

Assessing allele-specific expression reveals genes with recurrent cis-regulatory changes that are enriched in glioblastoma stem cells, including SLFN11, which modulates chemotherapy resistance and susceptibility to the oncolytic Zika virus.
Abstract
Glioblastoma is the most prevalent primary malignant brain tumor in adults and is characterized by poor prognosis and universal tumor recurrence. Effective glioblastoma treatments are lacking, in part due to somatic mutations and epigenetic reprogramming that alter gene expression and confer drug resistance. To investigate recurrently dysregulated genes in glioblastoma, we interrogated allele-specific expression (ASE), the difference in expression between two alleles of a gene, in glioblastoma stem cells (GSC) derived from 43 patients. A total of 118 genes were found with recurrent ASE preferentially in GSCs compared with normal tissues. These genes were enriched for apoptotic regulators, including schlafen family member 11 (SLFN11). Loss of SLFN11 gene expression was associated with aberrant promoter methylation and conferred resistance to chemotherapy and PARP inhibition. Conversely, low SLFN11 expression rendered GSCs susceptible to the oncolytic flavivirus Zika. This discovery effort based upon ASE revealed novel points of vulnerability in GSCs, suggesting a potential alternative treatment strategy for chemotherapy-resistant glioblastoma.
Significance:
Assessing allele-specific expression reveals genes with recurrent cis-regulatory changes that are enriched in glioblastoma stem cells, including SLFN11, which modulates chemotherapy resistance and susceptibility to the oncolytic Zika virus.
Introduction
Glioblastoma ranks among the most lethal of human malignancies with current therapies only offering palliation (1). Reasons for treatment failure are myriad, with tumor heterogeneity at the genetic and transcriptional levels contributing to the malignancy of glioblastoma (2, 3). Glioblastoma displays a functional cellular hierarchy with stem-like, self-renewing glioblastoma stem cells (GSC) residing at the apex (4, 5). GSCs contribute to resistance to chemotherapy and radiotherapy, neoangiogenesis, invasion into normal brain, and escape from the immune system (6–8). Therefore, targeting GSCs may improve current glioblastoma management and extend the lives of patients.
Although glioblastoma is one of the most deeply characterized solid tumors, precision medicine has not significantly benefited most neuro-oncology patients. Most studies and targeted therapeutic strategies have thus far focused on protein-coding mutations. However, many important mutations lie in noncoding DNA where they function by perturbing gene regulation. Noncoding mutations likely help drive glioblastoma tumorigenesis and drug resistance but are more challenging to identify and impact gene regulation in many different ways. Gene dysregulation can be caused by copy-number alterations (CNA; refs. 9, 10), as well as by mutations that affect splice sequences (11), untranslated regions (UTR; ref. 12), insulators (13, 14), promoters (15, 16), and enhancers (12, 17). Moreover, genes are often regulated by multiple enhancers, which can be located hundreds of kilobases away from their targets (18). Regulatory mutations are, therefore, diverse and frequently spread over very large regions of the genome. As a result, standard recurrence analyses that identify driver mutations in coding sequences are unlikely detect many important regulatory mutations in cancer genomes. Alternate approaches to discover regulatory mutations can by stymied by the myriad of noncoding mutations whose function is difficult to predict from sequence alone. Rather than relying on interpretation of noncoding mutations, unbiased identification of genes with altered regulation can pinpoint functionally important genes that are unlikely to be discovered through other methods.
Here, we leveraged allele-specific expression (ASE) as a new approach to interrogate recurrently dysregulated genes in glioblastoma. ASE is the difference in expression between two alleles of a gene and can be estimated from mapped RNA sequencing (RNA-seq) reads that overlap heterozygous variants within exons (19, 20). Specifically, by counting the number of RNA-seq reads that contain the reference or alternate allele, the relative expression of each copy of the gene can be measured. Unlike standard RNA-seq expression levels, ASE is particularly sensitive to cis-acting mutations because it is generally unaffected by trans-acting or environmental effects that impact both alleles equally. Heterozygous sites within a gene's exons are required to obtain a readout of ASE; however, these heterozygous sites are not necessarily the cause of a gene's ASE. For example, the effect of a distal cis-regulatory mutation on gene expression can be measured using heterozygous variants within a gene, even when the distal mutation has not been directly observed by sequencing. Thus, a major advantage of ASE is that it can identify genes that are dysregulated by cis-acting regulatory mutations, even when the specific identities of the regulatory mutations are unknown. However, ASE is not solely caused by somatic mutations and can also result from common germline polymorphisms (21), imprinting (22), or random monoallelic expression (23). Therefore, to discover genes that are dysregulated by cis-acting factors such as regulatory mutations in cancer, the frequency of ASE in disease samples must be compared with a panel of normal samples. This approach has recently been used to identify new pathogenic genetic variants in muscle disease (20) and oncogenic mutations in T cell–acute lymphoblastic leukemia that would be difficult to identify using traditional techniques (14).
Based on this background, we hypothesized that a discovery effort based upon ASE could reveal novel points of fragility in the most resistant tumor cells, the GSCs. GSCs maintained in serum-free conditions maintain both genetic and transcriptional signatures found in the tumors from which they were derived, while removing the contaminating nontransformed cells that complicate genetic discovery. Here, we interrogated ASE in 43 patient-derived GSCs and compare the frequency of ASE to normal tissues to reveal novel dysregulated molecular targets that promote drug resistance and confer therapeutic vulnerabilities.
Materials and Methods
Derivation and maintenance of GSCs
Glioblastoma samples were obtained from surgical resection from patients at Duke University or Case Western Reserve University with informed consent in accordance with the Cleveland Clinic Institutional Review Board-approved protocol 090401. Prior to use, all samples were reviewed and verified by a neuropathologist. All patient studies were conducted in accordance with the Declaration of Helsinki. GSC23 was acquired via a material transfer agreement from The University of Texas MD Anderson Cancer Center (Houston, TX). GSCs were cultured in Neurobasal media (Invitrogen) supplemented with B27 without vitamin A (Invitrogen), EGF, and bFGF (20 ng/mL each; R&D Systems), sodium pyruvate, and glutamax. Short tandem repeat analyses were performed to authenticate the identity of each tumor model used in this article both annually and prior to high-throughput sequencing. Cells were stored at −160°C when not being cultured. To minimize cell culture–based artifacts, patient-derived xenografts were produced and propagated as a renewable source of tumor cells for study. Cultured cells underwent PCR testing for mycoplasma routinely every 6 months, and all RNA-seq data were evaluated for mycoplasma content during quality control.
Variant calling
Exome-seq reads were aligned to the GRCh37 (hg19) assembly of the reference genome using BWA-MEM with default parameters. Mapped reads were filtered for mapping quality score ≥ 30 and duplicate reads were removed using samtools (1.9, RRID:SCR_00215; ref. 24). Genotypes were generated for each individual using GATK's HaplotypeCaller and jointly processed using the GenotypeGVCFs function in GATK (4.1.1, RRID:SCR_001876). Following genotyping, single-nucleotide variants (SNV) were extracted and filtered using the variant quality score recalibration (VQSR) method in GATK.
RNA-seq alignment and processing
RNA-seq reads were aligned end-to-end to the GRCh37 (or hg19) assembly of the reference genome using STAR (2.5.3a, RRID:SCR_004463). Mapped reads were filtered using mapping quality score ≥ 20 and duplicate reads were removed using samtools (1.9). Read counts for GENCODE (v28) genes were computed using FeatureCounts (1.6.3, RRID:SCR_012919; ref. 25) and fragments per kilobase per million (FPKM) values were estimated using DESeq2 (1.14.1; ref. 26). For downstream analysis, the FPKM values were quantile normalized, and then converted to z-scores by mean-centering and standardizing across samples.
Estimating allele-specific expression
RNA-seq read alignments were corrected for mapping bias and allele-specific read counts at heterozygous positions were collected using WASP (27). Heterozygous sites covered by at least 10 RNA-seq reads were retained for allele-specific expression analysis.
Misclassification of heterozygous sites can occur due to incorrect genotyping. To control for genotyping errors, we calculate the genotyping error rate, εG, directly from genotype quality scores (GQ) from GATK:
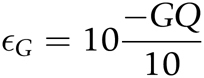 |
Incorrect allele-specific read counts can be caused by sequencing errors in RNA-seq reads. To control for sequencing errors, we approximate the sequencing error rate, εS, using the count of “other” reads, which do not match reference or alternate allele. To account for the fact that only 2/3 of sequencing errors will be observed in the “other” reads and the other 1/3 will match the alternate or reference allele, we scale the sequencing error rate estimate, by 3/2, such that
 |
where XO is the other read count, XR is the read count for the reference allele, and XA is the read count for the alternate allele.
Next, we assume a heterozygous site is equally likely to be misclassified as homozygous reference or alternate. Thus, conditional upon a genotyping error having occurred, we define the likelihood at site i as
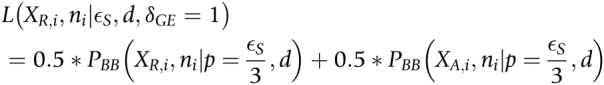 |
and conditional on no genotyping error, the likelihood is
where PBB is the Beta binomial probability distribution function, 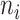 is the total count of reads matching the reference or alternative allele i.e., ni = XR,i + XA,i at heterozygous site
is the total count of reads matching the reference or alternative allele i.e., ni = XR,i + XA,i at heterozygous site 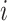 , p is the reference allele proportion (
, p is the reference allele proportion (
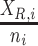 ), a is the allelic imbalance parameter defined over the range [−0.5, 0.5], d is the dispersion parameter, and δGE is an indicator variable that is 1 when a genotyping error has occurred and 0 otherwise. We estimate d by maximum likelihood over all heterozygous sites overlapping exons (setting a to 0).
), a is the allelic imbalance parameter defined over the range [−0.5, 0.5], d is the dispersion parameter, and δGE is an indicator variable that is 1 when a genotyping error has occurred and 0 otherwise. We estimate d by maximum likelihood over all heterozygous sites overlapping exons (setting a to 0).
Finally, a single gene might contain multiple heterozygous sites that need to be combined to estimate the allele imbalance for a gene. We define the likelihood of the read counts for the first heterozygous site within a gene as
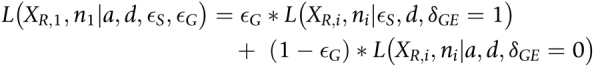 |
For subsequent heterozygous sites in the same gene, we do not know the phase of the alleles with respect to the first heterozygous site. We assume that the reference and alternative alleles are equally likely to be on the same haplotype as the reference allele at the first site. Thus, the combined likelihood of all sites within a gene is then defined as
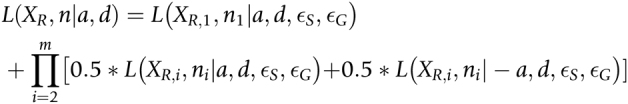 |
We estimate a for each gene by maximum likelihood under the alternative model of allele imbalance. Then we use a likelihood ratio test to compare the alternative model to the null model of no allele imbalance (i.e., with a fixed to a = 0). We correct the P values from the likelihood ratio test for multiple testing using the Benjamini–Hochberg method. To make it clear we are referring to allele imbalance in RNA-seq reads, we refer to a as aRNA in the rest of the manuscript.
Enrichment compared with a normal whole-blood and brain tissues from GTEx
To generate a reference profile of ASE in normal samples, we obtained RNA-seq data for 369 whole blood and 216 brain samples distributed across 13 brain regions from the GTEx consortium (19) and analyzed these data using our ASE algorithm described in the previous section. To discover genes that were enriched for ASE in GSCs, we compared the frequency of samples with significant ASE for each gene between GSCs and whole blood using Fisher exact test (FET) and adjusted the resulting P values using the Benjamini–Hochberg procedure. We only analyzed genes that were testable for ASE (i.e., had ≥ 1 heterozygous site with ≥10 reads) in both GSCs and whole-blood tissues. For further analysis of ASE frequency for individual genes, we also compared estimated allele imbalance from our model (aRNA) among whole blood, 13 brain tissues, and GSCs. Manhattan plots for enriched genes were generated using ggbio (1.30). Gene ontology enrichment analysis for genes showing recurrent ASE in GSCs was carried out using topGO (2.34, RRID:SCR_014798; ref. 28).
Association between DNA methylation and gene expression
We downloaded precomputed genome-wide methylation data for 43 GSCs from Gene Expression Omnibus (GSE119774, RRID:SCR_005012; ref. 29). These methylation data were generated using the Illumina Infinium Epic Methylation Array. In this assay, DNA methylation levels at CpG sites are represented by β, which is the ratio of the methylated (C) to unmethylated (T) signal. We annotated the CpG probe positions based on GENCODE (v28, RRID:SCR_014966) genes and computed the mean β values for promoter regions (i.e., 1 kb upstream to 500 bp downstream of annotated transcription start sites; βpromoter). To discover ASE genes that may be dysregulated by aberrant DNA methylation, we computed Spearman rank correlation between βpromoter and normalized gene expression. We corrected correlation P values for multiple testing using the Benjamini–Hochberg procedure. For this analysis, we only considered genes with ≥ 3 CpG probes mapping to their promoter regions.
Analysis of H3K27ac chromatin immunoprecipitation and sequencing data
We downloaded H3K27ac chromatin immunoprecipitation and sequencing (ChIP-seq) data for 35 GSCs from Gene Expression Omnibus (GSE119755, RRID:SCR_005012; ref. 29). ChIP-seq reads were aligned to the GRCh37 (hg19) assembly of the reference genome using BWA-MEM with default parameters. The mapped reads were filtered using mapping quality score of ≥20, and duplicate reads were removed using samtools (1.9). H3K27ac peaks were called using MACS2 in paired-end mode with custom parameters (–nomodel –extsize 200 –qvalue 0.05; ref. 30). To generate a unified set of test regions, we divided the genome in 1 kb nonoverlapping genomic bins and kept the bins that overlapped MACS2.0 peaks in at least one GSC. We recounted the reads mapping to these genomic bins using exomeCopy (1.28.0, RRID:SCR_001276) and calculated FPKM using DESeq2 (1.22.2, RRID:SCR_000154; ref. 26). FPKM measurements were quantile normalized and mean-centered for downstream analysis.
The 1 kb genomic bins generated from H3K27ac ChIP-seq peaks were annotated using the ChIPSeeker (1.18.0) package. To discover cis-regulatory elements (CRE), we selected all distal intergenic and intronic genomic bins located within 100 kb of promoters (i.e., 1 kb upstream and 500 bp downstream of transcription start sites) of ASE genes. We performed a Spearman correlation analysis between normalized coverage for bins and normalized expression for genes to identify CREs. We corrected the P values for multiple testing using the Benjamini–Hochberg procedure.
Quantitative RT-PCR
TRIzol reagent (Sigma-Aldrich) was used to isolate total cellular RNA from cell pellets, and the qScript cDNA Synthesis Kit (Quanta BioSciences) was used for reverse transcription. Quantitative real-time PCR was performed using SYBR-Green PCR Master Mix (Thermo Fisher Scientific) on an Applied Biosystems 7900HT cycler.
Western blotting
Cells were collected and lysed in RIPA buffer (50 mmol/L Tris-HCl, pH 7.5; 150 mmol/L NaCl; 0.5% NP-40; 50 mmol/L NaF with protease inhibitors) and incubated on ice for 30 minutes. Lysates were centrifuged at 4°C for 15 minutes at 14,000 rpm, supernatant was collected, and protein concentration was confirmed using a Pierce BCA protein assay kit (Thermo Scientific, cat #23225). Equal amounts of protein samples were mixed with SDS Laemmli loading buffer, boiled for 10 minutes, electrophoresed using NuPAGE Bis-Tris gels and transferred onto PVDF membranes. Membranes were blocked for 1 hour with TBS-T plus 5% nonfat dry milk, then incubated in primary antibodies overnight at 4°C. Blots were washed 3 times for 5 minutes each with TBS-T and then incubated in TBS-T plus 5% milk for 1 hour with appropriate secondary antibodies. Blots were imaged using Bio-Rad Image Lab software and processed using Adobe Illustrator (RRID:SCR_010279) to create the figures. The following antibodies were used for Western blot: SLFN11 (Santa Cruz Biotechnology; cat #SC-374339, RRID:AB_10989536) and HRP-conjugated GAPDH (Proteintech; cat #HRP-60004, RRID:AB_2737588).
Lentiviral transduction
Lentiviral constructs expressing shRNAs directed against SLFN11 (Sigma TRCN, TRCN0000152057) or a nontargeting control shRNA (TRCN0000231489) with no targets in the human genome were obtained from Sigma-Aldrich. The SLFN11 expression vector was obtained from VectorBuilder along with an empty vector control with the same lentiviral backbone. 293T cells (ECACC; cat #12022001, RRID:CVCL_0063) were used to generate lentiviral particles by cotransfection of packaging vectors psPAX2 (RRID:Addgene_12260) and pMD2.G (RRID:Addgene_12259) using a standard PEI transfection method in DMEM plus 1% penicillin/streptomycin. GSCs were transduced with the lentiviral constructs, and selection was started 48 hours later using 1 μg/mL of puromycin for 72 hours, at which times, cells were assayed for SLFN11 expression.
In vitro treatment and cell viability
For in vitro cell viability assays, 2,000 cells/well for individual or 5,000 cells/well for combinatorial drug studies were plated in a 96-well plate. Cells were then treated 24 hours later with temozolomide (Selleck Chem; cat #S1237), olaparib (Selleck Chem; #S1060), both drugs, or DMSO at an equivalent percent volume to the highest drug concentration. Cell viability was assayed 4 days later following a 12-hour incubation with alamarBlue (Thermo Fisher; cat #DAL1025) and detected using a fluorescence-based plate reader. For Zika virus studies, 5,000 cells/well were infected with Dakar 41519 strain ZIKV at a multiplicity of infection of 5 (MOI = 5; ref. 31). Viability was assayed at 3 days of post infection using CellTiter-Glo according to the manufacturer's instructions.
Statistical analysis
Tests for ASE were performed using likelihood ratio tests as described above. Frequencies of samples with significant ASE were compared using FET, as described above. For analysis of all in vitro data, comparisons between more than two samples were performed using ANOVA followed by Tukey HSD or using Student t test. The number of replicates and specific statistical tests used are indicated in the figure legends.
Data availability statement
RNA-seq (GSE119834), H3K27ac ChIP-seq (GSE119755), and human methylation 450K array data (GSE119774) for GSCs used in the paper were previously published and can be downloaded from GEO (GSE119776; ref. 29).
Results
Glioblastoma target gene discovery leveraging recurrent allele-specific expression
To discover genes with ASE, we implemented a statistical model to estimate allele imbalance for every gene (Fig. 1A). Our systematic methodology utilizes RNA-seq reads overlapping heterozygous sites and reduces false positives by accounting for technical sources of variation, including sequencing errors, genotyping errors, and overdispersion of RNA-seq read counts. We quantified ASE with RNA allelic imbalance (aRNA), which is the difference between the reference allele proportion and the expected value of 0.5. We applied our model to RNA-seq data from 43 patient-derived GSCs representing a diverse population of patients (age, biological sex, etc.) and tumor features (genetics, transcriptional subgroups, etc.) that we previously reported (29). We identified 5,808 genes with significant ASE in at least one GSC under a false discovery rate (FDR) of 10% (Supplementary Table S1). ASE was restricted to a single sample for most genes (3,948 out of 5,808). However, 1,860 genes showed ASE in 2 or more GSCs, and 298 genes showed highly recurrent ASE that was present in 5 or more GSCs (Fig. 1B).
Figure 1.

Discovery of genes with recurrent allele-specific expression in GSCs. A, Schematic of approach. ASE is the higher expression of one allele of a gene compared with the other allele and can be used to detect the effects of cis-regulatory mutations, even when the cis-regulatory mutation is unobserved. ASE is estimated from RNA-seq reads that overlap heterozygous variants within exons. We identify genes that exhibit ASE in GSCs more frequently than in normal tissues. B, Recurrence of ASE in GSCs. The histogram indicates the number of GSCs with ASE (FDR corrected P ≤ 10%) across 42 patient-derived GSCs, for genes that have ASE in at least one sample. C, Estimated ASE (aRNA) for GSCs and normal brain and whole-blood samples from GTEx for a known imprinted gene, H19 (top) and RHOB (bottom). Each point is a sample. Points are colored based on the significance of ASE (likelihood ratio test FDR ≤ 10%). D, GO analysis of 118 genes that are enriched for ASE in GSCs compared with normal samples. The figure shows the top 10 GO categories.
To discover genes that showed recurrent ASE specific to GSCs relative to normal tissues, we compared the frequency of ASE for each gene in GSCs to both normal whole-blood and normal brain samples from the Genotype Tissue Expression (GTEx) project using the FET. Whole blood has, by far, the largest number of samples in GTEx, so we used it as the initial reference tissue; however, we performed subsequent comparisons with ASE in 13 different brain tissues to account for tissue-specific imprinting, as described below. Under an FDR of 10%, 118 genes displayed significantly enriched ASE in GSCs (Supplementary Table S2). To illustrate the power of this approach, we examined the ASE patterns of the noncoding RNA gene H19, which is maternally imprinted (32), and RHOB, which is important for glioblastoma tumorigenesis (33, 34). Imprinting causes loss of expression of the allele inherited from one parent, and as expected for an imprinted gene, H19 showed ASE in almost all normal and tumor samples (Fig. 1C, top). In contrast, ASE of RHOB was restricted to GSCs (Fig. 1C, bottom).
Recurrent ASE in cancer genomes can be caused by frequent CNAs or loss-of-heterozygosity. In the presence of large CNAs, many genes are expected to exhibit ASE, but these genes would be clustered into the genomic regions that undergo frequent CNAs. However, in our data set, recurrent ASE genes were distributed throughout the genome, suggesting that their dysregulation was not due to large-scale CNAs, but was instead caused by localized cis-regulatory mutations or epigenetic changes (Supplementary Fig. S1A).
To examine the biological function of the 118 genes with recurrent ASE in GSCs, we performed Gene Ontology (GO) enrichment analysis, revealing overrepresentation of recurrent ASE genes involved in regulation of cell cycle and apoptosis (FDR ≤ 5%). Twenty-eight genes with recurrent ASE were associated with the Biological Process GO category “programmed cell death,” whereas only 13 genes were expected by chance (Fig. 1D). These genes included the kinase IP6K2 (35, 36), which exhibited a marked enrichment of ASE in GSCs compared with both whole-blood (FET P = 0.001; FDR-adjusted P = 0.06) and brain tissues from GTEx (Supplementary Fig. S1B and Supplementary Table S2).
Allele-specific gene expression is associated with H3K27ac marks at distal regulatory elements
Cis-acting pathogenic variants impact gene expression by disrupting regulatory sequences, such as enhancers (17, 37). To illuminate the cis-acting mechanisms that underlie ASE in GSCs, we tested whether the expression of the 118 ASE genes was associated with the activity of nearby CREs. We identified putative CREs within 100 kb of the ASE gene promoters using histone H3 lysine 27 acetyl (H3K27ac) chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) data that we previously generated for the GSCs (29). We divided the genome into 1 kb genomic bins and labeled bins that overlapped H3K27ac peaks in at least one GSC as putative CREs.
To connect CREs with genes, we correlated normalized H3K27ac levels with gene expression, focusing on distal intergenic and intronic elements. We used gene expression rather than ASE for this purpose, because ASE can only be estimated in samples that carry at least one heterozygous SNP within exons. Furthermore, ASE does not detect regulatory changes that affect both alleles equally. Of the 118 ASE genes, 56 had a significant Spearman rank correlation with the activity of a distal CRE (FDR ≤ 5%). In many cases, a single gene was associated with the activity of multiple CREs, such that 227 CRE bins were associated with the expression of these 56 genes (Supplementary Table S3). Thus, gene expression of many ASE genes was associated with activity levels of distal regulatory elements, as measured by H3K27ac, suggesting that cis-acting mutations within these CREs are a plausible and potentially common mechanism for the dysregulation of these genes in glioblastoma.
One example of an ASE gene that is associated with multiple CREs is NOTCH1. ASE of NOTCH1 was enriched in GSCs compared with both whole-blood (FET P = 5.09e−5; FDR-adjusted P = 0.0094) and brain samples, and we observed extreme biases in reference and alternate allele proportions at all heterozygous sites (Fig. 2A and B). NOTCH1 gene expression correlated with H3K27ac levels of 29 nearby CREs (Fig. 2C and D; Supplementary Table S3). To confirm that these CREs were more strongly associated with NOTCH1 expression than expected by chance, we created an empirical null distribution for each CRE by correlating H3K27ac levels with the expression of 1,000 randomly selected genes. Using these null distributions, 75% (22 out of 29) CREs remained significantly correlated with NOTCH1 gene expression (empirical P ≤ 5%). These regions may contain cis-regulatory noncoding mutations that lead to NOTCH1 gene dysregulation and may be excellent targets for future regulatory screens dissecting the regulation of NOTCH1 expression in GSCs.
Figure 2.

NOTCH1 exhibits recurrent allele-specific expression and association with multiple CREs. A, ASE estimates (aRNA) of NOTCH1 in GSC and normal brain and whole-blood samples from GTEx. B, The proportion of NOTCH1 RNA-seq reads matching the reference and alternate alleles at heterozygous sites in four GSCs with significant NOTCH1 ASE. Reads at these heterozygous variants are used to estimate ASE, but these variants are not necessarily the cause of ASE, which, for example, may be due to unobserved cis-regulatory mutations. C, The H3K27ac profile around NOTCH1. Samples are arranged in increasing order of gene expression, with the highest expression samples at the bottom. Blue bars below each track indicate H3K27ac peaks detected using MACS2. Putative CREs that are correlated with NOTCH1 gene expression (empirical P ≤ 0.05) are highlighted in gold. D, Spearman correlation between H3K27ac and NOTCH1 expression for two putative CREs. Each point is a sample. Samples are blue if they have significant ASE, red if they do not have significant ASE, and uncolored if ASE cannot be estimated (due to absence of a heterozygous variant).
Promoter methylation is associated with the expression of ASE genes
Aberrant DNA methylation of gene promoters is associated with widespread gene expression changes and chemotherapy resistance in brain tumors. In gliomas, MGMT CpG-rich promoter methylation is associated with decreased expression and improved response to temozolomide (TMZ) treatment (38). To determine whether genes with recurrent ASE were associated with aberrant DNA methylation, we analyzed the CpG methylome of the GSCs (29). We estimated the promoter methylation of the 118 ASE genes (βpromoter) and computed Spearman rank correlations with normalized gene expression. We identified 30 genes that displayed correlation between promoter methylation and gene expression at FDR ≤ 10% (Supplementary Table S4), of which, 16 overlapped with the 56 genes identified above with correlated gene expression and H3K27ac levels at CREs. Thus, 70 of the 118 ASE genes were associated with promoter methylation, CRE activity or both, suggesting possible mechanisms for their dysregulation.
SLFN11 promoter methylation associates with its gene expression
One of the 30 genes with correlated gene expression and promoter methylation was SLFN11 (rho = −0.68, FDR corrected P = 0.0001; Fig. 3A–C). SLFN11 is notable because it inhibits DNA replication and promotes cell death in response to DNA damage (39, 40). Loss of SLFN11 causes resistance to poly ADP ribose polymerase (PARP) inhibitors in small cell lung cancer, suggesting that it may be an important marker of chemotherapy resistance (41). ASE of SLFN11 was highly enriched in GSCs with 4 out of 10 testable GSCs exhibiting ASE, compared with 0 out of 159 testable normal whole blood tissues from GTEx (FET P = 6.4e−6; FDR-adjusted P = 2.2e−3; Supplementary Table S2). Although this gene showed enrichment of ASE in GSCs compared with whole blood samples, we detected ASE in a small number of normal brain tissue samples (4 out of 224; Fig. 3D). This suggests that rare germline variants or somatic events, such as mutations or DNA methylation, may affect SLFN11 expression in some phenotypically normal individuals. In the four GSCs with significant SLFN11 ASE the RNA-seq read counts show strong allelic bias at multiple SNVs at different locations within the gene, which rules out the possibility that the observed ASE is due to genotyping errors or read mapping artifacts that would be more likely to affect a single site (Fig. 3E).
Figure 3.

SLFN11 gene expression and allele-specific expression are correlated with promoter methylation. A, Scatterplot showing correlation between gene expression and mean promoter methylation (βpromoter) in GSCs. Samples are blue if they have significant SLFN11 ASE, red if they do not have significant ASE, and uncolored if ASE could not be estimated due to lack of a heterozygous variant. B, Correlation between ASE and promoter methylation or gene expression. ASE is positively correlated with promoter methylation but negatively correlated with gene expression. C, CpG methylation around the promoter region of SLFN11. Samples are arranged by increasing order of gene expression, with the highest expression sample at the bottom. D, Estimated ASE for SLFN11 in GSCs compared with normal brain and whole-blood samples from GTEx. E, The proportion of SLFN11 RNA-seq reads from the reference and alternate alleles at heterozygous sites in four GSCs with significant SLFN11 ASE.
Based on the promoter methylation and gene expression of SLFN11, GSCs can be divided in three distinct classes: (i) GSCs with high methylation and low expression; (ii) GSCs with intermediate methylation and intermediate expression; and (iii) GSCs with low methylation and high expression (Fig. 3A). Four GSC samples with ASE of SLFN11 had intermediate methylation, which is consistent with methylation and reduced expression of one allele (Fig. 3A and B). However, samples like GSCs 2907 and 007B had high promoter methylation levels (>60%) and very low expression of SLFN11 (Fig. 3A). Under these circumstances, genes would not be found by ASE because the expression of both alleles is reduced. These results demonstrate that low expression of SLFN11 in GSCs is associated with increased promoter methylation and that the samples with detectable ASE likely have methylation of one allele, but not the other.
SLFN11 augments chemotherapy resistance in GSCs
SLFN11 regulates cellular responses to DNA damaging agents (39, 40). Therefore, we hypothesized that SLFN11 expression in GSCs would be associated with chemotherapeutic resistance to an alkylating agent, TMZ, and a PARP inhibitor, olaparib. To test this hypothesis, we utilized four patient-derived GSCs: two with high SLFN11 expression and no evidence for ASE (839 and MNK1) and two with low SLFN11 expression and strong ASE (2012 and 1552). We confirmed both SLFN11 mRNA and SLFN11 protein abundance by RT-PCR (Fig. 4A) and immunoblot (Fig. 4B). We treated cells with drug concentrations ranging from 0 to 1,000 μmol/L for TMZ and 0 to 50 μmol/L for olaparib, then measured drug effects on cell survival to generate a concentration–response matrix where an effect of 100% corresponds to complete killing of all cells and 0% corresponds to no difference in cell survival (Fig. 4C and D; Supplementary Fig. S2). We estimated synergy between the two drugs using SynergyFinder 2.0 (42). Two GSCs with low expression and ASE of SLFN11 had reduced responses to drug treatment compared the GSCs with high expression of SLFN11. The maximum response for both ASE GSCs ranged from 40% to 50%, whereas the GSCs with high expression of SLFN11 had responses ranging from ∼70% to 80% (Fig. 4D). The two ASE GSCs also had lower drug synergy scores for the combination of the drugs (Fig. 4E).
Figure 4.

SLFN11 expression is associated with chemotherapeutic resistance in GSCs. A, qPCR of SLFN11 expression in GSCs without ASE of SLFN11 (839 and MNK1; red) compared with GSCs with ASE of SLFN11 (2012 and 1552; blue). Gene expression is plotted as 2–ΔΔCt, with SLFN11 expression for each sample normalized to GAPDH expression and then compared with MNK1. Values <1 were transformed by taking the negative inverse. Data are represented as mean ± SD. B, Western blot of the same non-ASE (red) and ASE (blue) GSCs for SLFN11 and GAPDH expression. C, Cell viability of each GSC (left, non-ASE; right, ASE) following treatment with TMZ and olaparib. Cell viability relative to DMSO control is annotated on a blue–white–red scale, with blue indicating high viability, or minimal drug effect, and red indicating low viability, or strong drug effect. Doses are scaled categorically on the x-axis (olaparib) and y-axis (TMZ). D, Top, effect of the maximum combinatorial drug dose on cell viability for non-ASE (red) and ASE (blue) GSCs. Bottom, maximal effect achieved at any dose for each model. Percent effect on reduction of cell viability is measured on the y-axis. E, Synergy of TMZ and olaparib for each model, with red indicating high synergy and green indicating antagonism. C–D, average of two biological replicates per model.
To directly test whether SLFN11 expression affects chemotherapeutic drug sensitivity in GSCs, we also performed knockdown (KD) and overexpression (OE) experiments. Specifically, we performed KD of SLFN11 in MNK1 GSCs, which have high baseline expression of SLFN11 (Fig. 5A), using two nonoverlapping shRNAs. After confirming shRNA-mediated knockdown of SLFN11 in MNK1 GSCs, we measured cell survival in response to treatment with TMZ and olaparib. SLFN11 KD in MNK1 cells reduced cell death in response to drug treatment (Fig. 5A and B). Conversely, overexpression of SLFN11 in 2012 GSCs, which have low baseline expression of SLFN11, increased cell death in response to combinatorial drug treatment (Figs. 5C and D). Knockdown and overexpression of SLFN11 similarly changed sensitivity to single-drug treatments, although not significantly so (0.05 < P < 0.1; Supplementary Fig. S3). Synergistic drug response to TMZ and olaparib treatment decreased upon SLFN11 KD and increased with OE (Fig. 5E). Thus, SLFN11 expression is causally linked to sensitivity to the chemotherapeutic drugs TMZ and olaparib.
Figure 5.

Knockdown and overexpression of SLFN11 modulates chemotherapeutic resistance in GSCs. A, Left, qPCR of SLFN11 expression in MNK1 GSCs transduced with a nontargeting control shRNA (shCONT) or shRNAs targeting SLFN11 (shSLFN11.382 and shSLFN11.2648). Gene expression is plotted as 2–ΔΔCt normalized to actin expression. Right, Western blot of the same samples for SLFN11 and GAPDH expression. B, Cell viability of MNK1 GSCs transduced with shCONT (left), shSLFN11.382 (top right), or shSLFN11.2648 (bottom right) following treatment with TMZ and olaparib. Cell viability relative to DMSO control is annotated on a blue–white–red scale, with blue indicating high viability, or minimal drug effect, and red indicating low viability, or strong drug effect. Doses are scaled categorically on the x-axis (olaparib) and y-axis (TMZ). C, Left, qPCR of SLFN11 expression in GSC 2012 transduced with an empty vector or an SLFN11 overexpression vector (OE). Gene expression is plotted as 2–ΔΔCt normalized to GAPDH expression. Right, Western blot of the same samples for SLFN11 and GAPDH expression. D, Cell viability of 2012 GSCs transduced with an empty vector (left) or OE vector (right). E, Synergy of TMZ and olaparib for each model, with red indicating high synergy and green indicating antagonism. All drug response experiments were performed in triplicate. Error bars in A and C, mean ± SD. Asterisks indicate significant differences between control and treatment samples: ****, P < 0.0001.
GSCs with low expression of SLFN11 are sensitive to Zika virus
SLFN11 may be involved in cellular response to viral infection. SLFN11 is upregulated following virus-induced type I interferon response and restricts flavivirus replication in human tumor cell lines (43–45). Oncolytic viruses that infect the central nervous system can be leveraged to treat brain tumors (46). We recently demonstrated that Zika virus, which is a member of the flavivirus genus of RNA viruses, preferentially infects and kills GSCs compared with differentiated tumor cells and normal neuronal cells (47). Based on this background, we hypothesized that tumor cells with promoter methylation of SLFN11 would be more susceptible to oncolytic destruction by Zika because they are unable to increase SLFN11 expression in response to interferon stimulation (47). Under this hypothesis, interferon exposure would induce upregulation of most interferon-responsive genes but would fail to induce SLFN11 expression in cells with SLFN11 promoter methylation.
To test the above hypothesis, we examined the association between SLFN11 gene expression and type I interferon response by analyzing gene expression data from 669 glioblastoma tumors obtained from The Cancer Genome Atlas (48). To quantify interferon response in each sample, we computed a single-sample gene set enrichment analysis (ssGSEA) score using the interferon alpha response hallmark gene set (49, 50). Glioblastoma had higher gene expression of SLFN11 compared with grade II and III gliomas (Fig. 6A), and SLFN11 gene expression correlated with the interferon alpha (IFNα) ssGSEA score (R = 0.4, P < 2.2e−16), consistent with induction of SLFN11 expression in response to interferon signaling (Fig. 6B). Glioblastomas with higher expression of SLFN11 had gene set enrichment for activation of immune response, complement activation, defense response to virus, and response to interferon alpha compared with tumors with lower SLFN11 expression (Fig. 6C). Enrichment of these immune pathways likely reflects the constitutive activation of autocrine interferon signaling, which might facilitate immune escape for tumors (51).
Figure 6.

SLFN11 expression affects sensitivity of GSCs to Zika virus. A, Box plot of SLFN11 expression in The Cancer Genome Atlas database comparing grade II, III, and IV gliomas. Boxes are notched at the median and extend from the first to third quartiles, with whiskers extending from 5% to 95%. B, Correlation of SLFN11 expression with ssGSEA score for the Hallmark IFNα gene set by tumor. Grade II and III tumors are in blue and glioblastoma tumors are in red. C, GSEA plot of SLFN11 expression correlation with selected immune signatures from GO biological process or Reactome data sets. D, qPCR expression of IFNα-induced genes OAS1, IFITM, ISG20, and SLFN11 following 8 hours of treatment with 20 ng/mL of IFNα. Gene expression is plotted as 2–ΔΔCt normalized to GAPDH expression. E, Cell viability following incubation with Zika virus vs. empty control virus. Non-ASE samples, blue; ASE samples, red. Data were compared using two-way ANOVA for cell line and ASE status. F, Cell viability following incubation with Zika virus (blue) vs. control in 2012 GSCs transduced with an SLFN11 overexpression (OE) vector (left) or empty vector (right). Student t test was used to test for differences in expression. G, Cell viability following incubation with Zika virus (red) vs. control in SLFN11 KD vs. control 839 (left) or MNK1 (right) GSCs. Mean expression was compared using Student t test. For E–G, statistical testing was performed on normalized values as shown in the figure. Corresponding raw data are presented in Supplementary Fig. S4. Data in D are the average of two biological replicates (2012, 1552) or one experiment (839, MNK1), resulting in at least two biological replicates per condition (ASE vs. non-ASE). E and F, average of two biological replicates. Data in G are from one experiment per model, resulting in biological duplicates per condition (control vs. KD). Data are represented as mean ± SD. Asterisks indicate significant differences: *, P < 0.05; ****, P < 0.0001.
To more directly test whether interferon signaling induces SLFN11 gene expression, we treated GSCs with IFNα and measured the expression of SLFN11 and type 1 immune response genes OAS1, ISG20, and IFITM 8 hours after treatment. All four GSCs increased expression of OAS1, ISG20, and IFITM following IFNα treatment, although the level of induction in the GSC 839 was modest (2–3-fold) compared with the other GSCs (Fig. 6D). IFNα treatment also increased SLFN11 gene expression in MNK1 and 839 GSCs, which have high baseline expression of SLFN11 and no ASE. However, IFNα treatment did not increase expression of SLFN11 in 2012 and 1552 GSCs, both of which showed ASE of SLFN11 and high promoter methylation (>25% βpromoter). These results suggest that DNA methylation of the SLFN11 promoter blocks upregulation of SLFN11 by interferon signaling (Fig. 6D).
To determine if GSCs with promoter methylation and low expression of SLFN11 were more susceptible to killing by Zika, we treated the same four GSCs with Zika or saline control (Fig. 6E; Supplementary Fig. S4). GSCs with promoter methylation of SLFN11 (2012 and 1552) displayed decreased viability following Zika infection compared with GSCs without promoter methylation (839 and MNK1). To directly test whether SLFN11 expression affects susceptibility to Zika, we performed SLFN11 OE in 2012 GSCs (which have low baseline expression of SLFN11) and SLFN11 KD in the 839 and MNK1 GSCs (which have high baseline expression of SLFN11). SLFN11 OE (Fig. 5C) decreased susceptibility to Zika (Fig. 6F), whereas KD (Fig. 5A) increased susceptibility to Zika (Fig. 6G).
Although each Zika experiment was performed with only a single biological replicate per GSC and more biological replicates would be desirable, we note that experiments were performed in multiple GSCs, and each experiment consisted of 3–10 technical replicates. Furthermore, our conclusions are supported by multiple lines of evidence. First, Zika infection and control experiments were performed on two GSCs with high SLFN11 expression (MNK1 and 839) and two GSCs with low SLFN11 expression (2012 and 1552). As expected, both of the low expression cell lines had lower viability in the presence of Zika (Fig. 6E). Second, overexpression of SLFN11 in the 2012 GSC (with low baseline SLFN11 expression) increased resistance to Zika (Fig. 6F). Third, knockdown of SLFN11 in the MNK1 GSC (with high baseline SLFN11 expression) increased susceptibility to Zika (Fig. 6G). In combination, these results provide compelling support for the hypothesis that SLFN11 expression is important for cell viability following Zika infection.
Discussion
ASE analysis of tumor genomes is a new approach that can discover biomarkers and therapeutic targets by illuminating genes that are recurrently dysregulated. ASE analysis makes it possible to prioritize genes that are affected by cis-regulatory factors such as regulatory mutations, even when the precise identity of the mutational events that drive the transcriptional changes is unknown (52, 53). This differs from standard differential gene expression analysis, which identifies thousands of genes when comparing normal and tumor tissues. As gene expression is affected by the environment and by the activity of upstream factors, most of these differentially expressed genes are “passengers” that are downstream of the primary events that drive transcriptional changes. In contrast, by analyzing ASE we discover a relatively small number (118) of candidate disease genes that are recurrently dysregulated by cis-regulatory factors in GSCs, but not in normal tissues.
Many of the candidate genes identified by our ASE analysis have an established role in tumor biology. For example, IP6K2, a proapoptotic protein kinase showed ASE almost exclusively in GSCs. IP6K2 selectively binds to HSP90, which decreases its catalytic activity and inhibits apoptosis (36). Disruption of this interaction by cisplatin and novobiocin, chemotherapeutic compounds that bind to the C-terminus of HSP90, restores its catalytic function and promotes apoptosis (54). Furthermore, knockdown of IP6K2 in colorectal cancer cells has been demonstrated to selectively impair p53-mediated apoptosis, instead favoring cell-cycle arrest (55). These observations from previous studies suggest that IP6K2 may be an important tumor suppressor in glioblastoma.
NOTCH1 also exhibited ASE that was specific to GSCs. NOTCH1 regulates neural stem cell fate during neurogenesis and high expression of NOTCH1 has been reported in many high-grade gliomas (56–58). Notch1 signaling promotes invasion, self-renewal, and growth of GSCs (59, 60); NOTCH1-KD suppresses cell proliferation and induces apoptosis (61). Furthermore, inhibition of the Notch1 signaling pathway sensitized tumor cells to apoptosis induced by ionizing radiation, the death ligand TRAIL (tumor necrosis factor-related apoptosis-inducing ligand), or the Bcl-2/Bcl-XL inhibitor ABT-737 (62). These studies suggest that NOTCH1 may help maintain the stem cell–like behavior of GSCs and promote tumor progression. The multiple CREs correlated with NOTCH1 expression are potentially excellent targets for subsequent studies and cis-regulatory screens.
Recurrent ASE of SLFN11 is an important finding because this gene has recently emerged as a biomarker of drug sensitivity in cancer (63). We demonstrate that in GSCs, SLFN11 gene expression is associated with DNA methylation of its promoter and its expression is required for the antitumor activities of the DNA alkylating agent TMZ and the replication inhibitor olaparib. The current standard of care for glioblastoma patients is maximum safe surgical resection followed by concurrent TMZ and radiotherapy (64). Similar to MGMT promoter DNA methylation, SLFN11 promoter methylation may be a biomarker that predicts the efficacy of DNA damaging agents, such as TMZ and olaparib. In addition, promoter methylation and reduced expression of SLFN11 may reflect evolution of resistance to TMZ within tumor cells. In GSCs, SLFN11 gene expression was regulated by type 1 interferons and frequently upregulated in high-grade tumors, alongside constitutive activation of autocrine interferon signaling that facilitates immune evasion of GBM cancer cells (51). However, in the presence of promoter CpG methylation, SLFN11 is unresponsive to IFNα cytokine treatment, rendering GSCs vulnerable to killing by oncolytic viruses, such as Zika (47). Thus, tumors refractory to DNA damaging agents may be more amenable to treatment with genetically modified viruses. As GSCs with low SLFN11 expression are susceptible to Zika, but resistant to chemotherapy and vice versa, the combination of oncolytic viruses and chemotherapy may be a powerful treatment approach (Fig. 7).
Figure 7.

Model of SLFN11 expression and therapeutic response. A, Interferon alpha increases SLFN11 expression when its promoter is unmethylated. GSCs with high SLFN11 expression are sensitive to TMZ and olaparib because they undergo apoptosis in response to DNA damage. However, GSCs with high SLFN11 expression are resistant to Zika virus because SLFN11 restricts flavivirus replication. B, IFNα fails to induce SLFN11 expression when its promoter is methylated. Glioma stem cells with low expression of SLFN11 are resistant to chemotherapy, but sensitive to Zika virus.
One limitation of our study is that it does not incorporate the role of the tumor microenvironment in the model systems. Future efforts could investigate interactions between immune cells and tumor cells to elucidate how SLFN11 expression in tumor cells affects tumor maintenance and the antitumor immune responses.
Authors' Disclosures
J.N. Rich reports grants and personal fees from Synchronicity Pharma outside the submitted work. G. McVicker reports grants from NIH/NCI, Padres Pedal the Cause/RADY, and NIH/NHGRI during the conduct of the study. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
This research was supported by the NCI funded Salk Institute Cancer Center (NIH/NCI CCSG: P30 014195) by a grant from Padres Pedal the Cause/RADY #PTC2019 (G. McVicker), by a Pioneer Fund Postdoctoral Scholar Award (A. Sen), and the following NIH grants and fellowships: CA217066 (B.C. Prager), CA217065 (R.C. Gimple), HG011315 (G. McVicker), CA197718, CA238662, and NS103434 (J.N. Rich). G. McVicker was supported by the Frederick B. Rentschler Developmental Chair. Figures 1A and 7 were created with BioRender.com.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Contributions
A. Sen: Conceptualization, software, investigation, visualization, methodology, writing–original draft. B.C. Prager: Conceptualization, investigation, visualization, methodology, writing–original draft. C. Zhong: Validation, investigation. D. Park: Investigation. Z. Zhu: Investigation. R.C. Gimple: Investigation. Q. Wu: Investigation. J.A. Bernatchez: Investigation. S. Beck: Investigation. A.E. Clark: Investigation. J.L. Siqueira-Neto: Resources, supervision. J.N. Rich: Conceptualization, supervision, funding acquisition, project administration, writing–review and editing. G. McVicker: Conceptualization, supervision, funding acquisition, project administration, writing–review and editing.
References
- 1. Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol 2020;22:iv1–iv96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011;20:810–7. [DOI] [PubMed] [Google Scholar]
- 3. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344:1396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev 2015;29:1203–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012;488:522–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alvarado AG, Thiagarajan PS, Mulkearns-Hubert EE, Silver DJ, Hale JS, Alban TJ, et al. Glioblastoma cancer stem cells evade innate immune suppression of self-renewal through reduced TLR4 expression. Cell Stem Cell 2017;20:450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, et al. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ 2006;13:1238–41. [DOI] [PubMed] [Google Scholar]
- 8. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444:756–60. [DOI] [PubMed] [Google Scholar]
- 9. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell 2013;155:462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Munoz-Hidalgo L, San-Miguel T, Megias J, Monleon D, Navarro L, Roldan P, et al. Somatic copy number alterations are associated with EGFR amplification and shortened survival in patients with primary glioblastoma. Neoplasia 2020;22:10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cao S, Zhou DC, Oh C, Jayasinghe RG, Zhao Y, Yoon CJ, et al. Discovery of driver non-coding splice-site-creating mutations in cancer. Nat Commun 2020;11:5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015;526:519–24. [DOI] [PubMed] [Google Scholar]
- 13. Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016;529:110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu Y, Li C, Shen S, Chen X, Szlachta K, Edmonson MN, et al. Discovery of regulatory noncoding variants in individual cancer genomes by using cis-X. Nat Genet 2020;52:811–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science 2013;339:957–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013;339:959–61. [DOI] [PubMed] [Google Scholar]
- 17. Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014;346:1373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gasperini M, Hill AJ, McFaline-Figueroa JL, Martin B, Kim S, Zhang MD, et al. A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell 2019;176:377–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pirinen M, Lappalainen T, Zaitlen NA, Consortium GT, Dermitzakis ET, Donnelly P, et al. Assessing allele-specific expression across multiple tissues from RNA-seq read data. Bioinformatics 2015;31:2497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mohammadi P, Castel SE, Cummings BB, Einson J, Sousa C, Hoffman P, et al. Genetic regulatory variation in populations informs transcriptome analysis in rare disease. Science 2019;366:351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pastinen T. Genome-wide allele-specific analysis: insights into regulatory variation. Nat Rev Genet 2010;11:533–8. [DOI] [PubMed] [Google Scholar]
- 22. Buckberry S, Bianco-Miotto T, Hiendleder S, Roberts CT. Quantitative allele-specific expression and DNA methylation analysis of H19, IGF2 and IGF2R in the human placenta across gestation reveals H19 imprinting plasticity. PLoS One 2012;7:e51210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deng Q, Ramskold D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 2014;343:193–6. [DOI] [PubMed] [Google Scholar]
- 24. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- 26. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van de Geijn B, McVicker G, Gilad Y, Pritchard JK. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat Methods 2015;12:1061–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alexa A, Rahnenfuhrer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006;22:1600–7. [DOI] [PubMed] [Google Scholar]
- 29. Mack SC, Singh I, Wang X, Hirsch R, Wu Q, Villagomez R, et al. Chromatin landscapes reveal developmentally encoded transcriptional states that define human glioblastoma. J Exp Med 2019;216:1071–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sapparapu G, Fernandez E, Kose N, Bin C, Fox JM, Bombardi RG, et al. Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 2016;540:443–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rachmilewitz J, Goshen R, Ariel I, Schneider T, de Groot N, Hochberg A. Parental imprinting of the human H19 gene. FEBS Lett 1992;309:25–8. [DOI] [PubMed] [Google Scholar]
- 33. Ma Y, Gong Y, Cheng Z, Loganathan S, Kao C, Sarkaria JN, et al. Critical functions of RhoB in support of glioblastoma tumorigenesis. Neuro Oncol 2015;17:516–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baldwin RM, Parolin DA, Lorimer IA. Regulation of glioblastoma cell invasion by PKC iota and RhoB. Oncogene 2008;27:3587–95. [DOI] [PubMed] [Google Scholar]
- 35. Morrison BH, Bauer JA, Kalvakolanu DV, Lindner DJ. Inositol hexakisphosphate kinase 2 mediates growth suppressive and apoptotic effects of interferon-beta in ovarian carcinoma cells. J Biol Chem 2001;276:24965–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chakraborty A, Koldobskiy MA, Sixt KM, Juluri KR, Mustafa AK, Snowman AM, et al. HSP90 regulates cell survival via inositol hexakisphosphate kinase-2. Proc Natl Acad Sci U S A 2008;105:1134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Abraham BJ, Hnisz D, Weintraub AS, Kwiatkowski N, Li CH, Li Z, et al. Small genomic insertions form enhancers that misregulate oncogenes. Nat Commun 2017;8:14385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005;352:997–1003. [DOI] [PubMed] [Google Scholar]
- 39. Zoppoli G, Regairaz M, Leo E, Reinhold WC, Varma S, Ballestrero A, et al. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc Natl Acad Sci U S A 2012;109:15030–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mu Y, Lou J, Srivastava M, Zhao B, Feng XH, Liu T, et al. SLFN11 inhibits checkpoint maintenance and homologous recombination repair. EMBO Rep 2016;17:94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, et al. PARP inhibitor activity correlates with SLFN11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clin Cancer Res 2017;23:523–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ianevski A, Giri AK, Aittokallio T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res 2020;48:W488–W93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li M, Kao E, Gao X, Sandig H, Limmer K, Pavon-Eternod M, et al. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 2012;491:125–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Malone D, Lardelli RM, Li M, David M. Dephosphorylation activates the interferon-stimulated Schlafen family member 11 in the DNA damage response. J Biol Chem 2019;294:14674–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Valdez F, Salvador J, Palermo PM, Mohl JE, Hanley KA, Watts D, et al. Schlafen 11 restricts flavivirus replication. J Virol 2019;93:e00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991;252:854–6. [DOI] [PubMed] [Google Scholar]
- 47. Zhu Z, Gorman MJ, McKenzie LD, Chai JN, Hubert CG, Prager BC, et al. Zika virus has oncolytic activity against glioblastoma stem cells. J Exp Med 2017;214:2843–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 2017;32:42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yi M, Nissley DV, McCormick F, Stephens RM. ssGSEA score-based Ras dependency indexes derived from gene expression data reveal potential Ras addiction mechanisms with possible clinical implications. Sci Rep 2020;10:10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Silginer M, Nagy S, Happold C, Schneider H, Weller M, Roth P. Autocrine activation of the IFN signaling pathway may promote immune escape in glioblastoma. Neuro Oncol 2017;19:1338–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Delbare SYN, Clark AG. Allele-specific expression elucidates cis-regulatory logic. PLoS Genet 2018;14:e1007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mayba O, Gilbert HN, Liu J, Haverty PM, Jhunjhunwala S, Jiang Z, et al. MBASED: allele-specific expression detection in cancer tissues and cell lines. Genome Biol 2014;15:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shames DS, Minna JD. IP6K2 is a client for HSP90 and a target for cancer therapeutics development. Proc Natl Acad Sci U S A 2008;105:1389–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Koldobskiy MA, Chakraborty A, Werner JK Jr, Snowman AM, Juluri KR, Vandiver MS, et al. p53-mediated apoptosis requires inositol hexakisphosphate kinase-2. Proc Natl Acad Sci U S A 2010;107:20947–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Borghese L, Dolezalova D, Opitz T, Haupt S, Leinhaas A, Steinfarz B, et al. Inhibition of notch signaling in human embryonic stem cell-derived neural stem cells delays G1/S phase transition and accelerates neuronal differentiation in vitro and in vivo. Stem Cells 2010;28:955–64. [DOI] [PubMed] [Google Scholar]
- 57. Ables JL, Decarolis NA, Johnson MA, Rivera PD, Gao Z, Cooper DC, et al. Notch1 is required for maintenance of the reservoir of adult hippocampal stem cells. J Neurosci 2010;30:10484–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Purow BW, Haque RM, Noel MW, Su Q, Burdick MJ, Lee J, et al. Expression of notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res 2005;65:2353–63. [DOI] [PubMed] [Google Scholar]
- 59. Zhang XP, Zheng G, Zou L, Liu HL, Hou LH, Zhou P, et al. Notch activation promotes cell proliferation and the formation of neural stem cell-like colonies in human glioma cells. Mol Cell Biochem 2008;307:101–8. [DOI] [PubMed] [Google Scholar]
- 60. Man J, Yu X, Huang H, Zhou W, Xiang C, Huang H, et al. Hypoxic induction of vasorin regulates notch1 turnover to maintain glioma stem-like cells. Cell Stem Cell 2018;22:104–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hai L, Zhang C, Li T, Zhou X, Liu B, Li S, et al. Notch1 is a prognostic factor that is distinctly activated in the classical and proneural subtype of glioblastoma and that promotes glioma cell survival via the NF-kappaB(p65) pathway. Cell Death Dis 2018;9:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fassl A, Tagscherer KE, Richter J, Berriel Diaz M, Alcantara Llaguno SR, Campos B, et al. Notch1 signaling promotes survival of glioblastoma cells via EGFR-mediated induction of anti-apoptotic Mcl-1. Oncogene 2012;31:4698–708. [DOI] [PubMed] [Google Scholar]
- 63. Luan J, Gao X, Hu F, Zhang Y, Gou X. SLFN11 is a general target for enhancing the sensitivity of cancer to chemotherapy (DNA-damaging agents). J Drug Target 2020;28:33–40. [DOI] [PubMed] [Google Scholar]
- 64. Fernandes C, Costa A, Osorio L, Lago RC, Linhares P, Carvalho B, et al. Current standards of care in glioblastoma therapy. In: De Vleeschouwer S, editor. Glioblastoma. Brisbane (AU): Codon Publications; 2017. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq (GSE119834), H3K27ac ChIP-seq (GSE119755), and human methylation 450K array data (GSE119774) for GSCs used in the paper were previously published and can be downloaded from GEO (GSE119776; ref. 29).