

Abstract
In pig-to-human xenotransplantation, the transmission risk of porcine endogenous retroviruses (PERVs) is of great concern. However, the distribution of PERVs in pig genomes, their genetic variation among Eurasian pigs, and their evolutionary history remain unclear. We scanned PERVs in the current pig reference genome (assembly Build 11.1), and identified 36 long complete or near-complete PERVs (lcPERVs) and 23 short incomplete PERVs (siPERVs). Besides three known PERVs (PERV-A, -B, and -C), four novel types (PERV-JX1, -JX2, -JX3, and -JX4) were detected in this study. According to evolutionary analyses, the newly discovered PERVs were more ancient, and PERV-Bs probably experienced a bottleneck ~0.5 million years ago (Ma). By analyzing 63 high-quality porcine whole-genome resequencing data, we found that the PERV copy numbers in Chinese pigs were lower (32.0±4.0) than in Western pigs (49.1±6.5). Additionally, the PERV sequence diversity was lower in Chinese pigs than in Western pigs. Regarding the lcPERV copy numbers, PERV-A and -JX2 in Western pigs were higher than in Chinese pigs. Notably, Bama Xiang (BMX) pigs had the lowest PERV copy number (27.8±5.1), and a BMX individual had no PERV-C and the lowest PERV copy number (23), suggesting that BMX pigs were more suitable for screening and/or modification as xenograft donors. Furthermore, we identified 451 PERV transposon insertion polymorphisms (TIPs), of which 86 were shared by all 10 Chinese and Western pig breeds. Our findings provide systematic insights into the genomic distribution, variation, evolution, and possible biological function of PERVs.
Keywords: PERVs, Chinese and Western pigs, Copy number variation, Evolutionary history, Biological function prediction
INTRODUCTION
Xenotransplantation has become the primary medical method by which the shortage of human organs is mediated. Due to similarities in organ size and physiology to humans, as well as their rapid reproductive ability, pigs are the preferred animal source species for xenotransplantation (Cooper, 2003; Yang & Sykes, 2007). However, the risk of immune rejection and transmitting diseases from animals to humans are two major challenges to overcome in xenotransplantation. These problems of immune rejection and partial viral infection were largely resolved, but the risk of porcine endogenous retrovirus (PERV) infection remains a great concern (Denner & Tönjes, 2012; Sykes & Sachs, 2019).
Pigs originated from Island Southeast Asia (ISEA), then spread to and colonized almost the entire Eurasian continent (Frantz et al., 2013; Groenen, 2016). Pigs were domesticated in at least two locations, Anatolia and China (Larson et al., 2005). During the pig domestication process, which began ~10 000 years ago, both natural and artificial selection have shaped and stabilized distinct pig breeds, including two main pig lineages of Asian and European pigs. In Asia, China has the most pig breeds, accounting for more than one-third of the world’s pig breeds (Ai et al., 2015). Chinese indigenous pigs are known for their low growth rate, poor feed conversion efficiency, and early puberty. European pigs, which are distributed worldwide, including America, Africa, Australia, and Asia (Yang et al., 2017), originated from European pig breeds and are thus referred to as Western pigs. Western pig breeds, such as Large White Duroc, Duroc, Landrace, and Pietrain pigs, have fast growth rates, excellent feed efficiency, and a low fat-deposition ability.
A complete PERV contains three protein-coding genes, gag, pol, and env. PERVs are typically classified into three types, PERV-A, -B, or -C, based on env sequence differences. PERV-A and -C recombine to generate new PERV variants PERV-A/C, which infect human cells (Denner, 2008; Karlas et al., 2010; Wilson et al., 2000). Recently, another PERV type, PERV-IM, has been reported and its env protein shows low similarity to PERV-A, -B, and -C. It is located in the middle phylogenetic position of PERVs and is close to PERV-A and -C (Chen et al., 2020b). Long terminal repeats (LTRs) are located on both flanks of a PERV. Based on sequence structure and length differences, LTRs flanked to PERVs can be divided into two subfamilies, LTR-A (named in the RepeatMasker database as ERV1-2B_SSC-LTR) and LTR-B (ERV1-2_SSC-LTR). LTR-A consists of an R region, U5, and U3 without the 18 and 21 bp repeat structures, but with sequences homologous to the repeats. LTR-B consists of an R region, U5, and U3 with the 18 and 21 bp repeat structures (Scheef et al., 2001; Tönjes & Niebert, 2003). PERV is the only retrovirus that harbors two different LTRs; only PERV-A can be flanked by two different LTRs, both flanking LTRs of a PERV-A belong to the same subfamily (Tönjes & Niebert, 2003).
Multiple methods have been developed to estimate PERV copy numbers, including Southern blot, semi-quantitative PCR (Semi-qPCR), real-time PCR (RT-PCR), droplet digital PCR (ddPCR), fluorescence in situ hybridization (FISH), and genome-wide sequencing (Denner, 2016b). In early 1997, Southern blot was applied to detect PERVs in the cells of a pig kidney cell line (PK15) and in Landrace × Duroc, Meishan, and Pietrain pigs to roughly estimate PERV copy numbers (Le Tissier et al., 1997; Patience et al., 1997). Patience et al. (2001) estimated the PERV copy numbers in Landrace × Duroc F1 hybrid pigs by PCR titration (Patience et al., 2001). In 2002, Semi-qPCR was employed to measure the PERV copy numbers of 11 local Chinese pig breeds (Lian et al., 2002). The average PERV copy number was estimated to range from 27.1 (Bama Xiang (BMX) pig) to 62.9 (Meishan pig). By applying RT-PCR, 22–34, 17–27, 19–34, 9–23, 3–43, and 4–96 PERV copies were detected in Duroc, Landrace, Yorkshire, South Korean Jeju, Spanish Iberian, and Chinese miniature pigs, respectively (Lee et al., 2011; Liu et al., 2011; Quereda et al., 2012). A total of 32 PERV copies were detected in Western pigs via FISH (Lee et al., 2002). By employing ddPCR, 69 copies were detected in Aachen minipigs, 117 in Black minipigs, 59 in genetically modified pigs generated for xenotransplantation, 93 in Göttingen minipigs, and 3–69 in wild boars around Berlin (Fiebig et al., 2018; Krüger et al., 2019, 2020). By combining genome-wide sequencing and ddPCR, 25 PERV copies were confirmed in a Chinese BMX pig (Niu et al., 2017). Based on these previous estimates, it is clear that PERV copy numbers vary among pig breeds and different animals within the same breed. Most of the above methods detect a short region in PERVs, usually the conserved fragment in the pol gene (Yang et al., 2015). Because there are several different (at least three) PERV types, methods based on PCR or FISH may be biased, may not detect all PERVs, and could estimate copy numbers inaccurately in pigs. Previous studies have reported the PERV copy numbers in different pig lines (Denner & Tönjes, 2012; Denner, 2016b). However, comparisons of different PERV types among various Chinese and Western pig breeds remain sparse.
Regarding the potential infection risk of PERVs, PERV transmission has not been documented in pre-clinical trials of pig cells or organs transplanted into primates (Denner, 2018). Previously, it was reported that PERV-A and -B infected human cells in vitro (Le Tissier et al., 1997). Although PERV-C does not infect human cells, it can recombine with PERV-A to form a highly pathogenic virus, PERV-A/C, that infects human cells in vitro (Bartosch et al., 2004; Harrison et al., 2004; Karlas et al., 2010; Wilson et al., 2000). Furthermore, PERV-A and -B exist widely in all pigs and PERV-C has been observed in most pigs (Denner & Tönjes, 2012). Therefore, the potential risks of PERVs cannot be eliminated by using certain pathogen-free pigs as donor organ sources. The best way to reduce the potential risk of PERVs is by selecting pig breeds with fewer PERV copies or to completely remove PERVs by gene knockout (Denner, 2016b, 2018). Yang et al. inactivated 25 PERVs in BMX pigs via CRISPR/Cas9 and successfully produced PERV-inactivated pigs (Niu et al., 2017; Yang et al., 2015; Yue et al., 2021). Previous studies have found that some specific retrotransposons of human endogenous retroviruses contribute to transcription factor binding sites (Cohen et al., 2009; Wang et al., 2007). However, the biological function of PERVs and interaction mechanism between PERVs and their host remain unclear.
Therefore, it is necessary to comprehensively investigate different PERV types, their distribution in the Sus scrofa reference genome, estimate the copy number variation, and detect potential insertion sites in a variety of pig breeds. Here, we scanned all PERVs in the current pig reference genome (S. scrofa genome, assembly Build 11.1) by sequence similarity searching. Based on the whole-genome sequencing data, we introduced and applied four methods (mapping-to-genome, mapping-to-PERV, k-mer-based, and mapping-to-LTR) to identify PERV types, investigate their copy numbers, and detect PERV transposon insertion polymorphisms (TIPs) in 10 divergent Chinese and Western pig breeds. These findings will assist with the identification of pig breeds suitable for pig-to-human xenotransplantation. Furthermore, we performed analyses on PERV evolution and selection, and investigated their potential biological functions. Our findings will serve as a valuable reference for future studies on pig-to-human xenotransplantation.
MATERIALS AND METHODS
Retrieving PERVs in the pig reference genome
Three clearly classified PERVs, PERV-A (accession No.: AY099323.1), PERV-B (accession No.: AY099324.1), and PERV-C (accession No.: KY352351.1) were retrieved from the NCBI database and selected as reference sequences (Bartosch et al., 2002). These PERVs were mapped to the pig reference genome (assembly Build 11.1) using BLAT (Kent, 2002). We retrieved putative PERVs using the following parameters: sequence similarity >90%, sequence length >2 000 bp, and at least one flanking LTR. A total of 59 putative PERVs were obtained, including 36 long complete or near- complete PERVs (lcPERVs) with a sequence length >7 000 bp and 23 short incomplete PERVs (siPERVs) with a sequence length <6 000 bp and >2 000 bp (Supplementary Tables S1, S2). RepeatMasker v4.0.9 was used to clarify the boundaries of the sequences and annotate LTR types ( Chen, 2004). The gag, pol, and env open reading frames (ORF) were annotated by ORF-FINDER v0.4.3 (Rombel et al., 2002).
Determination of PERV types
Sequence homology alignment and topological location determination of the phylogenetic tree were employed to identify the types of 59 PERVs distributed across the pig reference genome. To determine the categories of 36 lcPERVs, MegaBLAST was used to align the lcPERVs without flanking LTR sequences to three reference PERVs (PERV-A-AY099323.1, PERV-B-AY099324.1, and PERV-C-KY352351.1) (Morgulis et al., 2008). PERVs that had >97% similarity with a reference PERV were classified as the same type as the reference PERV. Then, we used these classified PERV sequences as additional reference sequences for realigning unclassified PERVs. PERV sequences with >97% similarity with these reference PERVs were classified according to the same rule. We repeated this process of PERV classification until no PERV had >97% similarity with the references. Finally, seven lcPERV sequences with >90% and <97% similarity with the references were not classified into the known PERVs. A phylogenetic tree was constructed using the GTR + gamma DNA substitution model in BEAST v1.8.4 for the 36 lcPERVs ( Drummond & Rambaut, 2007). In the tree, the AKV murine leukemia virus (MLV) (accession No.: J01998.1) sequence was used as the outgroup. Based on the clustering position in the phylogenetic tree, a PERV with 95% similarity to PERV-A was named as a like type of PERV-A; the remaining six lcPERV sequences that had >90% and <97% similarity with the reference PERVs were defined as new PERVs. These six sequences were further divided into four types, PERV-JX1, -JX2, -JX3, and -JX4.
We also calculated the genetic distance of 36 lcPERVs without flanking LTR sequences and the genetic distance of their env genes to verify our classifications. First, we performed multi-sequence alignment of these 36 lcPERVs or their env genes using the muscle function implemented in MEGAX (Kumar et al., 2018). Then, we estimated the distance of the 36 lcPERVs without flanking LTR sequences and the distance of env genes using a maximum composite likelihood model with rate uniformity and pattern homogeneity using MEGAX.
For classification of the 23 siPERVs, each siPERV was aligned to 10 reference sequences (PERV-A-AY099323.1, PERV-A-1-262.2M, PERV-A-X-73.7M, PERV-A- Y-20.1M, PERV-B-AY099324.1, PERV-C-KY352351.1, PERV-JX1-7-21.2M, PERV-JX2-X-71.4M, PERV-JX3-3-17.8M, and PERV-JX4-2-76.6M). Similar to the lcPERV classification rules, siPERVs with >97% similarity to a reference sequence were classified as the same type as the reference PERV, siPERVs with >95% and <97% similarity to a reference sequence were classified as the like type of the reference PERV (siPERV-like), and siPERVs with >90% and <95% similarity to any reference sequence were classified as an unclear PERV type (siPERV-UC).
Estimating PERV and LTR divergence times
BEAST v1.8.4 was used to reconstruct the tree topology of the lcPERVs and their flanking LTRs, and estimate their divergence times under a strict molecular clock model (Drummond & Rambaut, 2007). In addition to the 36 lcPERVs, we included three PERV reference sequences (PERV-A-AY099323.1, PERV-B-AY099324.1, and PERV-C-KY352351.1) and one MLV sequence (MLV-J01998.1) downloaded from the NCBI Genbank database. For the PERV analyses, we employed MAFFT to make multiple alignments of these 39 PERV sequences without their flanking LTRs and one MLV sequence. Then, BEAST was used to reconstruct their phylogenetic tree and estimate their divergence times. We set the divergence time between MLV (MLV-J01998.1) and PERV (PERV-A-AY099323.1) as 96 million years ago (Ma), which is the divergence time of pig and mouse from the TimeTree database (Katoh & Standley, 2013; Kumar et al., 2017). For the LTR analyses, we chose the 5' LTR sequences flanking the above PERVs to build their phylogenetic tree. In the LTR tree, the 5' LTR sequence of MLV was used as the outgroup and the divergence time of the LTRs between MLV and PERV was the same as in the above PERV analyses. We used the BEAUti program in BEAST v1.8.4 to set up the XML file with the following parameters: selecting GTR + gamma as DNA substitution model, enabling link trees, strict clock, and specifying tree prior as constant size of coalescence. Each MCMC chain was run for 10 million steps. Logging trees and model parameters were generated every 2 000 steps and the first 10% of steps was discarded as burn-in. Posterior distributions of tree likelihoods and other estimated parameters were analyzed using Tracer v1.7.1 (https://github.Com/beast-dev/tracer /releases) to ensure an estimated sample size (ESS) >200 for each statistic. Finally, the results were summarized using the TreeAnnotator program implemented in BEAST. Finally, the tree was visualized by iTOL v4 ( Letunic & Bork, 2019).
Resequencing data of Eurasian pigs
We collected next-generation whole-genome resequencing data of 63 pigs from 10 breeds, including five Chinese pig breeds (six South Chinese wild boar (CNWB), six BMX, eight Erhualian (EHL), six Laiwu (LWU), and six Tibetan (TB) pigs) and five Western pig breeds (five European wild boar (EUWB), eight White Duroc (WD), seven Landrace (LR), five Large White (LW), and six Duroc (DRC) pigs). Among these data, 52 were previously published by our research team and 11 (six DRC pigs and five EUWB) were downloaded from data published by Wageningen University (Ai et al., 2015, 2021; Chen et al., 2020a; Frantz et al., 2015; Yan et al., 2018). All data were mapped to the reference genome using BWA-MEN to evaluate the sequencing depth and genome coverage (Li & Durbin, 2009). The average depth of EUWB data was 16.9×, DRC data was 17.2×, and other data were >20×. The coverage of all data was >0.97 (for more details, see Supplementary Table S3).
Calculation of PERV copy numbers
We employed two methods to calculate PERV copy number using the above sequencing data. In method 1 (mapping-to-genome), the sequencing data were aligned to the pig reference genome using bowtie2 v2.3.5.1 with the parameter “--end-to-end -k 1 --very-fast” (Langmead & Salzberg, 2012). The PERV copy number of each individual was calculated using the following formula:
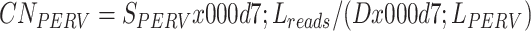 |
1 |
where CNPERV represents the PERV copy number of an individual, SPERV represents the summation of reads mapped to all 59 PERVs located on the reference genome, Lreads represents the average length of mapped reads at PERV regions, D represents the average read depth of the individual, and LPERV represents the average length of PERVs detected in the pig reference genome, which was set to 6 000.
The LTR copy number of each animal was calculated using the following formula:
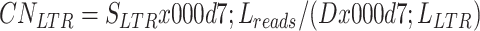 |
2 |
where CNLTR represents the LTR copy number of an individual, SLTR represents the summation of reads mapped to all 247 LTRs located on the reference genome, Lreads represents the average length of mapped reads at LTR regions, D represents the average read depth of the individual, and LLTR represents the average length of LTRs found in the pig reference genome, which was set to 600.
In method 2 (mapping-to-PERV), 59 PERV sequences without flanking LTRs were used as reference sequences for calculating the PERV copy number; 247 LTR sequences were used as reference sequences for calculating the LTR copy number. Whole-genome sequencing data were mapped to these PERV or LTR reference sequences using bowtie2 v2.3.5.1 with the parameter, “--local --ma 1 --very-sensitive-local --no-overlap --no-contain --no-unal.” CNPERV and CNLTR were calculated using the two formulas above. However, in method 2, SPERV represents the summation of reads mapped to all 59 PERV reference sequences and SLTR represents the summation of reads mapped to all 247 LTR reference sequences.
T-test of PERV copy number differences between domestic pigs and wild boar
To compare the differences in PERV copy numbers between domestic pigs and wild boar, Student’s t-tests were performed on the PERV copy numbers between European domestic pigs (LW, LR, DRC, and WD) and EUWB (P=0.0094), and Chinese domestic pigs (BMX, EHL, LWU, and TB) and CNWB (P=0.041).
Prediction of PERV types in Eurasian pigs
We developed a pipeline (k-mer-based method) to predict which PERV types were included in an individual among the 59 known PERVs using the resequencing data of individuals based on unique k-mers of the known PERVs (Figure 1). First, the k-mers (k=31) of a known PERV and reference genome were generated using the KMC v3.1.1 software and their single-copy k-mers were selected (Deorowicz et al., 2015). We defined the intersection of single-copy k-mers between a PERV and reference genome as the unique k-mer of the PERV. The k-mers of an individual were generated using the resequencing data of an individual. We counted the k-mer types in the intersection between all k-mers of an individual and unique k-mers of the PERV, then calculated the ratio of unique k-mers of the PERV in an individual to the total unique k-mers of the PERV. Finally, we set a threshold to determine whether the known PERVs existed in an individual. We tested the thresholds from 5%–30% and found that when the threshold was 18%, the correlation between the PERV copy numbers estimated by the pipeline and method 1 at the section of “Calculation of copy number of PERVs” was the largest (>0.85). Therefore, if the ratio of the unique k-mers of the PERV in an individual to the total unique k-mers of the PERV was >18%, the PERV was determined to be present in the individual. However, if the ratio was <18%, the individual did not contain the PERV.
Figure 1.
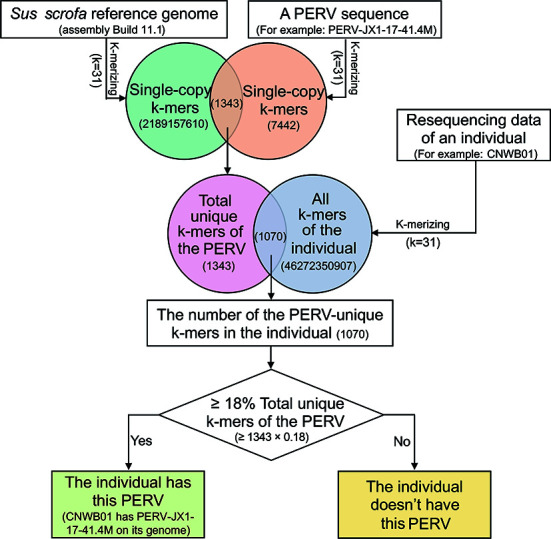
Prediction pipeline of PERV types in an individual using the resequencing data based on the unique k-mers
An example was given to determine whether the individual CNWB01 contained the PERV (PERV-JX1-17-41.4M). The result indicated CNWB01 had PERV-JX1-17-41.4M in its genome.
Comparison of PERV sequence diversity in Eurasian pigs
We employed the k-mer spectra approach to compare the sequence diversity of PERVs between European and Chinese pigs. First, the resequencing data of 63 Eurasian pigs were mapped to a PERV reference panel containing 36 lcPERVs without flanking LTRs using bowtie2 v2.3.5.1 with the parameter, “--end-to-end --very-sensitive”. To avoid potential bias of k-mer spectra caused by sequencing depth and read length, we consistently and randomly selected the same total length (2 400 000 bp) of resequencing reads per individual. The mapped reads of 31 Western and 32 Chinese pigs were collected and k-merized (k=31) using KMC v3.1.1. We filtered the k-mers whose frequency was <3.0 due to sequencing errors and counted the number of remaining k-mer types. To compare the PERV sequence diversity between CNWB and EUWB, we selected five individuals from both CNWB and EUWB. We repeated the random selection of resequencing data and performed the above analyses 10 times. All results were consistent, indicating that the methods were valid and our results were reliable.
Detecting PERV TIPs in Eurasian pigs
To detect PERV TIPs in Eurasian pigs using the next-generation sequencing data, we developed a new method, method 3 (mapping-to-LTR). First, we mapped all paired reads to two LTR reference sequences, ERV1-2_SSc-LTR (668 bp) and ERV1-2B_SSc-LTR (742 bp), using BWA-MEM v0.7.17 with the parameter, “-M”. We recorded the IDs of the mapped reads and extracted their mate reads. We kept the mate reads that were not mapped to the two LTR reference sequences. Next, these unmapped mate reads were aligned to the pig reference genome using BWA-MEM v 0.7.17. We retained the reads mapped to the pig reference genome with a mapping quality (MAPQ) >30 for further analysis. The locations of the mapped reads were merged using BEDTools merge v2.27.1 ( Quinlan & Hall, 2010). A candidate TIP was determined if at least two reads were found with a distance <2 000 bp. We tested different MAPQ thresholds (from 0 to 60) and read distances (from 1 000 to 2 500 bp) to compare the correlation between the number of TIPs identified by method 3 and PERV copy numbers detected by method 1 or 2. The highest correlation (method 1: r 2=0.774, P=9.603×10-14; method 2: r2=0.795, P=7.064×10-15) was observed when MAPQ was set to 30 and the distance of reads was set to 2 000 bp. Chromosomal TIP sites were visualized by the R package, “karyoploteR” (Gel & Serra, 2017).
ClueGO enrichment analysis
Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were enriched using the default options of the ClueGO plug-in of Cytoscape v3.5.0 (Bindea et al., 2009; Shannon et al., 2003). Human orthologous EnsemblGeneIDs were set as the gene list and the human GO database as the query database.
RESULTS
PERV landscape in the S. scrofa reference genome
A total of 36 lcPERVs with sequence lengths >7 000 bp and 23 siPERVs with sequence lengths >2 000 bp and <6 000 bp were identified in the current S. scrofa reference genome, which mainly originated from a female DRC pig (Figure 2A; Table 1; Supplementary Tables S1, S2). At least one PERV per S. scrofa chromosome (SSC) was detected, except in SSC10. PERV-A/C was not detected in the current S. scrofa reference genome. Among the 36 lcPERVs, 18 PERV-As (including a PERV-Alike) were detected on SSC1, -3, -5, -7, -8, -12, -13, -15, -17, -X, and -Y, and two unplaced contigs, NW_018084965 and NW_018085136. Eleven PERV-Bs were located on SSC3, -4, -8, -9, -11, -14, -15, and -16, and NW_018085086, NW_018085255, and NW_018085331. Only one PERV-C was located on SSC14. Six newly identified PERVs were located on SSC2, -3, -7, -17, and -X, and NW_018085293 (Figure 2A).
Figure 2.

59 PERVs in the S. Scrofa reference genome
A: The positions of PERVs in the reference genome. NW965: NW_018084965; NW136: NW_018085136; NW086: NW_018085086; NW255: NW_018085255; NW331: NW_018085331; NW293: NW_018085293; NW154: NW_018085154; NW193: NW_018085193; NW355: NW_018085355. siPERV: short incomplete PERV. B: Maximum-likelihood (ML) phylogenetic tree of 36 long complete or near-complete PERVs (lcPERVs) without LTR. In the tree, 3 complete PERVs (PERV-A: AY099323.1, PERV-B: AY099324.1, and PERV-C: KY352351.1) were included, and an MLV (MLV-J01998.1) was set as the outgroup. The color on the outer circle represents the detection rate of PERVs on the genome by 10 breeds (n=63). The bluer the color, the more pigs have this PERV copy, and the redder the color, the fewer pigs. C: Maximum-likelihood (ML) phylogenetic tree of 36 lcPERVs with LTRs. The color on the outer circle represents LTR types. D: ORF prediction of 62 PERVs and their LTRs. 59 PERVs were identified from the swine reference genome Build 11.1, and 3 complete PERVs (PERV-A: AY099323.1, PERV-B: AY099324.1, and PERV-C: KY352351.1) were downloaded from the NCBI Genbank database. ORFs of gag, pol, env were predicted in three forward reading frames. For example, gag1 means gag ORF predicted in forward frame 1; gag2 means gag ORF predicted in forward frame 2; gag3 means gag ORF predicted in forward frame 3. The sequences of pol-probe and env-probe were downloaded from the reference (Yang et al., 2015).
Table 1. Classification and descriptive statistics of all 59 PERVs in the Sus scrofa reference genome (assembly Build 11.1) .
| No. | Category1 | Subclass | PERV name | Chromosome / unplaced contig | Strand | Position | Length2 | Unique k-mer3 | Detection rate4 |
| 1: LcPERV indicates long and complete or near-complete PERV; siPERV indicates short incomplete PERV. 2: The pol gene of sPERV-JX3like-15-116.3M was inserted by a 6 302 bp long sequence containing LINE1 transposon and (T)n simple repeat, which resulted in the length of the PERV increasing to 13 105 bp. 3: The number of unique k-mer (k=31) of PERV on pig reference genome. 4: The detection proportion of the PERV in the 63 Chinese and Western pigs; since the PERV sPERV-Clike-Y-25.29M has no unique k-mers, it cannot be detected in the tested pigs. /: Not available. | |||||||||
| 1 | lcPERV | PERV-A | PERV-A-1-132.0M | 1 | + | 132020281–132028361 | 8 081 | 253 | 0.57 |
| 2 | lcPERV | PERV-A | PERV-A-1-262.2M | 1 | + | 262166347–262175262 | 8 916 | 35 | 0.38 |
| 3 | lcPERV | PERV-A | PERV-A-3-10.6M | 3 | − | 10660619–10669533 | 8 915 | 647 | 0.24 |
| 4 | lcPERV | PERV-A | PERV-A-5-92.1M | 5 | − | 92185133–92194050 | 8 918 | 226 | 0.56 |
| 5 | lcPERV | PERV-A | PERV-A-7-105.7M | 7 | + | 105710884–105719193 | 8 310 | 119 | 0.17 |
| 6 | lcPERV | PERV-A | PERV-A-8-51.6M | 8 | + | 51570546–51579460 | 8 915 | 466 | 0.94 |
| 7 | lcPERV | PERV-A | PERV-A-12-28.2M | 12 | + | 28221374–28230287 | 8 914 | 95 | 0.43 |
| 8 | lcPERV | PERV-A | PERV-A-13-142.0M | 13 | − | 142030847–142039759 | 8 913 | 451 | 1 |
| 9 | lcPERV | PERV-A | PERV-A-13-146.7M | 13 | − | 146750532–146759521 | 8 990 | 520 | 1 |
| 10 | lcPERV | PERV-A | PERV-A-15-66.8M | 15 | − | 66864153–66873044 | 8 892 | 500 | 0.44 |
| 11 | lcPERV | PERV-A | PERV-A-17-3.7M | 17 | − | 3794010–3802709 | 8 700 | 411 | 1 |
| 12 | lcPERV | PERV-A | PERV-A-17-33.1M | 17 | + | 33062883–33071800 | 8 918 | 930 | 0.41 |
| 13 | lcPERV | PERV-Alike | PERV-Alike-U965-14.7kb | NW_018084965 | + | 14674–22827 | 8 154 | 1 933 | 0 |
| 14 | lcPERV | PERV-A | PERV-A-U136-0.5M | NW_018085136 | − | 563793–572725 | 8 933 | 1 205 | 0.86 |
| 15 | lcPERV | PERV-A | PERV-A-X-73.7M | X | − | 73752986–73761903 | 8 918 | 577 | 0.51 |
| 16 | lcPERV | PERV-A | PERV-A-Y-20.1M | Y | − | 20194782–20203572 | 8 791 | 983 | 0.46 |
| 17 | lcPERV | PERV-A | PERV-A-Y-21.8M | Y | − | 21867301–21876035 | 8 735 | 920 | 0.38 |
| 18 | lcPERV | PERV-A | PERV-A-Y-25.2M | Y | + | 25245606–25253905 | 8 300 | 11 | 0.63 |
| 19 | lcPERV | PERV-B | PERV-B-3-51.1M | 3 | − | 51108601–51117360 | 8 760 | 197 | 0.86 |
| 20 | lcPERV | PERV-B | PERV-B-4-45.5M | 4 | − | 45599494–45608252 | 8 759 | 132 | 0.62 |
| 21 | lcPERV | PERV-B | PERV-B-8-15.3M | 8 | + | 15319428–15328187 | 8 760 | 177 | 0.76 |
| 22 | lcPERV | PERV-B | PERV-B-9-138.8M | 9 | − | 138895584–138904340 | 8 757 | 51 | 0.46 |
| 23 | lcPERV | PERV-B | PERV-B-11-38.2M | 11 | + | 38201361–38210116 | 8 756 | 386 | 0.86 |
| 24 | lcPERV | PERV-B | PERV-B-14-119.7M | 14 | + | 119667621–119676299 | 8 679 | 486 | 0.46 |
| 25 | lcPERV | PERV-B | PERV-B-15-110.9M | 15 | − | 110958945–110967782 | 8 838 | 374 | 0.44 |
| 26 | lcPERV | PERV-B | PERV-B-16-59.6M | 16 | + | 59571885–59580646 | 8 762 | 141 | 0.44 |
| 27 | lcPERV | PERV-B | PERV-B-U086-42.1kb | NW_018085086 | − | 42158–50876 | 8 719 | 564 | 0.81 |
| 28 | lcPERV | PERV-B | PERV-B-U255-1.9kb | NW_018085255 | + | 1949–10743 | 8 795 | 242 | 0.54 |
| 29 | lcPERV | PERV-B | PERV-B-U331-13.8kb | NW_018085331 | + | 13836–22597 | 8 762 | 85 | 0.87 |
| 30 | lcPERV | PERV-C | PERV-C-14-62.5M | 14 | − | 62550397–62557645 | 7 249 | 652 | 0.78 |
| 31 | lcPERV | PERV-JX1 | PERV-JX1-7-21.2M | 7 | + | 21236160–21244976 | 8 817 | 1 387 | 1 |
| 32 | lcPERV | PERV-JX1 | PERV-JX1-17-41.4M | 17 | − | 41467519–41476101 | 8 583 | 1 343 | 1 |
| 33 | lcPERV | PERV-JX1 | PERV-JX1-U293-0.8M | NW_018085293 | + | 820148–829192 | 9 045 | 2 015 | 1 |
| 34 | lcPERV | PERV-JX2 | PERV-JX2-X-71.4M | X | + | 71402222–71409912 | 7 691 | 1 472 | 0.49 |
| 35 | lcPERV | PERV-JX3 | PERV-JX3-3-17.8M | 3 | + | 17778858–17788086 | 9 229 | 1 460 | 1 |
| 36 | lcPERV | PERV-JX4 | PERV-JX4-2-76.6M | 2 | − | 76648002–76655788 | 7 787 | 859 | 1 |
| 37 | siPERV | PERV-A | sPERV-A-6-10.4M | 6 | + | 10402614–10407657 | 5 044 | 20 | 0.57 |
| 38 | siPERV | PERV-A | sPERV-A-X-55.1M | X | + | 55079139–55083211 | 4 073 | 168 | 0.86 |
| 39 | siPERV | PERV-B | sPERV-B-1-38.5M | 1 | + | 38459013–38465006 | 5 994 | 61 | 0.48 |
| 40 | siPERV | PERV-B | sPERV-B-X-112.8M | X | − | 112842765–112847657 | 4 893 | 108 | 0.76 |
| 41 | siPERV | PERV-C | sPERV-C-5-29.4M | 5 | + | 29356367–29361638 | 5 272 | 639 | 0.78 |
| 42 | siPERV | PERV-C | sPERV-C-11-29.1M | 11 | − | 29084884–29090328 | 5 445 | 611 | 0.57 |
| 43 | siPERV | PERV-C | sPERV-C-13-146.75M | 13 | − | 146759522–146764990 | 5 469 | 19 | 0.56 |
| 44 | siPERV | PERV-C | sPERV-C-13-146.76M | 13 | − | 146764392–146769858 | 5 467 | 34 | 0 |
| 45 | siPERV | PERV-C | sPERV-C-X-81.5M | X | − | 81543240–81545899 | 2 660 | 178 | 0.56 |
| 46 | siPERV | PERV-Alike | sPERV-Alike-U293-0.9M | NW_018085293 | + | 909902–914028 | 4 127 | 729 | 0 |
| 47 | siPERV | PERV-Clike | sPERV-Clike-U154-3.2kb | NW_018085154 | + | 3178–6483 | 3 306 | 505 | 0 |
| 48 | siPERV | PERV-Clike | sPERV-Clike-U193-4.0kb | NW_018085193 | + | 3949–7250 | 3 302 | 232 | 0.57 |
| 49 | siPERV | PERV-Clike | sPERV-Clike-U355-19.3kb | NW_018085355 | + | 19304–23938 | 4 635 | 604 | 0.81 |
| 50 | siPERV | PERV-Clike | sPERV-Clike-Y-25.269M | Y | − | 25269797–25273827 | 4 031 | 16 | 0.24 |
| 51 | siPERV | PERV-Clike | sPERV-Clike-Y-25.29M | Y | − | 25299007–25302057 | 3 051 | 0 | / |
| 52 | siPERV | PERV-JX2like | sPERV-JX2like-12-23.0M | 12 | − | 22986893–22991220 | 4 328 | 704 | 1 |
| 53 | siPERV | PERV-JX3like | sPERV-JX3like-15-116.3M | 15 | + | 116321677–116334781 | 13 105 | 1 206 | 1 |
| 54 | siPERV | PERV-JX3like | sPERV-JX3like-17-9.5M | 17 | − | 9536784–9541504 | 4 721 | 854 | 1 |
| 55 | siPERV | PERV-JX3like | sPERV-JX3like-X-72.2M | X | + | 72244018–72249170 | 5 153 | 907 | 0.49 |
| 56 | siPERV | PERV-UC | sPERV-UC1-9-63.2M | 9 | − | 63235598–63238136 | 2 539 | 405 | 0.63 |
| 57 | siPERV | PERV-UC | sPERV-UC2-13-21.3M | 13 | + | 21308535–21310732 | 2 198 | 422 | 0.78 |
| 58 | siPERV | PERV-UC | sPERV-UC3-Y-24.4M | Y | + | 24407502–24411155 | 3 654 | 644 | 0.24 |
| 59 | siPERV | PERV-UC | sPERV-UC4-Y-25.261M | Y | + | 25261084–25264796 | 3 713 | 728 | 0.16 |
We constructed a maximum likelihood (ML) tree using the 36 lcPERVs from the swine reference genome and three complete PERVs (PERV-A: AY099323.1, PERV-B: AY099324.1, and PERV-C: KY352351.1) deposited to the NCBI Genbank database (Bartosch et al., 2002). In the tree, all 19 PERV-As clustered together, all 12 PERV-Bs formed their own clade, and two PERV-Cs grouped closely together. The six newly discovered PERVs were scattered on branches between the PERV-As and -Bs; they did not cluster into any clade of PERV-A, -B, or -C, and were classified into four types named, PERV-JX1 (three copies), -JX2 (one copy), -JX3 (one copy), and -JX4 (one copy) (Figure 2B; Supplementary Figure S1). These PERVs had genetic distances > 0.04 (the largest genetic distance within each PERV type) with PERV-A, -B, -C, or each other, as well as their env genes or env homologous sequences (Supplementary Table S4 and Figures S2–S5). The PERV-JX1s were located close to the PERV-Bs, and PERV-JX2, -JX3, and -JX4 were located near the root of the ML tree. Based on their close position next to the root of the phylogenetic tree, we speculated that these newly discovered PERVs were more ancient than the well-known types, PERV-A, -B, and -C.
All 39 lcPERVs were flanked by LTRs on both sides. Among these lcPERVs, 23 PERVs were flanked by LTR-B (subfamily type, ERV1-2_SSC-LTR), including all 12 PERV-Bs, five -As, three -JX1s, one -Alike, one -JX2, and one -JX3. The remaining 16 lcPERVs were flanked by LTR-A (subfamily type, ERV1-2B_SSC-LTR), including 13 PERV-As, two -Cs, and one -JX4 (Figure 2C). A total of 247 LTRs were detected in the swine reference genome, including 117 LTRs flanked to PERVs and 130 solo LTRs (Supplementary Table S5). No PERV was detected on SSC10, but one solo LTR-A was found at 29.4 Mb on SSC10, suggesting that a PERV may have not been inserted or a PERV would have the opportunity to transpose on SSC10.
We predicted the ORF structures of the gag, pol, and env genes for all 59 PERVs identified from the swine reference genome and three complete PERVs deposited to the NCBI Genbank database (Figure 2D). The ORF structures of the three complete PERVs from the NCBI Genbank database and seven lcPERVs (PERV-A-3-10.6M, PERV-A-5-92.1M, PERV-A-17-33.1M, PERV-A-Y-25.2M, PERV-B-3-51.1M, PERV-B-16-59.6M, and PERV-B-U331-13.8kb) from the swine reference genome were complete and had not been truncated. This finding suggests that these lcPERVs with complete ORF structures had the ability to encode viral proteins. Roughly one-third (14) of the 36 lcPERVs encoded reverse transcriptase and were potentially active, as they contained a complete pol ORF. Half of the PERV copies (18) contained a complete env ORF, which encodes the most critical envelope protein for forming virus particles. The env gene of PERV-C on the reference genome was fragmented, suggesting that PERV-C does not produce viral particles (Figure 2D). The ORF structures of the newly discovered PERVs had been truncated many times and did not encode viral proteins. For example, PERV-JX1-17-41.4M, PERV-JX1-U293-0.8M, and PERV-JX3-17.8M were inserted by other transposons (Supplementary Figure S7D). These findings inferred that these newly discovered PERVs may have existed in the pig genome for a longer period of time and had more opportunities to be truncated or mutated than the known types with viral activity, PERV-A, -B, and -C.
Based on their sequence identities with the seven types of PERVs (PERV-A, -B, -C, -JX1, -JX2, -JX3, and -JX4), 23 siPERVs divided into the following eight siPERV types: siPERV-A (two copies), siPERV-B (two copies), siPERV-C (five copies), siPERV-Alike (one copy), siPERV-Clike (five copies), siPERV-JX2like (one copy), siPERV-JX3like (three copies), and siPERV-UC (four copies) (Figure 2A; Supplementary Table S2). Among the lcPERVs, PERV-As and -Bs accounted for the majority. However, among the siPERVs, siPERV-Cs and -Clikes had the greatest number of copies. Almost all the siPERV gene structures were incomplete (Figure 2D). The env structures of siPERVs were severely lost or completely disappeared, and some LTRs flanked to the siPERVs were broken or missing. For example, siPERV-C-5-29.4M had no 5' LTR, siPERV-Clike-U293-0.9M had no 3' LTR, and there was a large-fragment deletion in the LTRs of siPERV-Clike-U154-3.2kb, siPERV-C-U193like-4.0kb and siPERV-UC4-Y-25.261M. Notably, chimeric events caused by the insertion of other transposons occurred frequently in the siPERVs. Six siPERVs (siPERV-Clike-U355-19.3kb, siPERV-Clike-Y-25.269M, siPERV-JX3like-15-116.3M, siPERV-JX3like-17-9.5M, siPERV-UC3-Y-24.4M, and siPERV-UC4-Y-25.261M) were chimeric. Surprisingly, the pol gene of siPERV-JX3like-17-9.5M was inserted by a 6 302 bp transposon of LINE1 and simple (T)n repeat sequences. As a result, the length of siPERV-JX3like-17-9.5M reached 13, 104 bp and exceeded the length of a normal complete PERV.
Interestingly, we detected a region at 146.7 Mb on SSC13 that harbored three PERV copies, which was characterized by multiple insertions of different PERVs at the same genome site (Supplementary Figure S6). The sequence structure analysis suggested that PERV-A with LTR-Bs was first inserted into the position, then a subsequent insertion event of two tandem PERV-Cs with LTR-As occurred and broke the right flanking LTR of the first PERV-A.
Evolutionary history of PERVs and LTRs
The 36 lcPERVs in the reference genome were relatively complete and had a long length of consensus sequence, while the 23 siPERVs had many uneven deletions and shared few common sequences. Therefore, we estimated the evolutionary history of PERVs and LTRs using only the 36 lcPERVs in the reference genome and three complete typical PERVs deposited to the NCBI Genbank database. First, we ran Bayesian phylogeny analyses for the sequences of the 39 lcPERVs without flanking LTRs, identified as ERV1-2_SSc-I using Repeatmasker, and their flanked LTRs using BEAST (Figure 3) (Drummond & Rambaut, 2007). In the analyses, the divergence time of 96 Ma between pig and mouse, retrieved from the TimeTree database, was set as the split time between MLV-J01998.1 and PERV-A-AY099323.1 to account for the mutation rate (Kumar et al., 2017). Our results showed that the time of the most recent common ancestor (TMRCA) of all 39 PERVs was estimated to be 6.9 Ma (HPD 95% (5.96, 7.85)). Interestingly, the PERVs containing complete gene structures were usually younger than those with broken gene structures. We also found that most PERVs with full gag, pol, and env ORFs originated within the last 500 000 years (Figure 3).
Figure 3.

Evolutionary histories of 39 lcPERVs and their 5' LTRs
The evolutionary tree of lcPERVs is on the left. Yellow triangle: PERV contains full gag, pol and env ORFs; Yellow circular: the pol ORF is complete but the other ORFs are incomplete. The phylogenetic tree was estimated using 40 ERV internal sequences (including 3 PERV reference sequences and one outgroup MLV) by BEAST. We set the divergence time between MLV and PERV-A-AY099323.1 to be 96 Ma with confidence intervals from 91 to 101 Ma same as the divergence time of pig and mice in TimeTree database. The LTR tree is on the right. 5’ LTRs of 39 lcPERVs were used, and 5' LTR of MLV was used as an outgroup. The divergence time was the same as above. PERV and its corresponding LTR are connected with a solid line.
There were generally two lineages of PERVs, one containing PERV-JX3, -JX4, -C, and -As, and the other containing PERV-Bs, -JX1s, and -JX2. PERV-As clustered into two independent clades in the first PERV lineage, one where PERV-As were flanked by LTR-A and the other where PERV-As were flanked by LTR-B. The TMRCA of these two PERV-As was estimated to be 3.2 Ma (HPD 95% (2.74, 3.67)). PERV-Cs were found to belong to this PERV lineage and were close to the PERV-As flanked by LTR-A. Therefore, we speculated that PERV-C was more likely to recombine with this type of PERV-A to form the highly infectious and human-tropic PERV-A/C. PERV-JX4 diverged from the PERV-As flanked by LTR-A around 3.74 Ma (HPD 95% (3.17, 4.31)). PERV-JX3 was the most ancient and located at the root of this PERV lineage, having diverged from all PERV-As and -Cs around 5.5 Ma (HPD 95% (4.70, 6.30)). In the second PERV lineage, PERV-Bs were predominant. To our surprise, the TMRCA of PERV-Bs was very short, around 0.5 Ma, far smaller than the PERV-As in the first lineage. Accordingly, the nucleotide diversity (Pi) of the PERV-Bs was calculated as 0.0059, also much lower than the PERV-As (0.0125) (Supplementary Figure S7). These findings suggested that PERV-As and -Bs were under different selection pressures during the process of PERV evolution, and PERV-Bs possibly experienced a bottleneck around 0.5 Ma. PERV-JX1s were the closest to the PERV-Bs, which diverged around 2.0 Ma. PERV-JX2 was the most ancient PERV in the second lineage and diverged from all PERV-Bs and -JX1s around 5.4 Ma (HPD 95% (4.62, 6.24)).
In the Bayesian phylogenetic analysis of the LTRs, we used the common sequences of the 5' LTR flanking lcPERVs, as there were more deletions in the 3' LTR than 5' LTR of the 36 lcPERVs. The divergence time of 96 Ma between pig and mouse was also set as the split time between the LTRs of MLV-J01998.1 and PERV-A-AY099323.1. We found that the TMRCA of the LTR-As and -Bs was approximately 24 Ma, far larger than the TMRCA of all PERVs (6.9 Ma). The TMRCA of the LTR-As was roughly 8.7 and 8.0 Ma for the LTR-Bs (Figure 3). This deep divergence suggested that there were significant genetic differences and independent evolutionary histories between these two LTRs.
We compared the phylogenetic topologies of the PERVs and their related LTRs. Generally, PERV-As corresponded to two subfamilies of their own LTRs, while PERV-Bs, -Cs, -JX1s, and -JX2 matched with their own LTRs. However, when looking at the details within a specific local tree, the corresponding relationships between PERVs and their LTRs were not good, no matter whether the PERVs were young or ancient. For example, the phylogenetic tree of the PERV-Bs did not match well with their LTRs; if we focused on the phylogenetic tree of the newly identified PERVs (PERV-JX1s, -JX2, -JX3, and -JX4), we could observe that PERV-JX3 was farther away from PERV-JX2, while their LTRs were much closer to each other in the tree (Figure 3).
PERV and LTR Copy number variations in Eurasian pigs
We used two different methods (see Materials and Methods) to calculate the PERV and LTR copy numbers in 10 Eurasian breeds (five Chinese and five Western pig breeds), including six South CNWB, five EUWB, six BMX , eight EHL, six LWU, six TB, six DRC, eight WD, seven LR, and five LW pigs (Supplementary Table S3). The correlation coefficient of the PERV copy numbers estimated via the two methods was 0.993 and one of the LTR copy numbers estimated via the two methods was 0.975, indicating that both methods were robust and the estimated PERV and LTR copy numbers were reliable (Supplementary Figure S8). Due to the high correlation between the two methods, we present the results of method 1 only. Generally, Chinese pigs had a lower average PERV copy number (32.0±4.0) than Western pigs (49.1±6.5) (Figure 4; Table 2; Supplementary Table S6). Specifically, the PERV copy number in LR was 52–63, 41–54 in LW, 36–51 in DRC, 45–57 in WD, 39–50 in EUWB, 30–39 in EHL, 30–36 in TB, 23–37 in BMX, 31–38 in LWU, and 27–35 in CNWB. The average PERV copy number in LR was 56, the highest of the European pigs, while EUWB had the lowest average number of 43. The average PERV copy number (34) in LWU was the highest of the Chinese pigs. Notably, among the investigated Chinese pig breeds, BMX had the lowest average PERV copy number of 27.8, indicating that BMX pigs were the most ideal pig donors for pig-to-human xenotransplantation. Additionally, we found that there was a significant difference between domestic and wild pigs (P<0.05), and both EUWB and CNWB had a lower PERV copy number than European and Chinese domestic pigs, respectively.
Figure 4.
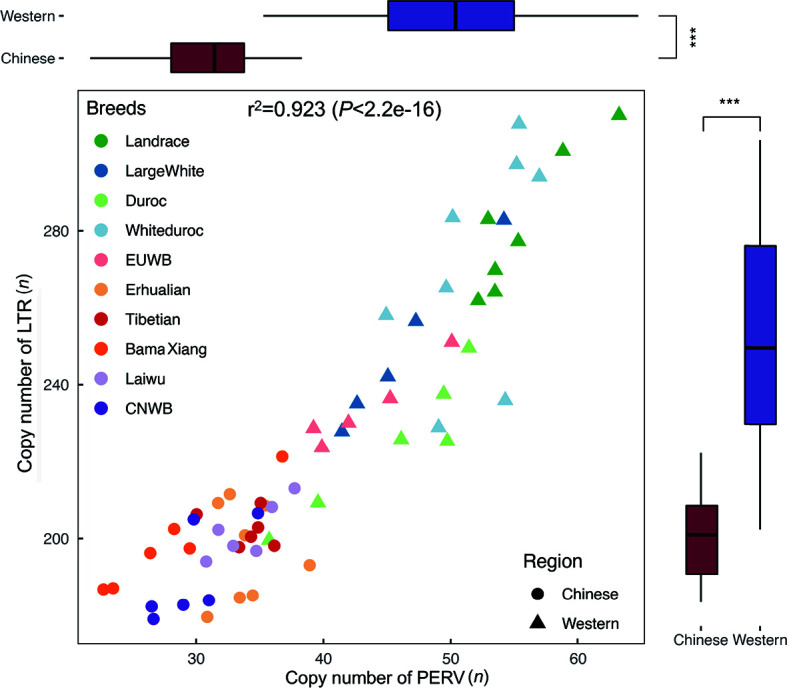
Correlation between PERV copy number and LTR copy number measured by method 1 (mapping-to-genome method)
The correlation between the copy number of PERV and LTR in ten pig breeds was shown in the scatter plot. The box plot at the top shows the difference of PERV copy number between Chinese and Western pig breeds, and the box plot at the right shows the difference of LTR copy number between Chinese and Western pig breeds. ***: P<0.0001.
Table 2. Average PERV and LTR copy numbers in ten pig breeds.
| Pig breed | Average PERV copy No. (n, Method 1) | Average PERV copy No. (n, Method 2) | Average LTR copy No. (n, Method 1) | Average LTR copy No. (n, Method 2) |
| 1: P-value indicates the comparison result of PERV/LTR copy numbers between Chinese and Western pig breeds by student’s t-test. Number in bold indicates the minimum copy number detected by this method. | ||||
| Western pigs | 49.1±6.5 | 48.2±6.3 | 254.8±30.0 | 204.7±26.7 |
| Duroc | 45.3±6.3 | 44.0±6.2 | 224.5±18.2 | 172.6±16.8 |
| European
wild boar |
43.3±4.5 | 42.5±4.5 | 234.0±10.6 | 192.0±8.8 |
| Landrace | 55.6±4.0 | 54.6±4.0 | 280.9±18.4 | 230.4±16.1 |
| Large White | 46.1±5.0 | 45.7±4.2 | 248.8±21.7 | 197.0±15.0 |
| White Duroc | 51.9±4.1 | 50.7±4.0 | 271.3±29.1 | 219.1±23.0 |
| Chinese pigs | 32.0±4.0 | 32.0±4.2 | 197.8±10.9 | 160.4±13.6 |
| Bama Xiang | 27.8±5.1 | 28.5±5.4 | 198.5±12.7 | 164.0±13.6 |
| Chinese
wild boar |
29.6±3.1 | 29.6±3.5 | 189.9±12.4 | 150.7±18.2 |
| Erhualian | 33.9±2.5 | 33.8±2.5 | 196.5±12.6 | 159.9±11.8 |
| Laiwu | 34.0±2.6 | 33.9±3.4 | 202.0±7.3 | 163.6±11.3 |
| Tibetian | 34.0±2.1 | 34.1±3.4 | 202.4±4.6 | 163.9±12.3 |
| P-value1 | 6.63×10-12 | 7.90×10-11 | 1.01×10-07 | 3.64×10-06 |
In swine genomes, LTRs have many more copies than PERVs. In European pigs, the average LTR copy number was 224 in DRC, 234 in EUWB, 281 in LR, 249 in LW, and 271 in WD. In Chinese pigs, the average LTR copy numbers of BMX, CNWB, EHL, LWU, and TB were 199, 190, 197, 202, and 202, respectively. The LTR copy number of CNWB was the lowest. These results indicated that Chinese pigs had a lower likelihood of PERV insertion. Interestingly, we found that the LTR copy numbers in Eurasian pigs highly correlated with the PERV copy numbers (Figure 4), suggesting that there was a high degree of coevolution between PERVs and LTRs in Eurasian pigs.
Inferred PERV types in Eurasian pigs based on unique k-mers
We developed a pipeline (see Materials and Methods) to infer the PERV types in 63 Eurasian individuals based on unique k-mers (k=31) of the 59 identified PERV sequences in the reference genome. When the determination threshold of the unique k-mer rate was 18%, the correlation between the PERV copy numbers estimated by the pipeline and method 1 was the largest (r2=0.87, P<2.2×10-16). Therefore, we used the determination threshold of 18% for PERV inferring in Eurasian pigs.
Information regarding the unique k-mers of the 59 known PERVs is presented in Table 1 and Supplementary Table S7. Among these PERVs, one siPERV (siPERV-Clike-Y-25.29 M) had no unique k-mer and could not be inferred in Eurasian pigs. Another siPERV (siPERV-C-13-146.76M) contained 34 unique k-mers, but none of these unique k-mers were detected in the 63 Eurasian individuals, suggesting that siPERV-C-13-146.76M was the unique PERV in the pig reference genome that originated from a DRC female pig. Three PERVs, PERV-Alike-U965-14.7kb, siPERV-Alike-U293-0.9M, and siPERV-Clike-U154-3.2kb, whose unique k-mers were partially detected in Eurasian pigs, did not exceed the 18% threshold. This result suggested that these three PERVs were not detected, but similar types possibly existed in the 63 Eurasian pigs.
Of the 59 known PERVs, 54 PERV types were inferred in the tested Eurasian pigs. In summary, the siPERVs accounted for 34% of the total in Eurasian pigs and lcPERVs accounted for 66%. Among the lcPERVs, the three known types, PERV-A, -B, and -C, accounted for 50% and the newly identified PERVs, PERV-JX1, -JX2, -JX3, and -JX4, accounted for 16%. Among the well-known types, PERV-A accounted for 28%, PERV-B accounted for 20%, and PERV-C accounted for 2% (Figure 5A). The copies of PERV-A and -B made up the majority of the total PERV copies in each pig. In European pigs, the PERV-A copy number was the largest and accounted for 30.9%, while PERV-B accounted for 17.2%. In Chinese pigs, the PERV-A copy number (23.7%) was roughly the same as PERV-B (24.1%). There were significant differences between Chinese and European pigs in the copy numbers of PERV-A, PERV-JX2, siPERV-B, siPERV-C, siPERV-A, siPERV-JX3like, and siPERV-UC (P<0.0001) (Figure 5B, C). A total of 49 individuals contained lcPERV-C (Supplementary Table S8). The high frequency of PERV-C occurrence indicated that the problem of PERV infection in pig xenotransplantation should be a research focus. All newly discovered PERVs, except PERV-JX2, were detected in each Chinese and Western pig (Figure 2B). The number of unique k-mers of PERV-JX2 in Western pigs passed the determination threshold. In all Chinese pigs, although the number of unique k-mers of PERV-JX2 was >100, it was still below the unique k-mer rate determination threshold. This result suggested that a PERV similar to PERV-JX2 possibly exists in Chinese pigs. With the exception of PERV-JX2, other PERV-JXs were detected in all Eurasian individuals, indicating that they were inserted in the genome of pigs before European and Asian pigs diverged. Additionally, the insertions of PERV-A, -B, and -C occurred continuously until relatively recently. Ancient PERV-JXs may have experienced more insertion/deletion or recombination events, which could explain why their gene structure was incomplete. This investigation of these PERV structures enhanced our understanding of PERV evolution.
Figure 5.

PERV copy number and PERV types in 63 Chinese and Western pigs
A: The percentage of different PERV types in 63 Chinese and Western pigs. B: Copy number difference of each PERV type between Chinese and Western pig breeds. ***: P<0.0001. C: Heatmap of PERVs with a significant difference in copy number between Chinese and Western pig breeds.
We counted the detection rate of the 59 identified PERVs in each Eurasian individual. The PERV detection rate in WD and LR were 80% and 73.4%, respectively, which were the highest among all Eurasian pig breeds, followed by DRC, LW, and EUWB with detection rates of 72.9%, 67.2%, and 64.4%, respectively. Chinese pig breeds had much lower detection rates of the identified PERVs. The largest PERV detection rate (54.2%) was in LWU and the smallest (42.9%) was in CNWB.
PERV sequence diversity in Eurasian pigs
PERV is polymorphic not only in copy number but also in internal sequence. For example, PERVs at the same position may differ in their sequence among different individuals, like PERV-JX2 mentioned above. Therefore, we evaluated the diversity of PERV sequences in Eurasian pigs by comparing the total number of k-mer (k=31) types of PERVs in Chinese and Western pig breeds. The results showed that the diversity of PERV sequences in Western pigs was higher than Chinese pigs (Figure 6A). We further analyzed the diversity of PERV sequences in Chinese and Western domesticated breeds and found that the diversity was higher in European domestic pigs (Figure 6A). In contrast, the PERV sequence diversity of CNWB was higher than EUWB (Figure 6A). In each dataset, the common k-mer of Chinese and European pig breeds accounted for only ~1/3 and each of their unique k-mers accounted for ~1/3 as well. The low number of shared k-mers in Eurasian pigs indicated that the PERV sequences in Chinese and European pig genomes varied after divergence.
Figure 6.

Differences in PERV sequence diversity, copy number of PERVs with transposable potentiality and TIPs between Chinese and Western pig breeds
A: Sequence diversity of PERVs without LTRs in Eurasian pig breeds. The number of k-mer types in the reads aligned to PERVs without LTRs can reflect the sequence diversity of PERVs. The left venn diagram shows the numbers of specific k-mer types of PERVs not shared and common k-mer types of PERVs shared between all Chinese and Western pigs; the middle one shows the relation between Chinese and Western domestic pigs; the right one shows the relation between Chinese and Western wild boars. B: The percentage of PERVs with transposable potentiality in 63 Chinese and Western pigs. The above bar plot with pink background shows the percentage of seven PERVs with complete gag, pol, and env ORF in all Chinese (32) and Western (31) pigs. The bar plot below with green background shows the percentage of four PERVs with incomplete gene structure but with complete pol ORF in all Chinese and Western pigs. C: Total copy number of eleven PERVs with transposition potentialities in different pig breeds. D: PERV TIPs in ten Chinese and Western pig breeds. The left horizontal bar plot shows the total number of PERV TIPs in each pig breed. The top bar plot shows the number of PERV TIPs for each intersection. Only 86 all breed-common, 6 Chinese pig breed-common, 6 Western pig breed-common, and 150 breed-specific TIP loci are showed.
We further calculated the intersections of PERV k-mers (k=31) in each breed and 59 PERV k-mers (k=31) from the reference genome (Supplementary Figure S9). The results showed that there were more shared PERV k-mers between Western pigs and the reference genome than Chinese pigs. When the PERV k-mer pool of the 59 PERVs intersected with the PERV k-mer pool of Chinese pig breeds, the number of PERV unique k-mers was greater than Western pigs. Eurasian pigs exhibited a different PERV landscape, revealing that the PERV copy numbers and locations were related to their unique lineages and habitation environments.
Copy number of PERVs with transposable potentiality in Eurasian pigs
Among the 59 PERVs identified in the pig reference genome, 11 PERVs (six PERV-As and five PERV-Bs) were a research focus as they contained at least one complete pol ORF, suggesting that they have potential transposition activity. Of the 11 PERVs, seven had the potential to form virus particles due to the integrity of their gag, pol, and env ORFs. These seven PERVs were PERV-A-3-10.6M, PERV-A-5-92.1M, PERV-A-17-33.1M, PERV-A-Y-25.2M, PERV-B-3-51.1M, PERV-B-16-59.6M, and PERV-B-U331-13.8kb. The four PERVs with a complete pol ORF only were PERV-A-15-66.8M, PERV-A-13-146.7M, PERV-B-4-45.5M, and PERV-B-9-138.8M.
We investigated the distribution of these PERVs with potential transposition activity in Chinese and Western pigs (Figure 6B; Supplementary Table S6). Among the six PERV-As, one PERV (PERV-A-13-146.7M) existed in all Chinese and Western pigs; the other five PERV-As more frequently occurred in Western pigs than Chinese pigs. For the five PERV-Bs, the occurrence frequency in Chinese pigs was similar to Western pigs. The average numbers of these 11 PERVs with potential transposition activity were 9.9 in WD, 9.0 in DRC, 8.7 in LR, 8.0 in LW, 5.8 in EUWB, 5.9 in EHL, 5.7 in LWU, 5.7 in TB, 5.2 in BMX, and 4.7 in CNWB (Figure 6C). This result showed that the number of PERVs with potential transposition activity was smaller in Chinese pigs than Western pigs and the potential risk of PERV transposition was lower in Chinese pigs than Western pigs.
TIPs of PERV
We used two methods to estimate PERV copy numbers; however, neither method could obtain accurate positions of the PERVs in the reference genome. Therefore, we developed a new method based on a previously described approach to detect PERV transposon insertion sites (Carpentier et al., 2019). To improve its accuracy, we modified the approach (see Materials and Methods) by narrowing the read alignment range for one TIP to 2 kb instead of 10 kb. Reads located within 2 kb downstream of the start position of any read alignment were determined to support the same site as the first reads. The advantage of this modification was that every TIP could be identified when there was more than one potential PERV insertion site within a 10 kb range.
In total, we identified 451 PERV TIP loci (Supplementary Table S9), among which 86 were shared by all 10 Chinese and Western pig breeds. A total of 116 loci were unique to Chinese pig breeds, six of which were shared by all Chinese pig breeds. A total of 131 loci were found only in Western pig breeds, among which all Western pig breeds shared six of these loci (Figure 6D). The positions of all TIPs in the genome are shown in Supplementary Figure S10. Overall, the average number of PERV TIPs in Western pigs (n=208) was larger than Chinese pigs (n=183). Among them, WD had the largest number of PERV TIPs with a total of 242 TIPs and EHL had the least with a total of 171 TIPs, which were detected in eight individuals.
According to the overlap of TIP loci and genes, we annotated 248 TIP loci, including 86 all breed-common, six Chinese pig breed-common, six Western pig breed-common, and 150 breed-specific. We found 99 genes that overlapped with these TIPs (Supplementary Table S10), suggesting that these genes may be affected by PERV transpositions. A total of 30 genes overlapped with the common TIPs shared by all 10 Eurasian breeds, of which 29 overlapped with the PERVs or solo LTRs in the reference genome. Two genes overlapped with the Chinese pig breed-common TIPs, which were shared by all five Chinese breeds, but were not detected in any Western breed. Notably, the specific TIPs overlapped with genes that varied in each breed. There were 14, 8, 7, 5, and 1 breed-specific TIPs that potentially affected genes in CNWB, TB, BMX, EHL, and LWU, respectively. Among the PERV TIPs shared by all Western pig breeds, but not Chinese breeds, there were two TIPs that overlapped with genes, which corresponded to a solo LTR and a PERV-A (PERV-A-13-146.7M). There were 13, 7, 6, 2, and 1 genes that were found to be potentially affected by the TIPs specific to LR, EUWB, WD, DRC, and LW, respectively. Most of the proteins encoded by these genes belong to transmembrane signal receptors, ion channels, and other families (Supplementary Table S10), which play essential roles in maintaining normal bodily life activities.
Additionally, we used a total of 30 genes that overlapped with the 86 common TIPs shared by all 10 breeds to perform GO and KEGG pathway enrichment analyses using ClueGO (Bindea et al., 2009). No significant KEGG pathways were enriched. A total of 11 GO pathways were predominantly enriched in ruffle, growth cone, ion gated channel activity, regulation of synapse organization, mitotic sister chromatid segregation, positive regulation of synapse assembly, sodium ion transport, transcription by RNA polymerase I, and isomerase activity (Supplementary Table S11). The ClueGO enrichment results indicated that PERV TIPs may contribute to the progression of morphology and biological function.
DISCUSSION
The pig reference genome has been dramatically improved due to the continuous development of sequencing technologies and improvement of genome assembly methods (Groenen et al., 2012; Warr et al., 2020). Currently, the swine reference genome (assembly Build 11.1) is the best and most commonly used genome. Here, we scanned this high-quality pig reference genome to identify PERVs by BLAT searching and obtained a total of 59 PERVs. Based on their sequence similarity and phylogenetic topology, we systematically classified these PERVs. Among the 59 PERVs, 36 were long and complete or near-complete and 23 were short and incomplete. We classified the 36 lcPERVs into eight types, including three well-known PERVs (PERV-A, -B, and -C), one PERV-Alike, and four novel PERVs (named PERV-JX1, -JX2, -JX3, and -JX4). The 23 siPERVs were also classified into eight types: siPERV-A, -B, -C, -Alike, -Clike, -JX2like, -JX3like, and -UC. These siPERVs were believed to have originated from complete PERVs and formed by recombination and insertion/deletion during the pig evolution process. It is a common phenomenon that incomplete proviruses exist in host genomes. For example, the incomplete provirus of human endogenous retrovirus, HC2, exists in humans and the incomplete endogenous mammary tumor virus exists in mice (Kabát et al., 1996; Kozak, 1984).
Among the lcPERVs, PERV-As and -Bs were predominant. PERV-C and the newly identified PERVs had fewer copies, most of which had only one copy detected in the reference genome. However, among the siPERVs, siPERV-C and -Clike had the most copies, indicating that PERV-Cs or -Clikes had been predominant, but were now destructed or fragmented in modern pig genomes. Previously, according to its intermediate position in the phylogenetic tree, a new PERV lineage close to PERV-A and -C was reported and named, PERV-IM (Chen et al., 2020b). Based on their locations in the reference genome, we determined that these PERV-IMs corresponded to PERV-JX3 and siPERV-JX3like. Therefore, we speculated that the newly identified PERVs (PERV-JX1, -JX2, -JX3, and -JX4) were more ancient than PERV-A, -B, or -C due to their closer positions to the root (Figures 2B, 3). And most of PERV-JXs were detected in all Eurasian pigs (Figure 2B). Identification of these new PERVs will enhance our understanding of PERV evolution.
In the present study, we scanned one high-quality genome. This genome mainly originated from a DRC pig belonging to a European domestic pig breed, which cannot totally represent Asian pig breeds, wild boars, or other Suids. Therefore, we cannot rule out the possibility that more novel PERV types could be identified in the genomes of Suidae animals. In the future, more high-quality pig genomes should be investigated to potentially observe more PERV types.
Moreover, a strict molecular clock model (GTR + gamma nucleotide substitution) was employed to estimate the divergence time of PERVs and their LTRs using BEAST in this study (Drummond & Rambaut, 2007). The analyses were calibrated by setting normally distributed priors as the time of the most recent common ancestor of pig and mouse, retrieved from the TimeTree database. This correction method of the molecular clock by using the divergence time between two species with distant relationships has been widely used in the evolutionary inference of family genes (Matos et al., 2021; Premachandra et al., 2017). Here, the TMRCA of PERVs was estimated to be 6.9 Ma, which was similar to previous estimations of 6.6 and 7.6 Ma (Chen et al., 2020b; Tönjes & Niebert, 2003). A previous study about PERV evolutionary spread in Suiformes showed that PERVs were completely absent in Pecari tajacu and Babyrousa babyrussa samples, both being the most distantly related to modern pigs, and that the first PERV was detected in Phacochorus africanus originated from the late Miocene epoch (3.5–7.5 Ma) (Niebert & Tönjes, 2005). Our TMRCA estimation of the PERVs was consistent with these previous findings.
The common ancestor of the PERV-As and -Bs was traced back to 6.9 Ma, but the TMRCA of the PERV-As was estimated to be 3.2 and 0.5 Ma for the PERV-Bs, both of which were less than previous estimates of the earliest PERV-A and -B origin times (6.6 and 6.4 Ma) (Chen et al., 2020b; Tönjes & Niebert, 2003). These findings could be explained by the fact that we applied a time estimation method that differed from previous studies and filtered out siPERVs with sequence lengths <7 000 bp. As for PERV-C, we found that it split from its closest relative, PERV-JX4, a newly discovered PERV, around 3.7 Ma, which was roughly consistent with the previous prediction of 1.5–3.5 Ma ( Chen et al., 2020b; Niebert & Tönjes, 2005).
The branch points of the newly discovered PERVs (PERV-JX2 and -JX3) were closer to the root of the phylogenetic tree than the well-known PERVs (PERV-A, -B, and -C), indicating that these newly discovered PERVs, especially PERV-JX2 and -JX3, were more ancient or appeared earlier than PERV-A, -B, or -C. Additionally, among the newly discovered PERVs, PERV-JX4 was more similar to PERV-C, while the PERV-JX1s were the closest to the PERV-Bs. PERV-JX4 and -C converged into one branch around 3.7 Ma and the PERV-JX1s and -Bs converged into one branch around 2.9 Ma.
From the LTR phylogenetic tree, two LTR lineages were clearly observed, corresponding to the LTR-As and -Bs. According to our estimates, their common ancestor was traced back to 22.1 Ma. Our estimated TMRCA of the LTR-As was 6.5 Ma, which was similar to a previous origin time estimated for the LTR-As of PERV-A (6.6 Ma) (Chen et al., 2020b). However, the TMRCA of the LTR-Bs was estimated to be 11.8 Mya, far earlier than the previous origin time (6.4 Ma) (Chen et al., 2020b). This result was mainly due to the distant relationship between the LTR-Bs flanked to PERV-Cs and other LTR-Bs flanked to PERV-As. A previous study suggested that PERV-C originated from the recombination of PERV-A and an unknown ancestor (Niebert & Tönjes, 2005). According to our present LTR evolutionary tree, we hypothesized that the LTRs flanked to PERV-Cs were likely inherited from this unknown ancestor.
When the LTR-As flanking PERV-Cs were excluded, the common ancestor time of the LTR-As was similar to their corresponding PERVs, including PERV-JX4 and the PERV-As flanked by LTR-As, and both of their common ancestors were traced back to 4.2–4.5 Ma. The common ancestral time of the PERV-As flanked by LTR-As was similar to their corresponding LTR-As, which was around 2.0 Ma. The common ancestral time of the PERV-As flanked by LTR-Bs was similar to their corresponding LTR-Bs, which was about 3.1 Ma. Similarly, the common ancestor time of the LTR-Bs was 6.6 Ma, which was close to their corresponding PERVs (6.9 Ma), including PERV-Bs, -JX1s, -JX3s, and -As flanked by LTR-Bs. These relationships between PERVs and LTRs suggested that our estimation method for inferring the history of PERVs and LTRs was reliable and stable, and that PERV-As and their corresponding LTRs were in a coevolutionary state. Additionally, we found that the common ancestor time of the PERV-Bs was about half of their LTRs and the common ancestor time of PERV-JX1 was also about half of their LTRs. Thus, we speculated that the PERV-Bs may have experienced a bottleneck event due to their significantly lower Pi when compared to their LTRs and PERV-As. However, the underlying reason requires further investigation.
Previous studies have reported the PERV copy numbers in different pig breeds or one breed of different animals (Chen et al., 2020b; Denner, 2016b; Garkavenko et al., 2008; Groenen et al., 2012; Krüger et al., 2020; Le Tissier et al., 1997; Lee et al., 2002, 2011; Lian et al., 2002; Liu et al., 2011; Mang et al., 2001; Patience et al., 1997, 2001; Quereda et al., 2012; Yang et al., 2015; Yoon et al., 2015). However, the PERV copy number of the same pig breed varies considerably between studies, even within the same study. For example, Liu et al. (2011) reported PERV copy numbers ranging from 4–96 in Chinese experimental miniature pigs. Moreover, when different tissues were used, the extracted DNA lead to different results (Sypniewski et al., 2005; Zhang et al., 2010). Additionally, technical factors in the experiments, such as different detection methods, the DNA extraction method, DNA purity, PCR sensitivity, possible pollution, and artificial error, may lead to different results. Genome-wide sequencing detection could significantly reduce deviations in PERV copy number estimations caused by technical factors. Here, we developed several methods based on whole-genome resequencing data to identify different PERV types, as well as estimate and compare their copy numbers in 10 Eurasian pig breeds.
There are no reports on the PERV copy number in CNWB, but data on German wild boars have been recently reported (Krüger et al., 2020). Previous PERV copy number comparisons of Eurasian pig breeds were mainly between European and non-Chinese breeds (Lee et al., 2011). To our knowledge, this is the first study where comparisons of the PERV copy number between Western and Chinese pigs, including CNWB, domestic pigs, and wild boars, were conducted. Results revealed that Chinese pigs had a lower PERV copy number (32.0±4.0) than Western pigs (49.1±6.5). Additionally, we found that the PERV copy number of both CNWB and EUWB was lower than that of domestic pigs.
Few studies have been conducted on the PERV copy number in Chinese local pig breeds. In 2002, Lian et al. reported the PERV copy number of 11 Chinese local pig breeds, including EHL, BMX, and LWU (Lian et al., 2002). Among them, the PERV copy number of BMX was the lowest, which was consistent with our results. In this study, the average PERV copy number in BMX pigs was only 27.8. The lowest PERV copy number in BMX pigs was only 23, less than half of the copy number of PERVs in the reference genome. BMX has the characteristic of high inbreeding endurance (Zhang et al., 2018). Inbreeding would not lead to increased PERV copy numbers and may even decrease the number of PERV copies (Lian et al., 2002; Quereda et al., 2012), which could explain the low number of PERV copies in BMX pigs.
Our research showed that Chinese pigs had the advantage of low PERV copy numbers compared to Western pigs. However, the PERV copy number is vital for their use in xenotransplantation, as well as the number of PERVs with the potential to release infectious human-tropic virus. Although the presence of PERV provirus did not necessarily translate to a functional virus, PERVs with complete gag, pol, and env ORFs were more likely to produce virus particles. We found that the copy number of this type of PERV with transposition potentiality in Chinese pigs was lower than Western pigs. In our study, BMX had the lowest PERV copy number among the 10 Western and Chinese pig breeds. The copy numbers of PERVs with the potential to release infectious human-tropic virus in CNWB (4.7) and BMX (5.2) were the lowest. From the perspective of reducing PERV infection risk, our results suggested that the most suitable pig breed for xenotransplantation donors was BMX. Furthermore, Southern CNWB was identified as a possible xenotransplantation donor for the first time.
Several methods and software designed for copy number variations and TIPs are currently available (Fan et al., 2014; Hénaff et al., 2015; Lanciano & Cristofari, 2020). However, mapping sequencing reads to a reference genome or database is first required, which requires many computing resources and, accordingly, the computation time for large datasets is too long. Our new method was able to quickly detect PERV types with second-generation sequencing data, as the first mapping step was not required. For a 20× sample, it took ~1.5 hours of calculation by a single thread to obtain the results. We used the number of k-mers specific to PERVs in different individuals to determine the support rate of 59 PERV copies in 10 Chinese and Western pig breeds. Interestingly, we found that some PERVs located on the Y chromosome had specific k-mers in sows, indicating that the same PERV copy was not distributed at the same location across pig genomes. Thus, the PERV copy located on the Y chromosome could be distributed on other chromosomes in sows. Therefore, some PERV-derived reads were mapped at multiple positions in the genome, but their actual original positions could not be determined. By using the k-mer based method in this study, we determined whether the same copies as the reference sequences existed. However, our method was unable to detect sequences not available in the references or sequences without unique k-mers, nor could it detect their locations in the reference genome.
In order to avoid sequencing error effects on specific k-mer detection, we changed the thresholds of specific k-mers when predicting support rates of 59 PERVs in 10 breeds. We found that 18% was the lowest threshold that could guarantee the reliability of PERV prediction. When the threshold of 18% was applied to identify a PERV with several specific k-mers, we could not determine if an individual contained the PERV. For example, the individual that contained >50 specific k-mers of a PERV still could not reach the above threshold. Thus, we hypothesized that the PERV copy did exist in the individual in the above example for a period of time, but mutations, like structural variation, resulted in a new PERV that was only partially consistent with the original PERV. For instance, every European pig contained >1 300 unique k-mers of PERV-JX2-X-71.4M, while in all investigated Chinese pigs, the unique k-mer number was only ~100, indicating that PERV-JX2-X-71.4M in Chinese pigs mutated following the divergence from Eurasian pigs. Other PERVs, such as PERV-A-15-66.8M, siPERV-C-81.5M, and siPERV-JX3like-72.2M, also varied in their unique k-mer content between Chinese and Western pig breeds (Supplementary Figure S11). Additionally, we found that the unique k-mer number of siPERV-A-X-55.1M and siPERV-Clike-U355-19.3kb in southern Chinese pigs (BMX and CNWB) was significantly lower than Eastern and Northern Chinese pigs and European pigs. One possible explanation was that these two PERVs were associated with the climate. Specifically, the warm climate in Southern China corresponded to the type of PERV in BMX and CNWB. It has been reported that the environment affects retrotransposon activity ( Grandbastien, 1998; Morales et al., 2003). However, this phenomenon in pigs should be verified with more samples.
High support rate lcPERVs were closer to the root of the phylogenetic tree, such as PERV-JXs, PERV-A-13-142.0M, PERV-A-17-33.1M, and PERV-B-U331-13.8kb, and were retained by all Eurasian pig breeds (the PERV detection rate in 63 pigs was 100%). PERV-A-U136-0.5M, PERV-B-3-51.1M, PERV-B-11-38.2M, PERV-B-U331-13.8kb, and PERV-A-8-51.6M were detected in >85% of Eurasian pigs. Among these PERVs, PERV-A-17-33.1M, all gene ORFs of PERV-B-3-51.1M, and PERV-B-U331-13.8kb were complete. The pol and env ORFs of PERV-A-13-146.7M were complete and the env ORF of PERV-A-13-142.0M and PERV-A-8-51.6M was complete. Expression of env proteins in mammals, including human, mouse, cattle, sheep, cats, and dogs, allows for the generation of multinuclear syncytiotrophoblasts in the placenta as an outer cellular layer by the fusion of trophoblast cells (Denner, 2016a). The physiological function of PERV env remains unknown. Multi env-complete PERVs widely exist in different pig genomes. Thus, we speculated that PERV may also play a role in the development of pig placenta. This study excavated several shared env-complete PERVs in the genome of 10 Eurasian pig breeds from different regions, including wild boars, laying a foundation for follow-up studies on the PERV env function.
A total of 99 PERV TIPs were inserted in the genes and some genes were enriched in primary biological pathways. Therefore, PERVs could possibly influence these genes. Additionally, among the TIPs shared by all breeds, the genomic positions corresponding to 24 TIPs were annotated as PERV solo LTRs. Previous studies identified TEs with various regulatory functions (Cohen et al., 2009; Kunarso et al., 2010; Wang et al., 2007). These PERV solo LTRs fixed in different pig breeds may be the best materials for studying the function of PERVs and/or LTRs.
In summary, we identified four novel PERV types: PERV-JX1, -JX2, -JX3, and -JX4. Phylogenetic and evolutionary analyses indicated that these newly discovered PERVs were more ancient than the known PERV types. The TMRCA of PERVs was 6.9 Ma, far smaller than the LTRs that flank the sides of PERVs (22.1 Ma). We analyzed the differences in PERVs between Chinese and Western pigs, including copy number variation, type variation, sequence diversity polymorphism, and insertion position polymorphism. We found that Chinese pigs had a lower copy number (32.0±4.0) than Western pigs (49.1±6.5), and lower PERV sequence diversity than Western pigs. From the perspective of reducing PERV infection risk, our results suggested that the most suitable pig breed as a xenotransplantation donor was BMX. However, this conclusion should be verified by including more pig breeds, especially Chinese pig breeds.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
AUTHORS’ CONTRIBUTIONS
Conceptualization, L.S.H and H.S.A; formal analysis and experiment, J.Q.C; investigation, J.Q.C, M.P.Z, X.K.T, J.Q.L, Z.Z, F.H, H.P.D and M.Z.; methodology, J.Q.C, M.P.Z and H.S.A; resources, L.S.H; visualization, J.Q.C and M.P.Z; writing-original draft, J.Q.C; writing-review & editing, L.S.H and H.S.A. All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
The authors would like to thank colleagues in the State Key Laboratory for Swine Genetic Improvement and Production Technology, Jiangxi Agricultural University for their help in collecting samples and discussion during analysis.
Funding Statement
This study was supported by the National Swine Industry and Technology System of China (nycytx-009), and National Natural Science Foundation of China (31672383)
Contributor Information
Hua-Shui Ai, Email: aihsh@hotmail.com.
Lu-Sheng Huang, Email: lushenghuang@hotmail.com.
References
- 1.Ai HS, Fang XD, Yang B, Huang ZY, Chen H, Mao LK, et al Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing. Nature Genetics. 2015;47(3):217–225. doi: 10.1038/ng.3199. [DOI] [PubMed] [Google Scholar]
- 2.Ai HS, Zhang MP, Yang B, Goldberg A, Li WB, Ma JW, et al Human-mediated admixture and selection shape the diversity on the modern swine (Sus scrofa) Y chromosomes . Molecular Biology and Evolution. 2021;38(11):5051–5065. doi: 10.1093/molbev/msab230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartosch B, Stefanidis D, Myers R, Weiss R, Patience C, Takeuchi Y Evidence and consequence of porcine endogenous retrovirus recombination. Journal of Virology. 2004;78(24):13880–13890. doi: 10.1128/JVI.78.24.13880-13890.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartosch B, Weiss RA, Takeuchi Y PCR-based cloning and immunocytological titration of infectious porcine endogenous retrovirus subgroup A and B. Journal of General Virology. 2002;83(9):2231–2240. doi: 10.1099/0022-1317-83-9-2231. [DOI] [PubMed] [Google Scholar]
- 5.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, et al ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carpentier MC, Manfroi E, Wei FJ, Wu HP, Lasserre E, Llauro C, et al Retrotranspositional landscape of Asian rice revealed by 3000 genomes. Nature Communication. 2019;10(1):24. doi: 10.1038/s41467-018-07974-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen H, Huang M, Yang B, Wu ZP, Deng Z, Hou Y, et al Introgression of Eastern Chinese and Southern Chinese haplotypes contributes to the improvement of fertility and immunity in European modern pigs. Gigascience. 2020a;9(3):giaa014. doi: 10.1093/gigascience/giaa014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen N Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics. 2004;5(1):4.10.1–4.10.14. doi: 10.1002/0471250953.bi0410s25. [DOI] [PubMed] [Google Scholar]
- 9.Chen YC, Chen MY, Duan XY, Cui J Ancient origin and complex evolution of porcine endogenous retroviruses. Biosafety and Health. 2020b;2(3):142–151. doi: 10.1016/j.bsheal.2020.03.003. [DOI] [Google Scholar]
- 10.Cohen CJ, Lock WM, Mager DL Endogenous retroviral LTRs as promoters for human genes: a critical assessment. Gene. 2009;448(2):105–114. doi: 10.1016/j.gene.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 11.Cooper DKC Clinical xenotransplantion—how close are we? The Lancet. 2003;362(9383):557–559. doi: 10.1016/S0140-6736(03)14118-9. [DOI] [PubMed] [Google Scholar]
- 12.Denner J Recombinant porcine endogenous retroviruses (PERV-A/C): a new risk for xenotransplantation? Archives of Virology. 2008;153(8):1421–1426. doi: 10.1007/s00705-008-0141-7. [DOI] [PubMed] [Google Scholar]
- 13.Denner J Expression and function of endogenous retroviruses in the placenta. APMIS. 2016a;124(1-2):31–43. doi: 10.1111/apm.12474. [DOI] [PubMed] [Google Scholar]
- 14.Denner J How active are porcine endogenous retroviruses (PERVs)? Viruses. 2016b;8(8):215. doi: 10.3390/v8080215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denner J Why was PERV not transmitted during preclinical and clinical xenotransplantation trials and after inoculation of animals? Retrovirology. 2018;15(1):28. doi: 10.1186/s12977-018-0411-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denner J, Tönjes RR Infection barriers to successful xenotransplantation focusing on porcine endogenous retroviruses. Clinical Microbiology Reviews. 2012;25(2):318–343. doi: 10.1128/CMR.05011-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deorowicz S, Kokot M, Grabowski S, Debudaj-Grabysz A KMC 2: fast and resource-frugal k-mer counting . Bioinformatics. 2015;31(10):1569–1576. doi: 10.1093/bioinformatics/btv022. [DOI] [PubMed] [Google Scholar]
- 18.Drummond AJ, Rambaut A BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology. 2007;7(1):214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan X, Abbott TE, Larson D, Chen K BreakDancer: identification of genomic structural variation from paired-end read mapping. Current Protocols in Bioinformatics. 2014;45:15.6.1–11. doi: 10.1002/0471250953.bi1506s45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fiebig U, Fischer K, Bähr A, Runge C, Schnieke A, Wolf E, et al Porcine endogenous retroviruses: Quantification of the copy number in cell lines, pig breeds, and organs. Xenotransplantation. 2018;25(4):e12445. doi: 10.1111/xen.12445. [DOI] [PubMed] [Google Scholar]
- 21.Frantz LAF, Schraiber JG, Madsen O, Megens HJ, Bosse M, Paudel Y, et al. 2013. Genome sequencing reveals fine scale diversification and reticulation history during speciation in Sus. Genome Biology, 14(9): R107.
- 22.Frantz LAF, Schraiber JG, Madsen O, Megens HJ, Cagan A, Bosse M, et al Evidence of long-term gene flow and selection during domestication from analyses of Eurasian wild and domestic pig genomes. Nature Genetics. 2015;47(10):1141–1148. doi: 10.1038/ng.3394. [DOI] [PubMed] [Google Scholar]
- 23.Garkavenko O, Wynyard S, Nathu D, Muzina M, Muzina Z, Scobie L, et al Porcine endogenous retrovirus transmission characteristics from a designated pathogen-free herd. Transplantation Proceedings. 2008;40(2):590–593. doi: 10.1016/j.transproceed.2008.01.051. [DOI] [PubMed] [Google Scholar]
- 24.Gel B, Serra E karyoploteR: an R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics. 2017;33(19):3088–3090. doi: 10.1093/bioinformatics/btx346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grandbastien MA Activation of plant retrotransposons under stress conditions. Trends in Plant Science. 1998;3(5):181–187. doi: 10.1016/S1360-1385(98)01232-1. [DOI] [Google Scholar]
- 26.Groenen MAM A decade of pig genome sequencing: a window on pig domestication and evolution. Genetics Selection Evolution. 2016;48(1):23. doi: 10.1186/s12711-016-0204-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groenen MAM, Archibald AL, Uenishi H, Tuggle CK, Takeuchi Y, Rothschild MF, et al Analyses of pig genomes provide insight into porcine demography and evolution. Nature. 2012;491(7424):393–398. doi: 10.1038/nature11622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harrison I, Takeuchi Y, Bartosch B, Stoye JP Determinants of high titer in recombinant porcine endogenous retroviruses. Journal of Virology. 2004;78(24):13871–13879. doi: 10.1128/JVI.78.24.13871-13879.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hénaff E, Zapata L, Casacuberta JM, Ossowski S Jitterbug: somatic and germline transposon insertion detection at single-nucleotide resolution. BMC Genomics. 2015;16(1):768. doi: 10.1186/s12864-015-1975-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabát P, Tristem M, Opavský R, Pastorek J Human endogenous retrovirus HC2 is a new member of the S71 retroviral subgroup with a full-length pol gene . Virology. 1996;226(1):83–94. doi: 10.1006/viro.1996.0630. [DOI] [PubMed] [Google Scholar]
- 31.Karlas A, Irgang M, Votteler J, Specke V, Özel M, Kurth R, et al Characterisation of a human cell-adapted porcine endogenous retrovirus PERV-A/C. Annals of Transplantation. 2010;15(2):45–54. [PubMed] [Google Scholar]
- 32.Katoh K, Standley DM MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kent WJ BLAT-the BLAST-like alignment tool. Genome Research. 2002;12(4):656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kozak CA Differential expression of murine leukemia virus loci in chemically induced hybrid cells. Journal of Virology. 1984;51(3):876–879. doi: 10.1128/jvi.51.3.876-879.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krüger L, Kristiansen Y, Reuber E, Möller L, Laue M, Reimer C, et al A comprehensive strategy for screening for xenotransplantation-relevant viruses in a second isolated population of Göttingen minipigs. Viruses. 2019;12(1):38. doi: 10.3390/v12010038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krüger L, Stillfried M, Prinz C, Schroder V, Neubert LK, Denner J Copy number and prevalence of porcine endogenous retroviruses (PERVs) in German Wild Boars. Viruses. 2020;12(4):419. doi: 10.3390/v12040419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar S, Stecher G, Li M, Knyaz C, Tamura K MEGA X: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution. 2018;35(6):1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar S, Stecher G, Suleski M, Hedges SB TimeTree: a resource for timelines, timetrees, and divergence times. Molecular Biology and Evolution. 2017;34(7):1812–1819. doi: 10.1093/molbev/msx116. [DOI] [PubMed] [Google Scholar]
- 39.Kunarso G, Chia NY, Jeyakani J, Hwang C, Lu XY, Chan YS, et al Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nature Genetics. 2010;42(7):631–634. doi: 10.1038/ng.600. [DOI] [PubMed] [Google Scholar]
- 40.Lanciano S, Cristofari G Measuring and interpreting transposable element expression. Nature Reviews Genetics. 2020;21(12):721–736. doi: 10.1038/s41576-020-0251-y. [DOI] [PubMed] [Google Scholar]
- 41.Langmead B, Salzberg SL Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larson G, Dobney K, Albarella U, Fang MY, Matisoo-Smith E, Robins J, et al Worldwide phylogeography of wild boar reveals multiple centers of pig domestication. Science. 2005;307(5715):1618–1621. doi: 10.1126/science.1106927. [DOI] [PubMed] [Google Scholar]
- 43.Le Tissier P, Stoye JP, Takeuchi Y, Patience C, Weiss RA Two sets of human-tropic pig retrovirus. Nature. 1997;389(6652):681–682. doi: 10.1038/39489. [DOI] [PubMed] [Google Scholar]
- 44.Lee D, Lee J, Yoon JK, Kim NY, Kim GW, Park C, et al Rapid determination of perv copy number from porcine genomic DNA by real-time polymerase chain reaction. Animal Biotechnology. 2011;22(4):175–180. doi: 10.1080/10495398.2011.595294. [DOI] [PubMed] [Google Scholar]
- 45.Lee JH, Webb GC, Allen RDM, Moran C Characterizing and mapping porcine endogenous retroviruses in Westran pigs. Journal of Virology. 2002;76(11):5548–5556. doi: 10.1128/JVI.76.11.5548-5556.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Letunic I, Bork P Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Research. 2019;47(W1):W256–W259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lian ZX, Rogel-Gaillard C, Li N, Chardon P, Wu CX Copy number polymorphism of endogenous retrovirus sequence in Chinese local pig breeds by Semi-PCR. Acta Veterinaria et Zootechnica Sinica. 2002;33(6):521–524. [Google Scholar]
- 49.Liu G, Li Z, Pan M, Ge M, Wang Y, Gao Y Genetic prevalence of porcine endogenous retrovirus in Chinese experimental miniature pigs. Transplantation Proceedings. 2011;43(7):2762–2769. doi: 10.1016/j.transproceed.2011.06.061. [DOI] [PubMed] [Google Scholar]
- 50.Mang R, Maas J, Chen XH, Goudsmit J, Van Der Kuyl AC Identification of a novel type C porcine endogenous retrovirus: evidence that copy number of endogenous retroviruses increases during host inbreeding. Journal of General Virology. 2001;82(8):1829–1834. doi: 10.1099/0022-1317-82-8-1829. [DOI] [PubMed] [Google Scholar]
- 51.Matos MC, Pinheiro A, Melo-Ferreira J, Davis RS, Esteves PJ Evolution of Fc receptor-like scavenger in mammals. Frontiers in Immunology. 2021;11:590280. doi: 10.3389/fimmu.2020.590280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morales JF, Snow ET, Murnane JP Environmental factors affecting transcription of the human L1 retrotransposon. II. Stressors. Mutagenesis. 2003;18(2):151–158. doi: 10.1093/mutage/18.2.151. [DOI] [PubMed] [Google Scholar]
- 53.Morgulis A, Coulouris G, Raytselis Y, Madden TL, Agarwala R, Schäffer AA Database indexing for production MegaBLAST searches. Bioinformatics. 2008;24(16):1757–1764. doi: 10.1093/bioinformatics/btn322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Niebert M, Tönjes RR. 2005. Evolutionary spread and recombination of porcine endogenous retroviruses in theSuiformes. Journal of Virology, 79(1): 649–654.
- 55.Niu D, Wei HJ, Lin L, George H, Wang T, Lee IH, et al Inactivation of porcine endogenous retrovirus in pigs using CRISPR-Cas9. Science. 2017;357(6357):1303–1307. doi: 10.1126/science.aan4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patience C, Switzer WM, Takeuchi Y, Griffiths DJ, Goward ME, Heneine W, et al Multiple groups of novel retroviral genomes in pigs and related species. Journal of Virology. 2001;75(6):2771–2775. doi: 10.1128/JVI.75.6.2771-2775.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patience C, Takeuchi Y, Weiss RA Infection of human cells by an endogenous retrovirus of pigs. Nature Medicine. 1997;3(3):282–286. doi: 10.1038/nm0397-282. [DOI] [PubMed] [Google Scholar]
- 58.Premachandra HKA, La Cruz FLD, Takeuchi Y, Miller A, Fielder S, O’Connor W, et al Genomic DNA variation confirmed Seriola lalandi comprises three different populations in the Pacific, but with recent divergence . Scientific Reports. 2017;7(1):9386. doi: 10.1038/s41598-017-07419-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quereda JJ, Herrero-Medrano JM, Abellaneda JM, García-Nicolás O, Martínez-Alarcón L, Pallarés FJ, et al Porcine endogenous retrovirus copy number in different pig breeds is not related to genetic diversity. Zoonoses and Public Health. 2012;59(6):401–407. doi: 10.1111/j.1863-2378.2012.01467.x. [DOI] [PubMed] [Google Scholar]
- 60.Quinlan AR, Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rombel IT, Sykes KF, Rayner S, Johnston SA ORF-FINDER: a vector for high-throughput gene identification. Gene. 2002;282(1-2):33–41. doi: 10.1016/S0378-1119(01)00819-8. [DOI] [PubMed] [Google Scholar]
- 62.Scheef G, Fischer N, Krach U, Tönjes RR The number of a U3 repeat box acting as an enhancer in long terminal repeats of polytropic replication-competent porcine endogenous retroviruses dynamically fluctuates during serial virus passages in human cells. Journal of Virology. 2001;75(15):6933–6940. doi: 10.1128/JVI.75.15.6933-6940.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sykes M, Sachs DH Transplanting organs from pigs to humans. Science Immunology. 2019;4(41):eaau6298. doi: 10.1126/sciimmunol.aau6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sypniewski D, Machnik G, Mazurek U, Wilczok T, Smorag Z, Jura J, et al Distribution of porcine endogenous retroviruses (PERVs) DNA in organs of a domestic pig. Annals of Transplantation. 2005;10(2):46–51. [PubMed] [Google Scholar]
- 66.Tönjes RR, Niebert M Relative age of proviral porcine endogenous retrovirus sequences in Sus scrofa based on the molecular clock hypothesis . Journal of Virology. 2003;77(22):12363–12368. doi: 10.1128/JVI.77.22.12363-12368.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang T, Zeng J, Lowe CB, Sellers RG, Salama SR, Yang M, et al Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(47):18613–18618. doi: 10.1073/pnas.0703637104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Warr A, Affara N, Aken B, Beiki H, Bickhart DM, Billis K, et al An improved pig reference genome sequence to enable pig genetics and genomics research. Gigascience. 2020;9(6):giaa051. doi: 10.1093/gigascience/giaa051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wilson CA, Wong S, VanBrocklin M, Federspiel MJ Extended analysis of the in vitro tropism of porcine endogenous retrovirus. Journal of Virology. 2000;74(1):49–56. doi: 10.1128/JVI.74.1.49-56.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yan GR, Guo TF, Xiao SJ, Zhang F, Xin WS, Huang T, et al Imputation-based whole-genome sequence association study reveals constant and novel loci for hematological traits in a large-scale swine F2 resource population . Frontiers in Genetics. 2018;9:401. doi: 10.3389/fgene.2018.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang B, Cui LL, Perez-Enciso M, Traspov A, Crooijmans RPMA, Zinovieva N, Schook LB, et al Genome-wide SNP data unveils the globalization of domesticated pigs. Genetics Selection Evolution. 2017;49(1):71. doi: 10.1186/s12711-017-0345-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang LH, Güell M, Niu D, George H, Lesha E, Grishin D, et al Genome-wide inactivation of porcine endogenous retroviruses (PERVs) Science. 2015;350(6264):1101–1104. doi: 10.1126/science.aad1191. [DOI] [PubMed] [Google Scholar]
- 73.Yang YG, Sykes M Xenotransplantation: current status and a perspective on the future. Nature Reviews Immunology. 2007;7(7):519–531. doi: 10.1038/nri2099. [DOI] [PubMed] [Google Scholar]
- 74.Yoon JK, Choi J, Lee HJ, Cho Y, Gwon YD, Jang Y, et al Distribution of porcine endogenous retrovirus in different organs of the hybrid of a Landrace and a Jeju domestic pig in Korea. Transplantation Proceedings. 2015;47(6):2067–2071. doi: 10.1016/j.transproceed.2015.05.023. [DOI] [PubMed] [Google Scholar]
- 75.Yue YN, Xu WD, Kan YN, Zhao HY, Zhou YX, Song XB, et al Extensive germline genome engineering in pigs. Nature Biomedical Engineering. 2021;5(2):134–143. doi: 10.1038/s41551-020-00613-9. [DOI] [PubMed] [Google Scholar]
- 76.Zhang L, Huang YM, Si JL, Wu YJ, Wang M, Jiang QY, et al Comprehensive inbred variation discovery in Bama pigs using de novo assemblies . Gene. 2018;679:81–89. doi: 10.1016/j.gene.2018.08.051. [DOI] [PubMed] [Google Scholar]
- 77.Zhang P, Yu P, Wang W, Zhang L, Li SF, Bu H An effective method for the quantitative detection of porcine endogenous retrovirus in pig tissues. In Vitro Cellular & Developmental Biology - Animal volume. 2010;46(5):408–410. doi: 10.1007/s11626-009-9264-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data to this article can be found online.