, Patel AJ & Zoghbi HY Jak2-mediated phosphorylation of Atoh1 is critical for medulloblastoma growth. Elife 6, doi: 10.7554/eLife.31181 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lapidot T et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367, 645–648, doi: 10.1038/367645a0 (1994). [DOI] [PubMed] [Google Scholar]
- 99.Bonnet D & Dick JE Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3, 730–737, doi: 10.1038/nm0797-730 (1997). [DOI] [PubMed] [Google Scholar]
- 100.Singh SK et al. Identification of a cancer stem cell in human brain tumors. Cancer Res 63, 5821–5828 (2003). [PubMed] [Google Scholar]
- 101.Singh SK et al. Identification of human brain tumour initiating cells. Nature 432, 396–401, doi: 10.1038/nature03128 (2004). [DOI] [PubMed] [Google Scholar]
- 102.Garg N et al. CD133(+) brain tumor-initiating cells are dependent on STAT3 signaling to drive medulloblastoma recurrence. Oncogene 36, 606–617, doi: 10.1038/onc.2016.235 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Read TA et al. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell 15, 135–147, doi: 10.1016/j.ccr.2008.12.016 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ward RJ et al. Multipotent CD15+ cancer stem cells in patched-1-deficient mouse medulloblastoma. Cancer Res 69, 4682–4690, doi: 10.1158/0008-5472.CAN-09-0342 (2009). [DOI] [PubMed] [Google Scholar]
- 105.Vanner RJ et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 26, 33–47, doi: 10.1016/j.ccr.2014.05.005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Weng Q et al. Single-Cell Transcriptomics Uncovers Glial Progenitor Diversity and Cell Fate Determinants during Development and Gliomagenesis. Cell Stem Cell 24, 707–723 e708, doi: 10.1016/j.stem.2019.03.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Filbin MG et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335, doi: 10.1126/science.aao4750 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Venteicher AS et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, doi: 10.1126/science.aai8478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tirosh I et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313, doi: 10.1038/nature20123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Patel AP et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401, doi: 10.1126/science.1254257 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vladoiu MC et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature, doi: 10.1038/s41586-019-1158-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hovestadt V et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature, doi: 10.1038/s41586-019-1434-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Oliver TG et al. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development 132, 2425–2439, doi: 10.1242/dev.01793 (2005). [DOI] [PubMed] [Google Scholar]
- 114.Morrissy AS et al. Spatial heterogeneity in medulloblastoma. Nat Genet 49, 780–788, doi: 10.1038/ng.3838 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fults DW, Taylor MD & Garzia L Leptomeningeal dissemination: a sinister pattern of medulloblastoma growth. J Neurosurg Pediatr, 1–9, doi: 10.3171/2018.11.PEDS18506 (2019). [DOI] [PubMed] [Google Scholar]
- 116.Ramaswamy V et al. Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol 14, 1200–1207, doi: 10.1016/S1470-2045(13)70449-2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hill RM et al. Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell 27, 72–84, doi: 10.1016/j.ccell.2014.11.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Morrissy AS et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 529, 351–357, doi: 10.1038/nature16478 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zapotocky M et al. Differential patterns of metastatic dissemination across medulloblastoma subgroups. J Neurosurg Pediatr 21, 145–152, doi: 10.3171/2017.8.PEDS17264 (2018). [DOI] [PubMed] [Google Scholar]
- 120.Garzia L et al. A Hematogenous Route for Medulloblastoma Leptomeningeal Metastases. Cell 173, 1549, doi: 10.1016/j.cell.2018.05.033 (2018). [DOI] [PubMed] [Google Scholar]
- 121.Gajjar A et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol 7, 813–820, doi: 10.1016/S1470-2045(06)70867-1 (2006). [DOI] [PubMed] [Google Scholar]
- 122.Sharma T et al. Second-generation molecular subgrouping of medulloblastoma: an international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol 138, 309–326, doi: 10.1007/s00401-019-02020-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brodeur GM, Nichols KE, Plon SE, Schiffman JD & Malkin D Pediatric Cancer Predisposition and Surveillance: An Overview, and a Tribute to Alfred G. Knudson Jr. Clin Cancer Res 23, e1–e5, doi: 10.1158/1078-0432.CCR-17-0702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Romer JT et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/−)p53(−/−) mice. Cancer Cell 6, 229–240, doi: 10.1016/j.ccr.2004.08.019 (2004). [DOI] [PubMed] [Google Scholar]
- 125.Lee Y et al. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene 26, 6442–6447, doi: 10.1038/sj.onc.1210467 (2007). [DOI] [PubMed] [Google Scholar]
- 126.Robinson GW et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J Clin Oncol 33, 2646–2654, doi: 10.1200/JCO.2014.60.1591 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Robinson GW et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 8, 69295–69302, doi: 10.18632/oncotarget.20619 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kimura H, Ng JM & Curran T Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell 13, 249–260, doi: 10.1016/j.ccr.2008.01.027 (2008). [DOI] [PubMed] [Google Scholar]
- 129.Lee C et al. Lsd1 as a therapeutic target in Gfi1-activated medulloblastoma. Nat Commun 10, 332, doi: 10.1038/s41467-018-08269-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Morabito M et al. An autocrine ActivinB mechanism drives TGFbeta/Activin signaling in Group 3 medulloblastoma. EMBO Mol Med, e9830, doi: 10.15252/emmm.201809830 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gilbertson RJ & Ellison DW The origins of medulloblastoma subtypes. Annu Rev Pathol 3, 341–365, doi: 10.1146/annurev.pathmechdis.3.121806.151518 (2008). [DOI] [PubMed] [Google Scholar]
- 132.Gibson P et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 468, 1095–1099, doi: 10.1038/nature09587 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pei Y et al. An animal model of MYC-driven medulloblastoma. Cancer Cell 21, 155–167, doi: 10.1016/j.ccr.2011.12.021 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kawauchi D et al. Novel MYC-driven medulloblastoma models from multiple embryonic cerebellar cells. Oncogene 36, 5231–5242, doi: 10.1038/onc.2017.110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kawauchi D et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell 21, 168–180, doi: 10.1016/j.ccr.2011.12.023 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]