

Abstract
Trastuzumab targets the HER2 receptor on breast cancer cells to attenuate HER2-driven tumor growth. However, resistance to trastuzumab-based therapy remains a major clinical problem for women with HER2+ breast cancer. Breast cancer stem cells (BCSCs) are suggested to be responsible for drug resistance and tumor recurrence. Notch signaling has been shown to promote BCSC survival and self-renewal. Trastuzumab-resistant cells have increased Notch-1 expression. Notch signaling drives cell proliferation in vitro and is required for tumor recurrence in vivo. We demonstrate herein a mechanism by which Notch-1 is required for trastuzumab resistance by repressing PTEN expression to contribute to activation of ERK1/2 signaling. Furthermore, Notch-1-mediated inhibition of PTEN is necessary for BCSC survival in vitro and in vivo. Inhibition of MEK1/2-ERK1/2 signaling in trastuzumab-resistant breast cancer cells mimics effects of Notch-1 knockdown on bulk cell proliferation and BCSC survival. These findings suggest that Notch-1 contributes to trastuzumab resistance by repressing PTEN and this may lead to hyperactivation of ERK1/2 signaling. Furthermore, high Notch-1 and low PTEN mRNA expression may predict poorer overall survival in women with breast cancer. Notch-1 protein expression predicts poorer survival in women with HER2+ breast cancer. These results support a potential future clinical trial combining anti-Notch-1 and anti-MEK/ERK therapy for trastuzumab-resistant breast cancer.
Introduction
Breast cancer is a heterogeneous disease divided into several intrinsic subtypes based on molecular and pathologic profiles. These include Luminal A and B, epidermal growth factor receptor 2 (HER2 or ERBB2) amplified, and triple negative. Luminal A and B tumors express estrogen and/or progesterone receptors (ER/PR+). HER2+ breast cancers contain gene amplification of the ERBB2 proto-oncogene on chromosome 17. Triple negative breast cancers lack of expression of ER, PR, and amplification of ERBB2 [1]. Amplification of the ERBB2 gene is detected in 15–25% of breast cancers [2]. HER2+ breast cancers are aggressive and associated with worse overall prognosis [3]. Primary therapy for HER2+ breast cancer includes the humanized, monoclonal antibody trastuzumab. As a single agent, trastuzumab is modestly effective, but gains significant efficacy when combined with taxane-based chemotherapy and/or pertuzumab, an antibody targeting the HER2:HER3 heterodimer [4, 5]. Trastuzumab is effective in early stage breast cancer, but ~15% of women treated with trastuzumab go on to develop metastatic disease within the first year [6] and 20–50% develop trastuzumab resistance [7].
Trastuzumab-based therapy targets multiple pathways downstream of HER2 including phosphotidylinositol-4, 5-bisphosphate 3-kinase (PI3-K) and mitogen-activated protein kinase (MAPK) [8, 9]. HER2 activation requires phosphorylation of multiple tyrosine residues (PY) on its cytoplasmic domain. These PY residues are docking sites for SH2-containing proteins such as the p85 subunit of PI3K. Upon binding to PY residues of HER2, the p85 regulatory subunit recruits the p110 catalytic subunit, resulting in full activation of PI3-K. PI3-K then phosphorylates phosphatidylinositol (4, 5) bis-phosphate (PIP2) converting it to phosphatidylinositol (3, 4, 5) tris-phosphate (PIP3). PIP3 activates PDK1 which phosphorylates Akt on Threonine 308, resulting in partial activation of Akt [10]. Complete activation of Akt requires mTORC2-catalyzed phosphorylation of Serine 473. Trastuzumab treatment also induces expression of the tumor suppressor, phosphatase and tensin homolog on chromosome 10 (PTEN). PTEN is a lipid phosphatase that dephosphorylates PIP3 to PIP2, thus reducing Akt activity [11]. Increased PI3-K/Akt activity from PTEN downregulation or PIK3CA mutations predicts poor outcome in women treated with trastuzumab-based therapy [12], suggesting that increased PI3K/Akt activity may predict trastuzumab resistance [13]. In agreement, women with tumors expressing low PTEN were less likely to achieve a pathologic complete response (pCR) when treated with trastuzumab, compared to women with tumors expressing high levels of PTEN [14]. Somatic gene mutations of PTEN are found in 5% of HER2+ tumors resulting in increased Akt phosphorylation at threonine 308 and serine 473, as well as increased downstream phosphorylation of mTOR and p70S6K [15].
HER2 also activates the Ras-mitogen activated protein kinase (MAPK) pathway. Ras binds to and activates Raf (MAPKKK) to initiate a cascade of phosphorylation events leading to activation of kinases such as MEK1/2 (MAPKK) and downstream ERK1/2 (MAPK) [16]. Once phosphorylated and activated by MEK1/2, ERK1/2 enters the nucleus to phosphorylate transcription factors that mediate expression of genes which promote cell proliferation or attenuate apoptosis. PTEN also functions as a protein phosphatase by dephosphorylating ERK1/2 in the nucleus of breast cancer cells, thereby attenuating MAPK pathway activity [17, 18]. Ras signaling is frequently hyper-activated in breast cancer [19] and hyper-activation of the MAPK pathway correlates with increased enrichment of cancer stem cells and poor patient survival [20]. It was shown previously that Notch-1 is necessary for Ras-mediated transformation [21]. Recently, it has been reported that loss of PTEN in HER2+ breast cancer promotes dependency on the MAPK pathway through elevated MEK/ERK activity [9]. PTEN expression can be used as a predictor of response to anti-HER2 plus taxane-based chemotherapy, and its low expression is a potential indicator of resistance to anti-HER2 therapies [22]. Transcriptional downregulation of PTEN is the primary cause for low-PTEN expression in breast tumors [23–25]. Thus, understanding the mechanism by which PTEN expression is regulated could potentially lead to new targets for the treatment of HER2+ breast cancer.
Notch-1 expression and activity are increased in trastuzumab-resistant, HER2+ breast cancer cells in vitro and in vivo, thus providing a compensatory survival pathway [26, 27]. Notch signaling is activated by cell-cell contact, where a Notch receptor (Notch-1, −2, −3, or −4) on one cell engages with a Notch ligand (Jagged-1, −2, or Delta-Like-1, −4) on an adjacent cell to initiate a pulling force and subsequent dissociation and cleavage of the Notch receptor by ADAM10 [28, 29]. The extracellular and ecto-domain of the transmembrane portion is endocytosed into the ligand-bearing cell. The remaining transmembrane portion (NEXT) is highly susceptible to cleavage by the γ-secretase complex releasing the notch intracellular domain (NICD) [30]. NICD is translocated into the nucleus where it binds to the transcriptional repressor CSL (CSL/RBP-Jκ) and releases co-repressors and recruits co-activators such as MAML1 [31] and p300. Some classical genes activated by NICD include the hairy enhancer of split repressors (HES, HERP, HEY, and HEYL) and NRARP, as well as c-Myc [32].
Notch regulates cell fate [33], proliferation [34], apoptosis [35], as well as stem cell survival and self-renewal [33, 36, 37]. Increased expression of Notch-1 and its ligand Jagged-1 predicts poorer overall survival for women with breast cancer [38]. Notch signaling can be pharmacologically inhibited by γ-secretase inhibitors (GSIs) which have been shown to reduce HER2+ tumor growth and recurrence [27]. Notch-1 has been shown to be a compensatory survival signal in HER2+ breast cancer cells resistant to trastuzumab [39]. However, mechanisms by which Notch-1 contributes to drug resistance and cancer stem cell biology are not clear in HER2+ breast cancer. In T-ALL cells, Notch1 suppresses PTEN expression via its target HES1, a negative regulator bHLH transcription factor [40]. However, this mechanism may be cell context dependent.
Objectives of this study were to determine whether Notch-1 is a direct transcriptional repressor of PTEN in trastuzumab-resistant, HER2-positive breast cancer and to assess if Notch-1-mediated repression of PTEN activates MAPK signaling to promote breast cancer stem cell survival in vitro and in vivo.
Results
Notch-1 is required for repression of PTEN in HER2+ breast cancer cells
Two trastuzumab-resistant breast cancer cell models: acquired (BT474TR; trastuzumab-resistant), and intrinsic resistance (HCC1954) were used in the study [27, 41–44]. BT474TR cells have higher expression of total HER2 and phosphorylated forms of Akt-1 proteins (PSer473-Akt1 and PThr308-Akt1) compared to BT474TS cells, yet both have similar expression levels of tyrosine phosphorylated HER2 (PY-HER2) (Fig. 1a). Resistant cells have higher Notch-1 and lower PTEN protein expression compared to sensitive cells (Fig. 1b, left panel). Knockdown of Notch-1 in resistant cells resulted in increased PTEN protein (Fig. 1b, right panel) and transcript (Fig. 1e) expression. Similar results were observed in another cell line, HCC1954 (Fig. 1c, f). Results were confirmed by the use of a second Notch-1 siRNA in BT474TR cells (Fig. 1d). These studies were repeated to determine the cellular localization of PTEN by immunofluorescence. Figure 2a shows that BT474TR cells express lower levels of PTEN compared to BT474TS cells. PTEN protein expression was increased upon Notch-1 knockdown in BT474TR cells to similar levels observed in BT474TS cells. PTEN expression was localized both in the cytoplasm and the nucleus in BT474TS cells although relatively low levels of PTEN in BT474TR cells were found in the cytoplasm. Notch-1 knockdown in BT474TR cells appeared to increase overall PTEN expression in both the cytoplasm and the nucleus (Fig. 2a).
Fig. 1.
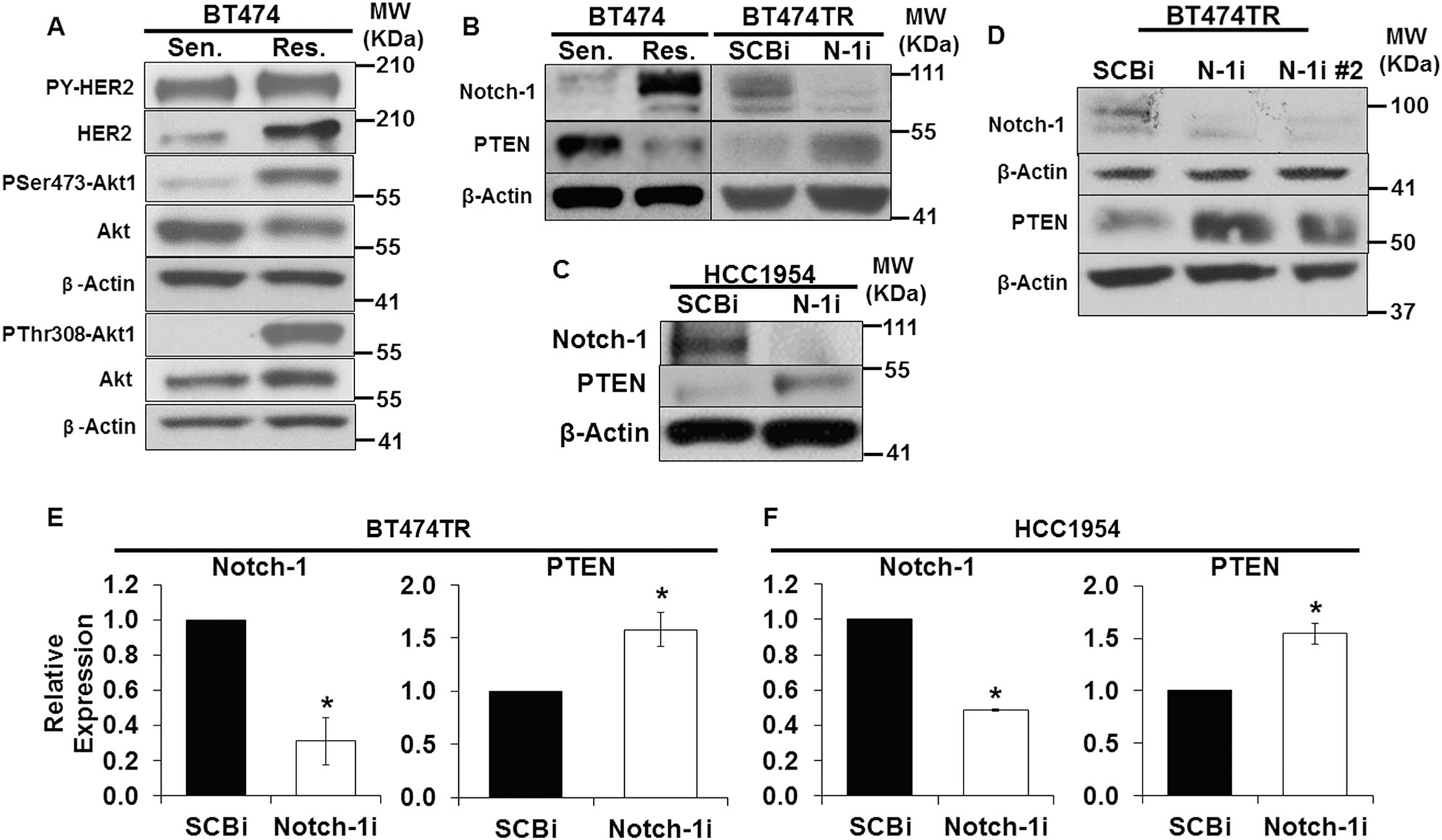
Notch-1 is required to repress PTEN in trastuzumab-resistant cells. a Total protein lysates from BT474TS [Trastuzumab Sensitive (Sen.)] and BT474TR [Acquired Trastuzumab Resistant (Res.)] cells underwent Western blot analysis to detect HER2, HER2 phosphorylation (PY-HER2), Akt phosphorylation (PSer473-Akt1, PThr308-Akt1), total Akt1, and β-Actin protein expression. b Total lysates from both BT474TS and BT474TR cells were subjected to Western blot analysis to detect Notch-1, PTEN, and β-Actin protein levels. BT474TR cells were transfected with scrambled control (SCBi) or Notch-1 siRNA (N-1i) for 48 h. Post transfection, total cellular Notch-1, PTEN, and β-Actin protein expression was analyzed by Western blot. c Intrinsically resistant HCC1954 cells were transfected with SCBi or Notch-1i for 48 h. Post transfection, total cellular Notch-1, PTEN, and β-Actin protein expression was analyzed by Western blot. d BT474TR cells were transfected with SCBi, N-1i, and a second Notch-1 siRNA (N-1i#2) for 48 h. Post transfection, total cellular Notch-1, PTEN, and β-Actin protein expression was analyzed by Western blot. BT474TR cells (e), or HCC1954 cells (f) were transfected with SCBi or Notch-1i for 48 h. Post transfection, relative expression of Notch-1 or PTEN mRNA was measured by RT-PCR. Asterisk (*) represents statistical significance compared to SCBi from three independent experiments using a Student’s t-test
Fig. 2.
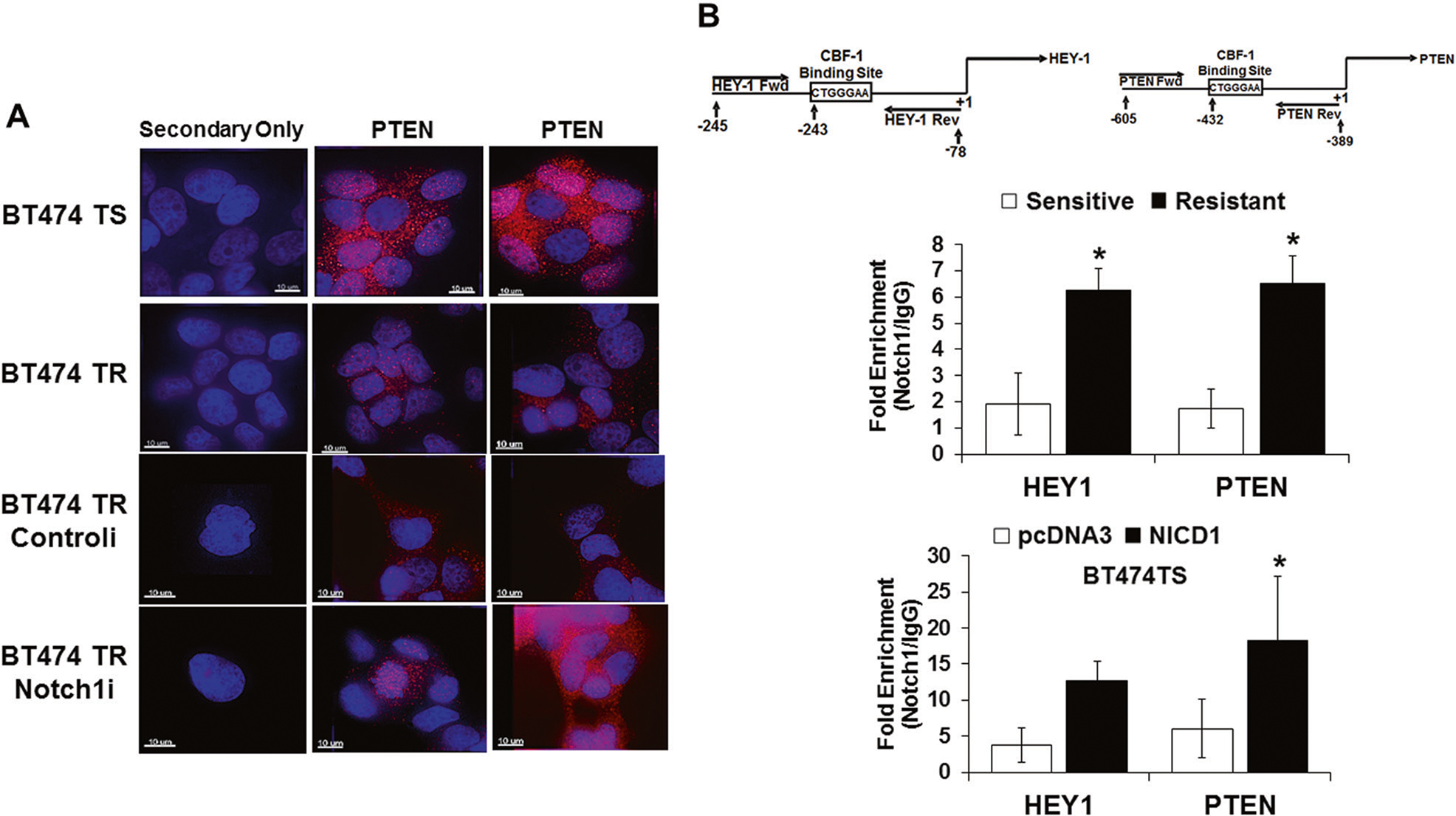
Localization and expression of PTEN in trastuzumab sensitive and resistant cells and recruitment of Notch-1 to the PTEN promoter. a Confocal immunofluorescence was performed on both BT474TS and TR cells to detect PTEN protein expression and cellular localization under conditions where BT474TR cells were transfected with a control siRNA or a Notch-1 siRNA. Scale bar = 10 μm. b BT474TR and BT474TS cells were fixed, fragmented, and chromatin was immunoprecipitated by a Notch-1 or control IgG antibody. Purified DNA from Notch-1 or IgG immunoprecipitated chromatin was used to quantify NICD1 enrichment on HEY-1 or PTEN promoters by real-time PCR using primers flanking a CSL site (maps above bar graphs) (upper graph). BT474TS cells were transfected with a vector control (pcDNA3) or NICD1-expression vector (NICD1) for 48 h. Post transfection, cells were collected, fixed, fragmented, immunoprecipitated by a C-terminal Notch-1 or control IgG antibody and NICD1 enrichment on HEY-1 or PTEN promoters was quantified by real-time PCR using HEY-1 or PTEN primers that flanked a CSL element (lower graph). Asterisk (*) represents statistical significance between sensitive and resistant (upper graph) or between pcDNA3 and NICD1 (lower graph) from three independent experiments using a Student’s t-test
To assess whether increased Notch-1 was sufficient to repress PTEN expression, the active form of Notch-1 (NICD1) was overexpressed in BT474TS cells, and PTEN mRNA and protein expression were measured. Results showed that NICD1 overexpression decreased PTEN protein and transcript expression compared to vector control (pcDNA3)-transfected BT474 sensitive cells (SFig. 1), suggesting that Notch-1 is both necessary and sufficient to decrease PTEN expression in HER2+ breast cancer cells.
Notch-1 is enriched at a CSL site on the PTEN promoter
To assess whether Notch-1 potentially directly represses PTEN, Notch-1 ChIP assays were performed. The HEY-1 specific promoter region at −243 bps upstream of the transcriptional start site (TSS) was selected as a positive control for NICD1 recruitment (Fig. 2b, upper left panel) [45]. A putative CSL binding sequence (CTGGGAA) −432 bps upstream of the TSS was identified on the PTEN promoter (Fig. 2b, upper right panel). Primers flanking the CSL binding sequences detected more than a 3-fold increase in endogenous NICD1 recruitment to the CSL region on both HEY-1 and PTEN promoter regions in resistant compared to sensitive cells (Fig. 2b, top graph). Similarly, ChIP assays in sensitive cells transfected with an NICD1-expression vector showed a 3-fold increase in NICD1 enrichment on HEY-1 and PTEN promoters, respectively, compared to the vector control (Fig. 2b, bottom graph). Results indicate that Notch-1 may attenuate PTEN expression by recruitment to its CSL-containing promoter in HER2+ breast cancer cells.
Notch-1-mediated inhibition of PTEN promotes trastuzumab-resistant cell proliferation
The biological significance of Notch-1-mediated repression of PTEN was determined using a knockdown approach followed by measurement of cell numbers and activation of the PI3K-Akt1 pathway. Cell proliferation assays were conducted by measuring the number of live cells at day 10 over the number of cells plated at day 0. The graphs depict a fold increase in live cell numbers. To confirm that HER2+ breast cancer cells used for the subsequent studies were resistant to trastuzumab, a proliferation assay was performed on BT474TR cells in response to increasing concentrations of trastuzumab. The results showed that proliferation of BT474TS cells was completely inhibited with only 2.5 μg/mL trastuzumab, although BT474TR cells continued to proliferate at concentrations up to 40 μg/mL (SFig. 2A). In addition, HCC1954 cells were confirmed to be intrinsically resistant to trastuzumab at concentrations from 5 to 40 μg/mL (SFig. 2B). Cell cycle analysis confirmed that BT474TS cells growth arrest in the G1 phase of the cell cycle in response to 20 μg/mL trastuzumab while BT474TR cells continue to cycle (SFig. 2C). To confirm previous findings that proliferation of these cells is dependent on Notch-1 [41], Notch-1 was knocked down (SFig. 2D) and cell cycle progression was analyzed. Results showed that upon Notch-1 knockdown, the G1-phase fraction increased from 58 to 72% and this is further enhanced with trastuzumab treatment (SFig. 2C). To address whether proliferation of BT474TR and HCC1954 cells was due to Notch-1-mediated repression of PTEN expression, both Notch-1 and PTEN were simultaneously knocked down. Results showed that proliferation of BT474TR (Fig. 3a, b) and HCC1954 (Fig. 3c, d) cells requires Notch-1 regardless of trastuzumab treatment. Notch-1 knockdown significantly reduced the fold increase in number of live cells of both BT474TR and HCC1954 compared to scrambled control. PTEN knockdown alone using a SmartPool of three siRNAs had a modest growth inhibitory effect on proliferation of BT474TR cells, while having no effect on HCC1954 cell proliferation (Fig. 3a–d). To determine whether growth inhibitory effects of Notch-1 knockdown were due to increased PTEN expression, both BT474TR and HCC1954 cells were transfected with Notch-1 and PTEN siRNAs and fold increase in live cell numbers was assessed. Combined knockdown of Notch-1 and PTEN partially rescued proliferation of BT474TR cells by 2-fold compared to Notch-1 knockdown alone (Fig. 3a, b) while completely rescuing proliferation of HCC1954 cells (Fig. 3c, d). To determine possible off-target effects of either the Notch-1 or PTEN siRNAs, three independent PTEN siRNAs (A, B, C) were tested individually in cell proliferation studies to reveal that one PTEN siRNA (B) in the SmartPool reduced proliferation (SFig. 3A). Proliferation studies were repeated with one PTEN siRNA (A) and a second Notch-1 siRNA [46] in which combined PTEN and Notch-1 inhibition rescued cell proliferation compared to Notch-1 knockdown alone (SFig. 3B). These results using two distinct Notch-1 and PTEN siRNAs were similar to previous studies shown in Fig. 3a.
Fig. 3.
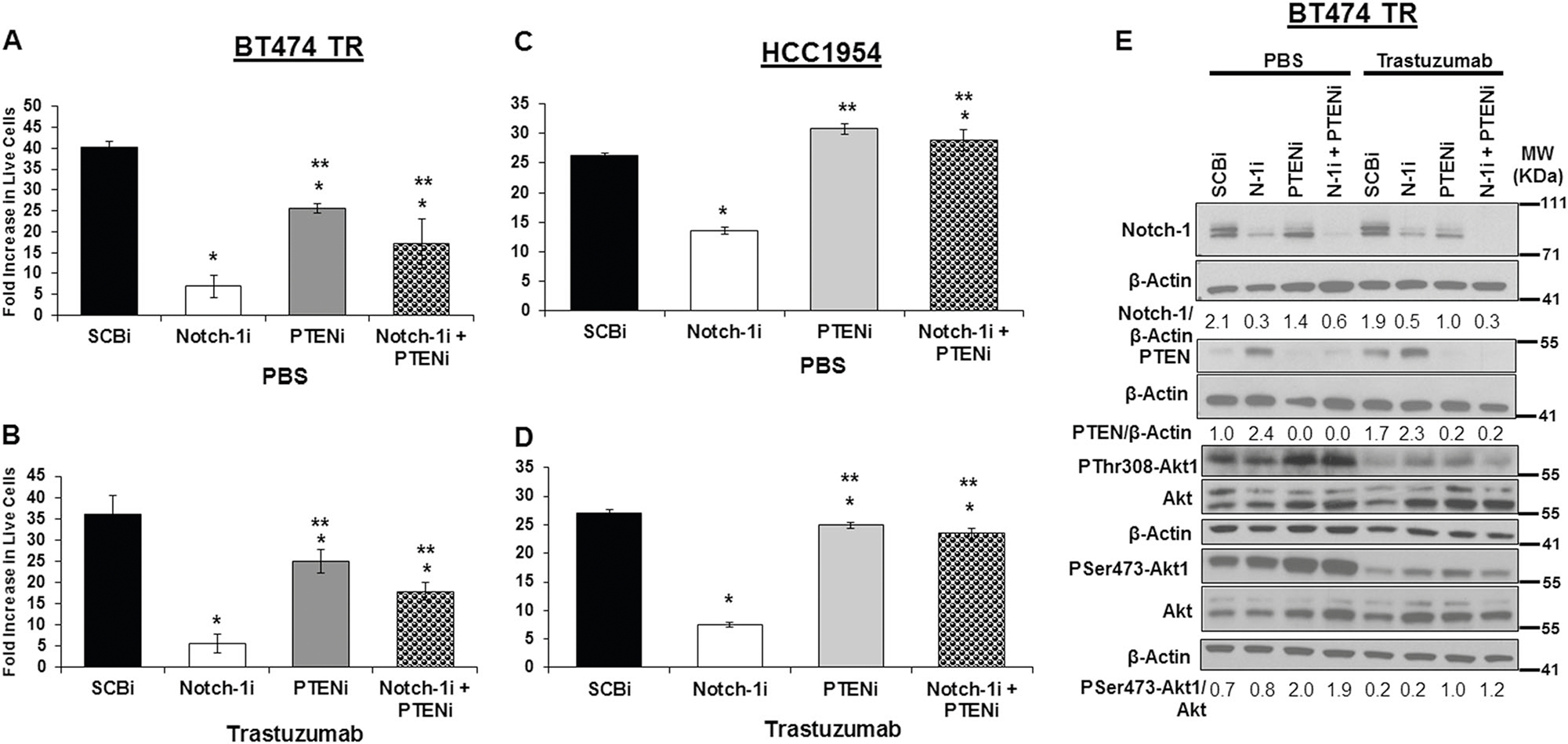
Notch-1-mediated inhibition of PTEN is necessary for trastuzumab-resistant bulk cell proliferation a–d. BT474TR or HCC1954 cells were transfected with control siRNA (SCBi), Notch-1 siRNA, and/or PTEN siRNA for 48 h. Post transfection, cells were seeded at 100,000 cells/well in a 6 well plate and treated daily with PBS (a, c) or 20 μg/mL trastuzumab (b, d) for 10 days. Post treatment, cells were collected and counted. Mean fold increase in live cells was calculated as the number of live cells harvested at day 10/number of live cells seeded at day 0. Asterisk (*) denotes statistical significance compared to SCBi or (**) Notch-1i as calculated using a one-way ANOVA with an overall statistical significance of p < 0.0001 from three independent experiments. e Total cellular Notch-1, PTEN, Akt, β-Actin, phosphorylation of Akt-1 at threonine308 (PThr308-Akt1) and serine473 (PSer473-Akt1) protein expression from the BT474 TR lysates in a, b post 48 h PBS (left) or trastuzumab (right) treatment was detected by Western blot. Mean values from densitometric analysis of Notch-1, PTEN, and PSer473-Akt1 normalized to β-Actin or Akt, respectively, are displayed below their respective blots, and quantitated from three independent experiments using Image J
To determine effects of Notch-1 and/or PTEN knockdown on the PI3K-Akt1 signaling pathway, Western blot analysis to detect protein levels of Notch-1, PTEN, P-Akt, total Akt, and Actin from lysates of transfected resistant cells was performed. Results showed that Notch-1 or PTEN knockdown was efficient in BT474TR cells (Fig. 3e). PTEN knockdown alone or when combined with Notch-1 resulted in increased Akt-1 phosphorylation at Ser473 and Thr308, indicating Akt-1 activation. Interestingly, Notch-1 knockdown alone did not result in decreased Akt-1 phosphorylation (Fig. 3e). Similar results were observed in HCC1954 cells (SFig. 4). Moreover, overall Akt-1 phosphorylation was decreased by trastuzumab treatment in both BT474TR and HCC1954 cells regardless of Notch-1 or PTEN knockdown, suggesting that there could be a molecular disconnect between HER2 effects on Akt signaling and effects on cell proliferation once cells become resistant to trastuzumab.
The role of ERK1/2 signaling in trastuzumab-resistant breast cancer
Thus far, results indicate that Notch-1 is necessary to repress PTEN levels, but that cell proliferation of trastuzumab-resistant cells may not be mediated solely through increased PI3K/Akt pathway activation. It was shown previously that PTEN can function as a protein phosphatase when localized in the nucleus to attenuate P-ERK1/2 activity [47, 48]. As observed in Fig. 2a, PTEN is localized in both the cytoplasm and nucleus when Notch-1 is knocked down in BT474TR cells. Furthermore, BT474TR cells have increased ERK1/2 phosphorylation compared to sensitive cells (Fig. 4a, left panel), which is decreased upon Notch-1 knockdown (Fig. 4a, right panel). On the basis of this finding, we asked whether Notch-1 promoted trastuzumab-resistant cell proliferation by activation of ERK1/2, and whether this was dependent on PTEN repression. Results showed that Notch-1 knockdown decreased ERK1/2 phosphorylation and this decrease was rescued upon PTEN knockdown. The MEK1/2 inhibitor, U0126, completely inhibited ERK1/2 phosphorylation regardless of Notch-1 and/or PTEN knockdowns (Fig. 4b) resulting in reduced resistant cell proliferation compared to the vehicle control DMSO (Fig. 4c). Importantly, the MEK1/2 inhibitor U0126 completely inhibited cell proliferation under conditions where Notch-1, PTEN, or the combination of Notch-1 and PTEN were knocked down (Fig. 4c). These results suggested that Notch-1-mediated repression of PTEN was necessary to at least maintain or activate ERK1/2 via MEK1/2 to drive cell proliferation. To test this hypothesis, a constitutively active MEK1 (MEK1DD)-expression vector was expressed in BT474TR cells to determine whether persistent activation of ERK1/2 could rescue the inhibition of cell proliferation by Notch-1 knockdown. Results demonstrated that Notch-1 knockdown decreased ERK1/2 phosphorylation, but that this was not evident when MEK1DD was overexpressed (Fig. 4d). In agreement, Notch-1 knockdown decreased BT474TR cell proliferation and this decrease was partially rescued by overexpression of MEK1DD (Fig. 4e). Altogether, these results suggest that Notch-1-mediated repression of PTEN activated the ERK1/2 pathway to, at least in part, promote trastuzumab-resistant, HER2+ cell proliferation.
Fig. 4.
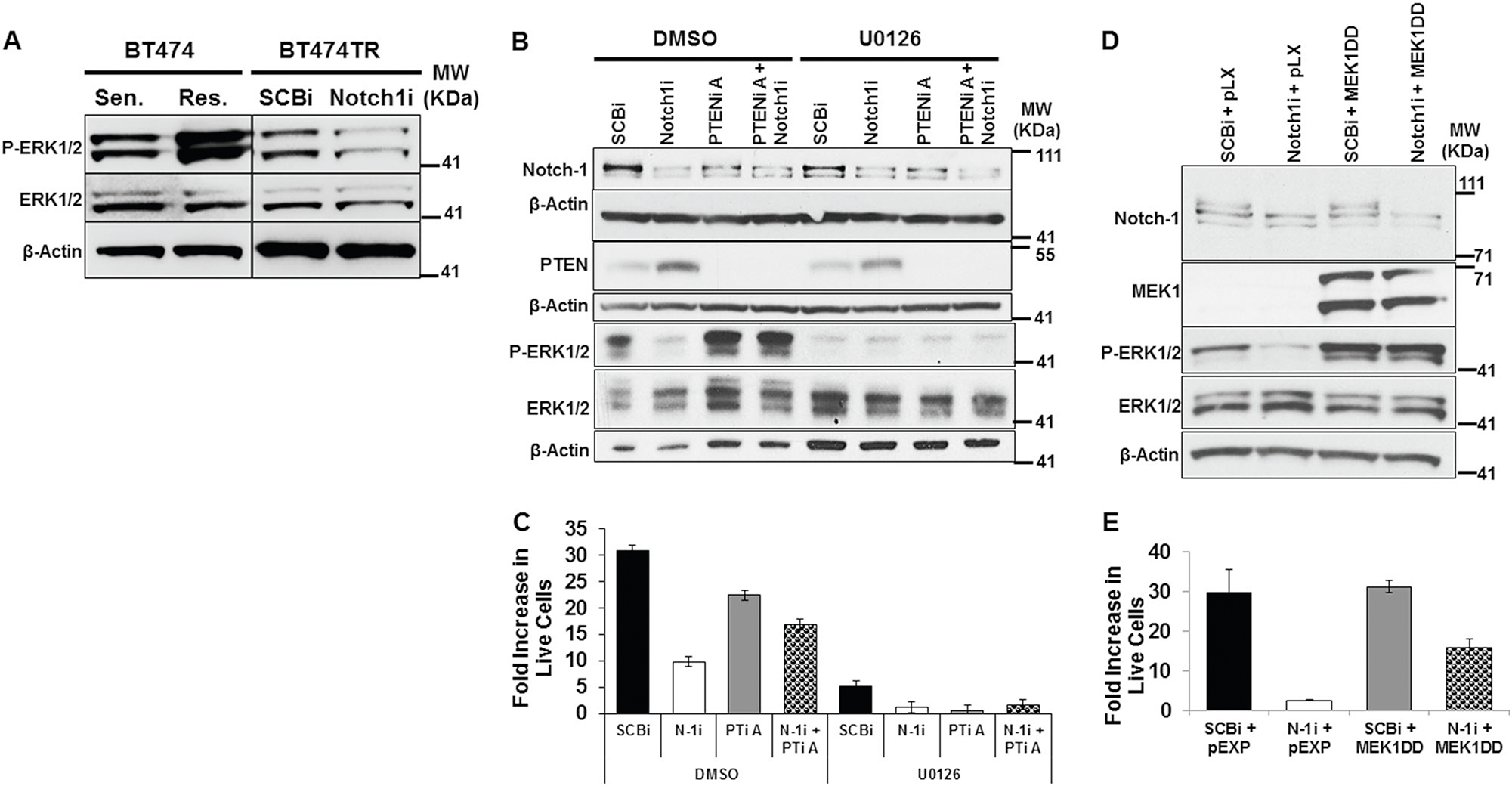
Notch-1 is required to maintain ERK1/2 signaling and bulk cell proliferation by repressing PTEN. a Total protein lysates from BT474TS (Sen.) and BT474TR (Res.) (left panel) or BT474TR cells post 48 h SCBi or Notch-1i transfection (right panel) underwent Western blot analysis to detect P-ERK1/2, ERK1/2, and β-Actin protein expression. b BT474TR cells were transfected with SCBi, Notch-1i, and/or PTENi-A siRNA for 48 h. Post transfection, 100,000 cells/well were plated in a 6 well plate and treated daily with 10 μM U0126 (left) or DMSO (right) for 10 days. Post treatment, cells were harvested and total cellular Notch-1, PTEN, P-ERK1/2, ERK1/2 and β-Actin protein expression was detected by Western blot. c Cells were counted after 10 days of treatment. Mean fold increase in live cells was calculated as the number of live cells harvested at day 10/ number of cells seeded at day 0 from triplicate wells. d BT474TR cells were transfected with SCBi or Notch-1i siRNAs and MEK1DD or pEXP expression vectors for 48 h. Total cellular Notch-1, MEK1, P-ERK1/2, ERK1/2 and β-Actin protein expressions were analyzed by Western blot. e Post transfection, cells were harvested and seeded at 100,000 cells/well of a six well plate. After 10 days, the cells were harvested and counted. Mean fold increase in live cells was calculated as the number of live cells harvested at day 10/number of cells seeded at day 0 from triplicate wells. Graphs show mean plus or minus standard deviations of three replicate assays
Notch-1-mediated inhibition of PTEN promotes trastuzumab-resistant BCSC survival and self-renewal in vitro
To determine whether the trastuzumab-resistant, HER2+ breast cancer cells were enriched for BCSCs, we performed a series of primary and secondary mammosphere forming assays [49]. First, BT474 resistant cells were compared to sensitive cells. BT474TR cells displayed an increase in size and mammosphere forming efficiency (%MFE) compared to sensitive cells indicating that after long term trastuzumab treatment, resistant cells have increased BCSCs (SFig. 5). To determine if Notch-1 is necessary for BCSC survival and self-renewal in the resistant cell lines (BT474 TR and HCC1954), Notch-1 was knocked down and %MFE was determined. Results showed that Notch-1 knockdown reduced primary and secondary %MFE of BT474TR (Fig. 5a, b) and HCC1954 cells (Fig. 5c, d) by nearly 3-fold compared to control, regardless of trastuzumab treatment. PTEN knockdown alone had little effect on %MFE (Fig. 5). However, PTEN knockdown prevented the reduction in % MFE mediated by Notch-1 knockdown alone in both cell lines (Fig. 5). To verify that knockdown of Notch-1 on % MFE was not due to off-target effects by a Notch-1 siRNA, comparable %MFE results were observed using a second Notch-1 siRNA (SFig. 6).
Fig. 5.
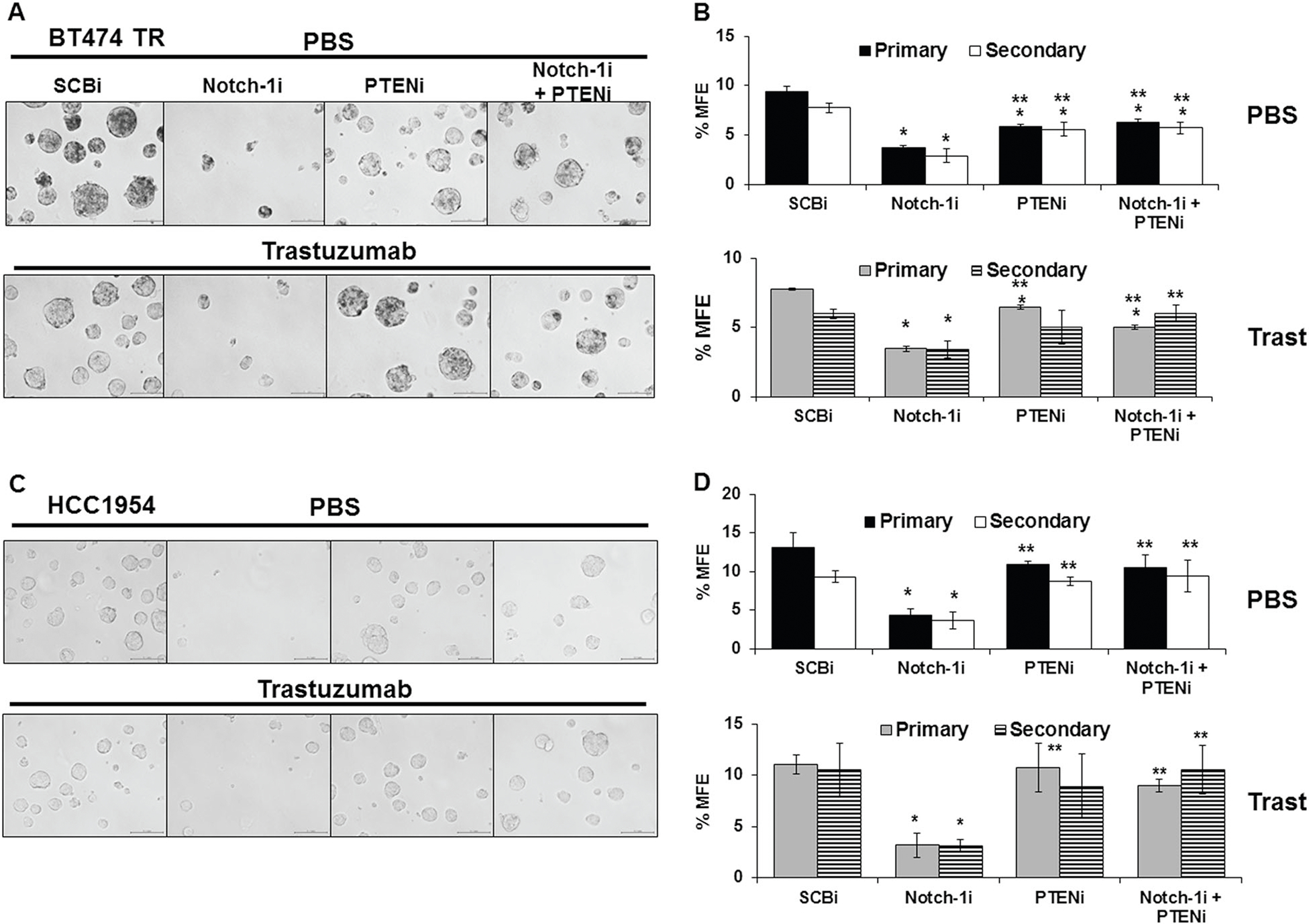
Notch-1-mediated inhibition of PTEN is necessary for survival and self-renewal of breast cancer stem cells (BCSCs) in vitro. BT474TR (a, b) or HCC1954 (c, d) cells were transfected with SCBi, Notch-1i, and/or PTENi siRNAs for 48 h. Post transfection, cells were collected and 100,000 cells/well were seeded into PBS (upper panel) or trastuzumab (20 μg/mL) (lower panel) inoculated mammosphere forming media in a 6 well plate. Mammospheres were incubated for 10 days to form primary (1°) mammospheres then harvested, disaggregated, and 100,000 live cells from the primary mammospheres were seeded into PBS or trastuzumab inoculated mammosphere forming media. The cells were incubated for 10 days to form secondary (2°) mammospheres. Before harvesting, representative photographs of primary mammosphere were taken on day 10 at ×20 magnification. Mammospheres were harvested in PBS and mammospheres over 50 μm were counted. b, d Primary (black bars) and secondary (white bars) percent mammosphere forming efficiency (% MFE) was calculated as: [(number of mammospheres counter/well)/number of cell seeded×100]. Asterisk (*) denotes statistical significance compared to SCBi or (**) Notch-1i mean %MFE as calculated using a one-way ANOVA with an overall statistical significance of p < 0.01 from three independent experiments. Scale bar = 100 μm
To assess whether Notch-1 is required for BCSC survival by maintaining ERK1/2 phosphorylation, %MFE was repeated using the MEK1/2 inhibitor, U0126 compared to vehicle DMSO. Figure 6 demonstrated that Notch-1 knockdown reduced %MFE, as previously shown. PTEN knockdown partially prevented the decrease in %MFE by Notch-1 knockdown. The MEK1/2 inhibitor, U0126, decreased %MFE of BT474TR cells transfected with control siRNA by >50% and completely inhibited %MFE in cells transfected with Notch-1, PTEN, or Notch-1 and PTEN siRNA compared to the vehicle DMSO (Fig. 6a, b). Therefore, Notch-1 is necessary for BCSC survival at least in part through PTEN inhibition and ERK1/2 activation.
Fig. 6.
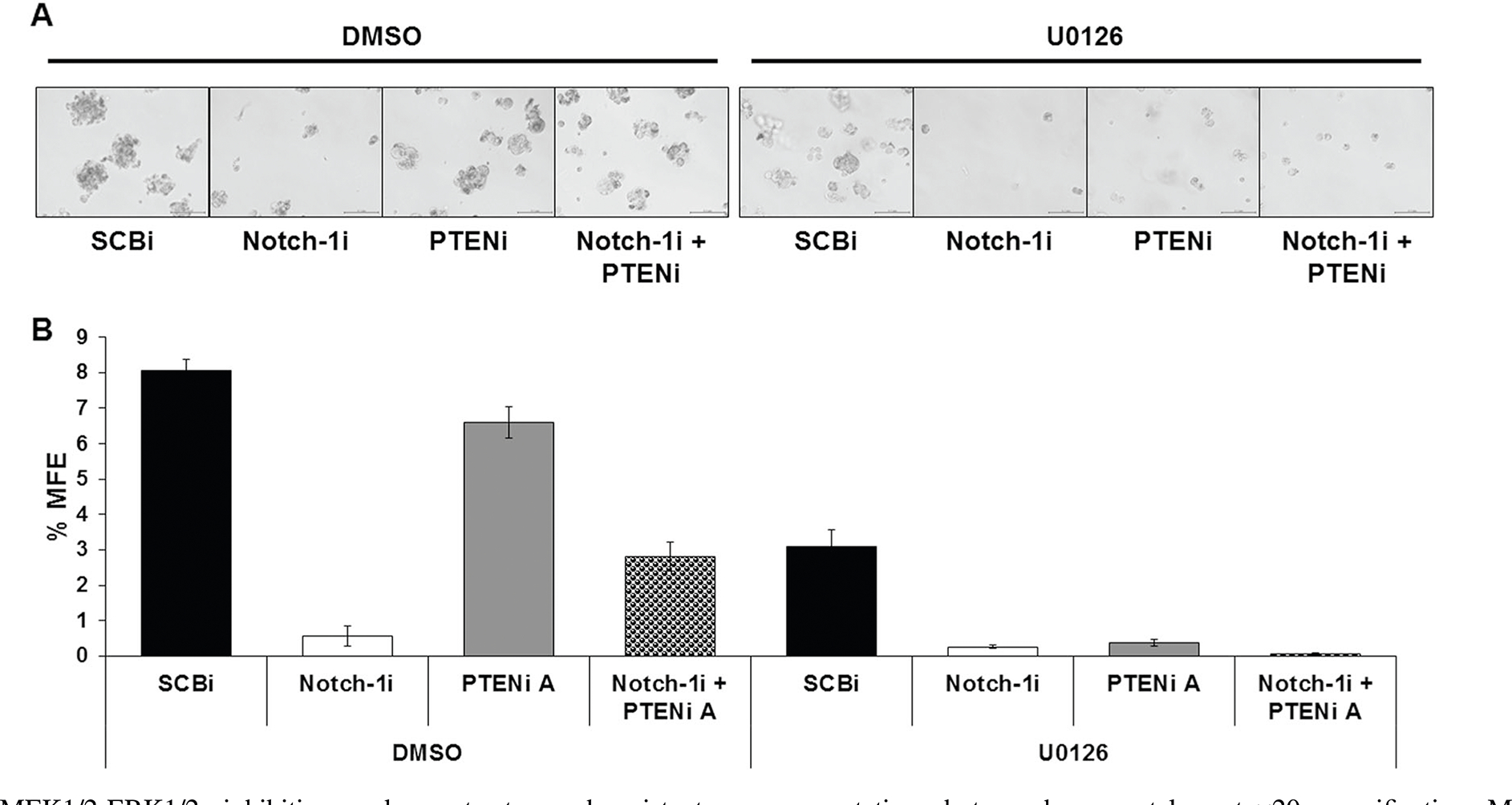
MEK1/2-ERK1/2 inhibition reduces trastuzumab-resistant BCSC survival. a BT474TR cells were transfected with SCBi, Notch-1i, and/or PTENi-A siRNAs for 48 h. Post transfection, cells were harvested and 100,000 live cells/well were seeded into DMSO (left) or 10 μM UO126 (right) inoculated mammosphere forming media in a 6 well plate and incubated for 10 days. On day 10, representative photographs were taken at ×20 magnification. Mammospheres were harvested in PBS and counted if they were greater than 50 μm. b Mean percent mammosphere forming efficiency (% MFE) was calculated as [(number of mammospheres counted/number of cells seeded)×100] from triplicate wells. Scale bar = 100 μm
Notch-1 mediated inhibition of PTEN promotes trastuzumab-resistant breast tumor formation in vivo
To determine if Notch-1-mediated inhibition of PTEN is required for trastuzumab-resistant tumor initiation, we tested the effect of Notch-1 and/or PTEN inhibition on the rate of tumor onset and stem cell frequency using the Extreme Limiting Dilution Assay (ELDA). Primary mammosphere-derived cells at 10,000 and 100,000 cell numbers were injected into female nude mice. Notch-1 knockdown prevented tumor initiation of 10,000 injected cells up to 12 weeks (Fig. 7a–c). PTEN knockdown alone resulted in tumor initiation at a similar rate to the control siRNA-transfected cells (Fig. 7a–c). Combined Notch-1 and PTEN knockdown resulted in similar rate of tumor initiation compared to control, Notch-1, or PTEN siRNA (Fig. 7a–c). Stem cell frequency estimate was calculated using the following website (http://bioinf.wehi.edu.au/software/elda/). Figure 7d shows that Log fraction nonresponding over Dose (# of cells injected) plot for the four groups. Notch-1 knockdown decreased the stem cell frequency estimate by nearly two fold compared to control. PTEN knockdown alone increased the stem cell frequency estimate by >9 fold. The combined Notch-1 and PTEN knockdown resulted in a similar stem cell frequency estimate compared to control (Fig. 7d). The limiting dilution data entered, confidence intervals for 1/stem cell frequency, and overall test for differences between groups are provided in supplementary Table 1. These results together suggest that Notch-1 is necessary for trastuzumab-resistant BCSC-driven tumor initiation through PTEN inhibition.
Fig. 7.
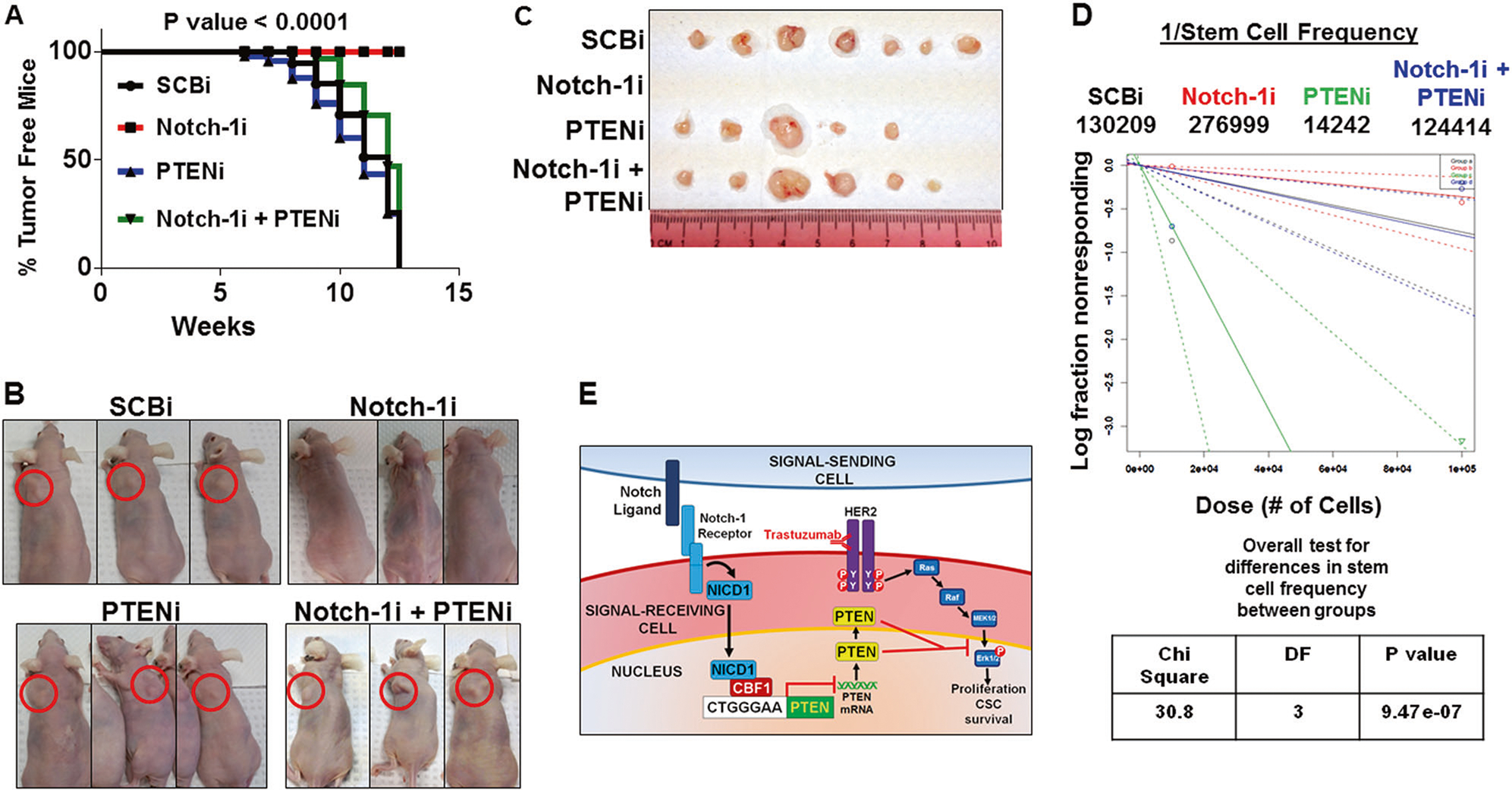
Notch-1 mediated inhibition of PTEN is required for trastuzumab-resistant breast cancer stem cell xenograft tumor formation in vivo. a BT474TR cells were transfected with SCBi, Notch-1i, and/or PTENi for 48 h. Post transfection, cells were collected and seeded in mammosphere medium at 100,000 cells/well in a 6 well plate for 10 days. After 10 days, primary mammospheres were harvested and trypsinized into single cells. In total 10,000 or 100,000 live cells in 50 μL of PBS were mixed with 50 μl of Matrigel and injected bilaterally into the mammary fat pads of female athymic nude mice. Mice were monitored for tumors weekly for 12 weeks. The Kaplan–Meier curves and log rank, Mantel–Cox test (p < 0.0001) displays percent tumor free mice injected with 10,000 cells from SCBi, Notch-1i, PTENi, and Notch-1i plus PTENi siRNA transfections. b Representative photographs of SCBi, Notch-1i and/or PTENi treated BCSC tumor formation (red circles) in nude mice. c SCBi, Notch-1i, PTENi, and Notch-1i+PTENi derived tumors excised from nude mice. d Extreme Limiting Dilution Assay results to estimate stem cell frequency for 10,000 and 100,000 cells injected bilaterally into 6–8 mice for four groups is plotted as Log fraction nonresponding on the Y-axis and Dose (# of cells injected) on the X-axis. Results from the overall test for differences in stem cell frequency between groups was calculated using Chi-Square analysis. e Schematic of Notch-1 mediated inhibition of PTEN in trastuzumab-resistant cells promotes activation of the MAPK/ERK1/2 pathway to drive cell proliferation and breast cancer stem cell survival
Expression of Notch-1 and PTEN transcripts and protein in HER2+ breast tumors and their association with overall survival outcome
On the basis of laboratory results, we sought to examine patterns of Notch-1 and PTEN co-expression in human breast cancer tissues. Using the oncomine database (oncomine.org), three cohorts of microarray data sets were identified that expressed Notch-1 and PTEN [50–53]. Table 1A shows that there is a modest inverse correlation between Notch-1 and PTEN mRNA expression in two of the three cohorts. The correlations were negative and approached statistical significance in the Curtis and Pereira and Bonnefoi cohorts. We focused on the largest cohort, the Curtis breast dataset, to assess the overall survival outcome associated with Notch-1 and PTEN expression. To this end, we fitted a Cox proportional hazards model using Notch-1 and PTEN mRNA expression as two predictors for overall survival in all breast cancer cases (Table 1B) and HER2+ only (Table 1C). Table 1B shows that Notch-1 and PTEN expression influenced the survival time in opposite directions, and their effects as a whole were statistically significant (p-value = 0.001262). Similar results were found in the HER2+ subset with a p-value of 0.022 (Table 1C). These results demonstrated a trend that higher Notch-1 and lower PTEN mRNA expression was associated with poorer overall survival outcome in women with breast cancer.
Table 1.
Inverse expression of Notch-1 and PTEN in breast cancer patient tumors is associated with poorer outcome
| A. Co-Expression correlations of Notch-1 and PTEN | ||
| Rank correlation | p-value | |
| Curtis | −0.139 | 0.051 |
| Bonnefoi | −0.1537 | 0.052 |
| Hatzis | −0.03651 | 0.412 |
| B. Cox Proportional Hazards Model (all BC) | ||
| Hazard Function Estimate | p-value | |
| Notch-1 Coefficient | 0.112 | 0.0636 |
| PTEN Coefficient | −0.227 | 0.0038 |
| C. Cox Proportional | Hazards Model (HER2+) | |
| Hazard Function Estimate | p-value | |
| Notch-1 Coefficient | 0.107 | 0.2344 |
| PTEN Coefficient | −0.292 | 0.0133 |
| HER2 Coefficient | 0.540 | 8.85 × 10−5 |
A Correlation of Notch-1 and PTEN co-expression in each of the three breast microarray data sets as calculated by Spearman’s correlation coefficient
B Co-expression of high Notch-1 and low PTEN regardless of HER2 status (all BC) correlates with an overall decrease in patient survival time in the Curtis Breast dataset as determined by the Cox proportional hazards model. The log-rank test p-value = 0.001262
C In the HER2+ dataset, the overall significance of Notch-1, PTEN, and HER2 is 3.11 × 10−5 by the Log rank test. The overall significance of Notch-1 and PTEN based on HER2+ status is 0.022, by the likelihood ratio test
To determine whether Notch-1 and PTEN mRNA expression correlated with protein expression, we screened the well characterized Nottingham cohort data set with associated outcome of stage II–III primary invasive HER2+ breast tumor arrays for expression of Notch-1 and PTEN proteins and correlations to survival outcomes. Results showed that high membrane Notch-1 protein expression was associated with poorer cumulative survival in women with HER2+ breast cancers (SFig. 7 and Stable 2). PTEN protein expression alone was not found to be significantly correlated with outcome (data not shown). In addition, the number of HER2+ breast tumor samples were too small to achieve statistical power to assess whether Notch-1 and PTEN protein co-expression would predict better or worse survival outcomes (Suppl Fig. 8–10).
Altogether these results indicate that Notch-1-mediated repression of PTEN is necessary to promote survival of HER2+, trastuzumab-resistant bulk cells, breast CSCs, and breast tumors. Further, in vitro results suggest the survival of both bulk and CSC cells at least in part is mediated through MEK1/2-ERK1/2 signaling. Finally, Notch-1 RNA and protein expression are associated with poorer survival for women with HER2+ breast cancer. Additional prospective studies are needed to determine the clinical impact of Notch-1 and PTEN co-expression in trastuzumab-resistant, HER2+ disease.
Discussion
A working model based on findings from this study is summarized in Fig. 7e. Trastuzumab-resistant breast cancer cells increase Notch-1 expression, which represses PTEN levels. This decrease in PTEN results in hyper-phosphorylation and activity of the ERK1/2 signaling pathway. Notch-1, by keeping PTEN expression low, allows for increased ERK1/2 signaling that promotes proliferation of resistant cells, and maintains the survival and tumor-initiating potential of BCSCs. Of note, Notch-1 was not required to maintain Akt1 activity in trastuzumab-resistant cells. It is possible that Notch-1 could be necessary to maintain activity of other isoforms of Akt (Akt2 or Akt3). Alternatively, chronic trastuzumab treatment may upregulate compensatory pathways in HER2+ cells in order to maintain cell survival and proliferation. A shift from dependence on the Akt pathway to the MAPK pathway may be partially facilitated by a novel mechanism whereby Notch-1 represses PTEN.
Cytoplasmic PTEN functions as a lipid phosphatase, dephosphorylating PIP3 and leading to attenuated Akt activity, upregulation of cell cycle inhibitor p27kip1 and pro-apoptotic caspase 3/7, which in turn promotes apoptosis [17]. Nuclear PTEN has been shown to function as a protein phosphatase to dephosphorylate ERK1/2 in the nucleus, causing downregulation of the MAPK pathway and reduced cyclin D1 expression [54]. Nuclear localization of PTEN may be dependent on the presence of the Major Vault Protein (MVP) [54] that can bind to PTEN at two nuclear localization signals (NLS) in a calcium-dependent fashion [18]. In addition, the subcellular localization of PTEN can be modulated by a mutation or masking of the NLS or nuclear export signals (NES) [55] in the PTEN protein or by deubiquitinylation of PTEN by HAUSP as seen in PML [56]. Notch-1 may tip the balance of PTEN subcellular localization towards the nucleus or the cytoplasm as a delicate balance between PTEN stability, and degradation may also be pertinent to its location and activity in trastuzumab-resistant cells. These mechanistic studies are currently underway.
Translocation of PTEN to the nucleus may enable a shift from Akt inhibition and apoptosis of trastuzumab sensitive cells to MAPK inhibition and quiescence in trastuzumab-resistant cells. This shift from Akt to MAPK dependency may allow dormant BCSC to escape trastuzumab treatment and generate a subset of cells dependent on the MAPK pathway to proliferate despite trastuzumab-mediated Akt inhibition. Deregulation of PTEN by Notch-1 may be an early event in the development of trastuzumab resistance as overexpression of Notch-1 (ICD) in sensitive cells repressed PTEN but was not sufficient to promote resistance to trastuzumab (data not shown). These results indicate that other signaling pathways in conjunction with Notch-1 are necessary to promote trastuzumab resistance.
PTEN transcription is regulated by a number of different mechanisms and pathways particularly during development and tumorigenesis. PTEN transcription is regulated by p53, SMADs, and NF-κB for proper organogenesis and immune function [57, 58]. Mutation of PTEN is infrequent in breast cancer, yet PTEN loss of heterozygosity regularly occurs [59]. Here we show Notch-1 represses PTEN transcript expression possibly through a direct transcriptional mechanism. Previous work in T-ALL demonstrated that PTEN inhibition by Notch-1 occurs through Notch-1-mediated upregulation of the transcriptional repressor, HES-1, but this was not the case in our model (data not shown) [40]. Our data support a direct repression of the PTEN promoter, similarly to what is observed in NSCLC cells [60]. The mechanism by which Notch-1 may inhibit transcription could be due to formation of an incomplete Notch-1 transcriptional complex or interaction with negative co-repressors. For instance, p53 can inhibit Notch-mediated transcriptional activation by binding to the Notch transcriptional complex [61]. Notch-1 may promote p65 cytoplasmic sequestration thereby blocking p65 nuclear translocation and failure to transcribe PTEN as demonstrated by metadherin-mediated inhibition of the NF-κB/p65 pathway in trastuzumab-resistant cells [62]. Notch-1 has been shown to upregulate NF-κB activity in TNBC [63] and HER2-negative BCSCs [64] yet its role in mediating PTEN inhibition through NF-κB activation in trastuzumab-resistant cells has yet to be determined. Inhibition of PTEN nuclear translocation is facilitated by sirtuin-1 (SIRT1). Co-expression of low SIRT1 and high Notch-1 correlates with a poor prognosis in breast cancer patients [65] suggesting potential crosstalk between SIRT1 and Notch-1 to regulate PTEN. Notch-1 could repress PTEN through Sal4/NuRD-mediated hyper-methylation of the PTEN promoter [66] or by mediating upregulation of the microRNA, miR-205, to suppress PTEN translation as seen in BCSCs [67, 68].
Surprisingly, the phosphorylation status of Akt1 is unaltered, when Notch-1 is depleted indicating that Notch-1 is not necessary for Akt1 activation in trastuzumab-resistant, HER2+ breast cancer cell lines. However, as stated previously, this does not preclude a role for Notch-1 on Akt2 or Akt3. Increased PTEN levels in response to Notch-1 depletion may be redirected towards maintaining ERK1/2 phosphorylation in the nucleus and thus not available to attenuate PI3-K-Akt1 signaling in the cytoplasm. Alternatively, hyper-phosphorylation of Akt1 in resistant cells could be due reduced expression of PP2A rather than low PTEN levels [69, 70] and thus increased Akt1 activity would be independent of Notch-1.
In conclusion, we describe a mechanism by which Notch-1 is necessary to maintain trastuzumab-resistant, HER2+ breast cancer cell bulk proliferation and BCSC survival by repressing PTEN to promote ERK1/2 activity (summarized in Fig. 7e). Further, co-expression of Notch-1 and PTEN mRNA could serve as better predictive biomarkers of response to trastuzumab in women with HER2+ breast cancer. Interestingly, high expression of membrane Notch-1 protein in untreated, primary invasive, HER2+ breast cancer tissue is associated with poorer survival outcomes, suggesting that Notch-1 is expressed and ready to become activated upon trastuzumab treatment to promote resistance. These findings provide a rationale for a clinical prospective trial in the neoadjuvant setting with matched pre-treated and post-treated tissue to precisely determine whether changes in Notch-1 and PTEN co-expression are more accurate predictors of response to trastuzumab and importantly a resistant phenotype.
Materials and methods
Cell culture and reagents
BT474TS (trastuzumab sensitive) and HCC1954 (trastuzumab-resistant) breast cancer cells were purchased from American Type Culture Collection (ATCC). BT474TR (trastuzumab-resistant) cells were generated by treating parental BT474TS cells with increasing concentrations of trastuzumab for 6 months and maintained in trastuzumab (10 μg/mL in PBS) [22]. BT474TR cells have been characterized reproducibly and confirmed to be resistant to trastuzumab compared to their parental sensitive cells (BT474TS) by our laboratory and others [26, 27, 42–44]. Cells were cultured in Dulbecco’s minimal essential media with high glucose, [1 × DMEM (Cat. MT-10–013-CV, Fisher Scientific, Hampton, NH)] 10% fetal bovine serum (FBS), 100 μM L-Glutamine, and 100 μM nonessential amino acid. All cell lines were authenticated by STR allelic profiling (DCC Medical).
Proliferation assay
In total 3 × 106 BT474TR, BT474TS, or 1 × 106 HCC1954 cells were plated in 10 cm2 plates and transfected with scrambled control (Cat. sc-37007, Santa Cruz, Dallas, TX), Notch-1 (Cat. sc-36095, Santa Cruz), Notch-1 #2 (Cat. HSS107248, Dharmacon, Lafayette, CO), and/or PTEN (Cat. sc-29459, A, B, C, Santa Cruz) siRNA with RNAiMax (Cat. 13778150, Thermo Fisher, Waltham, MA) at a 1:1 ratio in Opti-MEM (Cat. 31985-010, Life Technologies, Carlsbad, CA) for 48 h. BT474TS or BT474R cells were transfected with pcDNA3 (Invitrogen, Carlsbad, CA), pEXP304-LacZ/V5, pEXP304-MEK1DD/V5 [MEK1 S218D/S222D phosphomutant (generous gift from Dr. Takeshi Shimamura at Loyola University Chicago)], or pcDNA3.1-NICD1 (generous gift from Dr. Lucio Miele at Louisiana State University) expression vector using polyethylenimine (Cat. 23966-2, PolySciences, Niles, IL) in Opti-MEM for 48 h. Post transfection, cells were seeded into 6-well plates at a density of 100,000 BT474 cells/well or 50,000 HCC1954 cells/well and then treated everyday with phosphate-buffered saline (PBS) or trastuzumab (20 μg/mL in PBS) for 10 days. Cells were counted using the countess automated cell counter (Cat. C10310, Life Technologies) by trypan blue staining. Fold increase in live cell numbers was calculated as the total number of live cells divided by the number of cells seeded at day 10 post treatments.
Real-time PCR
Total RNA was extracted from cells using the RiboPure™ Kit (Cat. 1924, Thermo Fisher, Waltham, MA). Total cDNA was reverse transcribed using random hexamers with the TaqMan Reverse Transcriptase Kit (Cat. N8080234, Applied Biosystems, Ford City, CA). Real time PCR was performed using Syber green to detect relative expression of transcripts by specific primers (STable 3). Relative expression was calculated using the 2−ΔΔCt method [71]. HPRT primers were used to detect endogenous control gene transcripts as loading controls. Statistical significance was calculated using ΔCt values.
Western blot assay
Cells were lysed in cold lysis buffer [Triton-X (50 mM HEPES, pH 7.4, 1% Triton X-100, 150 mM NaCl, 5 mM EDTA, 1 mM Na3VO4, 10 mM NaF, 1 mM PMSF, 1 protease inhibitor cocktail tablet) or Radioimmunoprecipitation Assay (RIPA) buffer [(0.1 % SDS, 50 mM Tris–HCl, 1% NP-40, (pH to 7.0)] 0.5% Na deoxycholate, 1 mM Na3VO4, 1 mM NaF, 1 mM PMSF, 1 protease cocktail tablet)]. A 20 μg/mL of protein lysate was subjected to SDS–PAGE on a 7% Tris-Acetate pre-cast gel (Cat. EA0355BOX, Invitrogen, Carlsbad, CA) in 1 × Tris-Acetate SDS running buffer (Cat. LA0041, Invitrogen) for 1 h at 150 volts using the XCell SureLock © MiniCell apparatus (Cat. EI0001 Thermo Fisher, Waltham, MA). The gel was transferred to a polyvinylidene fluoride membrane using 1 × transfer buffer [50 mL of 10 × NuPAGE © transfer buffer (Cat. NP0006-1, Invitrogen), 200 mL methanol, 750 mL water] at 40 volts for 2 h using the XCell II™ Blot Module (Cat. EI9051, Thermo Fisher). The membrane was blocked in 5% non-fat milk (Cat. 170-6404, Bio-Rad, Hercules, CA) or Roche blocking solution [Roche buffer (Cat. 10473300, Roche, Basel, Switzerland)] in 1 × Tris Buffered Saline with Tween 20 (TBST) [(10 × TBST (12.1 g Tris Base, 87 g sodium chloride, ph to 8.0 in 1 L) 0.2% Nonidet P-40, 0.05% Tween-20] or 1× Tris Buffered Saline [(TBS) no Tween-20 added], respectively, for 1 h at room temperature under constant agitation. The membranes were incubated with a primary antibody at 4 °C overnight under constant agitation. The following day, the membrane was washed three times in 1× TBST solution for 10 min, while under constant agitation at room temperature then the appropriate secondary antibody was added. The secondary antibody was diluted in the appropriate blocking solution and incubated for 1 h at room temperature under agitation. Membranes were washed three times in 1× TBST for 10 min, Enhanced Chemiluminesence (Cat. 32106, Thermo Fisher) or Super-Signal West Extended Duration substrate (Cat. 34075, Thermo Fisher) was used to develop the membranes on radiography film (MedSci). Band intensities were measured by densitometry using ImageJ software; β-Actin or Akt signals served as internal normalizers. PTEN (9559S), Phosphorylated Akt1 (Ser473, 9271S and Thr308, 9275S), Pan Akt (4691S), Total p44/42 ERK1/2 (4695), Histone H3 (4620) and Phosphorylated p44/42 ERK1/2 (Tyr202/Tyr204, 4370) were purchased from Cell Signaling Technologies (Danavers, MA). Notch-1 (C-20, sc-6014), MEK-1 (C-18, sc-219), and normal rabbit IgG (sc-2027) were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). Total HER2 [(ErbB2) MS0730-P] was purchased from Thermo Fisher Scientific (Waltham, MA). Phosphorylated HER2 at tyrosine 1248 [PY1248-ErbB2 (AF1768)] was purchased from R and D Systems (Minneapolis, MN). Beta-Actin (A5441) was purchased from Sigma Aldrich (St. Louis, MO) and used as an endogenous control for all Western blots. Secondary antibodies used include donkey anti-rabbit (1:1000, sc-2313) and donkey anti-mouse (1:5000, sc-2314) purchased from Santa Cruz Biotechnologies (Santa Cruz, CA).
Immunofluorescence
PTEN staining was performed using 1 μg/ml of the rabbit anti-human PTEN antibody (9559 S) from Cell Signaling and 5 μg/ml of AlexaFluor 594 labeled anti-rabbit IgG (Cat. A31572, Life Technologies, Grand Island, NY, USA) as previously described according to the manufacturer’s procedure. VectaShield mounting media containing DAPI (Cat. H1200, Vector Laboratories, Burlingame, CA, USA) was used to mount the slides after staining for specific protein was completed. PTEN (red) and nucleus (blue) were detected with a Zeiss LSM-510 confocal microscope (Carl Zeiss Microscopy, Jena, Germany).
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using the SimpleChIP© Plus kit (Cat. 9005, Cell Signaling, Danvers, MA). Cells were trypsinized and cross-linked in 1% formaldehyde (Cat. F8775, Sigma Aldrich, St. Louis, MO) for 30 min, then quenched with 1.25 M glycine. Cells were lysed, nuclei pelleted by centrifugation at 3000 RPM for 5 min at 4 °C, digested with 2 μl (4000 gel units/μl) of micrococcal nuclease for 30 min at 37 °C, sonicated three times for 20 s at 20% amplitude, then immunoprecipitated with 1 μl (200 μg/0.5 μl) of normal rabbit IgG, 10 μl (200 μg/mL) of C-terminal Notch-1, or 10 μl of an anti-Histone3 antibody (S Table 3). Chromatin was incubated under rotation at 4 °C overnight. The next day, 30 μl protein G magnetic beads were added to each immunoprecipitation and the mixture was incubated for 2.5 h at 4 °C under rotation, chromatin was washed with salt buffers [(1 × ChIP Buffer (#7008), (1 × ChIP Buffer (#7008) and 5 M NaCl], then eluted from the antibody/protein/magnetic breads by shaking at 65 °C for 2 h. Cross-link was reversed by adding 6 μl of 5 M NaCl (Cat. S3014, Sigma-Aldrich) and 2 μl (20 mg/mL) of Proteinase K to each immunoprecipitate and incubated at 65 °C for 2 h. DNA was purified and diluted (1:5) in deionized water. Quantitative PCR was used to detect immunoprecipitated DNA using ChIP primers that were designed to flank putative CSL binding elements [72] (S Table 3).
Mammosphere assay
In total 100,000 BT474 or 50,000 HCC1954 cells were plated in 2 mL of mammosphere-forming media [(DMEM/F12 (Cat. 21041025, Life Technologies, Carlsbad, CA), 4 g methyl cellulose (Cat. 274429, Sigma Aldrich, St. Louis, MO), 10 mL B27 (Cat. 12587-010, Life Technologies), 1 μL recombinant EGF (Cat. E-9644, Sigma Aldrich)] in low attachment 6-well plates (Cat. 229506, Dot Scientific, Burton, MI) and harvested on day 10. 2 mL of mammosphere-forming media was inoculated with PBS, trastuzumab (20 μg/mL in PBS), dimethylsulfoxide (DMSO), or the MEK inhibitor, U0126 (10 μg/mL in DMSO) per well. Mammospheres were harvested by dissolving the mammosphere media. In total 2 mL of PBS was added to each well and incubated for 30 min at 37 °C. After 30 min, mammospheres were harvested and the processes was repeated three times in total. Harvested mammospheres in PBS:mammosphere media solution were pelleted by centrifugation at 1200 RPM for 2 min, aspirate the PBS: Mammosphere mixture, and resuspend the sensitive or resistant mammospheres in 3 or 5 mL of PBS, respectively. In total 50 μL of the mammospheres were plated in a 96-well plate and at least four to five fields were photographed. Mammospheres reaching a size greater that 50 μm were counted, and percent mammosphere forming efficiency (% MFE) was calculated as follows: number of mammospheres counted/number of cells seededx100. Photographs were taken at ×20 magnification of mammospheres using a Nikon Diaphot TMD Fluorescence Phase Contrast Inverted microscope (Nikon, Tokyo, Japan) before and at ×4 magnification after harvesting mammospheres. Primary mammospheres were trypsinized into single cell suspensions and re-plated into secondary mammospheres as described previously.
Trastuzumab resistant xenografts
BT474TR cells were transfected with a control, Notch-1, and/or PTEN siRNA for 48 h and then plated at a density of 100,000 cells/well in low-attachment 6-well plates containing mammosphere forming media. The mammospheres were grown for 10 days and then disaggregated into 10,000 or 100,000 viable cells in 50 μL of PBS and suspended in a 1:1 ratio of Matrigel (Corning, Corning, NY): PBS. Cells were bilaterally injected into the mammary fat pads of 5–6 week old, ovariectomized, FoxN1nu/nu athymic, nude female mice (N = 6–8 mice per group) (Charles River NIH, Bethesda, MD) followed by implantation of a 17β-estradiol-containing silastic release capsule of 0.3 cm in length for a constant release of 83–100 pg/mL, as previously described [73]. Mice were recognized by an ear tag and monitored for tumor growth once a week. Area (l×w) of tumors was measured weekly using Vernier calipers. Date and number of tumors formed, as well as cross sectional area [(l×w)π/4)] of tumors were calculated and graphed. All animal study protocols were approved by Loyola University’s Institutional Animal Care and Use Committee. The number of mice was determined using the previously published protocol [74]. To ensure unbiased measurement of tumor size and tumor incidence, cages were numbered and tumors monitored by an independent scientist.
Notch-1 and PTEN co-expression in human breast tumors
The assessment of the overall survival outcome associated with Notch-1 and PTEN RNA expression in the Curtis breast (2136 patient size) was also done using R. We used the Cox proportional hazards model h(t) = h0(t)exp(βNxN) + βPxP, where h(t) is the hazard function, h0(t) is the baseline hazard, xN and xP are the expression for Notch-1 and PTEN, respectively, and βN and βP are the corresponding coefficients. The estimated coefficients and p-values are shown in Table 1. By using the log-rank test, the overall significance of the effect of Notch-1 and PTEN was obtained, which is also shown in Table 1. This was done for all breast cancer cases (N = 997) and the HER2+ subset (N = 110).
Immunohistochemical staining for Notch-1 and PTEN in HER2+ breast tumors
Approximately 157–159 primary, stage II–II invasive HER2+ breast tumor tissue were adhered by placing the tissue microarray (TMA) sections in an oven at 58–60 °C. Xylene was used for de-paraffinizing the tissue followed by rehydration using ethanol, and washed with PBS 1× Reveal treatment in a Decloaking Chamber (Biocare Medical) for antigen retrieval. Once the TMA sections were rinsed by PBS for 15 min, sections were treated using 3%H2O2 in PBS for 20 min to quench endogenous peroxidase activity. Sections were incubated for 1 h in 3% normal rabbit serum (Vector Laboratories) in PBS at room temperature to block non-specific binding. Sections were then incubated with primary antibody (anti-Notch-1 or anti-PTEN) prepared in PBS containing 1.5% normal rabbit serum and were incubated for 1 h in a hydrated chamber at room temperature. TMA sections incubated with 1 μg/ml normal goat IgG (Santa Cruz Biotechnologies, CA) were used as negative controls. Following multiple washing, antigen–antibody complexes were detected using the Vectastain Elite ABC kit (Vector Laboratories, CA) as per the manufacturer’s protocol. Staining was performed with ImmPactTM DAB peroxidase substrate kit (Vector Laboratories, CA). Sections were then counterstained in Gill’s hematoxylin and dehydrated in ascending grades of ethanol before clearing in xylene and mounting under a coverslip using Cytoseal XYL. Levels of Notch-1 and PTEN protein expression in each specimen were scored per the extent (percent of stained cells) and intensity of staining. The score for the extent of the IHC stained area was scaled as 0 for no IHC signal at all and 1 for 10–80 tumor cells stained. Stained slides were sent to Nottingham, England where they were scanned. High-resolution images were uploaded to the Nottingham web-accessible scoring site, and were scored by two independent investigators (Alexandra Filopovic and Lucio Miele). Notch-1 intensity scores (0–1), PTEN scores (1–3) percent staining scores and H-Scores were uploaded as Excel spreadsheets and survival analysis was performed using SPSS.
Statistical analysis
Statistical analysis of at least three independent experiments was performed. Means±standard deviations were calculated and graphed using Excel software (Microsoft) or GraphPad. A two-sided Student’s t-test was used to compare two groups. A one-way ANOVA with Tukey’s range test was used to compare multiple groups. Asterisk (*) represents statistical significance between control groups and sensitive cells and (**) represents statistical significance between Notch-1 inhibition compared to other treatment groups. A Kaplan–Meier curve for tumor formation was generated by Prism Version 6 (GraphPad Software) and analyzed with a log-rank (Mantel-Cox) test. P-values < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgements
This work was supported by the American Cancer Society (RSG-11-181-01-TBE) awarded to Dr. Clodia Osipo, the Arthur J. Schmitt Fellowship to Dr. Andrew Baker, and in part by the Breast Cancer Research Foundation to Drs. Kathy Albain and Clodia Osipo. We thank Ianina Bognini for assistance during animal studies.
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
Electronic supplementary material The online version of this article (https://doi.org/10.1038/s41388-018-0251-y) contains supplementary material, which is available to authorized users.
References
- 1.Ellis MJ, Perou CM. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013;3:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wachsman W, Cann AJ, Williams JL, Slamon DJ, Souza L, Shah NP, et al. HTLV x gene mutants exhibit novel transcriptional regulatory phenotypes. Science. 1987;235:674–7. [DOI] [PubMed] [Google Scholar]
- 3.Ross JS, Slodkowska EA, Symmans WF, Pusztai L, Ravdin PM, Hortobagyi GN. The HER-2 receptor and breast cancer: ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist. 2009;14:320–68. [DOI] [PubMed] [Google Scholar]
- 4.Buzdar AU, Ibrahim NK, Francis D, Booser DJ, Thomas ES, Theriault RL, et al. Significantly higher pathologic complete remission rate after neoadjuvant therapy with trastuzumab, paclitaxel, and epirubicin chemotherapy: results of a randomized trial in human epidermal growth factor receptor 2-positive operable breast cancer. J Clin Oncol. 2005;23:3676–85. [DOI] [PubMed] [Google Scholar]
- 5.Untch M, Fasching PA, Konecny GE, Hasmuller S, Lebeau A, Kreienberg R, et al. Pathologic complete response after neoadjuvant chemotherapy plus trastuzumab predicts favorable survival in human epidermal growth factor receptor 2-overexpressing breast cancer: results from the TECHNO trial of the AGO and GBG study groups. J Clin Oncol. 2011;29:3351–7. [DOI] [PubMed] [Google Scholar]
- 6.Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353:1659–72. [DOI] [PubMed] [Google Scholar]
- 7.Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol. 2007;18:977–84. [DOI] [PubMed] [Google Scholar]
- 8.Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S, Arteaga CL. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects onp27, cyclin D1, and antitumor action. Cancer Res. 2002;62:4132–41. [PubMed] [Google Scholar]
- 9.Ebbesen SH, Scaltriti M, Bialucha CU, Morse N, Kastenhuber ER, Wen HY, et al. Pten loss promotes MAPK pathway dependency in HER2/neu breast carcinomas. Proc Natl Acad Sci USA. 2016;113:3030–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. [DOI] [PubMed] [Google Scholar]
- 11.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loibl S, Darb-Esfahani S, Huober J, Klimowicz A, Furlanetto J, Lederer B, et al. Integrated analysis of PTEN and p4EBP1 protein expression as predictors for pCR in HER2-positive breast cancer. Clin Cancer Res. 2016;22:2675–83. [DOI] [PubMed] [Google Scholar]
- 13.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. [DOI] [PubMed] [Google Scholar]
- 14.Stern HM, Gardner H, Burzykowski T, Elatre W, O’Brien C, Lackner MR, et al. PTEN loss is associated with worse outcome in HER2-amplified breast cancer patients but is not associated with trastuzumab resistance. Clin Cancer Res. 2015;21:2065–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dittrich A, Gautrey H, Browell D, Tyson-Capper A. The HER2 signaling network in breast cancer–like a spider in its web. J Mammary Gland Biol Neoplasia. 2014;19:253–70. [DOI] [PubMed] [Google Scholar]
- 17.Chung JH, Eng C. Nuclear-cytoplasmic partitioning of phosphatase and tensin homologue deleted on chromosome 10 (PTEN) differentially regulates the cell cycle and apoptosis. Cancer Res. 2005;65:8096–8100. [DOI] [PubMed] [Google Scholar]
- 18.Chung JH, Ostrowski MC, Romigh T, Minaguchi T, Waite KA, Eng C. The ERK1/2 pathway modulates nuclear PTEN-mediated cell cycle arrest by cyclin D1 transcriptional regulation. Hum Mol Genet. 2006;15:2553–9. [DOI] [PubMed] [Google Scholar]
- 19.Mittal S, Subramanyam D, Dey D, Kumar RV, Rangarajan A. Cooperation of Notch and Ras/MAPK signaling pathways in human breast carcinogenesis. Mol Cancer. 2009;8:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mittal S, Sharma A, Balaji SA, Gowda MC, Dighe RR, Kumar RV, et al. Coordinate hyperactivation of Notch1 and Ras/MAPK pathways correlates with poor patient survival: novel therapeutic strategy for aggressive breast cancers. Mol Cancer Ther. 2014;13:3198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weijzen S, Rizzo P, Braid M, Vaishnav R, Jonkheer SM, Zlobin A, et al. Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat Med. 2002;8:979–86. [DOI] [PubMed] [Google Scholar]
- 22.Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–27. [DOI] [PubMed] [Google Scholar]
- 23.Saal LH, Johansson P, Holm K, Gruvberger-Saal SK, She QB, Maurer M, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci USA. 2007;104:7564–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dave B, Migliaccio I, Gutierrez MC, Wu MF, Chamness GC, Wong H, et al. Loss of phosphatase and tensin homolog or phosphoinositol-3 kinase activation and response to trastuzumab or lapatinib in human epidermal growth factor receptor 2-overexpressing locally advanced breast cancers. J Clin Oncol: Off J Am Soc Clin Oncol. 2011;29:166–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong C, Yao Y, Wang Y, Liu B, Wu W, Chen J, et al. Upregulation of miR-21 mediates resistance to trastuzumab therapy for breast cancer. J Biol Chem. 2011;286:19127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osipo C, Patel P, Rizzo P, Clementz AG, Hao L, Golde TE, et al. ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a gamma-secretase inhibitor. Oncogene. 2008;27:5019–32. [DOI] [PubMed] [Google Scholar]
- 27.Pandya K, Meeke K, Clementz AG, Rogowski A, Roberts J, Miele L, et al. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br J Cancer. 2011;105:796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bozkulak EC, Weinmaster G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol. 2009;29:5679–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet. 2002;11:2615–24. [DOI] [PubMed] [Google Scholar]
- 30.Kopan R Notch signaling. Cold Spring Harbor Perspect Biol. 2012;4:a011213:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu L, Sun T, Kobayashi K, Gao P, Griffin JD. Identification of a family of mastermind-like transcriptional coactivators for mammalian notch receptors. Mol Cell Biol. 2002;22:7688–7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194:237–55. [DOI] [PubMed] [Google Scholar]
- 33.Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004;6:R605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cohen B, Shimizu M, Izrailit J, Ng NF, Buchman Y, Pan JG, et al. Cyclin D1 is a direct target of JAG1-mediated Notch signaling in breast cancer. Breast Cancer Res Treat. 2010;123:113–24. [DOI] [PubMed] [Google Scholar]
- 35.Meurette O, Stylianou S, Rock R, Collu GM, Gilmore AP, Brennan K. Notch activation induces Akt signaling via an autocrine loop to prevent apoptosis in breast epithelial cells. Cancer Res. 2009;69:5015–22. [DOI] [PubMed] [Google Scholar]
- 36.D’Angelo RC, Ouzounova M, Davis A, Choi D, Tchuenkam SM, Kim G, et al. Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol Cancer Ther. 2015;14:779–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70:709–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005;65:8530–7. [DOI] [PubMed] [Google Scholar]
- 39.Baker AT, Zlobin A, Osipo C. Notch-EGFR/HER2 bidirectional crosstalk in breast cancer. Front Oncol. 2014;4:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13:1203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Osipo C, Patel P, Rizzo P, Clementz AG, Hao L, Golde TE, et al. ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a γ-secretase inhibitor. Oncogene. 2008;27:5019–32. [DOI] [PubMed] [Google Scholar]
- 42.Das S, Sondarva G, Viswakarma N, Nair RS, Osipo C, Tzivion G, et al. Human epidermal growth factor receptor 2 (HER2) impedes MLK3 kinase activity to support breast cancer cell survival. J Biol Chem. 2015;290:21705–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nunes J, Zhang H, Angelopoulos N, Chhetri J, Osipo C, Grothey A, et al. ATG9A loss confers resistance to trastuzumab via c-Cbl mediated Her2 degradation. Oncotarget. 2016;7:27599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pandya K, Wyatt D, Gallagher B, Shah D, Baker A, Bloodworth J, et al. PKcalpha attenuates jagged-1-mediated notch signaling in ErbB-2-positive breast cancer to reverse trastuzumab resistance. Clin Cancer Res: Off J Am Assoc Cancer Res. 2016;22:175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Katoh M, Katoh M. Integrative genomic analyses on HES/HEY family: notch-independent HES1, HES3 transcription in undifferentiated ES cells, and Notch-dependent HES1, HES5, HEY1, HEY2, HEYL transcription in fetal tissues, adult tissues, or cancer. Int J Oncol. 2007;31:461–6. [PubMed] [Google Scholar]
- 46.Chu D, Zhang Z, Zhou Y, Wang W, Li Y, Zhang H, et al. Notch1 and Notch2 have opposite prognostic effects on patients with colorectal cancer. Ann Oncol. 2011;22:2440–7. [DOI] [PubMed] [Google Scholar]
- 47.Chetram MA, Hinton CV. PTEN regulation of ERK1/2 signaling in cancer. J Recept Signal Transduct Res. 2012;32:190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weng LP, Smith WM, Brown JL, Eng C. PTEN inhibits insulin-stimulated MEK/MAPK activation and cell growth by blocking IRS-1 phosphorylation and IRS-1/Grb-2/Sos complex formation in a breast cancer model. Hum Mol Genet. 2001;10:605–16. [DOI] [PubMed] [Google Scholar]
- 49.Shaw FL, Harrison H, Spence K, Ablett MP, Simoes BM, Farnie G, et al. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J Mammary Gland Biol Neoplasia. 2012;17:111–7. [DOI] [PubMed] [Google Scholar]
- 50.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonnefoi H, Potti A, Delorenzi M, Mauriac L, Campone M, Tubiana-Hulin M, et al. Validation of gene signatures that predict the response of breast cancer to neoadjuvant chemotherapy: a substudy of the EORTC 10994/BIG 00–01 clinical trial. Lancet Oncol 2007;8:1071–8. [DOI] [PubMed] [Google Scholar]
- 53.Hatzis C, Pusztai L, Valero V, Booser DJ, Esserman L, Lluch A, et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA. 2011;305:1873–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chung JH, Ginn-Pease ME, Eng C. Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) has nuclear localization signal-like sequences for nuclear import mediated by major vault protein. Cancer Res. 2005;65:4108–16. [DOI] [PubMed] [Google Scholar]
- 55.Denning G, Jean-Joseph B, Prince C, Durden DL, Vogt PK. A short N-terminal sequence of PTEN controls cytoplasmic localization and is required for suppression of cell growth. Oncogene. 2007;26:3930–40. [DOI] [PubMed] [Google Scholar]
- 56.Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, et al. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature. 2008;455:813–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Knobbe CB, Lapin V, Suzuki A, Mak TW. The roles of PTEN in development, physiology and tumorigenesis in mouse models: a tissue-by-tissue survey. Oncogene. 2008;27:5398–415. [DOI] [PubMed] [Google Scholar]
- 58.Luyendyk JP, Schabbauer GA, Tencati M, Holscher T, Pawlinski R, Mackman N. Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J Immunol. 2008;180:4218–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feilotter HE, Coulon V, McVeigh JL, Boag AH, Dorion-Bonnet F, Duboue B, et al. Analysis of the 10q23 chromosomal region and the PTEN gene in human sporadic breast carcinoma. Br J Cancer. 1999;79:718–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Graziani I, Eliasz S, De Marco MA, Chen Y, Pass HI, De May RM, et al. Opposite effects of Notch-1 and Notch-2 on mesothelioma cell survival under hypoxia are exerted through the Akt pathway. Cancer Res. 2008;68:9678–85. [DOI] [PubMed] [Google Scholar]
- 61.Yun J, Espinoza I, Pannuti A, Romero D, Martinez L, Caskey M, et al. p53 modulates notch signaling in MCF-7 breast cancer cells by associating with the notch transcriptional complex via MAML1. J Cell Physiol. 2015;230:3115–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Du C, Yi X, Liu W, Han T, Liu Z, Ding Z, et al. MTDH mediates trastuzumab resistance in HER2 positive breast cancer by decreasing PTEN expression through an NFkappaB-dependent pathway. BMC Cancer. 2014;14:869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thery JC, Spano JP, Azria D, Raymond E, Penault Llorca F. Resistance to human epidermal growth factor receptor type 2-targeted therapies. Eur J Cancer. 2014;50:892–901. [DOI] [PubMed] [Google Scholar]
- 64.Mao J, Song B, Shi Y, Wang B, Fan S, Yu X, et al. ShRNA targeting Notch1 sensitizes breast cancer stem cell to paclitaxel. Int J Biochem Cell Biol. 2013;45:1064–73. [DOI] [PubMed] [Google Scholar]
- 65.Cao YW, Li WQ, Wan GX, Li YX, Du XM, Li YC, et al. Correlation and prognostic value of SIRT1 and Notch1 signaling in breast cancer. J Exp Clin Cancer Res. 2014;33:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu J, Jeong HW, Kong N, Yang Y, Carroll J, Luo HR, et al. Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS ONE. 2009;4:e5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adachi R, Horiuchi S, Sakurazawa Y, Hasegawa T, Sato K, Sakamaki T. ErbB2 down-regulates microRNA-205 in breast cancer. Biochem Biophys Res Commun. 2011;411:804–8. [DOI] [PubMed] [Google Scholar]
- 68.Greene SB, Gunaratne PH, Hammond SM, Rosen JM. A putative role for microRNA-205 in mammary epithelial cell progenitors. J Cell Sci. 2010;123:606–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baldacchino S, Saliba C, Petroni V, Fenech AG, Borg N, Grech G. Deregulation of the phosphatase, PP2A is a common event in breast cancer, predicting sensitivity to FTY720. EPMA J 2014;5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McDermott MS, Browne BC, Conlon NT, O’Brien NA, Slamon DJ, Henry M, et al. PP2A inhibition overcomes acquired resistance to HER2 targeted therapy. Mol Cancer. 2014;13:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rao X, Huang X, Zhou Z, Lin X. An improvement of the 2^(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat Bioinforma Biomath. 2013;3:71–85. [PMC free article] [PubMed] [Google Scholar]
- 72.Persson LM, Wilson AC. Wide-scale use of Notch signaling factor CSL/RBP-Jkappa in RTA-mediated activation of Kaposi’s sarcoma-associated herpesvirus lytic genes. J Virol. 2010;84:1334–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Regan RM, Cisneros A, England GM, MacGregor JI, Muenzner HD, Assikis VJ, et al. Effects of the antiestrogens tamoxifen, toremifene, and ICI 182,780 on endometrial cancer growth. J Natl Cancer Inst. 1998;90:1552–8. [DOI] [PubMed] [Google Scholar]
- 74.Farnie G, Clarke RB, Spence K, Pinnock N, Brennan K, Anderson NG, et al. Novel cell culture technique for primary ductal carcinoma in situ: role of Notch and epidermal growth factor receptor signaling pathways. J Natl Cancer Inst. 2007;99:616–27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.