

Abstract
A series of allosteric kidney-type glutaminase (GLS) inhibitors possessing a mercaptoethyl (-SCH2CH2-) linker were synthesized in an effort to further expand the structural diversity of chemotypes derived from bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES), a prototype allosteric inhibitor of GLS. BPTES analog 3a with a mercaptoethyl linker between the two thiadiazole rings was found to potently inhibit GLS with an IC50 value of 50 nM. Interestingly, the corresponding derivative with an n-propyl (-CH2CH2CH2-) linker showed substantially lower inhibitory potency (IC50 = 2.3 μM) while the derivative with a dimethylsulfide (-CH2SCH2-) linker showed no inhibitory activity at concentrations up to 100 μM, underscoring the critical role played by the mercaptoethyl linker in the high affinity binding to the allosteric site of GLS. Additional mercaptoethyl-linked compounds were synthesized and tested as GLS inhibitors to further explore SAR within this scaffold including derivatives possessing a pyridazine as a replacement for one of the two thiadiazole moiety.
Graphic Abstract
1. Introduction
Given the central importance of glutamine in reprogrammed energy metabolism of malignant cells, kidney-type glutaminase (GLS) has gained growing interest as a therapeutic target for the treatment of cancer.1, 2 Since the discovery of bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES, Fig. 1),3 the first reported allosteric inhibitor of GLS,4 extensive structural optimization efforts have been collectively made by a number of research groups with a focus towards clinical translation.5–7 This led to the discovery of CB-839 (Fig. 1),8 a first-in-class GLS inhibitor, currently investigated in clinical trials.
Fig. 1.
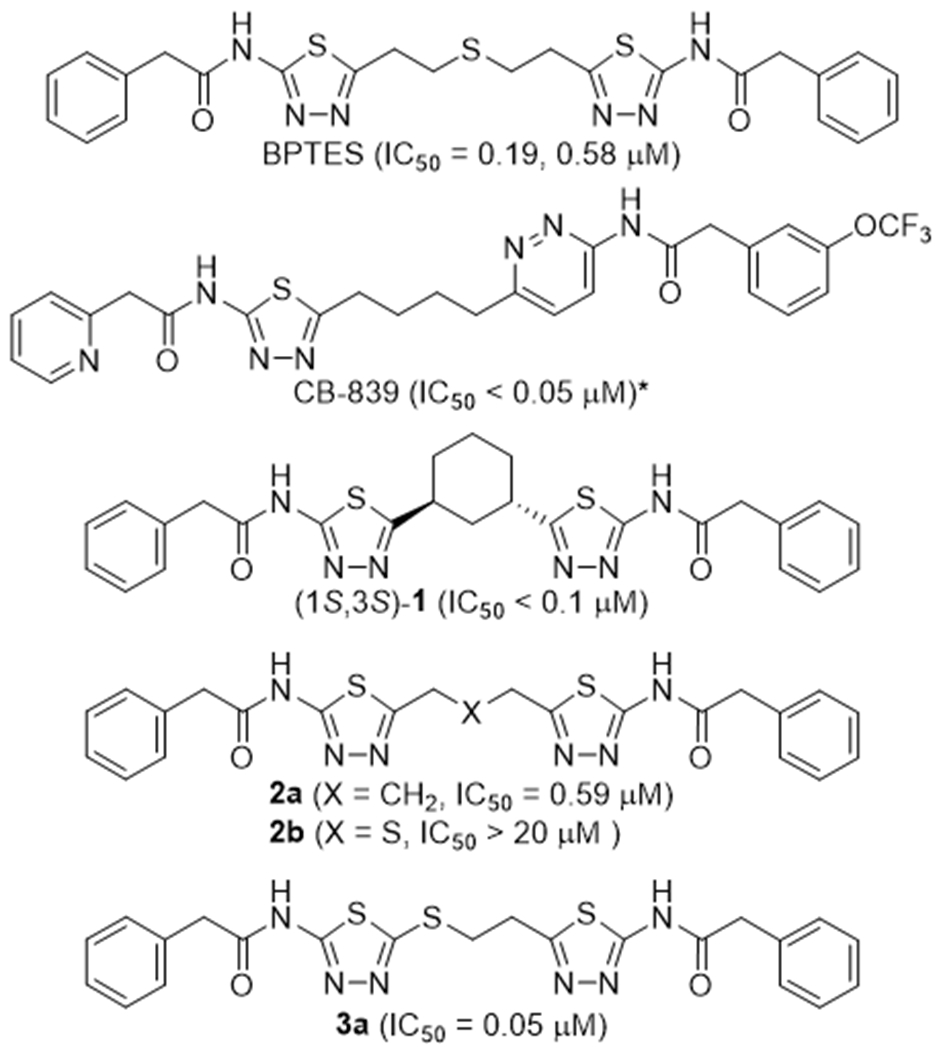
Representative allosteric GLS inhibitors and compound 3a containing a mercaptoethyl linker. The IC50 values are taken from the original publications3, 8–10 except for compound 3a. *After 60-min preincubation.
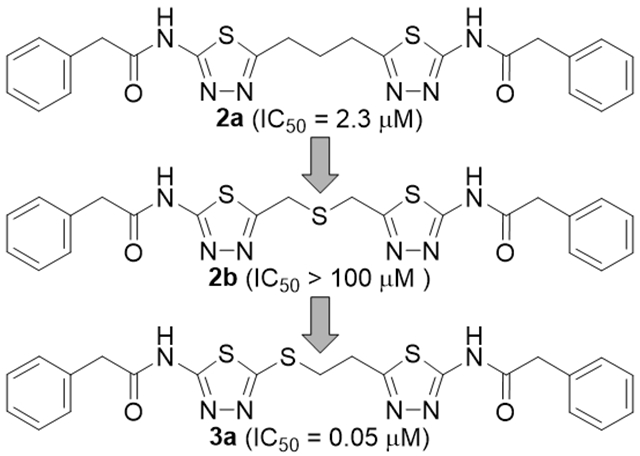
Examination of the SAR data for BPTES-derived compounds accumulated to date suggests that the allosteric sites of GLS tolerate many forms of linker that connect the two heterocycles. For instance, while BPTES has a diethylsulfide (-CH2CH2SCH2CH2-) linker connecting the two thiadiazole rings, two heterocycles in CB-839 are connected by an n-butyl (-CH2CH2CH2CH2-) linker devoid of the central sulfide group. Furthermore, incorporation of a cyclic system into the linker created further diversity in the structure of GLS inhibitors derived from BPTES,5 including compound (1S,3S)-1 containing a 1,3-cyclohexane linker.9 The co-crystal structure of GLS in complex with BPTES (PDB: 3UO9) suggests that the acyclic diethylsulfide (-CH2CH2SCH2CH2-) linker of BPTES is somewhat compressed to fit the allosteric binding site of GLS.10 This explains why compound (1S,3S)-1 with a substantially shorter linker can retain high GLS inhibitory potency. Indeed, the co-crystal structure of GLS in complex with (1S,3S)-1 (PDB: 5JYP)11 displayed the same network of hydrogen bonds as seen with BPTES (PDB: 3UO9). Subsequently, other cyclic systems have also been found to serve as effective linkers in GLS inhibitors.12, 13 Potent inhibition shown by compound (1S,3S)-1 indicates that an n-propyl (-CH2CH2CH2-) linker can also be very effective. Indeed, the corresponding BPTES analog 2a (Fig. 1) was found to inhibit GLS with submicromolar potency (IC50 = 0.59 μM).14 In contrast, compound 2b (Fig. 1) possessing a dimethylsulfide (-CH2SCH2-) linker showed substantially lower inhibitory activity (IC50 > 20 μM).14 These findings prompted us to explore other short acyclic linkers in the pursuit of a new series of GLS inhibitors derived from BPTES. To our surprise, BPTES analog 3a containing a mercaptoethyl (-SCH2CH2-) linker was found to potently inhibit GLS with an IC50 value of 0.05 μM, providing a new scaffold for the development of potent GLS inhibitors. Herein, we report the discovery of a series of mercaptoethyl-linked derivatives of BPTES as a new subclass of allosteric GLS inhibitors.
2. Results and discussion
As shown in Scheme 1, some of the mercaptoethyl-linked compounds were synthesized using 5-amino-1,3,4-thiadiazole-2-thiol 4 as a starting material. Reaction of 4 with tert-butyl acrylate in the presence of triethylamine gave the Michael adduct 5. After HATU-mediated N-acylation of 5 with phenylacetic acid, the tert-butyl ester of the resulting compound 6 was hydrolyzed by TFA, providing S-substituted 3-mercatopropionic acid 7. The carboxylic acid of 7 was converted into a 2-amino-1,3,4-thiadiazole moiety by treating with thiosemicarbazide and POCl3 to give compound 8. N-Acylation of 8 with phenylacetic acid mediated by HATU gave compound 3a, a mercaptoethyl-linked derivative of BPTES. Intermediate 8 was also coupled with 2-(pyridin-2-yl)acetic acid, 2-(3-(trifluoromethoxy)phenyl)acetic acid, and 2-(4-(dimethylamino)phenyl)acetic acid, affording 3b, 3c, and 3d, respectively.
Scheme 1.
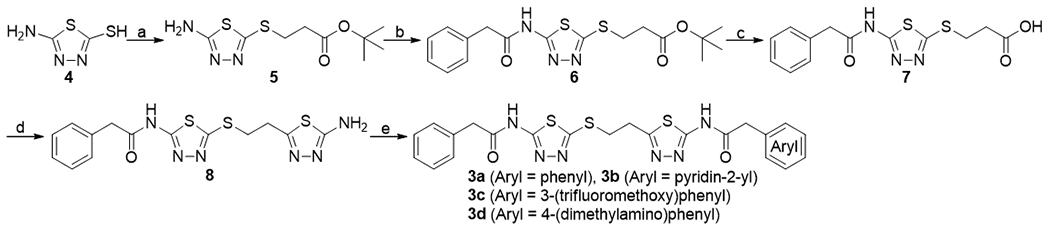
Synthesis of compounds 8 and 3a-3d. aReagents and conditions: (a) triethylamine, tert-butyl acrylate, MeOH, rt to 65 °C, 20%; (b) diisopropylethylamine, phenylacetic acid, HATU, DMF, 0 °C to rt, 100%; (c) trifluoroacetic acid, DCM, rt, 99%; (d) thiosemicarbazide, POCl3, 90 °C, 93%; (e) diisopropylethylamine, arylacetic acid, HATU, DMF, 0 °C to rt, 45% for 3a, 28% for 3b, 79% for 3c, and 42% for 3d.
As shown in Scheme 2, various acyl groups can be also introduced to the 5-amino-1,3,4-thiadiazol-2-yl)mercapto side of the scaffold by starting from 3-(benzyloxy)propanenitrile 9. The nitrile group of 9 was converted into a 2-amino-1,3,4-thiadiazole moiety by treating with thiosemicarbazide in TFA to give compound 10. N-Acylation with phenylacetyl chloride gave compound 11. After removal of the benzyl group with BBr3, the resulting primary alcohol 12 was transformed to the corresponding mesylate ester 13, which was subsequently reacted with 5-amino-1,3,4-thiadiazole-2-thiol 4 to give compound 14. Coupling of 14 with (pyridin-2-yl)acetic acid was most effectively achieved by using propylphosphonic anhydride (T3P) and triethylamine to afford the desired product 15a. 3-(Trifluoromethoxy)phenylacetamide derivative 15b was synthesized by HATU-mediated coupling of 14 and 3-(trifluoromethoxy)phenylacetic acid.
Scheme 2.
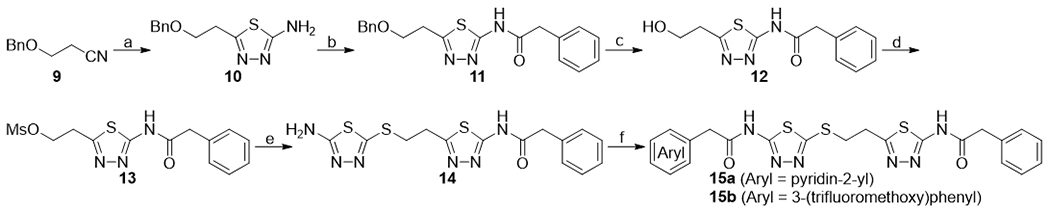
Synthesis of compounds 14 and 15a-15b. aReagents and conditions: (a) thiosemicarbazide, TFA, 65 °C, 57%; (b) triethylamine, phenylacetyl chloride, DCM, rt, 100%; (c) boron tribromide, DCM, rt; (d) mesyl chloride, DCM, 0 °C, 65% from 11; (e) 5-amino-1,3,4-thiadiazole-2-thiol, K2CO3, DMF, rt, 52%; (f) (pyridin-2-yl)acetic acid or 3-(trifluoromethoxy)phenylacetic acid, T3P, triethylamine, DMF, 0 °C, 23% for 15a, 26% for 15b.
We also synthesized derivatives in which one of the two thiadiazole rings was replaced by a pyridazine ring. As shown in Scheme 3, copper-catalyzed coupling of (6-iodopyridazin-3-yl)-2-phenylacetamide 16 with 1715 produced compound 18, which was subsequently reacted with phenylacetyl chloride to afford compound 19.
Scheme 3.
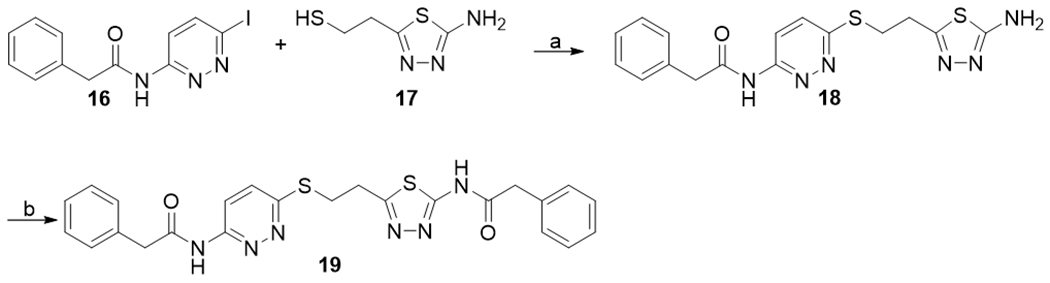
Synthesis of compounds 18 and 19. aReagents and conditions: (a) CuI, K2CO3, HO(CH2)2OH, i-PrOH, 80 °C, 40%; (b) triethylamine, phenylacetyl chloride, DCM, rt, 66%.
The other thiadiazole ring was also successfully replaced by a pyridazine ring (Scheme 4). Palladium-catalyzed Suzuki coupling of 16 with allylboronic acid pinacol ester 20 provided compound 21. Ozonolysis of 21 followed by treatment with sodium borohydride afforded primary alcohol 22. Compound 22 was converted into the corresponding mesylate 23, which was subsequently reacted with 5-amino-1,3,4-thiadiazole-2-thiol 4 to give compound 24. N-Acylation of 24 with phenylacetic acid mediated by HATU gave the desired product 25.
Scheme 4.
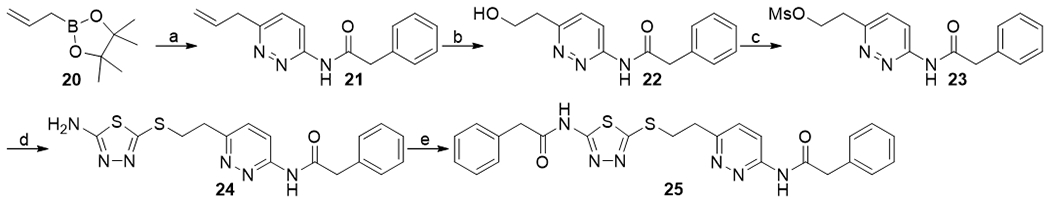
Synthesis of compounds 24 and 25. aReagents and conditions: (a) 16, Pd(Ph3)4, potassium carbonate, dioxane, 120 °C, 42%; (b) (i) O3, DCM, −78 °C; (ii) sodium borohydride, −78 to 0 °C, 14%; (c) mesyl chloride, triethylamine, DCM, 0 °C, (d) 4, potassium carbonate, ACN, 50 °C; (e) phenylacetic acid, diisopropylethylamine, HATU, DMF, 0 °C to rt, 11% (from 22).
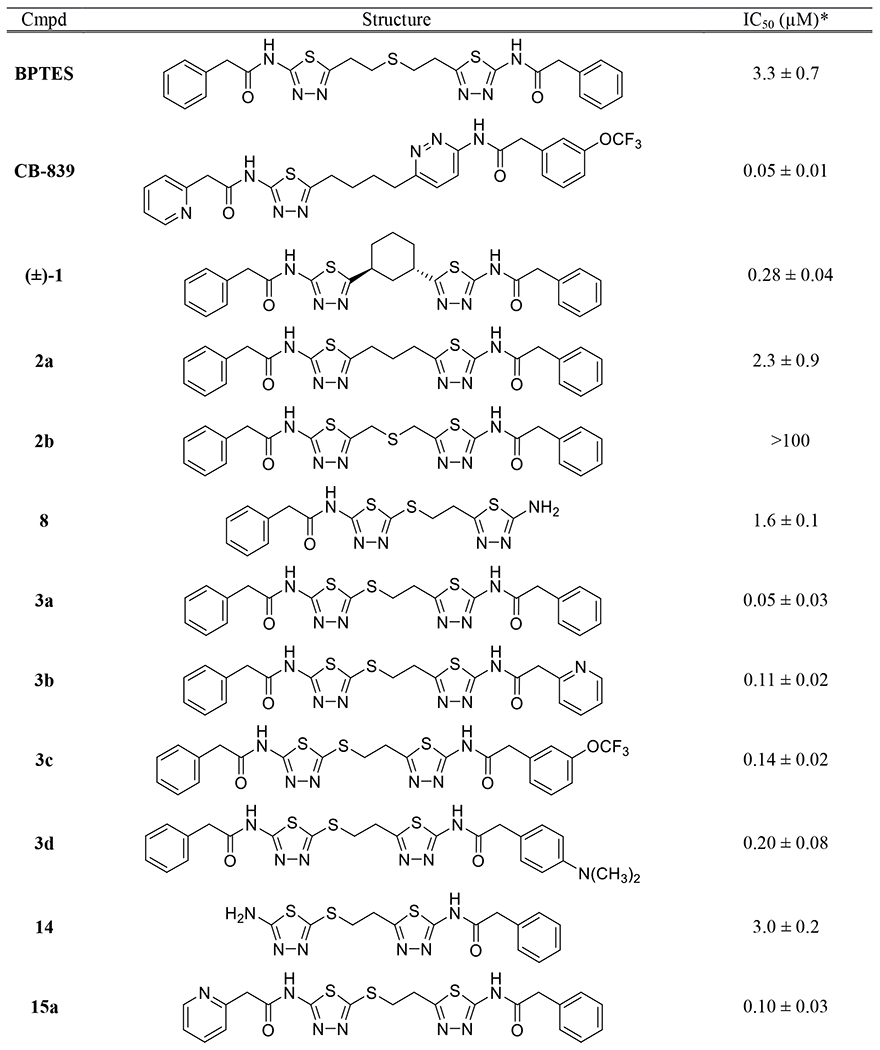
All new synthesized compounds were tested for their ability to inhibit GLS using L-[3H]-glutamine as substrate and human kidney-type glutaminase (hKGA124-669).16 Since IC50 values of GLS inhibitors are known to be highly assay-dependent, we used some of the representative inhibitors shown in Fig. 1 as controls in our assay, enabling a head-to-head comparison with the new compounds tested. In our assay, BPTES and CB-839 inhibited GLS with IC50 values of 3.3 and 0.05 μM, respectively. Compounds (±)-1 and 2a exhibited IC50 values of 0.28 and 2.3 μM, respectively, while compound 2b showed no inhibitory activity at concentrations up to 100 μM.
Compound 8 containing a mercaptoethyl linker was found to inhibit GLS with an IC50 value of 1.6 μM. As seen with compound 3a, incorporation of an additional phenylacetyl group into 8 resulted in substantial improvement of GLS inhibitory potency with an IC50 value of 50 nM.
It is worth noting that the corresponding analog containing an n-propyl (compound 2a) linker showed a nearly 50-fold reduction in inhibitory potency and compound 2b displayed negligible activity despite its structural similarity to compound 3a. Although the precise reason for the high inhibitory potency of 3a remains to be investigated, it is conceivable that noncovalent sulfur interaction plays an important role in the binding of 3a to the allosteric binding site of GLS. Among various forms of noncovalent sulfur interactions,17 the sulfur-aromatic interaction appears to be most relevant to our case given that Tyr394 and/or Phe322 is anticipated to be in the vicinity of the sulfide moiety of 3a bound to the allosteric site of GLS based on its co-crystal structures with various BPTES-derived GLS inhibitors.10–12, 18 Computational model studies have indicated that certain configurations for the complexes of dimethylsulfide with aryl rings lead to greater stabilization compared to the corresponding propane-containing complexes.19 Although speculative, these findings may explain the much higher GLS inhibitory potency of compound 3a containing a mercaptoethyl linker as opposed to compound 2a with an n-propyl linker. Besides the potent inhibitory GLS activity, compound 3a exhibited excellent phase I metabolic stability (87% remaining after 60-min incubation) in mouse liver microsomes fortified with NADPH, providing a promising molecular template for further structural optimization.
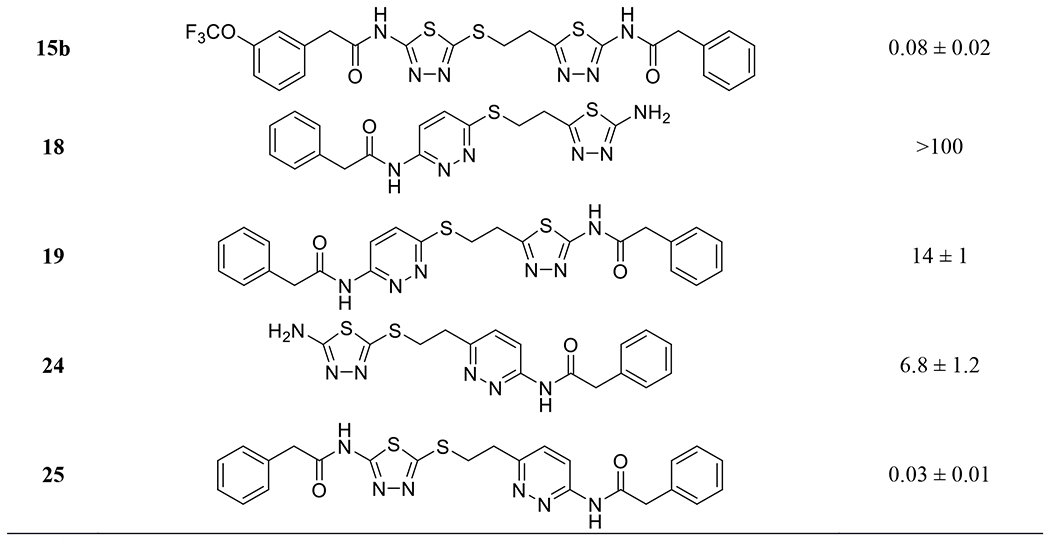
Additional analogs containing a mercaptoethyl-linked bis(thiadiazole) consistently showed submicromolar inhibitory potency ranging from 0.1 to 0.2 μM. One exception is compound 14 lacking one phenylacetyl group, which inhibited GLS with an IC50 value of 3.0 μM, similar to that of compound 8 devoid of a phenylacetyl group on the other side of compound 3a.
As demonstrated by CB-839, a pyridazine ring is one of the most effective alternatives to the thiadiazole ring found in many GLS inhibitors.8 Compounds 18 and 19 possess a pyridazine ring on the sulfur end of the mercatoethyl linker. These compounds displayed substantially weaker potency toward GLS compared to their thiadiazole counterparts 8 and 3a. In contrast, compounds 24 and 25 containing a pyridazine ring on the carbon side of the mercaptoethyl linker showed GLS inhibitory potency comparable to those of compounds 8 and 3a. These findings suggest that 2-mercapto-1,3,4-thiadiazole moiety is uniquely essential for the potent GLS inhibitory activity shown by our new series of GLS inhibitors.
Because of its highly lipophilic molecular properties, BPTES is known to have very poor aqueous solubility.16 Given its overall structural similarity to BPTES, compound 3a was expected to have an equally poor solubility. Indeed, our attempt to assess the antiproliferative effects of compound 3a at final concentrations of 1-10 μM failed due to precipitation that occurred upon addition of the stock solution of 3a to cell culture media. Our efforts to incorporate a pyridazine ring as an alternative to one of the two thiadiazole rings in compound 3a were motivated by its solubility-enhancing effect observed in other cases.20 Disappointingly, however, there was no improvement in solubility of compound 25 with a saturation concentration below 1 μM despite the presence of a pyridazine ring.
3. Conclusion
Allosteric GLS inhibitors derived from BPTES show a great deal of structural diversity as a result of continuous medicinal chemistry efforts made by many during the past decade. Structural modification of the linker component of GLS inhibitors was found to be particularly well tolerated, generating many variations in this region. The mercaptoethyl moiety represents another promising linker that can be utilized in BPTES-derived GLS inhibitors. As demonstrated by our SAR studies, GLS inhibitors containing the mercaptoethyl linker consistently showed potent GLS inhibitory activity as long as the mercapto side is attached to a 1,3,4-thiadiazole ring. One major challenge to be addressed is poor aqueous solubility which limits further pharmacological characterization. It should be noted that BPTES-derived inhibitors can incorporate a variety of acyl groups to both sides without substantial loss in GLS inhibitory potency. As such, introduction of a soluble moiety to these sites might represent one way to improve aqueous solubility while maintaining the potent inhibitory activity within this series of GLS inhibitors. Another interesting direction for further structural medications would be to replace the 1,3,4-thiadiazole with a 1,3,4-selenadiazole ring, which was reported to serve as a very effective moiety in GLS inhibitors.21
4. Experimental
4.1. Chemical synthesis
4.1.1. General
All solvents were reagent grade or HPLC grade. Unless otherwise noted, all materials were obtained from commercial suppliers and used without further purification. The preparation of 5,5’-thiobis(methylene)bis(1,3,4-thiadiazol-2-amine) has been previously reported.14 Melting points were obtained on a Mel-Temp apparatus and are uncorrected. 1H NMR spectra were recorded at 400 or 500 MHz. 13C NMR spectra were recorded at 100 or 125 MHz. In some cases, signals of quaternary carbons in 13C NMR were not observable. Ozone was obtained from oxygen in a GE60/FM100 tabletop generator (Ozone Services). The HPLC solvent system consisted of deionized water and acetonitrile, both containing 0.1% formic acid. Preparative HPLC purification was performed on an Agilent 1200 series HPLC system equipped with an Agilent G1315D DAD detector using a Phenomenex Luna 5 μm C18 column (21.2 mm × 250 mm, 5 μm). Analytical HPLC was performed on an Agilent 1200 series HPLC system equipped with an Agilent G1315D DAD detector (detection at 220 nm) and an Agilent 6120 quadrupole MS detector. The analytical HPLC conditions involve either (A) 20% acetonitrile/80% water for 0.25 min followed by gradient to 85% acetonitrile/15% water over 1.5 min and continuation of 85% acetonitrile/15% water for 2.25 min with an Agilent Eclipse Plus C18 column (2.1 mm × 50 mm, 3.5 μm) at a flow rate of 1.25 mL/min; or (B) 5% acetonitrile/95% water for 0.25 min followed by gradient to 40% acetonitrile/60% water over 1.5 min and continuation of 85% acetonitrile/15% water for 2.25 min with an Agilent Eclipse Plus C18 column (2.1 mm × 50 mm, 3.5 μm) at a flow rate of 1.25 mL/min. Unless otherwise noted, all final compounds biologically tested were confirmed to be of ≥ 95% purity by the HPLC methods described above.
4.1.2. N,N’-(5,5’-Thiobis(methylene)bis(1,3,4-thiadiazole-5,2-diyl))bis(2-phenylacetamide) (2b)
To a solution of 5,5’-thiobis(methylene)bis(1,3,4-thiadiazol-2-amine)14 (1.20 g, 4.61 mmol) in DMA (20 mL) was added triethylamine (2.43 g, 24.0 mmol) and phenylacetyl chloride (1.40 g, 9.06 mmol), and the mixture was stirred overnight at rt. The solvent was removed in vacuo and the resultant residue was triturated with 10% NaHCO3 (75 mL), with hot MeOH (75 mL), and then dried to afford 900 mg of 2b as an off-white solid (40% yield); 1H NMR (DMSO-d6) δ 3.81 (s, 4H), 4.13 (s, 4H), 7.25-7.28 (m, 2H) 7.31 (m, 8H), 12.76 (s, 2H). 13C NMR (DMSO-d6) δ 28.6, 41.5, 126.9, 128.4, 129.3, 134.5, 159.3, 162.0, 169.5. Analytical HPLC (Method A); retention time 1.91 min, m/z 497.1 [M + H]+.
4.1.3. tert-Butyl 3-(5-amino-1,3,4-thiadiazol-2-ylthio)propanoate (5)
To a solution of 5-amino-1,3,4-thiadiazole-2-thiol 4 (17.5 g, 131 mmol) in MeOH (160 mL) was added triethylamine (15.9 g, 157 mmol) and tert-butyl acrylate (20.2 g, 158 mmol) by addition funnel. The mixture was stirred at rt for 1 h and then stirred at 65 °C for 18 h. The solvent was removed in vacuo and the residue was partitioned between DCM (200 mL) and H2O (100 ml). The organic layer was dried over MgSO4, filtered, and concentrated to ~10 mL volume when precipitation occurs. The resultant solid was collected by vacuum filtration and washed with Et2O to afford 7.01 g of 5 as an off-white solid (20% yield); 1H NMR (DMSO-d6) δ 1.40 (s, 9H), 2.63 (t, J = 6.8 Hz, 2H), 3.19 (t, J = 6.8 Hz, 2H), 7.33 (brs, 2H).
4.1.4. tert-Butyl 3-(5-(2-phenylacetamido)-1,3,4-thiadiazol-2-ylthio)propanoate (6)
To a solution of tert-butyl 3-(5-amino-1,3,4-thiadiazol-2-ylthio)propanoate 5 (7.00 g, 26.8 mmol) in DMF (100 mL) was added DIEA (13.8 g, 107 mmol) and phenylacetic acid (4.01 g, 29.5 mmol). HATU (11.2 g, 29.5 mmol) was added at 0 °C and the resulting mixture was warmed to room temperature overnight. The solvent was removed in vacuo, and the residue was taken up in DCM (200 mL) and washed with 10% KHSO4 (100 mL), sat’d NaHCO3 (100 mL), and brine (100 mL). The organic layer was then dried over MgSO4 and concentrated. The resultant solid was triturated with 20% ethyl acetate/hexanes and then Et2O to afford 10.2 g of 6 as a white solid (100% yield); 1H NMR (CDCl3) δ 1.42 (s, 9H), 2.75 (t, J = 7.0 Hz, 2H), 3.45 (t, J = 7.0 Hz, 2H), 4.01 (s, 2H), 7.28-7.45 (m, 5H), 12.32 (s, 1H).
4.1.5. 3-(5-(2-Phenylacetamido)-1,3,4-thiadiazol-2-ylthio)propanoic acid (7)
To a solution of 6 (10.2 g, 27.0 mmol) in DCM (50 mL) was added TFA (50 mL). The mixture was stirred at rt for 2 h and then concentrated to give 8.68 g of 7 as an off-white solid that was used in the subsequent step without further purification (99% yield). 1H NMR (DMSO-d6) δ 2.70 (t, J = 6.7 Hz, 2H), 3.36 (t, J = 6.7 Hz, 2H), 3.82 (s, 2H), 7-26-7.35 (m, 5H), 12.90 (brs, 1H).
4.1.6. N-(5-(2-(5-Amino-1,3,4-thiadiazol-2-yl)ethylthio)-1,3,4-thiadiazol-2-yl)-2-phenylacetamide (8)
To a solution of 3-(5-(2-phenylacetamido)-1,3,4-thiadiazol-2-ylthio)propanoic acid 7 (8.68 g, 26.8 mmol) in POCl3 (50 mL) was added thiosemicarbazide (7.34 g, 80.5 mmol), and the mixture was heated at 90 °C for 1.5 h. The reaction was cooled to room temperature, poured into 100 mL of ice and stirred vigorously. The resultant solid was washed with copious amounts of water to afford 9.43 g of 8 as a white solid (93% yield); 1H NMR (DMSO-d6) δ 3.24 (t, J = 6.8 Hz, 2H), 3.54 (t, J = 6.8 Hz, 2H), 3.82 (s, 2H), 7.21-7.35 (m, 5H), , 12.91 (brs, 1H). 13C NMR (DMSO-d6) δ 29.6, 32.3, 41.4, 127.0, 128.5, 129.3, 134.4, 155.5, 158.1, 158.8, 168.8, 169.6. HPLC (Method A); retention time 1.41 min, m/z 379.1 [M + H]+.
4.1.7. 2-Phenyl-N-(5-(2-(5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl)ethylthio)-1,3,4-thiadiazol-2-yl)acetamide (3a)
To a solution of N-(5-(2-(5-amino-1,3,4-thiadiazol-2-yl)ethylthio)-1,3,4-thiadiazol-2-yl)-2-phenylacetamide 8 (770 mg, 2.03 mmol) in DMF (30 mL) was added phenylacetic acid (304 mg, 2.24 mmol) and diisopropylethylamine (1.05 g, 8.14 mmol). The mixture was cooled to 0 °C and HATU (773 mg, 2.03 mmol) was added. The reaction was allowed to warm to rt overnight, and the solvent was removed in vacuo. The resultant residue was taken up in DCM (100 mL) and washed with 10% KHSO4, (50 mL), sat’d NaHCO3 (50 mL), and brine (50 mL). The organic layer was dried over MgSO4, filtered, and concentrated. The residual product was triturated with 20% ethylacetate/hexanes to afford 450 mg of 3a as a tan solid (45% yield); 1H NMR (DMSO-d6) δ 3.41 (t, J = 7.0 Hz, 2H), 3.60 (t, J = 7.0 Hz, 2H), 3.80 (s, 2H), 3.81 (s, 2H), 7.24-7.45 (m, 10H), 12.72 (brs, 1H), 12.87 (brs, 1H). 13C NMR (DMSO-d6) δ 29.1, 32.4, 41.4, 41.5, 126.9, 127.0, 128.4, 129.2, 129.3, 134.4, 134.6, 157.9, 158.7, 158.8, 161.4, 169.4, 169.6. HPLC (Method A); retention time 1.98 min, m/z 497.1 [M + H]+.
4.1.8. 2-Phenyl-N-(5-(2-(5-(2-(pyridin-2-yl)acetamido)-1,3,4-thiadiazol-2-yl)ethylthio)-1,3,4-thiadiazol-2-yl)acetamide (3b)
Compound 3b was prepared from 8 as was described for 3a except 2-(pyridin-2-yl)acetic acid hydrochloride was used in place of phenylacetic acid. The solvent was removed in vacuo, and the residual material was triturated with sat’d NaHCO3, water, and then methanol to give 3b as a tan solid (28% yield); 1H NMR (DMSO-d6) δ 3.42 (t, J = 6.8 Hz, 2H), 3.61 (t, J = 6.8 Hz, 2H), 3.81 (s, 2H), 4.01 (s, 2H), 7.26-7.33 (m, 6H), 7.39 (d, J = 8.0 Hz, 1H), 7.77 (td, J = 7.6, 2.0 Hz, 1H). 8.48-8.50 (d, J = 4.0 Hz, 1H), 12.76 (s, 1H), 12.92 (s, 1H). 13C NMR (DMSO-d6) δ 29.1, 32.4, 41.4, 44.1, 122.2, 124.2, 127.0, 128.5, 129.3, 134.5, 136.8, 149.2, 154.9, 158.0, 158.7, 158.9, 161.5, 168.6, 169.7. HPLC (Method A); retention time 1.93 min, m/z 498.1 [M + H]+.
4.1.9. 2-Phenyl-N-(5-(2-(5-(2-(3-(trifluoromethoxy)phenyl)acetamido)-1,3,4-thiadiazol-2-yl)ethylthio)-1,3,4-thiadiazol-2-yl)acetamide (3c)
Compound 3c was prepared from 8 as was described for 3a except 2-(3-(trifluoromethoxy)phenyl)acetic acid was used in place of phenyl acetic acid. The solvent was removed in vacuo, and the residual material was triturated with sat’d NaHCO3 and then water to afford the crude product which was then chromatographed with 2% to 15% MeOH/DCM to afford 3c as a white solid (79% yield); 1H NMR (DMSO-d6) δ 3.42 (t, J = 7.2 Hz, 2H), 3.61 (t, J = 7.2 Hz, 2H), 3.81 (s, 2H), 3.90 (s, 2H), 7.26-7.36 (m, 8H), 7.45-7.49 (m, 1H), 12.78 (brs, 1H), 12.91 (brs, 1H). 13C NMR (DMSO-d6) 29.1, 32.4, 40.9, 41.4, 120.0 (q, J = 258 Hz), 119.4, 121.9, 126.9, 128.4, 128.6, 129.3, 130.3, 134.4, 137.2, 148.3, 157.9, 158.9, 161.5, 168.9, 169.6. HPLC (Method A); retention time 2.65 min, m/z 581.1 [M + H]+.
4.1.10. 2-(4-(Dimethylamino)phenyl)-N-(5-(2-(5-(2-phenylacetamido)-1,3,4-thiadiazol-2-ylthio)ethyl)-1,3,4-thiadiazol-2-yl)acetamide (3d)
Compound 3d was prepared from 8 as was described for 3a except 2-(4-(dimethylamino)phenyl)acetic acid was used in place of phenylacetic acid. The solvent was removed in vacuo, and the residual material was triturated with sat’d NaHCO3, water, and then DCM to afford 8 as a tan solid (42% yield); 1H NMR (DMSO-d6) δ 2.85 (s, 6H), 3.41 (t, J = 6.8 Hz, 2H), 3.60 (t, J = 6.8 Hz, 2H), 3.63 (s, 2H), 3.81 (s, 2H), 6.67 (d, J = 8.8 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H), 7.24-7.35 (m, 5H), 12.62 (brs, 1H), 12.85 (brs, 1H). 13C NMR (DMSO-d6) δ 29.1, 32.4, 40.2, 40.7, 41.4, 112.5, 121.9, 126.9, 128.5, 129.3, 129.7, 134.5, 149.5, 157.9, 158.8, 158.9, 161.4, 169.6, 170.1. HPLC (Method A); retention time 1.69 min, m/z 540.3 [M + H]+.
4.1.11. 5-(2-(Benzyloxy)ethyl)-1,3,4-thiadiazol-2-amine (10)
A solution of 3-(benzyloxy)propanenitrile 9 (12.8 g, 79.4 mmol) and thiosemcarbazide (8.74 g, 96.0 mmol) in TFA (50 mL) under an atmosphere of nitrogen was heated at 65 °C for 3 h. The reaction was concentrated in vacuo, the residue was taken up in DCM (300 mL), washed with sat’d NaHCO3 (100 mL). The organic layer was dried over MgSO4, filtered, and concentrated. Trituration of the crude material provided 10.7 g of 10 as a white solid (57% yield); 1H NMR (CDCl3) δ 3.23 (t, J = 6.1 Hz, 2H), 3.78 (t, J = 6.1 Hz, 2H), 4.56 (s, 2H), 5.01 (s, 2H), 7.34-7.39 (m, 5H).
4.1.12. N-(5-(2-(Benzyloxy)ethyl)-1,3,4-thiadiazol-2-yl)-2-phenylacetamide (11)
To a solution of 5-(2-(benzyloxy)ethyl)-1,3,4-thiadiazol-2-amine 10 (1.20 g, 5.10 mmol) in DCM (75 mL) was added phenylacetyl chloride (867 mg, 5.60 mmol) and triethylamine (774 mg, 7.65 mmol) at room temperature. After 1 h, the reaction was partitioned between DCM (75 mL) and sat’d NaHCO3 (25 mL). The organic layer was washed with sat’d NaCl (75 mL), dried over MgSO4, and filtered to afford 1.80 g of 11 as a white solid (100% yield); 1H NMR (CDCl3) δ 3.33 (t, J = 5.93 Hz, 2H), 3.81 (t, J = 5.9 Hz, 2H), 4.04 (s, 2H), 4.58 (s, 2H), 7.29-7.45 (m, 10H).
4.1.13. N-(5-(2-Hydroxyethyl)-1,3,4-thiadiazol-2-yl)-2-phenylacetamide (12)
To a solution of N-(5-(2-(benzyloxy)ethyl)-1,3,4-thiadiazol-2-yl)-2-phenylacetamide 11 (540 mg, 1.53 mmol) in DCM at rt was added BBr3 (1.15 g, 4.58 mmol). After 0.5 h, the reaction was quenched by the addition of H2O (20 mL). The reaction mixture partitioned between DCM (25 mL) and H2O (25 mL). The organic layer was dried over MgSO4, filtered, and concentrated. The crude material was triturated with Et2O and afforded 300 mg of 12 as a crude product which was used in the next step without further purification or characterization.
4.1.14. 2-(5-(2-Phenylacetamido)-1,3,4-thiadiazol-2-yl)ethyl methanesulfonate (13)
To a solution of N-(5-(2-hydroxyethyl)-1,3,4-thiadiazol-2-yl)-2-phenylacetamide 12 (300 mg, 1.14 mmol) in DCM (20 ml) at 0 °C was added triethylamine (172 mg, 1.70 mmol) followed by methanesulfonyl chloride (131 mg, 1.14 mmol). After 45 min, the solution was partitioned between DCM (50 mL) and H2O (25 mL), the organic layer was removed and dried over MgSO4, filtered, and concentrated. The crude material was purified by trituration with Et2O and afforded 340 mg of 13 as a white solid (65% yield from 11); 1H NMR (CDCl3) δ 3.03 (s, 3H), 3.49 (t, J = 6.4 Hz, 2H), 3.97 (s, 2H), 4.62 (t, J = 6.4 Hz, 2H), 7.29-7.40 (m, 5H), 10.95 (s, 1H).
4.1.15. N-(6-(2-(5-Amino-1,3,4-thiadiazol-2-ylthio)ethyl)pyridazin-3-yl)-2-phenylacetamide (14)
To a solution of 2-(5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl)ethyl methanesulfonate 13 (340 mg, 1.00 mmol) in DMF (5 mL) was added potassium carbonate (276 mg, 2.00 mmol) and 5-amino-1,3,4-thiadiazole-2-thiol (160 mg, 1.2 mmol). After 30 min, the DMF was removed the material was taken up in DCM (50 mL), washed with H2O (25 mL), dried over MgSO4, filtered, and concentrated. The crude material was purified by trituration with MeOH/EtOAc to afford 200 mg of 14 as a white solid (52% yield); 1H NMR (DMSO-d6) δ 3.34 (m, 2H), 3.44 (m, 2H), 3.80 (s, 2H), 7.24-7.32 (m, 5H). 13C NMR (DMSO-d6) δ 29.3, 33.2, 41.6, 126.9, 128.5, 129.3, 134.7, 149.2, 158.7, 161.6, 169.5, 169.7. HPLC (Method B); retention time 2.35 min, m/z 378.9 [M + H]+.
4.1.16. 2-Phenyl-N-(5-(2-(5-(2-(pyridin-2-yl)acetamido)-1,3,4-thiadiazol-2-ylthio)ethyl)-1,3,4-thiadiazol-2-yl)acetamide (15a)
To a solution of N-(6-(2-(5-amino-1,3,4-thiadiazol-2-ylthio)ethyl)pyridazin-3-yl)-2-phenylacetamide 14 (50 mg, 0.13 mmol) and 2-(pyridin-2-yl)acetic acid (20.0 mg, 0.15 mmol) in DMF at 0 °C was added propylphosphonic anhydride (T3P,~50% solution in DMF, 84 mg, 0.13 mmol), followed by triethylamine (35 mg, 0.34 mmol). The solution was stirred at rt for 4 h, was quenched with ice water (2 mL) and concentrated. The material was purified by RP HPLC to afford 15 mg of 15a as a white solid (23% yield); 1H NMR (DMSO-d6) δ 3.42 (t, J = 6.8 Hz, 2H), 3.61 (t, J = 6.8 Hz, 2H), 3.80 (s, 2H), 4.02 (s, 2H), 7.24-7.35 (m, 5H), 7.40 (d, J = 8.0 Hz, 1H), 7.78 (td, J = 8.0, 2.0 Hz, 1H), 8.48-8.50 (m, 1H), 12.75 (brs, 1H), 12.94 (brs, 1H). 1H NMR (DMSO-d6) δ 29.1, 32.4, 41.5, 44.0, 122.2, 124.2, 126.9, 128.5, 129.3 134.6, 136.8, 149.1, 154.8, 157. 9, 158.7, 158.8, 161.5, 168.8, 169.4. HPLC (Method A); retention time 2.47 min, m/z 497.9 [M + H]+.
4.1.17. 2-Phenyl-N-(5-(2-(5-(2-(3-(trifluoromethoxy)phenyl)acetamido)-1,3,4-thiadiazol-2-ylthio)ethyl)-1,3,4-thiadiazol-2-yl)acetamide (15b)
To a solution of N-(6-(2-(5-amino-1,3,4-thiadiazol-2-ylthio)ethyl)pyridazin-3-yl)-2-phenylacetamide 14 (50 mg, 0.13 mmol) in DMF (3 mL) at 0 °C was added 2-(3-(trifluoromethoxy)phenyl)acetic acid (34.9 mg, 0.16 mmol), DIEA (68.2 mg, 0.53 mmol) and then HATU (60.3 mg, 0.16 mmol). The reaction was allowed to warm to rt overnight, was concentrated, partitioned between CHCl3 (25 mL) and H2O (15 mL). The organic layer was washed with sat’d NaHCO3 (15 mL) and then 10% KHSO4 (15 mL), dried over MgSO4, filtered, concentrated, and then purified by RP HPLC to afford 20 mg of 15b as a white solid (26% yield); 1H NMR (DMSO-d6) δ 3.42 (t, J = 6.8 Hz, 2H), 3.60 (t, J = 6.8 Hz, 2H), 3.80 (s, 2H), 3.90 (s, 2H), 7.24-7.35 (m, 8H), 7.47 (t, J = 8.0 Hz, 1H), 12.75 (brs, 1H), 12.94 (brs, 1H). 13C NMR (DMSO-d6) δ 29.1, 32.4, 40.8, 41.5, 119.4, 120.1 (q, J = 255 Hz), 122.0, 126.9, 128.4, 128.6, 129.2, 130.3, 134.6, 137.1, 148.3 (q, J = 1.5 Hz), 157. 9, 158.7, 159.0, 161. 5, 169.2, 169.4. HPLC (Method A); retention time 2.12 min, m/z 580.9 [M + H]+.
4.1.18. N-(6-(2-(5-Amino-1,3,4-thiadiazol-2-yl)ethylthio)pyridazin-3-yl)-2-phenylacetamide (18)
To a solution of S-(2-(5-amino-1,3,4-thiadiazol-2-yl)ethyl) ethanethioate (110 mg, 0.54 mmol) in MeOH-water (2.5 mL:5 mL) was added NaOH (43 mg, 1.08 mmol) at rt. The mixture was stirred at rt for 2 h and MeOH was removed under vacuum. The water layer was lyophilized to afford compound 17, which was directly used in next step without further purification. Cu(I) iodide (10.3 mg, 0.054 mmol), potassium carbonate (149.3 mg, 1.08 mmol), N-(6-iodopyridazin-3-yl)-2-phenylacetamide 16 (183 mg, 0.54 mmol), and compound 17 were added to a flask. 2-Propanol (5.0 mL), and ethylene glycol (67 mg, 1.08 mmol) were added by syringes at room temperature. The mixture was stirred at 80 °C for 24 h. The reaction mixture was then allowed to reach room temperature. The reaction mixture was then filtered and concentrated. The crude product was purified by flash column chromatography on silica gel to afford 80 mg of 18 as a yellow solid (40% yield); 1H NMR (DMSO-d6) δ 3.22 (t, J = 7.2 Hz), 3.55 (t, J = 7.2 Hz), 3.78 (s, 2H), 7.10 (brs, 2H), 7.24-7.36 (m, 5H), 7.61 (d, J = 9.2 Hz), 8.15 (d, J = 9.2 Hz), 11.30 (brs, 1H). 13C NMR (DMSO-d6) δ 28.69, 29.43, 42.86, 118.88, 126.71, 127.96, 128.37, 129.27, 135.41, 153.48, 156.01, 156.64, 168.67, 170.80. HPLC (Method A); retention time 1.17 min, m/z 373.1 [M + H]+.
4.1.19. 2-Phenyl-N-(6-(2-(5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl)ethylthio)pyridazin-3-yl)acetamide (19)
To a solution of N-(6-(2-(5-amino-1,3,4-thiadiazol-2-yl)ethylthio)pyridazin-3-yl)-2-phenylacetamide 18 (20 mg, 0.053 mmol) in DCM (5 mL) was added triethylamine (16 mg, 0.16 mmol) and phenylacetyl chloride (9.1 mg, 0.06 mmol). After being stirred at rt overnight, phenylacetyl chloride (4.5 mg, 0.03 mmol) was added and the mixture was stirred for another 1 h. The solvent was removed in vacuo and the residue was purified by silica gel chromatography (100% DCM to 10% MeOH/DCM) and afforded 18 mg 19 as a white solid (66% yield); 1H NMR (DMSO-d6) δ 3.39 (t, J = 6.8 Hz), 3.62 (t, J = 6.8 Hz), 3.77 (s, 2H), 3.80 (s, 2H), 7.23-7.36 (m, 10H), 7.62 (d, J = 9.2 Hz), 8.14 (d, J = 9.6 Hz), 11.30 (brs, 1H), 12.72 (brs, 1H) . 13C NMR (DMSO-d6) δ 28.8, 29.0, 41.6, 42.8, 118.9, 126.7, 126.9, 128.0, 128.4, 128.5, 129.3, 134.6, 135.4, 153.5, 156.4, 158.7, 162.0, 169.4, 170.8. HPLC (Method A); retention time 1.94min, m/z 491.2 [M + H]+.
4.1.20. N-(6-Allylpyridazin-3-yl)-2-phenylacetamide (21)
To a solution of 16 (1.10 g, 3.2 mmol) in dioxane was added potassium carbonate (1.10 g, 8.0 mmol), allylboronic acid pinacol ester 20 (790 mg, 4.7 mmol), and palladium-tetrakis(triphenylphosphine) (180 mg, 0.16 mmol). The mixture was heated in a sealed tube at 120 °C for 14 h. The reaction mixture was cooled and the solvent was removed in vacuo. The residue was taken up in EtOAc (100 mL) and washed with H2O (50 mL). The organic layer was washed with sat’d NH4Cl, dried over MgSO4, filtered, concentrated and then purified by silica gel chromatography (20% EtOAc/hex to 80% EtOAc/hex) to afford 330 mg of 21 as a yellow solid (42% yield);1H NMR (CDCl3) δ 3.72 (dt, J = 6.8, 1.3 Hz, 2H), 3.90 (s, 2H), 5.18 (m, 2H), 6.02 (m, 1H), 7.33.-7.43 (m, 6H), 8.45 (d, J = 9.2 Hz, 1H), 8.95 (s, 1H).
4.1.21. N-(6-(2-Hydroxyethyl)pyridazin-3-yl)-2-phenylacetamide (22)
To a solution of N-(6-allylpyridazin-3-yl)-2-phenylacetamide 21 (280 mg, 1.1 mmol) in DCM at −78 °C was bubbled O3 (at a rate of ~ 31 ml/min) for 15 min. The solution changed color from dark to light yellow at this time. The solution was flushed with N2 for 5 min and sodium borohydride (43 mg, 1.14 mmol) was added to the mixture at this same temperature. The reaction was maintained at this temperature for 60 min, and then warmed and maintained at 0 °C for 60 min. The mixture was partitioned between DCM and water. The DCM layer was dried over MgSO4, filtered, and concentrated in vacuo. The residual material was purified by silica gel chromatography (50% EtOAc/hex to 100% EtOAc to 30% MeOH/EtOAc) and afforded 40 mg of 22 as a light yellow solid (14% yield); 1H NMR (CDCl3) δ 3.13 (t, J = 5.6 Hz, 2H), 3.88 (s, 2H), 4.07 (4.07, t, J = 5.6 Hz, 2H), 7.34-7.42 (m, 6H), 8.47 (d, J = 9.0 Hz, 1H), 8.93 (s, 1H).
4.1.22. 2-(6-(2-Phenylacetamido)pyridazin-3-yl)ethyl methanesulfonate (23)
To a solution of N-(6-(2-hydroxyethyl)pyridazin-3-yl)-2-phenylacetamide 22 (50 mg, 0.19 mmol) in DCM (10 mL) at 0 °C was added triethylamine (29 mg, 0.29 mmol) followed by methanesulfonyl chloride (25 mg, 0.21 mmol). After 2 h, additional triethylamine (29 mg, 0.29 mmol) and methanesulfonyl chloride (25 mg, 0.22 mmol) was added and the mixture was stirred for additional 6 h. The reaction mixture was partitioned between DCM (25 mL) and H2O (15 mL). The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The residual material was used in the subsequent step without further purification or characterization.
4.1.23. N-(6-(2-(5-Amino-1,3,4-thiadiazol-2-ylthio)ethyl)pyridazin-3-yl)-2-phenylacetamide (24)
To a solution of 2-(6-(2-phenylacetamido)pyridazin-3-yl)ethyl methanesulfonate 23 (65 mg, 0.19 mmol) in acetonitrile (30 mL) was added 5-amino-1,3,4-thiadiazole-2-thiol (28 mg, 0.21 mmol) and potassium carbonate (54 mg, 0.39 mmol). The mixture was heated at 50 °C for 7.5 h and the solvent was removed in vacuo. A small portion of the crude material was purified by RP HPLC to obtain 24 as a colorless oil for the in vitro screening. 1H NMR (CD3OD w/ two drops of DMSO-d6) δ 3.34 (t, J = 7.0 Hz, 2H), 3.54 (t, J = 7.0 Hz, 2H), 3.82 (s, 2H), 7.29 (t, J = 7.0 Hz, 1H), 7.35-7.41 (m, 4H), 7.62 (d, J = 9.0 Hz, 1H), 8.35-8.37 (m, 1H). 13C NMR (CD3OD w/ two drops of DMSO-d6) δ 35.1, 36.3, 44.5, 120.7, 128.3, 129.8, 130.6, 136.59, 136.63, 153.2, 156.0, 159.4, 173.1. HPLC (Method A); retention time 2.29 min, m/z 373.1 [M + H]+. The remaining material was taken to the next step without further purification.
4.1.24. 2-Phenyl-N-(6-(2-(5-(2-phenylacetamido)-1,3,4-thiadiazol-2-ylthio)ethyl)pyridazin-3-yl)acetamide (25)
To a solution of N-(6-(2-(5-amino-1,3,4-thiadiazol-2-ylthio)ethyl)pyridazin-3-yl)-2-phenylacetamide 24 (70 mg, 0.19 mmol) in DMF (1.0 mL) at 0 °C, was added phenylacetic acid (33 mg, 0.24 mmol), diisopropylethylamine (146 mg, 1.13 mmol), and HATU (92 mg, 0.24 mmol). The mixture was warmed to rt overnight, concentrated in vacuo, taken up in EtOAc (40 mL), washed with sat’d NaHCO3 (15 mL) and 10% KHSO4, dried over MgSO4, and concentrated in vacuo. Purification by RP HPLC afforded 10 mg of 25 as a light pink oil (11% yield from 22). 1H NMR (CDCl3) δ 3.39 (t, J = 6.5 Hz, 2H), 3.69 (t, J = 6.5 Hz, 2H), 3.93 (s, 2H), 4.06 (s, 2H), 7.29-7.39 (m, 11H), 8.54 (d, J = 9.0 Hz, 1H), 10.7 (s, 1H), 12.7 (s,1H). 13C NMR (CDCl3) δ 34.3, 36.3, 42.7, 44.2, 119.6, 127.4, 127.5, 128.8, 128.9, 129.4, 129.5, 130.1, 133.7, 134.3, 154.6, 157.1, 159.5, 161.3, 169.6, 171.2. HPLC (Method B); retention time 3.08 min, m/z 491.1 [M + H]+.
4.2. in vitro GLS assay
A 96-well microplate-based glutaminase assay was used to determine the activity of various glutaminase inhibitors.16 The hKGAΔ1 construct was prepared from the hKGA cDNA26 by deleting the sequence encoded in exon 1 and cloning into pET15b as described for the rat KGAΔ1 construct.22 Purified human kidney glutaminase (hKGA124−669; 250 nM) was used as the source of enzyme and radiolabeled glutamine (L-[3H]-glutamine, Perkin Elmer, Boston, MA); 2 mM and at a specific activity of 0.91 μCi/μmol) was used as the substrate. The assay was conducted in the presence and absence of inhibitors, at room temperature (45 min incubation), in 45 mM phosphate buffer (pH 8.2). At the end of the reaction period, the assay was terminated upon the addition of 20 mM imidazole buffer (pH 7). 96-Well spin columns packed with strong anion ion-exchange resin (Bio-Rad, Hercules, CA) were used to separate the substrate and the reaction product. Unreacted [3H]-glutamine was removed by washing with imidazole buffer. [3H]-Glutamate, the reaction product, was then eluted with 0.1 M HCl and analyzed for radioactivity using Perkin-Elmer’s TopCount instrument in conjunction with their 96-well LumaPlates (Waltham, MA).
4.3. Metabolic stability assessment
The metabolic stability of compound 3a was evaluated using mouse liver microsomes as we have previously described.23 The reaction was carried out in 100 mM potassium phosphate buffer, pH 7.4, in the presence of NADPH regenerating system (1.3 mM NADPH, 3.3 mM glucose 6-phosphate, 3.3 mM MgCl2, 0.4 U/mL glucose-6-phosphate dehydrogenase, 50 μM sodium citrate). Reactions in triplicate were initiated by addition of mouse liver microsomes to the incubation mixture (compound final concentration was 1 μM; 0.2 mg/mL protein). Control reaction was carried out in the absence of NADPH to measure CYP-independent metabolism of the compound. After 60 min of incubation, aliquots of the mixture were removed and the reaction was quenched by addition of two times the volume of ice cold acetonitrile spiked with the internal standard. Compound disappearance was monitored over time using liquid chromatography and tandem mass spectrometry (LC/MS/MS) as described in section 4.4.
4.4. Aqueous solubility assessment
The aqueous solubility of compound 25 was determined by the shake flask method.24 Compound 25 was dissolved in PBS buffer (pH = 7.4) at 5 mg/mL concentration and incubated for 24 h at 37 °C on a thermomixer. Samples were subjected to constant mixing at a shaking speed of 1000 rpm to ensure saturation. After 24 hours the solution was filtered (0.45 μm PTFE) and the filtrate was diluted appropriately and solubility in the filtrate samples was determined against a calibration curve ranging 5-10000 nM. Samples were analyzed on a Dionex ultra-high-performance LC system coupled with Q Exactive Focus orbitrap mass spectrometer (Thermo Fisher Scientific Inc., Waltham MA). The separation of analytes was achieved using the Agilent Eclipse Plus column (100 × 2.1 mm i.d.; maintained at 35 °C) packed with a 1.8 μm C18 stationary phase. The mobile phase consisted of 0.1% formic acid in water and 0.1% formic acid in acetonitrile. Pumps were operated at a flow rate of 0.3 mL/min for 8 min using gradient elution. The mass spectrometer controlled by Xcalibur software 4.0.27.13 (Thermo Scientific) was operated with a heated electrospray ionization (HESI) ion source in positive ionization mode. The [M + H]+ ion transitions of 25 at m/z 491.1318 and that of the internal standard (losartan) at m/z 423.1695 were monitored in Full MS scan mode. Data were acquired and quantified with Xcalibur 2.2.
Supplementary Material
Table 1.
Inhibition of GLS by representative GLS inhibitors and compounds containing a mercaptoethyl linker
|
|
Values are mean ± SD of at least three experiments.
Acknowledgement
The authors of this manuscript have been supported by NIH grants P30MH075673 (B.S.S.), R01CA193895 (B.S.S.), R21NS074151 (T.T.), and F32CA200278 (S.C.Z.). The authors are also grateful for the support provided by the Bloomberg-Kimmel Institute for Cancer Immunotherapy at Johns Hopkins School of Medicine.
Footnotes
Appendix A. Supplementary Data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2020.115698
References
- 1.Katt WP, Cerione RA. Glutaminase regulation in cancer cells: a druggable chain of events. Drug Discov Today. 2014;19:450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li L, Meng Y, Li Z, et al. Discovery and development of small molecule modulators targeting glutamine metabolism. Eur J Med Chem. 2019;163:215–242. [DOI] [PubMed] [Google Scholar]
- 3.Newcomb RW. Selective inhibition of glutaminase by bis-thiadiazoles. 2002. US 6,451,828 B1.
- 4.Robinson MM, McBryant SJ, Tsukamoto T, et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J 2007;406:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zimmermann SC, Duvall B, Tsukamoto T. Recent Progress in the Discovery of Allosteric Inhibitors of Kidney-Type Glutaminase. J Med Chem. 2019;62:46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song M, Kim SH, Im CY, et al. Recent Development of Small Molecule Glutaminase Inhibitors. Curr Top Med Chem. 2018;18:432–443. [DOI] [PubMed] [Google Scholar]
- 7.Xu X, Meng Y, Li L, et al. Overview of the Development of Glutaminase Inhibitors: Achievements and Future Directions. J Med Chem. 2019;62:1096–1115. [DOI] [PubMed] [Google Scholar]
- 8.Gross MI, Demo SD, Dennison JB, et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther 2014;13:890–901. [DOI] [PubMed] [Google Scholar]
- 9.Lemieux RM, Popovici-muller J, Salituro FG, et al. Compounds and their methods of use. 2015. U.S. Pat. Appl 20150291576.
- 10.DeLaBarre B, Gross S, Fang C, et al. Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry. 2011;50:10764–10770. [DOI] [PubMed] [Google Scholar]
- 11.Ramachandran S, Pan CQ, Zimmermann SC, et al. Structural basis for exploring the allosteric inhibition of human kidney type glutaminase. Oncotarget. 2016;7:57943–57954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDermott LA, Iyer P, Vernetti L, et al. Design and evaluation of novel glutaminase inhibitors. Bioorg Med Chem. 2016;24:1819–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDermott L, Koes D, Mohammed S, et al. GAC inhibitors with a 4-hydroxypiperidine spacer: Requirements for potency. Bioorg Med Chem Lett. 2019;29:126632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Chen L, Goyal B, et al. Heterocyclic Inhibitors of Glutaminase. 2013. WO 2013078123 A1.
- 15.Reddy RK, Glinka T, Totrov M, et al. Boronic acid derivatives and therapeutic uses thereof. 2019. US 10,206,937 B2.
- 16.Shukla K, Ferraris DV, Thomas AG, et al. Design, Synthesis, and Pharmacological Evaluation of Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl Sulfide 3 (BPTES) Analogs as Glutaminase Inhibitors. J. Med. Chem 2012;55:10551–10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beno BR, Yeung KS, Bartberger MD, et al. A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J Med Chem. 2015;58:4383–4438. [DOI] [PubMed] [Google Scholar]
- 18.Thangavelu K, Pan CQ, Karlberg T, et al. Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc. Natl. Acad. Sci. U.S.A 2012;109:7705–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valley CC, Cembran A, Perlmutter JD, et al. The methionine-aromatic motif plays a unique role in stabilizing protein structure. J Biol Chem. 2012;287:34979–34991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walker MA. Improvement in aqueous solubility achieved via small molecular changes. Bioorg Med Chem Lett. 2017;27:5100–5108. [DOI] [PubMed] [Google Scholar]
- 21.Chen Z, Li D, Xu N, et al. Novel 1,3,4-Selenadiazole-Containing Kidney-Type Glutaminase Inhibitors Showed Improved Cellular Uptake and Antitumor Activity. J Med Chem. 2019;62:589–603. [DOI] [PubMed] [Google Scholar]
- 22.Kenny J, Bao Y, Hamm B, et al. Bacterial expression, purification, and characterization of rat kidney-type mitochondrial glutaminase. Protein Expr Purif. 2003;31:140–148. [DOI] [PubMed] [Google Scholar]
- 23.Slack RD, Ku TC, Cao J, et al. Structure-Activity Relationships for a Series of (Bis(4-fluorophenyl)methyl)sulfinyl Alkyl Alicyclic Amines at the Dopamine Transporter: Functionalizing the Terminal Nitrogen Affects Affinity, Selectivity, and Metabolic Stability. J Med Chem. 2020;63:2343–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou L, Yang L, Tilton S, et al. Development of a high throughput equilibrium solubility assay using miniaturized shake-flask method in early drug discovery. J Pharm Sci. 2007;96:3052–3071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.