

Abstract
Background.
The genomic features and transmission link of circulating Group A Streptococcus (GAS) strains causing different disease types, such as pharyngitis and invasive disease, are not well understood.
Methods.
We used whole-genome sequencing to characterize GAS isolates recovered from persons with pharyngitis and invasive disease in the Denver metropolitan area from June 2016 to April 2017.
Results.
The GAS isolates were cultured from 236 invasive and 417 pharyngitis infections. Whole-genome sequencing identified 34 emm types. Compared with pharyngitis isolates, invasive isolates were more likely to carry the erm family genes (23% vs 7.4%, P < .001), which confer resistance to erythromycin and clindamycin (including inducible resistance), and covS gene inactivation (7% vs 0.5%, P < .001). Whole-genome sequencing identified 97 genomic clusters (433 isolates; 2–65 isolates per cluster) that consisted of genomically closely related isolates (median single-nucleotide polymorphism = 3 [interquartile range, 1–4] within cluster). Thirty genomic clusters (200 isolates; 31% of all isolates) contained both pharyngitis and invasive isolates and were found in 11 emm types.
Conclusions.
In the Denver metropolitan population, mixed disease types were commonly seen in clusters of closely related isolates, indicative of overlapping transmission networks. Antibiotic-resistance and covS inactivation was disproportionally associated with invasive disease.
Keywords: antimicrobial resistance, genomic cluster, Group A streptococcus, invasive disease, pharyngitis
Group A Streptococcus ([GAS] or Streptococcus pyogenes) is a Gram-positive bacterium that causes diverse infections in humans, ranging from mild disease such as pharyngitis and impetigo to more severe invasive infections including bacteremia, necrotizing fasciitis, and streptococcal toxic shock syndrome (STSS). The mechanisms underlying such a broad range of illnesses caused by a single pathogen remain poorly understood, although both host and pathogen factors have been implicated in influencing disease severity [1]. The GAS genome is characterized by high diversity and contains a large array of virulence factors with variable presence in different strains [2, 3]. Furthermore, the circulating GAS strains in a host population can frequently change [4–6]. It is difficult to obtain isolates from patients with different disease manifestations in the same time and place to compare the bacterial genomes; therefore, our understanding of the genetic relationships among these GAS strains remains very limited.
Previous studies suggested that strains causing invasive GAS infections and pharyngitis were not 2 distinct populations [7]. This observation supports the World Health Organization near-term strategy for GAS vaccine, which proposes that targeting noninvasive infections in children would also prevent both invasive GAS disease and nonsuppurative long-term consequences such as acute rheumatic fever and rheumatic heart disease [8]. Nonetheless, it remains unclear whether invasive and noninvasive GAS disease isolates generally share a closely related transmission network. A better understanding of the transmission connections among different types of GAS disease could help inform vaccine development. In this study, we used whole-genome sequencing (WGS) to characterize concurrently circulating GAS strains causing pharyngitis and invasive infections in persons living in the Denver metropolitan area from June 2016 to April 2017. We aimed to determine genomic features associated with disease severity and to identify potential transmission link.
METHODS
Surveillance for Pharyngitis and Invasive Group A Streptococcus (GAS) Infections and Collection of GAS Isolates
Children with symptomatic pharyngitis who received a rapid antigen detection test at Children’s Hospital Colorado emergency department between June 2016 to April 2017 and lived in the 5-county Denver metropolitan area (Adams, Arapahoe, Denver, Douglas, and Jefferson counties) were enrolled in this study. Throat cultures of the enrolled children were collected, and GAS isolates identified from positive pharyngitis cultures were sent to the Centers for Disease Control and Prevention (CDC) for characterization. The project was approved by the Colorado Multiple Institutional Review Board and informed consent was not required.
Invasive GAS (iGAS) infections were identified through the Colorado Active Bacterial Core surveillance (ABCs) currently implemented in the same 5-county Denver metropolitan area. The ABCs is an active, laboratory- and population-based surveillance for iGAS as previously described [9]. An iGAS disease case was defined as illness with isolation of GAS from a normally sterile site or from a wound culture accompanied by necrotizing fasciitis or STSS in a resident of the surveillance area. Isolates collected from patients with iGAS infections between June 1, 2016 and April 30, 2017 in the ABCs’s Colorado site were included in this study. The ABCs’s case reporting and isolate collection were regarded as surveillance activities and were either exempt from institutional review or approved by institutional review boards. Informed consent is not required by institutional review boards at the CDC or individual surveillance site.
Whole-Genome Sequencing Analysis
Whole-genome sequencing of the pharyngitis and invasive GAS isolates were performed using previously described methods [10, 11]. In brief, genomic deoxyribonucleic acid was extracted from colony-purified isolate and was sequenced on the Illumina MiSeq platform. The WGS reads were analyzed by a validated bioinformatics pipeline as previously described [10, 11]. The pipeline output included isolate emm type, multilocus sequence type, predicted antimicrobial susceptibility of 13 antibiotics, presence of virulence-related factors, and draft whole-genome assembly. Table 1 shows the list of 27 binary strain features present in more than 5 isolates. These 27 strain features represent commonly seen GAS genetic variations that have been linked to virulence previously and are routinely screened by the bioinformatics pipeline at the CDC Streptococcus laboratory. In the main analysis, we aimed to test whether 1 or more of these 27 strain features were associated with disease type. In a secondary analysis, all genes in the accessory genome were extracted from the draft whole genome assemblies using the Roary software (version 3.11.2) [12], and the association between the presence of each accessory gene and disease type was assessed using the pyseer software (version 1.3.3) [13] with the linear mixed-effect model (LMM) to account for population structure. The bioinformatics pipeline scripts and associated sequence databases are available at https://github.com/BenJamesMetcalf. The FASTQ files of the study isolates were submitted to National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under the BioProject number PRJNA395240.
Table 1.
Prevalence of Virulence-Related Strain Features in Pharyngitis (n = 417) and Invasive (n = 236) GAS Isolates, Colorado, June 2016 to April 2017
| Strain Featurea | Description | Cramer’s Vb | Number (Percent) Pharyngitis Isolatesc | Number (Percent) Invasive Isolatesc | Fisher P Valued | CMH P Valuee |
|---|---|---|---|---|---|---|
|
| ||||||
| ERM | The erm genes associated with macrolide resistance | 0.7 | 31(7%) | 54 (23%) | <.001 | <.001 |
| TET | The tet genes associated with tetracycline resistance | 0.89 | 33 (8%) | 63 (27%) | <.001 | .005 |
| COVS_NULL | Inactivating mutations in the covS gene | 0.34 | 2 (0.5%) | 15 (7%) | <.001 | .008 |
| SOF | Serum opacity factor | 0.98 | 243 (58%) | 198 (84%) | <.001 | .689 |
| FBAA | Fibronectin-binding protein of group A streptococci type A | 0.99 | 287 (69%) | 225 (95%) | <.001 | 1 |
| SFB1 | Streptococcus pyogenes fibronectin-binding protein I | 0.98 | 304 (73%) | 138 (58%) | <.001 | .913 |
| PRTF2 | S pyogenes fibronectin binding protein | 1 | 246 (59%) | 166 (70%) | .004 | 1 |
| R28 | Homolog of a group B streptococcal adhesin | 0.98 | 75 (18%) | 25 (11%) | .013 | .879 |
| ENN | emm-like enn virulence gene | 0.97 | 248 (59%) | 196 (83%) | <.001 | .452 |
| MRP | emm-like mrp virulence gene | 0.99 | 249 (60%) | 202 (86%) | <.001 | 1 |
| SME_Z | Streptococcal mitogenic exotoxin | 0.96 | 350 (84%) | 162 (69%) | <.001 | .602 |
| SPE_K | Streptococcal pyrogenic exotoxin K | 0.91 | 98 (24%) | 17 (7%) | <.001 | .294 |
| SS_A | Streptococcal superantigen A | 0.99 | 132 (32%) | 15 (6%) | <.001 | .986 |
| SPE_A | Streptococcal pyrogenic exotoxin A | 0.94 | 79 (19%) | 22 (9%) | .001 | .053 |
| SPE_L | Streptococcal pyrogenic exotoxin L | 0.96 | 17 (4%) | 18 (8%) | .057 | .063 |
| SPE_M | Streptococcal pyrogenic exotoxin M | 0.96 | 17 (4%) | 18 (8%) | .057 | .063 |
| SPE_J | Streptococcal pyrogenic exotoxin J | 0.95 | 93 (22%) | 63 (27%) | .207 | 1 |
| SPE_H | Streptococcal pyrogenic exotoxin H | 0.94 | 78 (19%) | 36 (15%) | .265 | .239 |
| SPE_I | Streptococcal pyrogenic exotoxin I | 0.91 | 72 (17%) | 35 (15%) | .420 | .745 |
| SPE_C | Streptococcal pyrogenic exotoxin C | 0.83 | 302 (72%) | 169 (72%) | .824 | .028 |
| SPE_G | Streptococcal pyrogenic exotoxin G | 0.98 | 342 (82%) | 219 (93%) | <.001 | .979 |
| SDA1 | Virulence-associated DNAse | 0.89 | 94 (23%) | 36 (15%) | .026 | .627 |
| SIC | Streptococcal inhibitor of complement | 0.99 | 89 (21%) | 24 (10%) | <.001 | .899 |
| NADase 330-G | NADase 330-G is associated with active NADase. The G330D substitution is a common inactivating substitution. | 0.91 | 371 (89%) | 173 (73%) | <.001 | .713 |
| PNGA3 | Pnga 3 - Clade 3 up-regulated promoter of the nga operon | 0.98 | 265 (64%) | 74 (31%) | <.001 | .028 |
| CAPSULE | hasA hyaluronic acid synthetase operon determinant | 0.96 | 225 (54%) | 163 (69%) | <.001 | .374 |
| ROCA | rocA null mutations | 1 | 39 (9%) | 4 (2%) | <.001 | 1 |
Abbreviations: CMH, Cochran-Mantel-Haenszel; GAS, Group A Streptococcus.
A strain feature is either positive (presence) or negative (absence) in an isolate.
Association between strain feature and emm type.
Number (percent) of feature-positive isolates.
Fisher’s exact test.
Cochran-Mantel-Haenszel (CMH) test using emm type as the stratum.
Genomic Cluster Identification
For each individual emm type containing 2 or more isolates, we calculated the pair-wise single-nucleotide polymorphism (SNP) distance for every isolate pair using their draft genome assemblies and the NUCmer software that is part of the MUMmer package [14]. The SNPs within 1-kb of pair-wise alignment were removed to reduce the potential confounding effect of recombination on SNP distance. A pair-wise SNP distance matrix of all isolates was then generated and was subjected to a hierarchical cluster analysis (HCA) by using the “hclust” function in the R software [15]. The HCA initially assigned each isolate to its own cluster and then iteratively joined the 2 most similar clusters, continuing until there was just a single cluster represented by a dendrogram tree. The dendrogram tree produced by the HCA was then processed by the “cutree” function in the R software [15] with a cutoff value of 10 SNPs, which was consistent with observed mutation rates of GAS in human populations [16, 17]. The “cutree” function generated isolate groups in which SNP distance between any 2 isolates was no larger than the cutoff value. A group containing 2 or more isolates was defined as a genomic cluster. (We used the Streptococcus pyogenes multilocus sequence typing website (https://pubmlst. org/organisms/streptococcus-pyogenes/) sited at the University of Oxford.)
Proportion of Isolates Covered by Group A Streptococcus Candidate Vaccines
A 30-valent M protein-based vaccine candidate (StreptAnova) containing M protein fragments from 30 different emm types is currently under development [18]. An isolate was defined as covered by StreptAnova if the isolate was one of the 30 emm types. Another vaccine strategy under consideration currently incorporates M-related protein (Mrp vaccine) to expand coverage of the vaccine against different emm types [19]. An isolate was defined as covered by the Mrp vaccine if the isolate was positive for the mrp gene encoding the M-related protein.
Statistical Analysis
The proportion for each strain feature within pharyngitis and invasive isolates was calculated. The Fisher exact test and the Cochran-Mantel-Haenszel (CMH) test was used to assess association between strain feature and disease type before and after controlling for emm type, respectively. All P values are 2 sided and a P value less than a Bonferroni-corrected threshold was considered statistically significant. Strength of association between emm type and a strain feature was evaluated by Cramer’s V. All analyses were performed using the R software version 3.4.3 [15].
RESULTS
Demographic Description
During the 11-month study period, a total of 417 pharyngitis isolates (414 from the ABCs 5-county area and 3 from contiguous counties) and 236 invasive isolates were recovered and characterized (Figure 1A). The median age of pharyngitis patients was 9.3 years (range, 2.0 to 23.1 years) and 50% were male. The median age of invasive disease patients was 54 years (range, 4 months to 99 years) and 62% were male. Among the 236 invasive disease patients, 45 (19%) were people who injected drugs (PWID) and 23 (9.7%) were people experiencing homelessness (PEH); 57 (24%) were either PEH or PWID populations at high risk for iGAS infections [20].
Figure 1.
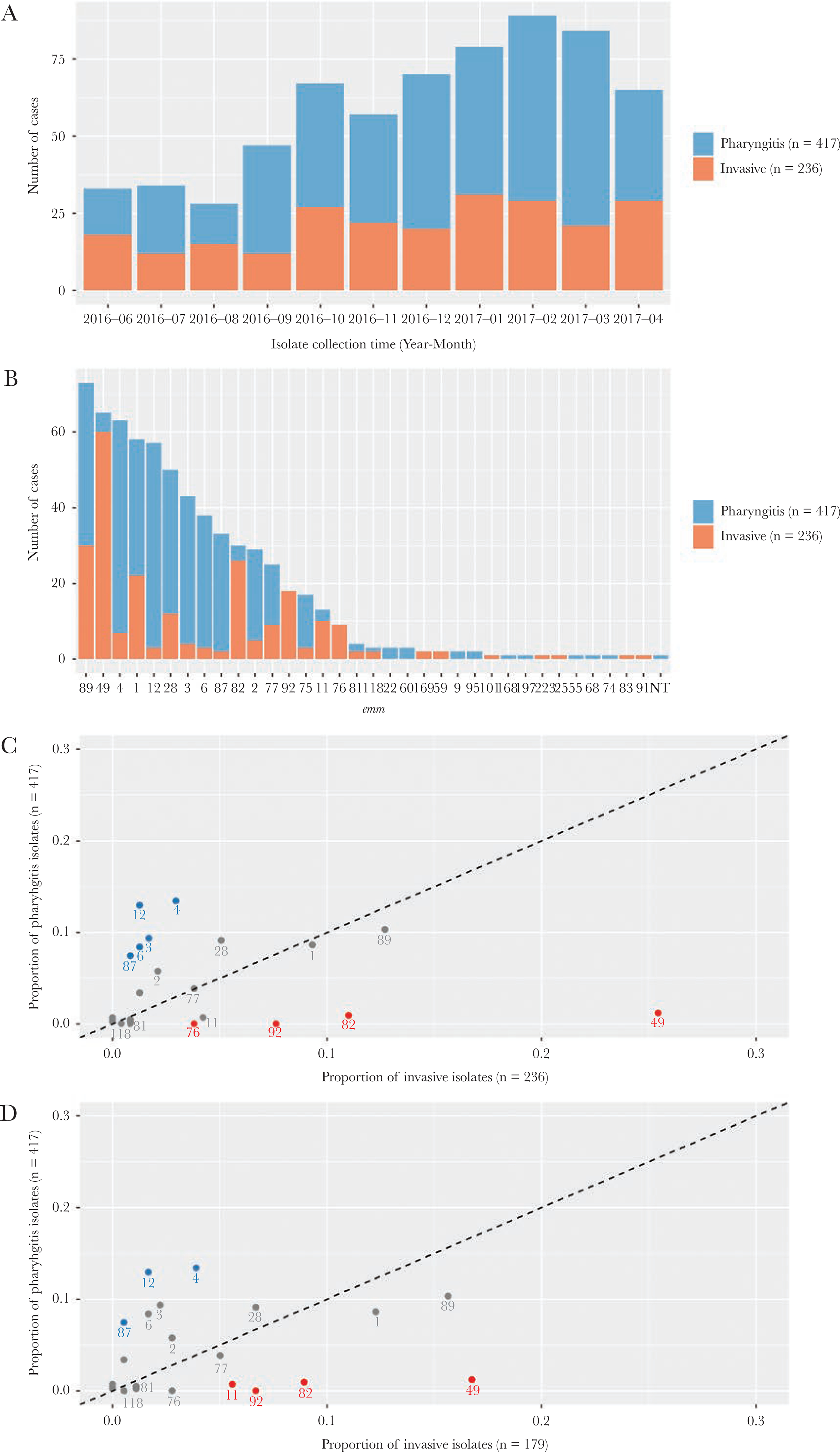
emm types identified in pharyngitis and invasive Group A Streptococcus isolates, Colorado, June 2016 to April 2017. (A) Number of isolates recovered from each month of the study period. (B) Number of isolates for each of the emm types identified. (C) Each point represents an emm type for which the x- and y-axis values indicate the proportion of invasive and pharyngitis isolates belonging to the emm type, respectively. The dotted line indicates where the 2 proportions are equal. Text labels under points are the emm types. emm types 49, 82, 92, and 76 (red) were significantly more common in invasive isolates, whereas emm types 4, 12, 3, 6, and 87 (blue) were significantly more common in pharyngitis isolates. (D) The same as (C) except that 57 invasive isolates that were recovered from people who injected drugs or who experienced homeless were removed.
Distribution of emm types
Whole-genome sequencing identified 34 emm types from 652 isolates and 1 isolate was nontypeable (Figure 1B). Compared with pharyngitis isolates, the invasive isolates had higher proportions of emm types 49 (25% vs 1%), 82 (11% vs 1%), 92 (7.6% vs 0), and 76 (3.8% vs 0) and lower proportions of emm types 4 (3.0% vs 13%), 12 (1.3% vs 13%), 3 (1.7% vs 9.4%), 6 (1.3% vs 8.4%), and 87 (0.8% vs 7.4%; all P values < Bonferroni-corrected threshold of 0.0015) (Figure 1C). After removing isolates obtained from the 57 high-risk invasive disease patients from analysis, the emm distribution between disease types did not alter substantially (Figure 1D).
Nineteen of the 30 emm types covered by the StreptAnova candidate vaccine were found in the 653 study isolates and accounted for 404 (97%) of the pharyngitis isolates and 219 (93%) of the invasive isolates. The M-related protein candidate vaccine target was identified in 249 (60%) of the pharyngitis isolates and 202 (86%) of the invasive isolates. The combination of M and Mrp antigens from both vaccine candidates covered more than 99% of isolates in each disease type.
Antimicrobial Resistance and Virulence-Related Factors
All isolates were susceptible to penicillin and other β-lactam antibiotics based on a PBP2x typing scheme used for groups A and B streptococci [21, 22]. The erm family genes, which confer resistance to erythromycin and clindamycin (including inducible resistance), were identified in 31 (7.4%) of pharyngitis isolates and 54 (23%) of the invasive isolates. Presence of the erm genes was strongly associated with emm type (Cramer’s V = 0.70). After adjusting for emm using the CMH test, erm remained significantly associated with disease type (CMH test P values < Bonferroni-corrected threshold of 0.0018). Of the 15 most frequent emm types in our study, the proportion of invasive isolates that were erm+ were higher than the proportion of pharyngitis isolates that were erm+ among emm types 4, 75, 77, 89, 2, 82, 28, and 1 (Figure 2A). A similar association of tet+ isolates with invasive isolates was seen (Figure 2B). The tet family genes confer tetracycline resistance and often colocalize with erm in the same mobile genetic element [23]. Other relatively uncommon antimicrobial resistance genes identified included the mef family genes, which conferred erythromycin resistance in 3 isolates (all pharyngitis), and gyrA/parC gene mutations conferred reduced susceptibility to levofloxacin in 5 isolates (3 pharyngitis and 2 invasive).
Figure 2.
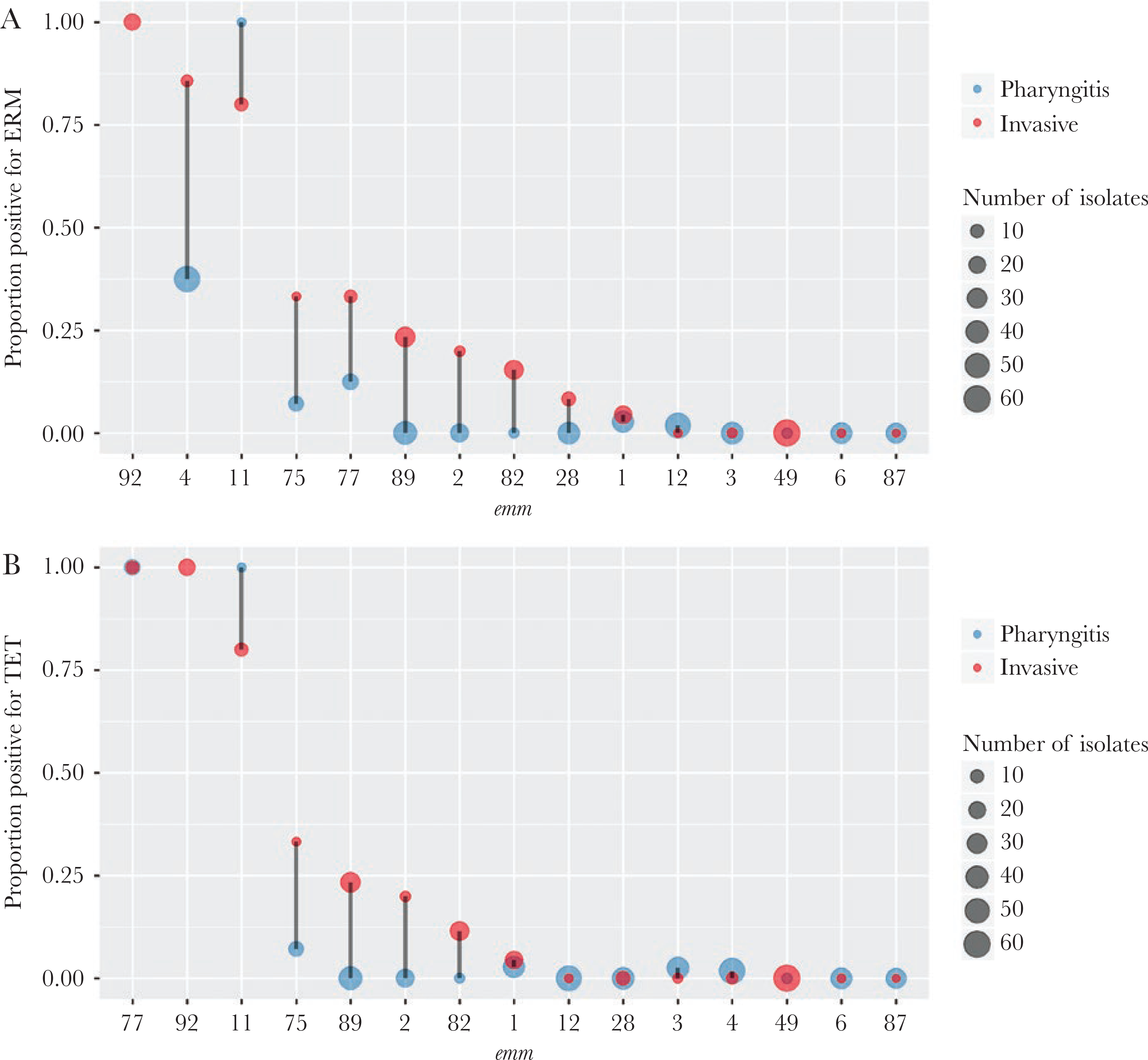
Distribution of antibiotic resistance genes by emm type. Within in individual emm type, the proportion of pharyngitis (blue) or invasive (red) isolates that were positive for the erm genes (A) or the tet genes (B) is shown. The x-axis is ordered by decreasing y-values of the invasive (red) group. The point size is proportion to isolate count. No pharyngitis isolates were emm 92; therefore, the corresponding proportion was absent. Only the 15 most frequent emm types each containing 10 or more isolates are shown.
Among the 25 virulence-related strain features routinely detected by our bioinformatics pipeline (Table 1 excluding EMR and TET), 18 were noted to be significantly associated with disease type (Fisher’s exact test P values < Bonferroni-corrected threshold of 0.0018) (Table 1). Most virulence-related strain features showed strong association with emm type (Cramer’s V > 0.85 except for COVS_NULL [0.34]) (Table 1). After controlling for emm using the CMH test, none of the 25 strain features showed a CMH P value that was less than the Bonferroni-corrected threshold of 0.0017. Nonetheless, covS gene inactivation, up-regulated nga-ifs-slo operon (encodes extracellular NADase, intracellular NADase inhibitor, and streptolysin O [1]), and lack of streptococcal pyrogenic exotoxin gene (speC) exhibited association with invasive isolates with CMH P < .05 (Table 1, Figure 3).
Figure 3.
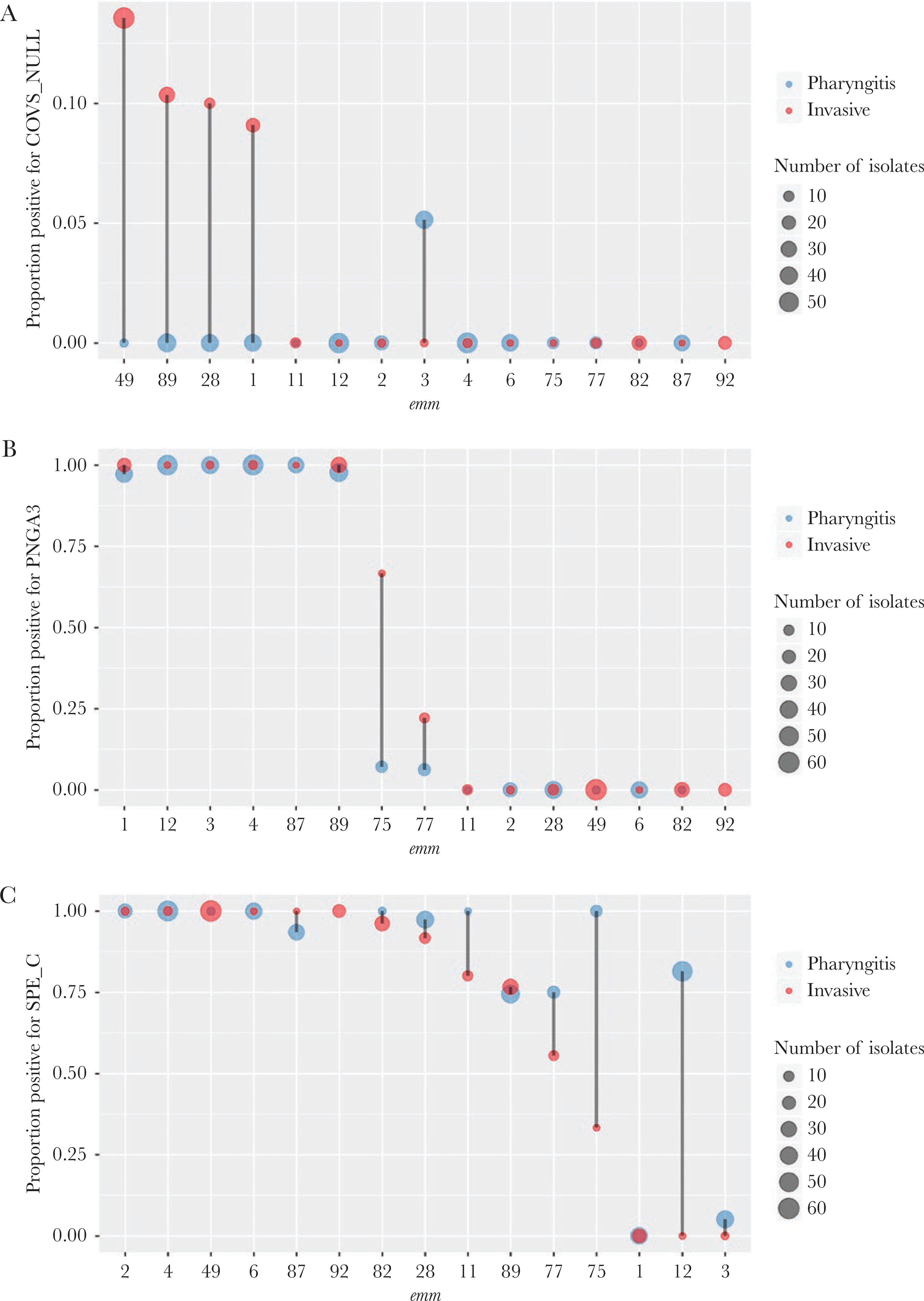
Distribution of virulence-related factors by emm type. The proportion of pharyngitis (blue) or invasive (red) isolates that were positive for inactivating mutations in the covS gene (COVS_NULL) (A), the Pnga 3 - Clade 3 up-regulated promoter of the nga operon (PNGA3) (B), and streptococcal pyrogenic exotoxin C (SPE_C) (C) is shown. The x-axis is ordered by decreasing y-values of the invasive (red) group. The point size is proportional to the isolate count. There were no emm92 pharyngitis isolates so no proportion existed. Only the 15 most frequent emm types each containing 10 or more isolates are shown.
The Roary software identified 1774 accessory genes that were present in >1% but <99% of isolates. None of the 1774 accessory genes showed significant association with disease type after controlling for multiple comparisons (all LMM test P values > Bonferroni-corrected threshold of 0.000028). Nonetheless, 160 accessory genes, including the erm and tet genes, exhibited association with invasive isolates with LMM P < .05 (Supplemental Table S1).
Genomic Clusters
From 24 emm types, each containing 2 or more isolates (totaling 642 [98%] isolates), cluster analysis identified 97 clusters of genomically closely related isolates (genomic clusters). The median cluster size was 3 isolates per genomic cluster (range, 2 to 65 isolates). Among the 642 isolates, 465 isolates (72%) belonged to one of the 97 genomic clusters (clustered isolates); the remaining 177 isolates (28%) were “nonclustered” isolates. The median SNP distance for a pair of isolates sampled from within a cluster was 3 SNPs (interquartile range, 1 to 4). In contrast, the median SNP distance for a pair nonclustered isolates within the same emm type was 57 SNPs (interquartile range, 47 to 91) (Figure 4A).
Figure 4.
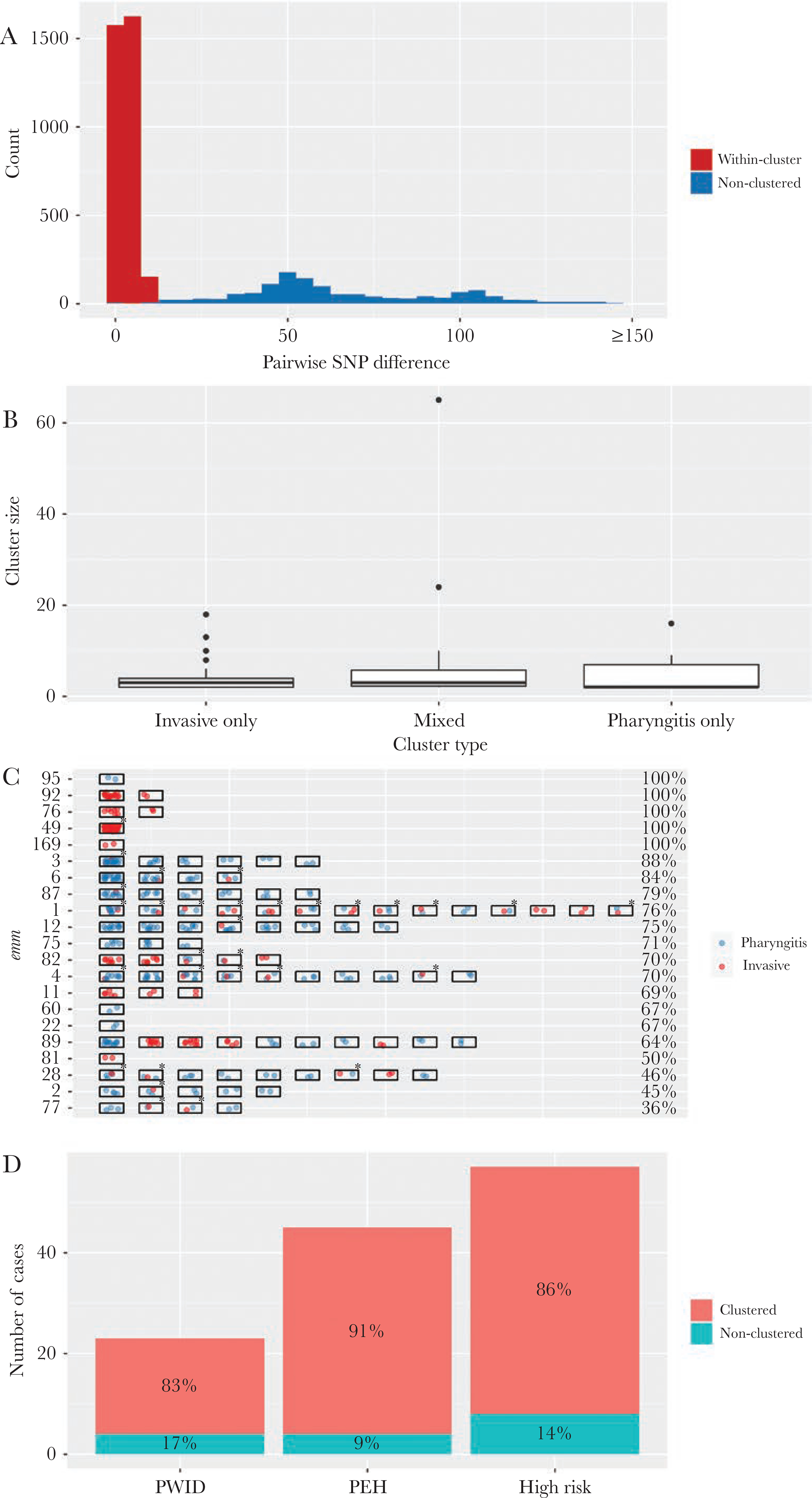
Clusters of genomically closely related pharyngitis and invasive Group A streptococcus isolates, Colorado, June 2016 to April 2017. (A) Distribution of single-nucleotide polymorphism (SNP) distance between a pair of emm-matched isolates that were within a same cluster (red, 3340 pairs) or nonclustered (blue, 1316 pairs). (B) Box-plot of cluster size (number of isolates within a cluster) by cluster type. (C) Clustered isolates within individual emm types. Each rectangle represents a cluster containing 2 or more isolates. Each spot within the rectangle indicates a pharyngitis isolate (blue) or an invasive isolate (red). An asterisk indicates a mixed cluster. The percentage of clustered isolates in each emm is shown at the end of the row. Only emm types in which at least 1 cluster was detected are shown. (D) Clustered isolates among invasive disease cases from people who injected drugs ([PWID] n = 45), who were experiencing homelessness (PEH, n = 23), and who had either of the 2 conditions (high risk, n = 57). Percentages of isolates that belong to a cluster among these patients are shown.
We categorized the 97 genomic clusters according to whether a cluster contained only pharyngitis isolates (pharyngitis-only cluster), only invasive isolates (invasive-only cluster), or both pharyngitis and invasive isolates (mixed cluster; an example is shown in Supplemental Figure S1). There were 48 pharyngitis-only clusters (n = 178 isolates), 19 invasive-only clusters (85 isolates), and 30 mixed clusters (200 isolates). Isolates belong to mixed clusters accounted for 31% of all isolates in the cluster analysis and were greater in number than those belonging to either of the other 2 cluster types. Individual cluster sizes did not differ significantly by cluster type (Kruskal test, P = .33) (Figure 4C).
For each cluster, the time- and county-resolved epi curve is shown in Supplemental Figure S2. These curves typically increased gradually and contained no obvious single peak. These observations are consistent with the notion that GAS is typically not associated with point-source outbreaks. Phylogenetic trees for individual emm types are shown in Supplemental Figure S3. In emm types 49, 89, and 1, multiple independent acquisitions of the covS_null allele were observed (Supplemental Figure S3). Likewise, independent introduction of the erm gene into multiple lineages was found in emm types 82 and 8 (Supplemental Figure S3). Thus, the loss of covS and gain of erm were consistent with signals of convergent evolution within emm types.
The proportion of clustered isolates within each emm type varied substantially (Figure 4D). For emm types 49, 76, 92, 95, and 169, all isolates within emm type corresponded to 1 or 2 genomic clusters, suggesting rapidly expanding lineages within the study population. For long-standing emm types in this region, such as 1, 3, 4, 12, and 89 among others, 64%–88% of isolates within an emm type were distributed into multiple clusters, whereas the remaining isolates were nonclustered. For emm types 2, 28, 77, and 81, 50% or less isolates within emm type were clustered.
Among the 57 high-risk patients (either PEH or PWID) with invasive disease, the proportion of clustered isolates (86%) was significantly higher than that among the remaining 179 invasive disease patients (73%; P = .048, Fisher’s exact test) (Figure 4E). These 57 isolates were emm types 49 (30), 82 (10), 92 (6), 76 (4), 89 (2), 75 (2), 87 (1), 59 (1), and 169 (1). The emm types that had a higher proportion of patients from high-risk population (such as emm types 49, 82, 92, and 76) tended to show a higher proportion of clustered isolates (Spearman’s correlation rho = 0.48, P = .003) and evidence of rapidly expanding lineages. After removing the 57 high-risk patients, clustered isolates still accounted for 414 isolates: 68 belonged to invasive-only clusters, 168 belonged to mixed clusters, and 178 belonged to pharyngitis-only clusters.
DISCUSSION
In this study, we used WGS to characterize GAS isolates recovered from children with pharyngitis and from patients of all ages with invasive GAS infections from the same 5-county geographic region during an 11-month period. Common emm types found in pharyngitis, such as 89, 1, and 28, were also most frequently found in invasive disease. Furthermore, it was commonly seen that pharyngitis and invasive isolates belonged to the same genomic cluster in multiple emm types. These findings suggested a common connection between the populations from which the pharyngitis and invasive GAS isolates were drawn. Pharyngitis isolates had a higher proportion of emm types 4, 12, 3, 6, and 87, 3 of which (emm 12, 3, 6) belonged to the emm pattern A-C with strong preference for infection at the throat over the skin [24, 25]; invasive isolates had a higher proportion of emm types 49, 82, 92, and 76, all of which belonged to the emm pattern E with no obvious tissue site preference [24, 25].
In this study, invasive isolates showed a higher proportion of erm+ isolates than pharyngitis isolates across multiple emm types, consistent with a previous report [26]. These erm+ isolates might be enriched in invasive disease patients due to their increased susceptibility to such strains or recent expansion of resistant lineages in this population. The increased prevalence of antibiotic resistance among invasive isolates could be a result of selection due to a prior history of antibiotic treatment. For example, a recent study characterized a set of 5 nearly isogenic iGAS recovered within a community [27] and found that only 2 of the 5 isolates shared a pbp2X point mutation associated with higher minimum inhibitory concentrations to ampicillin and amoxicillin, as well as an unlinked point mutation conferring fluoroquinolone resistance. Both patients had recent histories of extensive treatments with beta lactams and fluoroquinolones before onset of iGAS. Likewise, for virulence factors, prior studies have provided strong data indicating that covS inactivation is selected for during the transition from the noninvasive to invasive state [28, 29]. Multiple previous studies have shown loss of covS in GAS is associated with invasive disease over pharyngeal disease [29–33]. Because most virulence-related factors were strongly associated with specific emm types (and thus many other clonally linked strain features), caution should be taken in interpreting the significance of “invasiveness-associated” virulence-related factors.
In outbreak investigations, WGS is increasingly used to confirm close relatedness among isolates from a suspected disease cluster [34–36]. In this study, we systematically identified genomic clusters from population-based disease surveillance. Clustered isolates were presumably connected within a transmission network, representing either a recognized outbreak or unrecognized ongoing “cryptic” community transmission. In several emm types, rapid spread of strains with little genetic variation causing invasive disease has been previously reported [11, 37]. In contrast, nonclustered isolates were unlikely to have recent connections with each other or with the clustered isolates, representing sporadic disease cases or incomplete sampling due to not all pharyngitis cases accessing care. The results clearly showed that pharyngitis and invasive disease often share the same transmission network: 30 of the 97 genomic clusters, accounting for 31% of all isolates, contained both GAS disease types. The findings were consistent with a previous report showing pharyngitis and invasive strains were clustered tightly with one another among M3 strains collected at 6 sites in Ontario [7]. Still, this may be an underestimate, because pharyngitis isolates in this study were only obtained from 1 hospital and represented patients who presented at the emergency department. The most likely initial source of an invasive cluster may be a pharyngitis infection where a child comes into close contact with an adult that has predisposing conditions. This parallels invasive pneumococcal disease, where dramatic holiday spikes in adults over the holiday season are likely due to spread through upper respiratory secretions, especially those from young children [38]. Therefore, we hypothesize that in GAS disease, there is close contact between the 2 different isolation populations to generate the phylogenetic signals of closely connected transmission networks.
This study had several limitations. First, pharyngitis isolates were all recovered from children, whereas all but 4 invasive isolates were recovered from adults. Any strain features that differed by disease type could also reflect the difference in age group. It should be noted that pharyngitis is much more common among children than adults (peak age range is 4–9 years), and, in the United States, invasive disease is much more common among adults [39]. Second, the study was confined to a single large metropolitan area and lasted for a relatively short period (11 months). It is unknown whether the GAS genetic variants associated with disease type could be generalized to larger, more geographically and temporally diverse populations. Although invasive isolates were collected as part of comprehensive population-based surveillance, pharyngitis isolates were collected from a single children’s hospital in the area. Third, a substantial proportion (24%) of the invasive disease patients were people who injected drugs or experienced homelessness. This vulnerable population was probably less likely to be in contact with pediatric pharyngitis patients than the general population, and it has been shown that the most common strains in outbreaks among these high-risk populations are typically the less common emm types. This could lead to an underestimation of the transmission connection between pharyngitis and invasive disease. Nonetheless, a substantial proportion of isolates belonged to mixed clusters even after removing the high-risk group. We were also unable to assess the impact of skin infections on the noninvasive disease burden and to mixed disease type clusters because noninvasive skin infections were not under surveillance. Among injecting drug users and other disadvantaged groups, such noninvasive infections could have major roles. It is notable that pattern E emm types were most disproportionally associated with invasive infections of disadvantaged individuals, consistent with a previous report [20]. It is also probable that the number of disadvantaged individuals such as PWID and PEH are underestimated, given that these behaviors were identified when documented in a medical record only.
CONCLUSIONS
In conclusion, common emm types caused both pharyngitis and invasive disease in the study population. Within emm types, genomic clusters accounted for a large proportion of isolates. Mixed disease types were common in genomic clusters, indicating closely connected transmission networks for the 2 types of GAS diseases. There were clearly GAS lineages (eg, emm49 and emm92) that spread significantly among adult populations, especially PEH and injecting drug users, but caused little pharyngitis. A multifaceted approach addressing both pediatric pharyngitis and persons at high-risk for invasive disease is thus needed to limit GAS disease. The establishment of a GAS pharyngitis human adult infection study as a platform for vaccine evaluation is an important step in this direction [40].
Supplementary Material
Financial support.
This work was funded by the Centers for Disease Control and Prevention (CDC) as part of normal responsibilities. Major funding for this work was provided through support from the CDC’s Emerging Infection Program and the CDC Advanced Molecular Detection initiative.
Footnotes
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest.
Disclaimer. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
References
- 1.Walker MJ, Barnett TC, McArthur JD, et al. Disease manifestations and pathogenic mechanisms of Group A Streptococcus. Clin Microbiol Rev 2014; 27:264–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jespersen MG, Lacey JA, Tong SYC, Davies MR. Global genomic epidemiology of Streptococcus pyogenes. Infect Genet Evol 2020; 86:104609. [DOI] [PubMed] [Google Scholar]
- 3.Davies MR, McIntyre L, Mutreja A, et al. Atlas of group A streptococcal vaccine candidates compiled using large-scale comparative genomics. Nat Genet 2019; 51:1035–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu L, Olsen RJ, Nasser W, de la Riva Morales I, Musser JM. Trading capsule for increased cytotoxin production: contribution to virulence of a newly emerged clade of emm89 Streptococcus pyogenes. mBio 2015; 6:e01378–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lynskey NN, Jauneikaite E, Li HK, et al. Emergence of dominant toxigenic M1T1 Streptococcus pyogenes clone during increased scarlet fever activity in England: a population-based molecular epidemiological study. Lancet Infect Dis 2019; 19:1209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner CE, Abbott J, Lamagni T, et al. Emergence of a new highly successful acapsular group A Streptococcus clade of genotype emm89 in the United Kingdom. mBio 2015; 6:e00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shea PR, Beres SB, Flores AR, et al. Distinct signatures of diversifying selection revealed by genome analysis of respiratory tract and invasive bacterial populations. Proc Natl Acad Sci U S A 2011; 108:5039–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vekemans J, Gouvea-Reis F, Kim JH, et al. The path to group A Streptococcus vaccines: World Health Organization research and development technology roadmap and preferred product characteristics. Clin Infect Dis 2019; 69:877–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Centers for Disease Control and Prevention. Active Bacterial Core Surveillance Report, Emerging Infections Program Network, Group A Streptococcus, 2018. https://www.cdc.gov/abcs/reports-findings/survreports/gas18.html.
- 10.Chochua S, Metcalf BJ, Li Z, et al. Population and whole genome sequence based characterization of invasive group a streptococci recovered in the United States during 2015. mBio 2017; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y, Rivers J, Mathis S, et al. Genomic surveillance of Streptococcus pyogenes strains causing invasive disease, United States, 2016–2017. Front Microbiol 2020; 11:1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Page AJ, Cummins CA, Hunt M, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015; 31:3691–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lees JA, Galardini M, Bentley SD, Weiser JN, Corander J. pyseer: a comprehensive tool for microbial pangenome-wide association studies. Bioinformatics 2018; 34:4310–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurtz S, Phillippy A, Delcher AL, et al. Versatile and open software for comparing large genomes. Genome Biol 2004; 5:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2021. https://www.R-project.org/. [Google Scholar]
- 16.Davies MR, Holden MT, Coupland P, et al. Emergence of scarlet fever Streptococcus pyogenes emm12 clones in Hong Kong is associated with toxin acquisition and multidrug resistance. Nat Genet 2015; 47:84–7. [DOI] [PubMed] [Google Scholar]
- 17.Beres SB, Carroll RK, Shea PR, et al. Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics. Proc Natl Acad Sci U S A 2010; 107:4371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pastural É, McNeil SA, MacKinnon-Cameron D, et al. Safety and immunogenicity of a 30-valent M protein-based group a streptococcal vaccine in healthy adult volunteers: a randomized, controlled phase I study. Vaccine 2020; 38:1384–92. [DOI] [PubMed] [Google Scholar]
- 19.Courtney HS, Niedermeyer SE, Penfound TA, Hohn CM, Greeley A, Dale JB. Trivalent M-related protein as a component of next generation group A streptococcal vaccines. Clin Exp Vaccine Res 2017; 6:45–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valenciano SJ, Onukwube J, Spiller MW, et al. Invasive group A streptococcal infections among people who inject drugs and people experiencing homelessness in the United States, 2010–2017. Clin Infect Dis 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vannice KS, Ricaldi J, Nanduri S, et al. Streptococcus pyogenes pbp2x mutation confers reduced susceptibility to β-lactam antibiotics. Clin Infect Dis 2020; 71:201–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGee L, Chochua S, Li Z, et al. Multistate, population-based distributions of candidate vaccine targets, clonal complexes, and resistance features of invasive Group B Streptococci within the US: 2015–2017. Clin Infect Dis 2020; 72:1004–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brenciani A, Bacciaglia A, Vecchi M, Vitali LA, Varaldo PE, Giovanetti E. Genetic elements carrying erm(B) in Streptococcus pyogenes and association with tet(M) tetracycline resistance gene. Antimicrob Agents Chemother 2007; 51:1209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bessen DE, Lizano S. Tissue tropisms in group A streptococcal infections. Future Microbiol 2010; 5:623–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGregor KF, Spratt BG, Kalia A, et al. Multilocus sequence typing of Streptococcus pyogenes representing most known emm types and distinctions among subpopulation genetic structures. J Bacteriol 2004; 186:4285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanson MA, Macias OR, Shah BJ, et al. Unexpected relationships between frequency of antimicrobial resistance, disease phenotype and emm type in group A Streptococcus. Microb Genom 2019; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vannice KS, Ricaldi J, Nanduri S, et al. Streptococcus pyogenes pbp2x mutation confers reduced susceptibility to β-lactam antibiotics. Clin Infect Dis 2020; 71:201–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levin JC, Wessels MR. Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol Microbiol 1998; 30:209–19. [DOI] [PubMed] [Google Scholar]
- 29.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. Genome-wide analysis of group a streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog 2006; 2:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tran-Winkler HJ, Love JF, Gryllos I, Wessels MR. Signal transduction through CsrRS confers an invasive phenotype in group A Streptococcus. PLoS Pathog 2011; 7:e1002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin JN, Chang LL, Lai CH, Lin HH, Chen YH. Association between polymorphisms in the csrRS two-component regulatory system and invasive group A streptococcal infection. Eur J Clin Microbiol Infect Dis 2014; 33:735–43. [DOI] [PubMed] [Google Scholar]
- 32.Ikebe T, Ato M, Matsumura T, et al. Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog 2010; 6:e1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friães A, Pato C, Melo-Cristino J, Ramirez M. Consequences of the variability of the CovRS and RopB regulators among Streptococcus pyogenes causing human infections. Sci Rep 2015; 5:12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner CE, Bedford L, Brown NM, et al. Community outbreaks of group A Streptococcus revealed by genome sequencing. Sci Rep 2017; 7:8554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coelho JM, Kapatai G, Jironkin A, et al. Genomic sequence investigation Streptococcus pyogenes clusters in England (2010–2015). Clin Microbiol Infect 2019; 25:96–101. [DOI] [PubMed] [Google Scholar]
- 36.Nanduri SA, Metcalf BJ, Arwady MA, et al. Prolonged and large outbreak of invasive group A Streptococcus disease within a nursing home: repeated intrafacility transmission of a single strain. Clin Microbiol Infect 2019; 25:248.e1–7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fittipaldi N, Beres SB, Olsen RJ, et al. Full-genome dissection of an epidemic of severe invasive disease caused by a hypervirulent, recently emerged clone of group A Streptococcus. Am J Pathol 2012; 180:1522–34. [DOI] [PubMed] [Google Scholar]
- 38.Walter ND, Taylor TH Jr, Dowell SF, Mathis S, Moore MR; Active Bacterial Core Surveillance System Team. Holiday spikes in pneumococcal disease among older adults. N Engl J Med 2009; 361:2584–5. [DOI] [PubMed] [Google Scholar]
- 39.Nelson GE, Pondo T, Toews KA, et al. Epidemiology of invasive group A streptococcal infections in the United States, 2005–2012. Clin Infect Dis 2016; 63:478–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Osowicki J, Azzopardi KI, Baker C, et al. Controlled human infection for vaccination against Streptococcus pyogenes (CHIVAS): establishing a group A Streptococcus pharyngitis human infection study. Vaccine 2019; 37:3485–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.