

Abstract
The circadian clock provides cue-independent anticipatory signals for diurnal rhythms of baseline glucose levels and glucose tolerance. The central circadian clock is located in the hypothalamic suprachiasmatic nucleus (SCN), which is composed primarily of GABAergic neurons. The SCN clock regulates physiological diurnal rhythms of endogenous glucose production (EGP) and hepatic insulin sensitivity through neurohumoral mechanisms. Disruption of the molecular circadian clock is associated with the extended dawn phenomenon in type 2 diabetes, referring to hyperglycemia in the early morning without nocturnal hypoglycemia. The dawn phenomenon affects nearly half of the patients with diabetes, with poorly defined etiology and lack of targeted therapy. Here we review neural and secreted factors in physiological diurnal rhythms of glucose metabolism and their pathological implications in the dawn phenomenon.
Keywords: circadian rhythm, metabolism, diabetes, SCN, GABA, neuron
Molecular elements of the circadian clock
The circadian clock generates cue-independent daily rhythms in behaviors, hormones, and metabolism. The molecular circadian clock in mammals consists of interlocked transcriptional-translational feedback loops [1]. The transcription factors BMAL1 and CLOCK form heterodimers and bind to the E-box elements in the promoter/enhancer regions of the target genes and activate their transcription. Among BMAL1/CLOCK target genes are the clock genes Period (PER), Cryptochrome (CRY), and REV-ERB. PER and CRY proteins form heterodimers, interact with BMAL1/CLOCK, and counteract BMAL1/CLOCK-mediated transcriptional activation, while nuclear receptor REV-ERB represses BMAL1 transcription by binding to the ROR elements (RORE) in the promoter/enhancer regions of BMAL1. As a result of the negative feedback regulation, the levels of PER, CRY, and REV-ERB transcripts and proteins start to decay once they reach a certain threshold until it comes to a time point when their brake on BMAL1/CLOCK is released, followed by another wave of rising expression, with an oscillatory period of around 24 h. Clock genes also involve several other transcription factors in addition to the ones mentioned above [2]. Through their various target genes, the clock genes initiate many output pathways to confer anticipatory temporal cues to a variety of physiological processes in alignment with the diurnal environmental changes.
Neural outputs of the central clock
In mammals, the circadian clock is organized in a multi-organ system composed of the central clock in the hypothalamic suprachiasmatic nuclei (SCN) and peripheral clocks in other tissues [3]. The SCN central clock can entrain peripheral clocks through behavioral changes or neurohumoral signals, although the exact mechanisms remain partly unclear. In this article, due to space constraints, we will focus only on the central clock. The SCN contains primarily γ-aminobutyric acid (GABA) neurons. The spontaneous firing activity of SCN GABA neurons displays a robust, cell-autonomous, and synchronized circadian rhythm, with peak firing activity in the day and trough at night, in both diurnal and nocturnal animals. The phase of the SCN clock gene expression and SCN neural firing is kept in alignment with the external photic cues through light-sensing melanopsin-expressing intrinsically photoreceptive retinal ganglion cells (ipRGC) that project to the SCN via the retinohypothalamic tract (RHT) [4]. The RHT releases the excitatory neurotransmitter glutamate, stimulating both AMPA and NMDA receptors to directly increase the SCN spontaneous firing rate. Meanwhile, glutamate receptor activation increases intracellular Ca2+ levels, which activates cAMP-responsive element-binding protein (CREB) that binds to the promoter/enhancer regions of clock genes and regulates their expression phases so that the clock gene expression phases are synchronized with light.
The SCN relays the light-entrained temporal information to the rest of the brain through direct or indirect neural projections or secreting factors (Fig 1). The SCN innervates about 15 brain regions directly and additional regions indirectly, including the subparaventricular zone (SPZ), the paraventricular nucleus (PVN), the dorsomedial hypothalamus (DMH), and the arcuate nucleus (ARC) [5]. The PVN is a critical relay center that transmits SCN signals to the autonomous nervous system (ANS) through the intermediolateral cell column of the spinal cord (IML), a sympathetic nucleus, and the dorsal motor nucleus of the vagus (DMV), a parasympathetic nucleus [6,7]. Sympathetic nerve activity promotes glucose production, while parasympathetic nerve activity promotes hepatic glycogen synthesis in the liver. In pre-diabetes individuals and ahead of the development of insulin resistance, sympathetic activity is elevated compared to healthy individuals [8]. Pharmacological sympatholysis improved glycemic control in individuals with obesity [9]. In a thyrotoxicosis rat model with increased endogenous glucose production and impaired hepatic insulin sensitivity, hepatic sympathetic or parasympathetic denervation ameliorated the phenotype [10]. Nocturnal rodents are widely used mammal models for circadian rhythm studies, although the molecular mechanisms underpinning the difference between nocturnal and diurnal animals are largely unclear [11]. Except for melatonin, most neurohumoral signals downstream of the SCN are in the opposite phases between nocturnal and diurnal animals when described in terms of light/dark cycles. To avoid confusion, we describe times using their relationships with wake/sleep activities so that most neurohumoral regulations are conserved among all animals.
Figure 1. Pathways connecting the central clock activity and peripheral metabolic rhythms.
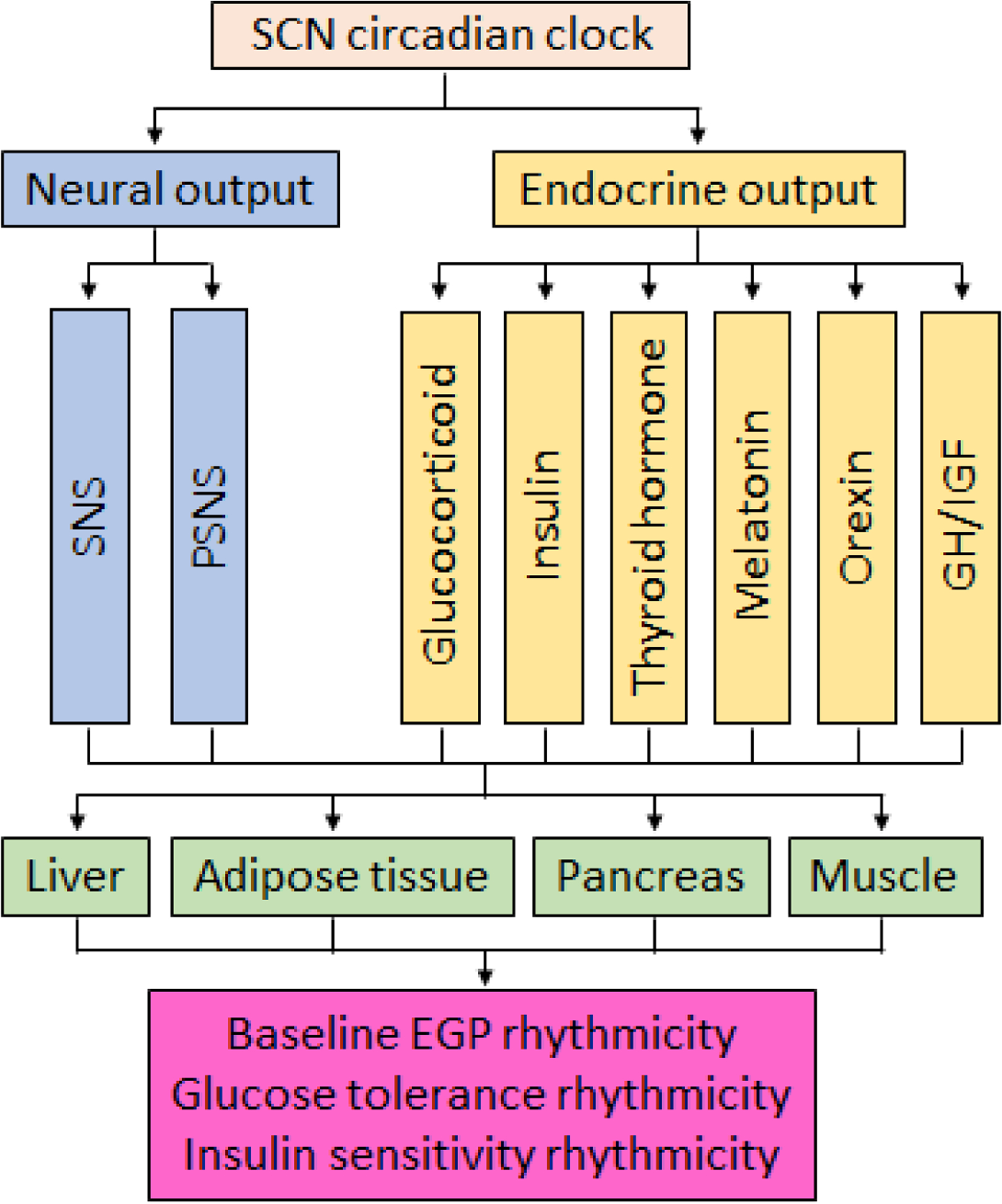
The schematic illustrates the key neurohumoral pathways connecting the SCN central clock with the peripheral metabolic organs in the regulation of diurnal rhythmicity in baseline endogenous glucose production (EGP), glucose tolerance, and insulin sensitivity. Abbreviations: SNS, sympathetic nervous system; PSNS, parasympathetic nervous system; GH, growth hormone; IGF, insulin-like growth factor.
The dawn phenomenon
The clinical dawn phenomenon (DP) refers to spontaneous early-morning hyperglycemia without nocturnal hypoglycemia or increased demand for insulin in the early morning to maintain normoglycemia [12]. Post-breakfast hyperglycemia is a more robust and reproducible feature of DP than fasting hyperglycemia and is referred to as the extended DP [13,14]. DP can be quantified by an absolute blood glucose elevation from the nocturnal nadir to pre- or post-breakfast glucose levels (ΔG pre-breakfast > 10–30 mg/dL or ΔG post-breakfast > 30–60 mg/dL), or by more than 20% increase in exogenous insulin requirement [15]. DP is a spontaneous pathophysiological event that occurs roughly in half of the patients with diabetes (Box 1).
Box 1. Prevalence of the dawn phenomenon.
The reported prevalence of DP ranged from 20% to 90% in type 1 diabetes (T1D) and 6% to 90% in type 2 diabetes (T2D) [17]. Earlier studies on DP are dependent on finger-tip glucose measurements every few hours, which might miss the DP time-points that caused the variations of the DP prevalence mentioned previously. Recent continuous glucose monitoring (CGM) allows more accurate evaluation of glucose levels throughout the day with high temporal resolution. Around 55% of patients with T1D show DP as monitored by overnight CGM [122]. Around 38% of T2D patients, including those treated with insulin sensitizers and insulin secretagogues, show DP [123]. Another study reported that more than 52% of the patients with T2D experience DP [124]. A multicenter CGM study reported the frequency of DP at 9%, 30%, and 52% in subjects with normal glucose tolerance, pre-diabetes, and newly diagnosed T2D, respectively, using ΔG pre-breakfast > 20 mg/dL as the criteria [16]. In a longitudinal CGM study of T1D patients, DP occurred at around 56% of the days in all tested subjects [125]. Such variations pose challenges for adjusting daily insulin administration doses in managing diabetes.
DP poses a significant challenge in glycemic management. In the overall T2D population, the 24h mean glucose levels and HbA1c levels in patients with DP are over 12 mg/dL and 0.4% (4 mM), respectively, higher than patients without DP, which are not mitigated by dietary interventions [13,16]. The current treatment of DP is limited to symptom management, for example, by long-acting insulin analogs administration before bedtime or by continuous subcutaneous insulin infusion (CSII) with special nocturnal programming [17]. In T2D, a combination of insulin secretagogues and insulin sensitizers can ameliorate DP. Notably, DP is different from pharmacological causes such as waning levels of hypoglycemic agents at night or antecedent hypoglycemia, known as the Somogyi effect that is preventable through optimizing the administration of the hypoglycemic agents. These three mechanisms may be concurrently present in patients with diabetes. Monitoring nighttime glucose levels with CGM and adjusting the dosage or timing of the anti-diabetic drug therapy can be used to distinguish them. There is no targeted therapy for DP, partly due to the incomplete understanding of its pathogenesis.
In patients with DP, although the insulin requirement rises in the early morning, the insulin levels or insulin clearance rate does not noticeably change [18,19], suggesting that DP is likely caused by insulin resistance rather than insulin secretion or clearance abnormalities. In particular, DP is associated with abnormally increased endogenous glucose production (EGP) from the liver in the morning [20,21]. T2D patients with the extended DP exhibit a different oscillatory expression pattern of REV-ERBα/β compared to T2D patients without DP [22]. Depletion of REV-ERBα/β in GABA neurons in the mouse brain (REV-GABA-KO) causes glucose intolerance and hepatic insulin resistance only at waking but not during sleep, which resembles the extended DP. These results suggest that the DP is likely due to the altered central circadian clock in the brain, which leads to failure to sufficiently elevate hepatic insulin sensitivity at waking and subsequent compromised glycemic responses to breakfast.
In line with the involvement of the central circadian clock in extended DP, poor sleep quality and peripheral leucocytes clock gene expression are independently correlated with the magnitude of DP in T2D [23]. Likewise, sleep disorders are associated with DP in T2D [24], although acute mild dim light at night can disturb sleep without impairing glucose homeostasis in healthy subjects [25], suggesting that chronic sleep disruption is probably needed to cause DP. Conversely, continuous positive airway pressure (CPAP) treatment of obstructive sleep apnea can alleviate DP in T2D [26]. In addition to sleep disruption, sedentary time in the morning and afternoon is associated with DP in T2D [27]. Conversely, acute light exercise, in a time-dependent manner, partially counteracts DP in T2D [28,29]. These human studies support the role of the central circadian clock in the DP of T2D. Since understanding normal physiology is a prerequisite for understanding disease states, and given that glucose metabolism displays physiological diurnal rhythms [30–32], in the next sections we first review the physiological rhythm of glucose metabolism, as well as the SCN-originated neurohumoral pathways in regulating the glucose metabolic rhythmicity, before turning to their implications in DP in T2D.
Baseline glucose diurnal rhythm
In the following, we use the term ‘baseline’ to refer to the pre-meal or mild fasting condition. The baseline pre-meal glucose levels in animals and healthy human subjects show diurnal rhythm under regular light/dark cycles, with the peak at waking and trough during sleep [33–35]. Such a diurnal rhythm is not due to eating behaviors because fasting at different times does not abolish the rhythm. Consistently, rats having identical meals once every 4 h retain the same glucose rhythm [33,36]. Isotopes tracer studies reveal a similar diurnal rhythm of endogenous glucose production (EGP) in healthy human subjects [37], suggesting that the baseline glucose rhythmicity is attributable to EGP. The liver is the major site of EGP from glycogen degradation or gluconeogenesis. Consistently, liver glycogen content displays the diurnal rhythm anti-phase to the baseline glucose levels in humans, while in mice, enzymes of gluconeogenesis in the liver oscillate in the consistent phase with the baseline glucose levels [38,39].
The diurnal rhythm of baseline glucose is disrupted in SCN-lesioned animals [31,33]. The liver gluconeogenic gene expression rhythm is also disrupted after SCN-lesion [40]. Injecting an antagonist of vasopressin, a neuropeptide involved in SCN function, into the PVN in rats abolishes the time-dependent decrease in the baseline EGP [41]. Pharmacological blockade of GABA receptors in the PVN in rats increases baseline blood glucose levels, while activation of GABA receptors in PVN decreases the glucose level [42]. The effects of GABA receptor antagonist administration into the PVN are only observed during sleeping [43], demonstrating a time-dependent effect of PVN GABA signaling on baseline glucose levels. SCN-lesion or hepatic sympathectomy, but not hepatic parasympathectomy, blocks the PVN GABA agonist-induced hyperglycemia [42]. In separate studies, hepatic sympathectomy or hepatic parasympathectomy were shown each to abolish the baseline glucose diurnal rhythm without affecting glucocorticoid levels [35,44]. However, complete denervation of both sympathetic and parasympathetic input to the liver had no effects [44], suggesting that PVN GABA signaling and the downstream ANS contribute to the baseline glucose rhythmicity. In addition to the PVN, other hypothalamic regions receiving projections from the SCN might also be involved. SCN lesion increased 2-deoxy-glucose (2DG)-induced c-Fos expression in ARC neurons and potentiated 2DG-induced hyperglycemia at waking, suggesting a possible role of the SCN-to-ARC projection in maintaining baseline glucose rhythm through promoting hypoglycemia-induced counter-regulation [45,46]. Compared to healthy subjects, individuals with pre-diabetes show an altered phase of fasting glucose oscillation, with the peak glucose level shifted to the evening [47]. Analysis of T2D patient post-mortem hypothalamic tissue showed that the SCN neuron numbers are decreased in T2D patients compared to healthy subjects [48], suggesting possible SCN disruption in T2D.
Diurnal rhythm of postprandial glucose and insulin sensitivity
The diurnal rhythm of postprandial (post meal) glucose is intriguingly anti-phase with the baseline pre-meal glucose levels. In healthy human subjects, an identical meal induces a higher glucose excursion at dinner than at breakfast [49–53]. Human studies using the oral glucose minimal model or hyperinsulinemic-euglycemic clamp show that insulin sensitivity peaks at waking and reaches the trough during sleep, with insulin-mediated suppression of EGP displaying robust diurnal changes [54,55,49]. Consistently, animal studies show that glucose tolerance peaks at waking and hits the trough during sleep [56–58,22]. Hyperinsulinemic euglycemic clamp studies in mice demonstrated that the heightened insulin sensitivity at waking is due to EGP suppression [22,58].
The SCN is required for the time-of-the-day variations in glycemic responses to meals since SCN-lesion abolishes the diurnal rhythm of glucose tolerance [56]. In SCN-lesioned mice, the altered insulin sensitivity is mainly due to EGP but not blood glucose clearance, although the increased body weight in SCN-lesioned mice can complicate data interpretation [59]. Disruption of the SCN neural firing rhythm by constant light disrupts the insulin sensitivity rhythm [58]. Suppression of SCN neural activity by injection of tetrodotoxin (TTX) into the SCN reduces hepatic insulin sensitivity but increases blood glucose clearance, while TTX injection at the PVN does not affect insulin sensitivity [41], suggesting that the PVN-independent mechanisms account for the diurnal rhythm of insulin sensitivity.
Normal expression of clock genes in the SCN is required for the diurnal rhythm of glucose tolerance and hepatic insulin sensitivity. Whole-body knockout of BMAL1 in mice abolishes the systemic insulin sensitivity rhythm [57], whereas liver-specific knockout of BMAL1 only mildly affects the diurnal rhythm of hepatic glycogen content [60]. Together, these findings suggest that the central SCN clock might be required. Whole-body knockout of RORγ in mice improves insulin sensitivity, particularly during sleep, via reduced EGP [38]. Depletion of REV-ERBα/β in GABA neurons in mice (REV-GABA-KO) causes glucose intolerance and hepatic insulin resistance only at waking but not during sleep, associated with abolished diurnal firing pattern of the SCNGABA neurons. Chemogenetic activation of SCNGABA neurons at waking, but not during sleep, causes glucose intolerance, whereas chemogenetic suppression of SCNGABA neurons at waking, but not at sleep, rescues glucose intolerance in REV-GABA-KO mice [22]. Inducible phase-specific re-expression of REV-ERBα in SCNGABA neurons in REV-GABA-KO mice rescues the time-dependent glucose intolerance, while overexpression of the REV-ERB target genes RGS16 or α7-Takusan in SCNGABA neurons recapitulates the time-dependent glucose intolerance [22]. Of note, mice with REV-ERB depleted in the retina and forebrain regions containing the SCN also exhibit glucose intolerance [61]. These genetic mouse models do not show abnormal locomotor rhythm differences under the regular light/dark cycles since light overrides the SCN clock in driving the behavioral rhythm, suggesting that the insulin sensitivity rhythm is independent of behavioral rhythms. Interestingly, REV-ERB knockout or chemogenetic manipulation of SCNGABA neurons does not abolish the baseline EGP rhythm or baseline glucose levels [22]. Therefore, REV-ERB-independent clock output pathways account for the baseline glucose rhythm.
The co-occurrence of high baseline glucose and high insulin sensitivity at waking seems counterintuitive but it makes sense, one could argue, from an evolutionary perspective. As the body goes through sleep, the demand for EGP to maintain euglycemia increases and peaks shortly after waking before a meal, as EGP is required to support cognitive and locomotor activities during food-seeking behaviors with an empty stomach. Once a meal is consumed, it is beneficial to suppress EGP efficiently to prevent postprandial hyperglycemia. Such an intricate regulation is accomplished by building up a potential for EGP to be suppressed by insulin even though the baseline EGP keeps rising during sleep. The arrival of insulin upon meal ingestion capitalizes on the potential and finishes the transition from high baseline EGP to low postprandial EGP. Thus, the SCN clock controls the anticipatory regulation of hepatic insulin sensitivity in alliance with the responsive insulin signaling to coordinate the diurnal rhythm of glucose metabolism under normal physiological conditions (Figure 2).
Figure 2. Endogenous glucose production (EGP) and its sensitivity to insulin-mediated suppression in healthy individuals and in the extended dawn phenomenon.
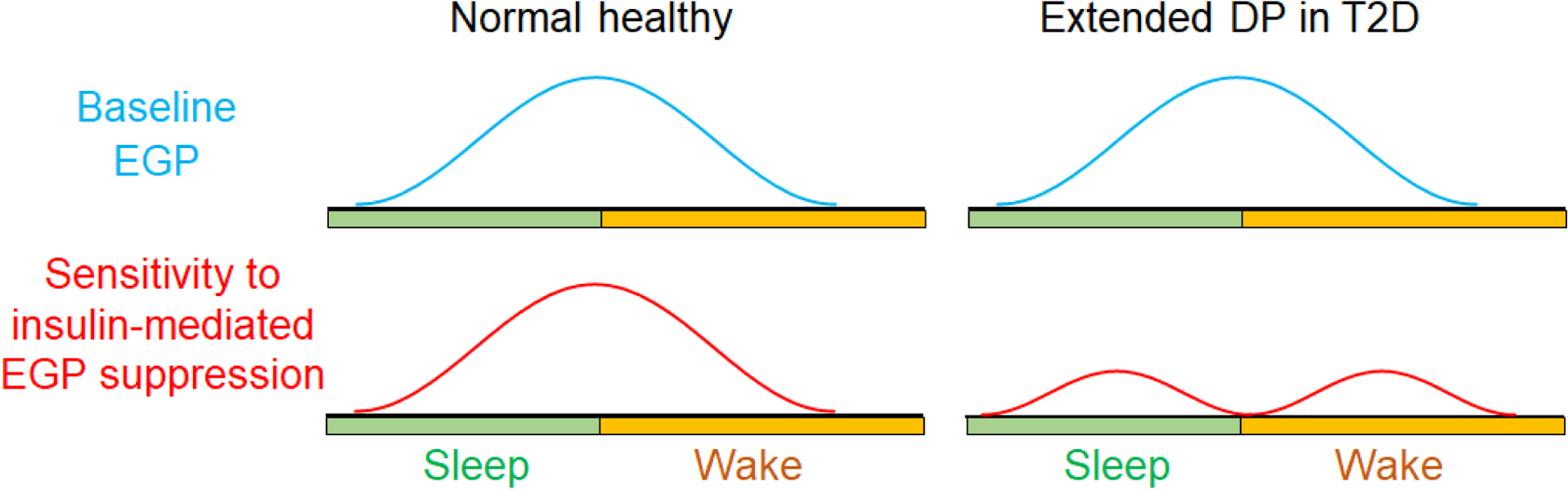
In normal physiology, baseline EGP and its sensitivity to insulin-mediated suppression both peak at wakening. Disruption of the clock-mediated insulin sensitivity rhythmicity at waking can lead to reduced insulin sensitivity at this time point and contribute to the extended dawn phenomenon in type 2 diabetes (T2D). The schematic illustrates one pattern of disruption out of many possible different ways of disruption.
The altered diurnal rhythm of glucose tolerance is an early sign of T2D. In patients with pre-diabetes, glycemic control in response to bolus glucose or meal intake is better in the evening than morning, in contrast to healthy subjects [62–64]. T2D patients with extended DP show altered temporal patterns of REV-ERB in peripheral blood mononuclear cells compared to patients without DP [22]. In combination with the waking-specific glucose intolerance in REV-GABA-KO mice, these results suggest that alteration of the molecular circadian clock may contribute to the extended DP (Figure 2). It should be noted that in addition to the roles of the ANS in regulating glucose rhythms as discussed above, the SCN can regulate the liver glucose rhythm through secreted factors (see Outstanding Questions).
Outstanding questions.
Which SCN-controlled neurohumoral pathways connect the central clock to the liver in regulating the diurnal rhythm of hepatic glucose production?
Which SCN-original neurohumoral pathways contribute to the dawn phenomenon?
What causes behavior-independent disruptions of the molecular circadian clock in diabetes patients?
Do the dawn phenomena in type 1 and type 2 diabetes share the same etiology?
Can neural or humoral factors be therapeutically targeted to alleviate the dawn phenomenon, and if so, how?
Are there sex differences in the diurnal glucose metabolic rhythmicity?
Glucocorticoid in glucose diurnal rhythm
The SCN projects to the PVN and regulates corticotropin-releasing hormone (CRH) release from the PVN, which evokes diurnal adrenocorticotropic hormone (ACTH) release from the corticotroph cells of the anterior pituitary. ACTH activates glucocorticoid release from the adrenal gland. The SCN can also regulate the glucocorticoid rhythm through the sympathetic nervous system (SNS) [65]. The peripheral clock in the adrenal gland regulates steroidogenesis and is another source of the temporal pattern of circulating glucocorticoid [66,67]. Glucocorticoid peaks at waking, and the diurnal rhythm persists under constant routine conditions but is lost after SCN lesion [67]. Glucocorticoid stimulates hepatic glucose production, increases lipolysis in white adipose tissues, inhibits blood glucose clearance, and mobilizes amino acids in skeletal muscles. Glucocorticoid promotes gluconeogenesis by inducing the expression of gluconeogenic genes in the liver. Glucocorticoid levels above the normal daily fluctuation range can impair insulin sensitivity, leading to diabetes and weight gain [68]. Meanwhile, glucocorticoid insufficiency causes hypoglycemia [69]. Given its peak in the morning and its role in glucose metabolism, it is tempting to speculate that it contributes to the high baseline glucose levels at waking. However, the blockade of glucocorticoid synthesis fails to inhibit the early morning increase of EGP in humans [70]. Further investigation is needed to fully address the role of glucocorticoid in the diurnal rhythm of baseline glucose levels or baseline EGP.
Insulin in glucose diurnal rhythm
In healthy human subjects, the insulin secretion in response to an identical meal or a glucose bolus is greater at waking than before or during sleep [71], which can contribute to the heightened glucose tolerance at waking in alliance with the abovementioned insulin sensitivity rhythmicity. Freshly isolated pancreatic islets from rats or mice show greater glucose-stimulated insulin secretion (GSIS) at waking than during sleep [71], suggesting an islet-autonomous mechanism. BMAL1 in β cell regulates the oscillation of genes encoding secretory machinery involved in insulin release, and BMAL1 ablation in adult mice causes glucose intolerance [72]. Conversely, β cell-specific overexpression of BMAL1 increases the amplitude of the islet oscillatory genes, enhances GSIS, and protects mice against obesity-induced glucose intolerance [73]. In addition to β cells, pancreatic α cells also have an oscillatory clock with distinct phases from β cells [74]. Aside from the pancreas-autonomous mechanisms, the hypothalamus regulates β cell insulin secretion. A subpopulation of oxytocin neurons in the PVN (PVNOXT) innervate the pancreatic islets via the SNS. Activation of PVNOXT neurons suppresses insulin secretion and causes hyperglycemia, while silencing of these neurons alters the secretory pathway in β cells, elevates insulin levels, and induces hypoglycemia in mice [75]. Depleting BMAL1 in the mouse PVN reduces insulin secretion and impairs glucose tolerance, probably through regulating vasopressin expression in the PVN and its release to the blood [76]. It is unclear whether and how the SCN regulates PVN-mediated insulin secretion rhythm. Clinical studies suggest that abnormal rhythmicity in insulin sensitivity, rather than insulin secretion, is associated with DP [18,19].
Thyroid hormone in glucose diurnal rhythm
Thyroid hormone synthesis and release are regulated by thyroid-stimulating hormone (TSH) from the anterior pituitary and thyrotropin-releasing hormone (TRH) from the hypothalamus [77]. TRH synthesized in the PVN and is secreted to the median eminence (ME) to reach the pituitary, where it binds to the cell membrane receptor on thyrotrophs and stimulates the synthesis of TSH. TSH stimulates the thyroid gland to release thyroid hormones, including thyroxine (T4) and triiodothyronine (T3) [78]. T4 and T3 suppress the synthesis and secretion of TRH and TSH, constituting the negative feedback loops of the hypothalamic-pituitary-thyroid axis [79]. Circulating thyroid hormone levels in healthy human subjects show a diurnal rhythm and peak around midnight [80,81]. Free T3 exhibits a similar nocturnal pattern as TSH, while free T4 shows much weaker rhythmicity [82]. Removing the pituitary gland in rats abolishes the thyroid hormones oscillation without affecting the clock genes in the thyroid, suggesting that the diurnal rhythm of thyroid hormone is governed by the SCN or the pituitary but not by the local thyroid clock [83]. Consistently, REV-ERB and its corepressor NCOR1 contribute to the circadian secretion of TSH and T3 in a thyrotroph cell line [84].
Hyperthyroidism is known to promote hyperglycemia, glucose intolerance, and insulin resistance, likely through elevated EGP and impaired pancreatic islet function [78]. Patients with hyperthyroidism Graves’ disease have a high prevalence of glucose intolerance and diabetes [85]. Even Graves’ disease patients with normal glucose metabolism exhibit higher basal and postprandial glucose levels in the morning compared to healthy subjects [86]. While single-case studies should be interpreted with caution, DP was observed in a patient with Graves’ disease complicated with T2D, and pharmacologically improving thyroid function alleviated DP in this patient [87], suggesting that hyperthyroidism can contribute to DP.
Melatonin in glucose diurnal rhythm
Melatonin is generated from the pineal gland in the evening and rises to the peak at midnight in both diurnal and nocturnal animals [88]. The nocturnal pattern of melatonin secretion is regulated by both the SCN clock and light-induced inhibition [89]. The activity of the key enzymes in melatonin synthesis is under the neural control from the SCN through the PVN and IML. The melatonin rhythm is abolished when the SCN-controlled input is disrupted [90]. Genetic variants in melatonin receptor MTNR1B are prominent genetic variants associated with T2D [91]. Pinealectomy in animals abolishes the melatonin oscillation and impairs diurnal rhythms of glucose tolerance and insulin sensitivity [89,92]. Most laboratory mouse strains do not produce melatonin [93], although the knockout of melatonin receptors in mice disrupts the baseline glucose diurnal rhythm [94]. How melatonin and its receptors regulate glucose metabolism remains controversial. Inconsistencies across studies in factors such as the timing of melatonin administration, food intake, and glycemic outcome measurements may contribute to the controversy [95]. It is unknown, as far as the authors are aware, whether disruption of the endogenous melatonin rhythmicity is involved in DP.
Orexin in glucose diurnal rhythm
Orexin is a neuropeptide involved in a number of functions including regulation of sleep/wake cycles. It is secreted by the lateral hypothalamus (LH) with peak levels in the cerebrospinal fluid during wake and trough levels during sleep [96–98]. In the mouse brain, gene expression of orexin receptors also shows some oscillation, with high levels during early sleep and low levels during the early awake period [97]. Orexin is present in the blood, but its blood levels do not show diurnal rhythm [97,98]. Orexin deficiency can cause sleep disorders, obesity, and glucose intolerance in both humans and mouse models [99]. In mice, intracerebroventricular administration of orexin during the awake period increases blood glucose levels and EGP acutely, but lowers glucose levels and EGP hours later during sleep. Hepatic parasympathectomy does not alter the acute effect but abolishes the delayed glucose-lowering effect of orexin [100]. Injection of GABA receptor antagonist into the LH increases EGP in rats, which is blocked by orexin receptor 1 (OX1R) antagonist or hepatic sympathectomy [101]. Therefore, orexin bidirectionally regulates EGP through a sympathetic pathway during wake and a parasympathetic pathway during sleep. These findings suggest that LH orexin signaling may regulate the baseline EGP rhythm, although the connection of this regulatory path with the SCN is unclear. It is also unknown whether disruption of endogenous orexin rhythmicity is involved in DP, pharmacologic targeting of the orexin system to treat insomnia can alleviate DP in patients with T2D [102].
GH and IGF in glucose diurnal rhythm
Growth hormone (GH) is released from the pituitary somatotrophs with diurnal and sex-specific ultradian rhythms [103,104]. Blood GH levels rise shortly after sleep onset and drop before waking. The diurnal rhythm of GH is regulated by the SCN and the local clock in the somatotrophs [105]. GH-releasing hormone (GHRH) and somatostatin (SST) regulate GH secretion. GHRH is primarily synthesized in the ARC and is also expressed in the PVN, dorsomedial hypothalamus (DMH), and ventromedial hypothalamus (VMH) [106]. SST is expressed in the periventricular nucleus (PeN), ARC-ME, SCN, medial preoptic area (MPOA), PVN, DMH, and VMH [107]. GHRH and somatostatin are released to the ME, enter the portal system, reach the pituitary, and bind to their respective membrane receptors on somatotroph cells in the pituitary [108]. GH decreases glucose utilization and increases lipolysis. The rise of GH during early sleep may contribute to the trough of glucose tolerance in the middle of the sleep in normal subjects, while an abnormal rise of GH before waking may contribute to the DP [17,109]. Some studies found that bedtime administration of SST or SST analogs in T1D patients alleviates nocturnal or early-morning hyperglycemia [110–112], but other studies did not observe beneficial effects and advised against the use of SST analogs due to side effects [113,114]. It remains incompletely understood how GH affects glycemic control in T2D.
The effects of GH on glucose metabolism are further complicated by insulin-like growth factor 1 (IGF-1). GH stimulates the production of IGF-1 in the liver, which inhibits GH secretion by stimulating SST release and inhibiting GHRH release from the hypothalamus. IGF-1 has overlapping functions with insulin in lowering blood glucose levels [115]. Mice with liver-specific knockout of GH receptor (GHR) exhibit reduced blood IGF-1, increased blood glucose and insulin levels, and impaired insulin tolerance [116]. Restoration of IGF-1 alleviates the metabolic changes due to GHR deficiency, suggesting a pivotal role of IGF-1 in mediating the metabolic effects of hepatic GH signaling. Blood IGF-1 levels in rats or mice show diurnal rhythm with high levels during sleep and low levels during wake [117,118]. In humans, blood IGF-1 levels display a nocturnal decrease and an increase in the morning [119]. T1D patients with DP display reduced free IGF-1 that does not bind IGF1 binding proteins (IGFBPs) in the morning, which may contribute to the DP [120,121].
Concluding remarks
In healthy subjects, baseline endogenous glucose production and its sensitivity to insulin-mediated suppression display similar diurnal rhythms, with the peak at waking. The central circadian clock governs the rise of hepatic insulin sensitivity in the early morning in anticipation of the meal. Disruption of clock-mediated anticipatory regulation could contribute to the extended dawn phenomenon in type 2 diabetes. Future studies are warranted to further elucidate neurohumoral mechanisms connecting the SCN clock with peripheral metabolic organs in orchestrating systemic insulin sensitivity.
Highlights.
Signals from the central circadian clock regulate diurnal rhythms of baseline glucose levels and glucose tolerance.
Baseline pre-meal blood glucose levels have a diurnal rhythm that peaks at waking.
Insulin-mediated suppression of hepatic glucose production also displays a diurnal rhythm that peaks at waking.
The diurnal glucose metabolic rhythms can be separated from sleep or eating behaviors.
The central circadian clock in the hypothalamus regulates the diurnal rhythm of hepatic insulin sensitivity.
The dawn phenomenon, referring to hyperglycemia limited to the early morning, affects nearly half of the diabetes patient population.
Disruption of the molecular circadian clock could contribute to the extended dawn phenomenon in type 2 diabetes.
Multiple neurohumoral mechanisms could contribute to the connection between the central circadian clock and peripheral metabolic organs.
Acknowledgment
We apologize to colleagues whose work we could not cite due to the space limitations. The authors’ laboratory was supported by the American Heart Association (AHA30970064) and NIH grants HL153320, DK111436, AG069966, AG070687, and ES027544. We also thank John S. Dunn Foundation, Mrs. Clifford Elder White Graham Endowed Research Fund, Cardiovascular Research Institute at Baylor, Dan L. Duncan Comprehensive Cancer Center (P30CA125123), the SPORE program (P50CA126752), the Gulf Coast Center for Precision Environmental Health (P30ES030285), and the Texas Medical Center Digestive Diseases Center (P30 DK056338).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no conflicts of interest.
References
- 1.Takahashi JS (2017) Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet 18, 164–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patke A et al. (2020) Molecular mechanisms and physiological importance of circadian rhythms. Nat. Rev. Mol. Cell Biol 21, 67–84 [DOI] [PubMed] [Google Scholar]
- 3.Hastings MH et al. (2018) Generation of circadian rhythms in the suprachiasmatic nucleus. Nat. Rev. Neurosci 19, 453–469 [DOI] [PubMed] [Google Scholar]
- 4.Colwell CS (2011) Linking neural activity and molecular oscillations in the SCN. Nat. Rev. Neurosci 12, 553–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morin LP (2013) Neuroanatomy of the extended circadian rhythm system. Exp. Neurol 243, 4–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buijs RM (2013) The autonomic nervous system: a balancing act. Handb. Clin. Neurol 117, 1–11 [DOI] [PubMed] [Google Scholar]
- 7.Okamura H (2007) Suprachiasmatic nucleus clock time in the mammalian circadian system. Cold Spring Harb. Symp. Quant. Biol 72, 551–556 [DOI] [PubMed] [Google Scholar]
- 8.Flaa A et al. (2008) Increased sympathetic reactivity may predict insulin resistance: an 18-year follow-up study. Metabolism. 57, 1422–1427 [DOI] [PubMed] [Google Scholar]
- 9.Chazova I et al. (2006) Moxonidine improves glycaemic control in mildly hypertensive, overweight patients: a comparison with metformin. Diabetes Obes. Metab 8, 456–465 [DOI] [PubMed] [Google Scholar]
- 10.Klieverik LP et al. (2008) Effects of thyrotoxicosis and selective hepatic autonomic denervation on hepatic glucose metabolism in rats. Am. J. Physiol. Endocrinol. Metab 294, E513–520 [DOI] [PubMed] [Google Scholar]
- 11.Smale L et al. (2003) Mammalian diurnality: some facts and gaps. J. Biol. Rhythms 18, 356–366 [DOI] [PubMed] [Google Scholar]
- 12.Schmidt MI et al. (1981) The dawn phenomenon, an early morning glucose rise: implications for diabetic intraday blood glucose variation. Diabetes Care 4, 579–585 [DOI] [PubMed] [Google Scholar]
- 13.Monnier L et al. (2013) Magnitude of the dawn phenomenon and its impact on the overall glucose exposure in type 2 diabetes: is this of concern? Diabetes Care 36, 4057–4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Neal TB and Luther EE (2020) Dawn Phenomenon. In StatPearls StatPearls Publishing; [PubMed] [Google Scholar]
- 15.Porcellati F et al. (2013) Thirty years of research on the dawn phenomenon: lessons to optimize blood glucose control in diabetes. Diabetes Care 36, 3860–3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li C et al. (2020) The dawn phenomenon across the glycemic continuum: Implications for defining dysglycemia. Diabetes Res. Clin. Pract 166, 108308. [DOI] [PubMed] [Google Scholar]
- 17.Carroll MF and Schade DS (2005) The dawn phenomenon revisited: implications for diabetes therapy. Endocr. Pract. Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol 11, 55–64 [DOI] [PubMed] [Google Scholar]
- 18.Boden G et al. (1996) Evidence for a circadian rhythm of insulin sensitivity in patients with NIDDM caused by cyclic changes in hepatic glucose production. Diabetes 45, 1044–1050 [DOI] [PubMed] [Google Scholar]
- 19.De Feo P et al. (1986) Studies on overnight insulin requirements and metabolic clearance rate of insulin in normal and diabetic man: relevance to the pathogenesis of the dawn phenomenon. Diabetologia 29, 475–480 [DOI] [PubMed] [Google Scholar]
- 20.Perriello G et al. (1997) Evidence of increased systemic glucose production and gluconeogenesis in an early stage of NIDDM. Diabetes 46, 1010–1016 [DOI] [PubMed] [Google Scholar]
- 21.Radziuk J and Pye S (2006) Diurnal rhythm in endogenous glucose production is a major contributor to fasting hyperglycaemia in type 2 diabetes. Suprachiasmatic deficit or limit cycle behaviour? Diabetologia 49, 1619–1628 [DOI] [PubMed] [Google Scholar]
- 22.Ding G et al. (2021) REV-ERB in GABAergic neurons controls diurnal hepatic insulin sensitivity. Nature 592, 763–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Y et al. (2017) Poor Sleep Quality Is Associated with Dawn Phenomenon and Impaired Circadian Clock Gene Expression in Subjects with Type 2 Diabetes Mellitus. Int. J. Endocrinol 2017, 4578973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ren H et al. (2015) [Association between sleep disorders and dawn phenomenon in patients with type 2 diabetes mellitus]. Zhonghua Yi Xue Za Zhi 95, 1209–1213 [PubMed] [Google Scholar]
- 25.Chamorro R et al. (2021) Acute mild dim light at night slightly modifies sleep but does not affect glucose homeostasis in healthy men. Sleep Med. 84, 158–164 [DOI] [PubMed] [Google Scholar]
- 26.Mokhlesi B et al. (2017) Effect of one week of CPAP treatment of obstructive sleep apnoea on 24-hour profiles of glucose, insulin and counter-regulatory hormones in type 2 diabetes. Diabetes Obes. Metab 19, 452–456 [DOI] [PubMed] [Google Scholar]
- 27.Paing AC et al. (2020) Diurnal patterns of objectively measured sedentary time and interruptions to sedentary time are associated with glycaemic indices in type 2 diabetes. J. Sci. Med. Sport 23, 1074–1079 [DOI] [PubMed] [Google Scholar]
- 28.Paing AC et al. (2019) Dose-response between frequency of interruption of sedentary time and fasting glucose, the dawn phenomenon and night-time glucose in Type 2 diabetes. Diabet. Med. J. Br. Diabet. Assoc 36, 376–382 [DOI] [PubMed] [Google Scholar]
- 29.Zheng X et al. (2020) Effects of Exercise on Blood Glucose and Glycemic Variability in Type 2 Diabetic Patients with Dawn Phenomenon. BioMed Res. Int 2020, 6408724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalsbeek A et al. (2014) Circadian control of glucose metabolism. Mol. Metab 3, 372–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.La Fleur SE (2003) Daily rhythms in glucose metabolism: suprachiasmatic nucleus output to peripheral tissue. J. Neuroendocrinol 15, 315–322 [DOI] [PubMed] [Google Scholar]
- 32.Van Cauter E et al. (1997) Roles of circadian rhythmicity and sleep in human glucose regulation. Endocr. Rev 18, 716–738 [DOI] [PubMed] [Google Scholar]
- 33.La Fleur SE et al. (1999) A suprachiasmatic nucleus generated rhythm in basal glucose concentrations. J. Neuroendocrinol 11, 643–652 [DOI] [PubMed] [Google Scholar]
- 34.Challet E et al. (2004) Daily variations of blood glucose, acid-base state and PCO2 in rats: effect of light exposure. Neurosci. Lett 355, 131–135 [DOI] [PubMed] [Google Scholar]
- 35.Cailotto C et al. (2005) The suprachiasmatic nucleus controls the daily variation of plasma glucose via the autonomic output to the liver: are the clock genes involved? Eur. J. Neurosci 22, 2531–2540 [DOI] [PubMed] [Google Scholar]
- 36.Kalsbeek A and Strubbe JH (1998) Circadian control of insulin secretion is independent of the temporal distribution of feeding. Physiol. Behav 63, 553–558 [DOI] [PubMed] [Google Scholar]
- 37.Bolli GB et al. (1984) Demonstration of a dawn phenomenon in normal human volunteers. Diabetes 33, 1150–1153 [DOI] [PubMed] [Google Scholar]
- 38.Takeda Y et al. (2014) Retinoic acid-related orphan receptor γ (RORγ): a novel participant in the diurnal regulation of hepatic gluconeogenesis and insulin sensitivity. PLoS Genet. 10, e1004331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iwayama K et al. (2020) Diurnal variation in the glycogen content of the human liver using 13 C MRS. NMR Biomed. 33, e4289. [DOI] [PubMed] [Google Scholar]
- 40.Nakagawa H and Okumura N (2010) Coordinated regulation of circadian rhythms and homeostasis by the suprachiasmatic nucleus. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci 86, 391–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foppen E et al. (2016) Suprachiasmatic Nucleus Neuropeptides and Their Control of Endogenous Glucose Production. J. Neuroendocrinol 28, [DOI] [PubMed] [Google Scholar]
- 42.Kalsbeek A et al. (2004) Suprachiasmatic GABAergic inputs to the paraventricular nucleus control plasma glucose concentrations in the rat via sympathetic innervation of the liver. J. Neurosci. Off. J. Soc. Neurosci 24, 7604–7613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalsbeek A et al. (2008) Circadian control of the daily plasma glucose rhythm: an interplay of GABA and glutamate. PloS One 3, e3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cailotto C et al. (2008) Daily rhythms in metabolic liver enzymes and plasma glucose require a balance in the autonomic output to the liver. Endocrinology 149, 1914–1925 [DOI] [PubMed] [Google Scholar]
- 45.Herrera-Moro Chao D et al. (2016) The Suprachiasmatic Nucleus Modulates the Sensitivity of Arcuate Nucleus to Hypoglycemia in the Male Rat. Endocrinology 157, 3439–3451 [DOI] [PubMed] [Google Scholar]
- 46.Rodríguez-Cortés B et al. (2022) Suprachiasmatic nucleus-mediated glucose entry into the arcuate nucleus determines the daily rhythm in blood glycemia. Curr. Biol. CB 32, 796–805.e4 [DOI] [PubMed] [Google Scholar]
- 47.Gubin DG et al. (2017) Disrupted circadian rhythms of body temperature, heart rate and fasting blood glucose in prediabetes and type 2 diabetes mellitus. Chronobiol. Int 34, 1136–1148 [DOI] [PubMed] [Google Scholar]
- 48.Hogenboom R et al. (2019) Loss of arginine vasopressin- and vasoactive intestinal polypeptide-containing neurons and glial cells in the suprachiasmatic nucleus of individuals with type 2 diabetes. Diabetologia 62, 2088–2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saad A et al. (2012) Diurnal pattern to insulin secretion and insulin action in healthy individuals. Diabetes 61, 2691–2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morris CJ et al. (2015) Endogenous circadian system and circadian misalignment impact glucose tolerance via separate mechanisms in humans. Proc. Natl. Acad. Sci 112, E2225–E2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leung GKW et al. (2017) Effect of meal timing on postprandial glucose responses to a low glycemic index meal: A crossover trial in healthy volunteers. Clin. Nutr. Edinb. Scotl DOI: 10.1016/j.clnu.2017.11.010 [DOI] [PubMed] [Google Scholar]
- 52.Takahashi M et al. (2018) Effects of Meal Timing on Postprandial Glucose Metabolism and Blood Metabolites in Healthy Adults. Nutrients 10, E1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davis R et al. (2020) Glycaemic response at night is improved after eating a high protein meal compared with a standard meal: A cross-over study. Clin. Nutr. Edinb. Scotl 39, 1510–1516 [DOI] [PubMed] [Google Scholar]
- 54.Schulz B et al. (1983) Diurnal rhythm of insulin sensitivity in subjects with normal and impaired glucose tolerance. Exp. Clin. Endocrinol 81, 263–272 [DOI] [PubMed] [Google Scholar]
- 55.Wu MS et al. (1986) Diurnal variation of insulin clearance and sensitivity in normal man. Proc. Natl. Sci. Counc. Repub. China B 10, 64–69 [PubMed] [Google Scholar]
- 56.la Fleur SE et al. (2001) A daily rhythm in glucose tolerance: a role for the suprachiasmatic nucleus. Diabetes 50, 1237–1243 [DOI] [PubMed] [Google Scholar]
- 57.Shi S et al. (2013) Circadian disruption leads to insulin resistance and obesity. Curr. Biol. CB 23, 372–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coomans CP et al. (2013) Detrimental effects of constant light exposure and high-fat diet on circadian energy metabolism and insulin sensitivity. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 27, 1721–1732 [DOI] [PubMed] [Google Scholar]
- 59.Coomans CP et al. (2013) The suprachiasmatic nucleus controls circadian energy metabolism and hepatic insulin sensitivity. Diabetes 62, 1102–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Udoh US et al. (2018) Genetic deletion of the circadian clock transcription factor BMAL1 and chronic alcohol consumption differentially alter hepatic glycogen in mice. Am. J. Physiol. Gastrointest. Liver Physiol 314, G431–G447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Adlanmerini M et al. (2021) REV-ERB nuclear receptors in the suprachiasmatic nucleus control circadian period and restrict diet-induced obesity. Sci. Adv 7, eabh2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee A et al. (1992) Diurnal variation in glucose tolerance. Cyclic suppression of insulin action and insulin secretion in normal-weight, but not obese, subjects. Diabetes 41, 750–759 [DOI] [PubMed] [Google Scholar]
- 63.Peter R et al. (2010) Daytime variability of postprandial glucose tolerance and pancreatic B-cell function using 12-h profiles in persons with Type 2 diabetes. Diabet. Med. J. Br. Diabet. Assoc 27, 266–273 [DOI] [PubMed] [Google Scholar]
- 64.Sonnier T et al. (2014) Glycemic control is impaired in the evening in prediabetes through multiple diurnal rhythms. J. Diabetes Complications 28, 836–843 [DOI] [PubMed] [Google Scholar]
- 65.Buijs RM et al. (1999) Anatomical and functional demonstration of a multisynaptic suprachiasmatic nucleus adrenal (cortex) pathway. Eur. J. Neurosci 11, 1535–1544 [DOI] [PubMed] [Google Scholar]
- 66.Li M-D et al. (2021) Circadian Clock-Controlled Checkpoints in the Pathogenesis of Complex Disease. Front. Genet 12, 721231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oster H et al. (2017) The Functional and Clinical Significance of the 24-Hour Rhythm of Circulating Glucocorticoids. Endocr. Rev 38, 3–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Suh S and Park MK (2017) Glucocorticoid-Induced Diabetes Mellitus: An Important but Overlooked Problem. Endocrinol. Metab. Seoul Korea 32, 180–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arshad MF and Debono M (2021) Current and future treatment options for adrenal insufficiency. Curr. Opin. Endocrinol. Diabetes Obes 28, 303–311 [DOI] [PubMed] [Google Scholar]
- 70.Bright GM et al. (1980) Failure of cortisol blockade to inhibit early morning increases in basal insulin requirements in fasting insulin-dependent diabetics. Diabetes 29, 662–664 [DOI] [PubMed] [Google Scholar]
- 71.Seshadri N and Doucette CA (2021) Circadian Regulation of the Pancreatic Beta Cell. Endocrinology 162, bqab089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perelis M et al. (2015) Pancreatic β cell enhancers regulate rhythmic transcription of genes controlling insulin secretion. Science 350, aac4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rakshit K and Matveyenko AV (2021) Induction of Core Circadian Clock Transcription Factor Bmal1 Enhances β-Cell Function and Protects Against Obesity-Induced Glucose Intolerance. Diabetes 70, 143–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Petrenko V et al. (2017) Pancreatic α- and β-cellular clocks have distinct molecular properties and impact on islet hormone secretion and gene expression. Genes Dev. 31, 383–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Papazoglou I et al. (2022) A distinct hypothalamus-to-β cell circuit modulates insulin secretion. Cell Metab. 34, 285–298.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nakata M et al. (2021) Circadian Clock Component BMAL1 in the Paraventricular Nucleus Regulates Glucose Metabolism. Nutrients 13, 4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ikegami K et al. (2019) Interconnection between circadian clocks and thyroid function. Nat. Rev. Endocrinol 15, 590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mullur R et al. (2014) Thyroid Hormone Regulation of Metabolism. Physiol. Rev 94, 355–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Philippe J and Dibner C (2015) Thyroid Circadian Timing: Roles in Physiology and Thyroid Malignancies. J. Biol. Rhythms 30, 76–83 [DOI] [PubMed] [Google Scholar]
- 80.Mazzoccoli G et al. (2004) The hypothalamic-pituitary-thyroid axis and melatonin in humans: Possible interactions in the control of body temperature. Neuro Endocrinol. Lett 25, 368–72 [PubMed] [Google Scholar]
- 81.Roelfsema F et al. (2014) Thyrotropin Secretion in Healthy Subjects Is Robust and Independent of Age and Gender, and Only Weakly Dependent on Body Mass Index. J. Clin. Endocrinol. Metab 99, 570–578 [DOI] [PubMed] [Google Scholar]
- 82.Russell W et al. (2008) Free Triiodothyronine Has a Distinct Circadian Rhythm That Is Delayed but Parallels Thyrotropin Levels. J. Clin. Endocrinol. Metab 93, 2300–2306 [DOI] [PubMed] [Google Scholar]
- 83.Fahrenkrug J et al. (2017) Hypophysectomy abolishes rhythms in rat thyroid hormones but not in the thyroid clock. J. Endocrinol 233, 209–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aninye IO et al. (2014) Circadian regulation of Tshb gene expression by Rev-Erbα (NR1D1) and nuclear corepressor 1 (NCOR1). J. Biol. Chem 289, 17070–17077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hage M et al. (2011) Thyroid Disorders and Diabetes Mellitus. J. Thyroid Res 2011, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gao G et al. (2019) Glycemic variation in uncontrolled Graves’ disease patients with normal glucose metabolism: Assessment by continuous glucose monitoring. Endocrine 64, 265–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Torimoto K et al. (2014) Glucose variability before and after treatment of a patient with Graves’ disease complicated by diabetes mellitus: assessment by continuous glucose monitoring. Endocr. J 61, 321–328 [DOI] [PubMed] [Google Scholar]
- 88.Tordjman S et al. (2017) Melatonin: Pharmacology, Functions and Therapeutic Benefits. Curr. Neuropharmacol 15, 434–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cipolla-Neto J and Amaral F.G. do (2018) Melatonin as a Hormone: New Physiological and Clinical Insights. Endocr. Rev 39, 990–1028 [DOI] [PubMed] [Google Scholar]
- 90.Maronde E and Stehle JH (2007) The mammalian pineal gland: known facts, unknown facets. Trends Endocrinol. Metab. TEM 18, 142–149 [DOI] [PubMed] [Google Scholar]
- 91.Karamitri A and Jockers R (2019) Melatonin in type 2 diabetes mellitus and obesity. Nat. Rev. Endocrinol 15, 105–125 [DOI] [PubMed] [Google Scholar]
- 92.la Fleur SE et al. (2001) Role for the pineal and melatonin in glucose homeostasis: pinealectomy increases night-time glucose concentrations. J. Neuroendocrinol 13, 1025–1032 [DOI] [PubMed] [Google Scholar]
- 93.Kennaway DJ (2019) Melatonin research in mice: a review. Chronobiol. Int 36, 1167–1183 [DOI] [PubMed] [Google Scholar]
- 94.Owino S et al. (2016) Melatonin Signaling Controls the Daily Rhythm in Blood Glucose Levels Independent of Peripheral Clocks. PloS One 11, e0148214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Garaulet M et al. (2020) Melatonin Effects on Glucose Metabolism: Time To Unlock the Controversy. Trends Endocrinol. Metab. TEM 31, 192–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chieffi S et al. (2017) Orexin System: The Key for a Healthy Life. Front. Physiol 8, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zeitzer JM et al. (2003) Circadian and homeostatic regulation of hypocretin in a primate model: implications for the consolidation of wakefulness. J. Neurosci. Off. J. Soc. Neurosci 23, 3555–3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Salomon RM et al. (2003) Diurnal variation of cerebrospinal fluid hypocretin-1 (Orexin-A) levels in control and depressed subjects. Biol. Psychiatry 54, 96–104 [DOI] [PubMed] [Google Scholar]
- 99.Tsuneki H et al. (2016) Sleep Control, GPCRs, and Glucose Metabolism. Trends Endocrinol. Metab. TEM 27, 633–642 [DOI] [PubMed] [Google Scholar]
- 100.Tsuneki H et al. (2015) Hypothalamic orexin prevents hepatic insulin resistance via daily bidirectional regulation of autonomic nervous system in mice. Diabetes 64, 459–470 [DOI] [PubMed] [Google Scholar]
- 101.Yi C-X et al. (2009) A major role for perifornical orexin neurons in the control of glucose metabolism in rats. Diabetes 58, 1998–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Toi N et al. (2019) Improvement of glycemic control by treatment for insomnia with suvorexant in type 2 diabetes mellitus. J. Clin. Transl. Endocrinol 15, 37–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang L et al. (2019) Rhythmic growth hormone secretion in physiological and pathological conditions: Lessons from rodent studies. Mol. Cell. Endocrinol 498, 110575. [DOI] [PubMed] [Google Scholar]
- 104.Wang W et al. (2021) The GH-IGF-1 Axis in Circadian Rhythm. Front. Mol. Neurosci 14, 742294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vakili H et al. (2016) Evidence for a Circadian Effect on the Reduction of Human Growth Hormone Gene Expression in Response to Excess Caloric Intake. J. Biol. Chem 291, 13823–13833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Müller EE et al. (1999) Neuroendocrine Control of Growth Hormone Secretion. 79, 97. [DOI] [PubMed] [Google Scholar]
- 107.Tan HY et al. (2013) Hypothalamic Distribution of Somatostatin mRNA Expressing Neurones Relative to Pubertal and Adult Changes in Pulsatile Growth Hormone Secretion in Mice. J. Neuroendocrinol 25, 910–919 [DOI] [PubMed] [Google Scholar]
- 108.Vijayakumar A et al. (2011) The Intricate Role of Growth Hormone in Metabolism. Front. Endocrinol 2, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shih K-C et al. (2013) Effect of growth hormone on dawn phenomenon in patients with type 2 diabetes. Growth Factors Chur Switz. 31, 66–73 [DOI] [PubMed] [Google Scholar]
- 110.Campbell PJ et al. (1985) Pathogenesis of the dawn phenomenon in patients with insulin-dependent diabetes mellitus. Accelerated glucose production and impaired glucose utilization due to nocturnal surges in growth hormone secretion. N. Engl. J. Med 312, 1473–1479 [DOI] [PubMed] [Google Scholar]
- 111.Navascues I et al. (1988) Effect of a long-acting somatostatin derivative SMS 201–995 (sandostatin) on glucose homeostasis in type I diabetes mellitus. Horm. Res 29, 92–94 [DOI] [PubMed] [Google Scholar]
- 112.Perriello G et al. (1990) Nocturnal spikes of growth hormone secretion cause the dawn phenomenon in type 1 (insulin-dependent) diabetes mellitus by decreasing hepatic (and extrahepatic) sensitivity to insulin in the absence of insulin waning. Diabetologia 33, 52–59 [DOI] [PubMed] [Google Scholar]
- 113.Johnston DG et al. (1986) Effects of SMS 201–995 on intermediary metabolism and endocrine status in normal and diabetic humans. Am. J. Med 81, 88–93 [PubMed] [Google Scholar]
- 114.Davies RR et al. (1989) Somatostatin analogues in diabetes mellitus. Diabet. Med. J. Br. Diabet. Assoc 6, 103–111 [DOI] [PubMed] [Google Scholar]
- 115.Livingstone C (2013) Insulin-like growth factor-I (IGF-I) and clinical nutrition. Clin. Sci. Lond. Engl 1979 125, 265–280 [DOI] [PubMed] [Google Scholar]
- 116.Liu Z et al. (2016) Growth Hormone Control of Hepatic Lipid Metabolism. Diabetes 65, 3598–3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bertani S et al. (2010) Circadian Profile of Peripheral Hormone Levels in Sprague-Dawley Rats and in Common Marmosets (Callithrix jacchus). In Vivo 24, 827–836 [PubMed] [Google Scholar]
- 118.Chaudhari A et al. (2017) Cryptochromes regulate IGF-1 production and signaling through control of JAK2-dependent STAT5B phosphorylation. Mol. Biol. Cell 28, 834–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Heuck C et al. (1999) Circadian Variation in Serum Free Ultrafiltrable Insulin-Like Growth Factor I Concentrations in Healthy Children. Pediatr. Res 45, 733–736 [DOI] [PubMed] [Google Scholar]
- 120.Kobayashi K et al. (1997) Role of IGF Binding Protein- 1 in the Dawn Phenomenon and Glycemic Control in Children and Adolescents With IDDM. Diabetes Care 20, 1442–1447 [DOI] [PubMed] [Google Scholar]
- 121.Kobayashi K et al. (2000) A Role of Free Insulin-Like Growth Factor-I in Dawn Phenomenon in Children and Adolescents with TYPE 1 Diabetes Mellitus. Endocr. J 47, S91–S93 [DOI] [PubMed] [Google Scholar]
- 122.Ostrovski I et al. (2020) Analysis of Prevalence, Magnitude and Timing of the Dawn Phenomenon in Adults and Adolescents With Type 1 Diabetes: Descriptive Analysis of 2 Insulin Pump Trials. Can. J. Diabetes 44, 229–235 [DOI] [PubMed] [Google Scholar]
- 123.Wu W et al. (2017) Self-Monitoring of Blood Glucose to Assess Dawn Phenomenon in Chinese People with Type 2 Diabetes Mellitus. Int. J. Endocrinol 2017, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Monnier L et al. (2012) Frequency and Severity of the Dawn Phenomenon in Type 2 Diabetes: Relationship to age. Diabetes Care 35, 2597–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bouchonville M et al. (2014) The Effectiveness and Risks of Programming an Insulin Pump to Counteract the Dawn Phenomenon in Type 1 Diabetes. Endocr. Pract. Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol DOI: 10.4158/EP14198.OR [DOI] [PubMed] [Google Scholar]