

Summary
JAM-A is a tight-junction-associated protein that contributes to regulation of intestinal homeostasis. We report that JAM-A interacts with NF2 and LATS1, functioning as an initiator of the Hippo signaling pathway, well-known for regulation of proliferation. Consistent with these findings, we observed increased YAP activity in JAM-A-deficient intestinal epithelial cells (IEC). Furthermore, overexpression of a dimerization-deficient mutant, JAM-A-DL1, failed to initiate Hippo signaling, phenocopying JAM-A-deficient IEC, whereas overexpression of JAM-A-WT activated Hippo signaling and suppressed proliferation. Lastly, we identify EVI1, a transcription factor reported to promote cellular proliferation, as a contributor to the pro-proliferative phenotype in JAM-A-DL1 overexpressing IEC downstream of YAP. Collectively, our findings establish a new role for JAM-A as a cell-cell contact sensor, raising implications for understanding the contribution(s) of JAM-A to IEC proliferation in the mammalian epithelium.
Subject areas: Biological sciences, Biochemistry, Cell biology
Graphical abstract
Highlights
-
•
JAM-A suppresses intestinal epithelial proliferation through the Hippo pathway
-
•
NF2 and LATS1 interact with the JAM-A signaling complex
-
•
JAM-A dimerization is a required signaling to initiate Hippo signaling
-
•
EVI1 is a driver of the pro-proliferative phenotype of JAM-A-deficient IEC
Biological sciences; Biochemistry; Cell biology
Introduction
Signaling events emanating from epithelial intercellular junctions are an integral part of the complex regulation of intestinal epithelial homeostasis, including coordination of intestinal epithelial cell (IEC) proliferation and migration. Intercellular junctions are specialized regions of contact between adjacent cells that include desmosomes, adherens junctions, and tight junctions (TJ) (Luissint et al., 2016). The TJ is the most apical intercellular contact composed of transmembrane proteins that seal the paracellular space and maintain apical-basal polarity (Zihni et al., 2016). Junctional adhesion molecule (JAM) proteins are among the major TJ-associated proteins and are type I transmembrane proteins characterized by extracellular immunoglobulin-like (Ig) loops that form homotypic and heterotypic adhesive interactions (Luissint et al., 2014). JAM-A, a prototypic family member, has been shown to regulate key barrier and proliferative epithelial cell functions (Laukoetter et al., 2007; Nava et al., 2011; Vetrano et al., 2008; Wang et al., 2018). JAM-A forms homodimers in cis and trans and has a short cytoplasmic tail that contains multiple tyrosine and serine phosphorylation sites and ends in a PDZ-binding motif (Kostrewa et al., 2001; Monteiro et al., 2014; Prota et al., 2003; Van Itallie and Anderson, 2018). JAM-A-dependent signal transduction has been shown to be mediated through its phosphorylation sites, resulting in functional consequences affecting TJ stability (Fan et al., 2019; Iden et al., 2012). The C-terminal PDZ-binding motif facilitates interaction between JAM-A and cytoplasmic scaffold proteins, enabling recruitment of kinases and small GTPases to assemble larger signaling complexes at the TJ, which has been reviewed elsewhere (Steinbacher et al., 2018).
In this study, we demonstrate that JAM-A is a previously unidentified initiator of Hippo pathway signaling. It is well appreciated that in confluent epithelial monolayers, Hippo signaling is stimulated, resulting in activation of the core pathway kinases, mammalian sterile 20-like 1 and 2 (MST1/2, alias serine threonine kinase) that phosphorylates large tumor suppressor kinase 1 (LATS1). Subsequently, LATS1 phosphorylates the Hippo transcriptional coactivators, Yes-associated protein 1 (YAP), and transcriptional coactivator with PDZ-binding motif (TAZ), sequestering them in the plasma membrane and cytoplasm, inhibiting proliferation. In contrast, in subconfluent epithelia, Hippo signaling is suppressed, and YAP/TAZ remain in a dephosphorylated active state and translocate to the nucleus to promote transcription of proliferation and antiapoptotic genes. We previously observed that loss of JAM-A resulted in enhanced IEC proliferation linked to Akt/β-catenin-dependent signaling (Nava et al., 2011); however, mechanisms delineating how JAM-A regulates downstream signaling events to suppress IEC proliferation have remained incompletely understood. Here, we report that JAM-A interacts with proteins involved in Hippo signaling, neurofibromatosis type 2 (NF2/Merlin) and LATS1, to function as a negative regulator of IEC proliferation. Consistent with this, we observed decreased NF2 and LATS1 activity and increased YAP activity in JAM-A-deficient IEC. Studies of dimerization-deficient JAM-A (JAM-A-DL1) overexpressing cells demonstrate that extracellular dimerization of JAM-A is a required signal to initiate Hippo signaling. Intriguingly, we also report that the transcription factor, ecotropic virus integration site 1 (EVI1/MECOM), drives IEC proliferation in JAM-A-DL1 overexpressing cells. Further investigation revealed that Evi1 transcription and protein levels are negatively regulated by the Hippo pathway, controlled by JAM-A. Collectively, these data establish JAM-A as a novel dimerization-dependent regulator of Hippo signaling in the mammalian intestinal epithelium.
This finding suggests that JAM-A functions as a sensor for cell-cell contact inhibition of proliferation, a key process in both homeostasis and disease.
Results
JAM-A regulates proliferation and associates with Hippo regulatory molecules LATS1 and NF2
We previously reported that total loss of JAM-A in mice results in a pro-proliferative IEC phenotype (Nava et al., 2011). To determine if this phenotype was epithelial specific, we assessed IEC proliferation in the colonic mucosa of inducible, epithelial-targeted JAM-A knockout mice (Villin-CreERT2; JAM-Af/f, termed JAM-AERΔIEC) compared with littermate controls (JAM-Af/f) by Ki67 staining. Significantly increased Ki67 staining of IEC was observed in the JAM-AERΔIEC mice compared with controls (Figure 1A). Moreover, we detected increased proliferation of enteroids derived from JAM-A null mice (JAM-A−/−) relative to wild-type (WT) controls using an EdU incorporation assay (Figure 1B). Given that multiple pathways have been shown to regulate proliferation in IEC, studies were performed to investigate possible JAM-A-dependent regulation of proliferation through evaluation of known binding partners. We previously demonstrated that JAM-A binds a PDZ domain of ZO-2 (Monteiro et al., 2013), and prior BioID studies had reported Hippo pathway molecules, LATS1 and NF2, as putative protein interactors of ZO-2 (Couzens et al., 2013; Hennigan et al., 2019). Because Hippo signaling is well appreciated to play critical roles in cellular proliferation, differentiation, and apoptosis (Hong et al., 2016), we performed co-immunolabeling and co-immunoprecipitation experiments to examine whether JAM-A interacts with NF2 and LATS1. ZO-2 was immunoprecipitated from IEC lysates and probed with an NF2 antibody, revealing positive co-association between NF2 and ZO-2. Conversely, co-association between HA-tagged JAM-A and NF2 was observed in immunoprecipitates of HA-tagged JAM-A from human IEC lysates probed with an NF2 antibody (Figure 1C). Importantly, p-NF2 S518, a suppressive modification (Rong et al., 2004; Surace et al., 2004), did not co-immunoprecipitate with JAM-A, suggesting that JAM-A only interacts with the active form of NF2 (Figure 1C). Co-immunolabeling of JAM-A and NF2 in WT murine colonic mucosa and a model human IEC line showed strong co-localization at TJs (Figures 1D and S1A).
Figure 1.

JAM-A regulates proliferation and associates with Hippo regulatory molecules LATS1 and NF2
(A) JAM-A-deficient mice exhibit increased colonic IEC proliferation. Representative confocal images of colon tissue from JAM-AERΔIEC and control (JAM-Afl/fl) mice stained for JAM-A (green), Ki67 (red), and DAPI/nuclei (blue). Increased levels of proliferating cells in the JAM-AERΔIEC mice compared with controls is shown in the graph plotting percentage of Ki67 positive cells/crypt with dots denoting individual crypts. Data are means ± SEM of crypts from JAM-Afl/fl and JAM-AERΔIEC mice, respectively. ∗∗∗p ≤ 0.001; two-tailed Student’s t test. Scale bar, 50 μm.
(B) EdU incorporation in 3D enteroids derived from JAM-A−/− and WT (JAM-A+/+) mice indicate that loss of JAM-A results in increased proliferation. Data are means ± SEM where buds counted from WT and JAM-A−/− enteroids and are denoted as dots on the graph. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001: two-tailed Student’s t test. Scale bar, 50 μm.
(C) Co-immunoprecipitation of ZO-2 and JAM-A with NF2. Whole-cell lysate of SKCO-15 IEC stably expressing a full-length HA-tagged JAM-A (HA-JAM-A) was immunoprecipitated with a ZO-2 (ZO-2 IP) or HA (HA IP) antibody followed by immunoblotting for NF2, ZO-2 and NF2, p-NF2 S518, and HA-JAM-A. p-NF2 S518 did not co-immunoprecipitate with HA-JAM-A.
(D) Representative immunostaining of NF2 (green) and JAM-A (red) detected by confocal microscopy in WT murine colon tissue demonstrate co-localization at the TJ. Scale bar, 50 μm.
(E) JAM-A co-immunoprecipitates with multiple Hippo pathway molecules. Immunoprecipitation of HA-tagged JAM-A from IEC lysates with an HA-specific antibody (HA IP) followed by western blot LATS1, YAP, p-YAP S127, 14-3-3, and JAM-A. Co-immunoprecipitation of these molecules with JAM-A is demonstrated.
(F) Endogenous JAM-A co-immunoprecipitation with LATS1, NF2, and YAP. Immunoprecipitation of LATS1 with LATS1 antibody (LATS1 IP) from IEC cell lysates followed by western blot for NF2, YAP, JAM-A, and LATS1. Co-immunoprecipitation is observed between these molecules.
(G) Representative immunofluorescence confocal images of LATS1 (green), JAM-A (red), and DAPI/nuclei (blue) in WT murine colon tissue. Scale bar, 50 μm. Lysate blots serve as input loading controls in (C), (E), and (F). Immunoprecipitation with a nonspecific IgG antibody served as a negative control (IgG IP) for (C), (E), and (F). All data are representative of three independent experiments. See also Figure S1.
In addition, co-association between HA-tagged JAM-A and LATS1, YAP, p-YAP S127, and the adaptor protein 14-3-3 was revealed in co-immunoprecipitation studies performed on lysates from IEC expressing an HA-tagged JAM-A that were probed with antibodies against these Hippo signaling elements (Figure 1E). Moreover, LATS1 immunoprecipitates probed for JAM-A, NF2, and YAP by western blot demonstrated interaction of these Hippo pathway constituents with endogenous JAM-A (Figure 1F). Identical to NF2, LATS1 exhibited strong co-localization with JAM-A at TJs of murine WT colonic IEC as well as model human IEC (Figures 1G and S1b). These data strongly support association of NF2 and LATS1 with JAM-A in a complex at the TJ, likely through the scaffold protein ZO-2.
Increased proliferation after loss of JAM-A is associated with suppressed Hippo pathway signaling
The interaction of JAM-A with NF2 and LATS1 suggested that JAM-A regulates cell proliferation, in part, through the Hippo pathway. Thus, we examined whether the loss of JAM-A modulates Hippo signaling. Consistent with results obtained from JAM-A−/− and JAM-AERΔIEC mice, SKCO-15 IEC silenced for JAM-A using siRNA (JAM-A KD) exhibited enhanced proliferation relative to nonsilencing control IEC, as determined by Ki67 staining (Figure 2A). Because initiation of Hippo signaling is characterized by sequential phosphorylation and activation of Hippo kinases to phosphorylate YAP and suppress its translocation to the nucleus (Meng et al., 2016), we conducted phosphoprotein-specific western blotting on cell lysates from JAM-A KD and control IEC. JAM-A KD IEC demonstrated significantly decreased p-LATS1 S909 levels relative to controls, consistent with decreased protein activity. In agreement with the observed decrease in LATS1 activity, p-YAP S127 levels were decreased in JAM-A KD IEC compared with controls (Figure 2B).
Figure 2.

Increased proliferation after loss of JAM-A is associated with suppressed Hippo pathway signaling
(A) Human JAM-A-deficient IEC exhibit a pro-proliferative phenotype. Ki67 staining was conducted in JAM-A knockdown (KD) human SKCO-15 IEC transfected with two siRNA targets (JAM-A siRNA-1, JAM-A siRNA-2). An average percentage of Ki67 positive cells from five fields of view were generated for three independent experiments (denoted by dots on the graph). JAM-A KD IEC also display enhanced proliferation like JAM-A−/− and JAM-AERΔIEC IEC.
(B) JAM-A KD IEC exhibited decreased Hippo pathway activity. Representative western blots for p-LATS S909, LATS1, p-YAP S127, YAP, and JAM-A in JAM-A KD IEC transfected with two JAM-A siRNA targets and one control (CTL siRNA). Calnexin served as a loading control. Graphs represent digital densitometry for p-LATS S909 and p-YAP S127. Activity of LATS1 is decreased, resulting in increased YAP activity JAM-A KD IEC.
(C) JAM-A loss results in increased YAP target gene transcription. qPCR was conducted on SKCO-15 JAM-A KD and CTL IEC to assess transcript expression of known YAP gene targets, CTGF and CYR61. Data represent gene transcript levels expressed as relative expression normalized to the control cells. β-actin served as the reference gene. Data represent means ± SEM of three independent experiments ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001: two-tailed Student’s t test.
These results suggest that JAM-A deficiency results in increased YAP activity, indicative of an attenuation in Hippo signaling activation. To provide a functional link between increased YAP activity in JAM-A-deficient IEC and downstream changes in transcription, we examined transcript levels of two well-known YAP target genes, connective tissue growth factor (CTGF) and cysteine-rich angiogenic inducer 61 (CYR61) (Zhang et al., 2011; Zhao et al., 2008). Expression of CTGF and CYR61 was significantly upregulated in JAM-A-deficient cells relative to control IEC (Figure 2C). Collectively, these results support the notion that JAM-A regulates upstream Hippo signaling through interaction with NF2 and LATS1, resulting in modulation of LATS1 activity, ultimately leading to an attenuation in Hippo signaling.
JAM-A is a regulator of Hippo signaling in primary cultured IEC
To corroborate findings obtained with well-characterized model human IEC lines, we performed complementary experiments with 2-dimensional (2D) primary murine colonoid cultures derived from JAM-AERΔIEC and JAM-Af/f mice. Co-immunolabeling for NF2 and JAM-A in 2D WT cultured murine colonoids showed strong co-localization similar to WT murine colon and human IEC (Figure 3A). LATS1 also colocalized with JAM-A at the level of the TJ in murine colonoids (Figure 3B). Because YAP activity is tightly controlled by its cellular localization, we examined localization of YAP in 2D colonoids derived from JAM-AERΔIEC and JAM-Af/f mice.
Figure 3.

JAM-A is a regulator of Hippo signaling in primary cultured IEC
(A) Representative confocal immunostaining of NF2 (green) and JAM-A (red) in WT murine 2D colonoids shows co-localization at the TJ. Scale bar, 20 μm.
(B) Representative confocal immunostaining of LATS1 (green) and JAM-A (red) associating at the TJ in WT 2D murine colonoids. Scale bar, 20 μm.
(C) JAM-AERΔIEC primary IEC exhibit prolonged nuclear YAP localization. Representative immunofluorescent images of YAP (green) and ZO-1 (red) in 2D colonoids cultured at low and high cell confluency derived from and JAM-AERΔIEC and JAM-Afl/fl mice. At low cell confluency YAP staining is visible in the nuclei of JAM-AERΔIEC and JAM-Afl/fl IEC. At high cell confluency, YAP localizes to the cytoplasm in control IEC but remains in the nuclei of JAM-AERΔIEC IEC, revealing a failure of Hippo signaling initiation. Graph depicts scoring of YAP localization (nuclear, cytoplasm, and nuclear + cytoplasm). Five fields of view for each condition were scored to generate an average percentage, and three independent experiments were performed (denoted by the dots). Scale bar, 20 μm.
(D) JAM-A-deficient primary IEC exhibit decreased Hippo signaling activity. Representative western blots for p-LATS1 S909, LATS1, p-NF2 S518, NF2, p-YAP S127, YAP, and JAM-A in subconfluent 2D colonoids derived from JAM-AERΔIEC and JAM-Afl/fl mice. Graphs represent digital densitometry for relative density of p-NF2 S518, p-LATS1 S909, and p-YAP S127. JAM-AERΔIEC colonoids exhibit decreased NF2 and LATS1 activity, resulting in increased YAP activity relative to JAM-Afl/fl controls. Calnexin serves as a loading control. Data are means ± SEM of three independent experiments. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001: two-tailed Student’s t test.
Colonoids were cultured at low (subconfluent) and high (confluent) cell density, followed by co-immunolabeling for YAP and ZO-1, a TJ protein. At low cell density, both JAM-AERΔIEC and JAM-Af/f colonoids exhibited nuclear localization of YAP, in support of a proliferative state, and inactivated Hippo signaling. At high cell density, JAM-AERΔIEC IEC failed to initiate Hippo signaling, as indicated by continued nuclear localization of YAP, whereas YAP was redistributed to the cytoplasm of JAM-Af/f IEC (Figure 3C). Consistent with our findings in human JAM-A KD IEC, phosphoprotein-specific western blotting revealed decreased levels of p-LATS1 S909 and p-YAP S127 in JAM-AERΔIEC colonoids relative to controls. In addition, we observed decreased NF2 activity in JAM-AERΔIEC colonoids, as detected by increased relative levels of p-NF2 S518 compared with controls (Figure 3D). These data confirm that Hippo pathway signaling is decreased by JAM-A deficiency in primary IEC and that JAM-A regulates this pathway upstream of LATS1 and NF2.
JAM-A dimerization serves as a cell-cell contact sensing mechanism to initiate Hippo signaling
Given a previous observation suggesting that dimerization of JAM-A plays a role in suppressing IEC proliferation (Nava et al., 2011), we investigated whether Hippo signaling is dependent on JAM-A dimerization. An exogenous overexpression system was employed to study effects of a dimerization-deficient JAM-A mutant (JAM-A-DL1) lacking the distal Ig domain on Hippo signaling and cell proliferation in SW480 IEC. This JAM-A-DL1 mutant has been reported to act in a dominant negative fashion, thus phenocopying the pro-growth phenotype of JAM-A-deficient IEC (Monteiro et al., 2014; Severson et al., 2008). Consistent with prior observations, overexpression of full-length JAM-A (JAM-A-WT) resulted in decreased proliferation relative to JAM-A-DL1 mutant IEC (Figure 4A). We then investigated whether Hippo signaling was regulated by formation of JAM-A dimers by phosphoprotein-specific western blotting of IEC lysates. JAM-A-WT overexpressing IEC exhibited high relative levels of p-LATS1 S909 and p-YAP S127. In contrast, JAM-A-DL1 mutant cells were unable to activate Hippo signaling and suppress proliferation, as evidenced by decreased levels of p-LATS1 S909 and p-YAP S127 (Figure 4B). Similar results were obtained with HEK293T cells that express very low levels of JAM-A protein, revealing decreased LATS1 activity and increased YAP activity in the JAM-A-DL1 relative to the JAM-A-WT cells (Figure S2). Collectively, these results support a model in which dimerization of JAM-A serves as a cell-cell contact sensing mechanism, initiating downstream Hippo signaling to suppress IEC proliferation.
Figure 4.

JAM-A dimerization serves as a cell-cell contact sensing mechanism to initiate Hippo signaling
(A) Overexpression of a dimerization-deficient JAM-A mutant (JAM-A-DL1) results in enhanced proliferation. Representative immunofluorescent images of EdU incorporation (green) and nuclei (blue) in SW480 IEC overexpressing JAM-A full-length (JAM-A- WT) or JAM-A-DL1. Graph represents average percentage of EdU positive cells from 10 fields of view for each experiment (JAM-A-WT or JAM-A-DL1) and three independent experiments performed, which are denoted by dots on the graph. Overexpression of JAM-A-WT resulted in a significant decrease in proliferating cells, whereas the JAM-A-DL1 mutant was unable to suppress proliferation. Scale bar, 20 μm.
(B) Evaluation of Hippo signaling activity in JAM-A-DL1 overexpressing IEC. Representative western blots of p-LATS1 S909, LATS1, p-YAP S127, YAP, and JAM-A on cell lysate from SW480 IEC overexpressing JAM-A-WT and JAM-A-DL-1. Beta-actin serves as a loading control. Graph represents digital densitometry of p-LATS1 S909 and p-YAP S127. JAM-A-WT overexpressing IEC exhibit increased LATS1 activity and decreased YAP activity characteristic of Hippo pathway activation. Relative to the JAM-A-WT overexpressing cells, JAM-A-DL1 cells exhibit decreased activation of Hippo signaling. All data are means ± SEM of three independent experiments. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001: two-tailed Student’s t test. See also Figure S2.
JAM-A regulates EVI1 through Hippo pathway molecules
To gain further insights into downstream mechanisms underlying the pro-proliferative effects observed with loss of JAM-A, we performed transcription factor PCR arrays on IEC isolated from JAM-A-deficient mice (JAM-A−/−, JAM-AERΔIEC) and their respective controls (WT, JAM-Af/f). In JAM-A−/− and JAM-AERΔIEC IEC, there was significant upregulation of Evi1, a well-reported oncogenic transcription factor in leukemia (Lugthart et al., 2008; Ogawa et al., 1996) and some epithelial tumors (Bei et al., 2010; Starr et al., 2009), that plays a critical role in cellular proliferation (Goyama et al., 2008; Hoyt et al., 1997). To corroborate PCR array results, qPCR analyses of Evi1 transcript expression in murine JAM-A-deficient IEC were performed. In agreement with array results, Evi1 transcript levels were significantly upregulated in IEC from JAM-A−/− and JAM-AERΔIEC mice relative to controls (Figures 5A and 5B). To understand how EVI1 upregulation might contribute to the pro-proliferative phenotype of dimerization-deficient JAM-A IEC, we transfected JAM-A-DL1 and JAM-A-WT overexpressing cells with EVI1 siRNA and assessed cellular proliferation by MTT assay. Under unstimulated conditions, JAM-A-DL1 mutant cells had increased proliferation; however, silencing of EVI1 ablated the increase in proliferation in JAM-A-DL1 cells relative to JAM-A-WT cells (Figure 5C). To confirm that EVI1 is a driver of IEC proliferation, an EdU incorporation assay and Ki67 staining was conducted in EVI1 KD IEC (siRNA). Indeed, EVI1 knockdown resulted in decreased cell proliferation compared with control IEC (Figures 5D and S3A). Because the pro-proliferative phenotype of JAM-A-deficient IEC is the result of decreased Hippo signaling, we investigated whether EVI1 transcription was modulated by Hippo signaling. Human IEC were silenced for NF2 (siRNA) and assessed for EVI1 protein levels by western blot. NF2 knockdown resulted in increased levels of EVI1 protein relative to control IEC, suggesting that EVI1 is a transcriptional target gene suppressed by Hippo signaling (Figure 5E). To further understand the link between EVI1 transcription and the Hippo pathway, IEC were treated with verteporfin, a YAP inhibitor that impedes YAP-TEAD binding and promotes sequestration of YAP in the cytoplasm (Brodowska et al., 2014; Liu-Chittenden et al., 2012; Wang et al., 2016). Such pharmacologic inhibition of YAP results in decreased transcription of gene targets CTGF and CYR61 (Figure S3B). Evaluation of EVI1 protein by western blot revealed that YAP inhibition resulted in nearly undetectable levels of EVI1 relative to untreated control IEC (Figure 5F). These observations indicate that increased transcription and protein expression of EVI1 observed in JAM-A-deficient IEC is dependent on YAP activity. Importantly, the increase in EVI1 protein level observed following the loss of JAM-A was ablated in response to YAP inhibition in JAM-A KD IEC (Figure 5G). To determine if YAP paired with TEAD transcription factors to target EVI1 transcription, all four TEAD family members present in IEC were knocked down (siRNA) and EVI1 protein levels assessed by western blotting. Indeed, loss of TEAD transcription factors resulted in significantly decreased EVI1 protein levels (Figure 5H). Together, these studies reveal that EVI1 functions as a downstream effector that significantly contributes to the observed JAM-A-DL1 pro-proliferative phenotype. Furthermore, these findings suggest that EVI1 is a previously unidentified downstream gene target of YAP that participates with TEAD transcription factors to induce its transcription. Collectively, these results highlight a model of dimerization-dependent JAM-A regulation of IEC proliferation through Hippo signaling and subsequent EVI1 expression (Figure 6).
Figure 5.

JAM-A regulates EVI1 through Hippo pathway molecules
(A) EVI1 transcript levels in IEC isolated from the ileum of JAM-A−/− and WT mice were measured by qPCR. Evi1 transcript is upregulated in JAM-A−/− relative to WT mice. Relative expression of Evi1 is normalized to Tbp. Dots denote individual animals with data pooled from three independent experiments.
(B) Evi1 transcript levels in IEC isolated from the ileum of JAM-AERΔIEC and JAM-Afl/fl mice were measured as in (A). Evi1 transcript is significantly upregulated in JAM-AERΔIEC mice relative to controls.
(C) EVI1 is a downstream effector that significantly contributes to the JAM-A dimerization-deficient pro-proliferative IEC phenotype. The cellular proliferation of HEK293T cells overexpressing JAM-A-DL1 and JAM-A-WT was assessed by MTT assay. JAM-A-DL1 cells exhibit increased proliferation relative to the JAM-A-WT cells. EVI1 knockdown (EVI1 siRNA) in JAM-A-DL1 overexpressing cells restored the proliferation to levels comparable to the JAM-A-WT cells. This result indicates that the increased proliferation observed in JAM-A-DL1 mutants is dependent on EVI1 activity.
(D) EVI1 knockdown (EVI1 siRNA) SKCO-15 IEC were subjected to an EdU incorporation assay to assess cell proliferation. Graph represents average percentage of EdU positive cells from 10 fields of view each for experiment. Three individual experiments were conducted, denoted by dots on the graph, which shows that EVI1 KD IEC exhibit decreased proliferation relative to control cells. Thus, EVI1 promotes proliferation in IEC. Representative western blot of SKCO-15 IEC transfected with EVI1 (EVI1) (2 targets: EVI1 siRNA-1, EVI1 siRNA-2) or non-targeting control (CTL) siRNA. EVI1 is appreciably depleted in the EVI1 silenced cells. Calnexin serves as a loading control.
(E) Evaluation of whether EVI1 is regulated by Hippo signaling. Representative western blots for EVI1 and NF2 in NF2 knockdown SKCO-15 cells (NF2 siRNA-1 and NF2 siRNA-2). Digital densitometry shows relative levels of EVI1 are increased by knockdown of NF2 relative to control IEC, confirming that the Hippo pathway suppresses EVI1. Actin served as a loading control.
(F) Assessment of EVI1 protein levels in response to YAP inhibition. Representative EVI1 and JAM-A western blots from SKCO-15 cells treated with 10 μM of the YAP inhibitor verteporfin. Graph represents digital densitometry for EVI1 relative levels. Treatment with verteporfin resulted in significant reduction in EVI1 protein, suggesting that EVI1 is a YAP gene target. Actin served as a loading control.
(G) Representative western blot for EVI1 in JAM-A KD and control SKCO-15 IEC treated with and without 10 μM of verteporfin. Graphs represent digital densitometry for EVI1. The upregulation of EVI1 under JAM-A silencing was ablated in response to YAP inhibition revealing that regulation of EVI1 is modulated by YAP activity. Actin served as a loading control.
(H) YAP pairs with TEAD transcription factors to target Evi1 transcription. Representative western blots for EVI1 and TEAD in TEAD1-4 KD (all four TEADs expressed in IEC were knocked down, two targets for each factor used: TEAD Target 1 and TEAD Target 2) SKCO-15 IEC. Actin serves as a loading control for western blot. Graph represents digital densitometry for EVI1, which is decreased by knockdown of TEAD transcription factors. Data for all experiments represent means ± SEM of three independent experiments. ∗p ≤ 0.05 ∗∗p ≤ 0.01 ∗∗∗p ≤ 0.001: two-tailed Student’s t test and two-way ANOVA. See also Figure S3.
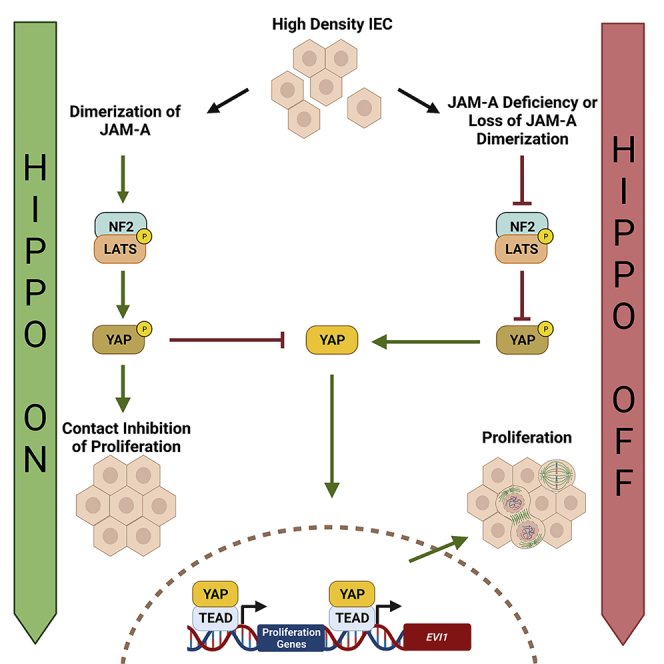
Figure 6.

Model of JAM-A regulation of Hippo pathway signaling through extracellular adhesive interactions
JAM-A senses cell-cell contacts through dimerization at the TJ, resulting in outside-in signaling that triggers changes in the intracellular JAM-A scaffold complex. These changes likely affect JAM-A cytoplasmic contacts mediated through its PDZ-binding motif with ZO-2. This interaction recruits TJ scaffold proteins that can facilitate interaction between NF2 and LATS1. The formation of this complex will activate Hippo core kinases, including LATS1, to mediate downstream Hippo signaling in which YAP is phosphorylated and subsequently sequestered by 14-3-3 in the cytoplasm. This results in decreased YAP target gene transcription and suppression of proliferation (Left panel).
When JAM-A is unable to dimerize, as in the case of the JAM-A-DL1 mutant, this signaling is lost. Subsequently, NF2 and LATS1 fail to be recruited to the TJ to activate Hippo signaling. Thus, YAP remains active, translocating to the nucleus, and interacts with TEAD to promote transcription of its pro-proliferative target genes, including EVI1 (Right panel).
Discussion
Results from a prior study suggested that JAM-A suppresses IEC proliferation through an Akt/β-catenin signaling axis (Nava et al., 2011). However, it is well appreciated that regulation of epithelial proliferation is complex and detailed molecular mechanisms of how JAM-A regulates cellular proliferation are incompletely understood. Previous studies identified two key Hippo regulatory molecules, LATS1 and NF2, as putative protein interactors of ZO-2 (Couzens et al., 2013; Hennigan et al., 2019), which we had previously established as a binding partner of JAM-A (Monteiro et al., 2013). In this study, we demonstrate interaction of NF2 and LATS1 with JAM-A and show this association links Hippo regulation of cellular proliferation to JAM-A. The co-immunoprecipitation and complementary co-immunolabeling studies in this report are the first to demonstrate that JAM-A interacts with LATS1, NF2, and YAP, as well as 14-3-3, a canonical p-YAP S127 binding protein. We propose that interaction of JAM-A with these Hippo molecules is bridged through PDZ domains of ZO-2, facilitating recruitment of other TJ scaffold proteins that can bind directly to NF2 and LATS1.
Previously, few mammalian transmembrane receptors have been identified as upstream initiators of Hippo signaling relative to studies in Drosophila melanogaster. In Drosophila, atypical Cadherins, Dachsous (mammalian DCHS1/2) and Fat (mammalian FAT2/4), as well as the polarity protein Crumbs, function as upstream regulators of the Hippo pathway, sensing cell density and initiating Hippo signaling (Chen et al., 2010; Ling et al., 2010; Staley and Irvine, 2012). However, studies of murine gene knockouts of DCHS1/2 and Fat4 phenocopy neither the growth phenotypes nor the increased YAP activation exhibited by Drosophila mutants (Kuta et al., 2016; Mao et al., 2011). In addition, the mammalian Crumbs homologue in IEC, Crumbs3 (CRB3), cannot sense cell density as in Drosophila, due to a shortened extracellular domain. Relative to the Drosophila Hippo pathway protein network, it is controversial which transmembrane proteins serve as counterparts for Dachsous, Fat, and Crumbs for sensing increasing cell density in mammalian epithelial cells. Importantly, Drosophila cell-cell adhesion differs from mammalian epithelial cells in that they lack TJs (Knust and Bossinger, 2002). Therefore, JAM-A and other TJ proteins are unique mammalian proteins that localize to cell-cell contacts during various physiological and pathological processes that could modulate Hippo signaling.
With evidence that JAM-A interacts with Hippo regulatory components, we conducted biochemical studies to investigate whether Hippo signaling is altered by the loss of JAM-A. Indicative of dampened Hippo signaling, these studies revealed decreased levels of p-LATS1 S909 and p-YAP S127 in JAM-A-deficient human IEC. Our findings in human IEC were confirmed in primary murine colonoids derived from JAM-AERΔIEC mice, demonstrating decreased Hippo signaling by phosphoprotein-specific western blotting and YAP localization studies. In addition to changes in LATS1 and YAP activity, we also observed increased levels of p-NF2 S518, a suppressive modification, in JAM-AERΔIEC IEC compared with controls. Our conclusions were further supported by significant upregulation of two established YAP target genes, CTGF and CYR61, in JAM-A-deficient IEC. Together, these studies show that JAM-A serves as an upstream regulator of Hippo signaling coordinating LATS1 and NF2 activity.
Given previous evidence indicating that dimerization of the extracellular Ig domain is necessary for JAM-A-dependent signaling for proliferation, we tested whether dimerization of JAM-A regulates Hippo signaling. Decreased LATS1 activity, increased YAP activity, and subsequent enhanced proliferation in the cells overexpressing JAM-A-DL1 demonstrate that dimerization of JAM-A is required to initiate Hippo signaling as cells reach confluency. Specifically, JAM-A extracellular dimerization at cell-cell contacts serves as a signal that enhances protein-protein interactions in an intracellular scaffold complex mediated, in part, through PDZ-dependent interactions (Mandell et al., 2005; Monteiro et al., 2014; Severson et al., 2009). JAM-A-binding partners, such as ZO-2 (Monteiro et al., 2013) and ZO-1 (Ebnet et al., 2000), likely coordinate with the polarity complex CRB3/PALS1/PATJ to directly recruit Hippo signaling scaffolding proteins Expanded and NF2 (Robinson et al., 2010) or recruit NF2 and LATS1 through interaction with Angiomotin (Li et al., 2015; Yi et al., 2011; Zhou et al., 2019). Alternatively, JAM-A dimerization may also directly recruit NF2 via the PAR3/PAR6/aPKC polarity complex (Roh et al., 2002). It is possible that JAM-A participates in more than one protein complex to initiate Hippo signaling, an idea raised by prior work showing that JAM-A interacts with multiple scaffold proteins at the TJ (Ebnet et al., 2000; Monteiro et al., 2013; Severson et al., 2009). The formation of such a scaffold complex would facilitate the recruitment and activation of Hippo kinases MST1/2 and LATS1/2 to mediate downstream Hippo signaling. Detailing the nature of the complex or complexes JAM-A participates in, including other protein constituents, the role of JAM-A cytoplasmic phosphorylation in initiating signaling, and interaction with MST1/2 requires further investigation. Another interesting avenue for further investigation is the possible role JAM-A plays in activating YAP through the stretch response, which was not studied in this work as cells were plated on the same surfaces throughout all cell culture experiments. Overall, these results are the first to identify JAM-A as a TJ-associated protein that acts as a cell-cell contact sensor through participation in extracellular adhesive interactions to initiate Hippo signaling. These findings pose significant implications to understanding mechanisms by which mammalian cells detect increasing cell density, triggering cell-cell contact inhibition of proliferation.
In addition to establishing JAM-A as a novel initiator of Hippo signaling, we identified EVI1 as a YAP target gene that is suppressed by JAM-A. EVI1, a pro-proliferative transcription factor, is shown to be upregulated in JAM-A-deficient IEC. Moreover, knockdown of EVI1 dampened increased proliferation exhibited by JAM-A-DL1 overexpressing cells, revealing EVI1 to be a major contributor to the pro-proliferative phenotype of these cells. We further demonstrate that EVI1 is targeted by YAP interactions with TEAD transcription factors, as TEAD knockdown resulted in decreased EVI1 protein levels. Further investigation is required to pinpoint which of the TEAD family members is the predominant TEAD responsible for targeting EVI1, as all four TEAD factors are expressed in IEC. Importantly, EVI1 is recognized as an oncogene in multiple solid tumors and leukemia, and high EVI1 expression correlates with poor clinical outcome (Barjesteh van Waalwijk van Doorn-Khosrovani et al., 2003; Koos et al., 2011; Lugthart et al., 2008). JAM-A expression has also been negatively correlated with clinical outcome in pancreatic (Fong et al., 2012), endometrial (Koshiba et al., 2009), gastric (Huang et al., 2014), and colorectal cancers (Lampis et al., 2021). The Hippo pathway is well appreciated to be deregulated in many cancers. Despite this, core Hippo signaling molecules are infrequently mutated in tumors (Harvey et al., 2013). Therefore, loss of JAM-A and subsequent targeting of genes that promote proliferation, including EVI1, could be a mechanism by which Hippo signaling becomes perturbed in cancer, contributing to oncogenesis. Future studies evaluating the connection between loss of JAM-A, deregulated Hippo signaling, and EVI1 may yield new insights into cancer progression and clinical outcomes.
Limitations of the study
In this study, we evaluate the role of JAM-A dimerization in activating Hippo signaling in primary murine IECs as well as human cancer IECs. The human cell lines used (SW480 and HEK293T) express endogenous JAM-A, which might affect the Hippo signaling initiated by JAM-A dimerization. An alternative approach to circumvent this limitation would be to conduct rescue experiments in which wild-type and JAM-A-DL1 are overexpressed in JAM-A null murine colonoids followed by assessing the effects of JAM-A dimerization in IEC. In addition, mutation(s) of JAM-A that result in loss of dimerization have not been described in humans. Therefore, it is not known if loss of JAM-A dimerization contributes to human disease. Of note, there is also controversy about the role of JAM-A in cancer. Some tumors show elevated protein levels (breast), whereas others show significant loss (colorectal, gastric, pancreatic, and endometrial), and both changes are associated with poor clinical outcomes. Therefore, an interesting future direction of this work would be to assess JAM-A-expressing human tumor samples for mutations that would ablate dimerization to determine if loss of dimerization contributes to tumor growth through deregulated Hippo signaling.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat Anti-JAM-A | R&D Systems | AF1077; RRID: AB_2100596 |
| Mouse Anti-JAM-A (Clone: J10.4) | In-House Reagent | Commercially Available; sc-53623; RRID: AB_784134 |
| Rabbit Anti-JAM-A | Invitrogen | 36-1700; RRID: AB_2533241 |
| Rabbit Anti-p-YAP S127 | Cell Signaling Technology | 4911; RRID: AB_2218913 |
| Rabbit Anti-YAP1 | Cell Signaling Technology | 14074; RRID: AB_2650491 |
| Rabbit Anti-Ki67 | Abcam | ab15580; RRID: AB_443209 |
| Mouse Anti-ZO-1 | Thermo Fisher Scientific | 33-9100; RRID: AB_2533147 |
| Rabbit Anti-EVI1 | Thermo Fisher Scientific | 711037; RRID: AB_2633107 |
| Rabbit Anti-LATS1 | Cell Signaling Technology | 3477; RRID: AB_2133513 |
| Rabbit Anti-p-LATS1 S909 | Cell Signaling Technology | 9157; RRID: AB_2133515 |
| Rabbit Anti-NF2 | Cell Signaling Technology | 6995; RRID: AB_10828709 |
| Rabbit Anti-NF2 | Cell Signaling Technology | 12888; RRID: AB_2650551 |
| Rabbit Anti-p-NF2/Merlin S518 | Cell Signaling Technology | 9163; RRID: AB_2149793 |
| Rabbit Anti-Calnexin | Sigma-Aldrich | C4731; RRID: AB_476845 |
| Mouse Anti-14-3-3 | BD Bioscience | 561465; RRID: AB_10896967 |
| Rabbit Anti-GAPDH | Sigma-Aldrich | G9545; RRID: AB_796208 |
| Rabbit Anti- ZO-2 | Life Technologies | 389100; RRID: AB_2533390 |
| Mouse Anti-ZO-2 | Thermo Fisher Scientific | 37-4700; RRID: AB_2533321 |
| Mouse Anti-HA | Sigma-Aldrich | H9658; RRID: AB_260092 |
| Rabbit Anti-HA | Sigma-Aldrich | H6908; RRID: AB_260070 |
| Rabbit Anti-pan-TEAD | Cell Signaling | 13295; RRID: AB_2687902 |
| Chemicals, peptides, and recombinant proteins | ||
| FuGENE 6 | Promega | E2691 |
| Lipofectamine™ 3000 | Thermo Fisher Scientific | L3000015 |
| Lipofectamine™ RNAiMAX reagent | Thermo Fisher Scientific | 13778075 |
| Verteporfin | Sigma-Aldrich | SML0534 |
| Tamoxifen | Sigma-Aldrich | T5648 |
| Corn oil | Sigma-Aldrich | C8267 |
| Matrigel | Corning | 365237; Lot 9112015 |
| Recombinant human EGF | R&D Systems | 236-EG |
| Antibiotics/antimycotic | Corning | 30-003-Cl |
| (Ζ)-4-hydroxytamoxifen | Sigma-Aldrich | H7904 |
| Protein A agarose beads | Thermo Fisher Scientific | 20333 |
| Protein G agarose beads | Thermo Fisher Scientific | 20398 |
| Protein A Dynabeads | Thermo Fisher Scientific | 1001D |
| Protein G Dynabeads | Thermo Fisher Scientific | 1003D |
| NuPAGE LDS Sample Buffer | Thermo Fisher Scientific | NP0007 |
| Prolong™ Gold anti-fade agent | Thermo Fisher Scientific | P36930 |
| Antigen unmasking buffer | Vector Laboratories | H-3300-250 |
| SsoAdvanced Universal SYBR Green Supermix | BioRad | 1725270 |
| Critical commercial assays | ||
| DC Protein Assay | Bio-Rad | 5000111 |
| Click-iT™ EdU Alexa Fluor™ 488 Imaging Kit | Invitrogen | C10337 |
| Experimental models: Cell lines | ||
| SKCO-15 | Gift from Enrique Rodriguez-Boulan, Sloan Kettering Institute | N/A |
| SW480 | ATCC | CCL-228™ |
| HEK293T | ATCC | CRL-3216™ |
| Enteroids derived from JAM-AERΔIEC mice | This paper | N/A |
| Enteroids derived from JAM-A-/- mice | This paper | N/A |
| Enteroids derived from JAM-Af/f mice | This paper | N/A |
| Enteroids derived from C57BL/6 mice | This paper | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | Jackson Laboratory | 000664; RRID: IMSR_JAX:000664 |
| JAM-A−/− mice | Gift from T. Sato, Cornell University | N/A |
| JAM-Af/f mice | Flemming et al., 2018 | N/A |
| Villin-CreERT2 mice | Gift from Sylvie Robine, Institut Curie-CNRS | N/A |
| JAM-AERΔIEC mice | This paper | N/A |
| Oligonucleotides | ||
| siRNA Targeting Sequence: JAM-A Target 1 GGGUGACCUUCUUGCCAAUU |
This paper | N/A |
| siRNA Targeting Sequence: JAM-A Target 2 CGGGUGACCUUCUUGCCAAUU |
This paper | N/A |
| siRNA Targeting Sequence: Evi1 Target 1 UCUAAGGCUGAACUAGCAGUUUU |
Adapted from Huang et al. 2016 | N/A |
| siRNA Targeting Sequence: Evi1 Target 2 GCUGAUUGCAGAACCCAAAUUUU |
Adapted from Huang et al. 2016 | N/A |
| siRNA Targeting Sequence: NF2 Target 1 | Thermo Fisher Scientific | s224112 |
| siRNA Targeting Sequence: NF2 Target 2 | Thermo Fisher Scientific | s224112 |
| siRNA Targeting Sequence: Non-Targeting Silencer™ Select Negative Control | Thermo Fisher Scientific | 4390843 |
| siRNA Targeting Sequence: TEAD1 Silencer™ Select | Thermo Fisher Scientific | 4392420; s13962 |
| siRNA Targeting Sequence: TEAD1 Silencer™ Select | Thermo Fisher Scientific | 4392420; s13963 |
| siRNA Targeting Sequence: TEAD2 Silencer™ Select | Thermo Fisher Scientific | 4392420; s16075 |
| siRNA Targeting Sequence: TEAD2 Silencer™ Select | Thermo Fisher Scientific | 4292420; s16076 |
| siRNA Targeting Sequence: TEAD3 Silencer™ Select | Thermo Fisher Scientific | 4292420; s13967 |
| siRNA Targeting Sequence: TEAD3 Silencer™ Select | Thermo Fisher Scientific | 4292420; s13968 |
| siRNA Targeting Sequence: TEAD4 Silencer™ Select | Thermo Fisher Scientific | 4292420; s13964 |
| siRNA Targeting Sequence: TEAD4 Silencer™ Select | Thermo Fisher Scientific | 4292420; s13965 |
|
qPCR Primer Sequence Pair: EVI1 (human) Forward: GACCAAGTTTTTCCTGATTTGC Reverse: CCCTCTCTTCAGTATGTGACAGC |
This paper | N/A |
|
qPCR Primer Sequence Pair: Evi1 (mouse) Forward: TCCTTGAAGTGACTGCCATTC Reverse: ATGCGTACTTTACAGAGATCCG |
This paper | N/A |
|
qPCR Primer Sequence Pair: TBP (human) Forward: TGCACAGGAGCCAAGAGTGAA Reverse: CACATCACAGCTCCCCACCA |
This paper | N/A |
|
qPCR Primer Sequence Pair: Tbp (mouse) Forward: GGAATTGTACCGCAGCTTCAAA Reverse: GATGACTGCAGCAAATCGCTT |
This paper | N/A |
|
qPCR Primer Sequence Pair: β-actin (human) Forward: TCCCTGGAGAAGAGCTACGA Reverse: AGCACTGTGTTGGCGTACAG |
This paper | N/A |
|
qPCR Primer Sequence Pair: CTGF (human) Forward: CCTGGTCCAGACCACAGAGT Reverse: TGGAG ATTTTGGGAGTACGG |
Fallah and Beaulieu, 2020 | N/A |
|
qPCR Primer Sequence Pair: CYR61 (human) Forward: TCCCTGTTTTTGGAATGGAG Reverse: GAGCACTGGGACCATGAAGT |
Fallah and Beaulieu, 2020 | N/A |
| Recombinant DNA | ||
| HA-tagged JAM-A pcDNA3.1 plasmid | Mandell et al. 2004 | N/A |
| JAM-A-WT pcDNA3.1 plasmid | Mandell et al. 2004 | N/A |
| JAM-A-DL1 pcDNA3.1 plasmid | Mandell et al. 2004 | N/A |
| Software and algorithms | ||
| GraphPad Prism version 8.01 | GraphPad Software, Inc. | https://www.graphpad.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Other | ||
| A1 Confocal Microscope | Nikon | N/A |
| Transwell filters | Costar | 3450 |
| Plastic Chamber Slides | Thermo Fisher Scientific | 177445 |
| Epoch microplate spectrophotometer | BioTek | N/A |
| RNeasy kit | Qiagen | 74106 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Charles A. Parkos (cparkos@med.umich.edu).
Materials availability
This study did not generate new unique reagents and all materials in this study are commercially available.
Experimental model and subject detail
Cell culture
SKCO-15 (male), SW480 (male), and HEK293T (female) cells were grown in high glycose DMEM supplemented with 10% fetal bovine serum, 100 IU of penicillin, 100μg/ml streptomycin, 2 mM L- glutamine, 15 mM HEPES, pH 7.4, and 1% non-essential amino acids. HA-tagged JAM-A, JAM- A-WT, and JAM-A-DL1 pcDNA3.1 constructs were described per our previous report (Mandell et al., 2004). Plasmids were transfected with FuGENE 6 (Promega, E2691) for HEK293T cells and Lipofectamine™ 3000 (Thermo Fisher Scientific, L3000015) for SKCO-15 and SW480 cells, according to the manufacturer’s protocols. For knockdown studies, subconfluent (∼20-30% confluent at time of transfection) SKCO-15 or HEK293T cells were transfected with 30pmol siRNA (siRNA sequences detailed in the key resources table) using Lipofectamine™ RNAiMAX reagent (Thermo Fisher Scientific, 13778075). IEC monolayers were harvested 48 hours after transfection (∼50% confluent) and knockdown was verified by Western blot. For Verteporfin studies, cells were treated in the dark for 6 hours with 10μM Verteporfin (Sigma-Aldrich, SML0534) diluted in cell media prior to harvest. For TEAD knockdown studies, 4 siRNA targets (120pmol total siRNA) for each TEAD were applied cells followed by harvest 48 hours later.
Animals
Animal studies were conducted in C57BL/6 wild-type (WT, JAM-A+/+) mice and JAM-A total knockout mice (JAM-A-/-). JAM-A−/− mice were a gift from T. Sato, Cornell University and WT mice were obtained from The Jackson Laboratory. JAMA−/− mice were genotyped as previously described (Cera et al., 2004). Tissue-targeted JAM-A-deficient mice under the control of the murine intestinal epithelial Villin promoter (JAM-AERΔIEC) and their respective littermate controls (JAM-Af/f) were generated by breeding JAM-Af/f mice with Villin-CreERT2 transgenic mice. Depletion of JAM-A was conducted by injecting 6-8-week-old mice intraperitoneally with 1mg/100μL of tamoxifen (Sigma-Aldrich, T5648) dissolved in sterile corn oil (Sigma-Aldrich, C8267) over five consecutive days followed by a 30-day rest period prior to use. Naïve, untreated male and female animals were used between 8-12 weeks of age and all mice were bred in the animal facilities at the University of Michigan. Mice were kept under specific pathogen-free (SPF) conditions with ad libitum normal chow and water. Littermates of the same sex were housed together with up to 5 individuals per cage. Colony rooms were kept on a 12- hour light/12-hour dark cycle in rooms ranging 18–23°C with 40–60% humidity. All experiments were approved and conducted in accordance with guidelines set by the University of Michigan Institutional Animal Care and Use Committee.
Enteroid and colonoid culture
Murine intestinal crypts were isolated from male and female JAM-AERΔIEC and JAM-A f/f mice (mice were sex-matched between genotypes for each experiment), embedded in Matrigel (Corning, 365237, Lot 9112015), and maintained in LWRN-conditioned media supplemented with 50 ng/ml recombinant human EGF (R&D Systems, 236-EG) and antibiotics/antimycotic (Corning, 30-003-Cl). Detailed protocols can be found in our previous report (Muraleedharan et al., 2021). Undifferentiated enteroids were used as 2D-differentiated cells do not proliferate. To acutely deplete JAM-A, enteroid and colonoid cultures were treated for 72 hours with 1μM (Ζ)-4- hydroxytamoxifen (Sigma-Aldrich, H7904) in complete media followed by passage and maintenance in Ζ-4-hydroxytamoxifen-free complete media. To generate 2D monolayers from murine 3D enteroids/colonoids, a single-cell suspension was obtained by disaggregation for 5 minutes with 0.05% Trypsin/0.5 mM EDTA and mechanical force. Dissociated cells were filtered through a 40μm cell strainer, then cultured in collagen type IV-coated 24-well tissue culture chamber slides or Transwell filters (Costar, 3450) until IEC monolayers reached the desired confluency. Murine 2D cultures were maintained in LWRN complete media.
Methods details
Immunoprecipitation and western blotting
For immunoprecipitation studies, SKCO-15 IEC were harvested 48 hours post transfection with siRNA or JAM-A constructs in 1% Triton X-100 lysis buffer (50 mM HEPES, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1% Triton X-100, 10% Glycerol) supplemented with a cocktail of protease and phosphatase inhibitors. Lysates were centrifuged and supernatants collected. Supernatants were pre-cleared with 50μl of 50% protein A or G agarose beads (Thermo Fisher Scientific, 20333, 20398) for one hour followed by 4°C incubation with rotation overnight in the presence of 5μg/mL antibodies or control IgG. Immune complexes were precipitated by 20μl of protein A or G Dynabeads (Thermo Fisher Scientific, 1001D and 1003D) for two hours. Immunoprecipitated complexes were washed three times with cold PBS + before boiling in 2X NuPAGE LDS Sample Buffer (Thermo Fisher Scientific, NP0007). Immunoprecipitates and 2% lysate loading controls were then run on an SDS-PAGE gel and subsequent immunoblotting conducted.
For Western blotting, in vitro cultured human IEC, murine IEC isolated from JAM-A-/- and WT control mice, or 2D primary cultured murine IEC monolayers were harvested using RIPA lysis buffer (10mM Tris-HCl, pH 8.0, 140mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, pH 7.4) supplemented with a cocktail of protease and phosphatase inhibitors. Lysates were cleared by centrifugation and protein concentrations were calculated by DC Protein Assay (Bio-Rad, 5000111) using an Epoch microplate spectrophotometer (BioTek). After boiling, protein samples were run on SDS-PAGE gels and immunoblotted by standard methods (Fan et al., 2019). Calnexin and β-actin served as protein loading controls.
Immunofluorescence and immunohistochemistry
IEC and 2D primary IEC monolayers were grown on Transwell filters (Costar, 3450) or plastic chamber slides (Thermo Fisher Scientific, 177445) and fixed with either 4% paraformaldehyde (PFA) or cold 100% ethanol. In some experiments, to visualize cytoplasmic and nuclear components, 4% PFA fixed monolayers were permeabilized with 0.5% Triton X-100 for 10 minutes. Monolayers were blocked with 2–3% goat or donkey serum in PBS+ with 0.05% Tween-20 blocking buffer for 30 minutes. Primary antibodies were diluted in blocking buffer and cells were incubated overnight at 4°C. Cells were washed with PBS with 0.05% Tween-20 and fluorescently labeled secondary antibodies were diluted in blocking buffer followed by incubation for one hour at room temperature. Cells were washed and mounted in Prolong™ Gold anti-fade agent (Thermo Fisher Scientific, P36930). For IHC staining, murine mucosa was fixed with 4% PFA, embedded in paraffin, and sectioned (7-10 μm) onto glass slides. Paraffin sections were prepared for staining by removing paraffin in xylene and rehydrating in ethanol. Antigen retrieval was performed in antigen unmasking buffer (Vector Laboratories, H-3300-250). 3,3′- Diaminobenzidine (DAB) staining was performed according to the manufacturer’s protocol (Vector Laboratories). Frozen ileal or colonic sections from JAM-A−/− and WT mice were fixed with 4% PFA and immunolabeling performed as described above. Fluorescent imaging was performed on Nikon A1 confocal microscope (Nikon) in the Microscopy & Image Analysis Laboratory Core of the University of Michigan.
qPCR analysis
Total RNA was isolated from murine IEC or SKCO-15 monolayers using a RNeasy kit (Qiagen, 74106) with DNAse I treatment according to the manufacturer’s protocol. For each sample, 1μg of total RNA were transcribed into cDNA using iScript™ Reverse Transcription Supermix (BioRad, 1708841,). Reactions with SsoAdvanced™ Universal SYBR Green Supermix (BioRad, 1725270) were performed in triplicate for each sample. Individual gene expression was detected by qPCR with a BioRad CFX Connect™ Cycler. The relative expression of the gene of interest was calculated using 2ΔΔCt and normalized to the housekeeping genes, TATA-box-binding Protein (TBP) or β-actin, and then normalized to the control condition. The primer sequences can be found in the key resources table.
EdU and MTT assays
Cell proliferation was assessed by EdU incorporation or MTT assay. For the EdU assay, the Click-iT™ EdU Alexa Fluor™ 488 Imaging Kit (Invitrogen, C10337) was used according to the manufacturer’s instructions. Briefly, cells were cultured at low confluency on 4-well chamber slides and incubated with 10μM EdU for two hours. Cells were then fixed, permeabilized, and incubated with the Click-iT™ reaction cocktail followed by staining for nuclei and subsequent imaging. For the MTT assay, HEK293T cells were plated at 50% confluence and then transfected with JAM-A-DL1 or JAM-A-WT constructs. The next day, cells were transfected with either EVI1 or control non-silencing siRNA. Following an overnight incubation, the cells were split into a 96-well plate, in triplicate, at a density of 10,000 cells/well. After 24 hours, the MTT reagent was added for three hours, the cells were then left to incubate overnight with the crystal dissolving solution. The plate was read with an Epoch microplate spectrophotometer (BioTek).
Quantification & statistical analysis
Microscopy data was analyzed using ImageJ. Data was visualized in, and statistical significance assessed by Student’s t-test (independent samples, equal variance, two-tailed), and two-way ANOVA using GraphPad Prism software (GraphPad). The data presented are from three individual experiments and bar graphs represent mean ± standard error of mean (SEM). All data were included in statistical analyses. Significance was set as ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001.
Acknowledgments
We thank the University of Michigan Microscopy Core for facilitating key microscopy experiments. All schematics were created with BioRender.com. This work was supported by the following National Institutes of Health grants: RO1-DK061379, R01-DK079392 (C.A.P.), and T32-DK094775 (M.S.S.).
Author contributions
S.F., M.S.S., J.K., and M.N.O. designed and conducted experiments and analyzed the data. S.F. and M.S.S. wrote the manuscript. C.A.P. and A.N. designed, supervised the project, and facilitated writing of this manuscript. All authors have reviewed and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104316.
Contributor Information
Asma Nusrat, Email: anusrat@med.umich.edu.
Charles A. Parkos, Email: cparkos@med.umich.edu.
Supplemental information
Data and code availability
-
•
Data: All data reported in this paper will be shared by the lead contact upon request.
-
•
Code: This paper does not report original code.
-
•
Additional information: Any additional information required to reanalyze the data reported in this paper can be made available by the lead contact upon request.
References
- Barjesteh van Waalwijk van Doorn-Khosrovani S., Erpelinck C Fau - van Putten W.L.J., van Putten Wl Fau - Valk P.J.M., Valk Pj Fau - van der Poel-van de Luytgaarde S., van der Poel- van de Luytgaarde S Fau - Hack R., Hack R Fau - Slater R., Slater R., Fau - Smit E.M.E., Smit Em Fau - Beverloo H.B., Beverloo Hb Fau - Verhoef G., et al. High EVI1 expression predicts poor survival in acute myeloid leukemia: a study of 319 de novo AML patients. Blood. 2003;101:837–845. doi: 10.1182/blood-2002-05-1459. [DOI] [PubMed] [Google Scholar]
- Bei J.X., Li Y., Jia W.H., Feng B.J., Zhou G., Chen L.Z., Feng Q.S., Low H.Q., Zhang H., He F., et al. A genome-wide association study of nasopharyngeal carcinoma identifies three new susceptibility loci. Nat. Genet. 2010;42:599–603. doi: 10.1038/ng.601. [DOI] [PubMed] [Google Scholar]
- Brodowska K., Al-Moujahed A., Marmalidou A., Meyer Zu Horste M., Cichy J., Miller J.W., Gragoudas E., Vavvas D.G. The clinically used photosensitizer Verteporfin (VP) inhibits YAP-TEAD and human retinoblastoma cell growth in vitro without light activation. Exp. Eye Res. 2014;124:67–73. doi: 10.1016/j.exer.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cera M.R., Del Prete A., Vecchi A., Corada M., Martin-Padura I., Motoike T., Tonetti P., Bazzoni G., Vermi W., Gentili F., et al. Increased DC trafficking to lymph nodes and contact hypersensitivity in junctional adhesion molecule-A-deficient mice. J. Clin. Invest. 2004;114:729–738. doi: 10.1172/JCI21231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.L., Gajewski K.M., Hamaratoglu F., Bossuyt W., Sansores-Garcia L., Tao C., Halder G. The apical-basal cell polarity determinant Crumbs regulates Hippo signaling in Drosophila. Proc. Natl. Acad. Sci. U S A. 2010;107:15810–15815. doi: 10.1073/pnas.1004060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couzens A.L., Knight J.D.R., Kean M.J., Teo G., Weiss A., Dunham W.H., Lin Z.Y., Bagshaw R.D., Sicheri F., Pawson T., et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal. 2013;6:rs15. doi: 10.1126/scisignal.2004712. [DOI] [PubMed] [Google Scholar]
- Ebnet K., Schulz C.U., Meyer Zu Brickwedde M.K., Pendl G.G., Vestweber D. Junctional adhesion molecule interacts with the PDZ domain-containing proteins AF-6 and ZO- 1. J. Biol. Chem. 2000;275:27979–27988. doi: 10.1074/jbc.M002363200. [DOI] [PubMed] [Google Scholar]
- Fallah S., Beaulieu J.F. The Hippo pathway effector YAP1 regulates intestinal epithelial cell differentiation. Cells. 2020;9:1895. doi: 10.3390/cells9081895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan S., Weight C.M., Luissint A.C., Hilgarth R.S., Brazil J.C., Ettel M., Nusrat A., Parkos C.A. Role of JAM-A tyrosine phosphorylation in epithelial barrier dysfunction during intestinal inflammation. Mol. Biol. Cell. 2019;30:566–578. doi: 10.1091/mbc.E18-08-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemming S., Luissint A.C., Nusrat A., Parkos C.A. Analysis of leukocyte transepithelial migration using an in vivo murine colonic loop model. JCI Insight. 2018;3:e99722. doi: 10.1172/jci.insight.99722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong D., Spizzo G., Mitterer M., Seeber A., Steurer M., Gastl G., Brosch I., Moser P. Low expression of junctional adhesion molecule A is associated with metastasis and poor survival in pancreatic cancer. Ann. Surg. Oncol. 2012;19:4330–4336. doi: 10.1245/s10434-012-2381-8. [DOI] [PubMed] [Google Scholar]
- Goyama S., Yamamoto G., Shimabe M., Sato T., Ichikawa M., Ogawa S., Chiba S., Kurokawa M. Evi-1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell. 2008;3:207–220. doi: 10.1016/j.stem.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Harvey K.F., Zhang X., Thomas D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer. 2013;13:246–257. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- Hennigan R.F., Fletcher J.S., Guard S., Ratner N. Proximity biotinylation identifies a set of conformation-specific interactions between Merlin and cell junction proteins. Sci. Signal. 2019;12:eaau8749. doi: 10.1126/scisignal.aau8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong A.W., Meng Z., Guan K.-L. The Hippo pathway in intestinal regeneration and disease. Nat. Rev. Gastroenterol. Hepatol. 2016;13:324–337. doi: 10.1038/nrgastro.2016.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt P.R., Bartholomew C., Davis A.J., Yutzey K., Gamer L.W., Potter S.S., Ihle J.N., Mucenski M.L. The Evil proto-oncogene is required at midgestation for neural, heart, and paraxial mesenchyme development. Mech. Dev. 1997;65:55–70. doi: 10.1016/s0925-4773(97)00057-9. [DOI] [PubMed] [Google Scholar]
- Huang J.F., Wang Y., Liu F., Liu Y., Zhao C.X., Guo Y.J., Sun S.H. EVI1 promotes cell proliferation in HBx-induced hepatocarcinogenesis as a critical transcription factor regulating lncRNAs. Oncotarget. 2016;7:21887–21899. doi: 10.18632/oncotarget.7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.Y., Xu Y.Y., Sun Z., Wang Z.N., Zhu Z., Song Y.X., Luo Y., Zhang X., Xu H.M. Low junctional adhesion molecule A expression correlates with poor prognosis in gastric cancer. J. Surg. Res. 2014;192:494–502. doi: 10.1016/j.jss.2014.06.025. [DOI] [PubMed] [Google Scholar]
- Iden S., Misselwitz S., Peddibhotla S.S., Tuncay H., Rehder D., Gerke V., Robenek H., Suzuki A., Ebnet K. aPKC phosphorylates JAM-A at Ser285 to promote cell contact maturation and tight junction formation. J. Cell Biol. 2012;196:623–639. doi: 10.1083/jcb.201104143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knust E., Bossinger O. Composition and formation of intercellular junctions in epithelial cells. Science. 2002;298:1955–1959. doi: 10.1126/science.1072161. [DOI] [PubMed] [Google Scholar]
- Koos B., Bender S., Witt H., Mertsch S., Felsberg J., Beschorner R., Korshunov A., Riesmeier B., Pfister S., Paulus W., Hasselblatt M. The transcription factor evi-1 is overexpressed, promotes proliferation, and is prognostically unfavorable in infratentorial ependymomas. Clin. Cancer Res. 2011;17:3631–3637. doi: 10.1158/1078-0432.CCR-11-0175. [DOI] [PubMed] [Google Scholar]
- Koshiba H., Hosokawa K., Kubo A., Tokumitsu N., Watanabe A., Honjo H. Junctional adhesion molecule: an expression in human endometrial carcinoma. Int. J. Gynecol. Cancer. 2009;19:208–213. doi: 10.1111/IGC.0b013e31819bc6e9. [DOI] [PubMed] [Google Scholar]
- Kostrewa D., Brockhaus M., D'Arcy A., Dale G.E., Nelboeck P., Schmid G., Mueller F., Bazzoni G., Dejana E., Bartfai T., et al. X-ray structure of junctional adhesion molecule: structural basis for homophilic adhesion via a novel dimerization motif. EMBO J. 2001;20:4391–4398. doi: 10.1093/emboj/20.16.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuta A., Mao Y., Martin T., Ferreira de Sousa C., Whiting D., Zakaria S., Crespo-Enriquez I., Evans P., Balczerski B., Mankoo B., et al. Fat4-Dchs1 signalling controls cell proliferation in developing vertebrae. Development. 2016;143:2367–2375. doi: 10.1242/dev.131037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampis A., Hahne J.C., Gasparini P., Cascione L., Hedayat S., Vlachogiannis G., Murgia C., Fontana E., Edwards J., Horgan P.G., et al. MIR21-induced loss of junctional adhesion molecule A promotes activation of oncogenic pathways, progression and metastasis in colorectal cancer. Cell Death Differ. 2021;28:2970–2982. doi: 10.1038/s41418-021-00820-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukoetter M.G., Nava P., Lee W.Y., Severson E.A., Capaldo C.T., Babbin B.A., Williams I.R., Koval M., Peatman E., Campbell J.A., et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J. Exp. Med. 2007;204:3067–3076. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Zhou H., Li F., Chan S.W., Lin Z., Wei Z., Yang Z., Guo F., Lim C.J., Xing W., et al. Angiomotin binding-induced activation of Merlin/NF2 in the Hippo pathway. Cell Res. 2015;25:801–817. doi: 10.1038/cr.2015.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling C., Zheng Y., Yin F., Yu J., Huang J., Hong Y., Hong Y., Wu S., Pan D. The apical transmembrane protein Crumbs functions as a tumor suppressor that regulates Hippo signaling by binding to Expanded. Proc. Natl. Acad. Sci. U S A. 2010;107:10532–10537. doi: 10.1073/pnas.1004279107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Chittenden Y., Huang B., Shim J.S., Chen Q., Lee S.-J., Anders R.A., Liu J.O., Pan D. Genetic and pharmacological disruption of the TEAD–YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugthart S., van Drunen E., van Norden Y., van Hoven A., Erpelinck C.A.J., Valk P.J.M., Beverloo H.B., Lowenberg B., Delwel R. High EVI1 levels predict adverse outcome in acute myeloid leukemia: prevalence of EVI1 overexpression and chromosome 3q26 abnormalities underestimated. Blood. 2008;111:4329–4337. doi: 10.1182/blood-2007-10-119230. [DOI] [PubMed] [Google Scholar]
- Luissint A.C., Nusrat A., Parkos C.A. JAM-related proteins in mucosal homeostasis and inflammation. Semin. Immunopathol. 2014;36:211–226. doi: 10.1007/s00281-014-0421-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luissint A.C., Parkos C.A., Nusrat A. Inflammation and the intestinal barrier: leukocyte-epithelial cell interactions, cell junction remodeling, and mucosal repair. Gastroenterology. 2016;151:616–632. doi: 10.1053/j.gastro.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell K.J., Babbin B.A., Nusrat A., Parkos C.A. Junctional adhesion molecule 1 regulates epithelial cell morphology through effects on β1 integrins and Rap1 activity. J. Biol. Chem. 2005;280:11665–11674. doi: 10.1074/jbc.M412650200. [DOI] [PubMed] [Google Scholar]
- Mandell K.J., McCall I.C., Parkos C.A. Involvement of the junctional adhesion molecule-1 (JAM1) homodimer interface in regulation of epithelial barrier function. J. Biol. Chem. 2004;279:16254–16262. doi: 10.1074/jbc.M309483200. [DOI] [PubMed] [Google Scholar]
- Mao Y., Mulvaney J., Zakaria S., Yu T., Morgan K.M., Allen S., Basson M.A., Francis-West P., Irvine K.D. Characterization of a Dchs1 mutant mouse reveals requirements for Dchs1-Fat4 signaling during mammalian development. Development. 2011;138:947–957. doi: 10.1242/dev.057166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z., Moroishi T., Guan K.-L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016;30:1–17. doi: 10.1101/gad.274027.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro A.C., Luissint A.C., Sumagin R., Lai C., Vielmuth F., Wolf M.F., Laur O., Reiss K., Spindler V., Stehle T., et al. Trans-dimerization of JAM-A regulates Rap2 and is mediated by a domain that is distinct from the cis-dimerization interface. Mol. Biol. Cell. 2014;25:1574–1585. doi: 10.1091/mbc.e14-01-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro A.C., Sumagin R., Rankin C.R., Leoni G., Mina M.J., Reiter D.M., Stehle T., Dermody T.S., Schaefer S.A., Hall R.A., et al. JAM-A associates with ZO-2, afadin, and PDZ-GEF1 to activate Rap2c and regulate epithelial barrier function. Mol. Biol. Cell. 2013;24:2849–2860. doi: 10.1091/mbc.E13-06-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraleedharan C.K., Mierzwiak J., Feier D., Nusrat A., Quiros M. Generation of murine primary colon epithelial monolayers from intestinal crypts. J. Vis. Exp. 2021;168:1–11. doi: 10.3791/62156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava P., Capaldo C.T., Koch S., Kolegraff K., Rankin C.R., Farkas A.E., Feasel M.E., Li L., Addis C., Parkos C.A., Nusrat A. JAM-A regulates epithelial proliferation through Akt/β-catenin signalling. EMBO Rep. 2011;12:314–320. doi: 10.1038/embor.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S., Kurokawa M., Tanaka T., Tanaka K., Hangaishi A., Mitani K., Kamada N., Yazaki Y., Hirai H. Increased Evi-1 expression is frequently observed in blastic crisis of chronic myelocytic leukemia. Leukemia. 1996;10:788–794. [PubMed] [Google Scholar]
- Prota A.E., Campbell J.A., Schelling P., Forrest J.C., Watson M.J., Peters T.R., Aurrand- Lions M., Imhof B.A., Dermody T.S., Stehle T. Crystal structure of human junctional adhesion molecule 1: implications for reovirus binding. Proc. Natl. Acad. Sci. U S A. 2003;100:5366–5371. doi: 10.1073/pnas.0937718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson B.S., Huang J., Hong Y., Moberg K.H. Crumbs regulates Salvador/Warts/Hippo signaling in Drosophila via the FERM-domain protein Expanded. Curr. Biol. 2010;20:582–590. doi: 10.1016/j.cub.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh M.H., Liu C.J., Laurinec S., Margolis B. The carboxyl terminus of zona occludens-3 binds and recruits a mammalian homologue of discs lost to tight junctions. J. Biol. Chem. 2002;277:27501–27509. doi: 10.1074/jbc.M201177200. [DOI] [PubMed] [Google Scholar]
- Rong R., Surace E.I., Haipek C.A., Gutmann D.H., Ye K. Serine 518 phosphorylation modulates merlin intramolecular association and binding to critical effectors important for NF2 growth suppression. Oncogene. 2004;23:8447–8454. doi: 10.1038/sj.onc.1207794. [DOI] [PubMed] [Google Scholar]
- Severson E.A., Jiang L., Ivanov A.I., Mandell K.J., Nusrat A., Parkos C.A. Cis-dimerization mediates function of junctional adhesion molecule A. Mol. Biol. Cell. 2008;19:1862–1872. doi: 10.1091/mbc.E07-09-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson E.A., Lee W.Y., Capaldo C.T., Nusrat A., Parkos C.A. Junctional adhesion molecule A interacts with afadin and PDZ-GEF2 to activate Rap1A, regulate β1 integrin levels, and enhance cell migration. Mol. Biol. Cell. 2009;20:1916–1925. doi: 10.1091/mbc.E08-10-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley B.K., Irvine K.D. Hippo signaling in Drosophila: recent advances and insights. Dev. Dyn. 2012;241:3–15. doi: 10.1002/dvdy.22723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T.K., Allaei R., Silverstein K.A.T., Staggs R.A., Sarver A.L., Bergemann T.L., Gupta M., O'Sullivan M.G., Matise I., Dupuy A.J., et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science. 2009;323:1747–1750. doi: 10.1126/science.1163040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbacher T., Kummer D., Ebnet K. Junctional adhesion molecule-A: functional diversity through molecular promiscuity. Cell Mol. Life Sci. 2018;75:1393–1409. doi: 10.1007/s00018-017-2729-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surace E.I., Haipek C.A., Gutmann D.H. Effect of merlin phosphorylation on neurofibromatosis 2 (NF2) gene function. Oncogene. 2004;23:580–587. doi: 10.1038/sj.onc.1207142. [DOI] [PubMed] [Google Scholar]
- Van Itallie C.M., Anderson J.M. Phosphorylation of tight junction transmembrane proteins: many sites, much to do. Tissue Barriers. 2018;6:e1382671. doi: 10.1080/21688370.2017.1382671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrano S., Rescigno M., Rosaria Cera M., Correale C., Rumio C., Doni A., Fantini M., Sturm A., Borroni E., Repici A., et al. Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology. 2008;135:173–184. doi: 10.1053/j.gastro.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Wang C., Zhu X., Feng W., Yu Y., Jeong K., Guo W., Lu Y., Mills G.B. Verteporfin inhibits YAP function through up-regulating 14-3-3σ sequestering YAP in the cytoplasm. Am. J. Cancer Res. 2016;6:27–37. [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Zheng J., Han Y., Zhang Y., Su L., Hu D., Fu X. JAM-A knockdown accelerates the proliferation and migration of human keratinocytes, and improves wound healing in rats via FAK/Erk signaling. Cell Death Dis. 2018;9:848. doi: 10.1038/s41419-018-0941-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C., Troutman S., Fera D., Stemmer-Rachamimov A., Avila J.L., Christian N., Persson N.L., Shimono A., Speicher D.W., Marmorstein R., et al. A tight junction-associated Merlin-angiomotin complex mediates Merlin's regulation of mitogenic signaling and tumor suppressive functions. Cancer Cell. 2011;19:527–540. doi: 10.1016/j.ccr.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Pasolli H.A., Fuchs E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc. Natl. Acad. Sci. U S A. 2011;108:2270–2275. doi: 10.1073/pnas.1019603108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B., Ye X., Yu J., Li L., Li W., Li S., Yu J., Lin J.D., Wang C.Y., Chinnaiyan A.M., et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P.J., Wang X., An N., Wei L., Zhang L., Huang X., Zhu H.H., Fang Y.X., Gao W.Q. Loss of Par3 promotes prostatic tumorigenesis by enhancing cell growth and changing cell division modes. Oncogene. 2019;38:2192–2205. doi: 10.1038/s41388-018-0580-x. [DOI] [PubMed] [Google Scholar]
- Zihni C., Mills C., Matter K., Balda M.S. Tight junctions: from simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016;17:564–580. doi: 10.1038/nrm.2016.80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: All data reported in this paper will be shared by the lead contact upon request.
-
•
Code: This paper does not report original code.
-
•
Additional information: Any additional information required to reanalyze the data reported in this paper can be made available by the lead contact upon request.