

Abstract
Deficiency in peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) expression or function is implicated in numerous neurological and psychiatric disorders. PGC-1α is required for the expression of genes involved in synchronous neurotransmitter release, axonal integrity, and metabolism, especially in parvalbumin-positive interneurons. As a transcriptional coactivator, PGC-1α requires transcription factors to specify cell-type-specific gene programs; while much is known about these factors in peripheral tissues, it is unclear if PGC-1α utilizes these same factors in neurons. Here, we identified putative transcription factors controlling PGC-1α-dependent gene expression in the brain using bioinformatics, and then validated the role of the top candidate in a knockout mouse model. We transcriptionally profiled cells overexpressing PGC-1α and searched for over-represented binding motifs in the promoters of upregulated genes. Binding sites of the estrogen-related receptor (ERR) family of transcription factors were enriched and blockade of ERRα attenuated PGC-1α-mediated induction of mitochondrial and synaptic genes in cell culture. Localization in the mouse brain revealed enrichment of ERRα expression in parvalbumin-expressing neurons with tight correlation of expression with PGC-1α across brain regions. In ERRα null mice, PGC-1α-dependent genes were reduced in multiple regions, including neocortex, hippocampus, and cerebellum, though not to the extent observed in PGC-1α null mice. Behavioral assessment revealed ambulatory hyperactivity in response to amphetamine and impairments in sensorimotor gating without the overt motor impairment characteristic of PGC-1α null mice. These data suggest that ERRα is required for normal levels of expression of PGC-1α-dependent genes in neurons, but that additional factors may be involved in their regulation.
INTRODUCTION
The transcriptional coactivator peroxisome proliferator activated receptor (PPAR) gamma coactivator 1-alpha (PGC-1α) interacts with a wide variety of transcription factors in different tissues. These transcription factors include, but are not limited to, members of the PPAR family, the nuclear respiratory factor family, estrogen receptor family, estrogen related receptor (ERR) family, and several non-nuclear receptor proteins (1–4). Interactions with these transcription factors largely mediate metabolic or mitochondrial gene programs that regulate the energy needs of that particular tissue (i.e. lipid metabolism versus oxidative phosphorylation).
While there is ample evidence for PGC-1α-interacting factors in peripheral tissues, not much is known about the mechanisms of PGC-1α-mediated transcriptional regulation in the brain, especially in discrete neuronal populations. In the brain, PGC-1α transcript is enriched in parvalbumin (PV)-positive interneurons (PV-INs), upregulated during PV-IN maturation, and required in adulthood for maintaining PV-IN-mediated circuit inhibition (5–7). Importantly, the silencing or activation of these interneurons is able to reduce or enhance cortical gamma oscillations and circuit function (8,9), highlighting the importance of understanding the transcriptional pathways that lead to their proper function. PV-INs are structurally complex with fast firing rates (10), making them inherently metabolically demanding. Accordingly, PV-INs have a higher number of mitochondria and enriched cytochrome c content relative to other neuronal populations (11) and PV-INs lacking PGC-1α show a reduction in intrinsic firing rate (5). The expression of PV itself is also dependent on PGC-1α and, within PV-INs, a loss of PGC-1α leads to a reduction in its previously identified dependent genes synaptotagmin II (Syt2), complexin 1 (Cplx1), neurofilament heavy chain (Nefh) (7,12), suggesting that PGC-1α facilitates the coordinated expression of genes involved in energy production and neurotransmission. As such, it is crucial to understand how these pathways are regulated by PGC-1α and to identify the transcription factors are responsible maintaining these pathways.
In this study, we use a combination of bioinformatics and cell culture studies to prioritize putative transcription factors which may mediate PGC-1α’s responses and then test whether the top candidate, ERRα, is required for PGC-1α-dependent gene expression in cell culture and the mouse brain.
METHODS
Cell Culture.
SH-SY5Y neuroblastoma cells were cultured in Dulbecco’s Modified Eagle’s Medium/DMEM and 2mM Glutamax (degradation-resistant L-glutamine) media with 10% fetal bovine serum and 1% penicillin-streptomycin. Cells were grown to 70% confluency in 10 cm plates in a humidified atmosphere containing 5% CO2 at 37°C. Cells were transfected for one hour with AdV-CMV-GFP (4.0x1012 particles/ml) or AdV-CMV-PGC-1α-ires-GFP (4.0x1012 particles/ml), originally provided by Bruce M. Spiegelman (Dana Farber Cancer Research Center, Harvard University) and further propagated at the University of Michigan Cancer Center Vector Core as in (7). For RNA-seq studies, cells were treated with AdV-CMV-GFP or AdV-CMV-PGC-1α-ires-GFP for 48 hours, total RNA isolated with Trizol and chloroform/isopropanol, and sent to the Hudson Alpha Genomics Services Lab (Hunstville, Alabama) for sequencing (3 plates pooled per sample; 3 samples per group). For studies involving blockade of ERRα activity, 24 hours following transduction, cells were treated with different doses of XCT790 (2–50 µM, cat#X4753, Sigma, St. Louis, MO, USA) or the vehicle, DMSO, and collected 48 hours later in Trizol for RNA isolation (see below).
RNA sequencing.
Libraries were constructed using the Poly A method, with the addition of a directional library preparation module to retain strand information. Sequencing was performed on an Illumina HiSeq 2500 (HI-PE50 v4), and FASTQ files were generated using DRAGEN Host Software v01.011.222.02.04.04.36875. Raw reads were aligned using STAR v2.6.0c, and transcript abundances were calculated using HTSeq-Count v0.10.0. Normalization and differential expression was performed using DESeq2 v1.20.0, using Gencode GRCh38 (p7 release 25) as a reference, with p and q values of < 0.05, as determined with Wald (p) and Benjamini-Hochberg (q) calculations. Data files have been deposited in the GEO NCBI database (GSE151499).
To assess overlap of genes across data sets, we used the SuperExactTest program (19). SuperExactTest analyzed the intersection between gene sets through a combinatorial algorithm that used fold enrichment, Fisher’s Exact Test and hypergeometric modeling to generate a p value and FDR for commonalities. This test is done by analyzing the gene lists as subsets of an overarching list, the proteome or the genome of the species, and then calculating the overlap as a derivative of the overarching list. For the Exact Test, overlap of multiple gene lists is assumed to be equal and then analyzed, creating a formula where there is a one-tailed probability that the overlap is greater than predicted.
Webgestalt: Identification of Transcriptional Regulators
To identify consensus binding sequences in the promoter of a set of genes, we used WEB-based GEne SeT AnaLysis 2017 (http://www.webgestalt.org/; (20). The set of genes of interest were input as Homo sapien gene symbols, as data were obtained from human neuroblastoma cells, with the method of interest set to Gene Set Enrichment Analysis (GSEA) and the functional database identified as “network: transcription factor target.” Default parameters were maintained; data was analyzed using Holm-Bonferroni and the top ten results were reported.
Secondary Analysis of Mouse Brain Single-cell Transcriptome Data.
To explore cell-type-specific expression patterns of putative PGC-1α-interacting factors and their correlation with PGC-1α distribution in the brain, we used data downloaded from Dropviz.org (18). Subcluster metacell data representing aggregate Unique Molecular Identifier (UMI) counts were downloaded and normalized to the total UMI count per subcluster, using the previously defined identity of subclusters as a guide. Additional classifications were made to specify fast-spiking interneurons, dopaminergic neurons (outside of the midbrain), dopaminergic neurons (in the midbrain), and medium spiny neurons. Clusters or subclusters with one or two datapoints per cell type were not included in the analyses. Datasets were visualized and analyzed in GraphPad Prism v.8 using volcano plots and a non-parametric one-way ANOVA test (Kruskal-Wallis test) followed by a Dunn’s multiple comparison test, with significance set at p < 0.05.
Animals.
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Southern Research and the University of Alabama at Birmingham and performed in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). ERRα null mice (Esrra−/−) were obtained from Jackson Labs (JAX#005801) and were designed with a beta-galactosidase gene inserted in place of exon 2. Breeding schemes were established using mice heterozygous for ERRα to generate littermate controls. All experiments were conducted using male and female mice between 3–5 months of age (unless otherwise stated), and the experimenter was blind to genotype. Mice were maintained on a C57BL6/J genetic background and housed two to seven per cage at 26 +/− 2°C room temperature with food and water ad libitum.
Small Molecule Fluorescent In situ Hybridization (SM-FISH)
SM-FISH was performed as previously described (21,22) using the RNAscope Multiplex Fluorescent assay (Advanced Cell Diagnostics, Newark, CA, USA) according to the manufacturer’s instructions. Mice were anesthetized with isoflurane and decapitated, and brains were rapidly removed and frozen with powdered dry ice. Brains were sectioned on a cryostat at 20 µm thickness and collected on SuperFrost Plus slides (Thermo Fisher Scientific) and kept frozen to maintain RNA integrity. For fluorescent in situ hybridization, sections were fixed in 4% pre-chilled paraformaldehyde followed by dehydration in ethanol. Due to the small length of transcript deleted in Esrra null mice, BaseScope (ACD, Newark, CA, USA) was used to detect Esrra transcript in these experiments. Probes were custom designed to recognize exons 1–3 (180–533bp) of Esrra (NM_007953, Advanced Cell Diagnostics/ACD, Newark, CA, USA). The protocol was conducted per manufacturer’s instructions. Sections were fixed and dehydrated then incubated in hydrogen peroxide followed by washes. Sections were then incubated in protease III (ACD) followed by incubation with probes for 2 hours at 40°C followed by amplification and incubation with BaseScope Fast RED A and B. BaseScope was followed by the RNAscope Multiplex Fluorescent assay (ACD) per manufacturer’s instructions for concurrent ISH. Tissues were treated with a mixture of probes for 2 hours at 40°C followed by fluorescent amplification. If BaseScope was not used, slides were pretreated with ethanol and protease IV (ACD); no hydrogen peroxide step was used for RNAscope alone. Probes used for RNAScope are as follows: Pvalb (Cat#421931), Slc17a7 (Cat#416631), Gja1 (Cat#486191). Slides were then coverslipped with Prolong Gold antifade mounting media containing DAPI (Thermo Fisher Scientific). The experimenter was blind to genotype for quantification of transcripts. Images were captured with a Nikon A1+ confocal microscope; all settings, including laser intensity, gain, offset, and zoom, were held constant across all groups for a given experiment. Image thresholding for individual channels and quantification of mean pixel density for a given area was performed using Image J (23). To generate representative images presented in figures, images were imported to Adobe Photoshop CS3 (Adobe, San Jose, CA) and adjustments to contrast, sharpness and brightness were held constant across experimental groups.
Western Blot
This protocol was conducted as previously described (7). Cortical and hippocampal tissue (n=3/genotype/region) were collected and frozen on dry-ice then stored at −80°C until use. For preparation, samples were placed in RIPA buffer (150 mM NaCl, 50 mM Tris, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholic acid, pH 8.0), and homogenized using the Omni Bead Ruptor Homogenizer (OMNI International). Total protein concentration was determined with a bicinchonicic acid protein assay kit (Thermo Fisher Scientific), and absorbance was measured at 540 nm. Protein was then denatured in sample buffer (62.5 mM Tris-HCl, 20% glycerol, 2% SDS, 5% β-mercaptoethanol, 1 mg/mL bromophenol blue; pH 6.8) at 95 °C and equivalent amounts of protein were loaded into 4–20% Mini-PROTEAN® TGX™ precast gels (Bio-Rad, Hercules, California, USA). Next, protein was transferred onto PVDF membranes which were then blocked for 1 h with 5% milk in Tris-buffered saline (TBS; pH 7.6) with 1% Tween (TBS-T). Following, membranes were probed with an antibody for ERRα (1:1000; ab76228, Abcam, Cambridge, UK) in 5% IgG-free bovine serum albumin (BSA; Jackson ImmunoResearch) in TBS-T overnight at 4 °C. Following incubation in HRP-conjugated secondary antibodies (Invitrogen) in 5% milk in TBS-T for 1 h at room temperature, membranes were placed in Clarity™ Western ECL HRP substrate (Bio-Rad) and imaged using the ChemiDoc MP Imaging System (Bio-Rad). Membranes were then washed and re-probed for actin (MAB1501, Millipore) in 5% milk in TBS-T for 1 h at room temperature and developed as before to confirm equal loading of protein in samples probed for ERRα. The optical density of bands was calculated after background subtraction using Image Studio Lite (LI-COR, Lincoln, NE, USA). All bands were normalized to the signal for actin from the corresponding lanes and expressed as mean optical density, ratio to actin.
Immunofluorescence
Animals were anesthetized with isoflurane and perfused intracardially with cold PBS and 4% PFA in PBS. Brains were removed, postfixed in 4% PFA for 24–72 h, and cryoprotected in 15% sucrose for 24 hours followed by 30% sucrose for 24 hours. Brains were then frozen in chilled 2-methyl-butane and stored at −80°C. Tissue was cryosectioned at 40 µm into 1:1 glycerol and PBS and stored at −20°C. For immunohistochemistry, sections were washed with PBS, permeabilized with 0.3% Triton-X100 (Sigma-Aldrich, St. Louis, MO, USA) in PBS, and then blocked with 10% serum from the host of the secondary antibody in PBS for 1 hour at room temperature. Following, sections were incubated in goat-anti-PV (1:1000; PVG213, Swant, Switzerland) and rabbit anti-ERRα antibodies (1:1000; ab76228, Abcam, Cambridge, UK) in 0.3% TxPBS and 5% serum from the host of the secondary antibody overnight at 4°C. On the second day, sections were washed with PBS, incubated with fluorescence-conjugated anti-goat and anti-rabbit secondaries (1:2000; Invitrogen, Thermo Fisher Scientific) for 2 hours at room temperature in 5% serum. Following washes, sections were coverslipped using Prolong Antifade Gold with DAPI (Invitrogen) and stored at 4 °C. Images were captured with a Nikon A1+ confocal microscope. All confocal settings, including laser intensity, gain, offset, and zoom, were held constant across all images.
Transcript Analyses
Quantitative PCR was performed as described (24). Mice were anesthetized with isoflurane prior to decapitation. Brains were rapidly removed, and microdissected brain regions were flash frozen on dry ice and stored at −80°C until use. Tissue was homogenized in TRIzol (Thermo Fisher Scientific, Pittsburg, PA, USA) using either a Tissue-Tearor homogenizer (Biospec, Bartlesville, OK, USA) or the Omni Bead Ruptor Homogenizer (OMNI International, Kennesaw, GA, USA), and RNA was isolated using the TRIzol/choloform-isopropanol method following the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). RNA concentration and purity were assessed using a NanoDrop One (Thermo Fisher Scientific, Pittsburg, PA, USA). Equivalent amounts of RNA (1 µg) were treated with DNase I (Promega, Madison, WI, USA) at 37°C for 30 minutes followed by DNase Stop solution at 65°C for 15 minutes. RNA was then reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Carlsbad, CA, USA). Transcripts were measured using mouse-specific primers from Applied Biosystems (Supplemental Table 1) and JumpStart Taq Readymix (Sigma, St. Louis, MO, USA) using a protocol with an initial ramp (2 minutes, 50°C; 10 minutes, 95°C) and 40 subsequent cycles (15 seconds, 95°C; 1 minute, 60°C). Relative concentration of transcript was calculated in comparison to a standard curve generated from pooled cDNA samples and then diluted (1:5, 1:10, 1:20, 1:40; calibrator method). These values were then normalized to beta-actin or 18S expression and reported as ratio to control samples. For cell culture studies, target transcript abundance was normalized to PGC-1α transcript abundance to take into account any changes in PGC-1α transcription caused by XCT790 treatment.
Behavioral analyses.
All behavioral testing was conducted between 10am and 3pm during the “lights-on” period (6am-6pm) with the exception of elevated plus maze and pre-pulse inhibition; these studies were conducted between 7 pm and 12 am in the dark. Initially, to measure anxiety, performance was assessed in the elevated plus maze as previously described (25). Animals were placed on an NIR backlit maze (MedAssociates, St. Albins, VT, USA) for 5 minutes. Data was analyzed and represented as an anxiety index which incorporates the time spent in the open arms as well as number of entries into open arms compared to the total maze time and total maze exploration; a higher index indicates more anxiety-like behaviors (26).
For assessment of baseline activity, animals were placed in a square apparatus (27.9 cm2) consisting of 48 infrared beams (MedAssociates, St. Albins, VT, USA) for 30 minutes. Data were collected with Open Field Activity Software (MedAssociates, St. Albins, VT, USA) in one-minute intervals over the test period. To evaluate responsivity to alterations in dopaminergic tone, animals were treated via intraperitoneal injection with 5mg/kg d-amphetamine and immediately placed in the open field for one hour. Data were collected with Open Field Activity Software (MedAssociates, St. Albins, VT, USA) in one-minute intervals over the test period.
Startle reflex and prepulse inhibition were measured as outlined (25) using the Acoustic Startle Reflex Package for mice (Med Associates, St. Albins, VT, USA). The startle response was recorded in response to 120dB and prepulse inhibition was recorded in response to 0, 4, 8, and 16dB pulses above a 65dB white noise background.
Statistics
All transcript data with three groups or more were analyzed using parametric one-way ANOVA followed by Tukey’s multiple comparisons following tests for normality using the Kolmogorov-Smirnov test and QQplots as well as variance across groups using the Bartlett’s test. These experiments included transcript data for both in vitro and in vivo experiments and behavioral assessment. In the event that these assumptions were not met, data were then analyzed using a Kruskal-Wallis followed by Dunn’s multiple comparisons. For analyses of two groups which included transcript analysis, sm-FISH experiments and western blot, data were analyzed using a two-tailed t-test unless assumptions of normality and variance were not met. In those instances, data were analyzed using a Mann-Whitney. For analysis of ambulation over time (open field) and prepulse inhibition, data were analyzed using a repeated measures two-way ANOVA using the Geisser-Greenhouse correction for sphericity followed by Tukey’s or Dunnett’s Multiple Comparison. Outliers for transcript data were identified using ROUT with Q set at 1%.
RESULTS
Identification of enriched promoter sequences in PGC-1α-dependent transcripts.
Overexpression of PGC-1α in SH-SY5Y leads to the upregulation of 887 transcripts compared to control through microarray data (7), and a number of these PGC-1α-responsive genes have been validated in multiple regions of PGC-1α null and cell-specific knockout lines (6,7,21,22). Here, we used RNA sequencing (RNA-seq) to comprehensively identify transcripts differentially regulated with PGC-1α overexpression; 2889 transcripts were upregulated whereas 874 transcripts were downregulated with PGC-1α overexpression compared to GFP control (n=3/group; Figure 1A). As predicted from our microarray data, the PGC-1α-dependent gene parvalbumin (PVALB) was robustly upregulated, being the second most-highly induced transcript identified by RNA sequencing (7,12).
Fig. 1.
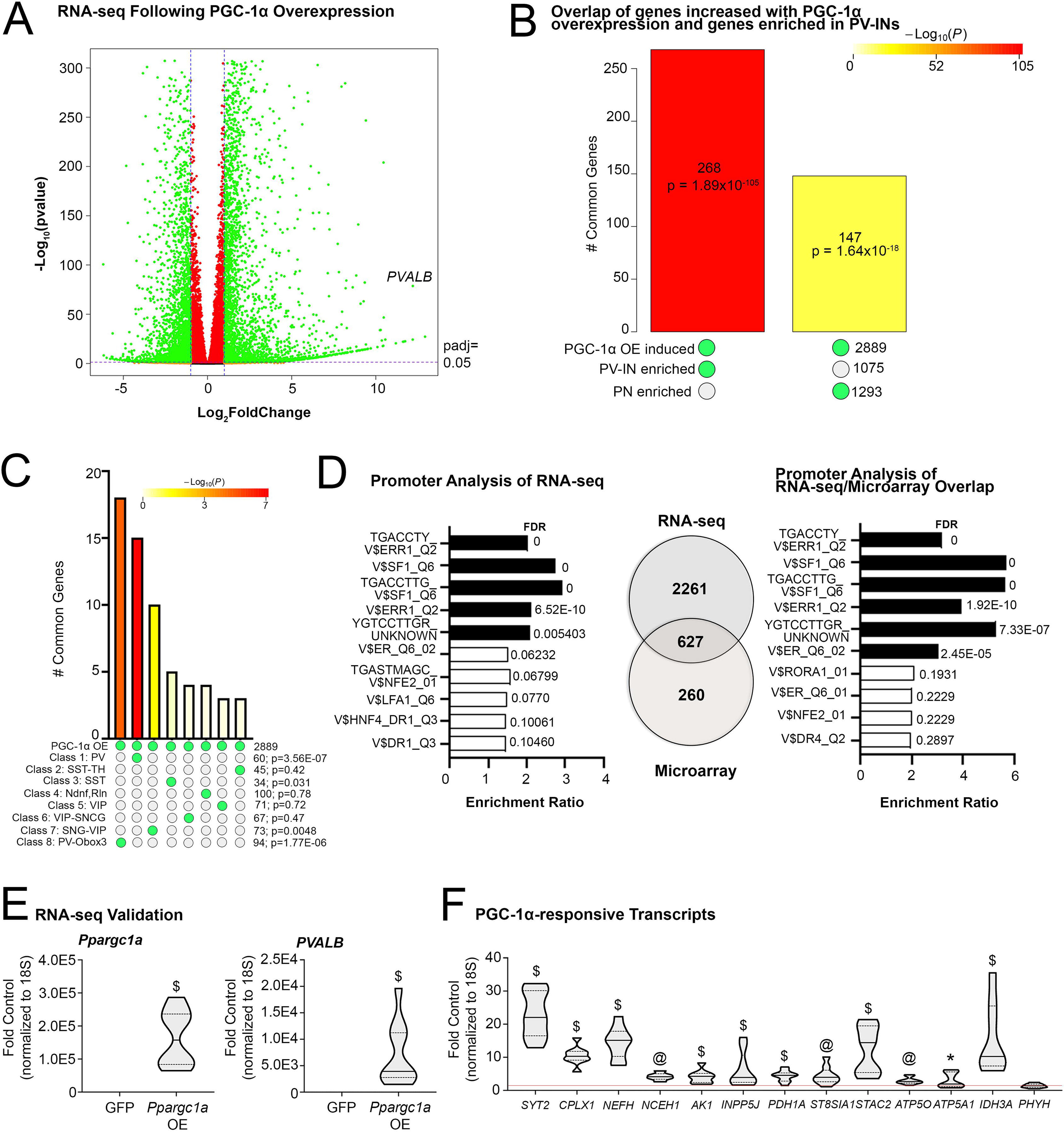
Transcriptional profiling of cells overexpressing PGC-1α reveals similarity to PV-IN profiles and identifies enriched transcription factor binding sequences in responsive genes. (A) RNA sequencing identified transcripts up- and down-regulated following AdV-CMV-Ppargc1a-ires-GFP (PGC-1αOE) or AdV-CMV-GFP (GFP) treatment of SH-SY5Y neuroblastoma cells. n = 3/group. (B) SuperExactTest of overlap between genes induced with PGC-1αOE in SH-SY5Y cells compared to genes enriched in either PV-INs or PNs (Mo et al., 2015). (C) SuperExactTest of overlap between genes induced with PGC-1αOE in SH-SY5Y cells with those enriched in eight distinct GABAergic subclusters. (D) WebGestalt enrichment analysis of consensus binding sites in the promoters of identified transcripts upregulated by PGC-1α overexpression. There was an overlap of 627 genes between these RNA sequencing data and previously published microarray data (Lucas et al., 2014). WebGestalt enrichment analysis for these overlapping genes are shown. (E) Ppargc1a and Pvalb transcript in samples treated with DMSO and either AdV-CMV-Ppargc1a-ires-GFP (PGC-1αOE) or AdV-CMV-GFP (GFP) to confirm overexpression. (F) Previously identified PGC-1α-responsive transcripts in the DMSO and PGC-1αOE as fold control. *p ≤ 0.05, @p ≤ 0.001, $p ≤ 0.0001.
We next compared the genes upregulated by PGC-1α overexpression in cell culture to previously identified genes found to be enriched in either PV-INs or pyramidal neurons (PNs; (27) in mice, to determine the utility of PGC-1α-overexpressing cells for understanding the biology of PV-INs. There was a significant overlap of genes increased in response to PGC-1α overexpression with those enriched in PV-INs (268, p = 1.89E10−105) versus those enriched in PNs (147, p = 1.64E10−18) as determined using the SuperExactTest (Figure 1B). The preferential expression of PGC-1α responsive targets in PV-INs was confirmed by SuperExactTest when compared across eight subcluster populations of human cortical inhibitory populations (28); overlap of PGC-1α-responsive targets was significant only in PV-INs (60, p = 3.56E10−07), PV-Obox3 (94, p = 1.77E10−06), and SST subtypes (34, p = 0.031); Figure 1C). These data show that the overexpression of PGC-1α can induce genes enriched in PV-INs and suggest that PGC-1α plays an important role in PV-INs versus other non-PV-expressing neuronal populations (7,12).
To identify putative transcription factors mediating the upregulation of PGC-1αresponsive genes, we used WebGestalt to detect enrichment of consensus binding sequences within the promoters of genes upregulated by PGC-1α. These factors included: splicing factor 1 (“SF1”), estrogen-related receptor alpha (“ERR1/ERRα”), estrogen receptor (“ER family”), lymphocyte function-associated antigen 1 (“LFA1/ITGB2”), nuclear factor erythroid 2 (“NFE2”), hepatocyte nuclear factor 4 (“HNF4”), and down-regulator of transcription 1 (“DR1”). The transcription factor highest in significance for enrichment in consensus binding sequences was ERR1/ERRα (Figure 1D). This information, in combination with previously published evidence for PGC-1α/ERRα interactions (13,15,16), implicated ERRα as a likely candidate for regulating PGC-1α-responsive genes.
We also compared these RNA-seq data to previously published microarray data and found an overlap of 627 genes as being upregulated by overexpression of PGC-1α. To identify if these overlap in genes yielded similar mechanisms of regulation, we again used WebGestalt to identify which transcription factor consensus binding sequences were enriched and again identified ERR1/ERRα as a candidate (Figure 1D). Importantly, we found that the RNA-seq data replicated previous findings with microarrays, with q-rt-PCR confirmation of PGC-1α expression and induction of Pvalb (Figure 1E) and other previously identified PGC-1αdependent genes with PGC-1α overexpression (Figure 1F). First, we confirmed with q-rt-PCR that PGC-1α was indeed overexpressed (OE) in cells treated with AdV-CMV-PGC-1α-ires-GFP (PGC-1αOE) (Mdn=157394) as compared to cells treated with AdV-CMV-GFP (GFP, Mdn=0.60; U=0, p<0.0001). We next measured Pvalb transcript; previous experiments have shown Pvalb transcript to be the most robust readout of PGC-1α activity and significantly increased transcript upregulated by PGC-1αOE (7,12). As expected, Pvalb transcript following PGC-1αOE (Mdn=3918) was significantly increased compared to those treated with GFP (Mdn=0.92; U=0, p<0.0001). Neuronally-enriched PGC-1α-dependent transcripts (7) including synaptotagmin II (Syt2), complexin I (Cplx1), and neurofilament heavy chain (Nefh) as well as ubiquitously-expressed metabolic transcripts were induced with overexpression, except Phyh. Syt2 (U=0, p<0.0001) and Cplx1 (U=0, p<0.0001) were both significantly upregulated with PGC-1αOE (Mdn=22.03 and 10.30, respectively) compared to GFP (Mdn=0.42 and 0.83, respectively). Nefh transcript responded similarly; there was a significant increase in transcript with PGC-1αOE (Mdn=15.11) compared to GFP (Mdn=1.20; U=0, p<0.0001). Nceh1 transcript was also upregulated with PGC-1αOE (Mdn=4.60) compared to GFP (Mdn=1.06; U=0, p=0.0003). PGC-1αOE significantly increased Ak1 (U=0, p<0.0001) and Inpp5j (U=0, p<0.0001) transcripts (Mdn=4.25 and 3.90, respectively) compared to GFP (Mdn=0.87 and 0.80, respectively). Pdh1a transcript was significantly increased in PGC-1αOE (Mdn=4.55) samples compared to GFP (Mdn=0.78; U=2, p<0.0001). St8sia1 (U=4, p=0.0005) and Stac2 (U=0, p<0.0001) transcripts were also significantly upregulated with PGC-1αOE (Mdn=3.96 and 14.42, respectively) compared to control (Mdn=0.99 and 0.91, respectively). PGC-1αOE upregulated Atp5o transcript compared to GFP (t17=4.40, p=0.0004). PGC-1αOE also increased Atp5a1 (Mdn=1.94; U=19, p=0.02) and Idh3a (Mdn=10.24; U=0, p<0.0001) transcripts compared to control (Mdn=0.69 and 1.08, respectively). There was no change in Phyh transcript between groups (t16=1.13, p=0.28).
Prioritization of Esrra based on neuroanatomical distribution similar to that of Ppargc1a in the mouse brain.
Previous studies show enrichment for Ppargc1a transcript and PGC-1α protein in GABAergic populations and document the transcriptional and functional consequences of its deletion in cortical PV-interneurons (PV-INs) (7,12,29). Based on these studies, we further prioritized candidates by exploring their neuroanatomical distributions with respect to the distribution of Ppargc1a using the single-cell RNA-sequencing database Dropviz.org (18). Three examples, Esrra, Nrf1, and Ppard are shown in Figure 2A with reference to Ppargc1a in all cell types in the database. Gene abundance data for all top hits from Figure 1 are included in Supplemental Figure 1 and descriptive statistics are located in Supplemental Table 2 for neuronal populations in the mouse cortex. Correlations of Ppargc1a to Esrra transcript in multiple neuronal populations (n=231 non-neuronal, n=152 glutamatergic, n=88 PV− GABAergic, n=33 PV+ GABAergic, n=16 medium spiny neuron, n=7+ cholinergic, n=13 DAergic) throughout the brain revealed a strong correlation (R2=0.64), with GABAergic PV+ populations showing the highest expression of both transcripts. In contrast, correlations between Ppargc1a and Nrf1 (R2=0.032) or Ppard (R2=0.0035) were weak (Figure 2A). When exploring transcription factor expression abundance in PV-IN populations (Supplemental Figure 1), Esrra and Esrrg were most highly expressed in PV-INs as compared to other transcription factors, with higher enrichment in PV-INs than in other neuronal populations. Together, the enrichment of ERR1/ERRα consensus binding sites in genes responsive to PGC-1α overexpression and localization data indicating Esrra enrichment in PV-INs suggested ERRα as a prime candidate for the regulation of PGC-1α-dependent transcription in the CNS, particularly in PV-expressing populations.
Figure 2. Inverse agonist of ERRα attenuates the induction of PGC-1α-responsive transcripts.
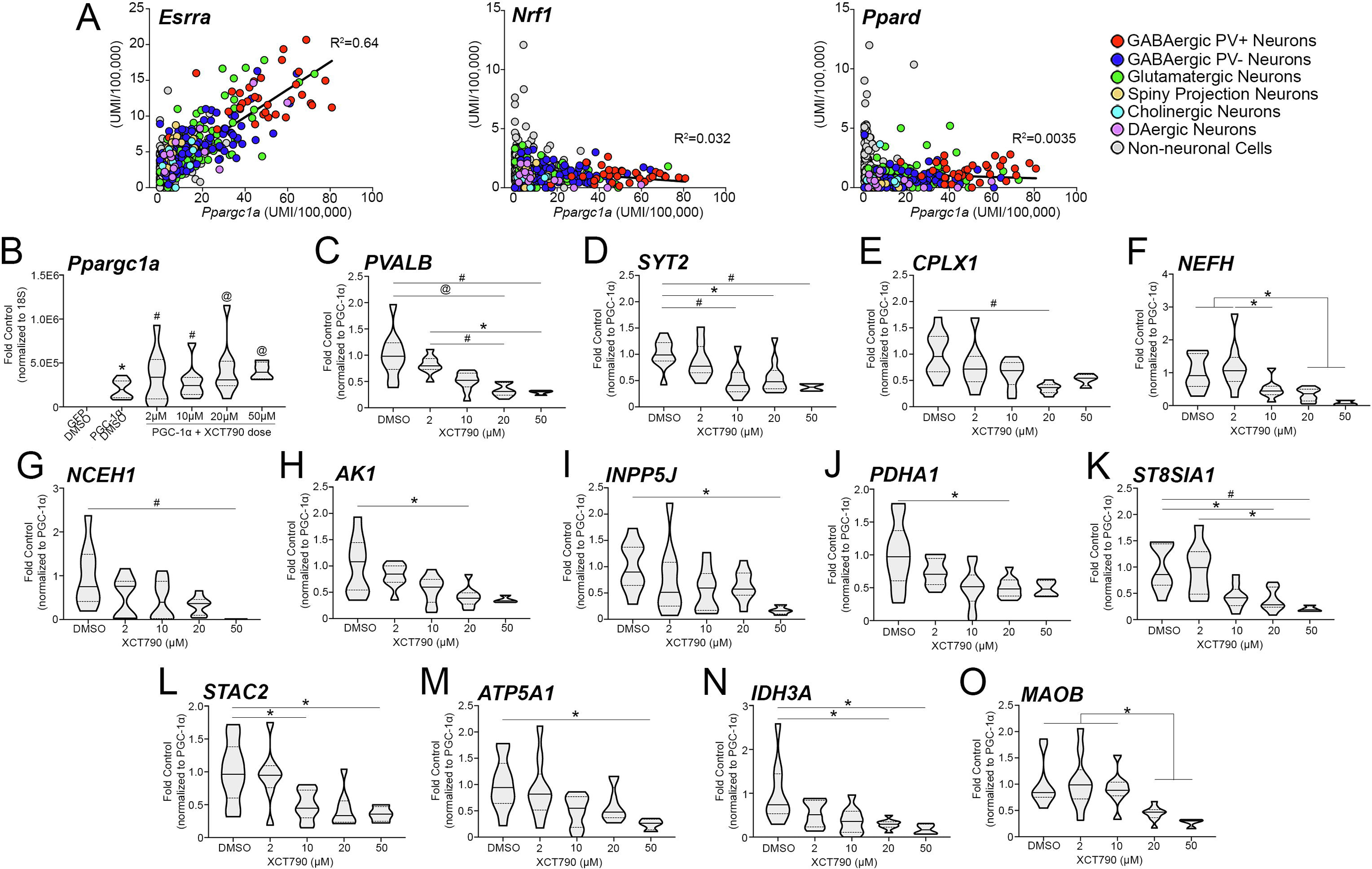
(A) Correlation of Esrra, Nrf1, and Ppard transcript abundance across neuronal and non-neuronal populations throughout the brain with that of Ppargc1a (Dropviz.org; n=231 non-neuronal, n=152 glutamatergic, n=88 PV− GABAergic, n=33 PV+ GABAergic, n=16 medium spiny neuron, n=7+ cholinergic, n=13 DAergic). (B) Significantly increased Ppargc1a transcript levels in DMSO and XCT790-treated SH-SY5Ys following PGC-1αOE compared with samples treated with DMSO and AdV-CMV-GFP. (C-F) Transcript for neuronally enriched genes PVALB, SYT2, CPLX1, and NEFH in the presence of both PGC-1αOE and increasing dose of XCT790. The expression of ubiquitous (G-M) and previously demonstrated direct targets of ERRα (N, O) was also measured. DMSO, n=8–10; 2 μM, n=8–10; 10 μM, n=8–10; 20 μM, n=8–10; 50 μM, n=3–4 XCT790. *p≤0.05, #p≤0.01, @p≤0.001, $p≤0.0001. Median is represented by the solid line, upper and lower quartiles are represented by dotted lines.
Parvalbumin induction by PGC-1α relies on ERRα activity
ERRα is a known binding partner of PGC-1α in peripheral cell types and tissues (13–17), and several studies have suggested its importance in neuronal function (32–34). To investigate the potential for ERRα to influence PGC-1α-responsive transcripts we used XCT790, an inverse agonist of ERRα (35,36). Notably, XCT790 not only disrupts PGC-1α interaction with ERRα, it also targets ERRα for proteosomal degradation. SH-SY5Y neuroblastoma cultures were treated with 2 μM (n=8–10), 10 μM (n=8–10), 20 μM (n=8–10), or 50 μM (n=3–4) XCT790 and compared to DMSO-treated control samples (n=9–10).
First, we verified that PGC-1αOE was maintained in the presence of the selected XCT790 doses compared to DMSO control (Figure 2B). There was a significant difference in the expression of Ppargc1a transcript across groups (H5=25.40, p=0.0001); when compared to dually treated DMSO and GFP control samples, post hoc analysis revealed that Ppargc1a transcript was significantly increased in PGC-1αOE samples treated with DMSO (p=0.03), 2 μM (p=0.0027), 10 μM (p=0.0045), 20 μM (p=0.0002), and 50 μM XCT790 (p=0.0006). To determine if XCT790-mediated blockade of ERRα could influence PGC-1α-responsive transcription, we first measured transcript for PVALB. PVALB transcript was significantly different between groups (H4=27.01, p<0.0001); post-hoc analysis revealed PVALB transcript to be significantly reduced at 20 μM (p=0.0002 and p=0.0048) and 50 μM (p=0.0062 and p=0.034), compared to DMSO and 2 μM XCT790 (Figure 2C). To confirm that knockdown of PVALB transcript was due to the interference of ERRα activity and not another known PGC-1α interacting factor, we measured PVALB transcript in response bisphenol A diglycidyl ether (BADGE), an antagonist of the known PGC-1α binding partner of PPARγ (37,38). First, we measured gene expression (n=3/group) of the PPARγ target, Glut4 (39). There were significant differences across BADGE dose for GLUT4 expression (F2,6=62.59, p>0.0001; data not shown); post-hoc analysis revealed significant reductions in 10 μM (0.54±0.0030) and 20 μM BADGE (0.34±0.011) compared to control (1.055±0.0030; p=0.0006 and p<0.0001, respectively) and a significant stepwise reduction from 10 μM and 20 μM (p=0.049). Notably, one-way ANOVA revealed no differences between control (1.00±0.075), 10 μM (0.82±0.087) and 20 μM BADGE (0.96±0.12) for PVALB transcript (F2,6=0.93, p=0.44; data not shown).
Blockade of ERRα in culture interferes with the ability of PGC-1α to induce neuron-enriched PGC-1α-responsive genes
We predicted that neuronally enriched transcripts for SYT2, CPLX1, and NEFH would be reduced in response to XCT790 in a manner similar to PVALB. SYT2 expression was significantly different across XCT790 doses (F4,36=5.99, p=0.0008); post-hoc analysis revealed significant reductions in expression at 10 μM (p=0.0053), 20 μM (p=0.022) and 50 μM (p=0.0088) doses of XCT790 compared to control (Figure 2D). CPLX1 transcript was also affected by XCT790 (H4=16.06, p=0.0029); post-hoc analysis showed that transcript was significantly reduced at 20 μM XCT790 compared to control (p=0.0012; Figure 2E). XCT790 also influenced NEFH expression (F4,33=6.36, p=0.0007); post-hoc analysis revealed a reduction in NEFH transcript at both 20 μM (p=0.041) and 50 μM (p=0.039) XCT790 compared to control. Additionally, expression levels at 10 μM (p=0.034), 20 μM (p=0.0056), and 50 μM (p=0.0092) treatments were significantly reduced compared to 2 μM XCT790 (Figure 2F). These data demonstrate that blockade of ERRα dramatically impedes the ability of PGC-1α to upregulate neuron-enriched PGC-1α-dependent transcripts in vitro.
Our lab has also identified several PGC-1α-responsive transcripts whose expression is not limited to neuronal populations, many of them with roles in metabolism (7). NCEH1 (H4=14.77, p=0.0052) and AK1 (H4=16.06, p=0.0029) were significantly affected by XCT790 treatment; 50 μM XCT790 significantly reduced NCEH1 expression compared to control (p=0.0018; Figure 2G) while AK1 was significantly reduced at 20 μM XCT790 compared to control (p=0.039, Figure 2H). One-way ANOVA revealed differences amongst conditions for INPP5J expression (H4=10.84, p=0.029); post-hoc analysis showed significant reductions in expression were between 50 μM and control (p=0.013, Figure 2I). PDHA1 expression was also influenced by ERRα inhibition (H4=0.014, p=0.014); post hoc analysis revealed differences at 20 μM XCT790 compared to control (p=0.037; Figure 2J).
Significant differences amongst XCT790 doses were also detected for ST8SIA1 expression (H4=20.96, p=0.0003); post-hoc analysis revealed reductions in ST8SIA1 expression at 20 μM (p=0.022) and 50 μM (0.0047) XCT790 compared to control. The reduction in expression at 50μM XCT790 was also significantly less compared to 2 μM XCT790 (p=0.012; Figure 2K). Significant reductions in STAC2 gene expression (F3,26=5.15, p=0.0063) were found between 10 μM (p=0.032) and 50 μM (p=0.035) XCT790 compared to control by post-hoc analysis (Figure 2L). ATP5A1 (F4,37=3.63, p=0.014; Figure 2M) was significantly reduced in expression in response to 50 μM XCT790 compared to control (p=0.036) as revealed by post hoc analysis. No differences were detected across XCT790 doses in ATP5O (F4,36=2.24, p=0.084) or PHYH (H4=2.50, p=0.64; Supplemental Figure 2) gene expression. All median and confidence intervals for in vitro data are included in Supplemental Table 3.
As positive controls for ERRα inhibition, we confirmed the reduction of two previously identified ERRα direct targets, isocitrate dehydrogenase 3a (Idh3a) and monoamine oxidase B (Maob) (32,40). Kruskal-Wallis revealed significant differences in IDH3A and MAOB expression across treatment groups (H4=15.25, p=0.0042 and H4=23.25, p=0.0001, respectively; Figure 2N, O). Post-hoc analysis showed a significant reduction in MAOB expression at 20 μM and 50 μM XCT790 doses compared to control (p=0.035 and p=0.015), 2 μM XCT790 (p=0.023 and p=0.011), and 10 μM XCT790 (p=0.011 and p=0.0058, respectively) and for IDH3A (H4=15.25, p=0.0042) at 50 μM XCT790 compared to control (p=0.036).
Expression of ERRα is concentrated in parvalbumin-positive neuron populations
Previous studies have demonstrated widespread expression of ERRα throughout the brain, particularly in cortex, hippocampus, thalamus, the mammillary nucleus, cerebellum and brainstem (33). To measure Esrra transcript in the brain in a cell-type-specific way, we designed a probe to specifically target exons 1–3 (180–533bp) of Esrra (NM_007953; ACDBio). The Esrra-specific probe shows expression in Esrra+/+ cortex and hippocampus and this signal is lost in these regions of the Esrra−/− brain, validating the utility of this probe in localization studies (n=2 sections/mouse; 2 mice/genotype; Figure 3A). Previous studies from our lab show enrichment by in situ hybridization for PGC-1α in cortical PV-INs compared to pyramidal neurons and astrocytes (21). To evaluate this enrichment pattern for Esrra, and confirm Dropviz.org data, we used fluorescent probes for Esrra, Pvalb, and Slc17a7 (glutamatergic marker) or Gja1 (astrocytic marker; Figure 3B) and quantified Esrra pixel density in these populations using ImageJ analysis (23,41). Within each experiment, Esrra pixel density in a given cell was normalized to that in Pvalb+ cells and then compared across cell-types (n=3 sections/mouse, 2 images/section, n=4 mice; n=1147–2006 Slc17a7+ cells/mouse, n=42–130 Pvalb+ cells/mouse, n=60–149 Gja1+ cells/mouse). There were significant differences amongst the cell-types (F2,9=22.28, p=0.0003); Esrra transcript was significantly higher in Pvalb+ cells compared to Slc17a7+ (p=0.0021) and Gja1+ (p=0.0003) cells (Figure 3C), largely replicating what is shown by Dropviz.org. Descriptive statistics are as follows: Pvalb+ cells (1±0.13; Q1=0.74, Q2=1.00, Q3=1.26), Slc17a7+ cells (0.40±0.062; Q1=0.27, Q2=0.42, Q3=0.51), Gja1+ cells (0.23±0.024; Q1=0.18, Q2=0.23, Q3=0.28).
Figure 3. Enrichment of Esrra in parvalbumin-expressing neuronal populations.
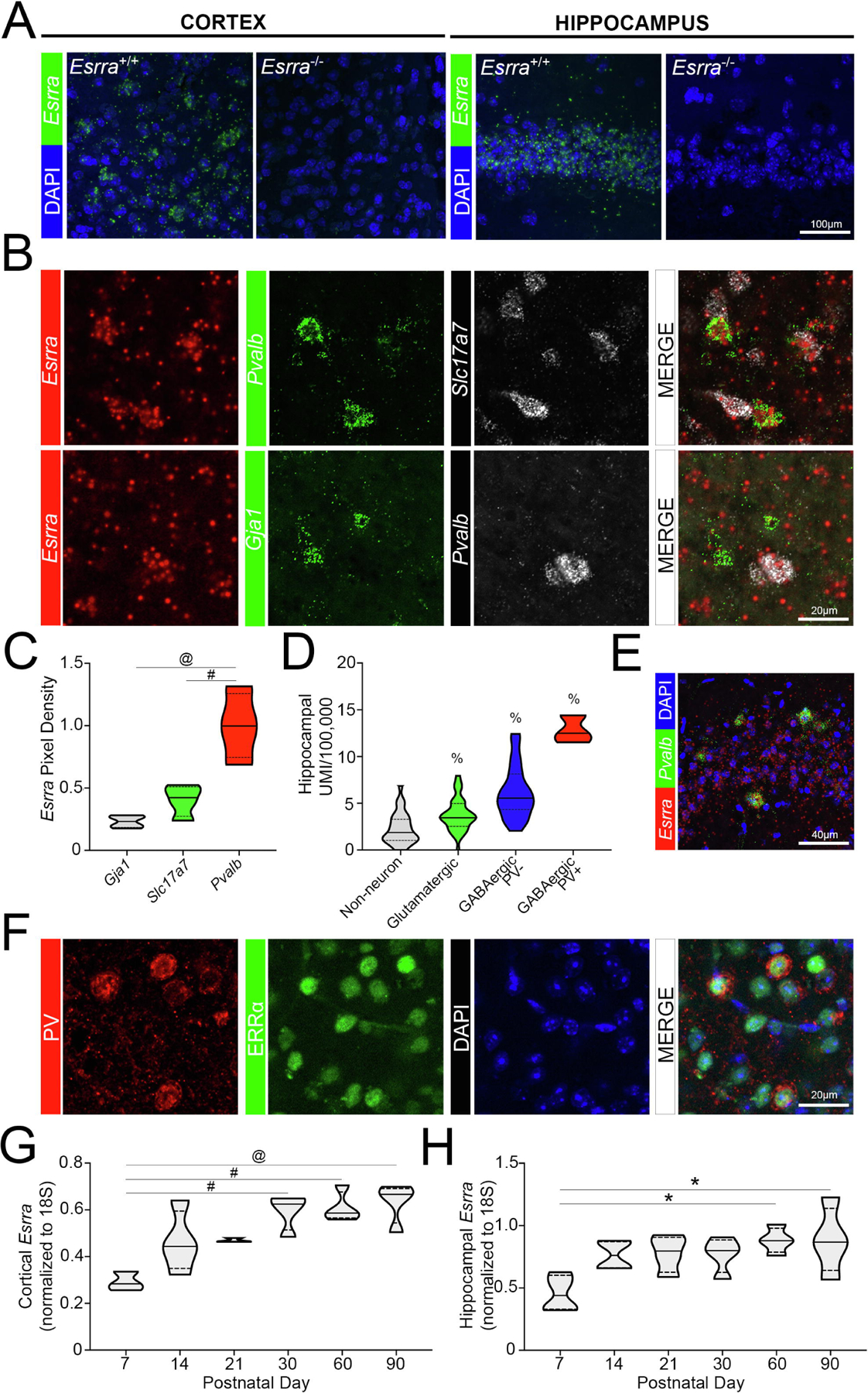
(A) Validation of SM-FISH probe in Esrra+/+ and Esrra−/− cortex and hippocampus (n=2 sections/mouse; 2 mice/genotype). (B) Distribution of Esrra transcript using SM-FISH in Pvalb+, Slc17a7+, and Gja1+ populations in the cortex of wildtype mice (n=3 sections/mouse, 2 images/section, n=4 mice; n=1147–2006 Slc17a7+ cells/mouse, n=42–130 Pvalb+ cells/mouse, n=60–149 Gja1+ cells/mouse). These data are quantified in (C) using ImageJ. (D) Localization data of Esrra obtained from Dropviz.org for hippocampus (n=3 PV+ GABAergic neurons, n=23 PV− GABAergic neurons, n=43 glutamatergic neurons, n=32 non-neuronal cells) and (E) represented by SM-FISH in wildtype (n=3 sections/mouse, 3 mice). (F) Confocal microscopy showing colocalization of ERRα protein and PV in murine cortex (n=3 mice). (G, H) Expression of Esrra in the wildtype cortex and hippocampus across age (n=3–4/group). *p≤0.05, #p≤0.01, @p≤0.001, $p≤0.0001, % indicates a significant difference from all other groups. Median is represented by the solid line, upper and lower quartiles are represented by dotted lines.
We used data obtained from the Dropviz.org database to determine if the enrichment for Esrra in PV-INs (n=3) was also seen in the hippocampus when compared to PV− GABAergic neurons (n=23), glutamatergic neurons (n=43) and non-neuronal cells (n=32). Indeed, there were significant differences across these populations (F3,97=36.79, p<0.0001; Figure 3D). Post-hoc analysis showed significant enrichment of Esrra transcript in PV-INs compared to PV− GABAergic and glutamatergic neurons and non-neurons (p<0.0001). PV− GABAergic neurons were also more enriched in Esrra transcript compared to glutamatergic neurons and non-neuronal populations (p<0.0001). Glutamatergic neurons were significantly higher in gene expression of Esrra compared to non-neuronal cells (p=0.011). Descriptive statistics are as follows: GABAergic PV+ cells (12.81±0.85; Q1=11.53, Q2=12.50, Q3=14.42), GABAergic PV− cells (6.15±0.58; Q1=4.34, Q2=5.55, Q3=8.16), glutamatergic cells (3.77±0.26; Q1=2.54, Q2=3.45, Q3=4.97), non-neuronal cells (2.29±0.29; Q1=1.05, Q2=1.87, Q3=3.29). These data are represented by SM-FISH in CA3 of the hippocampus (n=3 sections/mouse, 3 mice; Figure 3E). To confirm that protein mirrored enrichment for transcript in PV-INs, we colocalized ERRα protein with that for PV in wildtype cortex (n=3 mice); a similar localization pattern was observed (Figure 3F).
We hypothesized that the developmental timecourse of Esrra expression would correspond to that of PGC-1α, which increases from birth to adult levels at postnatal day 14 in cortex (29). We measured Esrra in the developing cortex and hippocampus at postnatal day 7 (P7, n=3,4 mice), P14 (n=4), P21 (n=3,4), P30 (n=4), P60 (n=4), and P90 (n=4). There was a significant difference in Esrra transcript across the ages in cortex (F5,16=8.29, p=0.0005; Figure 3G) and hippocampus (F5,18=3.67, p=0.018; Figure 3H). Post-hoc analysis revealed a significant increase in the expression of Esrra at P30 (p=0.0022), P60 (p=0.0014), and P90 (p=0.0007) compared to P7 in the cortex. Post hoc analysis similarly showed a significant increase in Esrra transcript at P60 (p=0.018) and P90 (p=0.018) compared to P7 in hippocampus. Descriptive statistics for cortical expression at these ages are as follows: P7 (0.29±0.024; Q1=0.26, Q2=0.28, Q3=0.34), P14 (0.46±0.066; Q1=0.35, Q2=0.44, Q3=0.60), P21 (0.47±0.0053; Q1=0.46, Q2=0.46, Q3=0.48), P30 (0.60±0.039; Q1=0.51, Q2=0.62, Q3=0.65), P60 (0.61±0.033; Q1=0.57, Q2=0.59, Q3=0.68), P90 (0.63±0.044; Q1=0.54, Q2=0.67, Q3=0.69). These data for hippocampal expression are as follows: P7 (0.46±0.073; Q1=0.33, Q2=0.44, Q3=0.60), P14 (0.77±0.058; Q1=0.66, Q2=0.76, Q3=0.87), P21 (0.78±0.073; Q1=0.63, Q2=0.80, Q3=0.91), P30 (0.77±0.071; Q1=0.63, Q2=0.80, Q3=0.89), P60 (0.88±0.051; Q1=0.79, Q2=0.88, Q3=0.98), P90 (0.88±0.13; Q1=0.64, Q2=0.87, Q3=1.14). These data indicate that Esrra expression increases through developmental maturation from P7 to its highest expression in adulthood, where it is maintained. A comparable developmental timeline is seen for Ppargc1a/PGC-1α (29) and some of its previously identified downstream targets (7).
ERRα is necessary in the cortex and hippocampus for the expression of PGC-1α-responsive neuron-enriched genes
Based on cell culture data and overlapping patterns of temporal and regional localization, we expected that mice lacking ERRα would have reduced gene expression of PGC-1α-dependent transcripts. Using qPCR, we measured the transcripts tested in vitro (Figure 2) in the murine cortex of Esrra null line (n=11 Esrra+/+, 6–7 Esrra+/−, 6 Esrra−/−). First, we confirmed knockdown of ERRα in the cortex Esrra+/− and Esrra−/− mice compared to control (Figure 4A). There were significant differences in Esrra transcript across genotypes (H2=19.90, p<0.0001); post-hoc analysis revealed significant reductions in transcript in Esrra+/− (p=0.025) and Esrra−/− (p=0.0001) compared to Esrra+/+. We also confirmed this loss of transcript in the hippocampus (n=10–11 Esrra+/+, 7 Esrra+/−, 5–6 Esrra−/−). There were significant differences in Esrra transcript across genotypes (H2=18.88, p<0.0001); post-hoc analysis showed a significant reduction in gene expression for Esrra in the knockout hippocampus compared to the Esrra+/+ (p=0.0001) and Esrra+/− (p=0.018) hippocampus. These transcript data corresponded to a loss of ERRα protein by western blot in the cortex and hippocampus (n=3/group; Figure 4B). There was a significant difference across genotypes in cortex (F2,6=185.4, p<0.0001) and hippocampus (F2,6=213.2, p<0.0001) for ERRα protein. There was a significant reduction in Esrra+/+ cortical protein compared to Esrra+/+ (p=0.0004) and a significant reduction in content in the Esrra−/− cortex compared to that of Esrra+/+ and Esrra+/− (p<0.0001). In the hippocampus, ERRα protein content was significantly reduced in both Esrra+/− and Esrra−/− compared to control (p<0.0001). ERRα protein was also significantly reduced in the Esrra−/− hippocampus compared to that of Esrra+/− (p<0.0002). Descriptive statistics for cortex as follows: Esrra+/+ (1.00±0.052; Q1=0.93, Q2=0.96, Q3=0.1.10), Esrra+/− (0.57±0.036; Q1=0.50, Q2=0.59, Q3=0.61), Esrra−/− (0.0046±0.0046; Q1=0, Q2=0, Q3=0.014). Descriptive statistics for hippocampus as follows: Esrra+/+ (1.00±0.042; Q1=0.92, Q2=1.03, Q3=1.05), Esrra+/− (0.47±0.042; Q1=0.39, Q2=0.51, Q3=0.52), Esrra−/− (0±0 6; Q1=0, Q2=0, Q3=0).
Figure 4. Mice lacking ERRα show a reduction in expression of PGC-1α-responsive transcripts in neocortex and hippocampus.
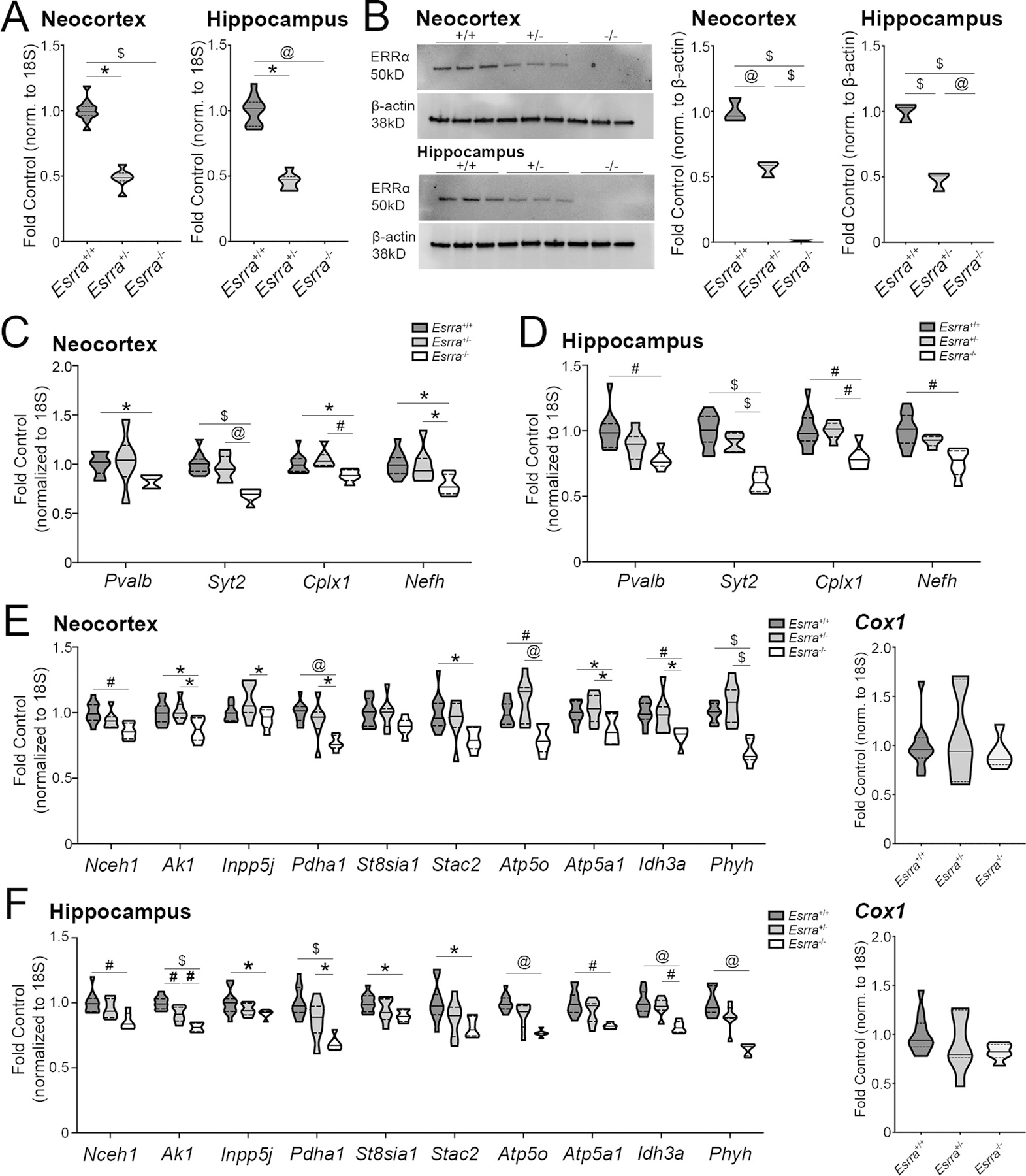
(A) Esrra transcript in Esrra+/+, Esrra+/−, and Esrra−/− cortex (n=11 Esrra+/+, 6–7 Esrra+/−, 6 Esrra−/−) and hippocampus (n=10–11 Esrra+/+, 7 Esrra+/−, 5–6 Esrra−/−). (B) Western blot and quantification of ERRα protein in cortex and hippocampus (n=3/genotype/region), with normalization to actin protein on the same membrane. (C, D) Expression of PGC-1α-responsive neuronal transcripts in the Esrra+/+, Esrra+/−, and Esrra−/− cortex and hippocampus. (E, F) Expression of remaining PGC-1α-responsive transcripts in cortex and hippocampus, including Cox1. *p≤0.05, #p≤0.01, @p≤0.001, $p≤0.0001. Median is represented by the solid line, upper and lower quartiles are represented by dotted lines.
We next measured transcripts that we have found to be responsive to PGC-1α and that are that are developmentally regulated and localized to PV-INs (7). In the cortex, there was a significant reduction in Pvalb transcript (H=7.84, p=0.02) in the Esrra−/− cortex compared to control (p=0.03). There were also significant differences amongst genotypes for the expression of Syt2 (F2,21=19.55, p<0.0001), Cplx1 (F2,21=6.24, p=0.0075), and Nefh (F2,21=5.26, p=0.014). Post hoc analysis revealed significant downregulation in Syt2, Cplx1, and Nefh transcripts in the Esrra−/− cortex compared to Esrra+/− (p=0.0003, p=0.0065, and p=0.036, respectively) and Esrra+/+ (p<0.0001, p=0.038, and p=0.016, respectively; Figure 4C). As seen in the cortex, neuronal transcripts were reduced in the hippocampus in the absence of ERRα (Figure 4D). As expected, there was a significant reduction in Pvalb transcript by across genotypes (F2,21=6.96; p=0.0048); post-hoc analysis showed a significant reduction in Pvalb expression in the Esrra−/− hippocampus compared to the Esrra+/+ hippocampus (p=0.0041). There were also significant differences amongst genotypes for Syt2 (F2,21=30.75; p<0.0001), Cplx1 (F2,21=7.50; p=0.0035) and Nefh (H2= 11.8, p=0.0027) transcript. Post-hoc analyses revealed reductions in Syt2 transcript in the Esrra−/− hippocampus compared to Esrra+/+ (p<0.0001) and Esrra+/− (p<0.0001) hippocampi. Likewise, Cplx1 transcript in the Esrra−/− hippocampus was reduced compared to Esrra+/+ (p=0.0045) and Esrra+/− (p=0.010) hippocampi. Nefh expression was reduced in the hippocampus of Esrra−/− mice compared to Esrra+/+ mice (p=0.0018).
We measured an additional set of genes in the cortex not restricted to neurons that are largely metabolic in function (Figure 4E). Post-hoc analyses found the following transcripts to be significantly reduced in the Esrra−/− cortex compared to the Esrra+/+ cortex: Nceh1 (p=0.002), Ak1 (p=0.02), Pdha1 (p=0.0005), Stac2 (p=0.02), Atp5o (p=0.008), Atp5a1 (p=0.03), Idh3a (p=0.008), Phyh (p<0.0001). Compared to the Esrra+/− cortex, the following transcripts were significantly reduced in the Esrra−/− cortex: Ak1 (p=0.02), Pdha1 (p=0.02), Atp5o (p=0.0006), Atp5a1 (p=0.01), Idh3a (p=0.03), Phyh (p<0.0001). St8sia1 transcript was unchanged between genotypes (F2,21=2.46, p=0.11).
As seen in cortex, the remaining transcripts responsive to PGC-1α were also reduced in Esrra−/− hippocampus (Figure 4F). Significant differences across genotypes were found for Nceh1 (F2,20=6.79, p=0.0056), Ak1 (F2,21=29.21, p=0.0001), Inpp5j (H2=6.087, p=0.048), Pdha1 (F2,21=14.18, p=0.0001), St8sia1 (F2,21=4.15, p=0.030), Stac2 (F2,21=4.41, p=0.025), Atp5o (H2=13.20, p=0.0014), Atp5a1 (H2=11.46, p=0.0032), Idh3a (F2,21=13.03, p=0.0002), Phyh (H2=14.92, p=0.0006). Those transcripts found by post-hoc analyses to be significantly downregulated in the Esrra−/− hippocampus compared to the Esrra+/+ hippocampus are as follows: Nceh1 (p=0.0041), Ak1 (p<0.0001), Inpp5j (p=0.042), Pdha1 (p<0.0001), Stac2 (p=0.025), Atp5o (p=0.0009), Atp5a1 (p=0.0022), Idh3a (p=0.0002), Phyh (p=0.0003). The following genes were also reduced in the Esrra−/− hippocampus compared to the Esrra+/− hippocampus: Ak1 (p=0.0034), Pdha1 (p=0.025), and Idh3a (p=0.0027). Ak1 expression was also reduced in the Esrra+/− hippocampus compared to wildtype (p=0.0039).
As a proxy for mitochondrial number, we assessed cytochrome c (Cox1) transcript in the cortex (Figure 4E) and hippocampus (Figure 4F). There was no significant difference across the genotypes for cortical Cox1 transcript (F2,19=0.33, p=0.72). Descriptive statistics are as follows: Esrra+/+ (1.00±0.082; Q1=0.87, Q2=0.96, Q3=1.08), Esrra+/− (1.08±0.17; Q1=0.63, Q2=0.94, Q3=1.68), Esrra−/− (0.93±0.078; Q1=0.81, Q2=0.86, Q3=1.08). Despite the reduction in Cox1 expression in the Esrra−/− hippocampus, there was no significant difference across genotypes (F2,21=1.48, p=0.25). Descriptive statistics are as follows: Esrra+/+ (1.00±0.060; Q1=0.87, Q2=0.94, Q3=1.11), Esrra+/− (0.91±0.11; Q1=0.76, Q2=0.79, Q3=1.25), Esrra−/− (0.82±0.035; Q1=0.76, Q2=0.82, Q3=0.90).
Considering the reduction in Pvalb gene expression in both the cortex and hippocampus, we measured glutamic acid decarboxylase 67 (Gad67/Gad1) and 65 (Gad65/Gad2) transcript as an indicator of potential neuron loss. In the cortical homogenate, Gad1 (F2,21=0.49, p=0.62) and Gad2 (H2=2.89, p=0.25) transcript was unchanged across genotypes. However, in hippocampus there was a significant difference across genotypes for Gad1 transcript (F2,19=3.67, p=0.045); post-hoc analyses revealed a significant reduction in the Esrra−/− hippocampus compared to control (p=0.036). In contrast, there was no difference detected across genotypes for Gad2 transcript (F2,18=1.28, p=0.30), indicating that a reduction in Gad1 may not necessarily represent a reduction in cell number but the expression of the transcript on a cell-by-cell basis. See Supplemental Table 4 for additional statistics.
Impact of ERRα knockout on PGC-1α-responsive transcript expression in the striatum and cerebellum
In addition to cortex and hippocampus, we assessed PGC-1α-dependent transcripts in striatal (n=9–12 Esrra+/+, 6–7 Esrra+/−, 3–6 Esrra−/−) and cerebellar (n=12 Esrra+/+, 7 Esrra+/−, 7–8 Esrra−/−) homogenates. First we assessed localization of Esrra using SM-FISH in the striatum, cerebellum and deep cerebellar nuclei (Figure 5A) and confirmed that Esrra is expressed in PV+ populations in these regions.
Figure 5. ERRα is required for the expression of a subset of PGC-1α-responsive transcripts in the striatum and cerebellum.
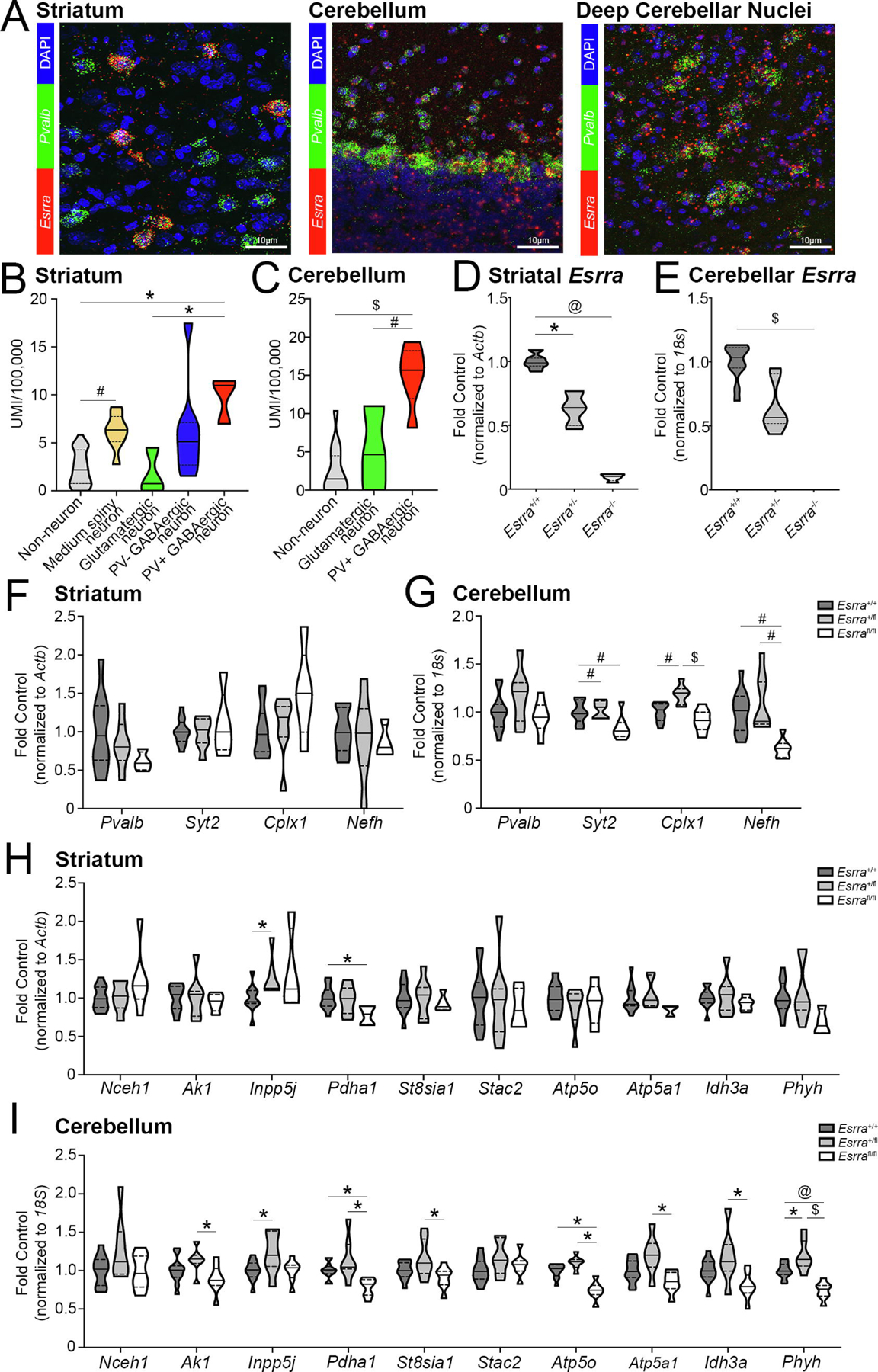
(A) Localization of Esrra by SM-FISH in the striatum, cerebellum and deep cerebellar nuclei. N=3 sections/mouse, 3 mice total. UMI enrichment for Esrra in the (B) striatum (n=3 PV+ GABAergic neurons, n=8 PV− GABAergic neurons, n=3 glutamatergic neurons, n=8 medium spiny neurons, n=27 non-neuronal populations) and (C) cerebellum (n=6 PV+, n=3 glutamatergic, and n=15 non-neuronal populations) obtained from dropviz.org. (D, E) Esrra transcript in Esrra+/+, Esrra+/−, and Esrra−/− striatum and cerebellum. Expression of PGC-1α-responsive transcripts in the Esrra+/+, Esrra+/−, and Esrra−/− (F, H) striatum and (G, I) cerebellum. Striatum: n=9–12 Esrra+/+, 6–7 Esrra+/−, 3–6 Esrra−/−. Cerebellum: n=12 Esrra+/+, 7 Esrra+/−, 7–8 Esrra−/−. *p≤0.05, #p≤0.01, @p≤0.001, $p≤0.0001. Median is represented by the solid line, upper and lower quartiles are represented by dotted lines.
We also explored the Dropviz.org localization data for Esrra in these regions. In the striatum (Figure 5B), we compared PV+ GABAergic neurons (n=3) to PV− GABAergic neurons (n=8), glutamatergic neurons (n=3), medium spiny neurons (n=8) and non-neuronal populations (n=27). There were significant differences across striatal cell type for Esrra abundance (H4=23.93, p<0.0001); post-hoc analysis revealed a significantly increased enrichment for Esrra expression in PV+ neurons compared to glutamatergic (p=0.045) and non-neuronal (p=0.013) cells. Esrra expression was also enriched in medium spiny neurons compared to non-neuronal cells (p=0.005). Descriptive statistics for striatal populations are as follows: GABAergic PV+ cells (9.81±1.41; Q1=7.00, Q2=10.98, Q3=11.45), GABAergic PV− cells (6.15±1.76; Q1=2.69, Q2=5.10, Q3=7.09), glutamatergic cells (1.74±1.39; Q1=0, Q2=0.74, Q3=4.48), MSNs (6.20±0.65; Q1=5.12, Q2=6.34, Q3=7.74), non-neuronal cells (2.39±0.35; Q1=0.75, Q2=2.18, Q3=4.26). We compared Esrra enrichment in PV+ populations (n=6) in the cerebellum to glutamatergic neurons (n=3) and non-neuronal cells (n=15; Figure 5C). There was a significant difference in Esrra abundance across populations (F2,21=25.50, p<0.0001); post-hoc analysis showed a significant enrichment of transcript in PV+ cells (14.99±1.62; Q1=11.95, Q2=15.67, Q3=18.22) compared to glutamatergic (5.21±3.19; Q1=0, Q2=4.64, Q3=10.99; p=0.0025) and non-neuronal (2.69±0.78; Q1=0, Q2=1.47, Q3=4.51; p<0.0001) cells.
We next confirmed knockdown of Esrra in these regions (Figure 5D, E). There was a significant difference in Esrra transcript in the striatum (H2=18.78, p<0.0001) and a significant reduction in transcript in the Esrra−/− (p=0.0001) and Esrra+/− (p=0.018) striata compared the Esrra+/+ striatum by post-hoc analysis. There was also a significant different between genotypes for Esrra transcript in the cerebellum (H2=20.76, p<0.0001); post hoc analysis revealed a significant reduction in the Esrra−/− cerebellum compared to wildtype (p<0.0001).
We then measured the neuronal transcripts in the striatum with the expectation that similar trends in these transcripts would be seen as in the cortex and hippocampus. There was no significant difference in Pvalb (F2,18=1.43, p=0.27), Syt2 (F2, 21=0.39, p=0.68), Cplx1 (F2, 20=2.56, p=0.10), or Nefh (F2,20=0.25, p=0.78) gene expression in the striatum (Figure 5F), contrary to what was observed in PGC-1α null striatum (22). In the cerebellum (Figure 5G), there was no difference in Pvalb transcript (F2,24=2.37, p=0.12), yet there was a difference in that of Syt2 (F2,24=7.06, p=0.0039), Cplx1 (F2,24=14.10, p<0.0001), and Nefh (H2=14.94, p=0.0006). Post-hoc analysis revealed a reduction in Syt2 expression in the Esrra−/− to Esrra+/− (p=0.0078) and Esrra+/+ (p=0.0092) cerebellum. For Cplx1 gene expression, there was a significant increase in the Esrra+/− cerebellum compared to Esrra−/− (p<0.0001) and Esrra+/+ (p=0.0021) cerebella. Finally, Nefh transcript was significantly reduced in the Esrra−/− cerebellum compared to Esrra+/− (p=0.0017) and Esrra+/+ (p=0.0028) cerebella.
We next measured the panel of PGC-1α-responsive metabolic transcripts in the striatum and cerebellum. In the striatum (Figure 5H), transcript for the following genes were unchanged: Nceh1 (H2=2.19, p=0.33), Ak1 (F2,21=0.22, p=0.80), St8sia1 (F2,21=0.25, p=0.78), Stac2 (F2,21=0.18, p=0.84), Atp5o (F2,18=0.60, p=0.56), Atp5a1 (H2=4.78, p=0.086), Idh3a (F2,21=0.60, p=0.56), and Phyh (F2,20=2.87, p=0.081). Significant differences were detected across genotypes for Inpp5j transcript (H2=7.02, p=0.022); post-hoc analysis revealed a significant increase in the Esrra+/− striatum compared to the Esrra+/+ striatum (p=0.030). There were also significant differences in Pdha1 transcript (F2,21=3.90, p=0.036); post-hoc analysis showed a significant reduction in the Esrra−/− striatum compared to wildtype (p=0.033). In the cerebellum (Figure 5I), there were no differences detected in transcript for Nceh1 (F2,24=2.73, p=0.086) or Stac2 (F2,24=2.04, p=0.15). There were significant differences detected amongst genotypes for Ak1 (F2,24=3.81, p=0.037); post-hoc analysis showed a significant reduction in the Esrra−/− cerebellum compared to Esrra+/− cerebellum (p=0.028). The same trend was seen for St8sia1 (F2,24=4.15, p=0.028), Atp5a1 (F2,24=7.30, p=0.0033), and Idh3a (F2,24=5.56, p=0.010); post-hoc analyses found reductions in the null cerebellum compared to Esrra+/− cerebellum for St8sia1 (p=0.023), Atp5a1 (p=0.0023) and Idh3a (p=0.0077). Significant differences in Inpp5j gene expression were detected amongst genotypes (F2,24=3.92, p=0.034); post-hoc analysis revealed a significant reduction in transcript in the Esrra+/+ cerebellum to Esrra+/− cerebellum (p=0.042). Differences in gene expression were also found across genotypes for Pdha1 (H2=12.90, p=0.0016), Atp5o (F2,24=29.32, p<0.0001), and Phyh (F2,24=21.31, p<0.0001). Pdha1 (p=0.021), Atp5o (p<0.0001), and Phyh (p=0.0009) transcripts were all reduced in the null cerebellum compared to wildtype by post-hoc analyses. Transcripts for Pdha1 (p=0.0017), Atp5o (p<0.0001), and Phyh (p<0.0001) transcripts were also reduced in the null cerebellum compared to Esrra+/− cerebellum. Phyh transcript was increased in the Esrra+/− cerebellum compared to wildtype cerebellum (p=0.017). Descriptive statistics for both striatum and cerebellum are include in Supplemental Table 5.
Similarly, we measured these transcripts in the midbrain. Differences in Esrra expression across genotypes were confirmed (H2=16.60, p=0.0002); by post-hoc analysis there was a significant reduction in the Esrra−/− midbrain compared to Esrra+/+ (p=0.0002). In homogenate, there was no difference between genotypes for Pvalb (F2,22=0.61, p=0.55), Syt2 (F2,22=1.13, p=0.34), Cplx1 (F2,22=0.30, p=0.74), or Nefh (F2,22=1.13, p=0.34). There were also no differences in the remaining measured PGC-1α-responsive transcripts: Nceh1 (H2=0.40, p=0.82), Ak1 (H2=1.34, p=0.51), Inpp5j (F2,22=0.78, p=0.47), Pdha1 (H2=2.18, p=0.34), St8sia1 (F2,22=0.66, p=0.53), Stac2 (F2,22=1.77, p=0.19), Atp5o (F2,22=0.78, p=0.47), Atp5a1 (H2=1.70, p=0.43), Idh3a (H2=2.94, p=0.23), Phyh (H2=3.69, p=0.16). Descriptive statistics are listed in Supplemental Table 6.
ERRα null mice display hyperactivity in response to amphetamine and impairment in sensorimotor gating.
To determine if motor behavior was influenced by a lack of ERRα, we measured ambulation in open field (n=17 Esrra+/+, n=14 Esrra+/−, n=12 Esrra−/−; Figure 6A). Though a one-way ANOVA indicated significant differences amongst the genotypes in overall activity (F2,40=4.73, p=0.014), post-hoc analysis revealed a significant reduction in just ERRα−/− mice compared to Esrra+/− mice (p=0.011). Descriptive statistics for ambulation are as follows: Esrra+/+ (9012±498; Q1=7222, Q2=9859, Q3=10522), Esrra+/− (10155±412.9; Q1=8870, Q2=10195, Q3=11305), Esrra−/− (7845±601.3; Q1=6084, Q2=7832, Q3=9351).
Figure 6. ERRα null mice show hyperactivity in response to amphetamine and deficits in sensory motor gating.
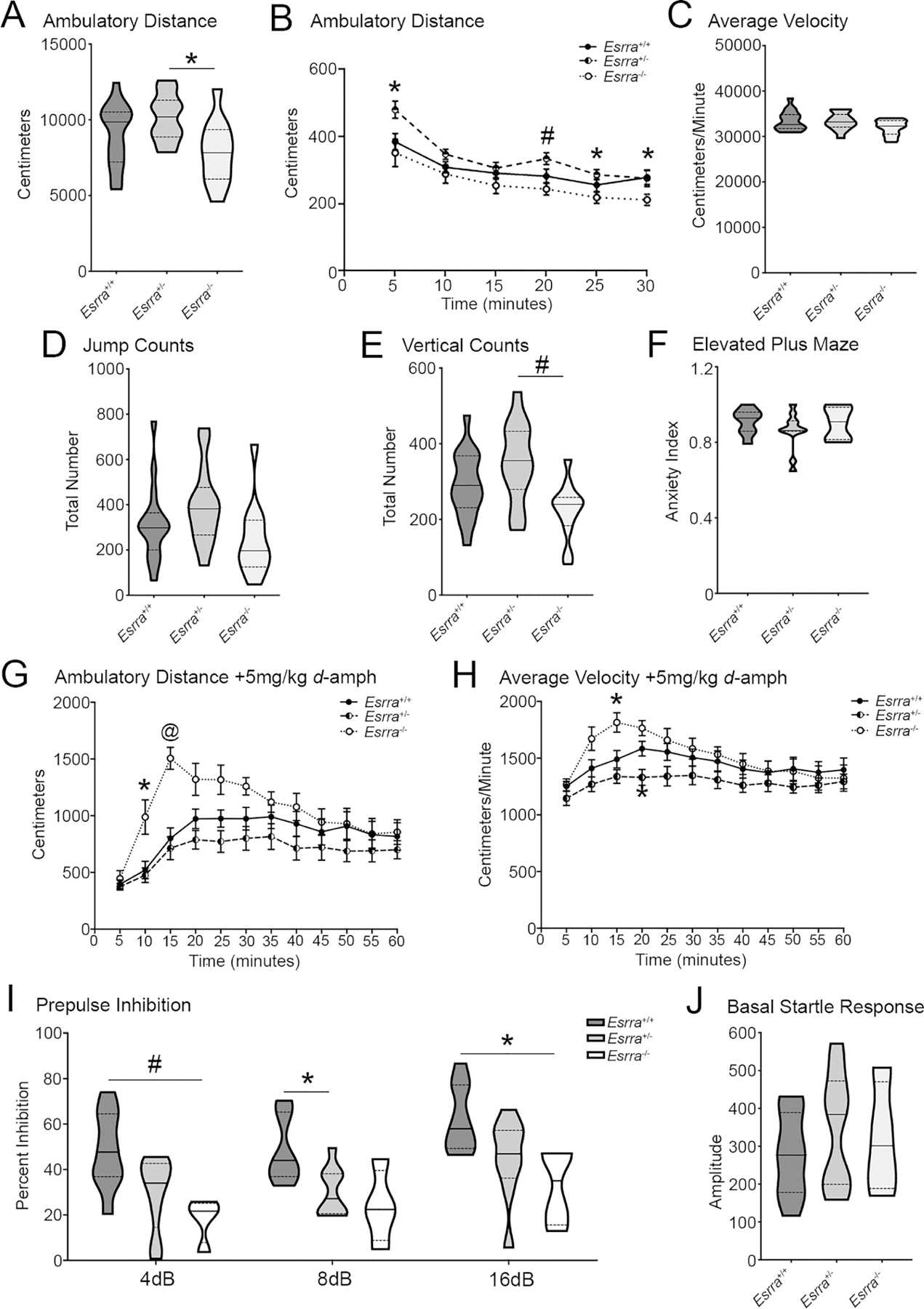
(A) Overall ambulatory distance and (B) across time, (C) average velocity, (D) jump counts, (E) and vertical counts in open field for Esrra+/+, Esrra+/−, and Esrra−/− mice. (n=17 Esrra+/+, n=14 Esrra+/−, n=12 Esrra−/−). (F) Anxiety index in elevated plus maze. (n=8 Esrra+/+, n=11 Esrra+/−, n=5 Esrra−/−). (G) Ambulatory distance and (H) average velocity in open field following intraperitoneal injection of 5mg/kg d-amphetamine. (n=10 Esrra+/+, n=9 Esrra+/−, n=7 Esrra−/−). (I) Prepulse inhibition (N=4–9/genotype) and (J) baseline startle response (n=9 Esrra+/+, n=9 Esrra+/−, n=4 Esrra−/−). (K, L) Maoa and Maob transcript in cortex, hippocampus, striatum, midbrain and cerebellum. (see previous regional data for samples sizes). *p≤0.05, #p≤0.01, @p≤0.001. Violin plots show median represented by the solid line, upper and lower quartiles are represented by dotted lines. Line graphs show mean ± SEM.
Since ambulatory distance was reduced in the Esrra−/− mice relative to control, we analyzed ambulation over time. A two-way repeated measures analysis revealed a significant effect of genotype on ambulatory distance (F2,40=4.73, p=0.014) with no interaction of genotype and time (F10,200=1.59, p=0.11). Post hoc analyses revealed significantly increased ambulation for Esrra+/− mice during the first five minutes compared to Esrra+/+ and Esrra−/− mice (p=0.03 and 0.041, respectively). Esrra+/− mice exhibited hyperactivity compared to Esrra−/− mice at 20 (p=0.0042) and 25 minutes (p=0.02) of the test session. By the end of the test session, Esrra+/+ mice were significantly more active than Esrra−/− mice (p=0.049; Figure 6B). There was no significant difference across genotypes in average velocity (F2,40=2.42, p=0.10; Figure 6C) and, though trending, there was no significant difference between genotypes for jump count (F2, 40=2.65, p=0.083; Figure 6D). Despite a significant difference amongst genotypes for vertical counts (F2,40=6.40, p=0.0039), post-hoc analyses only found a reduction in Esrra−/− mice compared to Esrra+/− mice (p=0.0026; Figure 6E). Together, these data show that a lack of ERRα does not lead to overt motor dysfunction in open field. Descriptive statistics for average velocity are as follows: Esrra+/+ (33425±514.1; Q1=31776, Q2=32633, Q3=34810), Esrra+/− (33313±502.8; Q1=32064, Q2=33217, Q3=34881), Esrra−/− (31929±493.2; Q1=30502, Q2=32298, Q3=33518). Descriptive statistics for jump count are as follows: Esrra+/+ (315.1±40.25; Q1=200, Q2=298, Q3=364), Esrra+/− (395±46.03; Q1=266.5, Q2=382, Q3=476.5), Esrra−/− (243.7±47.46; Q1=125.5, Q2=197, Q3=332). Descriptive statistics for vertical count are as follows: Esrra+/+ (291.5±20.91; Q1=231, Q2=290, Q3=368), Esrra+/− (350±28.15; Q1=279.5, Q2=355.5, Q3=432.8), Esrra−/− (223.2±21.64; Q1=183.5, Q2=240, Q3=258.3). To determine if a loss of ERRα confers an anxiety-like phenotype, we tested these mice in elevated plus maze (n=8 Esrra+/+, n=11 Esrra+/−, n=5 Esrra−/−). An anxiety index was calculated as previously described (26); no differences were identified across genotypes (H2=0.94, p=0.63; Figure 6E). Descriptive statistics for anxiety index are as follows: Esrra+/+ (0.91±0.024; Q1=0.86, Q2=0.93, Q3=0.96), Esrra+/− (0.86±0.031; Q1=0.86, Q2=0.86, Q3=0.91), Esrra−/− (0.90±0.039; Q1=0.82, Q2=0.91, Q3=0.99).
ERRα has been shown to regulate Maoa and Maob transcription (32,40). As such, a loss of ERRα would be expected to sensitize mice to dopamine. To test this, we enhanced dopaminergic signaling by intraperitoneally administering 5mg/kg d-amphetamine and assessing ambulatory activity in open field (n=10 Esrra+/+, n=9 Esrra+/−, n=7 Esrra−/−). There was a significant main effect of time (F2.05, 47.15=22.80, p<0.0001), genotype (F2,23=4.56, p=0.023), and interaction between the two (F22,253=3.29, p<0.0001). When compared to Esrra+/+ mice, post hoc analysis revealed a significant increase in ambulatory activity in the Esrra−/− mice at 10 minutes (p=0.041) and 15 minutes (p=0.0002) post injection (Figure 6G).
For average velocity, there was a significant main effect of time (F2.29, 52.68=11.74, p<0.0001) and a significant interaction between time and genotype (F22, 253=2.17, p=0.0023). There was no main genotypic effect (F2,23=3.03, p=0.068). When compared to Esrra+/+ mice, post hoc analysis revealed a significant increase in average velocity for the Esrra−/− mice at 15 minutes post injection (p=0.026) and in the Esrra+/− mice at 20 minutes post injection (p=0.033; Figure 6H). These data indicate that Esrra−/− mice are hypersensitive to dopamine release compared to controls, suggesting that a lack of ERRα may potentially be impacting transcripts involved in dopamine release, metabolism, or receptor responsivity and/or expression.
To evaluate if a loss of ERRα elicited deficits in sensorimotor gating, a phenotype observed in animal models (43–45) and patients with schizophrenia (46), we measured prepulse inhibition (PPI; n=9 Esrra+/+, n=9 Esrra+/−, n=4 Esrra−/−). A two-way ANOVA revealed a significant main effect of prepulse (F1.73, 32.87=20.56, p<0.0001) and a significant main effect of genotype (F2,19=6.90, p=0.0056) with no interaction between the two (F4,38=0.27, p=0.90). In comparison to control, there was a significant reduction in startle response for the Esrra−/− mice and a trending reduction for Esrra+/− mice at 4dB (p=0.0052 and p=0.057, respectively) and 16dB (p=0.049 and p=0.070, respectively). Inversely, there was a trending reduction in the Esrra−/− mice and significant reduction in the Esrra+/− mice at 8dB (p=0.078 and p=0.016, respectively; Figure 6I). There were no differences in basal startle responses (F2,19=0.55, p=0.59; Figure 6J). Considering the possibility that ERRα knockdown could be influencing dopamine metabolism by causing a deficiency in MAOB (suggested by SH-SY5Y data in Figure 2O) or MAOA (32,40), we measured Maoa and Maob transcript abundance in all assayed brain regions (cortex, n=6–11/genotype; hippocampus, n=5–11/genotype; striatum, n=5–11/genotype; cerebellum, n=7–12/genotype; and midbrain, n=5–11/genotype); we did not detect differences amongst any of the groups (Supplemental Figure 3; descriptive statistics are listed in Supplemental Table 7.
DISCUSSION
Studies have identified transcription factors which interact directly with PGC-1α to regulate metabolic programs in peripheral tissues. Similar efforts in the brain have been limited for numerous reasons, including the relatively restricted expression pattern of PGC-1α in the mouse brain and the cell and region-specific differences in PGC-1α-driven gene programs (4,6,7,12,21,22,24,42,47). The heterogeneity among neuronal populations within and across regions has only recently gained appreciation with help from cell-type-specific transcriptional and epigenomic profiling. This heterogeneity, along with the inability to isolate PGC-1α and its associated complexes via immunoprecipitation, has drastically impeded the ability to directly and accurately identify its binding partners in discrete neuronal populations.
We found an enrichment in consensus binding sites for ERRα in the promoters of PGC-1α-responsive genes in vitro. In peripheral tissues, ERRα is a key effector in the regulation of PGC-1α-dependent transcripts involved in mitochondrial biogenesis and function (13,14). Further, PGC-1α shows selective interactions with ERRα (13,15,16), upregulating and maintaining the expression of ERRα in a feed-forward manner (13,17) and potentiating its transcriptional activity (16). We found that ERRα transcript (Esrra) is enriched in PV+ populations in the cortex similar to Ppargc1a (PGC-1α), and ERRα is necessary for the expression of Syt2, Cplx1, Nefh and metabolic transcripts in the presence of PGC-1α both in vitro and in vivo. Furthermore, we identified behavioral abnormalities in the ERRα null mouse including hypersensitivity to changes in dopaminergic tone and deficits in prepulse inhibition. Together, our data suggest ERRα as an important driver of PGC-1α-dependent transcription in the CNS, particularly of transcripts enriched in PV-INs, and suggest that ERRα serves as a putative factor involved in PV-IN maturation, function, and metabolism.
Previously published data using ATAC-sequencing showed enrichment for ERRα in PV-INs relative to excitatory neurons and vasoactive intestinal peptide (VIP)-expressing interneurons in the cortex (27). As shown in Figure 3G–H, the temporal expression pattern for ERRa is similar to what has been shown for PGC-1a and PV. We and others (49) hypothesize that ERRα may be playing a large role particularly in this cell type. With an expansive post-synaptic network, PV-INs are largely responsible for oscillatory network activity in the gamma frequency (8,9,50,51) and the energy demand in maintaining the pacemaking activity characteristic of PV-INs is substantial (52). ERRα is largely known as a regulator of transcripts for the generation of cellular energy and responses to stress in peripheral tissues with high metabolic demand (13,14,48). While the brain has comparable energy demand (53) to peripheral tissues, genomic profiling of ERRα-dependent metabolic genes in this tissue are lacking.
Here we show that knockdown of ERRα prevents PGC-1α overexpression-mediated upregulation of downstream metabolic transcripts, including known ERRα targets Atp5o and Idh3a (48), in vitro and that ERRα null mice show deficiencies in Atp5o and Idh3a in vivo. Previous studies show that, in addition to blocking the PGC-1α-binding site on ERRα and causing ERRα degradation, XCT790 uncouples mitochondria and activates AMP-activated protein kinase (AMPK) in a dose-dependent manner below concentrations used to inhibit ERRα (54). In our study, transcriptional changes are normalized to overexpression of PGC-1α, a known mitochondrial regulator, and XCT790 at higher doses showed efficient blockade of known PGC-1α-responsive transcripts. As such, though it is possible that transcriptional changes may result from XCT790 independent of the effects of ERRα, it is likely in this case that the inability to upregulate transcripts by PGC-1α are due to effective blockade of ERRα, suggesting an important role of this transcription factor in the regulation of these transcripts. Additionally, the majority of tested transcripts are also reduced in ERRα null cortical and hippocampal homogenate. Notably, there were no significant differences in transcript for mitochondrial-encoded Cox1 in cortex and a trending decrease in hippocampus, suggesting that mitochondrial number is not reduced in these mice. It would be of interest to not only test cell-specific changes in transcript for Cox1 but to evaluate mitochondrial mass and structure in PV-INs via electron microscopy.
Generally, any metabolic compromise can lead to a multitude of negative consequences for PV-INs. For example, cytochcrome c oxidase ablation in PV-INs leads to deficits in circuit inhibition and impaired sensory gating and sociability (55), the latter two being documented in the ERRα null mouse. Further, PV-INs largely rely on PV to buffer calcium in order to maintain their pacemaking activity. A loss of PV itself significantly impacts their firing phenotype, shifting the excitatory/inhibitory balance towards overall circuit inhibition (56–59). Here, we show a significant decrease in Pvalb expression in the ERRα null cortex and hippocampus as well as decreased Syt2 and Cplx1 in these regions. Both Syt2 and Cplx1 are responsible for the synchronous release of neurotransmitter (60–62). A reduction in these and/or the above metabolic genes, many of which are reportedly enriched in PV-INs (63), would likely impact neuronal firing in the ERRα null brain. Notably, a loss of PGC-1α specifically in PV-INs leads to an overall reduction in synchronous release of neurotransmitter and an increase in the release of GABA (7), as expected. Previous studies indicate enhanced excitatory drive onto striatal medium spiny neurons in ERRα null mice (34). It is possible, in this model, that there is a shift in excitarory/inhibitory balance and that enhanced excitation in the corticostriatal circuit may be explained by reduced network inhibition by PV-INs in this region. As such, a reduction in PV would not solely account for such electrophysiological differences. ERRα expression is abundant in pyramidal neurons and this population would likely be affected by the absence of ERRα in the null mouse. Thus, it is equally plausible that a loss of ERRα in pyramidal neurons could in some way enhance their excitability and/or vesicular release. Further investigation into the cortical and hippocampal network activity in these mice and, more specifically, the effect of cell-specific deletion of ERRα in PV-INs versus pyramidal neurons would shed light on the downstream functional consequences of transcription deficiencies and their contributions to behavioral impairment. Considering the deficit in maturation related transcripts (i.e. Pvalb) and neurotransmission in PV-INs with a loss of PGC-1α, it is also of interest to evaluate the concurrent upregulation of Esrra expression in development. It is possible that PGC-1α and ERRα may work together to drive gene programs for PV-IN maturation and may be of importance for understanding PV-IN function in neurodevelopmental disorders in which PGC-1α dysregulation is implicated. Confirmation of their direct interaction in brain will require the development of selective antibodies for the immunoprecipitation of endogenous PGC-1α.
While we did not assess social behavior in this study, previous reports indicate that ERRα null mice exhibit sociability deficits (33). Notably, coordinated transcriptional control of metabolic and neuronal transcripts in response to social challenge is largely regulated by ERRα (64). Additionally, reduced transcriptional activity of ERRα is suggested to be a key contributor to deficits in motivation and reward-seeking behavior (33,34,65). In this same pathway, ERRα null mice exhibit behaviors reminiscent of psychiatric disorders including behavioral rigidity and increased compulsive tendencies (33). Here, we also report deficits in prepulse inhibition/sensory gating in these mice as well as hypersensitivity to amphetamine challenge. These cognitive and behavioral deficits are largely seen in mouse models of, and patients diagnosed with, neuropsychiatric disorders such as schizophrenia (SZ). Evidence implicates PV-INs at the junction between genetic and environmental stressors in cognitive dysfunction (66), SZ in particular. Insult to PV-INs in the form of oxidative stress, metabolic or mitochondrial deficit, or dysregulation of mitochondrial regulators, such as PGC-1α, is proposed to be a central effector in the onset of cognitive deficits in SZ (66).
As previously stated, PGC-1α is enriched in, and necessary for, normal PV-IN function. Notably, PGC-1α has been suggested as an etiological candidate in several psychopathologies including eating disorders (67), SZ, and bipolar disorder (68–72). Additionally, PGC-1α expression is reduced in mouse models that display SZ-like behaviors (66,73), and behavioral deficits in PGC-1α null mice are rescued by the antipsychotic haloperidol (74). In fact, the expression levels of Pvalb, Syt2, Cplx1, and Nefh, transcripts enriched in PV-INs (63) are significantly reduced in human postmortem cortex without a change in PGC-1α itself (47). These data suggest these transcriptional deficits are due to changes in posttranscriptional modifications, epigenetic modifications or alternations in the abundance or activation state of PGC-1α-interacting transcription factors. Reductions in Nrf1 in SZ cortical homogenate could contribute to the disassociation of PGC-1α from its targets, although transcriptional data from Dropviz.org (Figure 1) indicates that Nrf1 expression is low in neuronal populations. Others have shown that Esrra mRNA expression is reduced in human postmortem SZ, though this finding was not replicated (75). Further experiments are necessary to determine if reductions in ERRα expression occur specifically in PV-INs or the association between PGC-1α and ERRα is impaired.
We show that absence of ERRα enhances locomotor sensitivity to amphetamine. Previous studies identify ERRα as a regulator of Maoa and Maob transcription (32,40). Functionally, MAO-A and MAO-B are mitochondrially localized and deaminate biogenic amines, including dopamine. If Maoa and Maob were dependent on ERRα expression in vivo, it would be predicted that ERRα null mice would exhibit a hypersensitive response to dopaminergic transmission (76). Interestingly, MAO-A hypomorphic mice show perseverative behaviors and decreased sociability (77), behavior patterns others have revealed in the ERRα null mice. Hypersensitivity to amphetamine, along with several behavioral indicators of cognitive deficiencies, have been reported in mice lacking the dopamine receptor D2 (Drd2) specifically in PV-INs (78). Similar to what is seen in postmortem SZ, mice lacking Drd2 in PV-INs mice show reductions in both PV and Gad1 expression in the cortex and hippocampus without a loss in cell number (78). Considering the reduction in Pvalb and Gad1 gene expression in the ERRα null hippocampus as well as the increase dopaminergic sensitivity, it would be interesting to evaluate the effects on dopaminergic tone in the cortex and hippocampus following PV-IN specific ERRα deletion. Further, previous studies have demonstrated that the promoter region of Drd2 is rich in consensus binding sequences for ERRα and that overexpression as well as knockdown of ERRα in primary neuronal culture is able to modulate the expression of Drd2 expression (75). The cell-specific influence of ERRα modulation of Drd2 expression would be important to assess.
With regard to regulation of the MAOs by ERRα, our data suggests that there is not a dramatic change in expression of either MAO-A or MAO-B in homogenate of any regions tested. It would be important to evaluate these changes at a cell-specific level. However, it does raise the question of other mechanisms by which an increase in dopamine could elicit such hypersensitivity aside from receptor expression level including neurotransmitter release, packaging, and firing rate; the nigrostriatal circuit in these animals greatly warrants further investigation in the context of movement and motivation.
Of potential relevance for understanding alterations of dopamine responsivity in these mice, the E3-ubiquitin ligase and Parkinson Disease risk gene PARKIN associates directly with ERRα (40,79) and enhances both the transcription and turnover of ERRα (79). Consequently, the degradation of ERRα leads to a reduction in MAOs and associated oxidative stress (40). The Parkinson’s disease (PD)-linked mutation fails to adequately degrade ERRα, leading to an increase in MAOs and, consequently, increased oxidative stress and deamination of dopamine (40). Interestingly, nuclear translocation of PARKIN leads to an upregulation in PGC-1α (79). A relationship between PARKIN and PGC-1α in the midbrain and in the context of PD has been documented (80). Analysis of genes reduced in laser-captured nigral neurons of patients with symptomatic PD revealed that PGC-1α occupied the ERRE binding motif of several genes including Atp5a1 (81). In light of these data, the potential roles for ERRα in the maintenance of dopaminergic neuron function and viability should be determined.
Interestingly, analysis of the single-cell data (18) revealed an enrichment of estrogen-related receptor gamma (Esrrg/ERRγ) in PV-INs. ERRγ enrichment has been previously documented by others (27,63), though little is known about roles for this factor in PV-INs. Considering that the magnitude of reductions in PGC-1α-dependent gene expression in ERRα null mice did not reach the levels of genes in PGC-1α null mice (7,12), it is possible that ERRγ is also involved in gene regulation in PV-INs. ERRγ also binds the “AAGGTCA” consensus binding site in genes and can form heterodimers with ERRα (82). However, ERRγ is expressed earlier than ERRα in development (31), and ERRγ null mice show embryonic lethality (30), suggesting that ERRα and ERRγ have differential roles in the regulation of gene expression in neurons. In light of the developmental regulation of ERRγ, future experiments can explore the role for ERRγ in the induction of PGC-1a-dependent genes in development.
Supplementary Material
SIGNIFICANCE STATEMENT.
The transcription factors with which PGC-1α interacts determine specificity of the transcriptional program it drives across cell populations, but those mediating its functions in parvalbumin-expressing neurons are unknown. Relative to other PGC-1α-interacting transcription factors, ERRα is enriched in parvalbumin-expressing neurons and shows robust spatial and temporal correlation with PGC-1α expression throughout the brain. ERRα is also necessary for PGC-1α-dependent transcription both in vitro and in vivo for metabolic and neuronal transcripts. These data suggest that ERRα is an important player in cell-specific PGC-1α-dependent transcription in the CNS and may play a role in regulating parvalbumin-expressing neuron maturation and function.
HIGHLIGHTS:
ERR binding sites are enriched in the promoter region of PGC-1α-responsive genes.
ERRα antagonism impedes the ability of PGC-1α to upregulate genes in vitro.
ERRα and PGC-1α are colocalized in parvalbumin-expressing interneurons.
ERRα deletion leads to reductions in PGC-1α-dependent transcripts in vivo.
ERRα null mice exhibit schizophrenia-like behaviors.
Acknowledgements:
Evelyn F. McKnight
UAB Neurobiology Behavior Core
Funding Sources:
R01 NS101958
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: None
Bibliography
- 1.Zhang Y, Huypens P, Adamson AW, Chang JS, Henagan TM, Boudreau A, et al. Alternative mRNA splicing produces a novel biologically active short isoform of PGC-1alpha. J Biol Chem 2009. Nov 20;284(47):32813–32826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knutti D, Kralli A. PGC-1, a versatile coactivator. Trends Endocrinol Metab 2001. Oct;12(8):360–365. [DOI] [PubMed] [Google Scholar]
- 3.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest 2006. Mar;116(3):615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McMeekin LJ, Fox SN, Boas SM, Cowell RM. Dysregulation of PGC-1α-Dependent Transcriptional Programs in Neurological and Developmental Disorders: Therapeutic Challenges and Opportunities. Cells 2021. Feb 9;10(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dougherty SE, Bartley AF, Lucas EK, Hablitz JJ, Dobrunz LE, Cowell RM. Mice lacking the transcriptional coactivator PGC-1α exhibit alterations in inhibitory synaptic transmission in the motor cortex. Neuroscience 2014. Jun 20;271:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartley AF, Lucas EK, Brady LJ, Li Q, Hablitz JJ, Cowell RM, et al. Interneuron Transcriptional Dysregulation Causes Frequency-Dependent Alterations in the Balance of Inhibition and Excitation in Hippocampus. J Neurosci 2015. Nov 18;35(46):15276–15290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lucas EK, Dougherty SE, McMeekin LJ, Reid CS, Dobrunz LE, West AB, et al. PGC-1α provides a transcriptional framework for synchronous neurotransmitter release from parvalbumin-positive interneurons. J Neurosci 2014. Oct 22;34(43):14375–14387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 2009. Jun 4;459(7247):698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 2009. Jun 4;459(7247):663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu H, Gan J, Jonas P. Interneurons. Fast-spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function. Science 2014. Aug 1;345(6196):1255263. [DOI] [PubMed] [Google Scholar]
- 11.Gulyás AI, Buzsáki G, Freund TF, Hirase H. Populations of hippocampal inhibitory neurons express different levels of cytochrome c. Eur J Neurosci 2006. May;23(10):2581–2594. [DOI] [PubMed] [Google Scholar]
- 12.Lucas EK, Markwardt SJ, Gupta S, Meador-Woodruff JH, Lin JD, Overstreet-Wadiche L, et al. Parvalbumin deficiency and GABAergic dysfunction in mice lacking PGC-1alpha. J Neurosci 2010. May 26;30(21):7227–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villena JA, Kralli A. ERRalpha: a metabolic function for the oldest orphan. Trends Endocrinol Metab 2008. Oct;19(8):269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, et al. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci USA 2004. Apr 27;101(17):6472–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takacs M, Petoukhov MV, Atkinson RA, Roblin P, Ogi F-X, Demeler B, et al. The asymmetric binding of PGC-1α to the ERRα and ERRγ nuclear receptor homodimers involves a similar recognition mechanism. PLoS One 2013. Jul 9;8(7):e67810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schreiber SN, Knutti D, Brogli K, Uhlmann T, Kralli A. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha). J Biol Chem 2003. Mar 14;278(11):9013–9018. [DOI] [PubMed] [Google Scholar]
- 17.Laganière J, Tremblay GB, Dufour CR, Giroux S, Rousseau F, Giguère V. A polymorphic autoregulatory hormone response element in the human estrogen-related receptor alpha (ERRalpha) promoter dictates peroxisome proliferator-activated receptor gamma coactivator-1alpha control of ERRalpha expression. J Biol Chem 2004. Apr 30;279(18):18504–18510. [DOI] [PubMed] [Google Scholar]
- 18.Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018. Aug 9;174(4):1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang M, Zhao Y, Zhang B. Efficient Test and Visualization of Multi-Set Intersections. Sci Rep 2015. Nov 25;5:16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 2017. Jul 3;45(W1):W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McMeekin LJ, Bartley AF, Bohannon AS, Adlaf EW, van Groen T, Boas SM, et al. A Role for PGC-1α in Transcription and Excitability of Neocortical and Hippocampal Excitatory Neurons. Neuroscience 2020. May 21;435:73–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McMeekin LJ, Li Y, Fox SN, Rowe GC, Crossman DK, Day JJ, et al. Cell-Specific Deletion of PGC-1α from Medium Spiny Neurons Causes Transcriptional Alterations and Age-Related Motor Impairment. J Neurosci 2018. Mar 28;38(13):3273–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012. Jul;9(7):671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lucas EK, Dougherty SE, McMeekin LJ, Trinh AT, Reid CS, Cowell RM. Developmental alterations in motor coordination and medium spiny neuron markers in mice lacking pgc-1α. PLoS One 2012. Aug 14;7(8):e42878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kennedy AJ, Rahn EJ, Paulukaitis BS, Savell KE, Kordasiewicz HB, Wang J, et al. Tcf4 regulates synaptic plasticity, DNA methylation, and memory function. Cell Rep 2016. Sep 6;16(10):2666–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen H, Liu T, Kozlovsky N, Kaplan Z, Zohar J, Mathé AA. The neuropeptide Y (NPY)-ergic system is associated with behavioral resilience to stress exposure in an animal model of post-traumatic stress disorder. Neuropsychopharmacology 2012. Jan;37(2):350–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, et al. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 2015. Jun 17;86(6):1369–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods 2017. Oct;14(10):955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cowell RM, Blake KR, Russell JW. Localization of the transcriptional coactivator PGC-1alpha to GABAergic neurons during maturation of the rat brain. J Comp Neurol 2007. May 1;502(1):1–18. [DOI] [PubMed] [Google Scholar]
- 30.Alaynick WA, Kondo RP, Xie W, He W, Dufour CR, Downes M, et al. ERRgamma directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metab 2007. Jul;6(1):13–24. [DOI] [PubMed] [Google Scholar]
- 31.Pei L, Mu Y, Leblanc M, Alaynick W, Barish GD, Pankratz M, et al. Dependence of hippocampal function on ERRγ-regulated mitochondrial metabolism. Cell Metab 2015. Apr 7;21(4):628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Z, Chen K, Shih JC, Teng CT. Estrogen-related receptors-stimulated monoamine oxidase B promoter activity is down-regulated by estrogen receptors. Mol Endocrinol 2006. Jul;20(7):1547–1561. [DOI] [PubMed] [Google Scholar]
- 33.Cui H, Lu Y, Khan MZ, Anderson RM, McDaniel L, Wilson HE, et al. Behavioral disturbances in estrogen-related receptor alpha-null mice. Cell Rep 2015. Apr 21;11(3):344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Jesús-Cortés H, Lu Y, Anderson RM, Khan MZ, Nath V, McDaniel L, et al. Loss of estrogen-related receptor alpha disrupts ventral-striatal synaptic function in female mice. Neuroscience 2016. Aug 4;329:66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Busch BB, Stevens WC, Martin R, Ordentlich P, Zhou S, Sapp DW, et al. Identification of a selective inverse agonist for the orphan nuclear receptor estrogen-related receptor alpha. J Med Chem 2004. Nov 4;47(23):5593–5596. [DOI] [PubMed] [Google Scholar]
- 36.Lanvin O, Bianco S, Kersual N, Chalbos D, Vanacker J-M. Potentiation of ICI182,780 (Fulvestrant)-induced estrogen receptor-alpha degradation by the estrogen receptor-related receptor-alpha inverse agonist XCT790. J Biol Chem 2007. Sep 28;282(39):28328–28334. [DOI] [PubMed] [Google Scholar]
- 37.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998. Mar 20;92(6):829–839. [DOI] [PubMed] [Google Scholar]
- 38.Wright HM, Clish CB, Mikami T, Hauser S, Yanagi K, Hiramatsu R, et al. A synthetic antagonist for the peroxisome proliferator-activated receptor gamma inhibits adipocyte differentiation. J Biol Chem 2000. Jan 21;275(3):1873–1877. [DOI] [PubMed] [Google Scholar]
- 39.Armoni M, Harel C, Karnieli E. Transcriptional regulation of the GLUT4 gene: from PPAR-gamma and FOXO1 to FFA and inflammation. Trends Endocrinol Metab 2007. Apr;18(3):100–107. [DOI] [PubMed] [Google Scholar]
- 40.Ren Y, Jiang H, Ma D, Nakaso K, Feng J. Parkin degrades estrogen-related receptors to limit the expression of monoamine oxidases. Hum Mol Genet 2011. Mar 15;20(6):1074–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schindelin J, Rueden CT, Hiner MC, Eliceiri KW. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol Reprod Dev 2015. Aug;82(7–8):518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lucas EK, Reid CS, McMeekin LJ, Dougherty SE, Floyd CL, Cowell RM. Cerebellar transcriptional alterations with Purkinje cell dysfunction and loss in mice lacking PGC-1α. Front Cell Neurosci 2014;8:441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci 2010. Jan;13(1):76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Powell SB, Zhou X, Geyer MA. Prepulse inhibition and genetic mouse models of schizophrenia. Behav Brain Res 2009. Dec 7;204(2):282–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van den Buuse M, Garner B, Koch M. Neurodevelopmental animal models of schizophrenia: effects on prepulse inhibition. Curr Mol Med 2003. Aug;3(5):459–471. [DOI] [PubMed] [Google Scholar]
- 46.Giakoumaki SG. Cognitive and prepulse inhibition deficits in psychometrically high schizotypal subjects in the general population: relevance to schizophrenia research. J Int Neuropsychol Soc 2012. Jul;18(4):643–656. [DOI] [PubMed] [Google Scholar]
- 47.McMeekin LJ, Lucas EK, Meador-Woodruff JH, McCullumsmith RE, Hendrickson RC, Gamble KL, et al. Cortical PGC-1α-Dependent Transcripts Are Reduced in Postmortem Tissue From Patients With Schizophrenia. Schizophr Bull 2016;42(4):1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh BK, Sinha RA, Tripathi M, Mendoza A, Ohba K, Sy JAC, et al. Thyroid hormone receptor and ERRα coordinately regulate mitochondrial fission, mitophagy, biogenesis, and function. Sci Signal 2018. Jun 26;11(536). [DOI] [PubMed] [Google Scholar]
- 49.Saito K, Cui H. Emerging Roles of Estrogen-Related Receptors in the Brain: Potential Interactions with Estrogen Signaling. Int J Mol Sci 2018. Apr 5;19(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci 2007. Jan;8(1):45–56. [DOI] [PubMed] [Google Scholar]
- 51.Somogyi P, Klausberger T. Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol (Lond) 2005. Jan 1;562(Pt 1):9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kann O The interneuron energy hypothesis: Implications for brain disease. Neurobiol Dis 2016. Jun;90:75–85. [DOI] [PubMed] [Google Scholar]
- 53.Lord L-D, Expert P, Huckins JF, Turkheimer FE. Cerebral energy metabolism and the brain’s functional network architecture: an integrative review. J Cereb Blood Flow Metab 2013. Sep;33(9):1347–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eskiocak B, Ali A, White MA. The estrogen-related receptor α inverse agonist XCT 790 is a nanomolar mitochondrial uncoupler. Biochemistry 2014. Jul 29;53(29):4839–4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inan M, Zhao M, Manuszak M, Karakaya C, Rajadhyaksha AM, Pickel VM, et al. Energy deficit in parvalbumin neurons leads to circuit dysfunction, impaired sensory gating and social disability. Neurobiol Dis 2016. Sep;93:35–46. [DOI] [PubMed] [Google Scholar]
- 56.Albéri L, Lintas A, Kretz R, Schwaller B, Villa AEP. The calcium-binding protein parvalbumin modulates the firing 1 properties of the reticular thalamic nucleus bursting neurons. J Neurophysiol 2013. Jun;109(11):2827–2841. [DOI] [PubMed] [Google Scholar]
- 57.Vreugdenhil M, Jefferys JGR, Celio MR, Schwaller B. Parvalbumin-deficiency facilitates repetitive IPSCs and gamma oscillations in the hippocampus. J Neurophysiol 2003. Mar;89(3):1414–1422. [DOI] [PubMed] [Google Scholar]
- 58.Collin T, Chat M, Lucas MG, Moreno H, Racay P, Schwaller B, et al. Developmental changes in parvalbumin regulate presynaptic Ca2+ signaling. J Neurosci 2005. Jan 5;25(1):96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Orduz D, Bischop DP, Schwaller B, Schiffmann SN, Gall D. Parvalbumin tunes spike-timing and efferent short-term plasticity in striatal fast spiking interneurons. J Physiol (Lond) 2013. Jul 1;591(13):3215–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pang ZP, Melicoff E, Padgett D, Liu Y, Teich AF, Dickey BF, et al. Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J Neurosci 2006. Dec 27;26(52):13493–13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin M-Y, Rohan JG, Cai H, Reim K, Ko C-P, Chow RH. Complexin facilitates exocytosis and synchronizes vesicle release in two secretory model systems. J Physiol (Lond) 2013. May 15;591(10):2463–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang S, Reim K, Pedersen M, Neher E, Brose N, Taschenberger H. Complexin stabilizes newly primed synaptic vesicles and prevents their premature fusion at the mouse calyx of held synapse. J Neurosci 2015. May 27;35(21):8272–8290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paul A, Crow M, Raudales R, He M, Gillis J, Huang ZJ. Transcriptional architecture of synaptic communication delineates gabaergic neuron identity. Cell 2017. Oct 19;171(3):522–539.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saul MC, Seward CH, Troy JM, Zhang H, Sloofman LG, Lu X, et al. Transcriptional regulatory dynamics drive coordinated metabolic and neural response to social challenge in mice. Genome Res 2017. Mar 29;27(6):959–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cui H, Moore J, Ashimi SS, Mason BL, Drawbridge JN, Han S, et al. Eating disorder predisposition is associated with ESRRA and HDAC4 mutations. J Clin Invest 2013. Nov;123(11):4706–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang Z, Cowell RM, Nakazawa K. Convergence of genetic and environmental factors on parvalbumin-positive interneurons in schizophrenia. Front Behav Neurosci 2013. Sep 3;7:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pinheiro AP, Bulik CM, Thornton LM, Sullivan PF, Root TL, Bloss CS, et al. Association study of 182 candidate genes in anorexia nervosa. Am J Med Genet B, Neuropsychiatr Genet 2010. Jul;153B(5):1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Christoforou A, Le Hellard S, Thomson PA, Morris SW, Tenesa A, Pickard BS, et al. Association analysis of the chromosome 4p15-p16 candidate region for bipolar disorder and schizophrenia. Mol Psychiatry 2007. Nov;12(11):1011–1025. [DOI] [PubMed] [Google Scholar]
- 69.Christoforou A, McGhee KA, Morris SW, Thomson PA, Anderson S, McLean A, et al. Convergence of linkage, association and GWAS findings for a candidate region for bipolar disorder and schizophrenia on chromosome 4p. Mol Psychiatry 2011. Mar;16(3):240–242. [DOI] [PubMed] [Google Scholar]
- 70.Blackwood DH, He L, Morris SW, McLean A, Whitton C, Thomson M, et al. A locus for bipolar affective disorder on chromosome 4p. Nat Genet 1996. Apr;12(4):427–430. [DOI] [PubMed] [Google Scholar]
- 71.Dempster EL, Pidsley R, Schalkwyk LC, Owens S, Georgiades A, Kane F, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet 2011. Dec 15;20(24):4786–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Le Hellard S, Lee AJ, Underwood S, Thomson PA, Morris SW, Torrance HS, et al. Haplotype analysis and a novel allele-sharing method refines a chromosome 4p locus linked to bipolar affective disorder. Biol Psychiatry 2007. Mar 15;61(6):797–805. [DOI] [PubMed] [Google Scholar]
- 73.Jiang Z, Rompala GR, Zhang S, Cowell RM, Nakazawa K. Social isolation exacerbates schizophrenia-like phenotypes via oxidative stress in cortical interneurons. Biol Psychiatry 2013. May 15;73(10):1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brady LJ, Bartley AF, Li Q, McMeekin LJ, Hablitz JJ, Cowell RM, et al. Transcriptional dysregulation causes altered modulation of inhibition by haloperidol. Neuropharmacology 2016. Jul 29;111:304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Torretta S, Rampino A, Basso M, Pergola G, Di Carlo P, Shin JH, et al. NURR1 and ERR1 modulate the expression of genes of a DRD2 coexpression network enriched for schizophrenia risk. J Neurosci 2020. Jan 22;40(4):932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Holschneider DP, Chen K, Seif I, Shih JC. Biochemical, behavioral, physiologic, and neurodevelopmental changes in mice deficient in monoamine oxidase A or B. Brain Res Bull 2001. Nov 15;56(5):453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bortolato M, Chen K, Godar SC, Chen G, Wu W, Rebrin I, et al. Social deficits and perseverative behaviors, but not overt aggression, in MAO-A hypomorphic mice. Neuropsychopharmacology 2011. Dec;36(13):2674–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tomasella E, Bechelli L, Ogando MB, Mininni C, Di Guilmi MN, De Fino F, et al. Deletion of dopamine D2 receptors from parvalbumin interneurons in mouse causes schizophrenia-like phenotypes. Proc Natl Acad Sci USA 2018. Mar 27;115(13):3476–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shires SE, Quiles JM, Najor RH, Leon LJ, Cortez MQ, Lampert MA, et al. Nuclear parkin activates the errα transcriptional program and drives widespread changes in gene expression following hypoxia. Sci Rep 2020. May 22;10(1):8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shin J-H, Ko HS, Kang H, Lee Y, Lee Y-I, Pletinkova O, et al. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 2011. Mar 4;144(5):689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bakshi R, Mittal S, Liao Z, Scherzer CR. A Feed-Forward Circuit of Endogenous PGC-1α and Estrogen Related Receptor α Regulates the Neuronal Electron Transport Chain. Parkinsons Dis 2016. Mar 3;2016:2405176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huppunen J, Aarnisalo P. Dimerization modulates the activity of the orphan nuclear receptor ERRgamma. Biochem Biophys Res Commun 2004. Feb 20;314(4):964–970. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.