

Summary
Since 2005, genome-wide association (GWA) datasets have been largely biased toward sampling European ancestry individuals, and recent studies have shown that GWA results estimated from self-identified European individuals are not transferable to non-European individuals because of various confounding challenges. Here, we demonstrate that enrichment analyses that aggregate SNP-level association statistics at multiple genomic scales—from genes to genomic regions and pathways—have been underutilized in the GWA era and can generate biologically interpretable hypotheses regarding the genetic basis of complex trait architecture. We illustrate examples of the robust associations generated by enrichment analyses while studying 25 continuous traits assayed in 566,786 individuals from seven diverse self-identified human ancestries in the UK Biobank and the Biobank Japan as well as 44,348 admixed individuals from the PAGE consortium including cohorts of African American, Hispanic and Latin American, Native Hawaiian, and American Indian/Alaska Native individuals. We identify 1,000 gene-level associations that are genome-wide significant in at least two ancestry cohorts across these 25 traits as well as highly conserved pathway associations with triglyceride levels in European, East Asian, and Native Hawaiian cohorts.
Keywords: GWAS, multi-ancestry, enrichment analyses
Graphical abstract
Introduction
Over the past two decades, funding agencies and biobanks around the world have made enormous investments to generate large-scale datasets of genotypes, exomes, and whole-genome sequences from diverse human ancestries that are merged with medical records and quantitative trait measurements.1, 2, 3, 4, 5, 6, 7, 8 However, analyses of such datasets are usually limited to the application of standard genome-wide association (GWA) SNP-level association analyses in which SNPs are tested one-by-one for significant association with a phenotype9, 10, 11 (Table 1). Yet, even in the largest available multi-ancestry biobanks, GWA analyses fail to offer a comprehensive view of genetic trait architecture among human ancestries.
Table 1.
The three genomic scales and corresponding association tests used in this study
| Genomic scale | Association test | Model of genetic trait architecture | Relevant example |
|---|---|---|---|
| SNPs | standard univariate genome-wide association (GWA) test | the true mutation-level trait architecture is the same for all individuals | many inflammatory bowel disease mutations replicate across ancestries12 |
| SNP-sets/genes | gene-level association tests (e.g., gene-ε,13 SKAT14) | core genes are the same across all ancestries, with potentially varying causal SNPs | late-onset Alzheimer disease risk from ApoE4 allele is lower in African ancestry individuals15 |
| Pathways/networks | pathway enrichment and network propagation (e.g., Hierarchical HotNet,16 RSS17) | core genes differ across ancestries but are all in the same annotated pathway | skin pigmentation architecture in the same pathway differs between African and European ancestry individuals2 |
The models of genetic trait architecture corresponding to each genomic scale and statistical method that have been previously invoked in the literature (including relevant examples cited in the last column). These nested genomic scales should routinely be leveraged in multi-ancestry GWA studies to generate biologically interpretable hypotheses of trait architecture across ancestries.
SNP-level GWA results are difficult to interpret across multiple human ancestries because of a litany of confounding variables, including (1) ascertainment bias in genotyping,2,5 (2) varying linkage disequilibrium (LD) patterns,18,19 (3) variation in allele frequencies due to different selective pressures and unique population histories,19, 20, 21, 22, 23 and (4) the effect of environmental factors on phenotypic variation.24, 25, 26, 27 These confounders and the observed low transferability of GWA results across ancestries2,28,29 have generated an important call for increasing GWA efforts focused on populations of diverse, non-European ancestry individuals.
We also note, as other studies have,6,30 that the GWA SNP-level test of association is rarely applied to non-European ancestry individuals.31 There are two likely explanations for leaving non-European ancestry individuals out of analyses: (1) researchers are electing to not analyze diverse cohorts because of a lack of statistical power and concerns over other confounding variables (recently covered in Ben-Eghan et al.30) or (2) the analyses of non-European cohorts yield no genome-wide significant SNP-level associations. In either case, valuable information is being ignored in GWA studies or going unreported in resulting publications.30, 31, 32 Even when diverse ancestries are analyzed, GWA studies usually condition on GWA results identified with European ancestry cohorts to detect and give validity to other SNP-level associations.6 While this study design can also identify shared SNP-level associations in non-European ancestry cohorts that are underpowered for applications of the standard GWA framework, it will not identify ancestry-specific associations in non-European ancestry cohorts. In our own analysis of abstracts of publications between 2012 and 2020 with UK Biobank data, we found that only 33 out of 166 studies (19.87%) reported genome-wide significant associations in any non-European ancestry cohort (Figures S1 and S2). Focusing energy and resources on increasing GWA sample sizes without intentional focus on sampling of non-European populations will thus likely perpetuate an already troubling history of leaving non-European ancestry samples out of GWA analyses of large-scale biobanks such as the UK Biobank.30 However, we note that non-European ancestry GWA studies have—and will continue to have—smaller sample sizes than existing and emerging European-ancestry GWA cohorts, limiting the precision of effect size estimates in these studies. What has received less attention than the need to improve GWA study design is the potential of enrichment analyses to characterize genetic trait architecture in multi-ancestry datasets while accounting for variable statistical power to detect, estimate, and replicate genetic associations among cohorts.
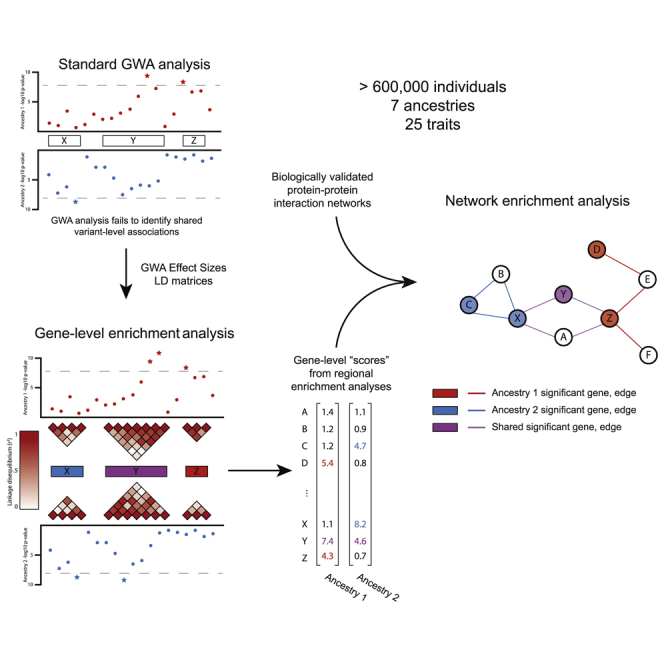
In this analysis, we illustrate that focusing solely on p values from the standard GWA framework is insufficient to capture the genetic architecture of complex traits. Specifically, we propose that expansion of association analyses to the genomic scale of genes and pathways generates robust and interpretable hypotheses about trait architecture in multi-ancestry cohorts. We define enrichment analyses as testing whether a user-specified set of SNPs, such as SNPs in a given gene or pathway, is enriched for trait associations beyond what is expected by chance based on the number of SNPs in the set and the LD structure among SNPs in the set.13,17,33 These enrichment analyses can 2increase the power to detect associated genes through the aggregation of SNPs of small effect (which explain the majority of the heritability of most traits34,35). Mathieson36 recently highlighted the pattern of homogeneity of direction of effect in multi-ancestry studies even when individual SNPs are not categorized as genome-wide significant in multiple ancestries. Gene-level and pathway-level enrichment analyses can prioritize biological regions where there is homogeneity in the direction of SNP-level signals of association, generating biologically interpretable hypotheses for the genetic architecture of complex traits in multiple ancestry cohorts. Gene and pathway enrichment analyses expand the existing opportunity for the characterization of conserved genetic architecture across multiple ancestries, or other partitions of samples in biobank datasets (e.g., by biological sex or age), through the identification of biologically interpretable associations.
In this study of 25 quantitative traits and more than 600,000 diverse individuals from the UK Biobank (UKB), BioBank Japan (BBJ), and the PAGE study data (Tables S1–S10), we detail biological insights gained from the application of gene and pathway level enrichment analyses to seven diverse ancestry cohorts. We perform genetic association tests for SNPs, genes, and pathways across multiple ancestry groups with a trait of interest. We test for significantly mutated subnetworks of genes by using known protein-protein interaction networks and the Hierarchical HotNet software.16 Enrichment analyses do not require generating additional information beyond standard GWA inputs (or outputs for methods that take in GWA summary statistics). We demonstrate that moving beyond SNP-level associations allows for a biologically comprehensive prioritization of shared and ancestry-specific mechanisms underlying genetic trait architecture.
Material and methods
Data overview
We performed statistical tests of association at the SNP, gene, and pathway level for 25 quantitative traits. These analyses were performed on data from seven ancestry cohorts drawn from the UK Biobank, BioBank Japan (BBJ), and PAGE consortia (Table S3). The number of samples included in each ancestry cohort ranged from 574 (American Indian/Alaska Native in the PAGE study data) to 349,411 (European in UK Biobank). The number of SNPs tested in each ancestry ranged from 578,320 (African in the UK Biobank) to more than 12 million (African American in the PAGE dataset). Full enumeration of the samples studied, including sample size, and number of SNPs for each ancestry cohort are given in Tables S1 and S5–S10. For an extensive description of each cohort from the three biobanks that we analyze in this study, see the supplemental information.
SNP-level GWA analyses
In the European, African, and South Asian ancestry cohorts from the UK Biobank, we performed GWA studies for each ancestry-trait pair in order to test whether the same SNP or SNPs are associated with a given quantitative trait in different ancestries. SNP-level GWA effect sizes were calculated with plink and the --glm flag.37 Age, sex, and the first twenty principal components were included as covariates for all traits analyzed.7 Principal-component analysis was performed with flashpca 2.038 on a set of independent markers derived separately for each ancestry cohort with the plink command --indep-pairwise 100 10 0.1. Using these parameters, --indep-pairwise removes all SNPs that have a pairwise correlation above 0.1 within a 100 SNP window and then slides forward in increments of ten SNPs genome wide. In the implementation of gene-ε, we assume pruning highly correlated SNPs still accurately captures the association signals identified in the standard GWA framework.14,39 Summary statistics for the 25 quantitative traits in the Biobank Japan, as well as available ancestry-trait pairs in the PAGE study data, were then compared with the results from the association analyses in the UK Biobank cohorts (same traits as listed in Table S5). In each analysis of an ancestry-trait pair, a separate Bonferroni-corrected significance threshold was calculated with the number of SNPs tested in that particular ancestry-trait pair. We elected to use a Bonferroni-corrected significance threshold to be conservative (compared to, say, an often-used GWA significance threshold of ; see Figure 1). We label a given SNP association as replicating among cohorts if the estimated effect size of that SNP surpasses the Bonferroni-corrected significance threshold in more than two ancestry cohorts analyzed here (Figure 1 and Table S11). We believe that the use of a conservative significance threshold, such as the Bonferroni correction, helps to illustrate the statistical challenges faced by multi-ancestry GWA studies due to very imbalanced sample sizes.
Figure 1.

Less stringent significance thresholds lead to a decrease in the proportion of replicated SNP-level associations and an increase in the proportion of gene-level associations among ancestries for each of the 25 traits analyzed
(A) Proportion of all SNP-level Bonferroni-corrected genome-wide significant associations in any ancestry that replicate in at least one other ancestry is shown on the x axis (see Table S11 for ancestry-trait-specific Bonferroni-corrected p value thresholds). On the y axis, we show the proportion of significant gene-level associations that were replicated for a given phenotype in at least two ancestries (see Table S16 for Bonferroni-corrected significance thresholds for each ancestry-trait pair). The black stars on the x- and y-axes represent the mean proportion of replicates in SNP and gene analyses, respectively. C-reactive protein (CRP) contains the greatest proportion of replicated SNP-level associations of any of the phenotypes.
(B) The x axis indicates the proportion of SNP-level associations that surpass a nominal threshold of p value in at least one ancestry cohort that replicate in at least one other ancestry cohort. The y axis indicates the proportion of gene-level associations that surpass a nominal threshold of p value in at least one ancestry cohort and replicate in at least one other ancestry cohort. Nominal p value thresholds tend to decrease the proportion of replicated SNP-level associations and tend to increase the proportion of replicated gene-level associations. The number of unique SNPs and genes that replicated in each cohort is given in Figure S18.
(C) The x axis indicates the proportion of SNP-level associations that surpass a nominal threshold of p value in at least one ancestry cohort that replicate in at least one other ancestry cohort. The y axis indicates the proportion of gene-level associations that surpass a nominal threshold of p value in at least one ancestry cohort and replicate in at least one other ancestry cohort. The number of unique SNPs and genes that replicated in each cohort is given in Figure S19.
(D) The x axis represents the proportion of variants that were significant in the European and at least one non-European ancestry cohort with an alternative significant threshold defined as 0.05 divided by the number of significant European variant associations. The y axis is the corresponding proportion of genes that were significant in at least one non-European ancestry cohort with the same threshold calculation for genes. Note that the axes in (D) are different from one another and the axes of the other panels. As shown in panels (B), (C), and (D) nominal p value thresholds tend to decrease the proportion of replicated SNP-level associations and tend to increase the proportion of replicated gene-level associations. Expansion of three letter trait codes are given in Table S2 and a version of this plot with all trait names displayed as text is shown in Figure S17. Figure S17 shows the same set of plots with all traits represented as text.
To further analyze our ability to accurately estimate GWA SNP-level effect sizes in each ancestry cohort given the imbalanced sample sizes of the datasets we studied, we performed theoretical power calculations across a range of values for both effect sizes and minor allele frequencies as described in Sham and Purcell.9 In this framework, we selected absolute value of effect sizes to be equal to 0.1, 0.5, and 1. We then performed power calculations for each of these when paired with a minor allele frequency of 0.01, 0.1, 0.25, and 0.5. In Figure S3, we plot the power of the standard GWA framework to detect SNP-level associations between a genotype and a quantitative trait of interest for sample sizes for up to 30,000 individuals by using a standard GWA significance threshold of .
Because multiple cohorts we analyzed (in particular the American Indian/Alaska Native and Native Hawaiian ancestry cohorts) lack power to estimate effect sizes accurately under the theoretical model described in Sham and Purcell,9 we also implemented two-stage GWA studies with a less stringent test of replication for SNP-level association signals. We used the European ancestry cohort as a discovery cohort for genome-wide significant SNP-level associations for each trait, and we used each non-European ancestry cohort as a validation cohort. We then calculated a nominal significance threshold for each trait as 0.05 divided by the number of significant variants for each trait in the European cohort that were tested in at least one other cohort. The corresponding nominal significance thresholds, number of SNPs that were significant in the European ancestry cohort, and the number of SNPs that surpassed the nominal significance threshold in at least one other ancestry cohort are given in Table S12. Replication counts and proportions are illustrated in Figure S4 and the ranges of significant effect sizes for each ancestry trait-pair are shown in Table S13. Finally, for each variant that was significant with the two-stage nominal threshold, we performed a variant-specific power calculation by using the marginal European-ancestry effect size and each non-European ancestry cohort’s minor allele frequency and sample size with the method outlined in Sham and Purcell.9 The maximum power achieved by a variant in each cohort-trait pairing is given in Table S14 and the number of variants with greater than 90% detection power are given in Table S15.
Gene-level association tests
In order to test aggregated sets of SNP-level GWA effect sizes for enrichment of associated mutations with a given quantitative trait, we applied gene-ε29 to each ancestry cohort we studied for each trait of interest, resulting in 125 sets of gene-level association statistics (Table S3). The gene-ε method takes two summary statistics as input: (1) SNP-level GWA marginal effect size estimates derived with ordinary least-squares and (2) an LD matrix empirically estimated from external data (e.g., directly from GWA study genotype data, a matrix estimated from a population with similar genomic ancestry to that of the samples analyzed in the GWA study). It is well-known that SNP-level effect size estimates can be inflated as a result of various correlation structures among genome-wide genotypes. gene-ε uses its input to derive regularized effect size estimates through elastic net penalized regression. gene-ε uses the LD matrix to test each gene for enrichment of SNP-level associations beyond what is expected by chance (given the number of SNPs in the gene and the LD among them), thereby identifying genes that are enriched for mutations associated with a trait of interest.13,14,17,33,40,41
In practice, gene-ε and other enrichment analyses14,41,42 can be applied to any user-specified set of genomic regions, such as regulatory elements, intergenic regions, or gene sets. These gene-level enrichment analyses enable identification of traits in which genetic architecture may be heterogeneous among individuals at the SNP level across individuals by increasing sensitivity to identify interacting mutations of moderate effect on a given trait. Applying gene-ε in multiple ancestry cohorts allows researchers to further test whether genes associated (i.e., enriched for SNP-level associations signals given the LD matrix) with a trait of interest are the same, or vary, across ancestries. gene-ε takes as input a list of boundaries for all regions to be tested for enrichment of associations. In our study, we applied gene-ε to all genes and transcriptional elements defined in Gusev et al.43 for human genome build 19.
In our gene-level analysis, SNP arrays included both genotyped and high-confidence imputed SNPs (information score 0.8) for each ancestry-trait pair. To compute the LD matrix, we first pruned highly linked SNPs so that the number of SNPs included for any chromosome was less than 35,000 SNPs—the computational limit of gene-ε due to the size of the LD matrix—by using the plink command --indep-pairwise 100 10 0.5. For the UK Biobank European, South Asian, and African ancestry cohorts, we then derived empirical LD estimates between each pair of SNPs for each chromosome in each cohort by using plink flag --r square applied to the empirical genotype and high-confidence imputed data. We then used the ancestry-specific SNP arrays to calculate 23,603 gene-level association statistics for the European ancestry cohort, 23,671 gene-level association statistics for the South Asian ancestry cohort, and 23,575 gene-level association statistics for the African ancestry cohort.
To calculate gene-level association statistics with Biobank Japan summary statistics, we first found the intersection between SNPs included in the analysis of each trait and SNPs included in the 1000 Genomes Project phase 3 data for the sample of 93 individuals from the Japanese in Tokyo (JPT) population. Note, this intersection was different among some traits, as the genotype data in the Biobank Japan were from different studies, which in turn used different genotyping arrays. We then pruned highly linked markers for each trait separately by using the plink flag --indep-pairwise 100 10 X where X was determined by finding the highest possible value that led to the inclusion of less than 35,000 SNPs on each chromosome for the trait. Because of the increased density of SNPs with effect size estimates for height, X was set to prune more conservatively at X = 0.15. For all other traits, X was set to 0.5. The number of regions for which a gene-ε gene-level association statistic was calculated for each trait is given in Table S5.
GWA summary statistics for the five cohorts in the PAGE study data were used as input to gene-ε for each available ancestry-trait combination. The array of markers for each ancestry cohort in the PAGE study data was pruned with plink flag --indep-pairwise 100 10 X. X was set to the maximum value in each ancestry that ensured no chromosome contained more than 35,000 markers: X was set to 0.05 for the African American cohort, 0.08 for the Hispanic and Latin American and American Indian and Alaska Native (AIAN) cohorts, and 0.25 for the Native Hawaiian cohorts. Finally, for each ancestry-trait combination, genes that passed the Bonferroni-corrected p value (p = 0.05/number of genes tested) were labeled as “significant” throughout this study (see Table S16 for specific thresholds).
We used LD estimates from reference panels for the ancestry cohorts where genotype data was unavailable (BioBank Japan and PAGE datasets). As discussed in other papers proposing enrichment analyses,17,44 the discordance between GWA summary statistics estimated from a large cohort and LD estimates from a small reference panel may lead to increased false discovery rate and power in the application of gene-ε. Finally, we additionally show that gene-ε detects a large proportion of the same significant genes in the European ancestry cohort with a more stringent threshold of 0.1 (see Table S17).
Pathway analysis and network propagation using Hierarchical HotNet
We tested for significantly mutated subnetworks of genes in each ancestry-trait pair by using the method Hierarchical HotNet.16 Briefly, Hierarchical HotNet uses hierarchical clustering of scores (such as gene-level association statistics41 or mutations in cancer genomes16,45) propagated on a protein-protein interaction network that are enriched for trait associations beyond what is expected by chance. The method enables identification of strongly connected components in the network, which are interpretable as subnetworks of interacting genes enriched for mutations associated with a trait of interest. Unlike using annotated gene lists,46 network propagation of association statistics enables identifying novel gene sets underwriting complex traits as well as crosstalk between annotated pathways. We identified subnetworks of interacting genes enriched for associations with each trait of interest by using network propagation of gene-ε gene-level association statistics as input to Hierarchical HotNet.16 In this study, these gene scores were set to -transformed p values of gene-ε gene-level association test statistics (see also Nakka et al.41,47). For each ancestry-trait combination, we assigned p values of 1 to genes with p values greater than 0.1 to make the resulting networks both sparse and more interpretable (again see Nakka et al.41,47). In addition, ancestry-trait pairs in which less than 25 genes produced gene-ε p values less than 0.1 were discarded as there were an insufficient number of gene-level statistics to populate the protein-protein interaction networks.
We used three protein-protein interaction networks: ReactomeFI 2016,48 iRefIndex 15.0,49 and HINT+HI.50,51 For the ReactomeFI 2016 interaction network, interactions with confidence scores less than 0.75 were discarded. The HINT+HI interaction network consists of the combination of all interactions in HINT binary, HINT co-complex, and HuRI HI interaction networks. We ran Hierarchical HotNet ( permutations) on the thresholded − -transformed gene-level p values for each ancestry-trait combination. We restricted our further investigation to the largest subnetwork identified in each significant ancestry-trait-interaction network combination (p < 0.05).
Results
Multiple recent studies have interrogated the extent to which SNP-level associations for a given trait replicate across ancestries, both empirically and under a variety of models (see Wojcik et al.,6 Durvasula and Lohmueller,28 Shi et al.,44 Carlson et al.,52 Liu et al.,12 Eyre-Walker,53 Shi et al.54). To extensively compare variant-level associations among the seven ancestry cohorts that we analyzed, we first examined the number of genome-wide significant SNP-level associations that replicated exactly on the basis of chromosomal position and reference SNP cluster ID (rsID) in multiple ancestries (see Figure S5A and Figure S5C, with Bonferroni-corrected thresholds provided in Table S11). Exact replication of at least one SNP-level association across two or more ancestries occurs in all 25 traits that were studied. The C-reactive protein (CRP) trait had the highest proportion of replicated SNP associations in multiple ancestries, with 18.95% replicating with the standard GWA framework in at least two ancestries, but has a relatively low number of unique GWA significant SNPs (2,734) when compared to other traits (Figure 1). This is most likely because the genetic architecture of CRP is sparse and highly conserved across ancestries, as is shown in Figure 2. We note that the concordance of genome-wide significant SNP-level association statistics for CRP among five ancestry cohorts is exceptional. In the other 24 traits we analyzed, we did not observe any SNP-level replication among five cohorts. C-reactive protein, which is encoded by the gene of the same name located on chromosome 1, is synthesized in the liver and released into the bloodstream in response to inflammation. In our standard GWA analysis of SNP-level association signal in each ancestry cohort with CRP, rs3091244 is genome-wide significant in a single ancestry cohort. rs3091244 has been functionally validated as influencing CRP levels55,56 and is linked to genome-wide significant SNPs in the other two ancestries for which genotype data are available. Interestingly, all GWA significant SNP-level associations for CRP in the Native Hawaiian ancestry cohort replicate in both the African American (PAGE) and the Hispanic and Latin American cohorts (these three cohorts were all genotyped on the same array).
Figure 2.

C-reactive protein is an exceptional trait where standard GWA analyses may be sufficient to identify shared genetic architecture among ancestry cohorts
(A) Manhattan plot for SNP-level associations with C-reactive protein levels. Ancestry-specific Bonferroni-corrected significance thresholds are shown with dashed horizontal gray lines and listed in Table S11. Note that the scale of the − -transformed p values on the y axis is different for each ancestry for clarity.
(B) Manhattan plot of SNP-level associations around the CRP gene located on chromosome 1 for each ancestry (zoomed in from A). Boundaries of the CRP gene are shown with vertical dashed black lines. All six ancestries contain genome-wide significant SNPs in the region. Black stars in the European, South Asian, and East Asian plots represent rs3091244, a SNP that has been functionally validated as contributing to serum levels of C-reactive protein.55,56
(C) Heatmap of Bonferroni-corrected significant genotyped SNPs replicated between each pair of ancestries analyzed. Here, we focus on SNPs in the 1 MB region surrounding the CRP gene. Entries along the diagonal represent the total number of SNP-level associations in the 1 MB region surrounding the CRP gene for each ancestry. The color of each cell is proportional to the percentage of SNP-level associations replicated out of all possible replications in each ancestry pair (i.e., the minimum of the diagonal entries between the pairs being considered). For example, the maximum number of genome-wide significant SNPs that can possibly replicate between the Hispanic and East Asian is 25, and 20 replicate, resulting in the cell color denoting 80% replication. A similar matrix, computed including imputed SNPs, is shown in Figure S20.
In the other 24 traits, the proportion of genome-wide SNP-level replications was below 10% (Figure 1A). For polygenic traits, replication of SNP-level GWA results is challenging to interpret considering the large number of GWA significant associations for the trait overall. For example, height contains the largest number of replicated SNP-level associations in our multi-ancestry analysis—but these only represent 8.90% of all unique SNP-level associations with height discovered in any ancestry cohort. A more comprehensive discussion of previously associated SNPs is available for both height and CRP in the supplemental information. The number of SNPs that replicate among cohorts vary by trait and, as a proportion of the total number of significant SNP-level association signals, is typically less than 10% (Figure S5).
We also explored how varying the SNP-level significance threshold for each ancestry cohort influences the replication of SNP-level associations by using the standard GWA framework (Figures 1B and 1C). We performed power calculations for a range of set effect sizes and minor allele frequencies (according to the design and discussion in Sham and Purcell9) to identify cohorts where GWA studies lack power to identify associations with a nominal significance threshold of . The two largest cohorts we studied here, the European ancestry cohort and East Asian ancestry cohort, are sufficiently large to identify true SNP associations regardless of choice of effect size or minor allele frequency (Figure S3). Conversely, in the two smallest cohorts we studied, the AIAN and Native Hawaiian cohorts, GWA studies would lack power to detect true SNP associations with either small effect sizes or low minor allele frequencies (MAF 0.05) (Figure S3).
We then applied a two-stage GWA study design with a less stringent nominal significance threshold for replication in the non-European ancestry cohorts; the resulting thresholds are shown in Table S12. The nominal significance threshold was calculated as 0.05 divided by the number of Bonferroni-corrected significant SNPs in the European ancestry cohort that were tested in at least one other ancestry cohort. Using the European ancestry cohort for discovery of associations, we calculated the proportion of associated SNPs that were nominally significant in at least one non-European ancestry cohort (Figure 1D). As expected, the proportion of SNP-level replication increased across all 25 traits that were studied (maximum percentage of replication was 94.23% for Basophil count, 49 of 52 SNPs; minimum percentage of replication was 40.39% for hemoglobin, 580 of 1,436 SNPs). Number and proportion of replicated SNP-level associations with this framework are shown in Figure S4. These results are in agreement with our results from the application of two fine-mapping methods, SuSiE17 and PESCA,44 to the SNP-level summary statistics of the European and East Asian ancestry cohorts; this analysis also indicated widespread homogeneity in direction of effect among significant SNPs (described in the supplemental information, Tables S18–S22).
Enrichment analyses aggregate SNP-level association statistics with predefined SNP sets, genes, and pathways to identify regions of the genome enriched for trait associations beyond what is expected by chance. Published enrichment analyses have demonstrated the ability to identify trait associations that go unidentified with standard SNP-level GWA analysis.13,14,17,57, 58, 59, 60 The standard GWA method is known to have a high false discovery rate (FDR),61,62 which enrichment analyses can mitigate. Our analyses in Figure S6 and Figure S7 illustrate that two methods—regression with summary statistics (RSS),17 a fully Bayesian method, and gene-ε29—control FDR particularly well both in the presence and absence of population structure. However, Figure S8 and Table S23 illustrate that both GWA study and gene-ε are limited by the sample size of the cohort of interest. Specifically, when the sample size is set to 2,000 individuals, power is low and FDRs are high for both the standard GWA framework and gene-ε. Enrichment methods increase power for identifying biologically interpretable trait associations in studies with smaller sample sizes and with heterogenous genetic architecture than do present-day GWA studies and therefore can be particularly useful when analyzing multi-ancestry genetic datasets with merged phenotype data. For example, Nakka et al.41 identified an association between ST3GAL3 and attention-deficit/hyperactivity disorder (ADHD) by using methods that aggregated SNP-level signals across genes and networks. ADHD is a trait with heritability estimates as high as 75% that had no known genome-wide significant SNP-level associations at the time; Nakka et al.41 studied genotype data from just 3,319 individuals with cases, 2,455 controls and 2,064 trios.63 A study by Demontis et al.64 later found a SNP-level association in the ST3GAL3 gene but was only able to do so with a cohort an order of magnitude larger (20,183 individuals diagnosed with ADHD and 35,191 controls, totaling 55,374 individuals).
Because non-European GWA ancestry cohorts usually have much smaller sample sizes compared to studies with individuals of European ancestry, enrichment analyses offer a unique opportunity to boost statistical power and identify biologically relevant genetic associations with traits of interest by using multi-ancestry datasets. In a simulation study with synthetic phenotypes generated from the European and African ancestry cohorts in the UK Biobank, we show that gene-ε is able to identify significantly associated genes even in smaller cohorts ( and in the European and African ancestry cohorts, respectively) without the inflated FDR that is often exhibited by the standard GWA framework (Figures S9 and S10). Additionally, in these simulations, gene-ε correctly identifies “causal” genes that are commonly associated in both cohorts (Figures S11 and S12). These simulations illustrate the utility of modeling LD (and in the case of gene-ε, additionally shrinking inflated effect sizes) information to identify enrichment of SNP-level associations in predefined SNP sets.
In an analysis performed by Ben-Eghan et al.30 on 45 studies analyzing UK Biobank data, the second most commonly stated reason for omitting non-European cohorts in applied GWA analyses was the lack of power for identifying SNP-level GWA signals. We tested for gene-level associations in each of the 25 complex traits in each ancestry cohort for which we had data (Tables S1–Table S10) and identified associations in genes and transcriptional elements shared across ancestries for every trait. All of our analyses discussed here used gene-ε (see performance comparison with other enrichment analyses in Cheng et al.13 and Figures S9–S12), an empirical Bayesian approach that aggregates SNP-level GWA summary statistics, where p values for each gene are derived by constructing an empirical null distribution based on the eigenvalues of a gene-specific partitioning of the LD matrix (for more details, see Cheng et al.13). Our analyses show that several hematological traits have a higher rate of significant gene-level associations that replicate across multiple ancestry cohorts than SNP-level associations that replicate across ancestry cohorts (Figure 1B). These include platelet count (PLC), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), hematocrit, hemoglobin, mean corpuscular volume (MCV), red blood cell count (RBC), and neutrophil count (Figure S5F). Focusing on platelet count as an example, we identify 65 genes that are significantly enriched for associations in multiple ancestries when tested with gene-ε (see Table S16 for details on Bonferroni thresholds we used to correct for the number of genes tested; Table S24 displays gene-ε for the shared significant genes in non-European ancestry cohorts).13 Fifty-five of these genes are significantly associated in both the European and East Asian ancestry cohorts, and the remaining ten all replicate in other pairs of ancestry cohorts. Overall, each of the six ancestry cohorts in our analysis share at least one significant gene with another ancestry cohort, as shown in Figure S13. Our analysis of platelet count illustrates how the implementation of gene-level enrichment analyses can lead to the identification of shared elements of genetic trait architecture among ancestry cohorts that would have not been identified with the standard SNP-level GWA framework alone. Additionally, gene-level enrichment analyses yield statistically significant results that are biologically interpretable across ancestry cohorts even when sample sizes were highly variable.
Results from gene-level enrichment analyses can be further propagated on protein-protein interaction networks to identify interacting genes enriched for association signals.45 Often, studies use network propagation as a way to incorporate information from multiple “omics” databases in order to identify significantly mutated gene subnetworks or modules contributing to a particular disease.65 An unexplored extension of network propagation is how it can be used with multi-ancestry GWA datasets to identify significantly mutated subnetworks that are shared or ancestry specific.47
To conduct network propagation of gene-level association results in our analyses, we applied the Hierarchical HotNet method16 to gene-ε gene-level association statistics for each trait-ancestry dataset. In Figure 3, we display the significant (p value ) network results for triglyceride levels in three ancestry cohorts: European, East Asian, and Native Hawaiian (networks separated by ancestry are available in Figure S14). In both the European and East Asian cohorts, we identify enrichment of mutations in a highly connected subnetwork of genes in the apolipoprotein family. In addition, we identify a gene subnetwork enriched for mutations in the East Asian and Native Hawaiian cohorts that interacts with the significantly mutated subnetwork identified in both the European and East Asian cohorts. For instance, beta-secretase 1 (BACE1) is a genome-wide significant gene-level association in the East Asian cohort but does not contain SNPs previously associated with triglycerides in any ancestry cohort in the GWAS Catalog. BACE1 has gene-ε significant p values in both the East Asian ancestry cohort (p = 3.57 × 10−13) and European ancestry cohort (p = 5.55 × 10−17). BACE1 was significant at the gene level but contained no previously associated SNPs in any cohort in the GWAS Catalog. BACE1 plays a role in the metabolism of amyloid beta precursor protein and is known to play a role in amyloid precursor protein (APP) metabolism.66 Additionally, both APOL1 and HBA1 were identified as significantly associated with triglycerides via gene-ε in our analysis of the Native Hawaiian ancestry cohort, and both genes were part of significant subnetworks identified by Hierarchical HotNet in the European and Native Hawaiian ancestry cohorts. It is important to underscore that the results from the Native Hawaiian ancestry cohort are based on a relatively small sample size ( 1,915). While small sample size can increase the FDR in the gene-ε (Figure S8), we highlight the networks identified as enriched for associations with triglycerides in the Native Hawaiian ancestry cohort due to their proximity and biological connection to the networks identified in the well-powered European and East Asian ancestry cohorts. Furthermore, there were no variant-level associations for triglyceride levels that were significant in both the European and Native Hawaiian cohorts (Tables S14 and S15). If analysis had stopped at the variant level, the shared signal of association would have gone unidentified. Details on replicated SNP-level and gene-level associations among ancestries for triglyceride levels are shown in Figure S15 and Figure S16, respectively. While the identification of these subnetworks is predicated on the use of LD panels derived from 1000 Genomes populations as references, we find widespread validation of SNP-level associations with triglyceride levels in many of the genes included in the significant subnetworks (see GWAS Catalog results in Table S25 and gene-ε p values for each gene included in the subnetwork in Table S26).
Figure 3.

A subnetwork of apolipoprotein genes is significantly enriched for mutations in European, East Asian, and Native Hawaiian ancestries associated with triglyceride levels
The largest significantly altered subnetwork (p value 0.05) for triglyceride levels contains overlapping gene subnetworks for each of the European, East Asian, and Native Hawaiian ancestries when analyzed independently with Hierarchical HotNet.16 Each node in the network represents a gene. The shading of each node indicates the statistical significance of the association of that gene with triglyceride levels in a particular cohort. Two genes are connected if their protein products interact based on the ReactomeFI 201648 (European, East Asian) or iRefIndex 15.049 (Native Hawaiian) protein-protein interaction networks. Several genes from the apolipoprotein gene family are significantly associated with triglyceride levels in both the European and East Asian cohorts (see data and code availability). Additionally, the interactions between them form a highly connected subnetwork. Smaller subnetworks identified in the Native Hawaiian cohort are distal modules that are connected to the subnetwork detected in the European cohort. Not all genes in the largest significantly altered subnetwork for the Native Hawaiian ancestry group are shown for visualization purposes (127 not pictured here). Genes that contain SNPs previously associated to triglyceride levels in a European cohort in the GWAS Catalog are indicated with †. Similarly, genes that contain SNPs previously associated with triglyceride levels in a non-European cohort in the GWAS Catalog are indicated with ‡. The studies identifying these associations are given in Table S25.
SNP-level and gene-level association results are further discussed for both platelet count and triglyceride levels in the supplemental information. Our results indicate that network propagation methods, such as Hierarchical HotNet, can identify subnetworks of genes on known protein-protein interaction networks that are enriched for significant gene-level associations beyond what is expected by chance. As is the case with triglyceride levels in the European, East Asian, and Native Hawaiian cohorts analyzed in this study, application of pathway enrichment analyses offers the potential to identify subnetworks of interacting genes that are enriched for gene-level association signals in multiple ancestry cohorts.
Discussion
Many recent studies have proposed changes to multi-ancestry GWA study design2,5,6,28,30, 31, 32,67, 68, 69. In this analysis, we have focused on the potential of methods to increase the insight gained into complex trait architecture from multi-ancestry GWA datasets via the generation of biologically interpretable hypotheses. We demonstrate the potential gains of moving beyond standard SNP-level GWA analysis by using 25 quantitative complex traits among seven human ancestry cohorts in three large biobanks: BioBank Japan, the UK Biobank, and the PAGE consortium database (Tables S1–S10). Ultimately, we believe that studying complex traits demands analysis across multiple genomic scales and ancestries in order to gain biological insight into complex trait architecture and ultimately achieve personalized medicine.
As has been previously noted,5,31 non-European ancestry cohorts are often excluded from GWA analyses of multi-ancestry biobanks; complementing the analyses of Ben-Eghan et al.,30 we find that 80.13% of UK Biobank studies over the last 9 years only report significant SNP-level associations in the white British cohort (Figures S1 and S2), despite the tens of thousands of individuals of non-European ancestry sampled in that dataset. Unless this practice is curbed by the biomedical research community, it will exacerbate already existing disparities in healthcare across diverse communities. There are undoubted benefits from increased sampling in a given ancestry for association mapping with the standard GWA framework, but it is still unknown the extent to which results from larger GWA and fine-mapping studies using European-ancestry genomes will generalize to the entire human population.2,26
As long as sample sizes of non-European ancestry cohorts in GWA studies remain relatively small, researchers tend to not analyze these data fully, making it imperative to consider alternative tests to the standard GWA framework in order to fully leverage the data available for studying the genetic basis of human complex traits. Two-stage GWA studies with nominal significance thresholds in replication cohorts offer one approach for identifying replicated associations. In addition, application of multi-ancestry fine-mapping methods such as PESCA44 support the results of recent analyses from Mathieson36 that even when SNP-level significance is not observed in multiple ancestry cohorts, the direction of effect is generally the same. These methods continue to rely on the statistical detection of association from individual SNPs. Given that multi-ancestry biobank datasets have variable power to detect SNP-level associations given differences in sample size and minor allele frequencies across cohorts (Figure S3), many researchers simply ignore non-European ancestry samples in GWA studies,(2,5,6,30) potentially perpetuating health disparities and limiting our understanding of the genetic basis of complex traits and any heterogeneity in genetic trait architecture.44,47 For these reasons, we strongly recommend drawing on enrichment analyses to study genetic associations with a trait of interest, as enrichment analyses offer the opportunity for generating interpretable hypotheses regarding the shared biological mechanisms underwriting complex traits by using large-scale multi-ancestry datasets with variable sample sizes. Enrichment analyses offer a biologically interpretable way of boosting power to detect genetic architecture without alterations to the original design of a study.
There are multiple other technical considerations for conducting multi-ancestry association analyses that we do not consider here, which future studies may explore further in the context of SNP-level association studies and enrichment analyses. First, because we are analyzing some datasets for which we did not have access to genotype data (BioBank Japan and PAGE study data), we relied on reference panels to estimate LD, which is not ideal for enrichment analyses (as has been explored by Zhu and Stephens,17 Shi et al.44). While we draw on the published literature to validate our findings of shared genetic architecture underlying the complex traits studied here (Table S25), our pipelines highlight the importance of managed access to individual-level genotype data for gaining insight into the genetic basis of complex traits. Second, we have not addressed the downstream consequences of using self-identified ancestry to define cohorts in large-scale GWA studies (but see Willer et al.,70 Lin et al.,71 Yang et al.,72 Urbut et al.73). Third, each sample we analyzed has also experienced environmental exposures that may influence the statistical detection of genetic associations, and some of those environmental exposures may be correlated with genomic ancestry.19,74, 75, 76 Interrogation of the influence of gene by environment interactions on complex traits must be done with highly controlled experiments, which can in turn help prioritize traits in which association studies will be interpretable and useful. Lastly, we underscore that increasing sample size in GWA studies alone will not resolve these fundamental biological questions: the proportion of phenotypic variance explained by associations discovered as sample sizes increase in GWA studies has largely reached diminishing returns,42 and gene-by-environment interactions are increasingly influential, and estimable, in large biobanks with cryptic relatedness.77,78
Many recent methodological advances that leverage GWA summary statistics have focused on testing the co-localization of causal SNPs (e.g., fine mapping79, 80, 81, 82), the non-additive effects of SNP-level interactions (i.e., epistasis83,84), and multivariate GWA tests.73,84, 85, 86 While these methods can be extended and applied to multi-ancestry GWA analyses, they still focus on SNP-level signals of genetic trait architecture (see also Brown et al.,87 Galinsky et al.88). Unlike the traditional GWA method, enrichment analyses increase statistical power by aggregating SNP-level signals of genetic associations and allowing for genetic heterogeneity in SNP-level trait architecture across samples as well as offering the opportunity for immediate insights into trait architecture with existing datasets. These methods have been comparatively underused in multi-ancestry GWA studies. Application of enrichment methods to small cohorts is prone to the same statistical limitations as the standard GWA framework, namely, less power to detect true associations and elevated false positive rates when applied to small sample sizes (Figure S8, Table S23). However, enrichment of associations can be assessed at multiple biological scales—genes, gene sets, and networks—thereby allowing for biologically informed insight into trait architecture when small sample sizes are studied in conjunction with better powered cohorts, generating targeted hypotheses for biological validation. Thus, comparison of results from enrichment analyses offer the opportunity to identify therapeutic targets in ancestries where sample sizes are limited.
While many studies note that differences in LD across ancestries affect transferability of effect size estimates,6,52,89, 90, 91 recent studies in population genetics have additionally debated how various selection pressures and genetic drift may hamper transferability of GWA results across ancestries (see for example, Edge and Rosenberg,21,22 Novembre and Barton,24 Harpak and Przeworski,26 Durvasula and Lohmueller,28 Mostafavi et al.29). Future GWA studies should be coupled with approaches from studies of how evolutionary processes shape the genetic architecture of complex traits.26,34,53,92
Two open questions must be tackled when studying complex trait architecture in the multi-ancestry biobank era: (1) to what extent is the true genetic trait architecture (causal SNPs and/or their effects on a trait of interest) heterogeneous across cohorts6,93 and (2) which components of GWA results (e.g., p values, estimated effect size, direction of effect sizes) are transferable across ancestries at any genomic scale? Continued application of the standard SNP-level GWA approach will not answer these questions. However, enrichment methods that aggregate SNP-level effects, test for effect size heterogeneity, and leverage genomic annotations and gene interaction networks offer opportunities to directly test these fundamental questions. Methods can and should play an important role as biomedical research shifts current paradigms to extend the benefits of personalized medicine beyond people of European ancestry.
Additionally, biomedical researchers should continue to pressure both funding agencies and institutions to diversify their sampling efforts in the name of inclusion and addressing—instead of exacerbating—genomic health disparities. In addition to those efforts, we believe existing and new methods can increase the return on investment in multi-ancestry biobanks, ensure that every bit of information from these datasets is studied, and prioritize biological mechanism above SNP-level statistical association signals by identifying associations that are robust across ancestries.
Acknowledgments
We thank Kirk Lohmueller and Alicia R. Martin for helpful comments on an earlier version of this manuscript as well as the Crawford and Ramachandran Labs for helpful discussions. This research was conducted in part with computational resources and services at the Center for Computation and Visualization at Brown University as well as with the UK Biobank Resource under application number 22419. The Population Architecture Using Genomics and Epidemiology (PAGE) program is funded by the National Human Genome Research Institute (NHGRI) with co-funding from the National Institute on Minority Health and Health Disparities (NIMHD). The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts 75N92021D00001, 75N92021D00002, 75N92021D00003, 75N92021D00004, and 75N92021D00005. The HCHS/SOL study was carried out as a collaborative study supported by contracts from the National Heart, Lung and Blood Institute (NHLBI) to the University of North Carolina (N01-HC65233), University of Miami (N01-HC65234), Albert Einstein College of Medicine (N01-HC65235), Northwestern University (N01-HC65236), and San Diego State University (N01-HC65237). S.P.S. was a trainee supported under the Brown University Predoctoral Training Program in Biological Data Science (NIH T32 GM128596). L.C. acknowledges the support of an Alfred P. Sloan Research Fellowship and a David & Lucile Packard Fellowship for Science and Engineering. This work was also supported by US National Institutes of Health R01 GM118652 and National Institutes of Health R35 GM139628 to S.R. S.R. acknowledges additional support from National Science Foundation CAREER Award DBI-1452622.
Declaration of interests
C.G. owns stock in 23andMe. E.K. and C.G. are members of the scientific advisory board for Encompass Bioscience. E.K. consults for Illumina.
Published: March 28, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.03.005.
Data and code availability
All scripts and publicly available data from GWA, gene, and pathway association tests are available at https://github.com/ramachandran-lab/multiancestry_enrichment. Results from PESCA analyses were provided through personal correspondence by Huwenbo Shi.
Web resources
Hierarchical HotNet, https://github.com/raphael-group/hierarchical-hotnet
Supplemental information
References
- 1.Nagai A., Hirata M., Kamatani Y., Muto K., Matsuda K., Kiyohara Y., Ninomiya T., Tamakoshi A., Yamagata Z., Mushiroda T., et al. BioBank Japan Cooperative Hospital Group Overview of the BioBank Japan Project: Study design and profile. J. Epidemiol. 2017;27(3S):S2–S8. doi: 10.1016/j.je.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin A.R., Gignoux C.R., Walters R.K., Wojcik G.L., Neale B.M., Gravel S., Daly M.J., Bustamante C.D., Kenny E.E. Human demographic history impacts genetic risk prediction across diverse populations. Am. J. Hum. Genet. 2017;100:635–649. doi: 10.1016/j.ajhg.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sankar P.L., Parker L.S. The Precision Medicine Initiative’s All of Us Research Program: an agenda for research on its ethical, legal, and social issues. Genet. Med. 2017;19:743–750. doi: 10.1038/gim.2016.183. [DOI] [PubMed] [Google Scholar]
- 4.Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J., et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin A.R., Kanai M., Kamatani Y., Okada Y., Neale B.M., Daly M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019;51:584–591. doi: 10.1038/s41588-019-0379-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wojcik G.L., Graff M., Nishimura K.K., Tao R., Haessler J., Gignoux C.R., Highland H.M., Patel Y.M., Sorokin E.P., Avery C.L., et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570:514–518. doi: 10.1038/s41586-019-1310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sohail M., Maier R.M., Ganna A., Bloemendal A., Martin A.R., Turchin M.C., Chiang C.W., Hirschhorn J., Daly M.J., Patterson N., et al. Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. eLife. 2019;8:e39702. doi: 10.7554/eLife.39702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berg J.J., Harpak A., Sinnott-Armstrong N., Joergensen A.M., Mostafavi H., Field Y., Boyle E.A., Zhang X., Racimo F., Pritchard J.K., Coop G. Reduced signal for polygenic adaptation of height in UK Biobank. eLife. 2019;8:e39725. doi: 10.7554/eLife.39725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sham P.C., Purcell S.M. Statistical power and significance testing in large-scale genetic studies. Nat. Rev. Genet. 2014;15:335–346. doi: 10.1038/nrg3706. [DOI] [PubMed] [Google Scholar]
- 10.Price A.L., Spencer C.C.A., Donnelly P. Progress and promise in understanding the genetic basis of common diseases. Proc. Biol. Sci. 2015;282:20151684. doi: 10.1098/rspb.2015.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Visscher P.M., Wray N.R., Zhang Q., Sklar P., McCarthy M.I., Brown M.A., Yang J. 10 years of gwas discovery: biology, function, and translation. Am. J. Hum. Genet. 2017;101:5–22. doi: 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J.Z., van Sommeren S., Huang H., Ng S.C., Alberts R., Takahashi A., Ripke S., Lee J.C., Jostins L., Shah T., et al. International Multiple Sclerosis Genetics Consortium. International IBD Genetics Consortium Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015;47:979–986. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng W., Ramachandran S., Crawford L. Estimation of non-null SNP effect size distributions enables the detection of enriched genes underlying complex traits. PLoS Genet. 2020;16:e1008855. doi: 10.1371/journal.pgen.1008855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu M.C., Lee S., Cai T., Li Y., Boehnke M., Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajabli F., Feliciano B.E., Celis K., Hamilton-Nelson K.L., Whitehead P.L., Adams L.D., Bussies P.L., Manrique C.P., Rodriguez A., Rodriguez V., et al. Ancestral origin of ApoE ε4 Alzheimer disease risk in Puerto Rican and African American populations. PLoS Genet. 2018;14:e1007791. doi: 10.1371/journal.pgen.1007791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reyna M.A., Leiserson M.D.M., Raphael B.J. Hierarchical HotNet: identifying hierarchies of altered subnetworks. Bioinformatics. 2018;34:i972–i980. doi: 10.1093/bioinformatics/bty613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu X., Stephens M. Bayesian large-scale multiple regression with summary statistics from genome-wide association studies. Ann. Appl. Stat. 2017;11:1561–1592. doi: 10.1214/17-aoas1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pritchard J.K., Przeworski M. Linkage disequilibrium in humans: models and data. Am. J. Hum. Genet. 2001;69:1–14. doi: 10.1086/321275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berg J.J., Coop G. A population genetic signal of polygenic adaptation. PLoS Genet. 2014;10:e1004412. doi: 10.1371/journal.pgen.1004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jakobsson M., Edge M.D., Rosenberg N.A. The relationship between F(ST) and the frequency of the most frequent allele. Genetics. 2013;193:515–528. doi: 10.1534/genetics.112.144758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edge M.D., Rosenberg N.A. Upper bounds on FST in terms of the frequency of the most frequent allele and total homozygosity: the case of a specified number of alleles. Theor. Popul. Biol. 2014;97:20–34. doi: 10.1016/j.tpb.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edge M.D., Rosenberg N.A. A general model of the relationship between the apportionment of human genetic diversity and the apportionment of human phenotypic diversity. Hum. Biol. 2015;87:313–337. doi: 10.13110/humanbiology.87.4.0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hormozdiari F., Zhu A., Kichaev G., Ju C.J., Segrè A.V., Joo J.W.J., Won H., Sankararaman S., Pasaniuc B., Shifman S., Eskin E. Widespread allelic heterogeneity in complex traits. Am. J. Hum. Genet. 2017;100:789–802. doi: 10.1016/j.ajhg.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Novembre J., Barton N.H. Tread lightly interpreting polygenic tests of selection. Genetics. 2018;208:1351–1355. doi: 10.1534/genetics.118.300786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenberg N.A., Edge M.D., Pritchard J.K., Feldman M.W. Interpreting polygenic scores, polygenic adaptation, and human phenotypic differences. Evol. Med. Public Health. 2018;2019:26–34. doi: 10.1093/emph/eoy036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harpak A., Przeworski M. The evolution of group differences in changing environments. PLoS Biol. 2021;19:e3001072. doi: 10.1371/journal.pbio.3001072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pereira L., Mutesa L., Tindana P., Ramsay M. African genetic diversity and adaptation inform a precision medicine agenda. Nat. Rev. Genet. 2021;22:284–306. doi: 10.1038/s41576-020-00306-8. [DOI] [PubMed] [Google Scholar]
- 28.Durvasula A., Lohmueller K.E. Negative selection on complex traits limits phenotype prediction accuracy between populations. Am. J. Hum. Genet. 2021;108:620–631. doi: 10.1016/j.ajhg.2021.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mostafavi H., Harpak A., Agarwal I., Conley D., Pritchard J.K., Przeworski M. Variable prediction accuracy of polygenic scores within an ancestry group. eLife. 2020;9:e48376. doi: 10.7554/eLife.48376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ben-Eghan C., Sun R., Hleap J.S., Diaz-Papkovich A., Munter H.M., Grant A.V., Dupras C., Gravel S. Don’t ignore genetic data from minority populations. Nature. 2020;585:184–186. doi: 10.1038/d41586-020-02547-3. [DOI] [PubMed] [Google Scholar]
- 31.Popejoy A.B., Fullerton S.M. Genomics is failing on diversity. Nature. 2016;538:161–164. doi: 10.1038/538161a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bustamante C.D., Burchard E.G., De la Vega F.M. Genomics for the world. Nature. 2011;475:163–165. doi: 10.1038/475163a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu X., Stephens M. Large-scale genome-wide enrichment analyses identify new trait-associated genes and pathways across 31 human phenotypes. Nat. Commun. 2018;9:4361. doi: 10.1038/s41467-018-06805-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyle E.A., Li Y.I., Pritchard J.K. An expanded view of complex traits: from polygenic to omnigenic. Cell. 2017;169:1177–1186. doi: 10.1016/j.cell.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sinnott-Armstrong N., Naqvi S., Rivas M., Pritchard J.K. GWAS of three molecular traits highlights core genes and pathways alongside a highly polygenic background. eLife. 2021;10:e58615. doi: 10.7554/eLife.58615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mathieson I. The omnigenic model and polygenic prediction of complex traits. Am. J. Hum. Genet. 2021;108:1558–1563. doi: 10.1016/j.ajhg.2021.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abraham G., Qiu Y., Inouye M. FlashPCA2: principal component analysis of Biobank-scale genotype datasets. Bioinformatics. 2017;33:2776–2778. doi: 10.1093/bioinformatics/btx299. [DOI] [PubMed] [Google Scholar]
- 39.Bulik-Sullivan B.K., Loh P.R., Finucane H.K., Ripke S., Yang J., Patterson N., Daly M.J., Price A.L., Neale B.M., Schizophrenia Working Group of the Psychiatric Genomics Consortium LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu M.C., Kraft P., Epstein M.P., Taylor D.M., Chanock S.J., Hunter D.J., Lin X. Powerful SNP-set analysis for case-control genome-wide association studies. Am. J. Hum. Genet. 2010;86:929–942. doi: 10.1016/j.ajhg.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakka P., Raphael B.J., Ramachandran S. Gene and network analysis of common variants reveals novel associations in multiple complex diseases. Genetics. 2016;204:783–798. doi: 10.1534/genetics.116.188391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y., Qi G., Park J.H., Chatterjee N. Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat. Genet. 2018;50:1318–1326. doi: 10.1038/s41588-018-0193-x. [DOI] [PubMed] [Google Scholar]
- 43.Gusev A., Ko A., Shi H., Bhatia G., Chung W., Penninx B.W., Jansen R., de Geus E.J., Boomsma D.I., Wright F.A., et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016;48:245–252. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi H., Burch K.S., Johnson R., Freund M.K., Kichaev G., Mancuso N., Manuel A.M., Dong N., Pasaniuc B. Localizing Components of Shared Transethnic Genetic Architecture of Complex Traits from GWAS Summary Data. Am. J. Hum. Genet. 2020;106:805–817. doi: 10.1016/j.ajhg.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leiserson M.D., Vandin F., Wu H.-T., Dobson J.R., Raphael B.R. Pan-cancer identification of mutated pathways and protein complexes. Cancer Res. 2014;74:5324. [Google Scholar]
- 46.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakka P., Archer N.P., Xu H., Lupo P.J., Raphael B.J., Yang J.J., Ramachandran S. Novel gene and network associations found for acute lymphoblastic leukemia using case–control and family-based studies in multiethnic populations. Cancer Epidemiol. Biomarkers Prev. 2017;26:1531–1539. doi: 10.1158/1055-9965.EPI-17-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fabregat A., Sidiropoulos K., Garapati P., Gillespie M., Hausmann K., Haw R., Jassal B., Jupe S., Korninger F., McKay S., et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2016;44(D1):D481–D487. doi: 10.1093/nar/gkv1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Razick S., Magklaras G., Donaldson I.M. iRefIndex: a consolidated protein interaction database with provenance. BMC Bioinformatics. 2008;9:405. doi: 10.1186/1471-2105-9-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Das J., Yu H. HINT: High-quality protein interactomes and their applications in understanding human disease. BMC Syst. Biol. 2012;6:92. doi: 10.1186/1752-0509-6-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rolland T., Taşan M., Charloteaux B., Pevzner S.J., Zhong Q., Sahni N., Yi S., Lemmens I., Fontanillo C., Mosca R., et al. A proteome-scale map of the human interactome network. Cell. 2014;159:1212–1226. doi: 10.1016/j.cell.2014.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carlson C.S., Matise T.C., North K.E., Haiman C.A., Fesinmeyer M.D., Buyske S., Schumacher F.R., Peters U., Franceschini N., Ritchie M.D., et al. PAGE Consortium Generalization and dilution of association results from European GWAS in populations of non-European ancestry: the PAGE study. PLoS Biol. 2013;11:e1001661. doi: 10.1371/journal.pbio.1001661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eyre-Walker A. Evolution in health and medicine Sackler colloquium: Genetic architecture of a complex trait and its implications for fitness and genome-wide association studies. Proc. Natl. Acad. Sci. USA. 2010;107(Suppl 1):1752–1756. doi: 10.1073/pnas.0906182107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi H., Gazal S., Kanai M., Koch E.M., Schoech A.P., Siewert K.M., Kim S.S., Luo Y., Amariuta T., Huang H., et al. Population-specific causal disease effect sizes in functionally important regions impacted by selection. Nat. Commun. 2021;12:1098. doi: 10.1038/s41467-021-21286-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szalai A.J., Wu J., Lange E.M., McCrory M.A., Langefeld C.D., Williams A., Zakharkin S.O., George V., Allison D.B., Cooper G.S., et al. Single-nucleotide polymorphisms in the C-reactive protein (CRP) gene promoter that affect transcription factor binding, alter transcriptional activity, and associate with differences in baseline serum CRP level. J. Mol. Med. (Berl.) 2005;83:440–447. doi: 10.1007/s00109-005-0658-0. [DOI] [PubMed] [Google Scholar]
- 56.Zhang S.-C., Wang M.-Y., Feng J.-R., Chang Y., Ji S.-R., Wu Y. Reversible promoter methylation determines fluctuating expression of acute phase proteins. eLife. 2020;9:e51317. doi: 10.7554/eLife.51317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Browning B.L., Browning S.R. Efficient multilocus association testing for whole genome association studies using localized haplotype clustering. Genet. Epidemiol. 2007;31:365–375. doi: 10.1002/gepi.20216. [DOI] [PubMed] [Google Scholar]
- 58.Liu J.Z., McRae A.F., Nyholt D.R., Medland S.E., Wray N.R., Brown K.M., Hayward N.K., Montgomery G.W., Visscher P.M., Martin N.G., Macgregor S., AMFS Investigators A versatile gene-based test for genome-wide association studies. Am. J. Hum. Genet. 2010;87:139–145. doi: 10.1016/j.ajhg.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Leeuw C.A., Mooij J.M., Heskes T., Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Q.S., Browning B.L., Browning S.R. Genome-wide haplotypic testing in a Finnish cohort identifies a novel association with low-density lipoprotein cholesterol. Eur. J. Hum. Genet. 2015;23:672–677. doi: 10.1038/ejhg.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Storey J.D., Tibshirani R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stephens M. False discovery rates: a new deal. Biostatistics. 2017;18:275–294. doi: 10.1093/biostatistics/kxw041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neale B.M., Medland S.E., Ripke S., Asherson P., Franke B., Lesch K.P., Faraone S.V., Nguyen T.T., Schäfer H., Holmans P., et al. Psychiatric GWAS Consortium: ADHD Subgroup Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J. Am. Acad. Child Adolesc. Psychiatry. 2010;49:884–897. doi: 10.1016/j.jaac.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Demontis D., Walters R.K., Martin J., Mattheisen M., Als T.D., Agerbo E., Baldursson G., Belliveau R., Bybjerg-Grauholm J., Bækvad-Hansen M., et al. ADHD Working Group of the Psychiatric Genomics Consortium (PGC) Early Lifecourse & Genetic Epidemiology (EAGLE) Consortium. 23andMe Research Team Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 2019;51:63–75. doi: 10.1038/s41588-018-0269-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cowen L., Ideker T., Raphael B.J., Sharan R. Network propagation: a universal amplifier of genetic associations. Nat. Rev. Genet. 2017;18:551–562. doi: 10.1038/nrg.2017.38. [DOI] [PubMed] [Google Scholar]
- 66.Perneczky R., Alexopoulos P., Alzheimer’s Disease euroimaging Initiative Cerebrospinal fluid BACE1 activity and markers of amyloid precursor protein metabolism and axonal degeneration in Alzheimer’s disease. Alzheimers Dement. 2014;10(5, Suppl):S425–S429, 429.e1. doi: 10.1016/j.jalz.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hindorff L.A., Bonham V.L., Brody L.C., Ginoza M.E.C., Hutter C.M., Manolio T.A., Green E.D. Prioritizing diversity in human genomics research. Nat. Rev. Genet. 2018;19:175–185. doi: 10.1038/nrg.2017.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sinnott-Armstrong N., Tanigawa Y., Amar D., Mars N., Benner C., Aguirre M., Venkataraman G.R., Wainberg M., Ollila H.M., Kiiskinen T., et al. FinnGen Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat. Genet. 2021;53:185–194. doi: 10.1038/s41588-020-00757-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peterson R.E., Kuchenbaecker K., Walters R.K., Chen C.Y., Popejoy A.B., Periyasamy S., Lam M., Iyegbe C., Strawbridge R.J., Brick L., et al. Genome-wide association studies in ancestrally diverse populations: opportunities, methods, pitfalls, and recommendations. Cell. 2019;179:589–603. doi: 10.1016/j.cell.2019.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Willer C.J., Li Y., Abecasis G.R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin D.-Y., Tao R., Kalsbeek W.D., Zeng D., Gonzalez F., 2nd, Fernández-Rhodes L., Graff M., Koch G.G., North K.E., Heiss G. Genetic association analysis under complex survey sampling: the Hispanic Community Health Study/Study of Latinos. Am. J. Hum. Genet. 2014;95:675–688. doi: 10.1016/j.ajhg.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang J., Zaitlen N.A., Goddard M.E., Visscher P.M., Price A.L. Advantages and pitfalls in the application of mixed-model association methods. Nat. Genet. 2014;46:100–106. doi: 10.1038/ng.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Urbut S.M., Wang G., Carbonetto P., Stephens M. Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions. Nat. Genet. 2019;51:187–195. doi: 10.1038/s41588-018-0268-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vrieze S.I., Iacono W.G., McGue M. Confluence of genes, environment, development, and behavior in a post-gwas world. Dev. Psychopathol. 2012;24:1195–1214. doi: 10.1017/S0954579412000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gage S.H., Davey Smith G., Ware J.J., Flint J., Munafò M.R. G= e: What gwas can tell us about the environment. PLoS Genet. 2016;12:e1005765. doi: 10.1371/journal.pgen.1005765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Borrell L.N., Elhawary J.R., Fuentes-Afflick E., Witonsky J., Bhakta N., Wu A.H.B., Bibbins-Domingo K., Rodríguez-Santana J.R., Lenoir M.A., Gavin J.R., 3rd, et al. Race and genetic ancestry in medicine - a time for reckoning with racism. N. Engl. J. Med. 2021;384:474–480. doi: 10.1056/NEJMms2029562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Young A.I., Frigge M.L., Gudbjartsson D.F., Thorleifsson G., Bjornsdottir G., Sulem P., Masson G., Thorsteinsdottir U., Stefansson K., Kong A. Relatedness disequilibrium regression estimates heritability without environmental bias. Nat. Genet. 2018;50:1304–1310. doi: 10.1038/s41588-018-0178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Young A.I., Benonisdottir S., Przeworski M., Kong A. Deconstructing the sources of genotype-phenotype associations in humans. Science. 2019;365:1396–1400. doi: 10.1126/science.aax3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hormozdiari F., Kostem E., Kang E.Y., Pasaniuc B., Eskin E. Identifying causal variants at loci with multiple signals of association. Genetics. 2014;198:497–508. doi: 10.1534/genetics.114.167908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hormozdiari F., van de Bunt M., Segrè A.V., Li X., Joo J.W.J., Bilow M., Sul J.H., Sankararaman S., Pasaniuc B., Eskin E. Colocalization of GWAS and eQTL Signals Detects Target Genes. Am. J. Hum. Genet. 2016;99:1245–1260. doi: 10.1016/j.ajhg.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.LaPierre N., Taraszka K., Huang H., He R., Hormozdiari F., Eskin E. Identifying causal variants by fine mapping across multiple studies. PLoS Genet. 2021;17:e1009733. doi: 10.1371/journal.pgen.1009733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang G., Sarkar A., Carbonetto P., Stephens M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. Series B Stat. Methodol. 2020;82:1273–1300. doi: 10.1111/rssb.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Crawford L., Zeng P., Mukherjee S., Zhou X. Detecting epistasis with the marginal epistasis test in genetic mapping studies of quantitative traits. PLoS Genet. 2017;13:e1006869. doi: 10.1371/journal.pgen.1006869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Turchin M.C., Stephens M. Bayesian multivariate reanalysis of large genetic studies identifies many new associations. PLoS Genet. 2019;15:e1008431. doi: 10.1371/journal.pgen.1008431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou X., Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012;44:821–824. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stephens M. A unified framework for association analysis with multiple related phenotypes. PLoS ONE. 2013;8:e65245. doi: 10.1371/journal.pone.0065245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brown B.C., Ye C.J., Price A.L., Zaitlen N., Asian Genetic Epidemiology Network Type 2 Diabetes Consortium Transethnic genetic-correlation estimates from summary statistics. Am. J. Hum. Genet. 2016;99:76–88. doi: 10.1016/j.ajhg.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Galinsky K.J., Reshef Y.A., Finucane H.K., Loh P.R., Zaitlen N., Patterson N.J., Brown B.C., Price A.L. Estimating cross-population genetic correlations of causal effect sizes. Genet. Epidemiol. 2019;43:180–188. doi: 10.1002/gepi.22173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bitarello B.D., Mathieson I. Polygenic scores for height in admixed populations. G3. 2020;10:4027–4036. doi: 10.1534/g3.120.401658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marnetto D., Pärna K., Läll K., Molinaro L., Montinaro F., Haller T., Metspalu M., Mägi R., Fischer K., Pagani L. Ancestry deconvolution and partial polygenic score can improve susceptibility predictions in recently admixed individuals. Nat. Commun. 2020;11:1628. doi: 10.1038/s41467-020-15464-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huang H., Ruan Y., Feng Y.-C.A., Chen C.-Y., Lam M., Sawa A., Martin A., Qin S., Ge T. Improving polygenic prediction in ancestrally diverse populations. Preprint at medRxiv. 2021 doi: 10.1101/2020.12.27.20248738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hayward L.K., Sella G. Polygenic adaptation after a sudden change in environment. Preprint at bioRxiv. 2019 doi: 10.1101/792952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McClellan J., King M.C. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All scripts and publicly available data from GWA, gene, and pathway association tests are available at https://github.com/ramachandran-lab/multiancestry_enrichment. Results from PESCA analyses were provided through personal correspondence by Huwenbo Shi.