

Abstract
The peptidyl-prolyl isomerase, Pin1, is exploited in cancer to activate oncogenes and inactivate tumor suppressors. However, despite considerable efforts, Pin1 has remained an elusive drug target. Here, we screened an electrophilic fragment library to identify covalent inhibitors targeting Pin1’s active site Cys113, leading to the development of Sulfopin, a nanomolar Pin1 inhibitor. Sulfopin is highly selective, as validated by two independent chemoproteomics methods, achieves potent cellular and in vivo target engagement and phenocopies Pin1 genetic knockout. Pin1 inhibition had only a modest effect on cancer cell line viability. Nevertheless, Sulfopin induced downregulation of c-Myc target genes, reduced tumor progression and conferred survival benefit in murine and zebrafish models of MYCN-driven neuroblastoma, and in a murine model of pancreatic cancer. Our results demonstrate that Sulfopin is a chemical probe suitable for assessment of Pin1-dependent pharmacology in cells and in vivo, and that Pin1 warrants further investigation as a potential cancer drug target.
Cancer cells rely on multiple signaling pathways to sustain proliferation and downregulate apoptotic signals, including the phosphorylation of serine/threonine-proline motifs1. This motif is specifically recognized and isomerized by the peptidyl-prolyl isomerase NIMA-interacting-1 (Pin1), which is the only known phosphorylation-dependent isomerase amongst the ~30 peptidyl-prolyl cis-trans isomerases (PPIases) in the human proteome2. Pin1-mediated isomerization impacts substrate stability3, activity4, subcellular localization5 and binding to interaction partners, including proline-directed kinases and phosphatases6.
Several lines of evidence suggest that aberrant Pin1 activation drives oncogenesis. Pin1 is overexpressed and/or overactivated in at least 38 tumor types7. While elevated Pin1 expression correlates with poor clinical prognosis8, polymorphisms that lower Pin1 expression are associated with reduced cancer risk9. Pin1 sustains proliferative signaling in cancer cells by upregulation of >50 oncogenes or growth-promoting factors10, including NF-κB11 and c-Myc12, while suppressing >20 tumor suppressors or growth-inhibiting factors. Furthermore, Pin1-null mice are resistant to tumorigenesis induced by mutant p53 (ref.13), activated HER2/RAS14 or constitutively expressed c-Myc15. Pin1 inhibition also sensitizes cancer cells to chemotherapeutics16 and radiation therapy17 and blocks the tumorigenesis of cancer stem cells18. However, as evidenced by the Cancer Dependency Map19, Pin1 is not essential for cellular viability and Pin1-null mice are viable, although they develop phenotypes characteristic of premature aging20.
Collectively, these studies suggest that pharmacological inhibition of Pin1 has the potential to block multiple cancer-driving pathways simultaneously21 with limited toxicity. Compounds that inhibit Pin1, such as juglone22, all-trans retinoic acid (ATRA)23, arsenic trioxide (ATO)24 and KPT-6566 (ref.25), exhibit anticancer activity and have been used to investigate the role of Pin1 in oncogenesis. Nevertheless, these compounds have been shown to lack specificity and/or cell permeability, making them unreliable tools for interrogation of the pharmacological inhibition of Pin1 in vivo26. While we recently developed a selective Pin1 covalent peptide inhibitor, BJP-06-005-3 (ref.27), it has poor stability in mouse liver microsomes and is unsuitable for in vivo applications.
The active site of Pin1’s PPIase domain contains a nucleophilic cysteine residue (Cys113) that is suitable for labeling with targeted covalent inhibitors27. Covalent inhibitors can offer several advantages over noncovalent inhibitors, and have been used successfully against traditional and challenging protein targets. To explore this strategy, we screened for compounds targeting Pin1 Cys113 using covalent fragment-based drug discovery (FBDD), which combines the advantages of FBDD with the improved potency conferred by covalent bond formation28. Optimization of screening hits led to the development of Sulfopin (1), a double-digit nanomolar, highly selective Pin1 inhibitor that engages Pin1 in cells and in vivo. We found that Pin1 inhibition induced modest viability effects in two-dimensional (2D) cancer cell culture after prolonged exposure and resulted in the downregulation of Myc-dependent target genes. In MYCN-driven zebrafish and murine models of neuroblastoma, Sulfopin significantly reduced tumor progression and provided an overall survival benefit. We saw similar results in a murine pancreatic ductal adenocarcinoma (PDAC) model. Sulfopin is therefore a selective Pin1 inhibitor suitable for the evaluation of Pin1 biology in vivo, and provides evidence that Pin1 warrants further exploration as an anticancer target.
Results
A covalent fragment screen identifies Pin1 binders.
We previously compiled a library of 993 electrophilic fragments featuring mildly reactive cysteine-targeting ‘warheads’28. We incubated this library with the purified catalytic domain of Pin1 (2 μM protein, 200 μM compound; 24 h at 4 °C), followed by intact protein liquid chromatography–mass spectrometry (LC–MS) to identify and quantify Pin1 labeling (Fig. 1). In total, 111 fragments irreversibly labeled Pin1 at >50% (Fig. 1b and Supplementary Dataset 1). Among the 48 top hits (labeling >75%), nine chloroacetamides shared a cyclic sulfone core, suggesting a structure–activity relationship (Fig. 1b; see Supplementary Fig. 1 and Supplementary Table 1 for a full list of hits containing this motif). Given that the identified sulfone-containing hits were nonpromiscuous in previous screens,28 we selected these for further development. To avoid undesired reactivity arising from the additional Michael acceptor in the 2-sulfolene fragments, we focused exclusively on sulfolane analogs.
Fig. 1 |. Discovery of a covalent Pin1-binding fragment.
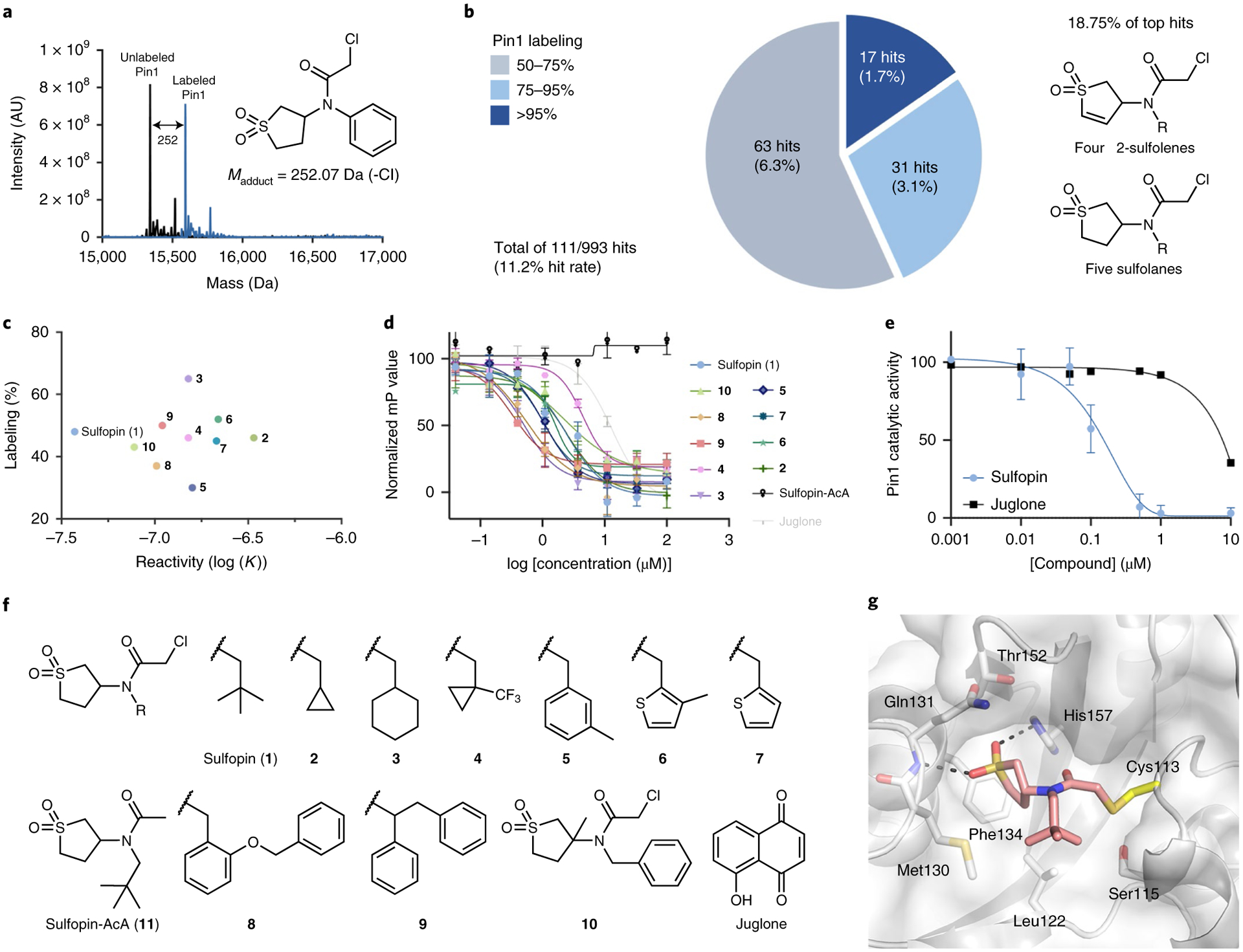
a, Intact protein LC–MS spectra of Pin1 (black) directly identify covalent binders (blue) in the electrophilic library screen (200 μM compound for 24 h). Madduct indicates the mass of the expected adduct for the indicated example. b, Distribution of hits in the Pin1 screening campaign and their corresponding labeling (%). Nine hits (18.75%) out of the 48 top hits that labeled Pin1 at >75% (dark and light blue) share sulfolene or sulfolane moieties. Labeling percentage calculated as previously described28. c, 2D analysis of the top ten optimized binders (structures shown in f); labeling percentage in the LC–MS assay plotted against reactivity (log (K)) suggests Sulfopin for further biological evaluation. d, Fluorescence polarization assay with the top ten binders, including juglone and a nonreactive control (Sulfopin-AcA), after 14 h of preincubation with Pin1. Data points are plotted as the average of n = 3 independent samples ± s.e.m., and are representative of n = 2 independent experiments. See Supplementary Table 3 for apparent Ki. mP represents the polarization value. e, PPIase substrate activity assay of Pin1 with Sulfopin (n = 3) and juglone (n = 2). Data points are plotted as the average of independent experiments ± s.e.m. for Sulfopin. f, Structures of the top ten binders in the Pin1-labeling LC–MS assay, the nonreactive control Sulfopin-AcA and juglone. g, X-ray crystal structure of Pin1 in complex with Sulfopin (1.4-Å resolution, PDB code 6VAJ). Pin1 (white) with relevant side chains in stick representation; Sulfopin is shown in pink. Hydrogen bonds are depicted as dashed lines. AU, arbitrary units.
Fragment optimization yields potent Pin1 binders.
We used covalent docking29 to guide compound optimization. Docking the sulfolane hits into various Pin1 structures yielded two plausible binding modes in which the lipophilic moiety (R in Fig. 1b) either protruded into the hydrophobic proline-binding pocket or interacted with a hydrophobic patch adjacent to Cys113 (Supplementary Fig. 2). Both poses suggested the diversification of the lipophilic moiety while leaving the sulfolane unaltered.
To optimize these original hits, we synthesized or purchased 25 sulfolane-containing compounds featuring a range of aliphatic, arylic or heterocyclic side chains (1–26; Supplementary Fig. 3). To identify high-affinity binders, we assessed their labeling of Pin1 under more stringent conditions (1:1 ratio of protein/compound, 2 μM compound; 1 h at room temperature). Remarkably, 22 of 25 second-generation compounds showed improved labeling whereas the original screening hits showed no labeling under these conditions (Supplementary Table 2). Overall, the second screen revealed that a wide range of lipophilic moieties were tolerated and that an additional methylene group between the amide and lipophilic side chain was crucial for Pin1 labeling (Supplementary Fig. 4). The top ten binders from this second screen (Fig. 1c,f) showed 35–65% Pin1 labeling. We evaluated these analogs in a competitive fluorescence polarization (FP) assay using a fluorescein isothiocyanate (FITC)-labeled substrate mimetic peptide inhibitor30. Following 14 h of incubation with recombinant full-length Pin1, all analogs competed in the FP to a greater extent than juglone, a frequently cited Pin1 inhibitor (Fig. 1d).
Identification and exclusion of overly reactive and potentially promiscuous compounds is critical in the development of covalent probes. Accordingly, we assessed the thiol reactivity of the top binders using a high-throughput assay that we previously applied to the entire fragment library28. We found no correlation between labeling efficiency and reactivity (Fig. 1c; Pearson R = 0.003; Supplementary Fig. 5). This was particularly evident when comparing the tert-butyl side chain-bearing 1 with the structurally similar 2, which has a cyclopropyl side chain. Both compounds showed similar Pin1 labeling (48 and 46%, respectively) but their reactivity varied by an order of magnitude, with 1 being dramatically less reactive.
We previously showed28 that electrophiles with reactivity rate constants >10−7 M−1 s−1 may exhibit nonselective cytotoxicity. We therefore evaluated the effects of selected Pin1 labelers on IMR90 lung fibroblast viability; 1 was the only compound with no toxicity up to 25 μM (Supplementary Table 3). With the lowest inherent reactivity of the top Pin1 labelers, 1 therefore offered the best balance of potency and selectivity. Consequently we selected 1, henceforth Sulfopin (1), for further evaluation.
Sulfopin potently binds and inhibits Pin1.
Sulfopin displayed potent Pin1 binding in the FP assay23 with an apparent Ki = 17 nM (after 14 h; Fig. 2). A corresponding noncovalent negative control (Sulfopin-AcA (11); Fig. 1f), which lacks the chloride leaving group, was inactive in the FP assay, suggesting that the binding affinity of Sulfopin is dependent on its electrophile (Figs. 1d and 2a). By performing the FP assay in a dose- and time-dependent manner, we determined the Kinact of Sulfopin to be 0.03 min−1, with a second-order rate constant (Kinact/Ki) of 84M−1 s−1 (Supplementary Fig. 6). Sulfopin also inhibited the catalytic activity of Pin1 with an apparent Ki of 211 nM measured at 12 h, as determined using a chymotrypsin-coupled peptidyl-prolyl isomerization assay31 (PPIase assay; Fig. 1e).
Fig. 2 |. Sulfopin engages Pin1 in cells and in vivo.
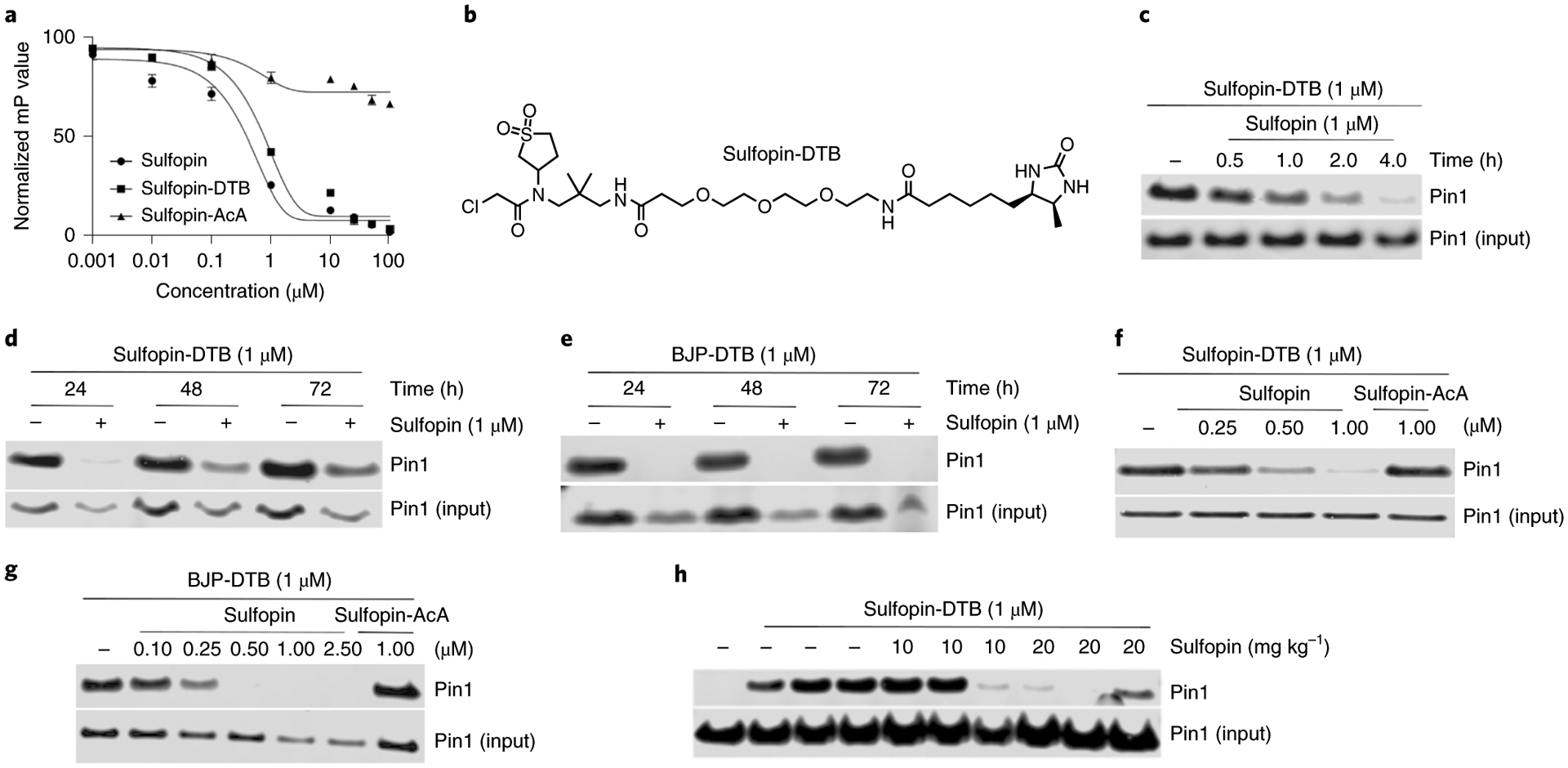
a, Fluorescence polarization assay showing that the DTB-labeled probe, Sulfopin-DTB, binds Pin1 with similar potency to Sulfopin following 14 h of incubation with Pin1. Data points are plotted as the average of n = 3 independent samples ± s.e.m., and are representative of n = 2 independent experiments. b, Chemical structure of Sulfopin-DTB. c, Sulfopin shows time-dependent engagement in PATU-8988T cells. PATU-8988T cells were treated with Sulfopin (1 μM) for the indicated time points followed by cell lysis, incubation with Sulfopin-DTB (1 μM), streptavidin pulldown and immunoblot analysis. d,e, Sulfopin shows long-term engagement of Pin1. PATU-8988T (d) or HCT116 (e) cells were incubated with or without Sulfopin for the indicated time points, followed by cell lysis, incubation with DTB probe, streptavidin pulldown and immunoblot analysis. Substantial engagement (>50%) was still evident after 72 h. f,g, Sulfopin fully engages Pin1 in PATU-8988T cells at 1 μM and in HCT116 cells at 0.5 μM (see Supplementary Fig. 9b for the structure of BJP-DTB). PATU-8988T (f) or HCT116 (g) cells were incubated with Sulfopin at the indicated concentrations for 5 h, followed by cell lysis, DTB probe incubation (1 h, 1 μM), streptavidin pulldown and immunoblot analysis. The noncovalent control, Sulfopin-AcA, is unable to outcompete Pin1 pulldown. c–g, Results are representative of n = 2 independent experiments. h, Sulfopin engages Pin1 in vivo. Mice were treated by oral gavage with the indicated amounts of Pin1 over 2 days for a total of three doses. Following this treatment, spleens were lysed for a competition pulldown experiment with Sulfopin-DTB. Results are representative of n = 2 independent pulldown experiments, starting from the same spleen lysates.
To evaluate the binding mode of Sulfopin, we determined the co-crystal structure of Pin1 in complex with Sulfopin at 1.4-Å resolution (Supplementary Table 4). The structure shows clear electron density to Cys113 in the 2FO–FC omit map, which confirmed a covalent interaction (Supplementary Fig. 7). In this structure, the sulfolane ring occupies the hydrophobic proline-binding pocket formed by Met130, Gln131, Phe134, Thr152 and His157 (Fig. 1g). Furthermore, the sulfonyl oxygens mediate hydrogen bonds to the backbone amide of Gln131, and the imidazole NH of His157, analogously to another Pin1 binder, arsenic trioxide24 (Supplementary Fig. 8). The tert-butyl group of Sulfopin covers a shallow hydrophobic patch comprised of Ser115, Leu122 and Met130 but is mostly solvent exposed, explaining the broad range of hydrophobic moieties that were tolerated at this position during the optimization campaign.
Despite being a very small ligand (heavy atom count, 17; calculated logP, 0.36), Sulfopin efficiently exploits interactions with Pin1, even in the absence of a negatively charged moiety, to interact with the phosphate-binding pocket, thus overcoming the cell permeability issues of previous Pin1 inhibitors, which are often highly anionic32.
Sulfopin engages Pin1 in cells and in vivo.
To evaluate the target engagement of Sulfopin in cells, we developed a desthiobiotin (DTB)-labeled probe for competition pulldown experiments. Based on the co-crystal structure of Sulfopin bound to Pin1, we derivatized the mostly solvent-exposed tert-butyl group of Sulfopin with a PEG-linked DTB (Sulfopin-DTB (27); Fig. 2b). Sulfopin-DTB maintained similar potency (apparent Ki = 38 nM in the FP assay; Fig. 2a) and successfully engaged Pin1 in PATU-8988T cell lysates, achieving robust pulldown at 1 μM following a 1-h incubation (Supplementary Fig. 9a).
To assess Sulfopin’s cellular target engagement, we performed live cell competition pulldown assays in PATU-8988T and HCT116 cells. A 1-μM treatment with Sulfopin achieved complete Pin1 engagement within 4 h (Fig. 2c) and maintained substantial engagement for up to 72 h, with target engagement monitored by loss of Sulfopin-DTB-mediated pulldown (Fig. 2d,e). Sulfopin exhibited dose-dependent Pin1 binding, with maximal engagement at 0.5–1 μM (Fig. 2f,g). This result was extensible to other cell lines, including IMR32 and MDA-MB-231 (Supplementary Fig. 9c,d).
Sulfopin exhibited encouraging metabolic stability in mouse hepatic microsomes (half-life, 41 min), prompting us to submit it for pharmacokinetic profiling. In three mice, oral administration of 10 mg kg–1 Sulfopin achieved an average cmax (peak concentration) of 11.5 μM and oral bioavailability (F%) of 30% (Supplementary Dataset 2), suggesting that Sulfopin is suitable for oral dosing in vivo. We next evaluated the toxicity of Sulfopin in an acute toxicity model in which mice were administered daily doses of 10, 20 or 40 mg kg–1 Sulfopin by intraperitoneal (IP) injection for 2 weeks. No adverse effects or weight loss were recorded, and a postmortem examination found no readily detectable pathologies.
Using Sulfopin-DTB, we were also able to assess Sulfopin’s in vivo Pin1 engagement. Over 2 days, mice were treated with three doses of vehicle or 10 or 20 mg kg–1 Sulfopin by oral gavage, followed by lysis of spleens for a competition pulldown experiment. Effective Pin1 engagement was observed in three out of three mice treated with 20 mg kg–1 Sulfopin (Fig. 2h). Based on these results, we chose a dose of 40 mg kg–1 for further mouse experiments to ensure complete Pin1 engagement.
Sulfopin is highly selective for Pin1 Cys113.
To evaluate the selectivity of Sulfopin in cells, we assessed its target profile using covalent inhibitor target-site identification (CITe-Id33; Extended Data Fig. 1a). This chemoproteomic platform enables identification and quantification of dose-dependent cysteine binding by covalent inhibitors on a proteome-wide scale. In this competition experiment, live PATU-8988T cells were incubated with Sulfopin (100, 500, 1,000 nM) for 5 h, followed by cell lysis and coincubation with Sulfopin-DTB (2 μM) for 18 h. Following trypsin digestion and avidin enrichment, the DTB-modified peptides were analyzed by multidimensional LC–tandem MS (LS–MS/MS). Of 162 cysteine residues labeled by Sulfopin-DTB, only Pin1 Cys113 (>2 s.d. from the median; Fig. 3a and Supplementary Dataset 3a) exhibited dose-dependent competition (Fig. 3b). Similar results were obtained following 12- and 24-h treatments (Supplementary Fig. 10 and Supplementary Dataset 3b), indicating the pronounced selectivity of Sulfopin.
Fig. 3 |. Sulfopin is highly selective for Pin1 C113 in cells.
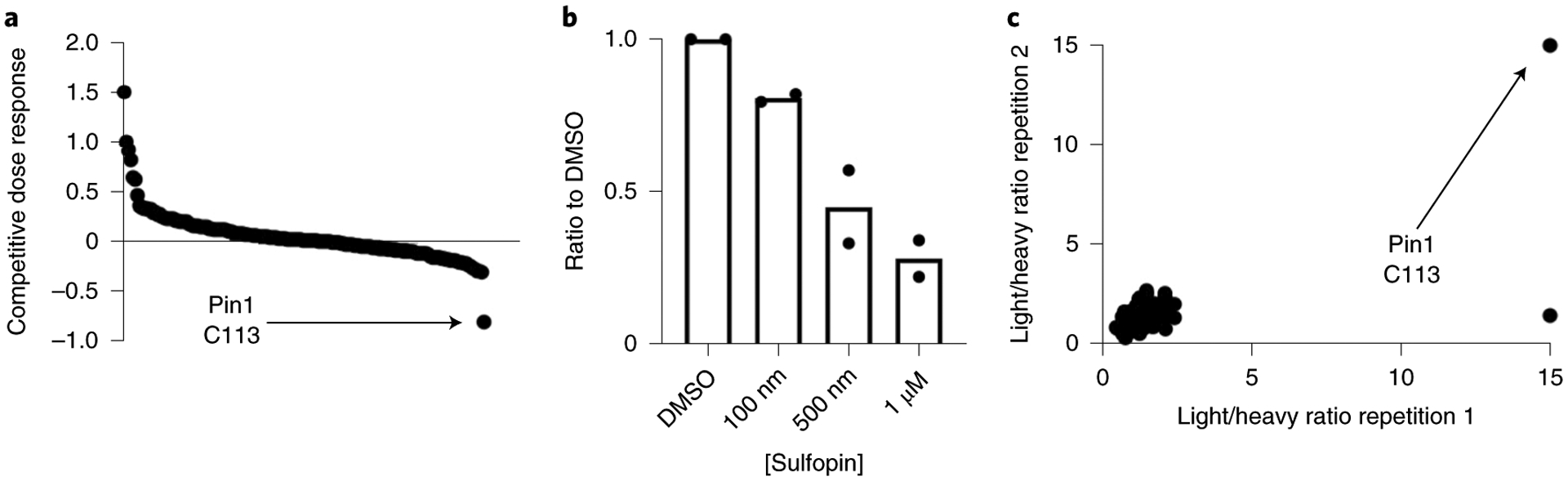
a, CITe-Id profiling results showing Sulfopin-DTB-labeled cysteine sites, rank ordered by competitive dose response to Sulfopin. Out of 162 cysteine residues reproducibly labeled by Sulfopin-DTB in n = 2 independent experiments, Pin1 C113 was the only site identified with a competitive dose response >2 s.d. from the mean value of the null. (see Supplementary Dataset 3a for a full list of identified peptides, and Supplementary Fig. 10 for results with 12/24-h treatment). b, Waterfall plot showing competitive dose dependency of Pin1 C113 labeling in the CITe-Id experiment. Bars represent mean of n = 2 independent experiments. c, Out of 2,134 cysteines identified in the rdTOP-ABPP experiment, only two showed a light/heavy ratio of >2.5 and, of these, one did not replicate and only Pin1 C113 showed the maximal ratio of 15 in both replicates.
To further profile Sulfopin’s proteome-wide cysteine selectivity, we used a complementary chemoproteomic method, rdTOP-ABPP (Extended Data Fig. 1b)34. This variant of the isoTOP-ABPP technique enables site-specific quantification of cysteine binding by label-free covalent inhibitors. In brief, MDA-MB-231 cells were treated with Sulfopin (5 μM, 2 h), lysed and labeled with a bio-orthogonal iodoacetamide-alkyne probe that was then conjugated to a cleavable biotin tag by copper-catalyzed azide−alkyne cycloaddition. After enrichment on beads, the peptides were isotopically derivatized by duplex reductive dimethylation, cleaved and analyzed by LC–MS/MS. After stringent filtering (s.d. of each sample < 2), we quantified 2,134 peptides. Of these, we identified Pin1 Cys113 as the top-ranked cysteine labeled by Sulfopin, with a competition ratio R = 15 across two biological replicates (Fig. 3c and Supplementary Dataset 3c). All other identified cysteines showed R < 2.5.
In summary, using two independent chemoproteomic techniques in two different cell lines, we demonstrated that Sulfopin has exquisite selectivity for Pin1 Cys113, making it a suitable probe for interrogation of Pin1 function.
Sulfopin treatment phenocopies Pin1 knockout.
We next assessed whether pharmacological inhibition of Pin1 by Sulfopin could recapitulate two previously reported phenotypes associated with Pin1 genetic knockout. First, Pin1 knockout is reported to abrogate phosphorylation of IRAK1 Thr209 and resensitize radioresistant cancer cells to irradiation17. Accordingly, we found that treatment of radioresistant HeLa cells with Sulfopin significantly resensitized them to irradiation in a dose-dependent manner (Extended Data Fig. 2a), and decreased IRAK1 Thr209 phosphorylation at concentrations as low as 100 nM (Extended Data Fig. 2b,c).
Second, Pin1-deficient mice are reported to display significant increases in the frequency of germinal center (GC) B cells in response to immunization35. GCs are sites where B cells proliferate and undergo somatic hypermutation in a BCL6- and Myc-dependent manner. To examine the effect of Sulfopin on GC B cells, we immunized the hind foot pads of 12 wild-type (WT) mice with ovalbumin coupled to 4-hydroxy-3-nitrophenylacetyl (NP-OVA) to induce GCs. The mice were injected with two doses of either Sulfopin (IP, 40 mg kg−1) or vehicle on days 7 and 9 after immunization, at the peak of GC response. On day 11 the mice were sacrificed and the frequency of GC B cells in lymph nodes was assessed by flow cytometry (Extended Data Fig. 2d). In accordance with a previous report35, Sulfopin-treated mice exhibited a 1.34-fold higher proportion of GC B cells compared to mice treated with vehicle (Extended Data Fig. 2e). Taken together, these data demonstrate that Sulfopin phenocopied the effects of Pin1 genetic deletion in the investigated models.
Viability effects of Sulfopin in cancer cell lines.
To broadly profile the antiproliferative activity of Sulfopin in a high-throughput fashion, we used the PRISM platform36 (Broad Institute) to evaluate its potency against 275 human cancer cell lines. Across almost all cell lines profiled, Sulfopin demonstrated limited to no antiproliferative activity after a 5-day treatment, with only nine cell lines showing half-maximal effective concentration (EC50) values <3 μM (Supplementary Dataset 4 and Supplementary Fig. 11). This result aligns with our initial cytotoxicity screening, as well as with data from the Cancer Dependency Map, in which Pin1 was not identified as a major genetic dependency in CRISPR–Cas9 and RNA interference screens across hundreds of cancer cell lines (https://depmap.org/portal/). These findings mirror our previous work with BJP-06-005-3, which was also nontoxic after 4–5-day treatments in diverse cancer cell lines27. These data suggest that the strong, single-agent cytotoxicity of previously published Pin1 inhibitors, such as juglone, is probably attributable to off-target effects37 (Supplementary Fig. 12).
Given our previous findings that BJP-06-005-3 reduces cell viability after extended treatments (up to 8 days)27, we assessed whether Sulfopin might similarly induce antiproliferative effects in a time-dependent manner. Matching the effects of BJP-06-005-3, we found that 6–8 days of Sulfopin treatment impacted the viability of PATU-8988T cells in a Pin1-dependent manner, having no effect on proliferation in corresponding Pin1 KO cells27 (Fig. 4a and Supplementary Fig. 12a).
Fig. 4 |. Sulfopin shows a Pin1-dependent viability effect following long-term exposure.
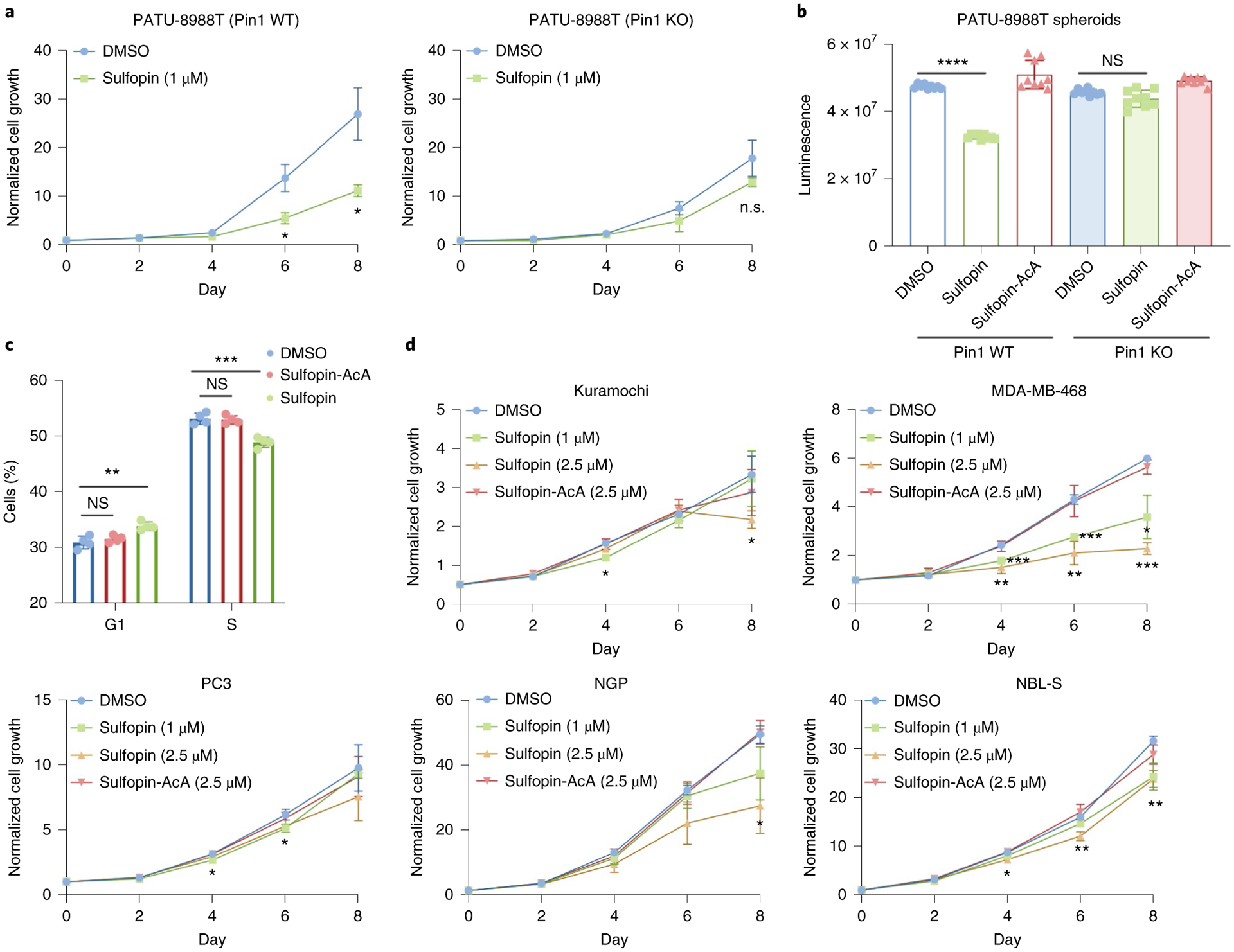
a, We previously27 generated a PATU-8988T Pin1 knockout (KO) cell line (Supplementary Fig. 12a). Sulfopin (1 μM) had a significant effect on cellular viability after 6 and 8 days (P = 0.01 and P = 0.01, respectively) in WT PATU-8988T cells (left), but showed no significant effect on viability in Pin1 KO cells (right); day 0-normalized growth rate for n = 3 biologically independent samples. b, Relative viability of PATU-8988T WT and Pin1 KO cells grown in 100% Matrigel domes following treatment with either Sulfopin (1 μM; n = 9 biologically independent samples; P = 1.24 × 10−18) or the noncovalent negative control, Sulfopin-AcA (1 μM; n = 9 biologically independent samples). Sulfopin-AcA showed no effect in any of the tested systems. c, Proportion of cells in various cell cycle stages as a function of Sulfopin treatment. The viability effects of Sulfopin are mediated by delayed cell cycle. PATU-8988T cells were treated with either DMSO, 2.5 μM Sulfopin or Sulfopin-AcA for 4 days. Cell cycle analysis was performed by BrdU and propidium iodide staining, followed by FACS analysis. Sulfopin treatment reduced the percentage of cells in S phase (P = 0.0004) and, in turn, increased the number of cells found in G1 phase (P = 0.003), while the noncovalent Sulfopin-AcA did not show this effect (n = 4; see Extended Data Fig. 3 for representative FACS analysis graphs and quantification of the results from two independent experiments). d, Cell culture growth curves. Sulfopin showed variation in antiproliferative effects across cancer cell lines Kuramochi, MDA-MB-468, NGP and NBL-S, with the most pronounced sensitivity observed in MDA-MB-468 cells (day 0-normalized growth rate for n = 3 biologically independent samples; P values for 2.5 μM Sulfopin after 4, 6 and 8 days were 0.007, 0.004 and 0.0004, respectively). Importantly we noted significant viability effects in Myc-high neuroblastoma cell lines NGP and NBL-S (P = 0.018 and 0.002, respectively for 2.5 μM Sulfopin after 8 days). Data points were plotted as the average of n = 3 biologically independent samples ± s.e.m. Statistical significance for all panels was calculated using one-tailed Student’s t-test with unequal variance (NS, not significant; P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Because three-dimensional (3D) culture models can better reflect in vivo results, we next evaluated the antiproliferative activity of Sulfopin in PATU-8988T WT Pin1 or Pin1 KO cells grown in 3D Matrigel domes. Following a 9-day treatment, Sulfopin demonstrated modest antiproliferative activity in PATU-8988T WT Pin1 cells, with no effects observed in PATU-8988T Pin1 KO cells, suggesting an on-target phenotype (Fig. 4b).
To understand why prolonged Sulfopin treatments were required to induce viability effects, we performed a cell cycle analysis. PATU-8988T cells were treated for 4 days with either DMSO (0.1%), Sulfopin (2.5 μM) or Sulfopin-AcA (2.5 μM) and stained with 5-bromodeoxyuridine (BrdU) and propidium iodide (Extended Data Fig. 3). This analysis showed a small but significant (P < 0.001) reduction in the number of synthesis (S)-phase cells and a corresponding increase of growth 1 (G1)-phase cells (Fig. 4c and Extended Data Fig. 3), but only in the Sulfopin treatment group. Additionally, we validated that Sulfopin does not induce apoptosis, even after treatments of up to 6 days, by immunoblot analysis of cleaved caspase 3 (Extended Data Fig. 4a and Supplementary Fig. 13a), and by AnnexinV-FITC and 7AAD staining followed by fluorescent activated cell sorter (FACS) analysis (Extended Data Fig. 4b and Supplementary Fig. 13b).
To evaluate whether this time-dependent growth phenotype is extensible to other cancer types, we performed additional experiments in breast (MDA-MB-468), prostate (PC3), ovarian (Kuramochi) and neuroblastoma (NGP, NBL-S) cancer cell lines, with MDA-MB-468 cells showing the most pronounced sensitivity to Sulfopin while PC3 and Kuramochi cells exhibited only slight effects (Fig. 4d). Collectively, these data suggest that Pin1 inhibition delays cell cycle progression and reduces the growth of cancer cell lines after extended treatments in 2D and 3D culture.
Sulfopin downregulates Myc transcriptional activity.
We and others have previously shown that Pin1 regulates the c-Myc oncoprotein38, affecting Myc protein stability39 as well as its DNA binding and transcriptional activity12,40. Pin1 physically interacts with c-Myc39, isomerizing P63 following phosphorylation of S62. We have shown that overexpression of Pin1 leads to an increase in the transcription of c-Myc target genes while knockdown of Pin1 decreases Myc-dependent transcription12. Matching reports that Pin1 promotes c-Myc turnover by stabilization of c-Myc protein levels39, and mirroring reported downstream effects of BJP-06-005-3 (ref.27), we found that Sulfopin treatment increased c-Myc protein levels (Supplementary Fig. 14).
To test whether Sulfopin affects Myc transcriptional output, we treated Mino B cells with either Sulfopin (1 μM, 6 h, in triplicate) or DMSO and performed a global RNA-seq analysis to detect differentially expressed genes: 206 genes were found to be significantly downregulated (Fig. 5a and Supplementary Dataset 5). Enrichr analysis of these transcripts, to identify transcription factors coordinating this response41, identified Myc target genes as the first and third most enriched sets in K562 and HeLa-S3 cells (adjusted P = 1.99 × 10−16 and 2.00 × 10−13, respectively; Fig. 5b), suggesting that Sulfopin downregulates Myc’s transcriptional signature. This finding matches the reported transcriptional effects of BJP-06-005-3 in PATU-8988T cells27. To further validate the effect of Sulfopin on Myc transcriptional activity, we cotransfected HEK293 cells with a Myc reporter construct (4× E-box luciferase) and Pin1. As expected, Pin1 expression increased Myc transcriptional activity while treatment with 2 μM Sulfopin for 48 h resulted in a significant reduction in relative luciferase activity (Fig. 5c). These results suggest that treatment with Sulfopin downregulates Myc target genes, making Myc-driven cancers natural candidates for its therapeutic application. Accordingly, we next evaluated Sulfopin using in vivo models of Myc-driven cancers.
Fig. 5 |. Sulfopin downregulates Myc transcription.

a, Results of an RNA-seq experiment comparing changes in RNA levels between Mino B cells treated with either Sulfopin (1 μM, 6 h, in triplicate) or DMSO. Each dot represents log2 fold change of a transcript (x axis) versus the P value for significance of that change (y axis; Wald test, as implemented in DESeq2). The dotted line indicates P = 0.05; 206 genes were significantly downregulated. b, Results of gene set enrichment analysis using Enrichr against the ENCODE TF chromatin immunoprecipitation–sequencing set. Two of the sets most enriched were Myc target genes from different cell lines. c, HEK293 cells were transfected with 4× E-box luciferase reporter for Myc transcriptional activity levels. Cotransfection with Pin1 increased reporter activity, while 48-h treatment with Sulfopin significantly (one-tailed Student’s t-test) reduced this activity compared to DMSO (n = 3; error bars indicate s.d.).
Sulfopin blocks neuroblastoma in zebrafish.
To evaluate the effects of Sulfopin on Myc-driven cancers, we turned to a zebrafish model of neuroblastoma42, a pediatric malignancy derived from the peripheral sympathetic nervous system (PSNS). During the development of the normal zebrafish embryo, neural crest-derived PSNS neuroblasts form the primordial superior cervical ganglia (SCG) and intrarenal gland (IRG) at the age of 3–7 days postfertilization (dpf) can be visualized using the dβh:EGFP fluorescent reporter42 (Fig. 6a). Overexpression of the MYCN oncogene, the oncogenic driver in approximately 20% of human high-risk neuroblastomas, results in the development of neuroblast hyperplasia in the PSNS of Tg(dβh:MYCN;dβh:EGFP) transgenic zebrafish (Fig. 6a, upper right). Neuroblast hyperplasia rapidly progresses to fully transformed tumors resembling human high-risk neuroblastoma42. When Sulfopin was added to the water containing Tg(dβh:MYCN;dβh:EGFP) zebrafish (at concentrations of 25–100 μM), neuroblastoma-initiating hyperplasia was significantly suppressed and fully transformed neuroblastoma did not develop over the treatment period (Fig. 6a,b). This indicates that Sulfopin blocked neuroblastoma initiation in our tumor model. Furthermore, no evidence of toxicity was observed in embryos treated with Sulfopin at these concentrations, further supporting our findings in mice that Sulfopin is well tolerated by healthy tissues in vivo.
Fig. 6 |. Sulfopin abrogates neuroblastoma and PDAC growth in vivo.
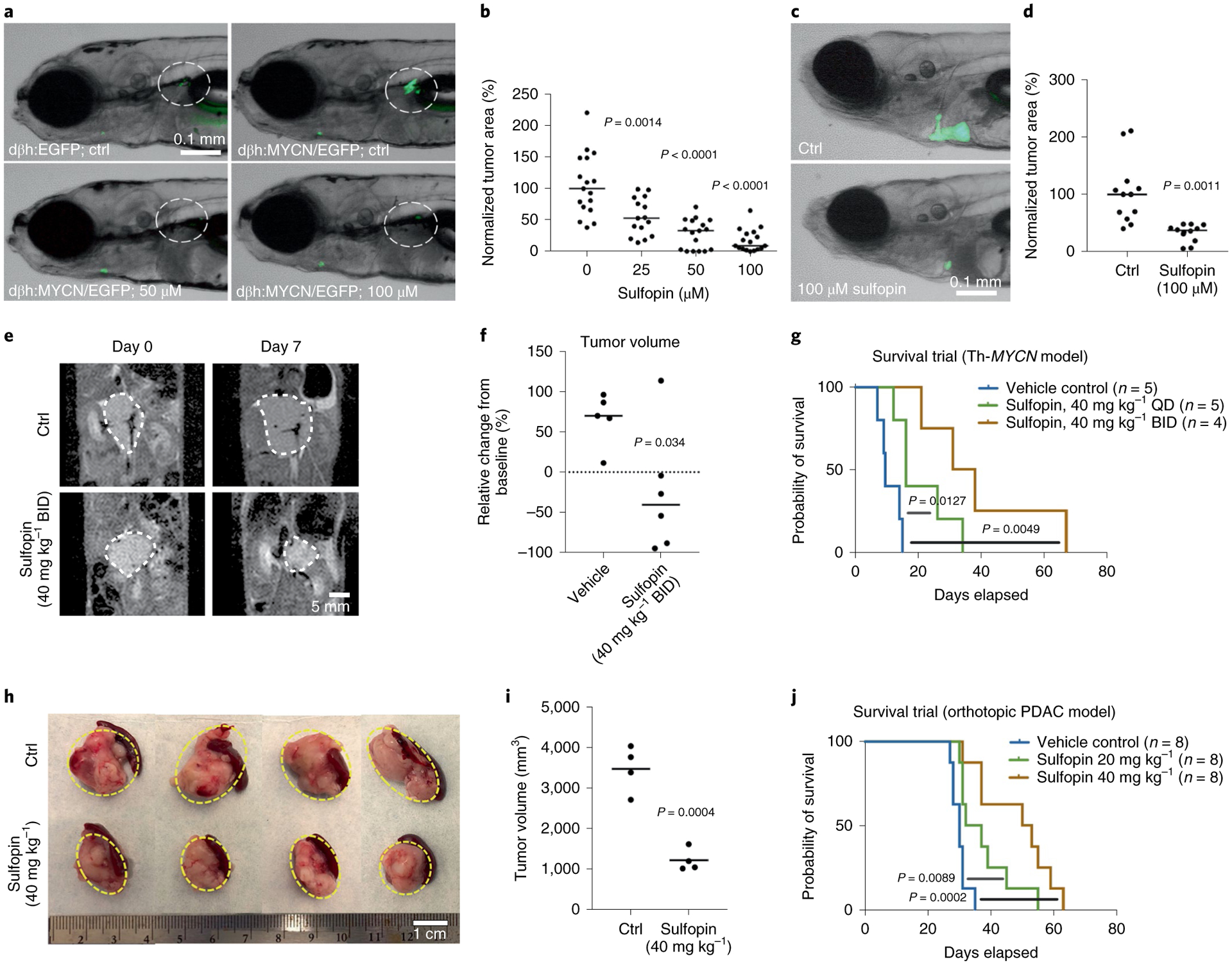
a, PSNS cells in the primordial SCG and IRG (highlighted by dotted circles) in representative embryos of Tg(dβh:EGFP) (upper left) and Tg(dβh:MYCN;dβh:EGFP) (upper right) transgenic zebrafish at 7 dpf. Representative 7-dpf Tg(dβh:MYCN;dβh:EGFP) zebrafish treated with 50 μM Sulfopin (bottom left) and 100 μM Sulfopin (bottom right). b, Quantification of green fluorescent protein (GFP)+ cells in primordial SCG and IRG of 7-dpf Tg(dβh:MYCN;dβh:EGFP) embryos treated with Sulfopin at multiple doses. A Mann–Whitney test with confidence intervals of 95% was used for analysis of significance (P value), and quantitative data are reported as median. c, Representative zebrafish embryos transplanted with neuroblastoma cells isolated from 4-month-old Tg(dβh:MYCN;dβh:EGFP) donor zebrafish and treated with DMSO control (upper) or 100 μM Sulfopin added to the water (lower). d, Quantification of enhanced GFP (eGFP)+ tumor area in zebrafish embryos treated with DMSO control and 100 μM Sulfopin added to the fish water. A Mann–Whitney test with confidence intervals of 95% was used for analysis of significance (P value), and quantitative data are reported as median. e, Representative MRI images of Th-MYCN mice pre- and post-7-day treatment with vehicle or 40 mg kg−1 BID Sulfopin. f, Significant differences in MRI-derived relative changes in tumor size of Th-MYCN mice after 7-day treatment with either vehicle (n = 5) or 40 mg kg−1 Sulfopin BID (n = 6) (P = 0.034, two-tailed Student’s t-test). Note the actual reduction in tumor size observed in five out of six treated mice. g, QD-treated mice showed significant (P = 0.0127) increase in overall survival, with an average increase of 10 days. BID-treated mice showed significant (P = 0.0049) increase in overall survival, with an average increase of 28 days. h, KPC mouse-derived pancreatic cancer cells were orthotopically transplanted into B6 mouse pancreas tail. One week after transplantation, mice were treated daily IP with either vehicle control, 20 mg kg–1 Sulfopin or 40 mg kg−1 Sulfopin. When maximum tumor length in control mice reached 2 cm, mice were euthanized and tumors collected and measured. i, Quantification of tumor volume (n = 4). Sulfopin-treated mice showed significant decrease in tumor volume (P = 0.0004, two-tailed Student’s t-test). j, Survival trial (Kaplan–Meier survival analysis, n = 8). Treated mice showed a significant increase in overall survival (P = 0.0002), with an average of 18.125 days. All mice survival significance data were evaluated using a Mantel–Cox test. Ctrl, control.
We then assessed the effects of Sulfopin on the maintenance of fully transformed neuroblastoma cells in primary-tumor-derived allograft models, constructed in transplanted zebrafish embryos. eGFP-labeled neuroblastoma cells were dissected from 4-month-old Tg(dβh:MYCN;dβh:EGFP) donor zebrafish, disaggregated, counted and 200–400 GFP-labeled tumor cells were injected intravenously into the duct of Cuvier (common cardinal vein) of 2-dpf zebrafish embryos43. One day later, 100 μM Sulfopin or DMSO was added to the fish water and, after a further 5 days, we quantified the area of eGFP-labeled tumor masses. While tumor masses in the DMSO-treated embryos grew larger, those in the Sulfopin-treated embryos decreased in size (Fig. 6c,d). Hence, Sulfopin treatment suppressed not only MYCN-driven neuroblastoma initiation but also the growth and survival of in vivo transplants of fully transformed primary neuroblastoma tumor cells.
Sulfopin regresses neuroblastoma in mice.
Following the encouraging results seen in zebrafish, we assessed the effects of Sulfopin in a murine model of neuroblastoma, Th-MYCN genetically engineered mice, in which human MYCN is expressed under the tyro-sine hydroxylase promoter44. The Th-MYCN model recapitulates the major molecular and histopathologic features of high-risk, MYCN-amplified neuroblastoma. We used both male and female hemizygous mice, which spontaneously developed palpable tumors at 50–130 days with 25% penetrance. Once tumors had become palpable, mice were randomly assigned to treatment groups and treated once (QD) or twice (BID) per day with either vehicle or 40 mg kg−1 Sulfopin. Tumor sizes were monitored over 7 days of treatment via magnetic resonance imaging (MRI; Fig. 6e). With the exception of one mouse, all tumors treated BID showed significant reduction in size, two of which showed near complete response (Fig. 6f). Sulfopin-treated QD mice showed a significant (P = 0.0127) average increase in survival of 10 days (Fig. 6g), while Sulfopin-treated BID mice showed an even more pronounced (P = 0.0049) average increase of 28 days. We note that mice in the BID arm received only 56 doses of compound (dose license limit).
Sulfopin inhibits pancreatic cancer progression in mice.
To investigate the efficacy of Sulfopin in an independent in vivo cancer model, we evaluated its effects in an orthotopic transplantation mouse model of pancreatic cancer. Pancreatic cancer cells derived from a KPC mouse (Pdx1- Cre, lsl-KrasG12D/+, lsl-tp53R172H/+) were orthotopically transplanted into the pancreatic tail of B6 mice. One week after transplantation, mice were treated daily with vehicle control or 20 or 40 mg kg−1 Sulfopin. Twenty-seven days after commencement of treatment and a tumor in the control group having grown to 2 cm, we euthanized the mice and evaluated tumor volume (n = 4). Sulfopin treatment had inhibited tumor growth and significantly decreased tumor volume (average tumor volume: control, 3,468.28 cm3 versus Sulfopin 40 mg kg−1, 1,210.91 cm3; P = 0.0004; Fig. 6h,i). In addition, Sulfopin treatment significantly extended survival with an average increase of 18 days (control, 30.0 days; 20 mg kg−1, 37.5 days; 40 mg kg−1, 48.125 days; P = 0.0002; Fig. 6j).
Discussion
Despite decades-long efforts to discover Pin1 inhibitors, no approach has yielded a compound capable of selectively blocking Pin1 in vivo. Here we describe the development and in vivo characterization of Sulfopin, a highly selective and potent covalent Pin1 inhibitor with low inherent reactivity and negligible toxicity. Sulfopin blocks tumor initiation in zebrafish and tumor progression in both neuroblastoma and pancreatic cancer mouse models, resulting in increased overall survival.
While inhibitors such as juglone, ATRA, ATO and KPT-6566 pioneered the investigation of Pin1 in cancer-related contexts, they all exert their anticancer effects in part through Pin1-independent mechanisms26 that include DNA damage25, induction of Pin1 degradation23 or direct blocking of transcription45. By contrast, Sulfopin is highly specific for Pin1 as we established here using multiple orthogonal experiments. Its selectivity allowed us to investigate Pin1 as a bona fide cancer drug target.
Pin1 has proven a challenging target for both ligand-30 and structure-based32,46 approaches, even in studies employing high-throughput screenings of up to one million compounds47. This is largely due to the shallow nature of the Pin1 active site and is further complicated by the phosphate-binding site, which favors negatively charged moieties. To overcome this hurdle, we screened a library of electrophilic fragments that can compensate for sparse protein interfaces by irreversibly labeling Pin1 Cys113. This screen resulted in a hit rate of 11% (111 compounds with >50% labeling), reflecting the high reactivity of Cys113 that has been found amongst the most reactive cysteines in several chemoproteomic campaigns48.
A major finding during our hit optimization campaign was that intrinsic warhead reactivity of Pin1 binders did not correlate with potency (Supplementary Fig. 5). Despite the structural similarity of the optimized compounds, their reactivities spanned >30-fold (Supplementary Table 2), with Sulfopin having one of the lowest reactivities nearing that of acrylamides (Supplementary Fig. 15). This low reactivity manifested in exquisite cellular selectivity. Two separate chemoproteomic approaches, performed in two different cell lines, both identified Pin1 Cys113 as the sole target of Sulfopin by a wide margin (Fig. 3). This result is rare for covalent inhibitors33 and even for Food & Drug Administration-approved covalent drugs.
Sulfopin induced negligible cytotoxicity across 275 cancer cell lines (PRISM screen), although it significantly impacted cancer cell growth after prolonged treatments (6–8 days; Fig. 4a,d) in 2D and 3D cell cultures (Fig. 4b), matching the reported effects of BJP-06-005-03. We also demonstrated that Sulfopin phenocopies Pin1 knockout in vivo (Extended Data Fig. 2). Furthermore, in a recent study Sulfopin was tested in Caenorhabditis elegans in which it inhibited PINN1, the worm Pin1 homolog, and successfully recapitulated genetic knockout phenotypes49. These results underscore the broad utility of Sulfopin as an in vivo tool compound.
Consistent with previous studies12,39, our RNA-seq experiment suggested that a major consequence of Pin1 inhibition was Myc target gene downregulation (Fig. 5a,b). However, given that Pin1 plays a central role in numerous signaling pathways, it is likely that Myc downregulation is not the only mechanism at play. Indeed, additional transcription factors showed significant downregulation in the RNA-sequencing (RNA-seq) dataset, including RNA Pol II (Fig. 5b and Supplementary Dataset 6) and RelA (Supplementary Dataset 6), which has been previously linked to Pin111 and may contribute to oncogenesis. RNA-seq experiments in additional cell lines may reveal other pathways regulated by Pin1.
While the viability effects of Sulfopin in cancer cell lines were modest, this does not preclude it from becoming an effective therapeutic agent. For example, inhibition of a validated cancer target such as KRASG12C has also shown only mild growth effects in 2D cell culture50. Further investigation is needed to establish the full therapeutic potential of Sulfopin. We never reached dose-limiting toxicity with Sulfopin in our experiments, indicating that treatment with higher doses, or in combination with other drugs, might enable more pronounced effects. Another aspect that warrants further investigation is the identification of biomarkers that render cell lines, including PATU-8988T, MDA-MB-468 and NGP, comparatively more sensitive to Pin1 inhibition (Fig. 4).
In summary, we present a potent and selective covalent Pin1 inhibitor with in vivo activity and no toxicity that facilitates investigation of Pin1 biology in physiologically relevant disease models. We envisage that the use of Sulfopin as a pharmacological tool will enable the study of the many diverse processes regulated by Pin1.
Methods
Electrophile library screening.
A total of 993 compounds (20 mM DMSO stocks) in 384-well-plate format were transferred to a 384-well plate working copy by combining 0.5 μl of five compounds per well. The catalytic domain of Pin1 (2 μM) in 20 mM Tris, 75 mM NaCl, pH 7.5 was incubated with 200 μM for each compound and moderately shaken for 24 h at 4 °C. The reaction was stopped by the addition of formic acid to 0.4% final concentration. The LC–MS runs were performed on Waters ACUITY ultra-performance LC (UPLC) class H, in positive ion mode using electrospray ionization. UPLC separation was performed using a C4 column (300 Å, 1.7 μM, 21 × 100 mm2); the column was held at 40 °C and the autosampler at 10 °C. Mobile solution A comprised 0.1% formic acid in water while mobile phase B was 0.1% formic acid in acetonitrile; run flow was 0.4 ml min−1. The gradient used for bovine serum albumin (BSA) was 20% B for 2 min, increasing linearly to 60% B for 4 min and holding for 2 min, changing to 0% B for 0.1 min and holding for 1.9 min. The gradient for the other proteins was 20% B for 2 min, increasing linearly to 60% B for 3 min and holding for 1.5 min, changing to 0% B for 0.1 min and holding for 1.4 min. Mass data were collected in the range 600–1300 m/z. The desolvation temperature was 500 °C, at a flow rate of 1,000 l h−1. The voltages used were 0.69 kV for the capillary and 46 V for the cone. Raw data were processed using openLYNX and deconvoluted using MaxEnt. Labeling assignment was performed as previously described28.
Covalent docking.
Covalent docking was performed using DOCKovalent 3.7 (ref.29) against 16 structures of Pin1. Protein Data Bank (PDB) codes: 1PIN, 2ITK, 2Q5A, 2XP3, 2ZQV, 2ZR4, 3IK8, 3KAB, 3KCE, 3NTP, 3ODK, 3OOB, 3TC5, 3TCZ, 3TDB, 3WH0. The docked compounds include seven sulpholane hits from the electrophilic library with the following IDs: PCM-0102138, PCM-0102178, PCM-0102105, PCM-0102832, PCM-0102313, PCM-0102760, PCM-0102755. The covalent bond length was set to 1.8 Å and the two newly formed bond angles to Cβ-Sγ-C, 109.5 ± 5° and Sγ-C-Ligatom, 109.5 ± 5°.
Thiol reactivity assay.
The compound 5,5’-dithio-bis(2-nitrobenzoic acid) (DTNB; 50 μM) was incubated with 200 μM tris(2-carboxyethyl)phosphine (TCEP) in 20 mM sodium phosphate buffer pH 7.4 and 150 mM NaCl for 5 min at room temperature, to obtain TNB2−. Next, 200-μM compounds were subsequently added to TNB2− followed by immediate ultraviolet (UV) absorbance measurement at 412 nm and 37 °C. UV absorbance was acquired every 15 min for 7 h. The assay was performed in a 384-well plate using a Tecan Spark10M plate reader. Background absorbance of compounds was subtracted by measuring the absorbance at 412 nm of each compound under the same conditions without DTNB. Compounds were measured in triplicate. The data were fitted to a second-order reaction equation such that the rate constant (K) is the slope of ln([A][B0]/[B][A0]), where [A0] and [B0] are the initial concentrations of the compound (200 μM) and TNB2− (100 μM), respectively, and [A] and [B] are the remaining concentrations as a function of time as deduced from spectrometric measurements. Linear regression using Prism was performed to fit the rate against the first 4 h of measurements.
Pin1 expression and purification.
A construct of full-length human Pin1 in a pET28 vector was overexpressed in Escherichia coli BL21 (DE3) in lysogeny broth medium in the presence of 50 mg ml−1 kanamycin. Cells were grown at 37 °C to an optical density of 0.8, cooled to 17 °C, induced with 500 μM isopropyl-1-thio-d-galactopyranoside, incubated overnight at 17 °C, collected by centrifugation and stored at −80 °C. Cell pellets were sonicated in buffer A (50 mM HEPES 7.5, 500 mM NaCl, 10% glycerol, 20 mM Imidazole and 7 mM β-mercaptoethanol (BME)), and the resulting lysate was centrifuged at 30,000g for 40 min. Ni-NTA beads (Qiagen) were mixed with lysate supernatant for 30 min and washed with buffer A. Beads were transferred to a fast-protein, LC-compatible column and the bound protein was washed with 15% buffer B (50 mM HEPES 7.5, 500 mM NaCl, 10% glycerol, 250 mM imidazole and 3 mM BME) and eluted with 100% buffer B. Thrombin was added to the eluted protein with incubation at 4 °C overnight. The sample was concentrated and passed through a Superdex 200 10/300 column (GE Healthcare) in a buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 5% glycerol and 1 mM TCEP. Fractions were pooled, concentrated to approximately 37 mg ml−1 and frozen at −80 °C.
Pin1 crystallization and soaking.
Apo protein, at a final concentration of 1 mM, was crystallized by sitting-drop (200 nl + 200 nl) vapor diffusion at 20 °C in the following crystallization buffer: 3 M NH4SO4, 100 mM HEPES pH 7.5, 150 mM NaCl, 1% PEG400 and 10 mM DTT. A volume of 200 nl of 1 mM Sulfopin was added directly to the crystals for soaking at 20 °C for 16 h. Crystals were transferred briefly into crystallization buffer containing 25% glycerol before flash-freezing in liquid nitrogen.
Crystallization data collection and structure determination.
Diffraction data from complex crystals were collected at beamline 24ID-C of the NE-CAT at the Advanced Photon Source, Argonne National Laboratory. Datasets were integrated and scaled using XDS51. Structures were solved by molecular replacement using the program Phaser52 and the search model PDB entry 1PIN. Iterative manual model building and refinement using Phenix53 and Coot54 led to the final models (Supplementary Table 4).
FP assay.
Binding affinity to Pin1 was determined using a FP assay to assess competition with an N-terminal fluorescein-labeled peptide (peptide core structure: Bth-D-pThr-Pip-Nal), which was synthesized by Proteintech. The indicated concentrations of candidate compounds were preincubated for 14 h at 4 °C with a solution containing 250 nM glutathione S-transferase (GST)-Pin1, 5 nM fluorescein-labeled peptide probe, 10 μg ml−1 BSA, 0.01% Tween-20 and 1 mM DTT in a buffer of 10 mM HEPES, 10 mM NaCl and 1% glycerol (pH 7.4). Measurements of FP were made in black, 384-well plates (Corning) using an EnVision reader. Ki values obtained from FP assay results were derived from the Kenakin Ki equation: Kenakin Ki = (Lb)(EC50)(Kd)/(Lo)(Ro) + Lb(Ro – Lo + Lb – Kd), where Kd (M) is Kd of the probe; EC50 (M) is the concentration of unlabeled compound resulting in 50% inhibition of binding (obtained from FP assay); total tracer Lo (M) is probe concentration in FP; bound tracer Lb (M) is 85%; and fraction of probe bound to Pin1, total receptor Ro (M) is Pin1 concentration in the FP assay, as described in ref.55.
Pin1 PPIase activity assay.
Inhibition of Pin1 isomerase activity was determined using the chymotrypsin-coupled PPIase assay with GST-Pin1 and Suc-Ala-pSer-Pro-Phe-pNA peptide substrate, as described previously31. GST-Pin1 (50 nM) was preincubated with the indicated concentrations of compound for 12 h at 4 °C in buffer containing 35 mM HEPES pH 7.8, 0.2 mM DTT and 0.1 mg ml−1 BSA. Immediately before the assay was started, chymotrypsin (final concentration, 6 mg ml–1) was added followed by the addition of the peptide substrate (Suc-Ala-pSer-Pro-Phe-pNA peptide substrate, final concentration 50 mM). The Ki value obtained from the PPIase assay was derived from the Cheng–Prusoff equation: Ki = IC50/(1 + S/Km), where Km is the Michaelis constant for the peptide substrate, S is the initial concentration of the substrate in the assay and IC50 is the half-minimal inhibitory concentration of the inhibitor.
Hepatic microsome stability.
In vitro hepatic microsomal stability was conducted by the DMPK core of the Scripps Research Institute, FL, USA. Sulfopin (1 μM) was incubated with 1 mg ml−1 hepatic microsomes in 100 mM KPi, pH 7.4. The reaction was initiated following the addition of 1 mM nicotinamide adenine dinucleotide phosphate (NADPH). Aliquots were collected at 0, 5, 10, 20, 40 and 60 min and added to acetonitrile (5× v/v) to stop the reaction. The NADPH dependence of the reaction was evaluated using no-NADPH control samples. Finally the samples were centrifuged through Millipore Multiscreen Solvinter 0.45-μm, low-binding polytetrafluoroethylene hydrophilic filter plates and analyzed by LC–MS/MS. The final data were log-transformed and the half-life calculated.
Cell culture and reagents.
PATU-8988T (DSMZ), MDA-MB-468 (ATCC) and HeLa (ATCC) cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS; Sigma) and 1% penicillin/streptomycin. PC3 (ATCC), IMR32 (ATCC) and Kuramochi (laboratory of P. A. Konstantinopoulos) cells were cultured in RPMI 1640 medium with l-glutamine, supplemented with 10% FBS and 1% penicillin/streptomycin. NGP (DSMZ) and NBL-S (DSMZ) cells were cultured in RPMI supplemented with 10% FBS and 1% penicillin/streptomycin. All cell lines were cultured at 37 °C in a humidified chamber in the presence of 5% CO2. All cell lines were tested for the absence of Mycoplasma infection on a monthly basis.
Immunoblotting.
Whole-cell lysates for immunoblotting were prepared by pelleting cells from each cell line at 4 °C (300 g) for 5 min. The resulting cell pellets were washed 1× with 5 ml of ice-cold PBS and then resuspended in the indicated cell lysis buffer. Lysates were clarified at 14,000 r.p.m. for 15 min at 4 °C before quantification by bicinchoninic acid (BCA) assay (Pierce, catalog no. 23225). Whole-cell lysates were loaded into Bolt 4–12% Bis-Tris Gels (Thermo Fisher, catalog no. NW04120BOX) and separated by electrophoreses at 95 V for 1.5 h. The gels were transferred to a nitrocellulose membrane using the iBlot Gel Transfer at P3 for 6 min (Thermo Fisher, catalog no. IB23001) and then blocked for 1 h at room temperature in Odyssey blocking buffer (LICOR Biosciences, catalog no. 927–50010). Membranes were probed overnight using antibodies against the relevant proteins at 4 °C in 20% Odyssey Blocking Buffer in 1× Tris buffered saline with Tween (TBST). Membranes were then washed three times with 1× TBST (at least 5 min per wash), followed by incubation with either IRDye goat anti-mouse (LICOR, catalog no. 926–32210) or goat anti-rabbit (LICOR, catalog no. 926–32211) secondary antibody (diluted 1:10,000) in 20% Odyssey Blocking Buffer/1× TBST for 1 h at room temperature. After three washes with 1× TBST (at least 5 min per wash), immunoblots were visualized using the ODYSSEY Infrared Imaging System (LICOR). Antibodies used against various proteins were as follows: Pin1 (1:1,000, Cell Signaling, catalog no. 3722), α-tubulin (1:1,000, Cell Signaling, catalog no. 3873), IRAK1 (Cell Signaling, catalog no. 4504), IRAK1 pT209 (Assay Biotech, catalog no. A1074), NFκB p65 (1:1,000, Cell Signaling, catalog no. 6956), c-Myc (1:1,000, Cell Signaling, catalog no. 5605) and ß-catenin (1:1,000, Cell Signaling, catalog no. 8480). For apoptosis experiments (Extended Data Fig. 4 and Supplementary Fig. 13) the cells were harvested, washed 1× with ice-cold PBS and lysed using RIPA buffer (Sigma). Lysates were clarified at 14,000 r.p.m. for 15 min at 4 °C, and protein concentration was determined using the BCA protein assay (Thermo Fisher Scientific). An aliquot of 50 μg was then loaded on a 4–20% Bis-Tris gel (SurePAGE, GeneScript) and proteins were separated by electrophoresis at 140 V followed by transfer to a nitrocellulose membrane (Bio-Rad) using the Trans-Blot Turbo system (Bio-Rad). The membrane was blocked using 5% BSA in TBST (w/v) for 1 h at room temperature, washed 3× for 5 min with TBST and incubated with the following primary antibodies: cleaved caspase 3 (Cell Signaling, catalog no. 9661, 1:500, overnight at 4 °C), Pin1 (Cell Signaling, catalog no. 3722, 1:1,000, overnight at 4 °C) and β-actin (Cell Signaling, catalog no. 3700, 1:1,000, 1 h at room temperature). The membrane was washed three times for 5 min with TBST and incubated with the corresponding horseradish peroxidase (HRP)-linked secondary antibody (Cell Signaling, mouse no. 7076/rabbit no. 7074) for 1 h at room temperature. The EZ-ECL Kit (Biological Industries) was used to detect HRP activity. The membrane was stripped using Restore stripping buffer (Thermo Fisher Scientific) after each primary antibody before blotting with the next one.
Apoptosis cytometry analysis.
PATU-8988T cells were seeded on a 10-cm2 plate and treated for 6 days with either DMSO (0.1%), Sulfopin (1.0 or 2.5 μM) or Sulfopin-AcA (2.5 μM). Cell medium was replaced with fresh medium after 3 days of treatment. Starutosporin (1 μM, 4 h) was used as a positive control. Cells were trypsinized, washed with ice-cold PBS and then stained using the FITC Annexin V Apoptosis Detection Kit with 7-AAD (BioLegend, catalog no. 640922) according to the manufacturer’s instructions, followed by FACS analysis using the LSRII instrument. FlowJo software was used for analysis of results. Live cells were defined as Annexin V−/7-AAD−, early apoptosis as Annexin V+/7-AAD– and late apoptosis as Annexin V+/7-AAD+.
Cell cycle analysis.
PATU-8988T cells were seeded on a 10-cm2 plate and treated for 4 days with either DMSO (0.1%), Sulfopin (2.5 μM) or Sulfopin-AcA (2.5 μM). Cell medium was replaced with fresh medium after 3 days of treatment. Cells were incubated with 10 μM BrdU for 1 h followed by trypsinization, washing with ice-cold PBS and fixation with 70% ethanol; they were then centrifuged at 300g for 3 min and the supernatant was removed. Cells were resuspended in 2 M HCl and 0.5% Triton X-100 in PBS, incubated for 30 min at room temperature, centrifuged at 600g for 3 min and the supernatant was removed. Cells were then resuspended in 0.1 M Na2B4O7 pH 8.5, centrifuged and the supernatant was again removed followed by staining with anti-BrdU-FITC antibody (Invitrogen, catalog no. 11-5071-42) overnight at 4 °C, with protection from light. Cells were washed once with ice-cold PBS, resuspended in PBS containing 50 μg ml−1 propidium iodide and 50 μg ml−1 RNase A (Sigma) and analyzed by FACS using the LSRII instrument. FlowJo software was used for analysis of the results.
Lysate pulldown with Sulfopin-DTB.
The indicated cells were lysed in 50 mM Hepes pH 7.4, 1 mM EDTA, 10% glycerol, 1 mM TCEP, 150 mM NaCl, 0.5% NP-40 and a protease inhibitor tablet (Roche, catalog no. 4693159001). After clarification (14,000 r.p.m. for 15 min), lysates were incubated with the indicated concentrations of Sulfopin-DTB for 1 h at 4 °C, using 500 μg of lysate per sample. Lysates were then incubated with streptavidin agarose resin (30 μl of 1:1 beads/lysis buffer slurry) (Thermo Scientific, catalog no. 20349) for 1.5 h at 4 °C. Beads were washed four times with 500 μl of buffer (50 mM Hepes pH 7.5, 10 mM NaCl, 1 mM EDTA, 10% glycerol) then pelleted by centrifugation and dried. Beads were boiled for 5 min at 95 °C in 30 μl of 2× lithium dodecyl sulfate (LDS) + 5% β-mercaptoethanol. Lysates were probed for specified proteins by immunoblotting using the Bolt system (Life Technologies).
Cellular target engagement and competition with Sulfopin-DTB.
The indicated cell lines were plated on 10-cm plates (2.5 million cells per plate) in 6 ml of medium. The day after plating, cells were treated with DMSO or the indicated concentrations of candidate inhibitor for the indicated time points. Cells were then washed two times with cold PBS (1 ml per 10-cm plate) and collected by scraping with a cell scraper. Cells were lysed in 50 mM Hepes pH 7.4, 1 mM EDTA, 10% glycerol, 1 mM TCEP, 150 mM NaCl, 0.5% NP-40 and a protease inhibitor tablet (Roche) using 210 μl of cell lysis buffer per 10-cm plate of cells. After clarification (14,000 r.p.m. for 15 min), 9 μl of each lysate sample was combined with 4× LDS + 10% β-mercaptoethanol (ratio 3:1), boiled for 5 min and set aside for input loading control (later to be loaded directly on the gel). Then, 200 μl of each lysate sample was incubated with 1 μM Sulfopin-DTB for 1 h at 4 °C and processed as in Lysate pulldown with Sulfopin-DTB.
Radiosensitization studies.
AlamarBlue-based cell viability assays were performed as previously described17. Briefly, HeLa cells were seeded at a density of 200 per well in a 96-well plate. After 16 h, cells were treated with Sulfopin and exposed to 7.5-Gy irradiation using an X-RAD 320 PRECISION X-RAY irradiator 1 h after drug treatment. At 3 days postirradiation, cells were incubated with AlamarBlue (Thermo Fisher) at a final concentration of 10%. At 4 days postirradiation, absorbance was measured at a wavelength of 570 nm with a 600-nm reference wavelength. Relative fluorescence was calculated using cell-free wells as a control reference, and percentage survival was calculated by comparison with DMSO-treated, nonirradiated controls. Sulfopin efficacy was assessed at 24 h postirradiation by immunoblot using antibodies anti-IRAK1 (Cell Signaling, catalog no. 4504) and anti-IRAK1 pT209 (Assay Biotech, catalog no. A1074).
In vivo GC evaluation.
Wild-type mice (C57BL/6) were provided by Harlan, Israel. All experiments with mice were approved by the Weizmann Institute IACUC committee. Mice were immunized by injection of OVA (25 μl) into the hind footpad, coupled to the hapten NP-OVA precipitated in alum (Imject Alum, Thermo Scientific). Single-cell suspensions were obtained by forcing popliteal lymph node through a 70-μm mesh into ice-cold FACS buffer (EDTA 1 mM and 2% serum in PBS). Cells were incubated with 2 μg ml−1 anti-16/32 (clone 93) for blockage of fragment crystallizable receptors, for 5–10 min. Cell suspensions were washed and incubated with fluorescently labeled antibodies (B220 V500, FAS FITC, CD38 Alexa fluor 700; Biolegend) for 20–40 min. Germinal center cells were gated as live/single, B220+ CD38Lo FASHi. Cell suspensions were analyzed using a Cytoflex (Beckman) flow cytometer.
Cell viability assay: growth over time in 2D-adherent monolayer cell culture.
The indicated cell lines were plated at a density of 500 per well (except for PATU-8988T cells, which were plated at 100 cells per well to avoid overconfluence by day 8) in 100 μl of medium in a 96-well, white, clear-bottom plate (Corning, catalog no. 3903), with one plate per time point (days 0, 2, 4, 6, 8). Cells were treated the day after plating with 1 μl of either DMSO, Sulfopin or Sulfopin-AcA to give the indicated concentrations, and were then incubated at 37 °C in 5% CO2. Every 48 h the medium was aspirated and replaced with fresh medium containing fresh compound or DMSO. When the indicated time points had been reached, cell viability was evaluated using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, catalog no. G7570) according to the manufacturer’s standards, measuring luminescence using an Envision plate reader. The day 0 time point plates were read the day after plating, before compound treatment. To normalize for any differences in plating density between cell lines, final absorbance values were divided by the average day 0 reading; n = 3 biological replicates were used for each treatment condition.
Cell viability assay: 5-day treatment.
PATU-8988T cells were plated in flat-bottom, 96-well plates (Corning, catalog no. 3903) at a density of 1,000 per well in 100 μl of medium, and were treated the next day with 1 μl of the indicated compounds in a threefold dilution series. Cells were incubated at 37 °C in 5% CO2 for 5 days. Antiproliferative effects were assessed using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, catalog no. G7570) according to the manufacturer’s standards, measuring luminescence using an Envision plate reader; n = 3 biological replicates were used for each treatment condition.
Assessment of antiproliferative activity in PATU-8988T 3D cell culture.
PATU-8988T (WT or Pin1−/−) Matrigel suspensions were prepared by resuspension of cells in 100% cold Matrigel (kept on ice). One dome was plated per well in a 24-well plate (Greiner CELLSTAR), with 50 μl of cells per Matrigel suspension per dome and 1,000 cells per dome. After plating the domes, the 24-well plate was placed on top of a T175 (previously filled with autoclaved water) in an incubator at 37 °C and 5% CO2, and left for 15 min for the domes to solidify. Keeping the 24-well plate on the T175 filled with water, the plate was then transferred to a tissue culture hood and 500 μl of cold DMEM (+10% FBS/1% penicillin/streptomycin) was added to each well. The plate was then returned to the incubator. The next day, either DMSO, Sulfopin (1 μM) or Sulfopin-AcA (1 μM) was added to the corresponding wells. Every 3 days the medium was carefully aspirated off (so as not to disrupt the Matrigel domes) and 500 μl of fresh medium/compound was added. After 9 days the medium was aspirated off and 300 μl of 3D CellTiter-Glo (Promega, catalog no. G9681) was added to each well. After shaking the plate for 1 h at room temperature, luminescence was measured using an Envision plate reader; n = 9 biological replicates were used for each treatment condition.
PRISM cancer cell viability screening.
Screening in the PRISM platform (performed as a service by the Broad Institute) followed the procedure described in ref.36. Briefly, Sulfopin was tested in an eight-point dose curve, fourfold dilutions with 20 μM as the top concentration, 5-day treatment, against pools of 24-nucleotide barcoded cancer cell lines, overall totaling 275 cell lines.
RNA-seq.
Mino cells (ATCC) were grown at 37 °C in a 5% CO2 humidified incubator and cultured in RPMI 1640 (Biological Industries), supplemented with 15% FBS (Biological Industries) and 1% penicillin/streptomycin solution (Biological Industries); 11 × 106 cells were incubated with either 1 μM Sulfopin (0.02% DMSO) or 0.02% DMSO in triplicate for 6 h. Total RNA was isolated with the RNeasy kit (Qiagen). RNA libraries were prepared from 2 μg of total RNA using the SENSE mRNA-Seq library prep kit v.2 (Lexogen). Total RNA and library quality were analyzed using Qubit fluorometric and TapeStation analysis (Agilent). Samples were sequenced using the NextSeq 500/550 High Output Kit v.2.5 (illumina) on NextSeq550.
RNA-seq reads were aligned to the human genome (hg19 assembly) using STAR56, and gene expression was determined using RSEM and RefSeq annotations. Differential expression was computed using DESeq2 (ref.57) with default parameters. Genes with baseMean (average normalized number of reads) >50 and downregulated with P < 0.05 were further analyzed using Enrichr41.
Profiling of Sulfopin-DTB reactive cysteines by CITe-Id.
PATU-8988T cells were cultured in DMEM supplemented with 10% fetal calf serum. DMSO (control) or Sulfopin was diluted in fresh medium (final DMSO concentration, <0.1%; final Sulfopin concentration, 100 nM, 500 nM and 1 μM) and added to subconfluent cultures. After 5 h of incubation at 37 °C, cells were harvested using a cell scraper and centrifuged at 300g for 3 min at 4 °C. Cell pellets were washed with ice-cold PBS and centrifuged again. A total of three washes were performed before freezing of cell pellets at −80 °C. This procedure was performed twice on cells independently cultured 1 week apart. Frozen pellets were then processed essentially as described33, except that precleared lysates were treated with 2 μM Sulfopin-DTB overnight at 4 °C. Protein desalting, digestion, enrichment of desthiobiotin-modified peptides, iTRAQ stable isotope labeling, peptide cleanup and multidimensional LC–MS/MS analysis were then performed exactly as previously described33. Data processing and a database search were performed as previously described33, except that spectral processing accounted for Sulfopin-DTB-specific fragment ions and Sulfopin-DTB labeling of cysteine was considered as a variable modification by Mascot (v.2.6.2). Inhibitor concentrations and ratios were used to generate a trendline for each labeled site, with the slope corresponding to the competitive dose response for each modified cysteine site. Similar experiments were performed, as for cells treated as above, for 12 or 24 h with DMSO or Sulfopin, except that peptides were labeled with TMT 10-plex reagents and analyzed on an Orbitrap Lumos mass spectrometer (Thermo Fisher Scientific). The mass spectrometer was operated in data-dependent mode such that the ten most abundant ions in each MS scan (resolution, 200 K; target, 5 × 10−6) were subjected to MS/MS (higher-energy collisional dissociation, image current detection collision energy 40%, resolution 30 K).
Profiling of Sulfopin-reactive cysteines by rdTOP-ABPP.
MDA-MB-231 cells were cultured at 37 °C under a 5% CO2 atmosphere in DMEM culture medium supplemented with 10% FBS and 1% PS (penicillin–streptomycin). Cells were grown to 70% confluence and incubated with either DMSO or 5 μM Sulfopin for 2 h in serum-free medium. Cells were harvested, lysed by sonication in ice-cold PBS containing 0.1% Triton X-100 and centrifuged at 100,000g for 30 min to remove cell debris. Protein concentrations were then determined by BCA protein assay. Proteomes were normalized to 2 mg ml−1 in 1 ml for each sample.
Each DMSO- and Sulfopin-incubated proteome was treated with 100 μM iodoacetamide-alkyne for 1 h at room temperature. Proteomes were then reacted with 1 mM CuSO4, 100 μM TBTA ligand, 100 μM biotin-acid-N3 tag and 1 mM TCEP for 1 h. After click reaction, proteomes were centrifuged at 8,000g for 5 min and precipitated proteins were washed two times with cold methanol. Proteomes were resuspended in 1.2% SDS/PBS and diluted to 0.2% SDS/PBS. Finally, the samples were prepared, analyzed on LC–MS/MS and quantified according to the published rdTOP-ABPP protocol34. Briefly, beads from trypsin digestion were washed and resuspended in 100 μl of TEAB buffer; 8 μl of 4% D13CDO or HCHO was added to the Sulfopin or DMSO sample, respectively. At the same time, 8 μl of 0.6 M NaBH3CN was added and the reaction was allowed to proceed for 2 h at room temperature. The beads were washed again and modified peptides were cleaved by 2% formic acid. LC–MS/MS data were analyzed by ProLuCID58 with static modification of cysteine (+57.0215 Da) and variable oxidation of methionine (+15.9949 Da). The isotopic modifications (+28.0313 and +34.0631 Da for light and heavy labeling, respectively) were set as static modifications on the N terminus of a peptide and lysines. Variable modification on cysteines was set at +322.23688 Da. The ratios were quantified by CIMAGE software.
Myc luciferase reporter assay.
HEK293 cells were maintained in DMEM supplemented with 10% standard FBS, 2.5 mM l-glutamine, nonessential amino acids and 1× penicillin/streptomycin. Cells were passaged to 80% confluence in six-well plates and transfected with 4× E-box-Luc and Pin1-Flag plasmids, as indicated, and β-galactosidase as an internal control12 using Lipofectamine 3000 (Invitrogen) following the manufacturer’s protocol. Sulfopin treatments were performed at the time of transfection and cells were harvested 48 h later.
Cell were washed with PBS and then lysed in 1× cell lysis buffer (Promega). Lysates were sonicated for ten pulses at an output of 1 and 10% duty (Branson), and incubated on ice for 20 min. Lysates were then cleared by centrifugation at 14,000 r.p.m. for 10 min at 4 °C. Luciferase activity was measured using the Promega Luciferase Assay Kit and Berthold luminometer (Bundoora) and normalized to β-galactosidase activity59.
Zebrafish neuroblastoma models.
All zebrafish studies and animal maintenance were performed in accordance with Dana-Farber Cancer Institute IACUC-approved protocol no. 02–107. For in vivo drug treatment, 3-day-old zebrafish embryos were placed in 48-well plates with five embryos per well and treated with either DMSO control or Sulfopin in standard egg water.
For neuroblastoma transplantation, zebrafish neuroblastoma cells were harvested by dicing 4-month-old Tg(dβh:MYCN;dβh:EGFP) transgenic zebrafish in PBS. The cell suspension was filtered with a Falcon 40-μm cell strainer (Corning) and loaded into thin-walled, borosilicate glass capillary needles (1.0 mm outer diameter, 0.75 mm inner diameter; World Precision Instruments). The recipients, 2-day-old Casper zebrafish embryos, were manually dechorionized and anaesthetized with 0.003% tricaine (Sigma) before positioning on a 10-cm Petri dish coated with 1% agarose. Intravenous tumor transplantation was performed as described43, with ~200–400 cells injected into the duct of Cuvier of each recipient. One day later, 3-day-old recipients were randomly divided into 48-well plates and treated with either DMSO control or Sulfopin in standard egg water for 5 days, with drug refreshment on the second day of treatment.
A Nikon SMZ1500 microscope equipped with a Nikon digital-sight DS-U1 camera was used for capture of both bright-field and fluorescent images from live zebrafish and embryos. For PSNS and neuroblastoma quantification, all animals in the same experiments were imaged under the same conditions and the acquired fluorescent images were quantified using ImageJ software by measuring the area of eGFP fluorescent tumor mass.
Mouse studies (pharmacokinetics/pharmacodynamics/toxicity).
Pharmacokinetic data were obtained as a fee-for-service from Scripps Florida. For the toxicity study, 18 mice were used for the following arms: three control mice injected with vehicle, three with 10 mg kg−1 Sulfopin injected once daily, three with 20 mg kg−1 Sulfopin injected once daily, three with 20 mg kg−1 Sulfopin injected once every other day, three with 40 mg kg−1 Sulfopin injected once daily and three with 40 mg kg−1 Sulfopin injected once every other day. The formulation was 5% N-methylpyrrolidone, 5% solutol and 20% DMSO). Mice were treated for 14 days (Supplementary Dataset 2).
The pharmacodynamics study was performed at the Dana-Farber Cancer Institute, and the procedure was approved by the IACUC under protocol no. 16–015. Mice were treated for three total doses spanning two consecutive days with either vehicle or Sulfopin (10 or 20 mg kg−1) by oral gavage, after which organs were harvested 4 h after the final dose. The spleen of each mouse was ground and lysed in 300 μl of 50 mM Hepes pH 7.4, 1 mM EDTA, 10% glycerol, 1 mM TCEP, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40 and a protease inhibitor tablet (Roche). After clarification (14,000 r.p.m. for 15 min), lysates were normalized by BCA and diluted to a final concentration of 2.5 μg μl−1. Next, 200 μl of 2.5 μg μl−1 of each spleen sample was then incubated with 1 μM Sulfopin-DTB for 1 h at 4 °C and processed (Lysate pulldown with Sulfopin-DTB). To prepare input samples, each original lysate sample (before pulldown) was combined with 4× LDS + 10% β-mercaptoethanol (at a ratio of 3:1), boiled for 5 min and 25 μg of each sample was then loaded on the gel.
Murine neuroblastoma models.
All experiments were approved by The Institute of Cancer Research Animal Welfare and Ethical Review Body, and performed in accordance with the UK Home Office Animals (Scientific Procedures) Act 1986, the United Kingdom National Cancer Research Institute Guidelines for the Welfare of Animals in Cancer Research and the guidelines of Animal Research: Reporting In Vivo Experiments.
Transgenic Th-MYCN mice were genotyped to detect the presence of the human MYCN transgene60. The study was performed using both male and female hemizygous mice that developed palpable tumors at 50–130 days with 25% penetrance. Transgenic Th-MYCN mice were genotyped to detect the presence of the human MYCN transgene. Tumor development was monitored weekly by palpation by an experienced animal technician. Mice with palpable tumors of size ≥4–5 mm were then enrolled into three groups. Group 1: all animals received Sulfopin at 40 mg kg−1, once per day by oral gavage. Group 2: all animals received Sulfopin at 40 mg kg−1, twice per day by oral gavage. Group 3: all animals received vehicle (5% NMP, 5% kolliphor, 20% DMSO) once per day by oral gavage. Changes in tumor volume in TH-MYCN mice were quantified using MRI on a 1 Tesla M3 small animal MRI scanner (Aspect Imaging). Mice were anaesthetized with isoflurane delivered via oxygen, and their core temperature was maintained at 37 °C. Anatomical T2-weighted coronal images were acquired through the mouse abdomen, from which tumor volumes were determined using segmentation from regions of interest drawn on each tumor-containing slice using a Horos medical image viewer. Mice were housed in specific-pathogen-free rooms in autoclaved, aseptic microisolator cages (maximum of four mice per cage). Mice were allowed access to sterile food and water ad libitum.
Extended Data
Extended Data Fig. 1 |. Chemoproteomic approaches to establish Sulfopin’s selectivity.
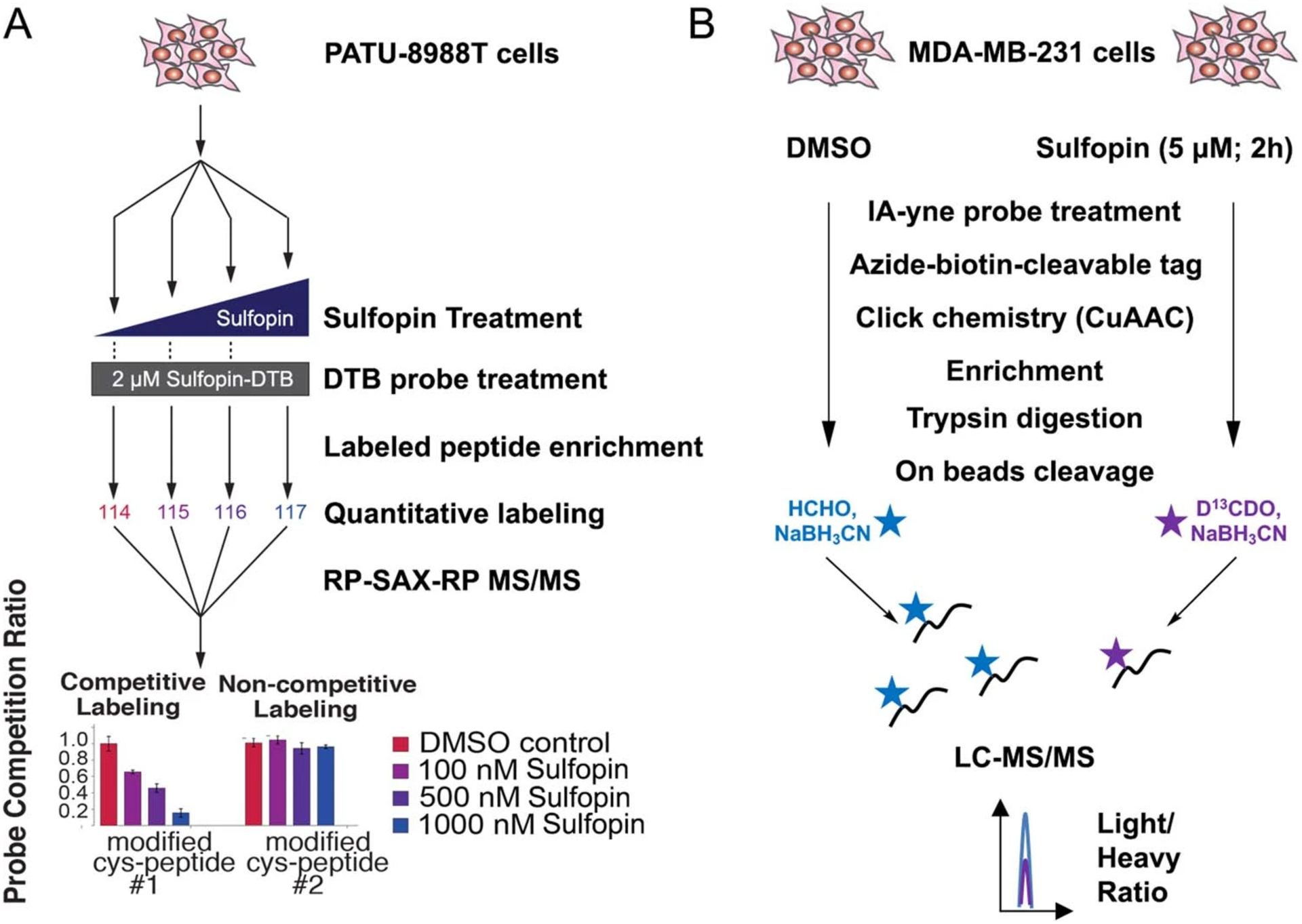
a, Schematic depiction of Covalent Inhibitor Target-Site Identification (CITe-Id) workflow, showing hypothetical results. CiTe-Id identifies Sulfopin-DTB modified sites across the proteome, and profiles competitively labeled cysteine residues following dose-response treatment with Sulfopin in live PATU-8988T cells. b, Schematic depicting the rdTOP-ABPP experimental workflow to assess Sulfopin proteomic selectivity in MDA-MB-231 cells.
Extended Data Fig. 2 |. Sulfopin phenocopies Pin1 knockout phenotypes.
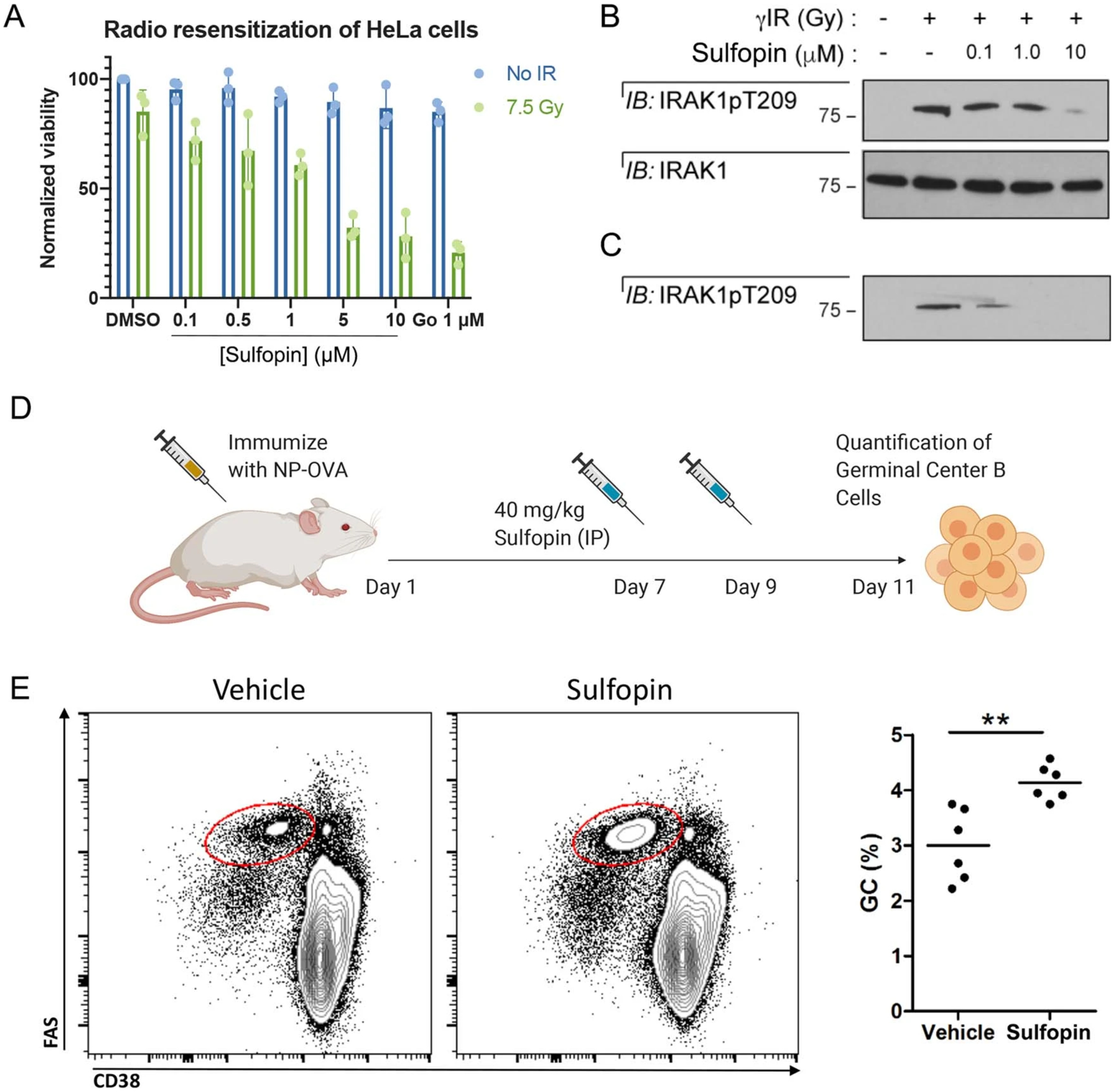
a, HeLa cells were treated with either DMSO, Sulfopin, or Go6976 (a Chk1 inhibitor) and exposed to 7.5 Gy IR 1 h after drug treatment. Viability was assessed 3 days post-IR. Sulfopin shows a dose dependent sensitization of the cells to irradiation (n=3; data are represented as mean values with standard deviation). b, Western blot analysis was performed 24 h post-IR, showing Sulfopin blocked phosphorylation of Thr209 of IRAK1. c, A shorter exposure shows that Sulfopin inhibits IRAK1 phosphorylation already at concentrations of 0.1 μM. d, A scheme for testing the effect of Sulfopin in vivo on germinal center B cells in response to immunization. e, Representative flow cytometric plots with Vehicle and Sulfopin (left) and quantification (right) of FASHi CD38− germinal center (GC) cells in WT mice 11 days after immunization with NP-OVA. ** p<0.01, two tailed Student’s t test.
Extended Data Fig. 3 |. Sulfopin affects PATu-8988T cell cycle.
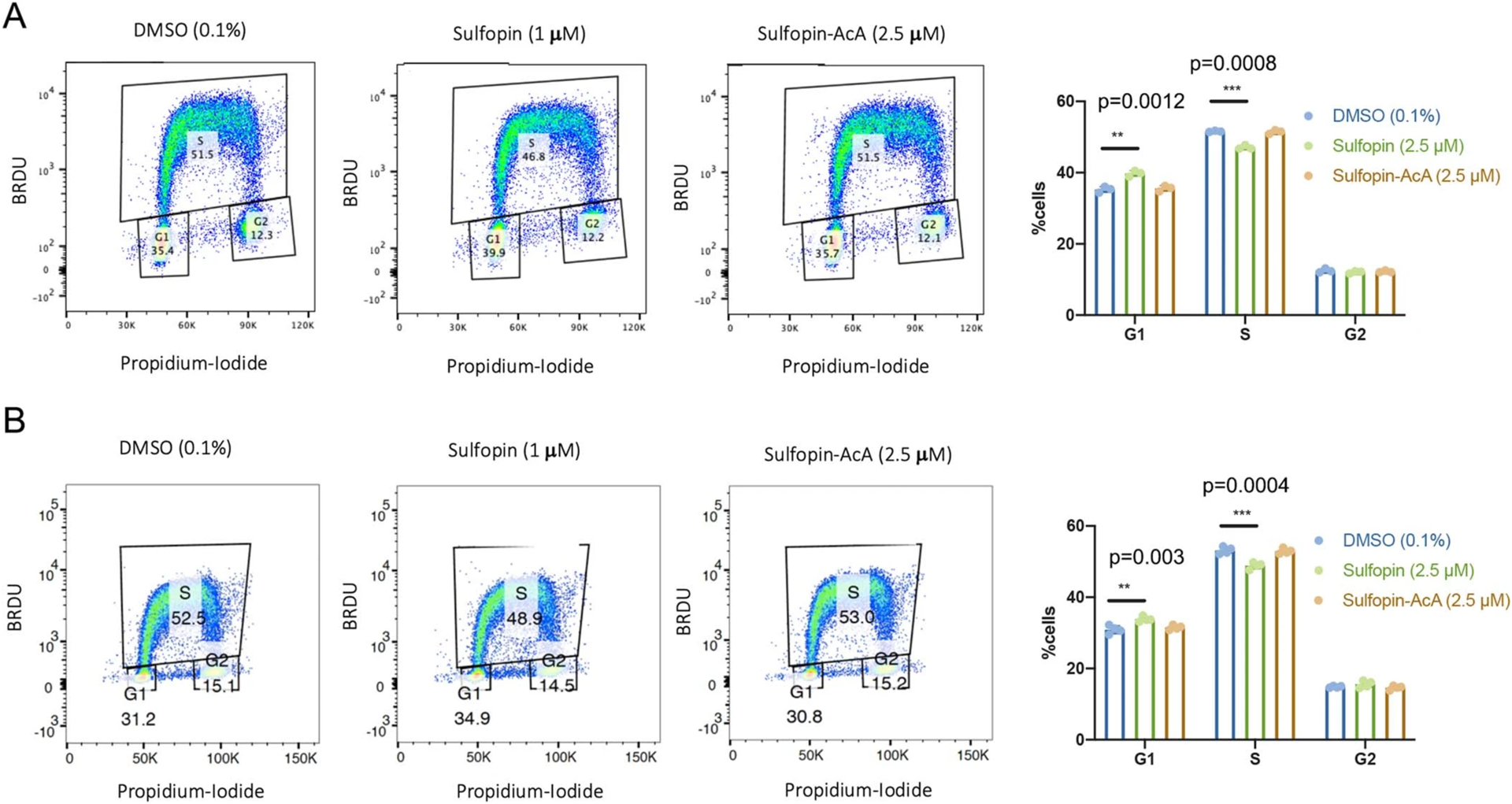
PATU-8988T cells were treated in triplicates or more for 4 days with either DMSO (0.1%), Sulfopin (2.5 M) or the non-covalent control Sulfopin-AcA (2.5 M). Cell cycle analysis was performed by BRDU and Propidium-Iodide staining, followed by FACS analysis. Sulfopin treatment reduces the % of cells in S-phase and in turn more cells are found in G1, while the non-covalent Sulfopin-AcA doesn’t show this effect. Representative FACS analysis graphs and a quantification of the results SD of two independent experiments are presented (A. n=3; B. n=4). Statistical significance was calculated using one-tailed Student’s t test (** = p < 0.01, *** = p < 0.001).
Extended Data Fig. 4 |. Sulfopin treatment does not induce apoptosis in cells.
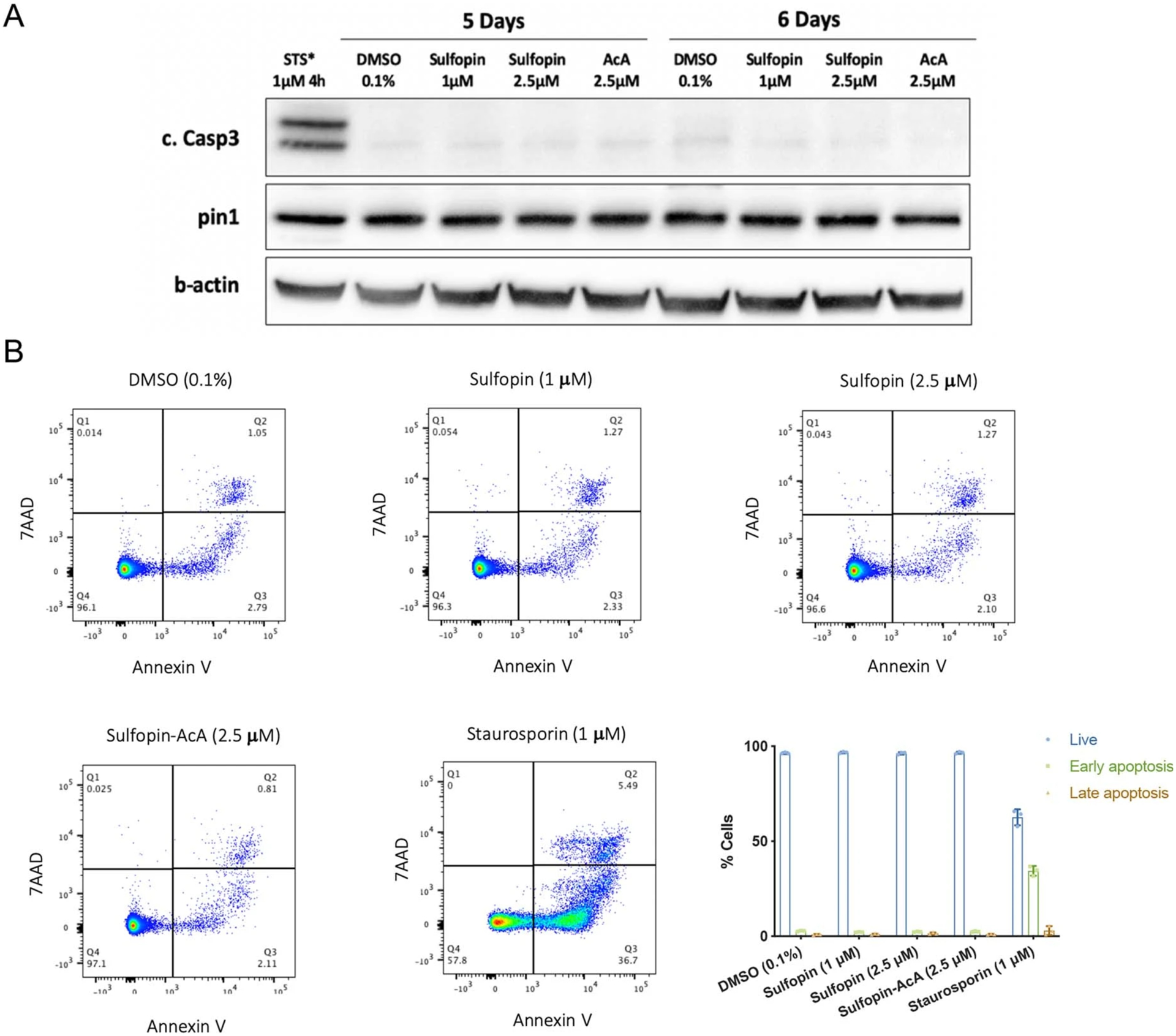
a, PATU-8988T cells were treated for 5 or 6 days with either DMSO (0.1%), Sulfopin (1 μM, 2.5 μM) or the non-covalent control Sulfopin-AcA (2.5 μM). The cells were lysed and activation of caspase 3 and Pin1 levels were analysed by Western blot. As a positive control for caspase 3 activation the cells were treated with Staurosporin (1 μM, 4h; STS). See Supplementary Fig. 13a for the results of an additional independent experiment. Caspase 3 was not activated and Pin1 levels were not changed by the treatment with Sulfopin. b, PATU-8988T cells were treated in triplicates for 6 days with either DMSO (0.1%), Sulfopin (1 M, 2.5 M) or the non-covalent control Sulfopin-AcA (2.5 M). The cells were then stained with AnnexinV-FITC/ 7AAD and analysed by FACS. Staurosporin treatment (1 M, 4h) was used as a positive control for apoptosis. Representative FACS analysis graphs and a quantification of the results (n=3; data are represented as mean values with standard deviation). See Supplementary Fig. 13b for the results of an additional independent experiment. Live cells were defined as AnnexinV−/7AAD−, early apoptosis AnnexinV+/7AAD− and late apoptosis AnnexinV+/7AAD+.
Supplementary Material
Acknowledgements
N.L. is the incumbent of the Alan and Laraine Fischer Career Development Chair.N.L. thanks the Israel Science Foundation for funding (grant no. 2462/19), The Rising Tide Foundation, The Israel Cancer Research Fund, the Israeli Ministry of Science and Technology (grant no. 3-14763) and the Moross integrated cancer center. N.L. is also supported by the Helen and Martin Kimmel Center for Molecular Design, Joel and Mady Dukler Fund for Cancer Research, the Estate of Emile Mimran and Virgin JustGiving and the George Schwartzman Fund. C.D. was supported by the Minerva Fellowship program of the Max Planck Society, funded by the German Federal Ministry for Education and Research. This work was supported in part by NIH grant no. R01CA205153 to K.P.L., N.S.G. and X.Z.Z. N.S.G. was also supported by the Hale Center for Pancreatic Research. Y.C. and C.W. thank the Computing Platform of the Center for Life Science at Peking University for supporting proteomic data analysis. S.D.-P acknowledges funding from the Linde Family Foundation. Part of this research was conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines (no. NIGMS P41 GM103403), and SBGrid compiled software (no. eLife 2013;2:e01456). J.A.M. acknowledges support from the National Institutes of Health (nos. CA233800 and TR002933) and the Mark Foundation for Cancer Research. B.J.P. was supported by the Ruth L. Kirschstein NRSA Individual Predoctoral Fellowship (no. F31 CA225066), the Training Grant in Pharmacological Sciences (no. NIH 5 T32 GM007306), the Training Grant in Chemical Biology (no. NIH 5 T32 GM095450-04) and the Chleck Foundation (also to Z.M.D.). B.N. was supported by an American Cancer Society Postdoctoral Fellowship (no. PF-17-010-01-CDD) and the Katherine L. and Steven C. Pinard Research Fund (also to N.S.G.). L.C. is supported by the Cancer Research UK Program Grant (nos. C34648/A18339 and C34648/A14610). Y.J. is supported by a Children with Cancer UK Research Fellowship (no. 2014/176). R.C.S. acknowledges funding support from NCI R01s (nos. CA196228 and CA186241) and foundation support from The Brenden-Colson Center for Pancreatic Care. We thank I. Ulitski for help with RNA-seq analysis, M. Kostic for critical reading of the manuscript, T.-M. Salame for help with FACS analysis and P. Gehrtz and I. You for helping with compound characterization.
Footnotes
Competing interests
N.S.G. is a Scientific Founder and member of the Scientific Advisory Board (SAB) of C4, Jengu, Inception, Larkspur, Syros, Soltego, Gatekeeper and Petra Pharmaceuticals and has received research funding from Novartis, Astellas, Taiho and Deerfield. N.L. is a member of the SAB of Totus medicines and Monte Rosa Therapeutics and has received research support from Teva and Pfizer. J.A.M. has received support through sponsored research agreements with AstraZeneca and Vertex. J.A.M. serves on the SAB of 908 Devices. C.M.B. is an employee of AstraZeneca. C.D., B.J.P., D.Z., S.H., X.L., K.P.L., X.Z.Z., T.L., N.S.G. and N.L. are inventors on a patent application related to the inhibitors described in this manuscript (no. PCT/IL2020/050043).
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41589-021-00786-7.
Reporting Summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41589-021-00786-7.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41589-021-00786-7.
Data availability
Covalent fragment screen data are provided as an Excel file (Supplementary Dataset 1). CITe-Id and rdTOP proteomics data are provided as an Excel file (Supplementary Dataset 3). RNA-seq differential expression data analysis and Enrichr results are provided as an excel file (Supplementary Datasets 5 and 6) and have been deposited with GEO (GSE166786) All structural data have been deposited in the PDB (PDB code 6VAJ). Source data are provided with this paper.
References
- 1.Pawson T & Scott JD Protein phosphorylation in signaling—50 years and counting. Trends Biochem. Sci 30, 286–290 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Lu KP & Zhou XZ The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol 8, 904–916 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Lam PB et al. Prolyl isomerase Pin1 is highly expressed in Her2-positive breast cancer and regulates erbB2 protein stability. Mol. Cancer 7, 91 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen M, Stukenberg PT, Kirschner MW & Lu KP The essential mitotic peptidyl-prolyl isomerase Pin1 binds and regulates mitosis-specific phosphoproteins. Genes Dev. 12, 706–720 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryo A, Nakamura M, Wulf G, Liou YC & Lu KP Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat. Cell Biol 3, 793–801 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Zhou XZ et al. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol. Cell 6, 873–883 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Bao L et al. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am. J. Pathol 164, 1727–1737 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan X et al. Pin1 expression contributes to lung cancer: prognosis and carcinogenesis. Cancer Biol. Ther 9, 111–119 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Li Q et al. The rs2233678 polymorphism in PIN1 promoter region reduced cancer risk: a meta-analysis. PLoS ONE 8, e68148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y et al. Prolyl isomerase Pin1: a promoter of cancer and a target for therapy. Cell Death Dis. 9, 883 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryo A et al. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell 12, 1413–1426 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Farrell AS et al. Pin1 regulates the dynamics of c-Myc DNA binding to facilitate target gene regulation and oncogenesis. Mol. Cell. Biol 33, 2930–2949 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Girardini JE et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 20, 79–91 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Wulf G, Garg P, Liou Y-C, Iglehart D & Lu KP Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. EMBO J. 23, 3397–3407 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D’Artista L et al. Pin1 is required for sustained B cell proliferation upon oncogenic activation of Myc. Oncotarget 7, 21786–21798 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boldetti A, Guarnaccia V, Rambaldi A & Parrella E Inhibition of the peptidyl-prolyl-isomerase Pin1 enhances the responses of acute myeloid leukemia cells to retinoic acid via stabilization of RARα and PML-RARα. Cancer Res. 69, 1016–1026 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Liu PH et al. An IRAK1-PIN1 signalling axis drives intrinsic tumour resistance to radiation therapy. Nat. Cell Biol 21, 203–213 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rustighi A et al. Prolyl-isomerase Pin1 controls normal and cancer stem cells of the breast. EMBO Mol. Med 6, 99–119 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsherniak A et al. Defining a cancer dependency map. Cell 170, 564–576 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee TH et al. Essential role of Pin1 in the regulation of TRF1 stability and telomere maintenance. Nat. Cell Biol 11, 97–105 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou XZ & Lu KP The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 16, 463–478 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Hennig L et al. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry 37, 5953–5960 (1998). [DOI] [PubMed] [Google Scholar]
- 23.Wei S et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med 21, 457–466 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kozono S et al. Arsenic targets Pin1 and cooperates with retinoic acid to inhibit cancer-driving pathways and tumor-initiating cells. Nat. Commun 9, 3069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campaner E et al. A covalent PIN1 inhibitor selectively targets cancer cells by a dual mechanism of action. Nat. Commun 8, 15772 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore JD & Potter A Pin1 inhibitors: pitfalls, progress and cellular pharmacology. Bioorg. Med. Chem. Lett 23, 4283–4291 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Pinch BJ et al. Identification of a potent and selective covalent Pin1 inhibitor. Nat. Chem. Biol 16, 979–987 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Resnick E et al. Rapid covalent-probe discovery by electrophile-fragment screening. J. Am. Chem. Soc 141, 8951–8968 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.London N et al. Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol 10, 1066–1072 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y et al. Structural basis for high-affinity peptide inhibition of human Pin1. ACS Chem. Biol 2, 320–328 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yaffe MB et al. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science 278, 1957–1960 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Guo C et al. Structure-based design of novel human Pin1 inhibitors (III): optimizing affinity beyond the phosphate recognition pocket. Bioorg. Med. Chem. Lett 24, 4187–4191 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Browne CM et al. A chemoproteomic strategy for direct and proteome-wide covalent inhibitor target-site identification. J. Am. Chem. Soc 141, 191–203 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang F, Gao J, Che J, Jia G & Wang C A dimethyl-labeling-based strategy for site-specifically quantitative chemical proteomics. Anal. Chem 90, 9576–9582 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Phan RT, Saito M, Kitagawa Y, Means AR & Dalla-Favera R Genotoxic stress regulates expression of the proto-oncogene Bcl6 in germinal center B cells. Nat. Immunol 8, 1132–1139 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Yu C et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol 34, 419–423 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fila C, Metz C & van der Sluijs P Juglone inactivates cysteine-rich proteins required for progression through mitosis. J. Biol. Chem 283, 21714–21724 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Pelengaris S, Khan M & Evan G c-MYC: more than just a matter of life and death. Nat. Rev. Cancer 2, 764–776 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Yeh E et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol 6, 308–318 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Su Y et al. Post-translational modification localizes MYC to the nuclear pore basket to regulate a subset of target genes involved in cellular responses to environmental signals. Genes Dev. 32, 1398–1419 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He S et al. Synergy between loss of NF1 and overexpression of MYCN in neuroblastoma is mediated by the GAP-related domain. eLife 5, e14713 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He S et al. Neutrophil-mediated experimental metastasis is enhanced by VEGFR inhibition in a zebrafish xenograft model. J. Pathol 227, 431–445 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chesler L & Weiss WA Genetically engineered murine models – contribution to our understanding of the genetics, molecular pathology and therapeutic targeting of neuroblastoma. Semin. Cancer Biol 21, 245–255 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chao SH, Greenleaf AL & Price DH Juglone, an inhibitor of the peptidyl-prolyl isomerase Pin1, also directly blocks transcription. Nucleic Acids Res. 29, 767–773 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dong L et al. Structure-based design of novel human Pin1 inhibitors (II). Bioorg. Med. Chem. Lett 20, 2210–2214 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Guo C et al. Structure-based design of novel human Pin1 inhibitors (I). Bioorg. Med. Chem. Lett 19, 5613–5616 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Backus KM et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salzberg Y et al. Synaptic protein degradation controls sexually dimorphic circuits through regulation of DCC/UNC-40. Curr. Biol 30, 4128–4141 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Janes MR et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172, 578–589.e17 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Kabsch W XDS. Acta Crystallogr. D Biol. Crystallogr 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCoy AJ et al. Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Auld DS et al. “Assay Interference by Aggregation” in Assay Guidance Manual (eds Sittampalam GS et al.) (Eli Lilly & Co. and the National Center for Advancing Translational Sciences, 2012). [PubMed] [Google Scholar]
- 56.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu T et al. ProLuCID: an improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J. Proteomics 129, 16–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sears R, Ohtani K & Nevins JR Identification of positively and negatively acting elements regulating expression of the E2F2 gene in response to cell growth signals. Mol. Cell. Biol 17, 5227–5235 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG & Bishop JM Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 16, 2985–2995 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Covalent fragment screen data are provided as an Excel file (Supplementary Dataset 1). CITe-Id and rdTOP proteomics data are provided as an Excel file (Supplementary Dataset 3). RNA-seq differential expression data analysis and Enrichr results are provided as an excel file (Supplementary Datasets 5 and 6) and have been deposited with GEO (GSE166786) All structural data have been deposited in the PDB (PDB code 6VAJ). Source data are provided with this paper.