

Summary
Background
Human cytomegalovirus (HCMV) infection has been actively implicated in complex neoplastic processes. Beyond oncomodulation, the molecular mechanisms that might underlie HCMV-induced oncogenesis are being extensively studied. Polycomb repressive complex 2 (PRC2) proteins, in particular enhancer of zeste homolog 2 (EZH2) are associated with cancer progression. Nevertheless, little is known about EZH2 activation in the context of HCMV infection and breast oncogenesis.
Methods
Herein, we identified EZH2 as a downstream target for HCMV-induced Myc upregulation upon acute and chronic infection with high-risk strains using a human mammary epithelial model.
Findings
We detected polyploidy and CMV-transformed HMECs (CTH) cells harboring HCMV and dynamically undergoing the giant cells cycle. Acquisition of embryonic stemness markers positively correlated with EZH2 and Myc expression. EZH2 inhibitors curtail sustained CTH cells’ malignant phenotype. Besides harboring polyploid giant cancer cells (PGCCs), tumorigenic breast biopsies were characterized by an enhanced EZH2 and Myc expression, with a strong positive correlation between EZH2 and Myc expression, and between PGCC count and EZH2/Myc expression in the presence of HCMV. Further, we isolated two HCMV strains from EZH2HighMycHigh basal-like tumors which replicate in MRC5 cells and transform HMECs toward CTH cells after acute infection.
Interpretation
Our data establish a potential link between HCMV-induced Myc activation, the subsequent EZH2 upregulation, and polyploidy induction. These data support the proposed tumorigenesis properties of EZH2/Myc, and allow the isolation of two oncogenic HCMV strains from EZH2HighMycHigh basal breast tumors while identifying EZH2 as a potential therapeutic target in the management of breast cancer, particularly upon HCMV infection.
Funding
This work was supported by grants from the University of Franche-Comté (UFC) (CR3300), the Région Franche-Comté (2021-Y-08292 and 2021-Y-08290) and the Ligue contre le Cancer (CR3304) to Georges Herbein. Zeina Nehme is a recipient of a doctoral scholarship from the municipality of Habbouch. Sandy Haidar Ahmad is recipient of a doctoral scholarship from Lebanese municipality. Ranim El Baba is a recipient of a doctoral scholarship from Hariri foundation for sustainable human development.
Keywords: Cytomegalovirus, Polyploid giant cancer cells, EZH2, Myc, Breast cancer, CTH cells
Research in context.
Evidence before this study
Beside the fact that the majority of the adult population worldwide are human cytomegalovirus (HCMV)-seropositive, HCMV infection and viral products meet the criteria of cancer hallmarks in which HCMV-viral products are being detected in multiple cancers. Most importantly, recent evidence reveals that HCMV gene products are found in more than 90% of tumors and metastases of breast cancers, and their augmented expression can be linked to a more aggressive phenotype. To intervene in the development and implementation of effective diagnostic, preventive, and therapeutic measures, we must imperatively identify HCMV's mechanism of action in breast cancer. Recent data demonstrated that polyploidy giant cancer cells play a critical role in breast cancer pathophysiology. Polyploid giant cancer cells (PGCCs) or tumorigenesis drivers were mostly detected in poorly differentiated and therapy resistant cancers. Previously, we showed that the infection with the two clinical strains HCMV-DB (KT959235) and HCMV-BL (MW980585) induces HMECs transformation into CMV-Transformed HMECs (CTH cells). CTH cells presented a polyploid phenotype, stemness, epithelial-to-mesenchymal plasticity which resulted in fast growing breast tumors in NSG mice.
Added value of this study
Given the oncogenic properties of enhancer of zeste homolog 2 (EZH2) and Myc especially in high-grade breast cancer, in the present study, we intended to assess the link between HCMV, PGCCs, EZH2, and Myc in the context of breast cancer development and progression. Originally, our findings show that high-risk HCMV strains can promote a polyploid phenotype and reveal the existence of a potential link between HCMV infection, Myc/EZH2 upregulation and polyploidy generation in vitro and in basal breast tumors. Altogether, our findings confirm the proposed tumorigenic properties of EZH2 and Myc in the context of HCMV infection.
Implications of all the available evidence
A better understanding of the potential link between HCMV and breast cancer will pave the way for new targeted therapies thus increasing the overall survival of breast cancer patients.
Alt-text: Unlabelled box
Introduction
Human cytomegalovirus (HCMV), or human herpesvirus 5 (HHV-5), is a ubiquitous DNA virus with a mid to high seroprevalence, reaching up to 95% of the adult population worldwide.1 Beyond the paradigm of oncomodulation, HCMV infection and viral products fulfill the criteria of cancer hallmarks updated by Hanahan and Weinberg to rationalize the complexities of neoplastic disease,2,3 with viral proteins and/or nucleic acids being detected in a multitude of cancer types.4 For instance, in the context of breast cancer, HCMV proteins were confined to neoplastic cells in primary tumors, sentinel lymph node5,6 and brain metastases, with shorter overall survival and time to tumor progression in patients having the highest expression of HCMV immediate early (IE) proteins.7 Recently, productive HCMV infection in breast tumor cells has been reported and could indicate that HCMV replication may play a role in breast cancer progression.8 This rationalizes the urgent need to actively dissect the exact etiological role of HCMV in the relevance of breast cancer in order to contribute to the development of novel diagnostic, preventive, or therapeutic strategies.
On the other hand, polyploid giant cancer cells (PGCCs) are recognized as a cancerous cellular subpopulation endowed with an enhanced tumorigenic properties, tumor maintenance and recurrence, as well as therapy resistance, the latter accentuated by tumor heterogeneity.9, 10, 11 Giant cancerous cells were described in a wide range of high-grade cancers, chemoresistant tumors and cancer cell lines, for instance glioma, breast, lung, prostate and colorectal cancer.12 Being greatly dedifferentiated with embryonic stem-like traits, PGCCs are able to replicate through an asymmetric division pattern, resulting in the establishment of a mitotically-competent progeny population.13 Remarkably, a shared characteristic among all malignancies triggered by oncoviruses is the presence of polyploidy, highlighting PGCCs as keystone phenotype in virally-induced tumors.14
Being the enzymatic subunit of polycomb repressive complex 2 (PRC2), enhancer of zeste homolog 2 (EZH2) is a histone-lysine N-methyltransferase responsible of transcriptional silencing.15 EZH2 aberrations are associated with the development and progression of a variety of cancers.16,17 Indeed, high EZH2 levels, notably in triple-negative breast cancer,18 are strongly associated with poor clinical outcome19 and correlate with aggressive pathologic features, including high nuclear grade and proliferative index.18 In parallel, EZH2 was shown to expand the stem cell pool and the breast tumor initiating cells, including luminal progenitor cells, hence enhancing accelerated breast cancer initiation, metastasis and growth.20, 21, 22 Interestingly, EZH2 is identified as a downstream target of Myc oncogene, the latter coordinately regulating EZH2 through transcriptional and post-transcriptional mechanisms during tumor initiation and disease progression.23,24 On the other hand, EZH2 was shown to be recruited to the major immediate early promoter (MIEP) in CD14+ monocytes where HCMV establishes latent infection in vivo.25 Further, EZH2 was demonstrated to be overexpressed in glioblastoma multiforme tissues harboring HCMV.26 Intriguingly, EZH2 is overexpressed in PGCCs,27,28 the latter being also triggered by HCMV infection,29 which points toward a potential link between HCMV, PGCCs, and EZH2.
Previously, we demonstrated that the infection with the clinical strains HCMV-DB (KT959235) and HCMV-BL (MW980585) induces HMECs transformation into CMV-Transformed HMECs (CTH), the latter displaying a polyploid phenotype, stemness, epithelial-to-mesenchymal (EMT) plasticity, and resulting in the appearance of fast growing breast tumors in NSG mice.29,30 Given the stated EZH2 oncogenic functions, we aimed to evaluate the presence of a potential link between the triad of HCMV, PGCCs and EZH2, as well as the potential interrelation with Myc in the context of breast carcinogenesis. Subsequently, in the present study, we assessed the activation of Myc, EZH2 and SUZ12 expression, as well as polyploidy levels following acute infection with low- or high-risk HCMV strains.31 This was complemented by deciphering the morphological and phenotypic characteristics of CTH cells undergoing the giant cell cycle and the potential implication of Myc and PRC2 proteins in both CTH cells and breast cancer biopsies.
Materials and methods
Cell lines and culture
Human primary mammary epithelial cells (HMECs, A10565) and embryonic lung fibroblasts (MRC5, 84002) were purchased from Life Technologies (Carlsbad, CA, USA) and RD-Biotech (Besançon, France), respectively. Human breast cancer cell lines MDA-MB231 and MCF7 were obtained from Institut Hiscia (Arlesheim, Switzerland). MRC5, MDA-MB231 and MCF7 cells were grown in Dulbecco's modified Eagle medium (DMEM) (PAN-Biotech) supplemented with fetal bovine serum (Dutscher) and penicillin/streptomycin (Life Technologies) (10% and 1% final concentration, respectively). HMECs were cultured in HMEC medium (Life Technologies) supplemented with HMEC supplement and bovine pituitary extract (Life Technologies) at 37 °C, 5% CO2 and 95% humidity. CTH cells emerging following chronic infection with HCMV-BL and HCMV-DB isolates were cultured in the same condition as HMECs. Mycoplasma monthly screening was performed to certify mycoplasma free cultures (VenorGem classic mycoplasma detection, Minerva biolabs).
HCMV clinical isolates growth and detection
HCMV-FS, HCMV-KM, HCMV-BL, and HCMV-DB clinical strains were isolated from patients hospitalized at Besançon University Hospital (France). The clinical and biological data for the four hospitalized patients from whom we isolated the HCMV-DB, BL, FS and KM strains were provided (Suppl. Table 1). Viral stocks were prepared and stored at -80°C as detailed previously.29 The presence of ULb’ region was detected in all the four strains as well as in the B544 and B693 strains isolated from the triple negative breast cancer (TNBC) tissues (see below), as measured by the detection of UL128/UL130/UL131 by qPCR (Suppl. Fig. 1). Infection of HMECs or MRC5 (1×106) cells with the clinical isolates was carried out at a multiplicity of infection (MOI) of 1. Briefly, cells were incubated at 37°C for 2 h, after which the inoculum was discarded, and the cellular monolayer was washed thoroughly three times with 1X PBS and covered with fresh medium. To determine viral kinetics, infectious cell-free supernatant was harvested at the indicated time points post-infection, DNA was isolated (EZNA Blood DNA Kit, D3392-02, Omega BIO-TEK) and real-time IE1 quantitative PCR (qPCR) was performed using a Stratagene Mx3005P thermocycler (Agilent), using IE1 primers (Suppl. Table 2). qPCR was performed using KAPA SYBR FAST Master Mix (KAPA BIOSYSTEMS, KK4601) and reactions were activated at 95°C for 10 min, followed by 50 cycles (15 s at 95°C and 1 min at 60°C). Results were collected and analyzed using MxPro qPCR software. Where indicated, HCMV lncRNA4.9 was determined by PCR assay by using DreamTaq DNA polymerase (EP0701, Thermo Scientific). PCR products were resolved by agarose gel electrophoresis and SYBR Green I (Roche) was used for visualization.
Microscopy
CTH cells were monitored using an Olympus optical microscope (Japan) and OPTIKA microscopy digital camera (Opticam, Italy). To study the giant cell cycle, confocal microscopy was used and DAPI (4′, 6′-diamidino-2- phenylindole) staining was performed according to the manufacturer's protocol. To assess the expression of EZH2, SUZ12, Myc, pp65, and HCMV late Ag, uninfected HMECs and CTH cells were washed with 1X PBS, fixed and permeabilized (BD Cytofix/Cytoperm, 554722). Primary antibodies targeting EZH2, SUZ12, Myc, pp65, and HCMV late Ag (Suppl. Table 3) were used in parallel to DAPI staining. After staining, slides were evaluated using 63X oil immersion objective lens with a Carl-Zeiss confocal microscope (Germany). Images were analyzed using ZenBlue Software (Carl-Zeiss Microscopy GmbH).
Western blotting
Western blotting was performed as described previously.30,32 Lysates from uninfected HMECs, CTH cells, MDA-MB231 and MCF7 were used to examine the expression of EZH2 and SUZ12. Lysates from uninfected HMECs and CTH cells were used to determine Myc expression. Antibodies used are listed in supplementary Table 3. Densitometry using ImageJ 1.40 software (National Institutes of Health, Bethesda, MA, USA) was employed to quantify protein levels. β-actin (A5316, St. Louis, MO, USA) was used as control to normalize sample loading.
Flow cytometry analysis
Uninfected HMECs and CTH cells collection, fixation, permeabilization, staining, and incubation were performed as described previously.29 Cytofluorometric analysis was performed using a BD LSRFortessa X-20 (BD Biosciences) flow cytometer. FACSDiva software (BD Biosciences) was used to collect and analyze data. Staining was performed for EZH2, SUZ12, Oct4, SSEA4, Nanog, Tra-1-60, SOX2, Akt, pAkt(Ser473), STAT3, pSTAT3, EpCAM, CD49f, Vimentin, E-Cadherin, CD24 and CD44. Antibodies used are listed in supplementary Table 3. Proliferation of CTH cells left untreated or treated with EZH2 inhibitors was assessed by measuring Ki67 antigen (Ag) expression by intracellular flow cytometry as described previously.33 For cell cycle analysis, uninfected HMECs or HCMV-infected HMECs were washed in 1X PBS, fixed in 70% ethanol and resuspended in 50 µg/ml propidium iodide (P3566, life technologies, Eugene, USA) with 0.1 mg/ml RNase (R4642, Sigma-Aldrich, Saint-Louis, MO, USA), then incubated at 37°C for 30 min as described previously.34
RNA cross-linking immunoprecipitation (RNA CLIP) assay
RNA Cross-linking Immunoprecipitation (RNA CLIP) was performed on CTH cells as previously reported.25
Semi-quantitative reverse-transcriptase polymerase chain reaction (RT-PCR)
Total RNA was extracted using E.Z.N.A.® Total RNA Kit I (Omega Bio-Tech, GA, USA) according to the manufacturer's instructions. SuperScript IV First-Strand Synthesis kit (Invitrogen, Carlsbad, CA, USA) was used for reverse transcription. Semi-quantitative PCR was performed for Oct4, SSEA-4 synthase, Nanog, Tra-1-60, SOX2 and β-globin expression. Primers used are listed in supplementary Table 2. Two μl target cDNA conversion mixture was amplified using DreamTaq DNA polymerase (EP0701, 83 Thermo Scientific). RT-PCR products were electrophoresed on a 2% agarose gel stained with SYBR Green I (Roche).
EZH2 inhibition assay
The EZH2 inhibitors EPZ6438 and GSK343 (Cayman Chemical, Ann Arbor, MI, USA) were dissolved in Dimethyl Sulfoxide (DMSO) (Sigma-Aldrich). CTH cells were left untreated or treated by EZH2 inhibitors at 0.1µM and fresh drugs were added every two days.
Soft agar colony formation and colorimetric MTT assays
CTH cells left untreated or treated with EZH2 inhibitors at 0.1µM and HMECs infected with HCMV-B544 and HCMV-B693 strains were seeded in soft agar (Colorimetric assay, CB135; Cell Biolabs Inc., San Diego, CA) as described previously.35 After 15 days of incubation, colonies quantification was performed using MTT (3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay as described previously30 and soft agar colonies were observed under an Olympus microscope (magnification 200×).
Mammosphere assay
CTH cells untreated or treated with EZH2 inhibitors were cultured in 2 mL of mammosphere media (CCM012, R&D Systems, Minneapolis, MN, USA) supplemented with 2U/ml heparin (2812, Bio-Techne, Minneapolis, MN, USA) and 0.5 µg/ml hydrocortisone (4093, Bio-Techne, Minneapolis, MN, USA), in an ultra-low attachment surface 96-well plate. Mammospheres were observed using an Olympus optical microscope (Japan).
Chromatin immunoprecipitation (ChIP) assay
ChIP was performed using a Chromatin Immunoprecipitation assay kit (Millipore, cat no. 17-295) as previously described.36 Chromatin cross-linking was achieved via a 10-min treatment of nuclear extracts with 1% formaldehyde at 37°C. Cross-linked lysates were sonicated to shear the DNA to an average length of 300 to 1000 base pairs. Following sonication, the lysates were pre-cleared via incubation with a 50% slurry of salmon sperm DNA/Protein A Agarose for 30 min. The pre-cleared supernatants were incubated with the primary antibodies anti-H3K27me3, anti-H3K4me3, and total anti-H3 (1:50 dilution, Suppl. Table 3) overnight and then with salmon sperm DNA/Protein A agarose beads at 4°C for 1 h. Following multiple washes, the DNA-protein complexes were eluted and the DNA was recovered by reversing the cross-linking with NaCl and proteinase K. The DNA was then extracted using the Qiaquick PCR Purification Kit (Qiagen, cat. no 28106) and analyzed by SYBR-Green real-time qPCR. The immunoprecipitated samples, along with the input DNA, were amplified for the HCMV MIEP enhancer (Enh) (Suppl. Table 3).
Genomic analysis of HCMV strains
The analysis of HCMV-DB and BL genomes and their comparison to other HCMV strains including the two laboratory adapted strains AD-169 and Towne in addition to Merlin, Toledo, TR, PH, VR1814, Davis, JP, and TB40/E (clinical strains with low passages in culture) was done using the NCBI nucleotide blast tool (https://blast. ncbi.nlm.nih.gov/BlastAlign.cgi). The phylogenetic analysis was performed as previously reported.37
Breast cancer biopsies
Tumor breast biopsies (luminal tumor biopsies n=10 and basal tumor biopsies n=9) and adjacent healthy breast biopsies (n=8) were provided by the Regional tumor bank (BB0033-00024 Tumorothèque Régionale de Franche-Comté). A written informed consent for participation was obtained from all patients. The study was authorized by the local ethics committees of Besançon University Hospital (Besançon, France) and the French Research Ministry (AC-2015-2496, CNIL n°1173545, NF-S-138 96900 n°F2015). Hematoxylin and eosin slides were used to detect PGCCs presence based on Zhang et al. description, with PGCCs being defined as a cancer cell with a nucleus at least three times larger than that of a diploid cancer cell.38 RNA was extracted from the biopsies using E.Z.N.A.® Total RNA Kit I (Omega Bio-Tech, GA, USA). Reverse transcription was performed using the SuperScript IV First-Strand Synthesis kit (Invitrogen, Carlsbad, CA, USA). Expression of EZH2, Myc, and GAPDH was assessed by real-time qPCR using KAPA SYBR FAST Master mix (KAPA BIOSYSTEMS, KK4601) and specific primers for EZH2, Myc and GAPDH (Suppl. Table 2) according to the manufacturer's protocol. Fold change expression was calculated using the delta-delta Ct method.39
The two strains, B544 and B693, were isolated from patients with TNBC. The treatment, clinical outcome and pathological data for these two TNBC patients were provided (Suppl. Table 4). HCMV strains, B544 and B693, were isolated by mechanical tissue disruption and filtration of the frozen biopsy through a 0.45 µm filter and initially grown on MRC5 cells. The supernatant from MRC5 culture was filtrated through a 0.45 µm filter and used to infect HMECs. The purity of our HCMV cultures was confirmed by ruling out the presence of other viruses (Epstein-Barr virus, human papillomavirus, Kaposi sarcoma herpesvirus and adenovirus).
Statistical analysis
All quantitative results are reported as mean ± SD of independent experiments and analyzed by using statistical software SPSS 23. Statistical analyses were performed using Mann-Whitney U test, and correlation analyses were done using Pearson and Spearman correlation tests. Differences with p-value≤0.05 were considered significant and all exact p-values were provided in each figure. Microsoft Excel was used to construct the plots and histogram data.
Role of funding source
The funders had no role in the study design, data collection, data analyses or interpretation, manuscript writing, or publication decisions.
Reagent validation
Relevant documentation on reagent validation (antibody, cell lines, viruses) has been provided in the Supplemental Data Reagent Validation file.
Results
Myc and EZH2 activation parallels polyploidy induction in HMECs acutely infected with high-risk HCMV
We previously reported the replication of the clinical strain HCMV-DB in HMECs in vitro.30 In the present study, we infected HMECs with three newly isolated HCMV clinical isolates that we previously sorted into low- (HCMV-KM, HCMV-FS) and high- (HCMV-DB, HCMV-BL) risk oncogenic strains,29,31 as well as the already described HCMV-DB strain. All strains productively replicated in HMECs with the peak viral titer detected between day 1 and 8 post-infection (Figure 1a) due to the conservation of an intact ULb’ region29 required for infection of epithelial/endothelial cells.40 As control, productive infection peak level was lower by 1 to 3 logs in HMECs compared to MRC5 fibroblasts (Figure 1b).
Figure 1.

Replication of HCMV clinical strains in HMECs with activation of oncogenic pathways and increased polyploidy. a. Time-course of the viral titer in supernatant of HMECs infected with the strains HCMV-FS, HCMV-KM, HCMV-BL and HCMV-DB, as measured by IE1 qPCR. b. Comparison of peak viral titer in supernatant of HMECs and MRC5 cells infected with the strains HCMV-FS, HCMV-KM, HCMV-BL and HCMV-DB, as measured by IE1 qPCR. c, d, e. Comparison of the expression of (c) Myc, (d) EZH2 and (e) SUZ12, in HMECs infected with the strains HCMV-FS, HCMV-KM, HCMV-BL and HCMV-DB. Histograms represent mean values ± SD of 3 independent experiments. f. Cell population repartition based on DNA content, as marked by PI staining of HMECs infected with the strains HCMV-FS, HCMV-KM, HCMV-BL and HCMV-DB. Cells are classified between <2N, 2-4N and >4N populations. Histograms represent the mean ±SD of 3 independent experiments. g. STAT3 and pSTAT3 expression in HMECs infected with HCMV-FS, HCMV-KM, HCMV-BL, and HCMV-DB strains, as measured by FACS. Histograms represent mean ±SD of 3 independent experiments. p-values were determined by Mann-Whitney U test.
Previously, activation of proto-oncogenes, including Myc was reported as an early event upon HCMV infection.41 We detected higher expression levels of Myc and EZH2 in HMECs infected with high-risk strains compared to low-risk ones (p-value=0.03 and 0.03, respectively; [Mann-Whitney U test]) (Figures 1c and d, Suppl. 2). No distinct pattern of SUZ12 expression variances was noted between low- and high-risk strains (Figures 1e, Suppl. 2). In parallel, we detected a higher percentage of <2N population in HMECs infected with HCMV-FS and HCMV-KM, versus a higher percentage of >4N population in HMECs infected with HCMV-BL and HCMV-DB (Figure 1f), pointing toward a higher polyploidy induction with the high-risk strains.
As we have previously demonstrated the activation of JAK/STAT3 axis in primary human hepatocytes and HepG2 cells infected with HCMV,33 we observed an increase in STAT3 phosphorylation and STAT3 protein in HMECs infected with the four strains at day 1 post-infection (Figure 1g). EZH2 inhibitors and ganciclovir partially blocked the upregulation of P-STAT3 and STAT3 observed in HMECs acutely infected with HCMV, mostly in cultures infected with the high-risk DB strain (Suppl. Figure 3).
CTH cells and giant cells cycling in HMECs chronically infected with high-risk HCMV strains
HMECs cultures were individually infected with the four isolates to monitor long-term cultures.30 As control, uninfected cells were used, the latter being discontinued after 50 days due to cellular senescence, as well as for HMECs infected with the low-risk HCMV-FS and HCMV-KM strains. Notably, CTH cells emerged only with HMECs infected with HCMV-BL and HCMV-DB isolates, with cells being heterogeneous in terms of shape and size (Figure 2). Compared to uninfected cultures that displayed typical epithelial-like morphology (Figure 2a1), we observed a mixture of spheroids and giant cells dispersed among small cells, with morphological heterogeneity raging from mesenchymal, epithelial, and fibroblastic-like structures to small spore-like cells (Figure 2). We were able to cluster the detected shapes into elongated cells, round dark cells, and big flat cells.
Figure 2.

Appearance of CTH and polyploid giant cancer cells following infection of HMECs with HCMV-DB and HCMV-BL strains. a. Presence of neuron-like elongated cells, displaying multiple nuclei, as observed under an inverted light microscope (1-3) and a fluorescence confocal microscope (4) using DAPI staining. Uninfected HMECs (1), CTH-DB (3 and 4) and CTH-BL (2). b. Presence of dense bodied cells, showing morula-like or blastomere-like structures, as observed under an inverted light microscope (1-6) and a fluorescence confocal microscope (7-8) using DAPI staining. CTH-DB (3, 5, 6, 7) and CTH-BL (1, 2, 4, 8). c. Presence of giant cells, showing granular appearance and blastocyst-like shapes and displaying intense polyploidy, as observed under an inverted light microscope (1-6) and a fluorescence confocal microscope (7-8) using DAPI staining. CTH-DB (1, 5) and CTH-BL (2, 3, 4, 6, 7, 8). Inverted light microscope scale bar represents 100µm; Confocal microscope scale bar represents 10µm.
Elongated spindle-like cells displayed neuron-like shapes, with some cells forming coral-like structures, long microvilli, and filopodia (Figure 2a2-3). Upon observation by confocal microscopy, multiple nuclei were detected within bodies and branches of elongated cells, from where small mononucleated daughter cells were budding (Figure 2a4, red arrow). In parallel, densely packed bodies with smooth edges were also detected (Figure 2b1-6). Some cells exhibited morula-like structure (Figure 2b5), while others were morphologically indistinguishable from blastomeres (Figure 2b1-4). Round cells with short microvilli and some small budding spherical bodies were also present (Figure 2b1, red arrows). Figure 2b7, 8 show asymmetric cell division pattern represented by budding of daughter cells from either mononucleated or multinucleated cells. On the other hand, giant cells appeared to be hypertrophic, irregular in shape with small apophyses and granular appearance (Figure 2c). Cells displayed ruffle shape (Figure 2c1) and multiple microvilli (Figure 2c2) with some cells exhibiting a blastocyst-like morphology (Figure 2c3). Intensive polyploidization with nuclei displaying irregular contours was detected, parallel to cells displaying a single giant nucleus (Figure 2c7, 8).
The described morphological heterogenicity confirmed the presence of PGCCs in our culture. As the latter are described to undergo the “giant cell cycle”, we monitored by confocal microscopy the fate of infected cells (Suppl. Figure 4). In response to HCMV infection (red arrow), a subset of cells enters the “giant cell cycle” (1), as evidenced by a nucleus size increase, possibly due to endoreplication. Some cells directly bud from giant mononucleated cells (2), whereas others form elongated ones (3), where DNA migrates horizontally into adjacent cells via the branch of cytoplasm, followed by budding (7). On the other hand, some cells continue endoreplication (4) to generate mononucleated or multinucleated PGCCs (5), followed by depolyploidization, as illustrated by daughter cells budding from multinucleated (6) or mononucleated (7) giant cells. Taken together, our data suggest that infected cells can enter a self-renewal state and grow as PGCCs, followed by depolyploidization, thus fulfilling the steps of the “giant cell cycle”.
CTH cells display upregulated EZH2 and Myc expression, EMT traits and stemness
As the deregulation of PRC2 proteins, particularly EZH2, has been linked to the initiation of tumorigenesis, and that Myc is identified as an EZH2 regulator during tumor initiation and progression23, we assessed the expression of EZH2, SUZ12 and Myc proteins in CTH cells. EZH2 and Myc expression was upregulated in CTH cells compared to uninfected HMECs (p-value=0.03 and 0.02, respectively; [Mann-Whitney U test]), with a slight increase in SUZ12 expression (p-value=0.11; [Mann-Whitney U test]) (Figure 3a). EZH2, SUZ12, and Myc proteins were positively stained in CTH cells as demonstrated by confocal microscopy imaging (Figure 3b). According to CTH cells subpopulation analysis on the basis of their size (FSC) and ploidy (N) (Figure 3a),29 PGCCs were shown to highly express EZH2, SUZ12 and Myc proteins, in contrast to small cells (SC). This upregulation was further confirmed by western blot, with a parallel enhanced expression of EZH2, SUZ12, and Myc in CTH cells (p-value=0.07, 0.07 and 0.02, respectively; [Mann-Whitney U test]) (Figure 3c).
Figure 3.

Myc, EZH2 and SUZ12 activation in CTH cells. a. Myc, EZH2, and SUZ12 expression in CTH cells and subpopulations, as measured by FACS. Histogram represents the mean ±SD of 3 independent experiments. PGCCs: polyploid giant cancer cells; LC: large cells; IC: intermediate cell; SC: small cells. b. Myc, EZH2 and SUZ12 expression in CTH cells as observed by fluorescence confocal microscope. Scale bar represents 10µm. c. Myc, EZH2, and SUZ12 expression in CTH cells as measured by western blot. Protein expression was measured by densitometry using ImageJ software; histogram represents the mean ±SD of 3 independent experiments. p-values were determined by Mann-Whitney U test.
PGCCs and SCs undergoing the giant cell cycle are described to have differential expression of proliferation, epithelial, mesenchymal and stemness markers.27,42 Hence, we assessed the expression of a panel of markers in PGCCs and SCs (Suppl. Figure 5). Compared to uninfected HMECs and SCs, PGCCs are highly proliferative, with high levels of Ki67 Ag, along with a high expression of the mesenchymal marker vimentin and the stemness markers CD49f and CD44, as well CD24, a marker overexpressed in breast cancer (Suppl. Figure 5). EpCAM downregulation was observed in both PGCCs and SCs, compared to uninfected HMECs (Suppl. Figure 5). A slight increase was seen with the epithelial marker E-cadherin in PGCCs, with a remarkable upregulation in SCs (Suppl. Figure 5).
As the acquisition of embryonic-like traits is activated in PGCCs,38,42 we detected the expression of the embryonic markers Oct4 and Nanog in CTH cells (Figure 4a). This was further confirmed by RT-PCR (Figure 4b) and flow cytometric analysis (Figure 4c), where an upregulation of Oct4, SSEA-4, Nanog, Tra-1-60, and SOX2 in CTH cells compared to uninfected HMECs was further demonstrated (p-value=0.03; [Mann-Whitney U test]), with PGCCs expressing the highest level of embryonic stemness compared to SCs (Figures 4d and Suppl. 6). Interestingly, high EZH2 and Myc expression in PGCCs positively correlated with embryonic stemness expression (Figure 4e and f), reinforcing the link between EZH2/Myc upregulation and stemness.
Figure 4.

Increased expression of embryonic markers in CTH cells compared to HMECs. a. Oct4 and Nanog expression in CTH cells and HMECs, observed by fluorescence confocal microscopy. b. Expression of Oct4, SSEA-4 synthase, Nanog, Tra-1-60 and SOX2 at mRNA levels in CTH cells and HMECs. Cellular DNA was used as positive control. Beta-globin was used as housekeeping control gene. RT: reverse transcriptase; NTC: non-treated control. c. Embryonic markers expression in CTH cells and HMECs, as measured by FACS. Histograms represent mean values ±SD of 3 independent experiments. p-values were determined by Mann-Whitney U test. d. EZH2 and embryonic markers expression for CTH cells subpopulations. Histograms represent mean ±SD of 3 independent experiments. PGCCs: polyploid giant cancer cells; LC: large cells; IC: intermediate cell; SC: small cells. e, f. Correlation between (e) EZH2 and (f) Myc expression and embryonic markers expression in subpopulations of CTH cells. p-values were determined by Spearman's correlation test.
EZH2 upregulation regulates proliferation, transformation, stemness, and HCMV replication in CTH cells
Viral presence in CTH cells was confirmed by detecting HCMV pp65 antigen (Figure 5a), which could indicate a potential link between the observed phenotype and HCMV. In addition to pp65 detection, HCMV-DB infected CTH cells were positive for HCMV late antigens (Figure 5a) and IE1 antigen as previously reported.29
Figure 5.

Presence of HCMV in CTH cells and enhanced viral replication upon EZH2 inhibition. a. HCMV pp65 protein and HCMV late Ag expression in CTH-DB cells, observed in fluorescence confocal microscopy. HCMV-infected cells were used as positive control. DAPI was used for nuclear staining. b. RNA4.9 gene presence in CTH-DB cells, as detected by conventional PCR. HCMV-infected HMECs and HCMV-DB genomic DNA were used as positive controls; MRC5 genomic DNA was used as negative control. Histogram represents the ratio of RNA4.9 signal to beta-globin signal as mean ±SD of 3 independent experiments. c. RNA-Immunoprecipitation of lncRNA4.9 using EZH2 antibody. d. Viral titer in supernatant of CTH-DB cells untreated or treated with EZH2 inhibitors GSK343 and EPZ6438, as quantified by IE1 qPCR. Histogram represents mean ±SD of 3 independent experiments. p-values were determined by Mann-Whitney U test.
As long non-coding RNAs (lncRNAs), in particular HCMV lncRNA4.9, interact with components of the polycomb repression complex and shape chromatin structure,25 lncRNA4.9 presence and interaction with EZH2 in CTH cells were confirmed by using conventional PCR and RNA cross-linking immunoprecipitation assay, respectively (Figure 5b and c). An increase in HCMV titer with EZH2 blockage was also detected, compared to untreated CTH cells (p-value=0.03; [Mann-Whitney U test]) (Figure 5d). The viral replication was upregulated in CTH cells treated with EZH2 inhibitors parallel to enhanced H3K4Me3 chromatin mark in the enhancer region of IE gene as measured by chromatin immunoprecipitation (Suppl. Figure 7).
Previously, it has been shown that PRC2 increases breast cancer proliferation and migration, with EZH2 being related to increased tumor cell proliferation43 and expansion of breast tumor-initiating cells while promoting a stem cell-like phenotype. Upon treatment with the EZH2 inhibitors (EPZ6438 and GSK343), EZH2 blockade significantly decreased the proliferation of treated CTH cells compared to untreated cells as assessed by Ki67 Ag expression levels (p-value=0.02; [Mann-Whitney U test]) (Figure 6a). In addition, EPZ6438 and GSK343 treatment significantly diminished CTH cells colony formation in soft agar (p-value=0.03; [Mann-Whitney U test]) (Figure 6b). Mammospheres formation was described to be inhibited upon EZH2 depletion.44 Unexpectedly, treatment with EZH2 inhibitors (EPZ6438 and GSK343) did not inhibit mammospheres formation in CTH cells with larger spheres formed with EZH2 inhibitors treatment (Figure 6c). Finally, EZH2, SUZ12, and Myc were highly expressed in HMECs infected with the supernatant which was harvested from CTH-DB cells as demonstrated by confocal microscopy imaging (Suppl. Figure 8).
Figure 6.

EZH2 inhibitors block CTH cell proliferation, colony formation in soft agar and enhance mammosphere formation. a. Ki-67 Ag expression in CTH untreated and treated with EZH2 inhibitors (GSK343 and EPZ6438), as measured by FACS. Histogram represents mean ±SD of 3 independent experiments. b. Soft-agar assay on CTH cells untreated and treated with EZH2 inhibitors (GSK343 and EPZ6438). Colony formation was assessed by inverted light microscope observation and quantified by MTT assay. Scale bar represents 100µm. Histogram represents mean ±SD of 3 independent experiments. c. Mammosphere formation assay on CTH cells untreated and treated with EZH2 inhibitors (GSK343 and EPZ6438). Mammospheres were observed under an inverted light microscope. Scale bar represents 100µm. p-values were determined by Mann-Whitney U test.
PGCCs, EZH2 and Myc upregulation are detected in breast cancer tissues that are HCMV-positive
Previously, PGCCs number in breast cancer was linked to invasion and metastasis.45 PGCCs with giant or multiple nuclei were present in human breast cancer biopsies, in particular triple negative breast cancer biopsies (Figure 7a). As expected, triple negative biopsies displayed higher tumor grade (p-value<0.001; [Mann-Whitney U test]), tubule formation (p-value=0.02; [Mann-Whitney U test]), nuclear pleomorphism (p-value<0.001; [Mann-Whitney U test]), and mitotic count (p-value=0.0017; [Mann-Whitney U test]) compared to luminal biopsies (Figure 7b). On the other hand, and compared to healthy biopsies (mean=2.4), tumor biopsies displayed an enhanced EZH2 expression (mean=31, p-value=0.02; [Mann-Whitney U test]), notably in the basal ones (mean=35.8) (Figure 7c). In this latter subset, EZH2 expression was prominent in biopsies harboring HCMV (mean=47.2) (Figure 7c), pointing toward a potential correlation between EZH2 upregulation and HCMV presence. Similar results were observed for Myc expression; Myc was highly expressed in HCMV-positive basal tumors (mean=17.9) compared to luminal ones (mean=2.2) (Figure 7d). High expression levels of EZH2 and Myc were measured in HCMV-positive basal biopsies compared to HCMV-negative basal biopsies and to all luminal biopsies independent of HCMV status (Figure 7c and d). This was further confirmed by the presence of a strong positive correlation between EZH2 and Myc expression in tumor biopsies, exclusively in the presence of HCMV (r=0.929, p-value <0.001; [Pearson's correlation test]) and particularly in basal samples (r=0.914, p-value=0.03; [Pearson's correlation test]) (Figure 7e). Further, PGCC count strongly correlated with EZH2 expression only in tumor biopsies harboring HCMV (r=0.750, p-value=0.012; [Pearson's correlation test]), notably in luminal ones (r=0.946, p-value=0.015; [Pearson's correlation test]) (Figure 7f). On the other hand, PGCC count strongly correlated with Myc expression in HCMV-positive tissues (r=0.733, p-value=0.009; [Pearson's correlation test]), remarkably in basal ones (r=0.757, p-value=0.13; [Pearson's correlation test]) (Figure 7g). All in all, two HCMV strains, namely B544 and B693, where detected in biopsies with high expression levels of EZH2 and Myc, and were classified as EZH2HighMycHigh basal breast tumors (Figure 7c and d).
Figure 7.

Detection of PGCCs in breast cancer biopsies and EZH2/Myc expression in healthy, tumor, luminal and basal biopsies. a. Presence of PGCCs in breast tumor biopsies (arrows). Tissue was stained using HES. Magnification: 80X. Scale bars are 25 µm. b. Comparison of histological characteristics of luminal and basal tumor biopsies. p-values were determined by Mann-Whitney U test. c, d. Scattered plots showing (c) EZH2 and (d) Myc expression in individual healthy, luminal, and basal HCMV-positive and negative biopsies. The cut-off for classifying high- and low-risk strains is represented by the horizontal red line. p-values were determined by Mann-Whitney U test. e, f, g. Correlation between the expression EZH2, Myc, and PGCC count in tumor biopsies in the presence or absence of HCMV. Correlation test between (e) EZH2 and Myc expression, (f) EZH2 expression and PGCC count, (g) Myc expression and PGCC count in all tumor, luminal and basal biopsies in the presence or absence of HCMV. *p-value≤0.05; ** p-value ≤0.01; p-values were determined by Pearson's correlation test.
Isolation of two oncogenic HCMV strains from EZH2HighMycHigh basal breast tumors
Two HCMV strains, B544 and B693, were isolated from EZH2HighMycHigh basal breast tumors by tissue disruption and filtration (Suppl. Figure 9a), and subsequently grown in MRC5 cells, showing a peak of viral load at day 20 and 19 post-infection, respectively (Suppl. Figure 9b). We measured the expression of EZH2, SUZ12, and Myc by FACS analysis, and lncRNA4.9 and IE1 transcripts by RT-PCR assay in HMECs acutely infected with B544 and B693 strains at day 1 post infection. We observed the upregulation of EZH2, SUZ12 and Myc expression in infected cells versus uninfected controls (Figure 8a). In parallel, lncRNA4.9 and IE1 transcripts were detected in HMECs acutely infected with the two strains (Figure 8a). Both B544 and B693 isolates of HCMV transformed infected HMECs toward CTH cells (Figure 8b). Thus, in the culture of CTH-B544 and CTH-B693, at day 105 post-infection, we detected large cells with blastomere-like morphology, numerous small round cells along with mesenchymal cells (Figure 8c). The previously described cell features are similar to that of CTH which were observed in HMEC cultures acutely infected with high-risk oncogenic strains DB and BL (Figure 2); these features could denote transformed cells which undergo different stages of the giant cell cycling. In agreement with the transformation of HMECs infected with B544 and B693 strains towards CTH cells, we observed the formation of colonies in soft agar seeded with HMECs infected with B544 and B693 strains (Figure 8d) indicating that they favor an anchorage-independent growth as previously observed for the high-risk DB and BL strains.29 Finally, we observed the upregulation of Myc and EZH2 expression in CTH-B693 and CTH-B544 cells (Figure 8e). Also, we detected lncRNA4.9 transcripts in CTH-B6544 and CTH-B693 cells (Figure 8e). In both CTH-B544 and CTH-B693 cells, we detected the IE1 and late HCMV Ag protein using confocal microscopy (Figure 8f).
Figure 8.
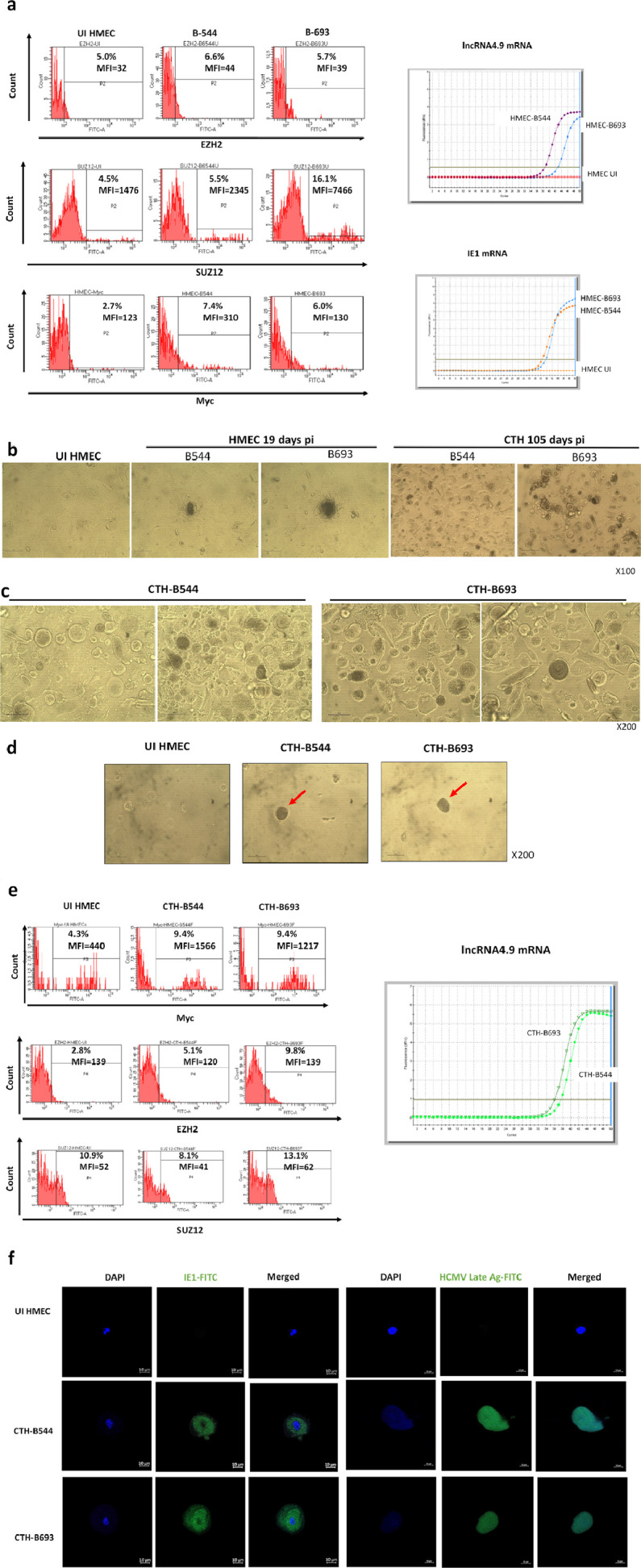
The acute transformation of HMECs infected with two HCMV strains which were isolated from EZH2HighMycHigh basal breast tumors toward CTH cells. a. Left Panel. Upregulation of EZH2, SUZ12 and Myc expression in HMECs infected with HCMV-B544 and HCMV-B693 strains at day 1 post infection versus uninfected controls, as measured by FACS. Right Panel. Detection of lncRNA4.9 and IE1 transcripts in HMECs infected with HCMV-B544 and HCMV-B693 strains at day 1 post infection, as measured by RT-qPCR. b. Time-course of appearance of CTH cells in HMEC cultures acutely infected with B544 and B693 HCMV strains. c. PGCCs structures obtained in CTH cells following the acute infection of HMECs with the two HCMV strains B544 and B693. d. Colony formation in soft agar seeded with HMECs infected with HCMV-B544 and HCMV-B693 strains. After 14 days in soft agar (day 15 post infection), colonies were observed under an Olympus microscope (magnification 200×). Results are representative of three independent experiments. e. Left Panel. Expression of Myc, EZH2 and SUZ12 in CTH-B693 and CTH-B544 cells, as measured by FACS. Right Panel. Detection of the lncRNA4.9 transcript in CTH-B544 and CTH-693 cells, as measured by RT-qPCR. f. HCMV IE1 protein and HCMV late Ag expression in CTH-B544 and CTH-B693 cells observed by fluorescence confocal microscopy. DAPI was used for nuclear staining.
Discussion
We previously reported the classification of HCMV clinical isolates into low- or high-risk transforming strains.29,31 Herein, we show that differential expression of Myc, EZH2 and SUZ12 proteins parallels polyploidy induction upon acute infection with low- or high-risk transforming strains. The activation of Myc/EZH2/PGCCs axis upon chronic infection with high-risk strains was further confirmed in CTH cultures, the latter being morphologically heterogeneous upon undergoing the “giant cell cycle”, with stemness and EMT traits. Further, CTH cells were shown to harbor HCMV, with a direct physical interaction between HCMV lncRNA4.9 and EZH2, the latter being implicated in proliferation, transformation potential, mammospheres formation and HCMV replication. Breast tumor biopsies were found to harbor PGCCs with enhanced EZH2 expression, a strong positive correlation between EZH2 and Myc expression on one hand, and a high correlation between PGCC count and EZH2/Myc expression on the other hand, exclusively in the presence of HCMV. Finally, two HCMV strains were isolated from EZH2HighMycHigh basal breast tumors which acutely transformed infected HMECs toward CTH cells, thereby indicating a direct link between high-risk HCMV strains and basal-like breast cancer.
Accumulated evidence highlight Myc and EZH2 as key players in both oncogenesis and stemness.46, 47, 48, 49 Upon acute infection with high-risk strains, a slight activation of Myc, EZH2 and polyploidy induction was noted (Figure 1). The activation of the described axis was further accentuated in CTH cells with a substantial upregulation of Myc and EZH2 proteins (Figure 3), and the appearance of PGCCs in culture (Figure 2). EZH2 overexpression was described to play a crucial role in breast cancer pathogenesis, where it represses early growth response 1 (EGR1) expression, resulting in the inhibition of EGR1-mediated tumor-suppressive signals.50 Furthermore, EZH2 mediates ribosomal DNA stability via silencing of PHACTR2-AS1, inducing genomic instability and promoting growth and metastasis in breast cancer,51 in line with the reduction in CTH cells proliferation observed upon EZH2 blockage (Figure 6a). Our results are also consistent with the reported GSK343-induced reduction in glioma cells proliferation.52 In this sense, one of the molecular mechanisms reported to trigger EZH2 overexpression is through transcriptional regulation by Myc.23 Indeed, Myc was shown to stimulate EZH2 expression by binding and activating the promoter of the latter,53,54 by alternatively repressing its negative regulator miR-26a,55 or by directly suppressing miR-137, where Myc-miR-137-EZH2 pathway was linked to cisplatin resistance in ovarian cancer.56 Further, bromodomain-4 protein (BRD4) was shown to positively regulate EZH2 transcription through Myc upregulation.57 Intriguingly, Myc activation was widely reported in breast cancer progression, particularly in triple-negative breast cancer and in tumors displaying drug resistant phenotype,11,47,58 similarly to aggressive medulloblastoma tumors where higher Myc levels were associated with increased EZH2 expression.59 On the other hand, by uncoupling DNA replication from mitosis, Myc overexpression can induce DNA replication, thus opening the door toward polyploidy.60 In this context, an intricate relationship between Myc, polyploidy and cancer was demonstrated,61 with Myc being correlated with nuclear pleomorphism in primary and metastatic renal cell carcinomas.62 Interestingly, we previously showed that the high-risk HCMV-BL robustly induced Myc expression, along with low p53 levels,29 which can enhance stem cells replicative potential and reprogramming of progenitors in breast cancer, as Myc was identified as a transcriptional target of p53 in mammary stem cells and is activated upon p53 loss.63 Besides, HCMV-DB strongly increased pRb expression,29 with the pRB-E2F pathway being described to regulate the expression of EZH2.64 Finally, we infected MCF-7 cells with HCMV-DB, BL, and TB40/E strains. We observed the upregulation of Myc expression following infection of MCF7 cells with the high-risk DB and BL strains, but not with TB40E (Suppl. Figure 10). The three strains slightly upregulated EZH2 and SUZ12 in MCF7 cells (Suppl. Figure 10). Previously, following the infection of HMECs with TB40/E strain, neither a significant activation of the molecular oncogenic pathways was observed in acutely infected HMEC nor CTH cells were detected in culture,31 especially when compared to the high-risk HCMV-DB and BL strains. Therefore, TB40/E strain should be classified as a low-risk HCMV strain. This data is consistent with the absence of colony formation in soft agar that had been seeded with HMECs infected with TB40/E strain.31 In addition, the phylogenetic analysis which was done using the whole genomic sequences of several HCMV strains corroborated the similarity of HCMV-DB and BL strains’ genomes; genomes of DB and BL strains were dissimilar to the genome of TB40-E strain (Suppl. Figure 11). This emphasizes the hypothesis that high-risk strains DB and BL differentially induce Myc upregulation, and consequently stimulate EZH2 overexpression as well as polyploidy induction, pointing toward the presence Myc/EZH2/PGCCs axis underlying the described results.
Though, the interrelationship between HCMV and EZH2 is further complexed by the detection of HCMV lncRNA4.9 gene in CTH cells (Figure 5b and c). Indeed, cellular lncRNAs such as HOX antisense intergenic RNA (HOTAIR) were described to interact with the PRC2 complex in breast cancer, and to increase cancer invasiveness and metastasis.65 In our present study, it was shown that HCMV-lncRNA4.9 interacts with EZH2 (Figure 5c), which is in line with Rossetto et al. report describing lncRNA4.9 interaction with EZH2 and SUZ12 proteins, suggesting a role of lncRNA4.9 in mediating gene suppression at the MIEP during latency.25 This is consistent with the increase in viral titer following EZH2 inhibition, possibly due to latency reversal (Figure 5d). In fact, the reactivation of HCMV under EZH2 inhibitors could be explained by at least two distinct mechanisms. First, EZH2 inhibitors could directly block the repressive H3K27 trimethylation in the HCMV promoter/enhancer and favor IE expression followed by viral reactivation.66 Second, the promoter of the MIEP transcriptional repressor, growth factor independence 1 (GFI1), is controlled by the EZH2-NDY1/KDM2B-JARID2 axis, therefore EZH2 inhibitors might result in an enhanced GFI1 expression which could block viral reactivation.36 To discriminate between the respective effects of EZH2 on GFI1/MIEP axis and histone methylation (H3K27, H3K4), we performed a ChIP assay. The repressive H3K27Me3 and activator H3K4Me3 marks on the enhancer of the MIEP were studied. The H3K4Me3 mark was shown to be strongly enhanced compared to a very limited increase in the H3K27Me3 chromatin mark on its enhancer at 24 hrs post treatment of CTH-DB (Suppl. Figure 7), consistent with an active transcriptional promoter. Interestingly, although the promoter of the MIEP transcriptional repressor GFI1 is controlled by the EZH2-NDY1/KDM2B-JARID2 axis and EZH2 inhibitors might therefore result in an enhanced GFI1 expression which could block viral replication in human foreskin fibroblasts acutely infected with HCMV,36 this was not observed in our CTH model. In fact, promoters occupied by GFI1 showed higher levels of the marks associated with active transcription such as H3K4me3.67 In addition, GFI1 associates with the chromodomain helicase DNA binding protein 4 (CHD4); GFI1/CHD4 complexes occupy active or bivalent promoters as well as active enhancers. These sites have a more closed chromatin configuration when CHD4 is present and a more open chromatin conformation when GFI1 is present.67 Thus, in CTH cells treated with EZH2 inhibitors, GFI1 might occupy an active transcriptional promoter MIEP with enhanced H3K4Me3 mark or a bivalent promoter, and thereby could stimulate the expression of the IE gene and viral replication. Further, the role of GFI1 on IE transcription and viral replication could depend on the type of HCMV infection (acutely infected cells versus “chronically-infected” CTH cells), the cell type involved (fibroblasts versus epithelial-derived CTH cells) and the differentiation state and lineage commitment of the infected cells.68 Altogether, the enhanced viral replication observed in CTH cells treated with EZH2 inhibitors could be mostly explained by the enhanced presence of GFI1 on the MIEP parallel to the enhanced H3K4Me3 rather than the decreased H3K27Me3 chromatin marks (Suppl. Figure 7). Finally, GFI1 may have additional non-transcriptional functions and interacts with a number of proteins involved in DNA repair.68 An E2F1-mediated DNA damage response contributes to the replication of HCMV69 making the role of GFI1 even more complex in HCMV replication. The role of GFI1 on HCMV replication definitively needs further studies.
Besides oncogenesis, recent studies have reinforced the importance of EZH2 in maintaining embryonic stemness,70,71 as well as its implication in cancer stem cells.72 This evidence correlates with the previously reported up-regulation of the stemness marker CD44 and mammospheres formation in CTH cells.29 Further, EZH2 was shown to promote breast tumor initiating cells expansion, including mammary stem and luminal progenitor cells,21,22,73 possibly through the activation of NOTCH1 signaling,20 prompting the hypothesis that EZH2 facilitates transformation by blocking differentiation.16 Interestingly, Myc controls the expression of developmental regulators in embryonic stem cells via the upregulation of the PRC2 complex.74 Myc inhibition depletes cancer stem-like cells in a dose-dependent manner in triple-negative breast cancer.75 Indeed, EZH2-mediated stemness could underlie not only the maintenance and expansion of PGCCs expressing high degree of embryonic stemness (Figure 4), but also their appearance in culture, as EZH2 expression in astrocytes induced their dedifferentiation toward stem-like cells expressing nestin, SOX2, and CD133.76 Contrary to the loss and compromised self-renewal of stem cells upon EZH2 deletion,71,77 we did not observe an inhibition of mammospheres formation upon EPZ6438 treatment; larger mammospheres were detected upon EZH2 blockage (Figure 6c). Indeed, this is in line with some reports describing an expansion of the stem and progenitor cell compartments upon EZH2 loss, coupled to JAK2-V617F axis activation.78 As EPZ6438 is a selective EZH2 inhibitor,79 the observed mammospheres expansion could be due to the expression of EZH1, a close homologue of EZH2 that can form an alternative PRC2 complex that partially compensates for the loss of EZH2.80 To note that, it would be possible that EZH2 depletion in luminal breast cancer cells such as MCF7 cells44 inhibits stemness and limits mammospheres formation by blocking the appearance of progenitor luminal cells. In this context, given the role of EZH2 in shifting progenitor luminal cells towards basal-like breast cancer cells, its blockade can result in the maintenance of progenitor cells, ultimately increasing mammospheres formation.81
A complex interrelation exists between Akt, STAT3 and EZH2. Certainly, STAT3 was described to act as a transcriptional factor that induces EZH2 upregulation by binding to the relative promoter region,82 justifying the higher EZH2 expression levels detected in HMECs infected with the high-risk strains compared to the low-risk ones (Figure 1d). In turn, EZH2 overexpression is sufficient for Akt activation, which can mediate breast cancer gene 1 (BRCA1) inhibition, aberrant mitoses with extra centrosomes, and genomic instability83. Alternatively, Akt phosphorylation of EZH2 was also reported, where the phosphorylation of the latter may exert pro-tumorigenic functions in a trimethylation-independent manner, as suggested by Xu et al.84 Phosphorylated EZH2 binds to and methylates STAT3, leading to enhanced STAT3 activity, highlighting the Akt-EZH2-STAT3 axis as a positive regulator of tumor malignancy.85 Interestingly, E2F8, an atypical transcription factor regulated by STAT3 signaling, was shown to be essential for polyploidization in mammalian cells,86 potentially participating in the induction of >4N population observed upon infection with the high-risk strains (Figure 1f). We previously described an increase in Akt activation in HCMV-DB and HCMV-BL infected HMECs which is consistent with the observed increase in EZH2 and STAT3 activation.29,30,87 Detection of a heterogeneous population in CTH cultures (Figure 2) is in line with the previous reports describing the evolution of mononuclear cells upon radio- or chemotherapy treatment into enlarged giant cells with single or multiple nuclei, followed by budding of small daughter cells that actively divide to generate their own progeny cells.88,89 The detection of asymmetric division in CTH cells confirmed previous reports’ findings in which they describe the return of PGCCs into non-polyploid state via the mechanisms of growth and division of simple organisms such as yeasts and other unicellular organisms.42,90 Indeed, this process of slow self-renewal (Suppl. Figure 4) that was referred to as the “giant cell cycle”9,38 not only coordinates the morphological dynamics observed in our cultures, but also explains the differential expression of proliferation, stemness and EMT markers between PGCCs and SCs (Suppl. Figure 5). In particular, the activation of embryonic-like stemness in CTH cells (Figure 4) points toward reprogramming and dedifferentiation as we and other researchers previously described.29,42,91
Further, we reported the detection of PGCCs in tumor biopsies displaying high EZH2 expression (Figure 7). Our results are in line with other findings describing strong EZH2 expression in the nucleus of invading stem-like PGCCs,28 as well as in cancer stem-cells population isolated from human breast cancer, xenograft tumor cells, and primary breast tumor cells.21 It is worth mentioning that the positive correlation detected between PGCC count and EZH2 or Myc expression in tumor biopsies is only established in the presence of HCMV (Figure 7), which indicates a potential role of the latter in the induction and/or maintenance of the observed phenotype. In fact, as our results are in accordance with a described significant positive correlation between Myc and EZH2 mRNA expression in primary prostate cancer specimens,23 it has been shown that the infection with Epstein-Barr virus (EBV), a closely related herpesvirus, markedly induced expression of both Myc, and EZH2 mRNA levels in the same experimental model.92 Since we did not detect EBV in our biopsy samples, this could emphasize a parallel role of HCMV in our model, where further experimental studies are certainly needed to comprehensively understand the exact role played by HCMV in the context of mammary tumors, in particular breast cancer molecular subtypes.
Using distinct methodological approaches, we detected the viral genes IE1 and lncRNA4.9, their transcripts, and most importantly viral proteins such as IE1, pp65, and the late HCMV antigens in CTH cells.29,31 The detection of early and late viral genes, transcripts and proteins in CTH cells suggested that a lytic cycle is taking place even at a low-level and that the whole genome (or at least most of it) of HCMV high-risk strains might be present in CTH cells. The lytic viral replication could be necessary, even if limited, as well as the viral latency, to maintain/favor the transformed state of CTH cells in culture. In fact, the other two oncogenic herpesviruses, namely EBV and Kaposi's sarcoma-associated herpesvirus (KSHV), require the presence of both lytic and latent viral stages to be oncogenic. Indeed, in addition to the role played by viral latency in EBV and KSHV-induced malignancies, lytic replication might also contribute to EBV-induced oncogenesis and KSHV-induced sarcoma, respectively.93,94 Further studies are required to clarify the role of HCMV lytic replication and latent dormancy in the described model and the potential contribution of individual lytic or latent HCMV genes to oncogenesis.
Lastly, since EZH2 and Myc have been implicated in tumor initiation, two HCMV strains (HCMV-B544 and HCMV-B693) were isolated from EZH2HighMycHigh basal tumors to assess their transforming potential. After infection of HMECs cells with these HCMV isolates, we were able to obtain CTH cells, with morphological features matching those of PGCC, giant cell cycling and the previously described CTH cells obtained with HCMV-DB and HCMV-BL strains.29, 30, 31 The detection of IE1 and HCMV late antigen proteins parallel to upregulated Myc, EZH2 expression and lncRNA4.9 transcript in cultures of CTH-B693 and CTH-B544 cells recapitulates the molecular phenotype observed in CTH-DB and CTH-BL cells. As a result, some high-risk HCMV strains are present in basal breast tumors in which they possess tumor-promoting abilities and therefore are considered as oncogenic strains.
In conclusion, our data indicate that high-risk HCMV strains can induce a polyploid phenotype with a distinctive cell cycle, tumor heterogeneity, epithelial to mesenchymal plasticity and embryonic-like stemness. Our findings highlight the presence of a potential link between HCMV infection, Myc/EZH2 upregulation and polyploidy induction in vitro and in human breast cancer biopsies, supporting the proposed tumorigenesis properties of EZH2 and providing new prospect of using EZH2 inhibitors in the context of breast cancer.95 A more detailed analysis of PRC2 target genes within CTH cells and their corresponding response to inhibitors may establish new avenues to understand the complex pathogenesis of breast cancer and open the door for targeted therapies in the future.
Contributors
Conceptualization, G.H.; formal analysis, S.H.A., S.P., Z.N., R.E.B, G.H.; investigation, S.H.A., S.P., R.E.B, Z.N.; writing—original draft preparation, Z.N., S.P., S.H.A., G.H.; writing—review and editing, Z.N., G.H.; directly accessed and verified the underlying data: Z.N., S.P., S.H.A., R.E.B., G.H.; visualization, S.H.A., S.P., Z.N.; supervision, G.H.; project administration, G.H.; funding acquisition, G.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the University of Franche-Comté (UFC) (CR3300), the Région Franche-Comté (2021-Y-08292 and 2021-Y-08290) and the Ligue contre le Cancer (CR3304) to Georges Herbein. Zeina Nehme is a recipient of a doctoral scholarship from the municipality of Habbouch. Sandy Haidar Ahmad is recipient of a doctoral scholarship from Lebanese municipality. Ranim El Baba is a recipient of a doctoral scholarship from Hariri foundation for sustainable human development.
Data availability statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Declaration of interests
The authors declare no conflict of interest.
Acknowledgments
We are grateful to the Pathology Department at the Besançon University Hospital for providing breast biopsies and data. We thank DImaCell Imaging Ressource Center, University of Bourgogne Franche-Comté, Faculty of Health Sciences, 25000 Besançon, France for technical support. This work was supported by grants from the University of Franche-Comté (UFC) (CR3300), the Région Franche-Comté (2021-Y-08292 and 2021-Y-08290) and the Ligue contre le Cancer (CR3304) to Georges Herbein. Zeina Nehme is a recipient of a doctoral scholarship from the municipality of Habbouch. Sandy Haidar Ahmad is recipient of a doctoral scholarship from Lebanese municipality. Ranim El Baba is a recipient of a doctoral scholarship from Hariri foundation for sustainable human development.
Footnotes
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.ebiom.2022.104056.
Appendix. Supplementary materials
References
- 1.Schottstedt V., Blümel J., Burger R., et al. Human cytomegalovirus (HCMV) – revised*. Transfus Med Hemother. 2010;37:365–375. doi: 10.1159/000322141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Herbein G. The human cytomegalovirus, from oncomodulation to oncogenesis. Viruses. 2018;10:408. doi: 10.3390/v10080408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michaelis M., Doerr H.W., Cinatl J. The story of human cytomegalovirus and cancer: increasing evidence and open questions. Neoplasia. 2009;11:1–9. doi: 10.1593/neo.81178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taher C., de Boniface J., Mohammad A.A., et al. High prevalence of human cytomegalovirus proteins and nucleic acids in primary breast cancer and metastatic sentinel lymph nodes. PLoS ONE. 2013;8:e56795. doi: 10.1371/journal.pone.0056795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El Shazly D.F., Bahnassey A.A., Omar O.S., et al. Detection of human cytomegalovirus in malignant and benign breast tumors in egyptian women. Clin Breast Cancer. 2018;18:e629–e642. doi: 10.1016/j.clbc.2017.10.018. [DOI] [PubMed] [Google Scholar]
- 7.Taher C., Frisk G., Fuentes S., Religa P., et al. High prevalence of human cytomegalovirus in brain metastases of patients with primary breast and colorectal cancers. Transl Oncol. 2014;7:732–740. doi: 10.1016/j.tranon.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Branch K.M., Garcia E.C., Chen Y.M., et al. Productive infection of human breast cancer cell lines with human cytomegalovirus (HCMV) Pathogens. 2021;10:641. doi: 10.3390/pathogens10060641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J. The dualistic origin of human tumors. Semin Cancer Biol. 2018;53:1–16. doi: 10.1016/j.semcancer.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Q., Zhang K., Li Z., et al. High migration and invasion ability of PGCCs and their daughter cells associated with the nuclear localization of S100A10 modified by SUMOylation. Front Cell Dev Biol. 2021;9 doi: 10.3389/fcell.2021.696871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y., Zhu C., Tang L., et al. MYC dysfunction modulates stemness and tumorigenesis in breast cancer. Int J Biol Sci. 2021;17:178–187. doi: 10.7150/ijbs.51458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White-Gilbertson S., Voelkel-Johnson C. Giants and monsters: unexpected characters in the story of cancer recurrence. Adv Cancer Res. 2020;148:201–232. doi: 10.1016/bs.acr.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amend S.R., Torga G., Lin K.C., et al. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate. 2019;79:1489–1497. doi: 10.1002/pros.23877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herbein G., Nehme Z. Polyploid giant cancer cells, a hallmark of oncoviruses and a new therapeutic challenge. Front Oncol. 2020;10 doi: 10.3389/fonc.2020.567116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao R., Wang L., Wang H., et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 16.Kim K.H., Roberts C.W.M. Targeting EZH2 in cancer. Nat Med. 2016;22:128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veneti Z., Gkouskou K., Eliopoulos A. Polycomb repressor complex 2 in genomic instability and cancer. Int J Mol Sci. 2017;18:1657. doi: 10.3390/ijms18081657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo S., Li X., Rohr J., et al. EZH2 overexpression in different immunophenotypes of breast carcinoma and association with clinicopathologic features. Diagn Pathol. 2016;11:41. doi: 10.1186/s13000-016-0491-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kleer C.G., Cao Q., Varambally S., et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez M.E., Moore H.M., Li X., et al. EZH2 expands breast stem cells through activation of NOTCH1 signaling. PNAS. 2014;111:3098–3103. doi: 10.1073/pnas.1308953111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang C.J., Yang J.Y., Xia W., et al. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-β-catenin signaling. Cancer Cell. 2011;19:86–100. doi: 10.1016/j.ccr.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu J., Crowe D.L. The histone methyltransferase EZH2 promotes mammary stem and luminal progenitor cell expansion, metastasis and inhibits estrogen receptor-positive cellular differentiation in a model of basal breast cancer. Oncol Rep. 2015;34:455–460. doi: 10.3892/or.2015.4003. [DOI] [PubMed] [Google Scholar]
- 23.Koh C.M., Iwata T., Zheng Q., Bethel C., Yegnasubramanian S., De Marzo A.M. Myc enforces overexpression of EZH2 in early prostatic neoplasia via transcriptional and post-transcriptional mechanisms. Oncotarget. 2011;2:669–683. doi: 10.18632/oncotarget.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuser-Abali G., Alptekin A., Cinar B. Overexpression of MYC and EZH2 cooperates to epigenetically silence MST1 expression. Epigenetics. 2014;9:634–643. doi: 10.4161/epi.27957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rossetto C.C., Tarrant-Elorza M., Pari G.S. Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14 (+) Monocytes and CD34 (+) cells. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahani N., Shirkoohi R., Rokouei M., Alipour Eskandani M., Nikravesh A. Overexpression of enhancer of zeste human homolog 2 (EZH2) gene in human cytomegalovirus positive glioblastoma multiforme tissues. Med Oncol. 2014;31:252. doi: 10.1007/s12032-014-0252-9. [DOI] [PubMed] [Google Scholar]
- 27.Zhang S., Mercado-Uribe I., Hanash S., Liu J. iTRAQ-based proteomic analysis of polyploid giant cancer cells and budding progeny cells reveals several distinct pathways for ovarian cancer development. PLOS ONE. 2013;8:e80120. doi: 10.1371/journal.pone.0080120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L., Ding P., Lv H., et al. Number of polyploid giant cancer cells and expression of EZH2 are associated with VM formation and tumor grade in human ovarian tumor. BioMed Res Int. 2014;2014 doi: 10.1155/2014/903542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nehme Z., Pasquereau S., Haidar Ahmad S., et al. Polyploid giant cancer cells, stemness and epithelial-mesenchymal plasticity elicited by human cytomegalovirus. Oncogene. 2021;40:3030–3046. doi: 10.1038/s41388-021-01715-7. [DOI] [PubMed] [Google Scholar]
- 30.Kumar A., Tripathy M.K., Pasquereau S., et al. The human cytomegalovirus strain DB activates oncogenic pathways in mammary epithelial cells. EBioMedicine. 2018;30:167–183. doi: 10.1016/j.ebiom.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haidar Ahmad S., Pasquereau S., El Baba R., Nehme Z., Lewandowski C., Herbein G. Distinct oncogenic transcriptomes in human mammary epithelial cells infected with cytomegalovirus. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.772160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan K.A., Coaquette A., Davrinche C., Herbein G. Bcl-3-regulated transcription from major immediate-early promoter of human cytomegalovirus in monocyte-derived macrophages. J Immunol. 2009;182:7784–7794. doi: 10.4049/jimmunol.0803800. [DOI] [PubMed] [Google Scholar]
- 33.Lepiller Q., Abbas W., Kumar A., Tripathy M.K., Herbein G. HCMV activates the IL-6-JAK-STAT3 Axis in HepG2 cells and primary human hepatocytes. PLOS ONE. 2013;8:e59591. doi: 10.1371/journal.pone.0059591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basheer F., Giotopoulos G., Meduri E., et al. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J Exp Med. 2019;216:966–981. doi: 10.1084/jem.20181276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borowicz S., Van Scoyk M., Avasarala S., et al. The soft agar colony formation assay. J Vis Exp. 2014:51998. doi: 10.3791/51998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sourvinos G., Morou A., Sanidas I., et al. The downregulation of GFI1 by the EZH2-NDY1/KDM2B-JARID2 axis and by human cytomegalovirus (HCMV) associated factors allows the activation of the HCMV major IE promoter and the transition to productive infection. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moussawi F.A., Kumar A., Pasquereau S., et al. The transcriptome of human mammary epithelial cells infected with the HCMV-DB strain displays oncogenic traits. Sci Rep. 2018;8:12574. doi: 10.1038/s41598-018-30109-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang S., Mercado-Uribe I., Xing Z., Sun B., Kuang J., Liu J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene. 2014;33:116–128. doi: 10.1038/onc.2013.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmittgen T.D., Livak K.J. Analyzing real-time PCR data by the comparative C T method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 40.Gerna G., Kabanova A., Lilleri D. Human cytomegalovirus cell tropism and host cell receptors. Vaccines (Basel) 2019;7:70. doi: 10.3390/vaccines7030070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boldogh I., AbuBakar S., Albrecht T. Activation of proto-oncogenes: an immediate early event in human cytomegalovirus infection. Science. 1990;247:561–564. doi: 10.1126/science.1689075. [DOI] [PubMed] [Google Scholar]
- 42.Niu N., Mercado-Uribe I., Liu J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene. 2017;36:4887–4900. doi: 10.1038/onc.2017.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fluge Ø., Gravdal K., Carlsen E., et al. Expression of EZH2 and Ki-67 in colorectal cancer and associations with treatment response and prognosis. Br J Cancer. 2009;101:1282–1289. doi: 10.1038/sj.bjc.6605333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J., Xi Y., Li W., et al. TRIM28 interacts with EZH2 and SWI/SNF to activate genes that promote mammosphere formation. Oncogene. 2017;36:2991–3001. doi: 10.1038/onc.2016.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fei F., Zhang D., Yang Z., et al. The number of polyploid giant cancer cells and epithelial-mesenchymal transition-related proteins are associated with invasion and metastasis in human breast cancer. J Exp Clin Cancer Res. 2015;34:158. doi: 10.1186/s13046-015-0277-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang C.J., Hung M.C. The role of EZH2 in tumour progression. Br J Cancer. 2012;106:243–247. doi: 10.1038/bjc.2011.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu J., Chen Y., Olopade O.I. MYC and breast cancer. Genes Cancer. 2010;1:629–640. doi: 10.1177/1947601910378691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumari K., Das B., Adhya A.K., Rath A.K., Mishra S.K. Genome-wide expression analysis reveals six contravened targets of EZH2 associated with breast cancer patient survival. Sci Rep. 2019;9:1974. doi: 10.1038/s41598-019-39122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao B., Liu X., Li Z., Zhao L., Pan Y. Overexpression of EZH2/NSD2 histone methyltransferase axis predicts poor prognosis and accelerates tumor progression in triple-negative breast cancer. Front Oncol. 2020;10 doi: 10.3389/fonc.2020.600514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guan X., Deng H., Choi U.L., et al. EZH2 overexpression dampens tumor-suppressive signals via an EGR1 silencer to drive breast tumorigenesis. Oncogene. 2020;39:7127–7141. doi: 10.1038/s41388-020-01484-9. [DOI] [PubMed] [Google Scholar]
- 51.Chu W., Zhang X., Qi L., et al. The EZH2–PHACTR2–AS1–ribosome axis induces genomic instability and promotes growth and metastasis in breast cancer. Cancer Res. 2020;80:2737–2750. doi: 10.1158/0008-5472.CAN-19-3326. [DOI] [PubMed] [Google Scholar]
- 52.Yu T., Wang Y., Hu Q., et al. The EZH2 inhibitor GSK343 suppresses cancer stem-like phenotypes and reverses mesenchymal transition in glioma cells. Oncotarget. 2017;8:98348–98359. doi: 10.18632/oncotarget.21311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nie Z., Guo C., Das S.K., et al. Dissecting transcriptional amplification by MYC. Elife. 2020;9:e52483. doi: 10.7554/eLife.52483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nie Z., Hu G., Wei G., et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151:68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sander S., Bullinger L., Klapproth K., et al. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112:4202–4212. doi: 10.1182/blood-2008-03-147645. [DOI] [PubMed] [Google Scholar]
- 56.Sun J., Cai X., Yung M.M., et al. miR-137 mediates the functional link between c-Myc and EZH2 that regulates cisplatin resistance in ovarian cancer. Oncogene. 2019;38:564–580. doi: 10.1038/s41388-018-0459-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu X., Liu D., Tao D., et al. BRD4 regulates EZH2 transcription through upregulation of C-MYC and represents a novel therapeutic target in bladder cancer. Mol Cancer Ther. 2016;15:1029–1042. doi: 10.1158/1535-7163.MCT-15-0750. [DOI] [PubMed] [Google Scholar]
- 58.Fallah Y., Brundage J., Allegakoen P., Shajahan-Haq A.N. MYC-driven pathways in breast cancer subtypes. Biomolecules. 2017;7:53. doi: 10.3390/biom7030053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Natsumeda M., Liu Y., Nakata S., et al. Inhibition of enhancer of zest homologue 2 is a potential therapeutic target for high-MYC medulloblastoma. Neuropathology. 2019;39:71–77. doi: 10.1111/neup.12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Q., Dang C.V. c-Myc overexpression uncouples DNA replication from mitosis. Mol Cell Biol. 1999;19:5339–5351. doi: 10.1128/mcb.19.8.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vazquez-Martin A., Anatskaya O.V., Giuliani A., et al. Somatic polyploidy is associated with the upregulation of c-MYC interacting genes and EMT-like signature. Oncotarget. 2016;7:75235–75260. doi: 10.18632/oncotarget.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kinouchi T., Saiki S., Naoe T., et al. Correlation of c-myc expression with nuclear pleomorphism in human renal cell carcinoma. Cancer Res. 1989;49:3627–3630. [PubMed] [Google Scholar]
- 63.Santoro A., Vlachou T., Luzi L., et al. p53 Loss in breast cancer leads to myc activation, increased cell plasticity, and expression of a mitotic signature with prognostic value. Cell Reports. 2019;26:624–638. doi: 10.1016/j.celrep.2018.12.071. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bracken A.P., Pasini D., Capra M., Prosperini E., Colli E., Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gupta R.A., Shah N., Wang K.C., et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dooley A.L., O'Connor C.M. Regulation of the MIE locus during HCMV latency and reactivation. Pathogens. 2020;9:E869. doi: 10.3390/pathogens9110869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Helness A., Fraszczak J., Joly-Beauparlant C., et al. GFI1 tethers the NuRD complex to open and transcriptionally active chromatin in myeloid progenitors. Commun Biol. 2021;4:1356. doi: 10.1038/s42003-021-02889-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beauchemin H., Möröy T. Multifaceted actions of GFI1 and GFI1B in hematopoietic stem cell self-renewal and lineage commitment. Front Genet. 2020;11 doi: 10.3389/fgene.2020.591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.E X., Pickering M.T., Debatis M., et al. An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog. 2011;7 doi: 10.1371/journal.ppat.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ezhkova E., Pasolli H.A., Parker J.S., et al. EZH2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell. 2009;136:1122–1135. doi: 10.1016/j.cell.2008.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang X.J., Wang X., Ma X., et al. EZH2 is essential for development of mouse preimplantation embryos. Reprod Fertil Dev. 2014;26:1166–1175. doi: 10.1071/RD13169. [DOI] [PubMed] [Google Scholar]
- 72.Yu J., Yu J., Rhodes D.R., et al. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res. 2007;67:10657–10663. doi: 10.1158/0008-5472.CAN-07-2498. [DOI] [PubMed] [Google Scholar]
- 73.Stefansson O.A., Esteller M. EZH2-mediated epigenetic repression of DNA repair in promoting breast tumor initiating cells. Breast Cancer Res. 2011;13:309. doi: 10.1186/bcr2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neri F., Zippo A., Krepelova A., Cherubini A., Rocchigiani M., Oliviero S. Myc regulates the transcription of the PRC2 gene to control the expression of developmental genes in embryonic stem cells. Mol Cell Biol. 2012;32:840–851. doi: 10.1128/MCB.06148-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang A., Qin S., Schulte B.A., Ethier S.P., Tew K.D., Wang G.Y. MYC inhibition depletes cancer stem-like cells in triple-negative breast cancer. Cancer Res. 2017;77:6641–6650. doi: 10.1158/0008-5472.CAN-16-3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sher F., Boddeke E., Copray S. EZH2 expression in astrocytes induces their dedifferentiation toward neural stem cells. Cell Reprogr. 2010;13:1–6. doi: 10.1089/cell.2010.0052. [DOI] [PubMed] [Google Scholar]
- 77.Collinson A., Collier A.J., Morgan N.P., et al. Deletion of the polycomb-group protein EZH2 leads to compromised self-renewal and differentiation defects in human embryonic stem cells. Cell Rep. 2016;17:2700–2714. doi: 10.1016/j.celrep.2016.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shimizu T., Kubovcakova L., Nienhold R., et al. Loss of Ezh2 synergizes with JAK2-V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J Exp Med. 2016;213:1479–1496. doi: 10.1084/jem.20151136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Knutson S.K., Kawano S., Minoshima Y., et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-hodgkin lymphoma. Mol Cancer Ther. 2014;13:842–854. doi: 10.1158/1535-7163.MCT-13-0773. [DOI] [PubMed] [Google Scholar]
- 80.Xie H., Xu J., Hsu J.H., et al. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell. 2014;14:68–80. doi: 10.1016/j.stem.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Granit R.Z., Gabai Y., Hadar T., et al. EZH2 promotes a bi-lineage identity in basal-like breast cancer cells. Oncogene. 2013;32:3886–3895. doi: 10.1038/onc.2012.390. [DOI] [PubMed] [Google Scholar]
- 82.Pan Y.M., Wang C.G., Zhu M., et al. STAT3 signaling drives EZH2 transcriptional activation and mediates poor prognosis in gastric cancer. Mol Cancer. 2016;15:79. doi: 10.1186/s12943-016-0561-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gonzalez M.E., DuPrie M.L., Krueger H., et al. Histone methyltransferase EZH2 induces akt-dependent genomic instability and BRCA1 inhibition in breast cancer. Cancer Res. 2011;71:2360–2370. doi: 10.1158/0008-5472.CAN-10-1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xu K., Wu Z.J., Groner A.C., et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim E., Kim M., Woo D.H., et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23:839–852. doi: 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pandit S.K., Westendorp B., Nantasanti S., et al. E2F8 is essential for polyploidization in mammalian cells. Nat Cell Biol. 2012;14:1181–1191. doi: 10.1038/ncb2585. [DOI] [PubMed] [Google Scholar]
- 87.Liao Y., Hung M.C. Physiological regulation of Akt activity and stability. Am J Transl Res. 2010;2:19–42. [PMC free article] [PubMed] [Google Scholar]
- 88.Niu N., Zhang J., Zhang N., et al. Linking genomic reorganization to tumor initiation via the giant cell cycle. Oncogenesis. 2016;5:e281. doi: 10.1038/oncsis.2016.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lin K.C., Torga G., Sun Y., et al. The role of heterogeneous environment and docetaxel gradient in the emergence of polyploid, mesenchymal and resistant prostate cancer cells. Clin Exp Metastasis. 2019;36:97–108. doi: 10.1007/s10585-019-09958-1. [DOI] [PubMed] [Google Scholar]
- 90.Zhang D., Wang Y., Zhang S. Asymmetric cell division in polyploid giant cancer cells and low eukaryotic cells. Biomed Res Int. 2014;2014:1–8. doi: 10.1155/2014/432652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Díaz-Carballo D., Saka S., Klein J., et al. A distinct oncogenerative multinucleated cancer cell serves as a source of stemness and tumor heterogeneity. Cancer Res. 2018;78:2318–2331. doi: 10.1158/0008-5472.CAN-17-1861. [DOI] [PubMed] [Google Scholar]
- 92.Ichikawa T., Okuno Y., Sato Y., et al. Regulation of epstein-barr virus life cycle and cell proliferation by histone H3K27 methyltransferase EZH2 in Akata cells. MSphere. 2018;3 doi: 10.1128/mSphere.00478-18. e00478-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Münz C. Latency and lytic replication in Epstein–Barr virus-associated oncogenesis. Nat Rev Microbiol. 2019;17:691–700. doi: 10.1038/s41579-019-0249-7. [DOI] [PubMed] [Google Scholar]
- 94.Ganem D. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Investig. 2010;120:939–949. doi: 10.1172/JCI40567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lehmann B.D., Colaprico A., Silva T.C., et al. Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes. Nat Commun. 2021;12:6276. doi: 10.1038/s41467-021-26502-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.