

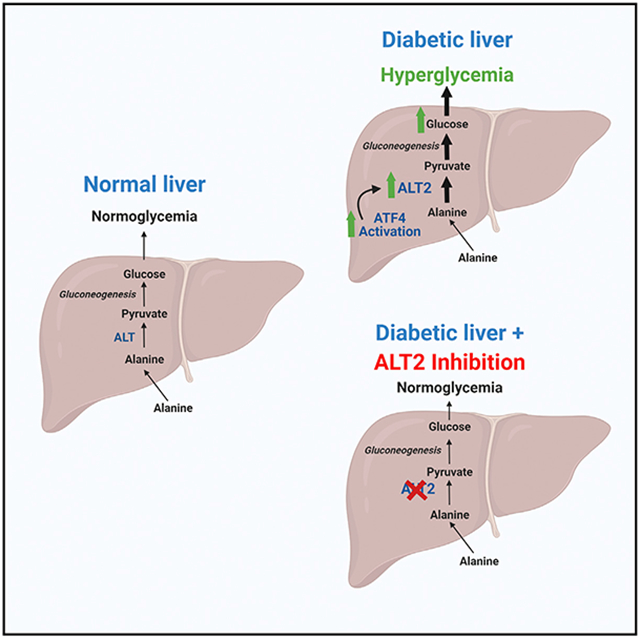
SUMMARY
Hepatic gluconeogenesis from amino acids contributes significantly to diabetic hyperglycemia, but the molecular mechanisms involved are incompletely understood. Alanine transaminases (ALT1 and ALT2) catalyze the interconversion of alanine and pyruvate, which is required for gluconeogenesis from alanine. We find that ALT2 is overexpressed in the liver of diet-induced obese and db/db mice and that the expression of the gene encoding ALT2 (GPT2) is downregulated following bariatric surgery in people with obesity. The increased hepatic expression of Gpt2 in db/db liver is mediated by activating transcription factor 4, an endoplasmic reticulum stress-activated transcription factor. Hepatocyte-specific knockout of Gpt2 attenuates incorporation of 13C-alanine into newly synthesized glucose by hepatocytes. In vivo Gpt2 knockdown or knockout in liver has no effect on glucose concentrations in lean mice, but Gpt2 suppression alleviates hyperglycemia in db/db mice. These data suggest that ALT2 plays a significant role in hepatic gluconeogenesis from amino acids in diabetes.
Graphical Abstract
In brief
Martino et al. find that alanine transaminase 2 (ALT2), which is encoded by Gpt2, is increased in liver of mice and people with obesity by activating transcription factor 4. Suppression of Gpt2 expression in obese, but not lean mice, lowers blood glucose by suppressing alanine-mediated gluconeogenesis.
INTRODUCTION
Glucose production from precursor substrates, such as amino acids or lactate/pyruvate (gluconeogenesis), is a critical adaptation to exercise and prolonged fasting, but dysregulated liver gluconeogenesis can contribute to hyperglycemia in diabetes. Indeed, the first-line antidiabetic agent, metformin, lowers blood glucose by suppressing liver glucose production (Foretz et al., 2010; Madiraju et al., 2014). In addition, genetic or chemical targeting of the mitochondrial pyruvate carrier (MPC) (Gray et al., 2015; McCommis et al., 2015) or pyruvate carboxylase (Cappel et al., 2019) in liver can suppress hyperglycemia in mouse models of obesity and diabetes by limiting the flux of pyruvate into new glucose. Amino acids, such as alanine or glutamine, are significant substrates for de novo synthesis of glucose, and gluconeogenesis from amino acids is known to be increased in diabetes (Chan et al., 1975; Snell and Duff, 1980; Yang et al., 2009) and obesity (Chevalier et al., 2006). However, relatively little is known about the effects of attenuating hepatic amino acid-mediated gluconeogenesis on hyperglycemia.
In conditions where gluconeogenic flux is high, skeletal muscle efflux of the gluconeogenic amino acids alanine and glutamine is increased disproportionate to their relative abundance in skeletal muscle protein (Felig et al., 1970; Felig and Wahren, 1971). Indeed, although some alanine is generated by proteolysis, much of the alanine released from muscle is generated by transamination of pyruvate; an alternative fate to pyruvate reduction to lactate. Muscle-synthesized alanine is subsequently delivered to the liver (Ruderman, 1975; Snell, 1980; Snell and Duff, 1980) where the alanine is re-converted to pyruvate, which can then enter the gluconeogenic pathway. This cycle is known as the Cahill cycle and serves to supply carbons from amino acids to the gluconeogenic pathway while disposing of the amino nitrogen in the urea cycle, since neither the gluconeogenic nor urea cycle pathways are operative in skeletal muscle (Felig et al., 1970).
Before alanine carbons can enter the gluconeogenic pathway, alanine must be converted to pyruvate by the alanine transaminase (ALT) enzymes, ALT1 and ALT2 (Figure 1A; DeRosa and Swick, 1975; Garcia-Campusano et al., 2009; Yang et al., 2009). These enzymes are also known as glutamic-pyruvic transaminases (GPTs) and are encoded by genes annotated as Gpt and Gpt2. The two ALT enzymes catalyze the bidirectional conversion of pyruvate and glutamate to alanine and α-ketoglutarate via transamination (Figure 1A; DeRosa and Swick, 1975; Felig, 1973). Adipose tissue, liver, skeletal muscle, and the intestines highly express ALT1, which is localized to the cytosolic compartment (Lindblom et al., 2007; Qian et al., 2015). Conversely, ALT2 is a mitochondrial matrix protein and is expressed in skeletal muscle, brain, heart, liver, and other tissues (Lindblom et al., 2007; Ouyang et al., 2016; Qian et al., 2015). Experimental determination of the Km values has suggested that ALT1 is important in the generation of alanine from pyruvate, while ALT2 favors the reverse reaction (DeRosa and Swick, 1975; Glinghammar et al., 2009). This would suggest that alanine is transported into the mitochondrion before conversion to pyruvate by ALT2. Consistent with this, chemical or genetic inhibition of the MPC in hepatocytes did not affect gluconeogenesis from alanine (Dieterle et al., 1978; McCommis et al., 2015).
Figure 1. ALT2 is expressed in human liver and is reduced after significant weight loss.

(A) Schematic of hepatic alanine metabolism. ALA, alanine; GLU, glutamine; MPC, mitochondrial pyruvate carrier; ALT, alanine transaminase; OAA, oxaloacetate; αKG, α ketoglutarate. Created with BioRender.com.
(B) Western blotting analysis for ALT2, ALT1, and HSP60 conducted using lysates from obese human liver (n = 2),Huh7 cell lysates (treated with scramble [scr] or GPT2 siRNA), and mouse liver lysates (WT or LS-Gpt2−/−).
(C) Western blot image for ALT2, ALT1, and tubulin from liver biopsies collected from cadaveric donor livers (n = 3) or during RYGBS (n = 2) from patients with obesity.
(D) Expression of mRNA for GPT and GPT2 in obese subjects before (pre) (n = 8) and 6 months after (post) (n = 8) bariatric surgery.
(E) Expression of the indicated genes encoding mitochondrial amino acid transporters in obese subjects during (pre) (n = 7) and 6 months after (post) (n = 7) bariatric surgery.
(D and E) Data are presented as mean ± SD. *p ≤ 0.05 for pre versus post.
Herein, we demonstrate that ALT2 is activated in obese liver and examine the role of hepatic ALT2 in gluconeogenesis and hyperglycemia by using liver-specific Gpt2−/− mice and Gpt2 shRNA. We found that loss of ALT2 had little effect on blood glucose concentrations in lean mice, but that ALT2 suppression in obese, db/db mice produced a robust glucose-lowering effect independent of changes in liver fat or insulin sensitivity. These findings are consistent with another recent study demonstrating that suppression of both ALT enzymes in liver lowered plasma glucose concentrations in mouse models of diabetes (Okun et al., 2021). Thus, although ALT activity is primarily considered a circulating biomarker for liver or muscle injury, these data collectively demonstrate that it also plays important roles in intermediary metabolism and may contribute to dysregulated glucose production by diabetic liver.
RESULTS
ALT2 is abundant in human liver and GPT2 expression is downregulated with marked weight loss
A recently published study suggested that hepatic expression of the gene encoding ALT2 (GPT2) is induced in people with type 2 diabetes and in mouse models of the disease (Okun et al., 2021), but other work has questioned whether ALT2 protein is expressed in human liver (Glinghammar et al., 2009). To confirm that ALT2 is abundant in human liver, hepatic protein lysates from biopsies collected during Roux-en-Y gastric bypass surgery (RYGBS) from patients with obesity were probed with antibodies for ALT1 and ALT2 and compared with lysates from mouse liver (wild-type [WT], mice with liver-specific Gpt2 knockout [KO]), and lysates from Huh7 human hepatoma cells (treated with siRNA against GPT2 or scrambled control). Western blotting demonstrated that, although both ALT proteins were more abundant in mouse liver compared with human liver, both were readily detectable in human liver specimens and that these antibodies are specific for ALT1 and ALT2, respectively (Figure 1B). We also examined the abundance of ALT1 and ALT2 in livers of lean cadaveric donor liver samples compared with the tissues collected from patients with obesity obtained during RYGBS. Qualitatively, ALT2 appeared to be much more abundant in liver of patients with obesity compared with lean controls (Figure 1C). However, this study was not powered to allow for a convincing conclusion.
To determine whether marked weight loss affected the expression of ALTs, the expression of the genes encoding ALT1 (GPT) and ALT2 (GPT2) were assessed in liver biopsies collected during RYGBS and in the same individuals after losing ~36% of pre-surgery BMI (collected by percutaneous biopsy). As expected, RYGBS-induced weight loss led to significant reductions in fasting glucose and insulin, as well as marked metabolic improvements in insulin sensitivity, as determined by both the HOMA-IR and HISI (Table S1). RYGBS-induced weight loss and metabolic improvement were associated with a marked reduction in the expression of GPT2, but not GPT (Figure 1D). Similarly, the expression of mitochondrial amino acid carriers (SFXN1, SLC25A2, and SLC25A39) was also decreased in the RYGBS patients after marked weight loss (Figure 1E), although expression of other transporters (SLC25A18, SLC25A38, and SLC25A40) was not affected. Taken together, these data demonstrate that ALT2 is present in human liver and that expression of the gene encoding this protein is reduced concordant with marked weight loss-induced metabolic improvements in people with obesity.
Obese mice exhibit higher expression of Gpt2 and ALT2 and are hyperglycemic after amino acid challenge
We then determined whether hepatic expression of Gpt/ALT enzymes was increased in rodent models of obesity. Livers of mice fed with a diet providing 60% of its calories as fat (high-fat diet [HFD]) for 23 weeks had greater ALT2 protein abundance than mice fed a control low-fat diet (LFD) (Figure 2A). The expression of the genes encoding ALT1 and ALT2 (Gpt and Gpt2) was increased in these same mice (Figures 2B and S1A). The expression of Sfxn1 and Slc25a39 was also increased in HFD-fed mice versus LFD controls (Figure S1A). Consistent with increased gluconeogenesis from amino acids, alanine (ATT) and glutamine (QTT) tolerance tests revealed that blood glucose concentrations were significantly elevated in HFD-fed mice compared with LFD-fed mice (Figures 2C and 2D) in both ATT and QTT analyses.
Figure 2. Diet-induced obese and db/db mice exhibit increased hepatic ALT abundance and exacerbated hyperglycemia from gluconeogenic amino acids.

(A and B) (A) Representative western blot images for ALT2 and GAPDH proteins and (B) expression of Gpt2 mRNA in livers of mice fed either a control low-fat diet (LFD) (n = 5) or a 60% high-fat diet (HFD) (n = 5).
(C and D) Blood glucose concentrations after an i.p. injection of L-alanine (C) or L-glutamine (D) in diet-induced obese (HFD; n = 5) and lean (LFD; n = 5) mice.
(E) Representative western blot image for ALT2 and mitochondrial complex III from either db/+ or db/db mouse liver homogenates.
(F) Expression of Gpt2 in liver RNA from db/+ (n = 8) and db/db (n = 5, 7) mice (bottom panel).
(G and H) Blood glucose concentrations after an i.p. injection of L-alanine (G) or L-glutamine (H) in db/+ (n = 7, 8) and db/db mice (n = 6, 7). For (A) and (E) densitometric quantification of ALT2/loading control band intensity is provided numerically between the blots. Data are presented as mean ± SEM. *p ≤ 0.05 for LFD versus HFD or db/+ versus db/db.
We also determined that leptin receptor-deficient db/db mice had a 3-fold elevation in hepatic ALT2 protein compared with lean, db/+ littermate control mice (Figure 2E). The expression of Gpt and Gpt2 was also increased in db/db mice compared with db/+ lean controls (Figures 2F and S1B). ATT and QTT conducted with 8-week-old db/db and lean db/+ mice demonstrated that glucose concentrations after amino acid challenge were markedly higher in db/db mice compared with lean db/+ controls (Figures 2G and 2H). While other factors can affect blood glucose in these tolerance tests, these data support the hypothesis that the increase in hepatic ALT2 protein that occurs with obesity may contribute to amino acid-fueled glucose production by the liver.
Activating transcription factor 4 regulates ALT2 expression in db/db liver
Previous work has suggested that Gpt2 expression is transcriptionally regulated by the ER stress-activated transcription factor ATF4 (activated transcription factor 4) (Hao et al., 2016) and that hepatic ER stress is increased in obesity and diabetes (Ozcan et al., 2004). In vivo treatment with an antisense oligonucleotide (ASO) to knockdown ATF4 for 3 weeks blocked the induction of Gpt2 expression and ALT2 protein abundance in db/db liver (Figure 3A). Conversely, the expression of Gpt was not affected following ATF4 knockdown in db/+ or db/db mice (Figure S1B). Interestingly, the expression of Gpt2 is more sensitive to ATF4 knockdown than several other known ATF4 target genes, including Asns, Fgf21, Trib3, Ddit4, and Psat1 (Figure S1B).
Figure 3. Gpt2 is regulated by the ER stress transcription factor ATF4.

(A) Liver tissue from db/+ and db/db mice treated with a control ASO or an ASO against ATF4 were used to assess gene expression (n = 8) and obtain lysates for western blotting for ALT2 and tubulin (n = 3).
(B) Expression of the indicated ATF4 target genes in obese subjects during (pre; n = 7) and 6 months after (post; n = 7) bariatric surgery. Data are presented as mean ± SD. *p ≤ 0.05 for pre versus post.
(C) Hepatocytes isolated from lean C57BL/6J mice were pretreated with a control ASO or an ASO against ATF4 for 24 h were treated with the ER stress-inducing agent tunicamycin (Tuni) or vehicle control (DMSO) for 6 h, after which gene expression was assessed (n = 11). Data are presented as mean ± SEM. *p ≤ 0.05.
Also consistent with a role for ATF4 in controlling GPT2 expression, we examined the expression of ATF4 target genes in the patients who underwent RYGBS. Consistent with reduced GPT2 expression after weight loss, the expression of ASNS, FGF21, TRIB3, and DDIT4 was concomitantly reduced and PSAT1 tended to be reduced (p = 0.058) (Figure 3B).
To determine whether ATF4 activation was sufficient to induce Gpt2 expression, we treated primary hepatocytes isolated from lean C57BL/6J mice with varying doses of an adenovirus to overexpress ATF4 or β-gal as a control. Overexpression of ATF4 resulted in a dose-dependent increase in expression of Gpt2 but had no effect on Gpt (Figure S1C). Similarly, induction of ER stress in hepatocytes by tunicamycin also resulted in increased Gpt2 expression (Figure 3C). This effect was prevented by pretreatment with ATF4 ASO (Figure 3C). ATF4 ASO treatment also suppressed the basal expression of Gpt2 in hepatocytes. Taken together, these data suggest that ALT2 is induced by ER stress in hepatocytes via a mechanism requiring the ATF4 transcription factor.
Loss of hepatic ALT2 reduces hepatic glucose production from alanine in vitro but not in vivo
Germline deletion of Gpt2 has previously been shown to lead to microcephaly and postnatal death before weaning (Ouyang et al., 2016). Morbidity and mortality were associated with impaired amino acid metabolism and anaplerotic flux to replenish tricarboxylic acid (TCA) cycle intermediates. We observed a similar lethal phenotype in homozygous constitutive Gpt2 knockout mice (data not shown), but mice with liver-specific loss of Gpt2 (LS-Gpt2−/−) were viable. Successful knockout of ALT2 protein in the liver was confirmed (Figure 4A).
Figure 4. Loss of ALT2 reduces hepatocyte alanine metabolism and alanine-mediated gluconeogenesis.

(A) Representative western blots for ALT2, ALT1, or GAPDH using liver lysates from WT and LS-Gpt2−/− mice demonstrating loss of ALT2 protein.
(B) Glucose concentrations in the media of hepatocytes isolated from WT or LS-Gpt2−/− mice stimulated with glucagon in the presence of no substrate, alanine, pyruvate, or glutamine. Some cells were also treated with the transaminase inhibitor β-chloroalanine (β-Cl) (n = 7). *Indicates significant differences (p < 0.05).
(C) Schematic depicting incorporation of 13C-alanine into pyruvate and TCA cycle intermediates. Black circles indicate 13C. White circles indicate 12C. Created with BioRender.com.
(D) Hepatocyte citrate enrichment from 13C-alanine is shown. *Significant differences (p < 0.05) between hepatocytes from different genotypes of mice.
(E) Hepatocyte M3 citrate enrichment from 13C-alanine is shown. *Significantly different (p < 0.05) from WT hepatocytes treated with vehicle. **Significantly different (p < 0.05) from all other groups.
(F) Media glucose enrichment from 13C-alanine is shown. *Significant differences (p < 0.05) between hepatocytes from different genotypes of mice. For (D)–(F), a representative experiment (of 3) performed in triplicate is shown. Data are presented as mean ± SEM.
(G) Blood glucose concentrations during ATT, QTT, and pyruvate tolerance test (PTT) analyses using lean WT or LS-Gpt2−/− mice.
We first assessed the ability of hepatocytes isolated from WT or LS-Gpt2−/− mice to produce glucose from alanine, pyruvate, and glutamine. Compared with WT mice, hepatocytes from LS-Gpt2−/− mice produced significantly less glucose in the presence of alanine (Figure 4B). Treatment of hepatocytes with β-chloro-alanine (β-Cl), an inhibitor of the ALT enzymes (Gray et al., 2015), reduced glucose production from alanine in WT hepatocytes, but not those from LS-Gpt2−/− mice (Figure 4B). When provided with pyruvate or glutamine as a gluconeogenic substrate, glucose production from these substrates was not different from WT hepatocytes (Figure 4B). These data suggest that ALT2 is essential for gluconeogenesis from alanine, but not other gluconeogenic substrates, in isolated hepatocytes.
To further assess alanine metabolism in hepatocytes from LS-Gpt2−/− mice, isolated hepatocytes were incubated with 13C-alanine or 13C-glutamine, and incorporation of 13C label was assessed by mass spectrometry (Figure 4C). Citrate M2, M3, and total enrichment from 13C alanine was reduced in hepatocytes from LS-Gpt2−/− mice (Figure 4D). However, enrichment was still quite substantial likely due to the activity of ALT1. Indeed, treating hepatocytes from LS-Gpt2−/− mice with UK5099 almost completely abolished 13C-alanine incorporation into citrate (Figure 4E). These findings are all consistent with the idea that alanine-pyruvate cycling is a compensatory mechanism in the Gpt2-deficient cells. Consistent with reduced contribution of alanine to gluconeogenesis, 13C-alanine enrichment incorporation into media glucose was also reduced in hepatocytes from LS-Gpt2−/− mice (Figure 4F). In contrast, deletion of Gpt2 did not affect the enrichment of 13C-glutamine in these intermediary metabolites or glucose (Figure S2A).
Next, we evaluated gluconeogenesis from amino acids in WT and LS-Gpt2−/− mice by performing a series of tolerance tests. Lean WT and LS-Gpt2−/− mice had similar increases in blood glucose concentrations during ATT and QTT analyses (Figures 4G and S2C). Knockout mice also exhibited normal glucose responses during a pyruvate tolerance test (Figures 4G and S2C). These data suggest that, although glucose production from alanine is impaired in isolated hepatocytes lacking ALT2, loss of this enzyme in the liver of lean nondiabetic mice does not constrain glucose production in vivo in response to amino acid challenge.
We also assessed the response to overnight fasting in WT and LS-Gpt2−/− mice. Fasting induces the hepatic expression of Gpt2 in WT mice (Figure 5A). We found that blood glucose concentrations were not different between genotypes in either the fed or fasted states (Figure 5B). We also quantified the abundance of various metabolites in liver and plasma of the fed and fasted WT and LS-Gpt2−/− mice. The effects of fasting on liver glycogen (Figure 5C) and a variety of organic and amino acids were not affected by liver-specific deletion of Gpt2 (Figure S3). Liver Gpt2 deficiency also did not affect plasma insulin, lipids, glycerol, or ketone body concentrations in fed or fasted mice (Figure 4). Plasma alanine and glutamate, two key substrates of the ALT2 enzyme, were not affected by loss of liver Gpt2, but plasma glutamine was increased in fed LS-Gpt2−/− mice compared with fed WT mice (Figure S4). Fasting increased the plasma concentrations of other gluconeogenic amino acids, including isoleucine, valine, and phenylalanine, in WT mice (Figure 5D). Interestingly, this increase was significantly blunted in LS-Gpt2−/− mice, suggesting altered utilization of other amino acids as metabolic substrates. Thus, while loss of Gpt2 constrains alanine metabolism in isolated hepatocytes, in vivo this does not seem to affect gluconeogenesis in lean mice; possibly reflecting compensatory mechanisms.
Figure 5. Loss of liver ALT2 does not affect blood glucose levels in lean mice.

(A) Gpt2 mRNA expression in liver from fed and fasted WT and LS-Gpt2−/− mice.
(B) Blood glucose concentrations of fed and fasted WT and LS-Gpt2−/− mice (n = 14–17 mice per group).
(C and D) (C) Liver glycogen concentrations and (D) plasma branched chain amino acids levels of fed and fasted WT and LS-Gpt2−/− mice.
(E and F) Bulk RNA sequencing analysis shows only Gpt2 is significantly altered by LS-Gpt2−/− knockout. Volcano plots of merged differential expression data from hepatic RNA of (E) fed WT versus LS-Gpt2−/− or (F) fasted WT versus LS-Gpt2−/− mice. Data were graphed as log2 fold change verses –log10 unadjusted p value. Gene expression changes were considered meaningful if they were >log2 fold change and had an adjusted p value of <0.05 versus WT mice. Graphical representation of pathway analysis showing the top 25 significant signal direction changes of KEGG Signaling and Metabolism pathways. The color of the pathway label indicates the degree and direction of the change and the p value is the x axis. Data are presented as mean ± SEM. *Significant differences (p < 0.05) between indicated groups.
Unlike Gpt2, the hepatic expression of Gpt was not increased by fasting and loss of Gpt2 did not affect the expression of Gpt (Figure S5A). We also did not detect compensation in the expression of several other genes encoding enzymes involved in gluconeogenesis (Pck1), the mitochondrial pyruvate carriers (Mpc1, Mpc2), glutamate/glutamine metabolism (Gdh1, Got1, and Got2), or mitochondrial amino acid transporters (Slc25a38, Slc25a39, Sfxn1) (Figure S5). To further interrogate the transcriptome potential compensatory changes, we performed bulk RNA-seq analyses of liver RNA from fed and fasted WT and LS-Gpt2−/− mice. Strikingly, the only transcript that was significantly different between genotypes was Gpt2 (Figure 5E), and this was true in both fed and fasted conditions. However, pathway analysis revealed significant alterations in the expression of some amino acid metabolic KEGG pathways (Figure 5E).
ALT2 deactivation in db/db mice attenuates hyperglycemia
To assess the contribution of ALT2 to hyperglycemia in obese, diabetic mice, db/+ and db/db mice were administered adenovirus-expressing shRNA to knockdown ALT2 (shGpt2) or control shRNA targeting LacZ (shLacZ). Six days later, Gpt2 RNA (Figure 6A) and ALT2 protein abundance (Figure 6B) were decreased by shGpt2 compared with the LacZ shRNA controls in both genotypes of mice. The expression of Sfxn1 and Slc25a39, which was increased in db/db mice versus db/+ controls, was not affected by shGpt2 (Figure S6A). The expression of other transporters (Slc25a2, Slc25a18, Slc25a38, and Slc25a40) was not affected by genotype or shGpt2 treatment. Knockdown of Gpt2 in db/db mice also reduced total hepatic ALT activity, which was increased in db/db mice versus lean controls (Figure 6C). Treatment with shGpt2 reduced fed blood glucose concentrations in db/db but not db/+ mice (Figure 6D). Plasma insulin concentrations were not different when comparing shLacZ versus shGpt2 groups in the db/+ genotype, but were lower with shGpt2 compared with shLacZ treatment in db/db mice (Figure 6E). These data indicate that ALT2 plays an important role in promoting pathologic elevations of glucose and insulin seen in type 2 diabetes, and that specifically knocking down ALT2 may be a strategy to improve blood glucose management.
Figure 6. Hepatic ALT2 knockdown results in decreased blood glucose concentration.

Mice (db/+ and db/db) were infected with an adenovirus expressing shRNA targeting LacZ or Gpt2.
(A) Gpt2 gene expression was assessed in adenovirus-treated db/+ (n = 11) and db/db (n = 9–13) mice 6 days post infection. *p < 0.05 versus indicated groups.
(B) Representative western blot image for ALT2 or mitochondrial complex III as a loading control using mouse liver lysates from adenovirus shRNA-treated db/+ or db/db mice 6 days after adenovirus administration.
(C) Total liver ALT activity in db/+ and db/db liver (n = 4–7 per group) after treatment with adenovirus expression shRNA against LacZ or Gpt2. *p < 0.05 versus indicated groups.
(D) Random fed blood glucose concentrations in adenovirus shRNA-treated db/+ or db/db mice 5 days after adenovirus injection (n = 13–18 per group). *p < 0.05 versus indicated groups.
(E) Plasma insulin concentrations in adenovirus shRNA-treated db/+ or db/db mice 7 days after adenovirus injection. Plasma was collected at sacrifice after a 4 h fast (n = 8–10 per group). *p < 0.05 versus indicated groups.
(F) Plasma amino acid concentrations in db/+ or db/db mice 7 days after administration of adenovirus expressing shRNA against LacZ or Gpt2. Plasma was collected at sacrifice after a 4 h fast (n = 4–5 per group). *p < 0.05 versus shLacZ db/db mice. **p < 0.05 versus db/+ mice.
Gpt2 knockdown alters plasma amino acid concentrations in db/db mice
To better understand how amino acid metabolism may be affected by the loss of hepatic ALT2, we measured plasma amino acid and organic acid concentrations in db/+ and db/db mice treated with shLacZ or shGpt2. Plasma alanine concentrations were not different in db/db versus db/+ mice but, compared with LacZ controls, shGpt2 treatment led to an increase in circulating concentrations of alanine in db/db mice (Figure 6F), suggesting impaired hepatic catabolism of alanine. Plasma glutamate concentrations were reduced in db/db mice compared with db/+ mice, and treatment with shGpt2 did not affect glutamate concentrations (Figure 6F). Consistent with previous reports in humans and rodents with diabetes (Newgard et al., 2009; Tai et al., 2010), plasma concentrations of phenylalanine, histidine, and the branched chain amino acids isoleucine, leucine, and valine, were increased and glycine was reduced in db/db mice versus db/+ mice (Figure 6F; Table S2). However, the effect of genotype on these amino acids was not corrected by shGpt2 treatment. Knockdown of Gpt2 resulted in an increase in plasma ornithine, an intermediate of the urea cycle, in both db/db and db/+ mice (Table S2). Compared with shLacZ-treated groups, shGpt2-treated mice exhibited higher concentrations of TCA cycle intermediates α-ketoglutarate, malate, and fumarate (Figure S6B).
Hepatic Gpt2 knockdown lowers plasma glucose without insulin sensitizing in db/db mice
Previous work has mechanistically linked hepatic steatosis, particularly accumulation of diacylglycerol (Petersen and Shulman, 2018) and ceramides (Summers et al., 2019), to hepatic insulin resistance and development of hyperglycemia. Knockdown of Gpt2 did not affect hepatic triglyceride content (Figure 7A) or plasma concentrations of triglyceride or cholesterol (Figure S7A) in mice of either genotype. Hepatic diacylglycerol and long chain saturated ceramides, which were increased in db/db mice compared with db/+ controls, were also not affected by Gpt2 knockdown (Figures 7B and 7C). This is interesting in light of previous data suggesting that alanine incorporation into hepatic lipids is increased in obese rats (Terrettaz and Jeanrenaud, 1990).
Figure 7. Loss of hepatic ALT2 lowers blood glucose in diabetic db/db mice without affecting hepatic steatosis or insulin sensitivity.

(A–C) Hepatic triglyceride (TAG) (A), diacylglycerol (DAG) (B), and ceramides (C) in liver extracts from db/+ and db/db mice collected 7 days after injection of adenovirus to knockdown Gpt2 or LacZ and after a 4 h fast. *p < 0.05 versus db/+ mice.
(D) Blood glucose area under the curve during insulin tolerance test (ITT) analyses in shLacZ or shGpt2 adenovirus-treated db/db mice 5 days post infection (n = 7).
(E) Western blots for AKT (phospho-S473, phospho-T308, or total) and ALT2 in db/db mice 7 days after administration of adenovirus expressing shLacZ or shGpt2. Mice were injected with insulin or saline 5 min before sacrifice as indicated.
(F) Blood glucose concentrations during an ATT in adenovirus-treated db/+ (n = 12) and db/db (n = 9–11) mice 6 days post adenovirus injection. *p < 0.05 versus indicated groups.
(G and H) Blood glucose area under the curve during QTT (G) or glucagon tolerance test (H) analyses in shLacZ or shGpt2 adenovirus-treated db/db mice 5 days post infection (n = 5). Data presented as mean ± SEM.
To determine if the observed improvements in blood glucose were due to differences in insulin sensitivity, we performed an insulin tolerance test with a separate cohort of db/db mice treated with shLacZ or shGpt2 adenovirus. Although initial glucose concentrations were lower in db/db mice injected with shGpt2 versus shLacZ, suppression of Gpt2 did not improve the response to insulin (area under the curve) in db/db mice when results were normalized to basal blood glucose concentrations in each group (Figure 7D). Similarly, a molecular correlate of insulin sensitivity, the phosphorylation of AKT in response to insulin bolus, was also not improved by Gpt2 knockdown in db/db mice (Figures 7E and S7B). Thus, despite significant improvements in plasma glucose concentrations, the data do not support the conclusion that insulin sensitivity is improved by Gpt2 knockdown.
Based on these data, and our findings that loss of Gpt2 reduced alanine-mediated gluconeogenesis in hepatocytes, we examined the effects of Gpt2 knockdown on alanine tolerance in db/db and db/+ mice. Total area under the curve for blood glucose concentrations in an ATT was significantly decreased in db/db mice treated with the shGpt2 adenovirus compared with LacZ shRNA-treated db/db mice (Figure 7F). In db/+ mice, Gpt2 deactivation did not affect blood glucose levels at baseline or after injection with L-alanine during an ATT compared with Ad-shLacZ-infected mice (Figure 7F). In QTT challenges, shGpt2 did not affect blood glucose concentration area under the curve in db/db or db/+ mice (Figure 7G), which is consistent with glutamine-stimulated gluconeogenesis not requiring ALT activity. Similarly, knockdown of Gpt2 did not affect the glucose area under the curve in a glucagon tolerance test, which would assess gluconeogenesis from multiple substrates (Figure 7H). These data are consistent with the idea that the glucose-lowering effects of Gpt2 knockdown in obese db/db mice are due to attenuated alanine-stimulated glucose production by the liver.
DISCUSSION
Use of alanine and other amino acids as gluconeogenic substrates is increased in diabetes and contributes to hyperglycemia (Andrikopoulos and Proietto, 1995; Chan et al., 1975; Song et al., 2001), but the molecular mechanisms involved remain understudied. Herein, we show that expression and protein abundance of ALT2 is increased in rodent and human models of obesity, and that, in db/db mice, increased Gpt2 expression is driven by the ER stress-activated transcription factor ATF4. We also demonstrate that suppression or genetic deletion of liver ALT2 does not affect glucose concentrations in lean mice but, in db/db mice, lowers blood glucose and suppresses metabolism of amino acids to fuel gluconeogenesis. One possible explanation is that the alanine-driven gluconeogenic pathway, which is activated in diabetic liver, is only critical in this state. Alternatively or in addition, it is possible that normal liver is more heavily reliant on cytosolic ALT1 activity to convert alanine to pyruvate, but shifts to the mitochondrial pathway in diabetes. Collectively, these data suggest that approaches to specifically target ALT activity in diabetic liver could lower blood glucose by impinging upon the flux of amino acids into gluconeogenesis.
The vast preponderance of previous work on the ALT enzymes has focused on their utility as clinical biomarkers for liver or muscle injury, but relatively little work has focused on their important metabolic functions. It is generally assumed that ALT is released by damaged hepatocytes or myocytes in an unregulated fashion. However, it is possible that increases in serum ALT may also reflect elevations in hepatic ALT expression, especially in the context of obesity and metabolic associated fatty liver disease. Consistent with the current data, there is also previous evidence that the ALT enzymes are transcriptionally induced with obesity and in steatotic liver (Jadaho et al., 2004; Liu et al., 2009; Okun et al., 2021), which could also contribute to the plasma ALT pool. Plasma ALT levels may also have prognostic value beyond liver injury since recent work has demonstrated a strong correlation between increased serum ALT levels and metabolic dysfunction in patients (Schindhelm et al., 2006) and is predictive of future development of diabetes (De Silva et al., 2019). This is often interpreted to suggest that nonalcoholic fatty liver disease may drive insulin resistance and development of type 2 diabetes. However, ALT-driven stimulation of gluconeogenesis from alanine may also directly promote development of hyperglycemia. Human mutations in GPT2 or germline deletion of Gpt2 in mice has profound effects on neurologic development (Ouyang et al., 2016). In addition, expression of Gpt2 is activated in skeletal muscle in response to thyroid hormone and Gpt2 knockout mice are insensitive to the anabolic effects of thyroid hormone on muscle mass after experimental denervation (Cicatiello et al., 2022). These observations illustrate important metabolic and biologic roles of ALT enzymes beyond their utility as biomarkers.
Also consistent with this, and while this manuscript was in preparation, Okun et al. (2021) demonstrated that ALT2 abundance was increased in liver of humans with type 2 diabetes and in several mouse models of obesity and diabetes. Consistent with the present studies, they showed that concomitant silencing of Gpt and Gpt2 in db/db liver attenuated hyperglycemia, and led to reduced blood glucose excursions in ATT and increased plasma alanine concentrations. The main findings of that work are remarkably congruent with the present studies. In addition to confirming several of their key findings, we show that marked weight loss leads to a reduction in Gpt2 expression in people with obesity and have developed and characterized the phenotype of mice with liver-specific deletion of Gpt2. We show, using tracer-based approaches, that loss of Gpt2 enzyme attenuates alanine utilization in hepatocytes but does not affect blood glucose concentrations in lean mice challenged with amino acid tolerance tests or overnight fasting. We also demonstrate that Gpt2 silencing produces a metabolic benefit without sensitizing db/db mice to the effects of insulin. Together, these two studies reproducibly demonstrate the effectiveness of silencing hepatic ALT activity as a potential therapeutic approach for hyperglycemia.
Our data suggest that the increase in hepatic Gpt2 expression that occurs in diabetes is mediated by the ER stress-activated transcription factor ATF4. These findings are consistent with previous work indicating that Gpt2 is a direct target gene of ATF4. Indeed, the proximal promoter of the mouse Gpt2 gene contains a canonical ATF4 binding motif (Han et al., 2013; Lee et al., 2015), which was bound by ATF4 in ChIP-seq studies (Han et al., 2013), and is directly activated by ATF4 in promoter-reporter analyses (Salgado et al., 2014). The regulation of Gpt2 by ATF4 is consistent with a coordinated regulation of amino acid catabolism by this transcription factor (Han et al., 2013), which could be an adaptive mechanism to dispose of amino acids released by proteolysis as part of the “unfolded protein response.” Previous work has indicated that ATF4 null mice are protected from diet-induced obesity and hyperglycemia (Seo et al., 2009), which is consistent with the effects of ATF4 on activating Gpt2 expression and gluconeogenesis from amino acids. On the other hand, Okun et al. (2021) found that glucocorticoid signaling was involved in the induction of Gpt2 expression in their studies. It remains to be determined whether the ATF4 and glucocorticoid receptor-mediated effects operate in parallel or in series to control Gpt2 expression.
Both ALT enzymes are bidirectional for the net production or consumption of alanine. In rat liver, some studies suggest that ALT1 is predominantly involved in alanine formation from pyruvate while ALT2 is more important for alanine catabolism (Dieterle et al., 1978; Glinghammar et al., 2009). This would suggest that alanine conversion to pyruvate occurs mostly in the mitochondrial matrix after alanine import into the mitochondrion. Previous research has shown that neutral amino acids are readily transported into the mitochondrion for further metabolism (Cybulski and Fisher, 1977). The present studies are consistent with this model since the disruption of the mitochondrial isoform of ALT suppressed alanine entry into the mitochondrial TCA cycle. However, we still found significant metabolism of alanine after Gpt2 knockout. This could be due to cytosolic metabolism of alanine by other enzymes, including ALT1. Although the Km of ALT1 for alanine is much higher than for pyruvate (DeRosa and Swick, 1975; Glinghammar et al., 2009), cytosolic alanine accumulation could affect the directionality of ALT1 and allow alanine to enter into the mitochondrion as pyruvate via the MPC (Figure 1A). Indeed, the MPC inhibitor UK5099 markedly reduced alanine enrichment in citrate in Gpt2-deficient hepatocytes, but had only modest effects in WT cells. Furthermore, some prior work suggests that much of the alanine entering the gluconeogenic pathway is converted to pyruvate in the cytosol, presumably by ALT1 (Patel and Olson, 1985). It is possible that both routes are important for alanine metabolism, but the relative importance depends upon the cell type, physiologic or pathophysiologic context, or the subcellular concentration of each substrate for the bidirectional interaction.
In conclusion, we show that Gpt2 expression is activated in diabetic liver in an ATF4-mediated manner. Gpt2 silencing in liver had no effect in lean mice, but alleviated alanine-induced hyperglycemia in db/db mice; likely by reducing the incorporation of alanine into newly synthesized glucose. Our results are consistent with a significant role for ALT2 in hepatic gluconeogenesis from amino acids and in the regulation of blood glucose levels in obesity and diabetes.
Limitations of the study
There are limitations to this study that should be discussed. Although our data indicate that GPT2 expression is decreased by marked weight loss after GBS, we were not able to compare the expression of this gene or the protein abundance between people with obesity or normal weight as we did in the mouse models. Obtaining liver biopsies from people with normal weight is extremely difficult to justify to the IRB due to risk of internal bleeding. Also, we have not yet examined the effect of genetic deletion of Gpt2 in livers of obese mice and it is possible that chronic deletion of the gene might lead to compensatory changes in metabolism, such as alanine-pyruvate cycling, which was observed in lean knockout mice. Finally, the therapeutic potential of inhibiting ALT activity is limited at this time due to lack of specific inhibitors for this transaminase reaction. Existing inhibitors, such as β-chloroalanine and amino-oxyacetate, likely inhibit other transaminases including aspartate aminotransferase.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Brian N. Finck bfinck@wustl.edu.
Materials availability
Adenoviral vectors and LS-Gpt2−/− mice are available upon request by academic researchers for non-commercial reasons after institutional material transfer agreement approval.
Data and code availability
Bulk RNA sequencing data were uploaded to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession number GSE199975.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
Eight subjects with a BMI of >35 kg/m2 (1 man and 7 women) between the ages of 35 and 52 years were recruited from the Bariatric Surgery Program at Barnes-Jewish Hospital (St. Louis, Missouri, USA). Written informed consent was obtained from all subjects before their participation in these studies, which were approved by the Institutional Review Board at Washington University School of Medicine in St. Louis, MO and registered in ClinicalTrials.gov (NCT00262964). All subjects completed a comprehensive screening evaluation, including a medical history and physical examination, and standard blood tests. Potential participants who had a history of liver disease other than NAFLD or consumed excessive amounts of alcohol (>21 units/wk for men and >14 units/wk for women) were excluded.
Animal studies
All experiments involving mice were approved by the Institutional Animal Care and Use Committee of Washington University in St. Louis and are consistent with best practices in the Guide for the Care and Use of Laboratory Animals. Mice that lack Gpt2 were generated by the Knockout Mouse Project Repository (KOMP) (project ID CSD24977) using the “knockout first” approach wherein the LacZ/Neo cassette constitutively blocks expression of the gene until it is removed by flp recombinase. This construct also contained exon 4 of the Gpt2 gene flanked by LoxP sites. We purchased frozen sperm from Gpt2 germline heterozygous mice from KOMP, established a colony of mice by in vitro fertilization, and then intercrossed these mice to generate germline knockouts. We also crossed germline heterozygotes with mice expressing flp recombinase in a global manner (B6.Cg-Tg(ACTFLPe)9205Dym/J mice; Jackson Laboratory stock number: 005703) to remove the LacZ and Neo cassettes and generate Gpt2 floxed mice. Gpt2 floxed mice were then crossed with transgenic mice expressing Cre under control of the albumin promoter (B6.Cg-Speer6-ps1Tg(Albcre)21Mgn/J; Jackson Laboratory stock number: 003574) to create LS-Gpt2−/− mice. Littermate mice not expressing Cre (fl/fl mice) were used as control mice in experiments involving LS-Gpt2−/− mice. Littermate db/db and heterozygous (db/+) (stock number: 000697) littermate mice were purchased from the Jackson Laboratory as well.
Mice were housed in a specific pathogen free animal facility at Washington University School of Medicine under a 12 h light/dark cycle (lights on from 0600-1800) and studies were conducted during the light cycle. Animals were given ad libitum access to food and water unless otherwise indicated. All animals were studied between 10 and 29 weeks of age and both male and female mice were used in studies as indicated in the text and Figure legends.
METHOD DETAILS
Human metabolic studies
Subjects were admitted to the Clinical Research Unit at Washington University School of Medicine in St. Louis in the evening on day 1 where they consumed a standard meal. After the evening meal was consumed subjects fasted except for water, until completion of the study the following day. On the morning of day 2, catheters were inserted into an arm vein for the infusion of stable isotopically labeled glucose and into a contralateral dorsal hand vein, which was heated to obtain arterialized blood samples. After baseline blood samples were obtained to assess background plasma glucose tracer enrichment, a primed (22.5 μmol/kg), constant infusion (0.25 μmol/kg/min) of [6,6-2H2]glucose (Cambridge Isotope Laboratories Inc.) was started and maintained for 210 min. Blood samples were drawn every 10 min during the last 30 min to assess plasma glucose and insulin concentrations and plasma glucose enrichment.
Human plasma measurements and insulin sensitivity calculations
Plasma glucose concentration was determined using the glucose oxidase method (Yellow Spring Instruments Co.). Plasma insulin concentrations were measured by using radioimmunoassay kits (Linco Research, St Louis, MO). Plasma proteins were precipitated with ice-cold acetone, and hexane was used to extract plasma lipids. The aqueous phase, containing glucose, was dried by speed-vac centrifugation (Savant Instruments, Farmingdale, NY). The plasma glucose tracer-to-tracee ratio (TTR) was determined by gas-chromatography/mass-spectrometry (MSD 5973 system with capillary column; Hewlett-Packard, Palo Alto, CA) after heptafluorobutyric (HFB) anhydride was used to form an HFB derivative of glucose. Glucose TTR were determined by selectively monitoring ions at mass-to-charge ratios (m/z) 519 and 521 (Korenblat et al., 2008). Hepatic insulin sensitivity was calculated as the inverse of the product of plasma insulin concentration and the endogenous glucose rate of appearance (Ra) into the systemic circulation, determined by dividing the glucose tracer infusion rate by the average plasma glucose TTR (Korenblat et al., 2008). The homeostasis model assessment of insulin resistance (HOMA-IR) was calculated by dividing the product of the plasma concentrations of insulin (in μU/mL) and glucose (in mmol/L) by 22.5 (Matthews et al., 1985).
Human liver sample collection
After completing baseline testing participants underwent Roux-en-Y gastric bypass surgery (RYGBS), according to standard clinical practice procedures. During the procedure, a liver biopsy was obtained under direct visualization. Samples were frozen immediately in liquid nitrogen and stored at −80°C until processing. About 1-year after surgery, the metabolism study was repeated with liver tissue also sampled by percutaneous needle liver biopsy in the radiology suite at Mallinckrodt Institute of Radiology at Washington University School of Medicine. Tissue obtained in these biopsies was used to quantify the expression of genes encoding ALT1 (GPT1) and ALT2 (GPT2) before and after RYGBS-induced weight loss. Tissue obtained from some subjects at time of surgery was used for protein isolation and western blotting analyses. Cadaveric donor liver samples were obtained from donor organs deemed unsuitable for transplantation.
High fat diet mouse studies
For high fat diet studies, male diet-induced obese mice in the C57BL/6J strain (stock number: 380050) or age matched lean controls (stock number: 380056) were purchased from Jackson Labs at 12 weeks of age after being on a 60 kcal% fat diet (#D12492 Research Diets, Inc.) or 10 kcal% low fat control diet (#D12450 Research Diets, Inc.), respectively, for 6 week. Alanine tolerance or glutamine tolerance tests were conducted 17 weeks of age and mice were sacrificed after 23 weeks on diet.
ATF4 ASO studies
To silence ATF4, male db/db and db/+ mice (purchased from Jackson Laboratories) received intraperitoneal injections of 25 mg/kg body weight antisense oligonucleotide (ASO) directed against ATF4 twice a week for 4 weeks. Control mice were injected with scrambled control ASO by the same route, dose, and frequency. The ASO against ATF4 (product ID: 489707; sequence GCAGCAGA GTCAGGCTTCCT) and the scrambled control ASO (product ID: 141923; sequence CCTTCCCTGAAGGTTCCTCC) were obtained from Ionis Pharmaceuticals (Carlsbad, CA). ASO treatment was initiated in db/+ and db/db mice at 8 weeks of age. After treatment with ASOs for 4 weeks, mice were sacrificed and tissues were harvested, frozen in liquid nitrogen, and stored at −80°C for further analysis.
Gpt2 shRNA studies
For experiments involving adenoviral mediated silencing of Gpt2, 6 week old male db/db and db/+ mice were anesthetized and infected by retro-orbital injection of ~200 μL high-titer adenovirus and experiments were performed 5 to 8 days post injection. Adenovirus expressing shRNA targeting mouse Gpt2 (shADV-260703) was obtained from Vector BioLabs and has been previously described (McCommis et al., 2015). The control adenovirus expressing shRNA targeting LacZ, as well as a GFP reporter, has been described (McCommis et al., 2015). Mice were administered between 1.0 and 1.5 × 10^13 viral particles per mouse for both viruses by intravenous injection.
Metabolic tolerance tests
L-alanine tolerance tests (ATT), L-glutamine tolerance tests (QTT), pyruvate tolerance tests (PTT), and glucagon tolerance tests were performed on mice fasted overnight (16 h) and housed on aspen chip bedding. For studies involving obese mice (DIO or db/db), lean body mass was determined by EchoMRI and all mice were dosed based on lean body mass (kg). Mice were injected intraperitoneally with 2 g/kg lean body mass L-alanine (ATT), 1 g/kg lean body mass L-glutamine (QTT), or 1 g/kg lean body mass pyruvate (PTT) dissolved in sterile saline. For glucagon tolerance tests, mice were injected i.p. with 1 mg/kg of lean body mass glucagon (MilliporeSigmaG2044) dissolved in 0.9% saline following a 16 h overnight fast. Insulin tolerance tests (ITT) were performed by injecting i.p. 0.75 U/kg lean body weight insulin (Humulin), after a 5 h fast. For studies involving db/db or DIO mice, lean body mass was determined by echo MRI prior to testing and doses were calculated using those values. Blood glucose was measured using a One-Touch Ultra glucometer (LifeScan) with a single drop of tail blood serially after challenge. For all tolerance tests, the total area under the curve (AUC) was calculated using the trapezoidal rule (Vigueira et al., 2014).
For overnight fasting studies, male and female mice were placed on aspen woodchip bedding and food was removed at the onset of the dark phase (18:00 h) and mice were sacrificed for tissue and blood collection 18 h later.
Blood and serum parameters
Blood samples for plasma insulin quantification were collected by puncture of the inferior vena cava and transferred to an EDTA coated Eppendorf tube. Plasma was isolated following centrifugation at 8,000 × G for 8 min at 4°C and frozen at −80°C. Insulin levels were measured using a Singulex mouse insulin assay according to the manufacturer’s instructions at the Core Laboratory for Clinical Studies, Washington University School of Medicine. Plasma triglycerides (Thermo Fisher, TR22421), total cholesterol (Thermo Fisher, TR13421), and glycerol (Millipore Sigma, F6428) were measured using commercially available colorimetric enzymatic assays. Plasma total ketone bodies were measured using a commercially available kinetic assay (Fujifilm WAKO, Autokit Total Ketone Bodies).
Plasma amino acid concentrations
Plasma amino acid samples were processed as previously described (Cappel et al., 2019). Briefly, plasma samples (25 μL) were mixed with a labeled amino acid internal standard (Cambridge Isotopes) and 5% perchloric acid. Samples were centrifuged to remove precipitate, and dried. Amino acids were then derivatized as previously described (Casetta et al., 2000). In short, the sample pellet was reconstituted in BuOH-HCL to form amino acid butyl esters. Amino acid derivatives were then separated using a reverse phase C18 column (Xbridge, Waters, Milford, MA′ 150 × 2.1 mm, 3.0 μm) with a gradient elution and detected using the MRM mode by monitoring specific transitions under positive electrospray on API 3200 triple quadrupole LC/MS/MS mass spectrometer (Applied Biosystems/Sciex Instruments). Data analysis and quantification involved comparisons of individual ion peaks to that of the internal standard for each amino acid.
Plasma organic acid concentrations
Organic acids were processed as previously described (Cappel et al., 2019). Briefly, thawed plasma samples (25 μL) were mixed with internal standard (Isotec), 0.8% sulfosalicylic acid and 5 M hydroxylamine-HCl. Samples were centrifuged, and 2 M KOH was added to neutralize the supernatant (pH 6-7), which was then heated (65°C) for 1 h. After incubation, 2 M HCL was added to acidify (pH 1-2) each sample before saturating with sodium chloride and extracting with ethyl acetate. The extract was dried and derivatized using acetonitrile and MTBSTFA as a silylation reagent while heating at 60°C for 1 h. Derivatives were then analyzed using both scan and SIM modes with an Agilent 7890A gas chromatography interfaced to an Agilent 5975C mass-selective detector (70 eV, electron ionization source). An HP-5ms GC column (30 m × 0.25 mm I.D., 0.25 μm film thickness) was used for all analyses (DesRosiers et al., 1994). Data analysis and quantification involved comparisons of individual ion peaks to that of the internal standard for each organic acid.
Liver RNA and protein analyses
Liver tissue was collected at sacrifice and snap frozen in liquid nitrogen. Total RNA from livers or hepatocytes was isolated using RNA-Bee (Tel-Test). Complementary DNA was made by using a reverse transcription kit (Invitrogen), and real-time quantitative PCR was performed using an ABI PRISM 7500 sequence detection system (Applied Biosystems) and a SYBR green master mix. Arbitrary units of target mRNA were normalized by the Comparative Ct Method (ΔΔCT Method) to levels of 36B4 mRNA. All sequences of the oligonucleotides can be found in the Tables S3 and S4.
Bulk RNA sequencing and analysis
Samples were prepared according to library kit manufacturer’s protocol, indexed, pooled, and sequenced on an Illumina HiSeq. Basecalls and demultiplexing were performed with Illumina’s bcl2fastq software and a custom python demultiplexing program with a maximum of one mismatch in the indexing read. RNA-seq reads were then aligned to the Ensembl release 76 primary assembly with STAR version 2.5.1a (Dobin et al., 2013). Gene counts were derived from the number of uniquely aligned unambiguous reads by Subread:featureCount version 1.4.6-p5 (Liao et al., 2014). Isoform expression of known Ensembl transcripts were estimated with Salmon version 0.8.2 (Patro et al., 2017). Sequencing performance was assessed for the total number of aligned reads, total number of uniquely aligned reads, and features detected. The ribosomal fraction, known junction saturation, and read distribution over known gene models were quantified with RSeQC version 2.6.2 (Wang et al., 2012).
All gene counts were then imported into the R/Bioconductor package EdgeR (Robinson et al., 2010) and TMM normalization size factors were calculated to adjust for samples for differences in library size. Ribosomal genes and genes not expressed in the smallest group size minus one samples greater than one count-per-million were excluded from further analysis. The TMM size factors and the matrix of counts were then imported into the R/Bioconductor package Limma (Ritchie et al., 2015). Weighted likelihoods based on the observed mean-variance relationship of every gene and sample were then calculated for all samples with the voomWithQuality-Weights (Liu et al., 2015). The performance of all genes was assessed with plots of the residual standard deviation of every gene to their average log-count with a robustly fitted trend line of the residuals. Differential expression analysis was then performed to analyze for differences between conditions and the results were filtered for only those genes with Benjamini-Hochberg false-discovery rate adjusted p-values less than or equal to 0.05.
For each contrast extracted with Limma, global perturbations in known KEGG pathways were detected using the R/Bioconductor package GAGE (Luo et al., 2009) to test for changes in expression of the reported log 2 fold-changes reported by Limma in each term versus the background log 2 fold-changes of all genes found outside the respective term. Perturbed KEGG pathways where the observed log 2 fold-changes of genes within the term were significantly perturbed in a single-direction versus background or in any direction compared to other genes within a given term with p-values less than or equal to 0.05.
Western blotting analyses
For western blotting, liver lysates were collected in lysis buffer (150 mM NaCl, 20 mM Tris (pH = 7.4), 1 mM EDTA, 0.2% NP-40, 10% glycerol) with protease inhibitors using a Tissuelyser. Lysates were normalized to protein concentration, denatured, and run on Criterion precast PAGE gels (BioRad). The antibodies used in this study were ALT2 (MilliporeSigmaHPA051514), ALT1 (GPT Abcam ab202083), Complex III (Oxphos Cocktail; Abcam ab110413), GAPDH (Invitrogen AM4300), AKT (Cell Signaling Technology 2920), pThr308 AKT (Cell Signaling Technology 2920), pSer473 AKT (Cell Signaling Technology 2920), or tubulin (MilliporeSigmaMonoclonal Anti-α- Tubulin Clone B-5-1-2, T51668).
Liver ALT assays
For ALT assays approximately 100 mg liver tissue was homogenized in 1 mL of homogenization buffer (25 mM HEPES, 5 mM EDTA, 0.1% CHAPS, pH 7.4) with protease inhibitors using a Tissuelyser. After centrifugation at 12,000 g for 5 min, the supernatants were diluted 1:10 in homogenization buffer. ALT activity was measured in the diluted supernatants using a kit from Teco Diagnostics according to the manufacturer’s instructions. The results were normalized to protein concentration and reported in standard units.
Liver glycogen assays
Frozen liver samples (30-90 mg) were hydrolyzed in 0.3 mL of 30% KOH solution in a boiling water bath for 30 min. Samples were vortexed at 10 and 20 min during the incubation to facilitate digestion. Samples were allowed to cool to room temperature before adding 0.1 mL of 1 M Na2SO4 and 0.8 mL of 100% EtOH. Next, samples were boiled for 5 min then centrifuged at 10,000G for 5 min. Liquid was aspirated and the remaining glycogen pellet was dissolved in 0.2 mL of water, followed by two additional ethanol washes. The final glycogen pellet was dried in a speed vacuum and dissolved in 0.2 mL of 0.3 mg/mL amyloglucosidase in 0.2 M sodium acetate buffer (pH 4.8) and incubated for 3 h at 40°C. The reaction mixture was diluted two-to fivefold with water. Glucose concentration was determined with a glucose assay kit (Sigma-Aldrich; cat# GAGO20).
Liver lipidomic analyses
Lipid species quantification was performed by the Washington University Metabolomics Facility. Mouse liver samples were homogenized in water (4 mL/g liver). TAG, DAG, and ceramide were extracted from 50 –L of homogenate using Blyth-Dyer lipid extraction method. TAG(17:1, 17:1, 17:1), d5-DAG(18:0, 18:0), and ceramide (17:0) were used as internal standards. Internal standards were added to the samples before extraction. Quality control (QC) samples were prepared by pooling the aliquots of the study samples and were used to monitor the instrument stability. The QC was injected six times in the beginning to stabilize the instrument, and was injected between every 5 study samples.
Measurement of TAG and ceramide was performed with a Shimadzu 20AD HPLC system coupled to an AB Sciex 4000QTRAP mass spectrometer operated in positive multiple reaction monitoring mode. Data processing was conducted with Analyst 1.6.3. Measurement of DAG was performed with a Shimadzu 10AD HPLC system and a Shimadzu SIL-20AC HT auto-sampler coupled to a Thermo TSQ Quantum Ultra mass spectrometer operated in positive selected reaction monitoring mode. Data processing was conducted with XCalibur 1.02. Only lipid species with CV < 15% in QC sample were reported. The relative quantification of lipids was provided, and the data were reported as the peak area ratios of the analytes to the corresponding internal standards. The relative quantification data generated in the same batch are appropriate to compare the change of an analyte in a test sample relative to other samples (e.g., control vs. treated). Lipidomic analyses were conducted by the Washington University Metabolomics Core.
Hepatocyte isolation and studies
Hepatocytes were isolated by perfusing livers of anesthetized mice with DMEM media containing collagenase from Clostridium histolyticum (MilliporeSigma) as reported before (McCommis et al., 2015). Briefly, hepatocytes were plated overnight in collagen coated 12 well-plates (200,000 cells/mL) with DMEM containing 10% FBS plus an antibiotic cocktail (penicillin/streptomycin and amphotericin) and washed twice with glucose-free Hank’s balanced salt solution (GF-HBSS) containing 127 mM NaCl, 3.5 mM KCl, 0.44 mM KH2PO4, 4.2 mM NaHCO3, 0.33 mM Na2HPO4, 1 mM CaCl2, 20 mM HEPES, pH 7.4.
In vitro ATF4 overexpression or knockdown studies
For experiments involving adenovirus mediated overexpression of ATF4, 1 μL (low) or 5 μL (high) of Ad-ATF4 was added to the media as indicated. Adenovirus overexpressing mouse ATF4 (ADV-253208) and the control β-gal-expressing adenovirus (catalog number: 1080) were obtained from Vector Biolabs. Hepatocytes treated with 10 μL of an adenovirus expressing β-gal served as a control. Cells remained in culture for 24 h following adenoviral transduction. For experiments involving ASO treatment, Lipofectamine RNAiMAX Transfection Reagent (ThermoFisher) was used per manufacturer instructions and treated hepatocytes remained in culture for 48 h. After 42 h of ASO treatment, 2 μg/mL of tunicamycin (Sigma) or an equal volume of DMSO (Sigma), which served as vehicle control; was added to the indicated wells for the remainder of the experiment. Following treatment of hepatocytes for the indicated times, media was removed and 1 mL of RNA Bee (Tel-Test) was added to each well to collect cells for RNA isolation. Complimentary DNA synthesis and real-time PCR were performed as described for liver tissue.
Hepatocyte glucose production assays
Hepatocyte glucose production assays were performed as described (McCommis et al., 2015). The morning after isolation, cells were washed 2X with PBS, and starved for 2 h in HBSS (containing 127 mM NaCl, 3.5 mM KCl, 0.44 mM KH2PO4, 4.2 mM NaHCO3, 0.33 mM Na2HPO4, 1 mM CaCl2, 20 mM HEPES, pH 7.4). HBSS was removed, cells were washed in fresh HBSS, and then treated for 3 h in HBSS containing glucagon (100 ng/mL) alone or with 5 mM alanine, glutamine, or sodium pyruvate. After the 3-h incubation, media was collected and glucose concentrations were measured using a glucose oxidase-based glucose assay kit (MilliporeSigma; cat# GAGO20). Glucose concentrations were normalized to cell protein amount measured by Micro BCA kit (ThermoFisher).
13C-amino acid tracer studies
For studies using 13C labeled tracer metabolites, the morning after isolation, cells were rinsed with PBS twice. No starving was done. Cells were treated with HBSS solution containing glucagon (100 ng/mL) alone (for background calculations) or 20 mM 100% uniformly 13C-labeled alanine or 5 mM 100% uniformly labeled 13C-labeled glutamine in 600 μL per well and allowed to incubate for 3 h. When indicated, cells were treated with 5 μM UK5099 MilliporeSigma (St. Louis, MO) or vehicle control, which was added at the time of 13C-labeled alanine provision. The media and cells were harvested and the extraction solvent (2:2:1 of methanol: acetonitrile: water) was mixed with media (1 part media: 9 parts extraction solvent) prior to being vortexed for 1 min prior and placed at −20°C for an hour. Samples were centrifuged at 14,000 X g and 4°C for 10 min, and the supernatant stored at −80°C until metabolite analysis. For cell harvest and extraction, cells were washed twice with PBS and twice with HPLC-grade water. Cold HPLC-grade methanol was used for quenching, and cells were scraped and the lysates transferred to sterile Eppendorf tubes. Samples were dried in a SpeedVac for 2-6 h. Dried samples were reconstituted in 1 mL of cold methanol:acetonitrile:water (2:2:1), and subjected to three cycles of vortexing, freezing in liquid nitrogen, and 10 min of sonication at 25°C. Samples were then stored at −20°C for 1 h. After this, samples were centrifuged at 14,000 X g and 4°C. The protein content of pellets was measured by Micro BCA kit (ThermoFisher). Supernatants were transferred to new tubes and dried by SpeedVac for 2-5 h. After drying, 1 μL of water:acetonitrile (1:2) was added per 2.5 μg of cell protein in pellets obtained after extraction. Samples were subjected to two cycles of vortexing and 10 min of sonication at 25°C. Next, samples were centrifuged at 14,000 X g and 4°C for 10 min, transferred supernatant to LC vials, and stored at −80°C until MS analysis.
Huh7 studies
For siRNA experiments, Huh7 cells were transfected with either GPT2 siRNA (Thermofisher; Cat# AM16708; ID# 112332) or negative control siRNA (Thermofisher; Cat# 4404021) using Lipofectamine RNAiMAX Transfection Reagent (Thermofisher) according to manufacturer’s instructions. After 72 h, cells were collected in lysis buffer for western blotting.
Metabolite analysis by LC/MS
Ultra-high performance LC (UHPLC)/MS was performed with a Thermo Scientific Vanquish Horizon UHPLC system interfaced with a Thermo Scientific Orbitrap ID-X Tribrid Mass Spectrometer (Waltham, MA). Hydrophilic interaction liquid chromatography (HILIC) separation was accomplished by using a HILICON iHILIC-(P) Classic column (Tvistevagen, Umea, Sweden) with the following specifications: 100 mm × 2.1 mm, 5 μm. Mobile-phase solvents were composed of A = 20 mM ammonium bicarbonate, 0.1% ammonium hydroxide (adjusted to pH 9.2), and 2.5 μM medronic acid in water:acetonitrile (95:5) and B = 2.5 μM medronic acid in acetonitrile:-water (95:5). The column compartment was maintained at 45°C for all experiments. The following linear gradient was applied at a flow rate of 250 μL min−1: 0-1 min: 90% B, 1-12 min: 90-35% B, 12-12.5 min: 35-25% B, 12.5-14.5 min: 25% B. The column was re-equilibrated with 20 column volumes of 90% B. The injection volume was 2 μL for all experiments.
Data were collected with the following settings: spray voltage, −3.5 kV; sheath gas, 35; auxiliary gas, 10; sweep gas, 1; ion transfer tube temperature, 275°C; vaporizer temperature, 300°C; mass range, 67-1500 Da, resolution, 120,000 (MS1), 30,000 (MS/MS); maximum injection time, 100 ms; isolation window, 1.6 Da. LC/MS data were processed and analyzed with the open-source Skyline software (Adams et al., 2020). Natural-abundance correction of 13C for tracer experiments was performed with AccuCor (Su et al., 2017).
QUANTIFICATION AND STATISTICAL ANALYSIS
Figures were prepared using Prism version 8.0.1 for Windows (GraphPad Software, La Jolla California USA, www.graphpad.com). All human data in Table S1 are presented as the mean ± SD. All animal data and human data in Figure 1 are presented as the mean ± SEM. All data sets were tested for normality by using Shapiro-Wilk’s test, and skewed data sets were log transformed for further analysis. Statistical significance was calculated using an unpaired Student’s t-test, two-way analysis of variance (ANOVA) with repeated measures, or one-way ANOVA with Tukey’s multiple comparisons test, with a statistically significant difference defined as p ≤ 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ALT2 | Sigma-Aldrich | cat#HPA051514; RRID:AB_2681516 |
| Rabbit monoclonal anti-GPT | Abcam | cat#ab202083 |
| Mouse monoclonal anti-OXPHOS Rodent Cocktail | Abcam | cat#ab110413; RRID:AB_2629281 |
| Mouse monoclonal anti-α-Tubulin | Sigma-Aldrich | cat#A11126; RRID:AB_2534135 |
| Mouse monoclonal anti-GAPDH | Invitrogen | cat#AM4300; RRID:AB_2536381 |
| Rabbit polyclonal anti-pSer473 AKT | Cell Signaling Technology | cat#9271; RRID:AB_329825 |
| Rabbit polyclonal anti-pThr308 AKT | Cell Signaling Technology | cat#9275; RRID:AB_329828 |
| Mouse monoclonal anti-AKT | Cell Signaling Technology | cat#2920; RRID:AB_1147620 |
| IRDye 800CW Goat Anti-Rabbit IgG Secondary Antibody | LI-COR | cat#926-32211; RRID:AB_621843 |
| IRDye 680RD Donkey Anti-Rabbit IgG Secondary Antibody | LI-COR | cat#926-68073; RRID:AB_10954442 |
| IRDye 680RD Goat Anti-Mouse IgG Secondary Antibody | LI-COR | cat#926-68070; RRID:AB_10956588 |
| Bacterial and virus strains | ||
| Mouse GPT2 shRNA silencing Adenovirus | Vector Biolabs | cat# shADV-260703 |
| Control LacZ- GFP Adenovirus | ThermoFisher | cat# K494100 |
| mouse ATF4 Adenovirus | Vector Biolabs | cat# ADV-253208 |
| b-gal/LacZ Adenovirus | Vector Biolabs | cat# 1080 |
| Biological samples | ||
| Human cadaveric donor liver tissue | Midwest Transplant Services | N/A |
| Pre/Post RYGBS liver samples | Bariatric Surgery Program at Barnes-Jewish Hospital (St. Louis, Missouri) | N/A |
| Blood samples | Clinical Research Unit, Washington University in St. Louis School of Medicine | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM - Dulbecco’s Modified Eagle Medium | ThermoFisher | cat#11960085 |
| Fetal Bovine Serum | Gibco | cat#26140-079 |
| Collagenase from Clostridium histolyticum | Millipore Sigma | cat#C5138 |
| Lipofectamine RNAiMAX | ThermoFisher | cat#13778075 |
| [6,6-2H]glucose | Cambridge Isotope Laboratories Inc. | cat#DLM-349-PK |
| Insulin (Humulin) | Eli Lilly and Company | cat#Humulin R U-100 |
| Glucagon | Millipore Sigma | cat#G2044 |
| RNA-Bee | Tel-Test | cat#CS-501B |
| Tunicamycin | Millipore Sigma | cat#T7765 |
| Penicillin-Streptomycin (10,000 U/mL) | Gibco | cat#15140122 |
| Amphotericin B | Gibco | cat#15290026 |
| L-Alanine | Millipore Sigma | cat#A7627 |
| L-Glutamine | Millipore Sigma | cat#G3126 |
| Sodium Pyruvate | Millipore Sigma | cat#P4562 |
| UK5099 | Millipore Sigma | cat#PZ0160 |
| Critical commercial assays | ||
| Glucose and Lactate Analyzer | Yellow Spring Instruments Co. | cat#YSI2300 or cat# YSI2900 analyzer |
| Human Insulin-Specific RIA Kit | Linco Research, Inc. | cat#EZHIASF-14K |
| One-Touch Ultra glucometer | LifeScan | N/A |
| Reverse transcription kit | Thermo Fisher (Applied Biosystems) | cat#4368814 |
| Power SYBR Green | Thermo Fisher (Applied Biosystems) | cat#4367659 |
| ALT (SGPT) Kinetic | Teco Diagnostics | cat#A524-150 |
| Erenna Immunoassay System | Singulex | N/A |
| Glucose Oxidase Activity Assay Kit | Millipore Sigma | cat#GAGO20 |
| Thermo Scientific Vanquish Horizon UHPLC | Thermo Scientific | cat#IQLAAAGABHFAPUMZZZ |
| Infinity Triglycerides | Thermo Scientific | cat#TR22421 |
| Infinity Cholesterol | Thermo Scientific | cat#TR13421 |
| Deposited data | ||
| RNA sequencing data | This paper | GSE199975 |
| Experimental models: Cell lines | ||
| Huh7 Cells | Glow Biologics | cat# GBTC-099H |
| Experimental models: Organisms/strains | ||
| Mouse - diet-induced obese mice in the C57BL/6J strain | Jackson Laboratories | Stock# 380050 |
| Mouse – lean controls for diet-induced obese mice in the C57BL/6J strain | Jackson Laboratories | Stock# 380056 |
| Mouse Littermate db/db and heterozygous (db/+) | Jackson Laboratories | Stock# 000697 |
| Mouse Gpt2tm1e(KOMP)Wtsi –“Gpt2 knockout” | Knockout Mouse Project Repository | project ID CSD24977 |
| Mouse B6.Cg-Speer6-ps1Tg(Alb-cre)21Mgn/J –“Alb-Cre” | Jackson Laboratories | Stock# 003574 |
| Mouse B6.Cg-Tg(ACTFLPe)9205Dym/J -“Actin-Flp” | Jackson Laboratories | Stock# 005703 |
| Mouse Gpt2 floxed - generated by crossing Gpt2 knockout and Actin-Flp | This paper | N/A |
| Mouse LS-Gpt2−/− generated by crossing Gpt2 floxed with Alb-Cre | This paper | N/A |
| Oligonucleotides | ||
| ATF4 ASO | Ionis Pharmaceuticals (Carlsbad, CA). | ID: 489707 |
| Scrambled ASO | Ionis Pharmaceuticals (Carlsbad, CA). | ID: 141923 |
| Human GPT2 siRNA | Thermo Fisher | Cat# AM16708; ID# 112332 |
| Human negative control siRNA | Thermo Fisher | Cat# 4404021 |
| PCR Oligonucleotides can be found in Table S3 | N/A | N/A |
| Software and algorithms | ||
| Prism Version 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Image Studio Lite Ver 5.2 | Licor | https://www.licor.com/bio/image-studio-lite/ |
Highlights.
ALT2 expression is increased in people and mice with obesity
ALT2 expression is regulated by activating transcription factor 4 in liver
Hepatic ALT2 deletion in lean mice has little effect on gluconeogenesis
Silencing liver ALT2 in obese mice reduces alanine-induced hyperglycemia
ACKNOWLEDGMENTS
This work was funded by NIH grant R01 DK117657 (to B.N.F.). The Core services of the Diabetes Research Center (P30 DK020579) and the Nutrition Obesity Research Center (P30 DK56341) at the Washington University School of Medicine also supported this work. N.K.H.Y. was supported by a training grant (T32 HL134635-05). K.S.M. was supported by (R00 HL136658). M.G.-A. was supported by Dirección General de Asuntos del Personal Académico, Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT) IA208121, UNAM, and UNAM-FQ-PAIP 5000-9171. K.H.H.L. is supported by the Dean’s Scholars Program which is in part funded from a grant from the Burroughs Wellcome Fund (1020047). S.C.B. and J.A.F. were supported by R01 DK078184 and a Robert A. Welch Foundation grant I-1804. Some metabolic analyses were supported by NIH grant R35 ES028365 (to G.J.P.). We thank Michael A. Cooper for assistance with animal experiments and Mitchell R. McGill with assistance in ALT assays.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110733.
DECLARATIONS OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure that the study questionnaires were prepared in an inclusive way. We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an under-represented ethnic minority in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
REFERENCES
- Adams KJ, Pratt B, Bose N, Dubois LG, St John-Williams L, Perrott KM, Ky K, Kapahi P, Sharma V, MacCoss MJ, et al. (2020). Skyline for small molecules: a unifying software package for quantitative Metabolomics. J. Proteome Res 19, 1447–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrikopoulos S, and Proietto J (1995). The biochemical basis of increased hepatic glucose production in a mouse model of type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 38, 1389–1396. [DOI] [PubMed] [Google Scholar]
- Cappel DA, Deja S, Duarte JAG, Kucejova B, Inigo M, Fletcher JA, Fu X, Berglund ED, Liu T, Elmquist JK, et al. (2019). Pyruvate-carboxylase-mediated anaplerosis promotes antioxidant capacity by sustaining TCA cycle and redox metabolism in liver. Cell Metab. 29, 1291–1305.e1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casetta B, Tagliacozzi D, Shushan B, and Federici G (2000). Development of a method for rapid quantitation of amino acids by liquid chromatography-tandem mass spectrometry (LC-MSMS) in plasma. Clin. Chem. Lab. Med 38, 391–401. [DOI] [PubMed] [Google Scholar]
- Chan TM, Young KM, Hutson NJ, Brumley FT, and Exton JH (1975). Hepatic metabolism of genetically diabetic (db/db) mice. I. Carbohydrate metabolism. Am. J. Physiol 229, 1702–1712. [DOI] [PubMed] [Google Scholar]
- Chevalier S, Burgess SC, Malloy CR, Gougeon R, Marliss EB, and Morais JA (2006). The greater contribution of gluconeogenesis to glucose production in obesity is related to increased whole-body protein catabolism. Diabetes 55, 675–681. [DOI] [PubMed] [Google Scholar]
- Cicatiello AG, Sagliocchi S, Nappi A, Di Cicco E, Miro C, Murolo M, Stornaiuolo M, and Dentice M (2022). Thyroid hormone regulates glutamine metabolism and anaplerotic fluxes by inducing mitochondrial glutamate aminotransferase GPT2. Cell Rep. 38, 110409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulski RL, and Fisher RR (1977). Mitochondrial neutral amino acid transport: evidence for a carrier mediated mechanism. Biochemistry 16, 5116–5120. [DOI] [PubMed] [Google Scholar]
- DeSilva NMG, Borges MC, Hingorani AD, Engmann J, Shah T, Zhang X, Luan J, Langenberg C, Wong A, Kuh D, et al. (2019). Liver function and risk of type 2 diabetes: bidirectional mendelian randomization study. Diabetes 68, 1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRosa G, and Swick RW (1975). Metabolic implications of the distribution of the alanine aminotransferase isoenzymes. J. Biol. Chem 250, 7961–7967. [PubMed] [Google Scholar]
- Des Rosiers C, Fernandez CA, David F, and Brunengraber H (1994). Reversibility of the mitochondrial isocitrate dehydrogenase reaction in the perfused rat liver. Evidence from isotopomer analysis of citric acid cycle intermediates. J. Biol. Chem 269, 27179–27182. [PubMed] [Google Scholar]
- Dieterle P, Brawand F, Moser UK, and Walter P (1978). Alanine metabolism in rat liver mitochondria. Eur. J. Biochem 88, 467–473. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felig P (1973). The glucose-alanine cycle. Metabolism 22, 179–207. [DOI] [PubMed] [Google Scholar]
- Felig P, Pozefsk T, Marlis E, and Cahill GF (1970). Alanine: key role in gluconeogenesis. Science 167, 1003–1004. [DOI] [PubMed] [Google Scholar]
- Felig P, and Wahren J (1971). Amino acid metabolism in exercising man. J. Clin. Invest 50, 2703–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, and Viollet B (2010). Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest 120, 2355–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Campusano F, Anaya VH, Robledo-Arratia L, Quezada H, Hernandez H, Riego L, and Gonzalez A (2009). ALT1-encoded alanine aminotransferase plays a central role in the metabolism of alanine in Saccharomyces cerevisiae. Can. J. Microbiol 55, 368–374. [DOI] [PubMed] [Google Scholar]
- Glinghammar B, Rafter I, Lindstrom AK, Hedberg JJ, Andersson HB, Lindblom P, Berg AL, and Cotgreave I (2009). Detection of the mitochondrial and catalytically active alanine aminotransferase in human tissues and plasma. Int. J. Mol. Med 23, 621–631. [DOI] [PubMed] [Google Scholar]
- Gray LR, Sultana MR, Rauckhorst AJ, Oonthonpan L, Tompkins SC, Sharma A, Fu X, Miao R, Pewa AD, Brown KS, et al. (2015). Hepatic mitochondrial pyruvate carrier 1 is required for efficient regulation of gluconeogenesis and whole-body glucose homeostasis. Cell Metab. 22, 669–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol 15, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Samuels Y, Li Q, Krokowski D, Guan BJ, Wang C, Jin Z, Dong B, Cao B, Feng X, et al. (2016). Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun 7, 11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadaho SB, Yang RZ, Lin Q, Hu H, Anania FA, Shuldiner AR, and Gong DW (2004). Murine alanine aminotransferase: cDNA cloning, functional expression, and differential gene regulation in mouse fatty liver. Hepatology 39, 1297–1302. [DOI] [PubMed] [Google Scholar]
- Korenblat KM, Fabbrini E, Mohammed BS, and Klein S (2008). Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology 134, 1369–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Oney M, Frizzell K, Phadnis N, and Hollien J (2015). Drosophila melanogaster activating transcription factor 4 regulates glycolysis during endoplasmic reticulum stress. G3 (Bethesda) 5, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lindblom P, Rafter I, Copley C, Andersson U, Hedberg JJ, Berg A-L, Samuelsson A, Hellmold H, Cotgreave I, and Glinghammar B (2007). Isoforms of alanine aminotransferases in human tissues and serum—differential tissue expression using novel antibodies. Arch. Biochem. Biophys 466, 66–77. [DOI] [PubMed] [Google Scholar]
- Liu R, Holik AZ, Su S, Jansz N, Chen K, Leong HS, Blewitt ME, Asselin-Labat ML, Smyth GK, and Ritchie ME (2015). Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res. 43, e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Pan X, and Whitington PF (2009). Increased hepatic expression is a major determinant of serum alanine aminotransferase elevation in mice with nonalcoholic steatohepatitis. Liver Int. 29, 337–343. [DOI] [PubMed] [Google Scholar]
- Luo W, Friedman MS, Shedden K, Hankenson KD, and Woolf PJ (2009). GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinf. 10, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, et al. (2014). Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510, 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, and Turner RC (1985). Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28, 412–419. [DOI] [PubMed] [Google Scholar]
- McCommis KS, Chen Z, Fu X, McDonald WG, Colca JR, Kletzien RF, Burgess SC, and Finck BN (2015). Loss of mitochondrial pyruvate carrier 2 in the liver leads to defects in gluconeogenesis and compensation via pyruvate-alanine cycling. Cell Metab. 22, 682–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, et al. (2009). A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 9, 311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun JG, Rusu PM, Chan AY, Wu Y, Yap YW, Sharkie T, Schumacher J, Schmidt KV, Roberts-Thomson KM, Russell RD, et al. (2021). Liver alanine catabolism promotes skeletal muscle atrophy and hyperglycaemia in type 2 diabetes. Nat. Metab 3, 394–409. [DOI] [PubMed] [Google Scholar]
- Ouyang Q, Nakayama T, Baytas O, Davidson SM, Yang C, Schmidt M, Lizarraga SB, Mishra S, Ei-Quessny M, Niaz S, et al. (2016). Mutations in mitochondrial enzyme GPT2 cause metabolic dysfunction and neurological disease with developmental and progressive features. Proc. Natl. Acad. Sci. U S A 113, E5598–E5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, and Hotamisligil GS (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461. [DOI] [PubMed] [Google Scholar]
- Patel TB, and Olson MS (1985). A reexamination of the role of the cytosolic alanine aminotransferase in hepatic gluconeogenesis. Arch. Biochem. Biophys 240, 705–711. [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, and Shulman GI (2018). Mechanisms of insulin action and insulin resistance. Physiol. Rev 98, 2133–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian K, Zhong S, Xie K, Yu D, Yang R, and Gong DW (2015). Hepatic ALT isoenzymes are elevated in gluconeogenic conditions including diabetes and suppressed by insulin at the protein level. Diabetes Metabol. Res. Rev 31, 562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderman NB (1975). Muscle amino acid metabolism and gluconeogenesis. Annu. Rev. Med 26, 245–258. [DOI] [PubMed] [Google Scholar]
- Salgado MC, Meton I, Anemaet IG, and Baanante IV (2014). Activating transcription factor 4 mediates up-regulation of alanine aminotransferase 2 gene expression under metabolic stress. Biochim. Biophys. Acta 1839, 288–296. [DOI] [PubMed] [Google Scholar]
- Schindhelm RK, Diamant M, Dekker JM, Tushuizen ME, Teerlink T, and Heine RJ (2006). Alanine aminotransferase as a marker of non-alcoholic fatty liver disease in relation to type 2 diabetes mellitus and cardiovascular disease. Diabetes Metabol. Res. Rev 22, 437–443. [DOI] [PubMed] [Google Scholar]
- Seo J, Fortuno ES 3rd, Suh JM, Stenesen D, Tang W, Parks EJ, Adams CM, Townes T, and Graff JM (2009). Atf4 regulates obesity, glucose homeostasis, and energy expenditure. Diabetes 58, 2565–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell K (1980). Muscle alanine synthesis and hepatic gluconeogenesis. Biochem. Soc. Trans 8, 205–213. [DOI] [PubMed] [Google Scholar]
- Snell K, and Duff DA (1980). Alanine and glutamine formation by muscle. Biochem. Soc. Trans 8, 501–504. [DOI] [PubMed] [Google Scholar]
- Song S, Andrikopoulos S, Filippis C, Thorburn AW, Khan D, and Proietto J (2001). Mechanism of fat-induced hepatic gluconeogenesis: effect of metformin. Am. J. Physiol. Endocrinol. Metab 281, E275–E282. [DOI] [PubMed] [Google Scholar]
- Su X, Lu W, and Rabinowitz JD (2017). Metabolite spectral accuracy on orbitraps. Anal. Chem 89, 5940–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers SA, Chaurasia B, and Holland WL (2019). Metabolic messengers: ceramides. Nat. Metab 1, 1051–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai ES, Tan ML, Stevens RD, Low YL, Muehlbauer MJ, Goh DL, II-kayeva OR, Wenner BR, Bain JR, Lee JJ, et al. (2010). Insulin resistance is associated with a metabolic profile of altered protein metabolism in Chinese and Asian-Indian men. Diabetologia 53, 757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrettaz J,and Jeanrenaud B (1990). Contribution of glycerol and alanine to basal hepatic glucose production in the genetically obese (fa/fa) rat. Biochem. J 270, 803–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigueira PA, McCommis KS, Schweitzer GG, Remedi MS, Chambers KT, Fu X, McDonald WG, Cole SL, Colca JR, Kletzien RF, et al. (2014). Mitochondrial pyruvate carrier 2 hypomorphism in mice leads to defects in glucose-stimulated insulin secretion. Cell Rep. 7, 2042–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Yang RZ, Park S, Reagan WJ, Goldstein R, Zhong S, Lawton M, Rajamohan F, Qian K, Liu L, and Gong DW (2009). Alanine aminotransferase isoenzymes: molecular cloning and quantitative analysis of tissue expression in rats and serum elevation in liver toxicity. Hepatology 49, 598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNA sequencing data were uploaded to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession number GSE199975.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.