

Abstract
Monosialodihexosylganglioside (GM3)-presenting lipid-coated polymer nanoparticles (NPs) that recapitulate the sequestration of HIV-1 particles in CD169+ virus-containing compartments (VCCs) of macrophages were developed as carriers for delivery and sustained release of a combination of two antiretrovirals (ARVs), rilpivirine (RPV) and cabotegravir (CAB). RPV and CAB were co-loaded into GM3-presenting lipid-coated polylactic acid (PLA) and poly(lactic-co-glycolic acid) (PLGA) NPs without loss in potency of the drugs. GM3-presenting PLA NPs demonstrated the most favorable release properties and achieved inhibition of HIV-1 infection of primary human macrophages for up to 35 days. Intracellular localization of GM3-presenting PLA NPs in VCCs correlated with retention of intracellular ARV concentrations and sustained inhibition of HIV-1 infection. This work elucidates design criteria of lipid-coated polymer NPs to utilize CD169+ macrophages as cellular drug depots for eradicating viral reservoir sites or to achieve long-acting prophylaxis against HIV-1 infection.
Keywords: Lipid-coated polymer nanoparticles, Biomimicry, HIV-1, Bio-nano interface, GM3
INTRODUCTION
Currently, the most effective treatment for human immunodeficiency virus-1 (HIV-1) is achieved by combinatorial antiretroviral therapy (cART).1–5 Despite tremendous progress in the development of effective cART, a cure remains elusive and lifelong suppression of the virus with cART remains standard practice.6–9 One key challenge for achieving HIV-1 eradication is believed to be its localization in tissue reservoirs that protect the virus from immune surveillance mechanisms as well as antiretrovirals (ARVs).10–13 Residual virus production, even in individuals on suppressive cART, might lead to chronic immune activation14 and could promote HIV-1 persistence by multiple mechanisms, including enhancing viral transcription in latently infected cells, increasing the number of activated target cells for residual infection, and/or by clonal amplification of infected cells. Thus, strategies to target tissue reservoirs of HIV-1 and minimize bystander immune activation are needed to achieve a sterilizing cure.
Long-lived, tissue resident CD169+ (Siglec1) macrophages that are located in the secondary lymphoid tissues (spleen and lymph nodes), liver, lungs, and the bone marrow, have recently attracted special attention because of their emerging role in HIV-1 dissemination.15–20 CD169+ macrophages collect HIV-1 particles in specialized, non-endolysosomal compartments, and these so-called virus containing compartments (VCCs) represent potential virus reservoir sites.16, 21, 22 Importantly, glycoprotein-independent sequestration of HIV-1 in VCCs can occur in myeloid cells via recognition and binding of the monosialodihexosylganglioside, GM3, in the viral membrane to CD169 on the cell surface.22–25 More specifically, the sialyllactose group in the ganglioside GM3 has been identified as the moiety responsible for the binding to CD169 receptors.26 CD169+ macrophages can facilitate dissemination of HIV-1 in secondary lymphoid tissues of humanized mice by capture of virus particles and trans-infection of CD4+ T cells.15 Additionally, CD169+ macrophages were also shown to aid virus spread via cell-free transmission through fluid flow–based dissemination.15 Our previous studies demonstrated that GM3-presenting lipid nanoparticles (NPs) can mimic ganglioside-mediated binding of HIV-1 to CD169,27 and show a similar segregation in non-endolysosomal compartments in CD169+ myeloid cells as observed for HIV-1.28–31 While both metal and polymer NPs were investigated as virus-mimicking NPs in these previous studies, only the polymer core can be loaded with ARVs. Thus, GM3-presenting polymer NPs present a viable strategy to inhibit virus replication in CD169+ myeloid cells and potentially bystander CD4+ T cells, and are therefore, a focus of this work. The polymer NP platform is compatible with cART and, importantly, facilitates the incorporation of multiple ARVs in one carrier. Delivery of multiple compounds combined in one NP ensures spatial colocalization of active compounds in tissue, provides control over relative drug concentrations, and could potentially maximize their antiretroviral efficacy in vivo.
In this work, we sought to develop lipid-coated polylactic acid (PLA) and poly(lactic-co-glycolic acid) (PLGA) NPs, that incorporate both the non-nucleoside reverse transcriptase inhibitor rilpivirine (RPV)32 and the integrase inhibitor cabotegravir (CAB),33 since an injectable combination of both compounds was previously demonstrated to achieve comparable viral suppression as a conventional regimen of an integrase inhibitor with two reverse transcriptase inhibitors.34, 35 Although a combination of long-acting NP formulations of CAB and RPV has been approved for use and is now commercially available under the brand name “Cabenuva”, long-acting nanoformulations that combine both compounds in one NP, for instance for targeting potential viral reservoir sites such as CD169+ myeloid cells, are still lacking. Here, we co-formulate RPV and CAB in a GM3-presenting polymer NP platform and demonstrate that the virus-mimicking NPs retain the antiviral efficacy of RPV and CAB in CD169+ monocyte-derived macrophages (MDMs) for a duration of 35 days. Both the properties of the polymer core (PLA versus PLGA) and the GM3 targeting functionality contribute to the performance of the NP platform. Long-term antiviral potency of GM3-presenting PLA NPs is found to be correlated with sustained intracellular and extracellular ARV (RPV and CAB) concentrations in CD169+ MDMs and an intracellular localization of GM3-presenting PLA NPs in CD169+ CD9+ VCCs. Given the role of CD169+ myeloid cells in HIV dissemination, targeting of these cells with ARV-loaded GM3-presenting PLA NPs provides opportunities for increasing tissue penetration of drugs in secondary lymphoid tissue to achieve sustained inhibition of viral replication.
RESULTS AND DISCUSSION
Fabrication and Characterization of ARV-loaded Lipid-coated Polymer NPs
RPV and CAB are hydrophobic compounds with poor aqueous solubility. Our strategy to deliver RPV and CAB to CD169+ macrophages is based on a biomimetic NP design, which contains a hydrophobic polymer core that serves as matrix for the lipophilic drugs encapsulated by a GM3 containing lipid monolayer, which we refer to as “membrane” throughout this manuscript. These NPs were prepared through a one-step synthesis via nanoprecipitation of polymers in the presence of lipids.27, 36, 37 RPV and CAB were added to the polymer solution (PLA or PLGA) before the nanoprecipitation process to ensure encapsulation.36 Hydrophobic interactions between the nascent polymer core and the aliphatic tails of the lipids result in the formation of the lipid membrane around the NP core. The membrane serves multiple purposes. It disperses the hydrophobic core in an aqueous medium and ensures a surface presentation of GM3 (or other suitable lipid targeting functionalities). Furthermore, the biomimetic lipid coating can minimize non-specific cellular interactions and impede the detection of NPs by host surveillance mechanisms (stealth delivery) if primarily zwitterionic lipids are chosen for the assembly of the membrane.30 Finally, the membrane can also have a role in regulating drug release from the NP core.36
The following lipid mixture was used in the assembly of lipid-coated PLA or PLGA polymer NPs: 57 mol% 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 40 mol% cholesterol, and 3 mol% GM3. This lipid composition was chosen as a minimalistic mimic of the lipid composition of a HIV-1 particle.28 The structure and composition of the ARV-loaded lipid-coated polymer NP is schematically depicted in Figure 1A–C. To fabricate negatively charged polymer NP controls without GM3 in the membrane, the membrane composition was modified to 50 mol% DPPC, 40 mol% cholesterol, and 10 mol% 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS). Negatively charged DOPS NPs can bind to scavenger receptors and phosphatidylserine-specific Tyro3, Axl, and Mer (TAM)38 and T cell/transmembrane, immunoglobulin, and mucin (TIM)39 family of receptors on macrophages. Small amounts of fluorescently labeled phosphatidylethanolamine (PE) lipid, as 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) ammonium salt, were also added to the lipid mixtures to facilitate a detection of lipid-coated NPs via their fluorescence signal. Throughout this manuscript, we refer to RPV and CAB loaded, GM3-presenting PLGA and PLA NPs as GM3 PLGA NPs and GM3 PLA NPs, respectively. RPV and CAB loaded PLA NPs containing DOPS instead of GM3 in the membrane are referred to as DOPS PLA NPs.
Figure 1.
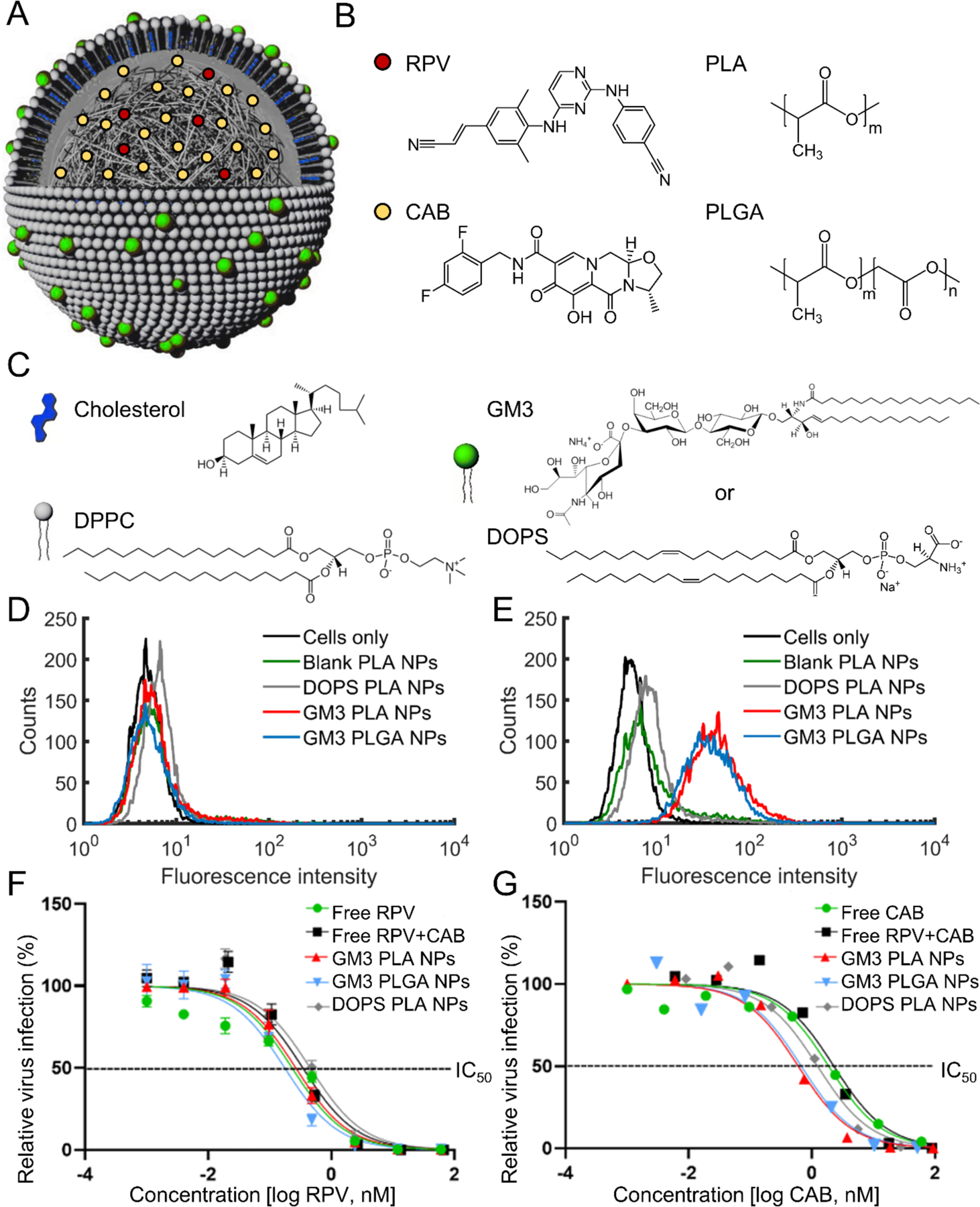
Structure, composition, and potency of ARV-loaded lipid-coated polymer NPs. A) Scheme of RPV and CAB loaded lipid-coated polymer NP structure. A lipid monolayer (“membrane”) around the NP core was formed due to hydrophobic interactions between NP polymer core and hydrocarbon chains of the lipids. The NP scheme in a is adapted from ref 24, Wiley-VCH. B) Chemical structure of the polymers in the NP core (PLA and PLGA) and drug molecules (RPV and CAB). C) Chemical structure of membrane components (DPPC, cholesterol, and GM3 or DOPS). D-E) Histograms of fluorescence intensities obtained after incubating blank PLA (with 60 mol% DPPC and 40 mol% cholesterol in the membrane but no GM3 or DOPS), DOPS PLA, GM3 PLA, or GM3 PLGA NPs for 10 min at 37 °C with THP-1 (D) or CD169+ THP-1 monocytes (E). F-G) Dose response curves of GM3 PLA NPs, DOPS PLA NPs, and GM3 PLGA NPs, and free ARVs in HIV-1 infected TZM-b1 cells over a concentration range of 0.0002 – 60 nM based on RPV concentration in (F) and CAB concentration in (G). The percentage of infection was normalized to the untreated (no ARV-exposed) cells. A non-linear regression model was used to generate the curve. Error bars represent standard error of the mean (SEM) of 3 replicates.
GM3 PLA, DOPS PLA, GM3 PLGA NPs had an average hydrodynamic diameter of 298 ± 38 nm, 255 ± 29 nm, 207 ± 38 nm as measured by dynamic light scattering (DLS) and a zeta potential of −32 ± 10 mV, −37 ± 10 mV, −29 ± 7 mV. Successful lipid coating of PLA and PLGA NPs through the fabrication strategy described in this work was recently provided through transmission electron microscopy (TEM) imaging, optical colocalization of the polymer core and the lipid coating, ganglioside quantification, differential scanning calorimetry (DSC), and biotin functionalization.27 The availability of GM3 on ARV-loaded polymer NPs was validated by specific binding of GM3 PLA and GM3 PLGA NPs, but not DOPS PLA NPs to CD169+ THP-1 monocytes (Figure 1D–E).
To determine the concentration of drugs loaded into NPs, the final NP products were dissolved in acetonitrile, and drug concentrations were then determined by HPLC with a diode array detector (DAD) following established procedures.40 The GM3-PLA NP formulations prepared in this work had average RPV and CAB concentrations of 37.5 ± 2.8 (mean ± standard error of the mean (SEM)) μg/mL and 532.1 ± 42.3 μg/mL, respectively. Similar RPV and CAB concentration were obtained for GM3 PLGA NPs (45.2 ± 1.5 μg/mL and 556.6 ± 45.3 μg/mL) and DOPS PLA NPs (50.8± 4.4 μg/mL and 402.1 ± 107.5 μg/mL). These values correspond to encapsulation efficiencies of 28.1 ± 2.1 (mean ± SEM) % for RPV and 8.0 ± 0.6 % for CAB in GM3 PLA NPs. The drug loadings were 1.5 ± 0.1 (mean ± SEM) % for RPV and 17.2 ± 1.9 % for CAB in GM3 PLA NPs. Encapsulation efficiencies and drug loadings of DOPS PLA and GM3 PLGA NPs were similar to those obtained with GM3 PLA NPs (Table S1). Overall, these values are in agreement with previously published drug loadings of polymer NPs generated by nanoprecipitation.36, 41, 42 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylterazolium bromide (MTT) viability assays confirmed no NP-associated cytotoxicity in CD169+ MDMs even at the highest dosage of NPs used in this study, corresponding to approximately 14 μM RPV and 148 μM CAB (Figure S1). The antiviral potencies of RPV and CAB loaded GM3 PLA NPs, DOPS PLA, and PLGA NPs were tested by challenging TZM-bl indicator cells with a VSV G pseudotyped single cycle HIV-1.
Cells were pre-treated with various concentrations of ARV-loaded NPs or free ARVs (soluble free drug mixture) prior to virus exposure. We observed a dose-dependent inhibition of HIV-1 infection of TZM-bl cells (Figure 1F–G). The half-maximal inhibitory concentrations (IC50) of GM3 PLA NPs, DOPS PLA NPs, GM3 PLGA NPs, free ARVs, and individual drugs are provided in Table S2. For GM3 PLA and PLGA NPs that did not contain any drugs, no inhibitory effect was detected. RPV and CAB co-formulated into the NPs yielded similar IC50 values as free RPV (0.3 ± 0.1 nM) (mean ± SEM) and CAB (1.8 ± 0.1 nM) drug mixtures, confirming that ARVs can be successfully co-encapsulated in a single NP and still retain their antiviral potency. The lack of a pronounced synergistic effect for a co-formulation of RPV and CAB (Figure S2) suggests that the lower RPV levels are rate limiting to virus infection in the TZM-bl cells used in the inhibition studies.
Quantification of RPV and CAB Release Kinetics in Lipid-coated Polymer NPs
We measured the release properties of RPV and CAB from GM3 PLA NPs in the temperature range relevant for virus inhibition and drug storage. Figure 2A–B shows the release of RPV and CAB from GM3 PLA NPs measured at 4°C, room temperature (RT), and 37 °C in 1 × PBS (Figure 2A–B) over 28 days. The percentages of RPV and CAB released at different temperatures are shown in Table S3–S4, and the corresponding remaining drug amounts in NPs are depicted in Table S5–S6. The size of the NPs was monitored throughout the duration of the experiments via DLS (Figure S3). The hydrodynamic diameter was constant at all investigated temperatures, indicative of excellent stability of GM3 PLA NPs.
Figure 2.
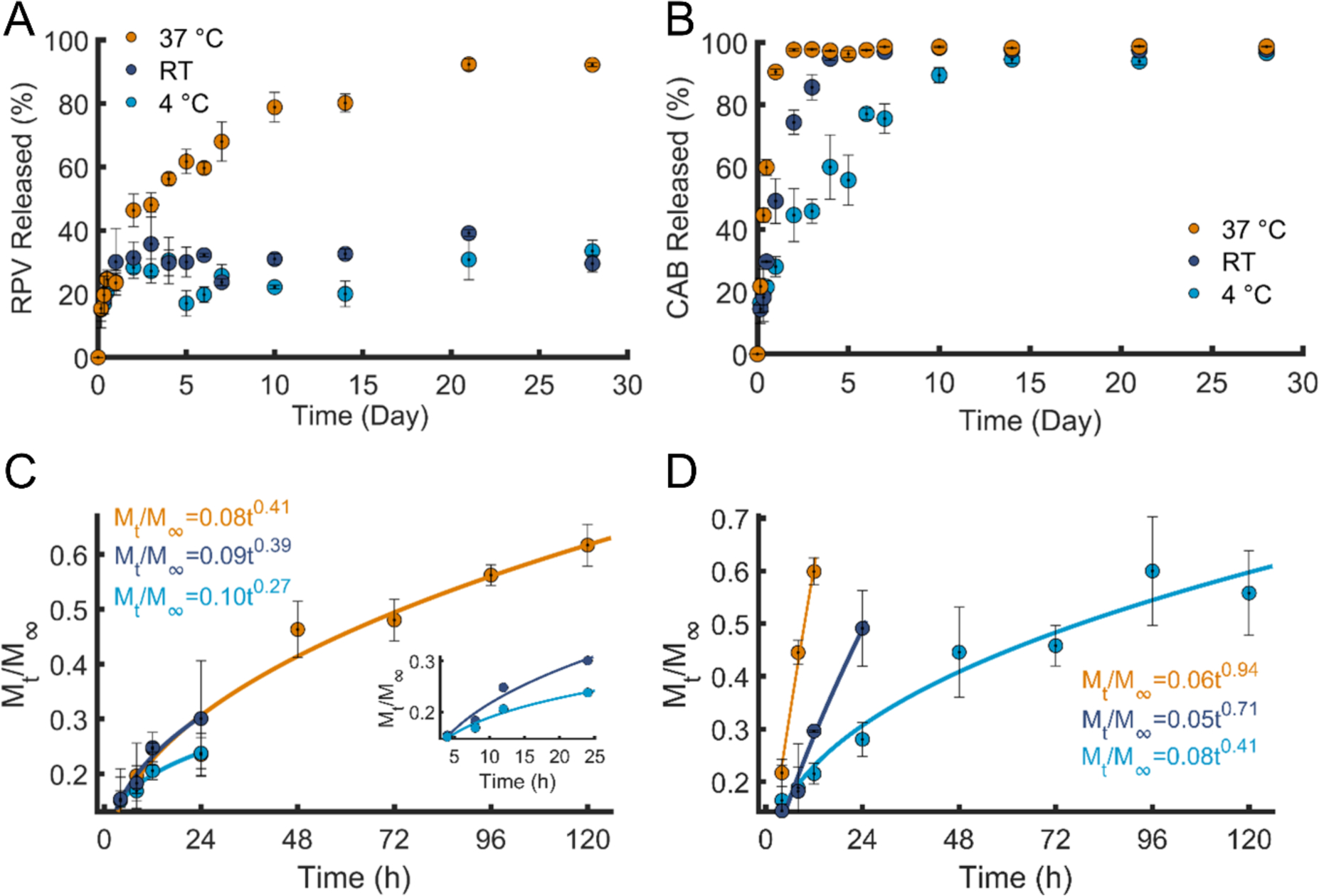
RPV and CAB release kinetics in GM3 PLA NPs. A) Percentage of the RPV released at 4°C, RT, and 37 °C in 1 × PBS over a duration of 28 days. B) Percentage of the CAB released at 4°C, RT, and 37 °C in 1 × PBS over a duration of 28 days. C) The Korsmeyer-Peppas fits and equations of the first 60% of RPV released, the insets shows the zoomed in view of RPV released at 4 °C and RT. The R2 of the corresponding fits are 0.9623 (4 °C), 0.9588 (RT), and 0.9564 (37 °C). D) The Korsmeyer-Peppas fits and equations of the first 60% of CAB released. The R2 of the corresponding fits are 0.9698 (4 °C), 0.9543 (RT), and 0.9926 (37 °C). Same colors in A and B are used in C and D. Error bars represent standard error of the mean (SEM) of 3 replicates.
In the case of RPV released from GM3 PLA NPs, a rapid burst followed by a plateau with essentially constant drug concentrations was observed at 4°C and RT, whereas at 37 °C a continuous release of RPV over a longer time was observed. The fraction of RPV released in the course of 28 days from GM3 PLA NPs increases from approximately 30 % at 4°C and 33 % at RT to 92 % at 37°C. The large difference in total drug release suggests that release occurs primarily from peripheral NP regions at 4°C and RT but that it extends to the entire NP volume at physiological temperatures. In the case of CAB, release from GM3 PLA NPs occurred at a faster rate than that observed for RPV at all temperatures. We attribute the distinct release behaviors of CAB and RPV from GM3 PLA NPs to differences in their molecular structure and properties, in particular their hydrophobicity and solubility. CAB is more hydrophilic than RPV, with calculated octanol-water partition coefficients (logP) values for RPV of 4.32 and 1.04 for CAB.43 The solubility of RPV 44 in water was reported to be 18.5 ± 1.1 μg/mL compared to 31.9 ± 14.4 μg/mL for CAB.45 The higher relative hydrophilicity and water solubility of CAB is consistent with a faster release from the hydrophobic GM3 PLA NPs than for RPV. It is important to point out, however, that although CAB is released faster than RPV, the absolute amount retained in the NPs after 28 days is still comparable to that of RPV due to a higher initial loading of CAB (Table S5–S6).
Next, RPV and CAB release data were fitted to different models, including zero- and first-order release, as well as to Higuchi and Korsmeyer-Peppas (power law) models.46, 47 The equations and R2 values of the zero- and first-order, and Higuchi fits are summarized in Table S7. The Korsmeyer-Peppas model is often applied to fit drug release from polymer NPs as it provides insight into the mechanism of release.48 In this model, the first 60% of the data are fit to equation 1:
| (1) |
In this expression is the fractional drug release, KKP is a kinetic constant characteristic of the polymer/drug system, t is the release time, and n is the diffusional exponent that characterizes the mechanism of release. Additional details on the fit parameters are provided in the Methods section.49–51 The Korsmeyer-Peppas fits of RPV and CAB at different temperatures are demonstrated in Figure 2C–D. For the RPV release from GM3 PLA NPs, we obtained n values of 0.27 (4°C), 0.39 (RT), and 0.41 (37°C) and KKP values of 0.10 (4°C), 0.09 (RT), and 0.08 (37°C) with the unit of (h −n) (Figure 2C). The measured n values indicate release by Fickian diffusion at RT and 37°C. Even at 4°C the n value is only slightly below the threshold (n = 0.30) for Fickian diffusion in a NP ensemble with finite size distribution.49–51 For CAB released from GM3 PLA NPs (Figure 2D), Korsmeyer-Peppas fits yielded n values of 0.41 (4°C), 0.71 (RT), and 0.94 (37°C) and KKP values of 0.08 (4°C), 0.05 (RT), and 0.06 (37°C) with the unit of (h −n). The n values for CAB were overall higher and showed a larger increase with temperature than for RPV. At 4 °C, the n value indicates Fickian diffusion, whereas the n values for RT and 37°C are more consistent with Non-Fickian and Case-II transport.
Similar release studies were performed for GM3 PLGA NPs, and Figure S4 provides a comparison of the RPV and CAB release curves of GM3 PLA and GM3 PLGA NPs at 37 °C. After 7 days, GM3 PLGA NPs have released 92% of the initial RPV compared with 68% in case of the GM3 PLA NPs. PLGA is in general more hydrophilic than PLA, and unlike the PLA NPs, which remain in the glassy state at 37 °C,27, 52 the PLGA NPs used in this work undergo a phase transition into the rubbery state under physiological conditions.27, 52 The higher hydrophilicity, the phase change into the rubbery state, and a faster degradation53–55 can all contribute to a faster release of RPV from GM3 PLGA NPs than from GM3 PLA NPs (Figure S4A). In the case of CAB at 37 °C, release from GM3 PLGA NPs is initially slower than for GM3 PLA NPs, however these differences diminish quickly with time. On day 2 both NPs have released over 94% of the total CAB, and on day 7 both NPs have released over 98% (Figure 4SB).
Long-term Inhibition of HIV-1 Infection in CD169+ MDMs
While CD169 is constitutively expressed on tissue-resident macrophages,56 its expression is low in MDMs. Since CD169 is a type I and type III interferon inducible protein,16 we induced CD169 expression on MDMs by exposure to type III IFN (IFN-λ, 5 ng/ml) for 48 h (Figure S5). Note that at these concentrations, IFN-λ displayed no antiviral effects (data not shown). We explored the long-term antiviral potential of RPV and CAB loaded NPs in CD169+ MDMs. Different NP conditions, including ARV-loaded GM3 PLA and PLGA NPs, as well as DOPS PLA NPs were compared, and a mix of free ARVs was added as control. CD169+ MDMs were incubated with NPs or free ARVs (ARV input normalized to 1 μM of RPV equivalent) for 3 h only once at the beginning of the viral inhibition study, extensively washed and returned to culture. Growth medium was changed every 3 days, and drug-exposed CD169+ MDMs were challenged weekly with a VSV G pseudotyped HIV-1 (Figure 3). As the TZM-bl infection assay indicated RPV as the rate-limiting compound of virus inhibition, the RPV concentration was kept constant between experiments using different NP preparations, resulting in varied CAB input concentrations between 6 – 33 μM. Note, no to low NP-associated cytotoxicity was observed for a duration of up to 35 days, as monitored via MTT assays (Figure S6).
Figure 3.
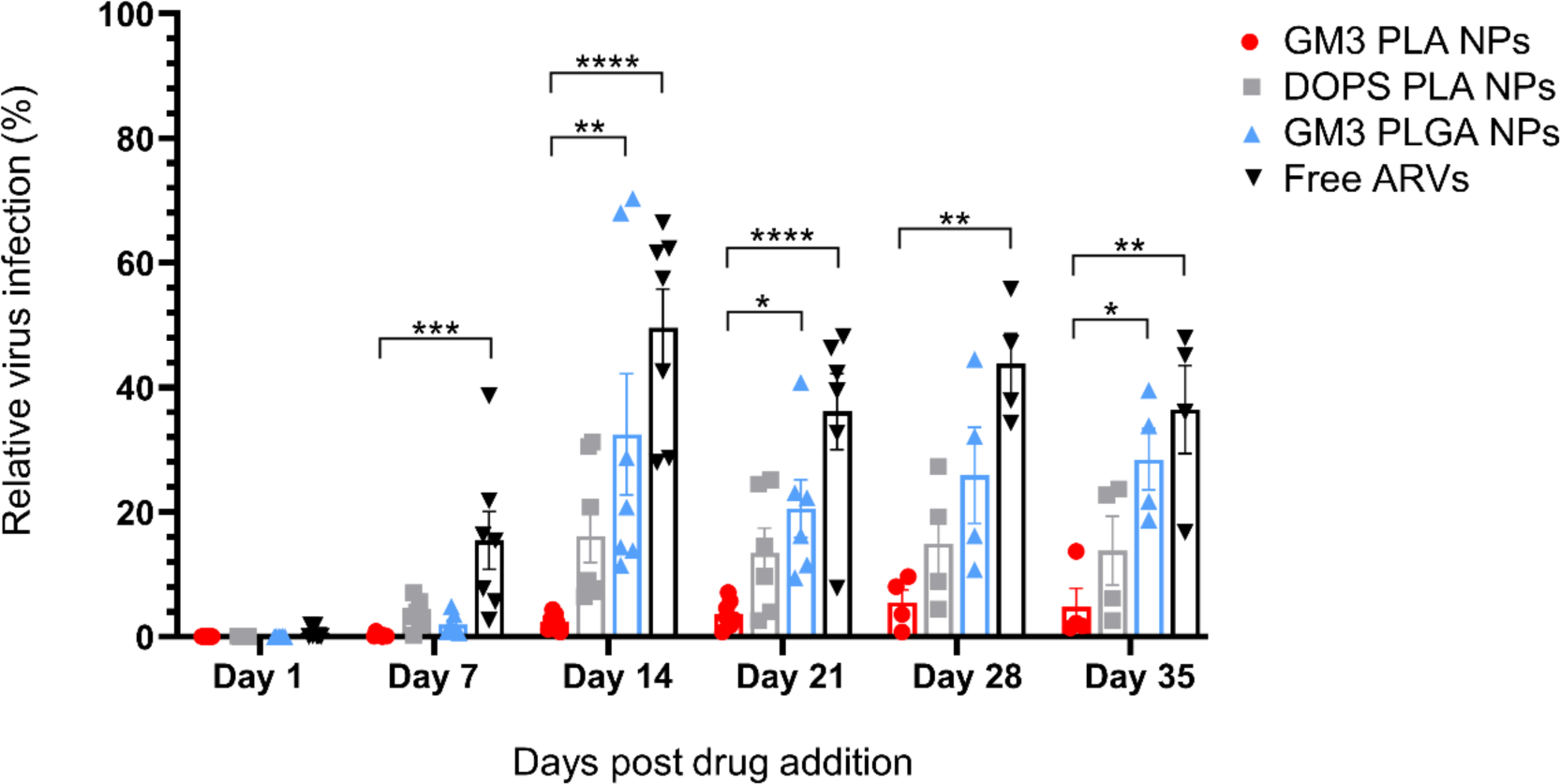
Long-term HIV-1 inhibition of RPV and CAB loaded NPs in CD169+ MDMs. CD169+ MDMs were pre-treated with GM3 PLA NPs, DOPS PLA NPs, GM3 PLGA NPs, or free ARVs at a fixed initial 1 μM RPV concentration for 3 h. Infections were carried with a pseudotyped HIV-1 virus on day 1, 7, 14, 21, 28, and 35 post drug addition. Infection data was collected 3 days post infection. The percentage of infection is normalized to untreated cells. Each data point represents CD169+ MDMs from a different donor, and CD169+ MDMs derived from at least 4 donors were used. Error bars represent SEMs. Results were analyzed by one-way ANOVA with Tukey multiple comparisons test, *P ≤ 0.05 **P ≤ 0.01 ***P ≤ 0.001 ****P ≤ 0.0001.
Complete inhibition of virus infection was achieved in CD169+ MDMs exposed to ARV-loaded NPs or soluble drugs on day 1 post ARV treatment, though by day 7, free ARVs were significantly attenuated in their antiviral effect compared to polymer NPs.
The inhibition achieved with free ARVs decreased with time and remained lower than for all the NP conditions during the entire experimental period. Interestingly, from day 14 onwards, the inhibitory effects of different polymer NP formulations began to differ. Importantly, GM3 PLA NPs maintained the highest antiviral effect in CD169+ MDMs throughout the duration of the experiment with an extremely low level of relative HIV-1 infection (4.7 ± 3.0%) (mean ± SEM) even at day 35 post NP addition. In contrast, higher levels of virus infection were observed in DOPS PLA and GM3 PLGA NPs treated CD169+ MDMs (13.8 ± 5.5% and 28.5 ± 4.9%, respectively), on day 35 post NP addition. The striking differences in the duration of the viral inhibition obtained with free and nanoformulated RPV and CAB as well as between the different RPV+CAB containing NPs confirm that nanoformulation of ARVs extends the inhibitory effect of ARVs over time.
Determination of RPV and CAB Concentrations in CD169+ MDMs
The distinct inhibition durations for the different nanoformulations might be due to differences in total drug uptake and/or metabolization of the drugs inside of the cells. An accurate assessment of the contributions of these two effects requires a precise quantification of the cellular drug content as a function of time for the different conditions. To allow for a precise quantification of cellular ARV concentrations for a total duration of at least four weeks by HPLC analysis, the RPV input concentrations and the number of cells were increased by a factor of 10 relative to the inhibition studies. The RPV concentration was, consequently, 10 μM with CAB concentrations varying between 50 to 165 μM for different preparations. CD169+ MDMs from 3 different donors were incubated with NPs and free ARVs for 3 h before the cells were washed and maintained in fresh medium. The total drug contents (NP-associated and free drugs inside the cells) were measured on day 0, 1, 3, 5, 10, 15, 20, and 28 post NP addition (Figure 4A–B).
Figure 4.
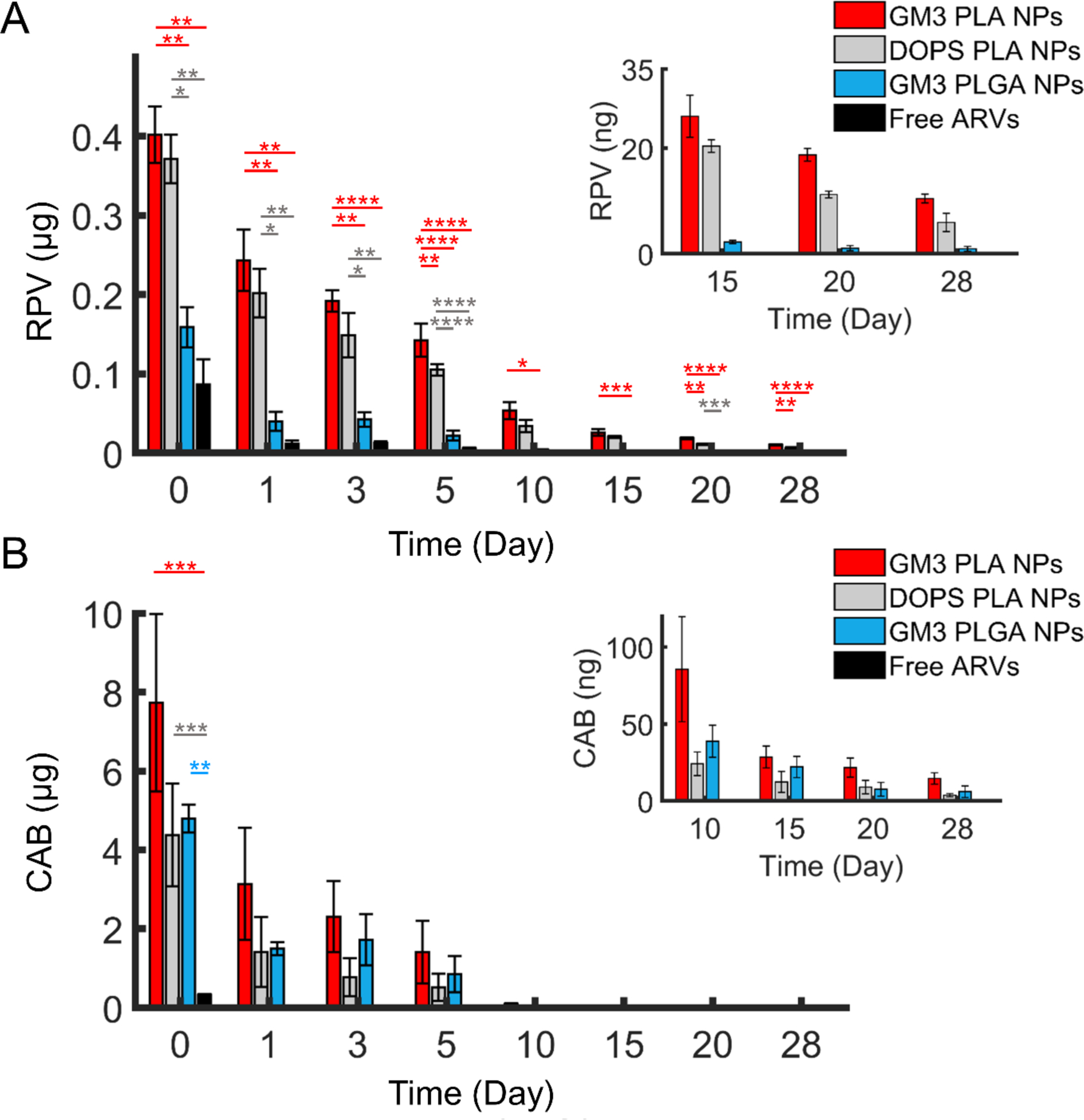
Quantification of RPV and CAB concentrations in CD169+ MDMs. A) RPV concentration in CD169+ MMDs over 28 days after 3 h incubation with GM3 PLA NPs, DOPS PLA NPs, GM3 PLGA NPs, or free ARVs with a fixed initial RPV concentration of 10 μM per 5 × 105 cells. The inset shows the RPV concentration after day 15. B) CAB concentration in CD169+ MDMs over 28 days in the same cells as in a The initial CAB concentrations were between 45 to 164 μM for different preparations. The inset shows the CAB concentration after day 10. The error bars represents the SEM of 3 different replicates performed with CD169+ MDMs derived from 3 independent donors. Statistical p-values determined using one-way ANOVA followed by a Tukey post-hoc test, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
The total internalized RPV contents in 5 × 105 cells at time 0 for GM3 PLA NPs, DOPS PLA NPs, GM3 PLGA NPs, and free ARVs are 401.5 ± 35.7 (mean ± SEM) ng, 371.0 ± 30.7 ng, 158.7 ± 25.3 ng, and 86.4 ± 32.0 ng, respectively. These results confirm that nanoformulation achieves an increased uptake of RPV in CD169+ MDMs and that for NPs, RPV uptake increases in the order of GM3 PLA ≥ DOPS PLA > GM3 PLGA.
The uptake of RPV obtained with GM3 PLA NPs was slightly higher than for DOPS PLA NPs, which could be due to a difference in the binding affinities of the two different ligands, GM3 and DOPS, presented on the respective NPs. However, RPV uptake for GM3 PLGA NPs was much lower than that observed with GM3 PLA NPs, despite the same GM3-CD169 binding mechanism. One possible explanation for these trends is provided by the difference in the glass transition temperature of the PLA and PLGA NPs used in this work and the related higher mechanical stiffness of PLA NPs at 37 °C.27, 52 Under otherwise identical conditions, stiff NPs are preferentially uptaken over soft NPs with comparable size.27, 57–60
The RPV concentration decreased as function of time for all experimental conditions, and by day 10 RPV was no longer detectable in CD169+ MDMs incubated with free ARVs. In contrast, RPV administered as a nanoformulation was detectable throughout the duration of the experiment for all NP conditions. After 28 days, the total RPV contents in CD169+ MDMs are 10.4 ± 0.8 (mean ± SEM) ng in GM3 PLA NPs, 5.9 ± 1.7 ng in DOPS PLA NPs, and 0.9 ± 0.5 ng in GM3 PLGA NPs.
The concentrations of CAB in CD169+ MDMs exceeded those of RPV due to higher initial CAB incorporation in polymer NPs (Figure 4B). The delivered CAB contents in 5 × 105 cells on day 0 are: 7.7 ± 2.3 (mean ± SEM) μg for GM3 PLA NPs, 4.4 ± 1.3 μg for DOPS PLA NPs, 4.8 ± 0.4 μg for GM3 PLGA NPs, and 319.5 ± 27.0 ng for the free ARVs. For all experiments, the NP input concentration was adjusted to keep the RPV input concentration constant across the experiments. Therefore, the error in the CAB concentrations was higher than for RPV. Though the resulting variability in the initial input concentration of CAB for the different experimental conditions precludes a systematic analysis of the relative intracellular concentrations of CAB achieved upon incubation with GM3 PLA, DOPS PLA or GM3 PLGA NPs, uptake of nanoformulated CAB was more efficient than that observed upon exposure of CD169+ MDMs to free ARVs. Consistent with a faster release of CAB from NPs (see Figure 2B), the rate of decrease of cellular CAB concentration was faster than that observed with RPV. However, due to the high initial loading, CAB was still detected even after 28 days for all experimental conditions. The total cellular CAB contents for the different NPs at day 28 are 14.6 ± 3.7 (mean ± SEM) ng for GM3 PLA NPs, 3.8 ± 1.0 ng for DOPS PLA NPs, and 6.0 ± 3.8 ng for GM3 PLGA NPs. As in the case of RPV, CAB was no longer detected on day 10 when administered as free ARVs.
In addition to cellular drug concentrations, RPV and CAB concentrations released from CD169+ MDMs into the medium were also quantified (Figure S7). The drug concentrations in extracellular medium show a decrease as a function of time that mirrors the same overall trend as observed with cell lysates. However, due to the higher dilution in medium, RPV and CAB were no longer detectable after day 20. At day 20, presence of RPV in medium was only detected for GM3 PLA NPs, while CAB is detected in medium of all NPs but with the highest concentration for GM3 PLA NP-exposed CD169+ MDMs.
Overall, the drug quantification reveals that GM3 PLA NPs achieve the highest concentrations of both RPV and CAB over the duration of the experiment in both cells and medium, underlining the potential of GM3 PLA NPs as long-acting nanocarriers for co-formulated RPV and CAB.
Confocal Mapping of RPV and CAB Loaded Lipid-coated Polymer NPs in CD169+ MDMs
CD169+ MDMs from different donors were imaged on day 1, 10, 15, 20, and 25 after treatment with ARV-loaded GM3 PLA, DOPS PLA, and GM3 PLGA NPs. Confocal z-stack images,61 corresponding to day 10 and 25 are shown in Figure 5A–B, and z-stacks recorded on days 1, 15 and 20 are depicted in Figure S8–S9. Especially at early time points, the z-stack images indicate a GM3 PLA NPs localization closer to the top cell plasma membrane, while DOPS PLA and GM3 PLGA NPs are preferentially located closer to the center of the cell (Figure 5A–B and Figure S8–S9). The localization of GM3 PLA NPs closer to the top surface of the cells is consistent with a collection of NPs in VCCs, as previous studies have demonstrated that VCCs are located closer to the cell surface.62, 63
Figure 5.
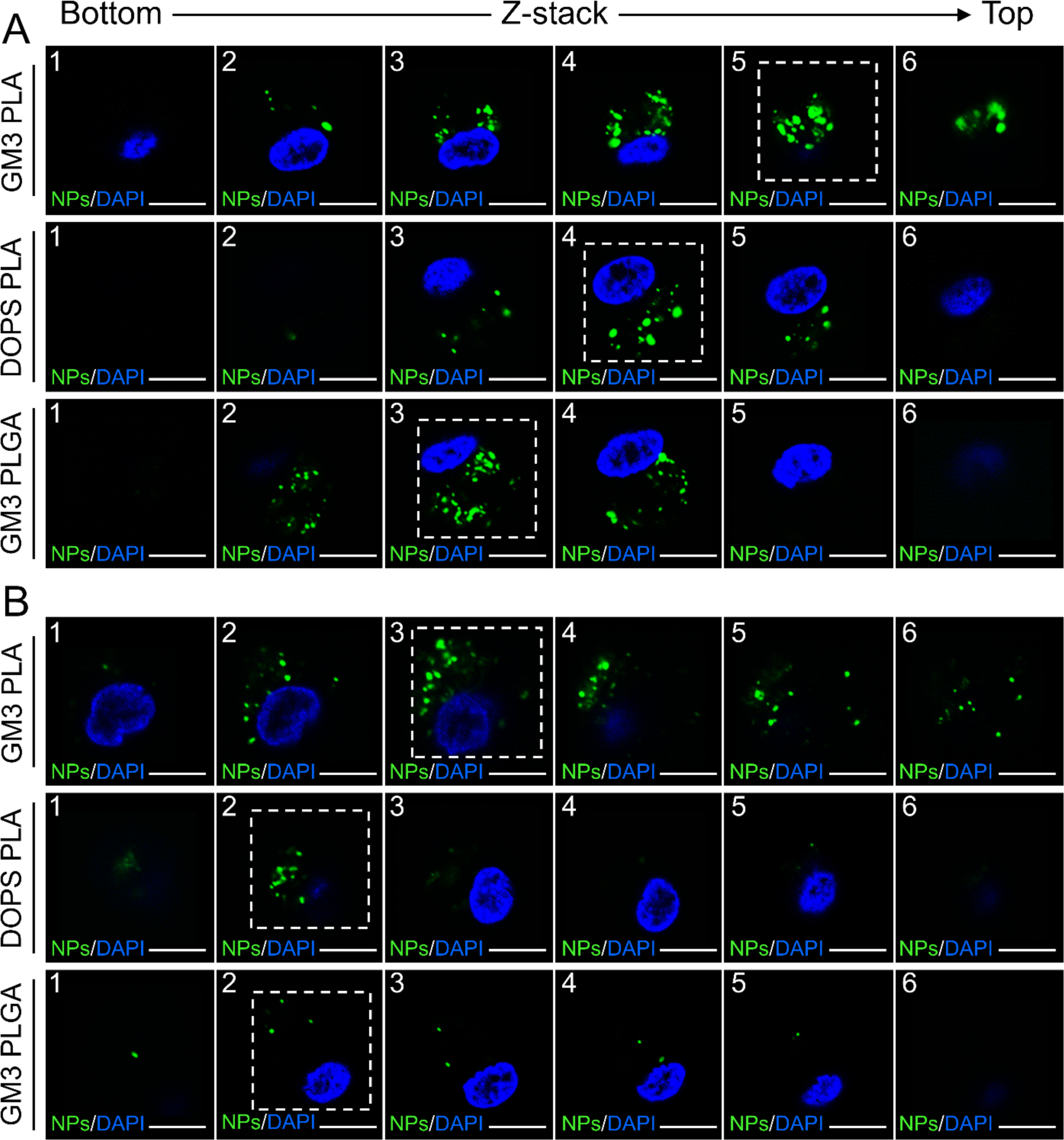
Localization of GM3 PLA, DOPS PLA, and GM3 PLGA NPs in regard to cell membrane of CD169+ MDMs. A) Confocal z-stack images of CD169+ MDMs 10 days post NP treatment. B) Confocal z-stack images of CD169+ MDMs 25 days post NP treatment. The dashed square is used to demonstrate the section with the highest NP-contained area. Z-stack images were collected at 3 μm steps. Scale bar = 10 μm.
Intriguingly, starting from day 15 the fluorescence intensities of DOPS PLA or GM3 PLGA NPs in the cells appear lower, whereas the degradation is less conspicuous for GM3 PLA NPs. In a THP-1 CD169+ macrophage model, it was previously observed that the intracellular fate of GM3 PLA NPs, DOPS PLA, and GM3 PLGA NPs, all with identical physico-chemical properties as used in this work, can differ.27 While DOPS PLA or GM3 PLGA NPs entered an endolysosomal pathway in CD169+ THP cells, a significant fraction of GM3 PLA NPs were found to localize outside of the lysosomes in VCC-like compartments. These distinct intracellular fates were attributed to binding via different receptors in the case of DOPS PLA and GM3 PLA NPs and to differences in the glass transition temperature and stiffness of the core in the case of GM3 PLGA and GM3 PLA NPs. It is conceivable that differences in the cellular processing contribute to differences in NP retention in CD169+ MDMs. To illustrate the intracellular fate of GM3 PLA NPs in CD169+ MDMs by confocal microscopy, GM3 PLA NPs treated CD169+ MDMs were immunolabelled for CD169, as well as for tetraspanin CD9 as a VCC marker,64 and for lysosomal-associated membrane protein 1 (LAMP-1) as a lysosome marker on days 1, 5, 10, 15, 20, and 25 (Figure 6). The optical colocalization of GM3 PLA NPs with CD169, CD9, and LAMP-1 was quantified by calculating Manders’ colocalization coefficients (MCCs)65 (Table S8). The optical colocalization analysis confirms an initial co-localization of GM3 PLA NPs with CD169 and CD9 but not with LAMP-1, which corroborates a localization of GM3 PLA NPs within non-endolysosomal, VCC-like compartments.
Figure 6.
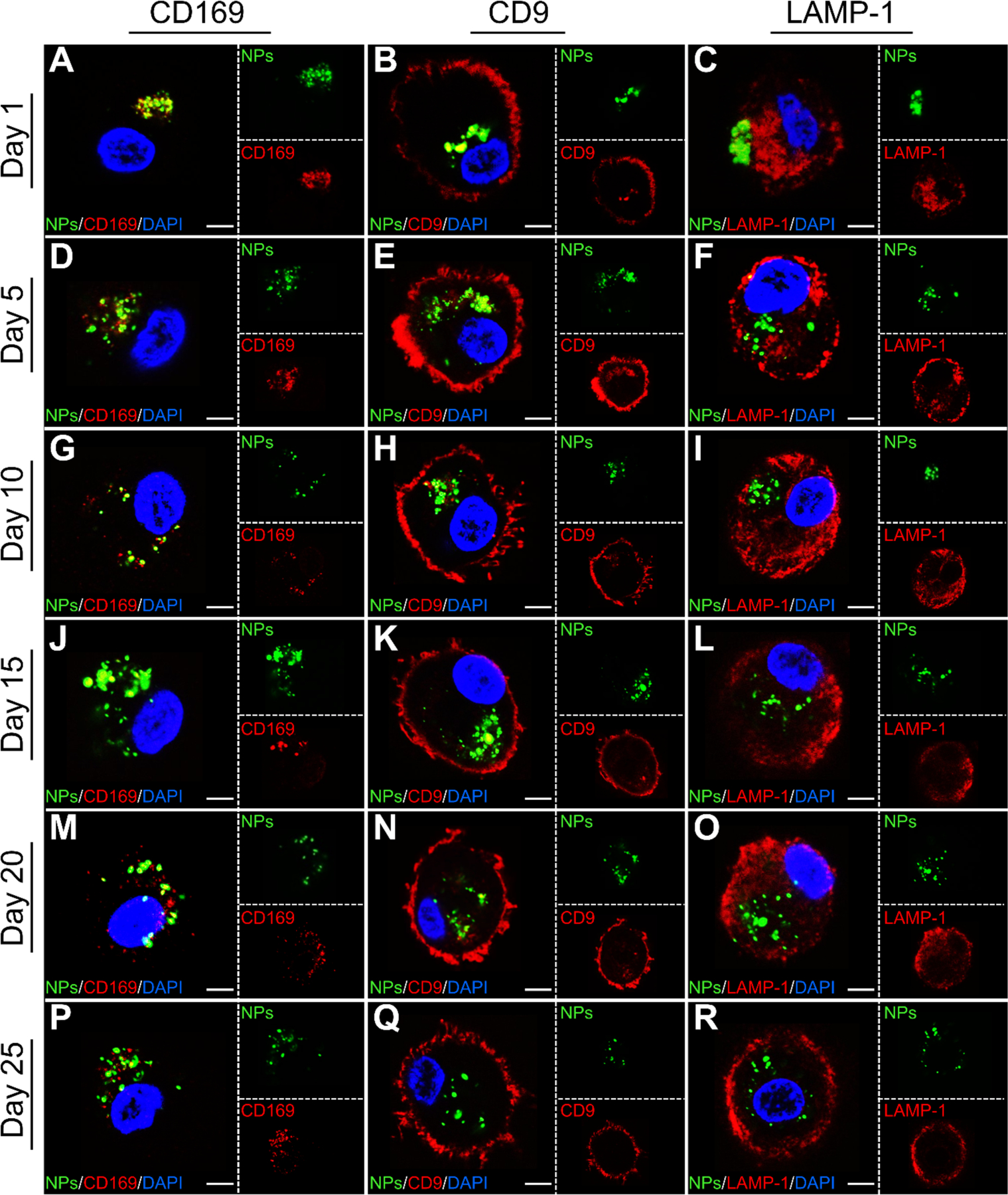
Characterization of intracellular fate of GM3 PLA NPs in CD169+ MDMs. Single confocal sections of CD169+ MDMs incubated with GM3 PLA NPs stained for CD169, CD9, and LAMP-1 on day 1 (A-C), 5 (D-F), 10 (G-I), 15 (J-L), 20 (M-O), and 25 (P-R). Scale bar = 5 μm.
In the case of negatively charged DOPS PLA NPs, the NPs were primarily located in lysosomes. A confocal section of CD169+ MDMs incubated with DOPS PLA NPs and stained for LAMP-1 on day 1 is shown in Figure S10. In contrast, GM3 PLGA NPs displayed variable co-localization with LAMP-1 in CD169+ MDMs with some donor-dependent variability (Figure S11).
Importantly, the avoidance of an endolysosomal trafficking pathway for GM3 PLA NPs in CD169+ MDMs is consistent with a slower degradation of these NPs and may contribute to the enhanced antiviral efficacy of this formulation observed at later time points in Figure 3.
CONCLUSION
In this study, we have demonstrated the successful co-encapsulation of RPV and CAB in HIV-1 mimicking GM3-presenting lipid-coated PLA and PLGA NPs as well as in DOPS-presenting PLA NPs. The PLA core in combination with GM3 as cell-targeting moiety showed overall the most advantageous delivery and release of RPV and CAB in CD169+ MDMs. Importantly, GM3 PLA NPs achieved detectable RPV and CAB concentrations in CD169+ MDMs for up to at least 28 days, and inhibited HIV-1 infection in CD169+ MDMs for up to 35 days post challenge with GM3 PLA NPs.
RPV and CAB loaded GM3 PLA, DOPS PLA, and GM3 PLGA NPs retained their potency upon encapsulation as demonstrated by similar IC50 values of NPs and free ARVs. Given the rapid release in the case of CAB, the similarity in IC50 between nanoformulation and free drug may not be surprising. However, in the case of the more slowly releasing RPV, similar IC50 values of nanoformulation and free drug suggest an improved intracellular ARV delivery for the nanoformulations, which was corroborated through quantification of the cellular ARV contents. Intriguingly, RPV and CAB loaded GM3 PLA, DOPS PLA, and GM3 PLGA NPs differed in their ability to achieve sustained long-term virus inhibition. RPV and CAB loaded GM3 PLA NPs achieved a nearly complete inhibition of HIV-1 infection in CD169+ MDMs for at least 35 days, but DOPS PLA NPs and GM3 PLGA NPs did not offer the same level of protection and the efficacy dropped in the order GM3 PLA > DOPS PLA > GM3 PLGA NPs > free ARVs. The relative efficiency of virus inhibition correlates with the intracellular ARV concentrations in CD169+ MDMs, which also decreased in the order GM3 PLA > DOPS PLA > GM3 PLGA> free ARVs.
The differences in the uptake and intracellular fate among NPs with different polymer core (PLA versus PLGA) and lipid membrane composition (GM3 versus DOPS) emphasize that the choice of polymer and membrane composition are important parameters for the design of virus-mimicking NPs for ARV delivery. The higher uptake and relative stability obtained with GM3 PLA NPs makes these NPs particularly attractive for engineering sustained drug release formulations and could be one of the factors that contribute to the higher antiviral efficacy of these NPs on longer time scales. Importantly, GM3 PLA NPs achieved a sustained release of RPV and CAB without the need for any chemical modifications of the drugs.
The superior performance observed for GM3 PLA NPs in our cell culture studies is expected to be even more relevant in vivo, where the GM3-CD169-mediated targeting of myeloid cells provides a viable strategy to enrich high local concentrations of both drugs in secondary lymphoid tissues. We postulate that virus-mimicking GM3 PLA NPs co-loaded with multiple ARVs can provide a new paradigm for targeted delivery of ARVs to CD169+ macrophages to enhance efficacy and sustained long-term antiviral potency and provide rationale for further development to deliver combination ARVs to tissue-resident reservoir sites of virus persistence. Additionally, future studies are warranted to test the in vivo efficacy of GM3 PLA NPs in animal models of chronic HIV infection and establish injectable ARV-loaded GM3 PLA NPs as a viable alternative to daily suppressive cART.
MATERIALS AND METHODS
Fabrication of ARV-loaded Lipid-coated Polymer NPs.
Drug loaded lipid-coated polymer NPs were fabricated via one-step nanoprecipitation procedure as described previously.27 0.15 mg of lipid mixture containing 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) (25 mg/mL), cholesterol (25 mg/mL), and GM3 Ganglioside (Milk, Bovine-Ammonium Salt) (2 mg/mL) in chloroform with mol % as specified in the text was added to the sterile, cell culture grade water (Gibco Distilled Water, Thermo Fisher Scientific, Waltham, MA). To prepare negatively charged lipid-coated polymer NPs, 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (sodium salt) (DOPS) (10 mg/mL) was added to the lipid mixture. The fluorescence marker 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) was added to the lipid mixture when NPs were subjected to the fluorescence microscopy or flow cytometry. 20 μg of RPV in acetonitrile (stock solution of 100 μg/mL) and 1 mg of CAB in dimethyl sulfoxide (DMSO) (stock solution of 10 mg/mL) were mixed with 1 mg polymer (2.5 mg/mL) in acetonitrile prior to nanoprecipitation. The best loading in polymer NPs with CAB+RPV is obtained using a constant 1:1 weight ratio of CAB : polymer at a RPV input corresponding to 2% of the polymer weight (Figure S12). A decrease in the CAB or further increase in the RPV input concentration resulted in a strong decrease of CAB loading. Next, using a lipid/polymer weight ratio of 15%, drug-included PLA (Poly(D,L-lactide) ester terminated, Resomer R207S, R207S with viscosity of 1.3–1.7 dL/g and molecular weight of 209,000)66 and PLGA (Poly(D,L-lactide-co-glycolide) lactide:glycolide 50:50, ester terminated, Mw 24,000–38,000, Resomer RG503) solution in acetonitrile was added dropwise to the aqueous solution. The final mixture was sonicated in a bath sonicator (Branson 5510, Branson Ultrasonics, Danbury, CT) for 5 min. Next, the drug loaded lipid-coated polymer NPs were purified via 3 washing cycles (4000g – 15 min) using an Amicon Ultra-4 centrifugal filter (MilliporeSigam, Burlington, MA) with a molecular weight cutoff of 10 kDa to remove organic solvent and free drug and lipid molecules. Polymers (PLGA and PLA), chloroform, acetonitrile, and DMSO were purchased from Sigma-Aldrich (St Louis, MO). Lipids were purchased from Avanti Polar Lipids (Alabaster, AL). Cabotegravir (GSK1265744) was purchased from Selleckchem (Houston, TX). Rilpivirine was obtained from the NIH AIDS Reagent Program.
Dynamic Light Scattering (DLS) and Zeta Potential Measurements.
Size and zeta potential measurements were determined by using Zetasizer Nano ZS90 (Malvern, Worcestershire, UK). To measure the size of NPs, Milli-Q water was used to dilute the NPs. 10 mM NaCl solution (pH 5) was used to determine the zeta potential of GM3 PLA, DOPS PLA, and GM3 PLGA NPs.
Determination of NP Concentrations through UV-Vis.
To determine the concentration of fluorescently labeled lipid-coated polymer NPs, their absorption spectra in Milli-Q water were recorded using a Spectronic 200 UV-Vis spectrometer (Fisher Scientific, Waltham, MA). Beer’s law using a molar absorptivity of ε = 73,000 M−1 cm−1 at the wavelength of 570 nm (Lambda max)27 was used to calculate the NP concentrations, and Milli-Q water was used for baseline correction.
Quantification of RPV and CAB Concentrations.
To determine the amount of the drugs (RPV and CAB) encapsulated in polymer NPs, NPs were dissolved in the acetonitrile, and the resulting free drugs form NPs were measured using an Agilent HPLC with a diode array detector (DAD) equipped with a Gemini C18 reversed-phase column (Phenomenex, Torrance, CA) with 5μm particle size, 4.5 mm internal diameter and 150 mm length.40 The temperature of the column oven was set at 35 °C. The mobile phase consisted of a 50/50 (v/v) ratio of 25 mM potassium dihydrogen phosphate (Sigma-Aldrich, St Louis, MO) in water and acetonitrile. The isocratic mode with the flow rate of 0.6 mL/min was used, with the total run time of 20 min. Atazanavir sulfate (ATZ) was used as the internal standard (IS) in all the HPLC measurements and was obtained from the NIH AIDS Reagent Program. CAB, RPV, and ATZ were detected at the wavelength detection of 290 nm and at average retention times of 5, 13, and 15 min, respectively. Calibration curve solutions were run with the samples for each time run of HPLC. Stock solutions of RPV (100 μg/mL), CAB (100 μg/mL), and ATZ (IS) (100 μg/mL) were prepared in acetonitrile. To prepare the calibration solutions, RPV and CAB stock solution was diluted with acetonitrile to achieve concentration of 125, 250, 500, 750, 1000, 1500, 2000 and 4000 ng/mL, whereas IS had a final concentration of 25 μg/mL. The encapsulation efficiency and drug loading percentages were determined using the following formulas:
| (2) |
| (3) |
RPV and CAB Release Kinetics Determination.
To measure the drug release profile, RPV+CAB encapsulated NPs were placed into Slide-A-Lyzer MINI dialysis microtubes with a molecular weight cutoff of 3500 Da (Thermo Fisher Scientific, Waltham, MA). The microtubes were dialyzed in 4 L of 1 × PBS buffer at 4, RT, and 37 °C. The PBS buffer was exchanged every 24 h during the first 10 days of the whole dialysis process, and then every 2 days till day 28. At each data point, NP solution from the microtubes were collected and mixed with acetonitrile to dissolve the NPs. The free RPV and CAB were determined via HPLC analysis as described above, and with addition of ATZ (IS) to the samples.
The release data were fitted using zero- and first-order, Higuchi, and Korsmeyer-Peppas (power law) models. The details of zero- and first-order, Higuchi fits and formulas can be found in the supporting information. For the Korsmeyer-Peppas model, the first 60% of the data were fit to equation 1, which contains n as the diffusional exponent that characterizes the mechanism of release. For ideal spherical polymer particles of a constant size made out of swellable polymers/devices, a n value of 0.43 indicates Fickian diffusion, n values in the range of 0.43 < n < 0.85 correspond to anomalous or non-Fickian diffusion, n = 0.85 is representative of Case-II transport, and n = 1 represents time-independent, zero-order release.51 For NP ensembles with different size distribution, the threshold n values need to be adjusted to 0.3 for Fickian diffusion and n = 0.45 for Case-II transport.51 Additional details of zero- and first-order, and Higuchi fits can be found in the Supplementary Information.
Cell Culture.
HEK293T (ATCC) and TZM-bl (NIH AIDS Reagent Program) were maintained in DMEM (Gibco) containing 10% heat-inactivated FBS (Gibco) and 1% pen/strep (Gibco).22, 67, 68 THP-1 (NIH AIDS Reagent Program) and CD169+ THP-1 cells69 were maintained in RPMI-1640 (Gibco) containing 10% FBS (Gibco, Catalog number: 16000044) and 1% pen/strep (Gibco, Catalog number: 15070063) and 2 mg/mL G418 for maintenance of CD169 expression. All cell lines have been tested for mycoplasma contamination and confirmed negative. Human MDMs were derived from positively isolated CD14+ peripheral blood monocytes by culturing in RPMI-1640 containing 10% heat-inactivated human AB serum (Sigma-Aldrich) and recombinant human macrophage colony stimulating factor (M-CSF) (20 ng/mL; PeproTech) for 5 days, as described previously.69 To induce CD169 expression on MDMs, cells were treated with IFN-λ (5 ng/ml) for 48 h, and cell surface expression of CD169 was confirmed by FACS using a Alexa 647-conjugated mouse anti-CD169 antibody (Biolegend). Cells were analyzed with BD LSRII (BD), and data was analyzed with FlowJo software (FlowJo).
Viruses.
Single-round-replication-competent HIV-1 pseudotyped with VSV-G, either expressing luciferase or not were generated from HEK293T cells via co-transfection of HIV-1Δenv/luc or HIV-1Δenv proviral plasmids and VSV-G expression plasmid.22 Virus-containing cell supernatants were harvested 2 days post-transfection, cleared of cell debris by centrifugation (300 × g, 5 min), passed through 0.45 μm filters, and concentrated by ultracentrifugation on a 20% sucrose cushion (24,000 rpm and 4°C for 2 h with a SW28 rotor (Beckman Coulter)). The virus pellets were resuspended in PBS, aliquoted and stored at −80 °C until use. The capsid content of HIV-1 was determined by a p24gag ELISA 22 and virus titer was measured on TZM-bl cells as previously described.70 In detail, single-cycle vesicular stomatitis virus G (VSV-G)-pseudotyped Bru Δenv virus particles were produced by co-transfection of HIV-1 env-deficient luciferase or GFP expressing proviral plasmid with a VSV-G expression plasmid (H-CMVG). Virus particles were generated by calcium phosphate-mediated transfections of HEK293T cells, as described previously.71 The p24gag content of all the virus stocks was quantitated by a previously described enzyme-linked immunosorbent assay (ELISA),72 with slight modifications. Briefly, p24gag was bound to HIV immunoglobulin (from NABI and National Heart Lung and Blood Institute)-coated wells and detected with an anti-p24gag monoclonal antibody (clone 183-H12-5C from the NIH AIDS Research and Reference Reagent Program, contributed by Bruce Chesebro) and horseradish peroxide (HRP)-conjugated goat anti-mouse secondary antibody (Sigma). To determine the infectious titer of the viruses, 104 TZM-Bl cells were infected with limiting dilutions of virus stocks. Cells were fixed and stained for β-galactosidase (β-Gal) activity 48 h post infection to quantify β-Gal+ (blue) cells as a measure of the virus infectious titer, as previously described.73
Specific Binding of GM3-presenting NPs and CD169 in THP-1 Cells.
THP-1 and CD169+ THP-1 cells (5 ×105) were exposed to 5 × 1011 ARV-loaded GM3 or DOPS or blank (without any GM3 or DOPS) PLA or GM3 PLGA NPs in RPMI/10% FBS. The mixture was incubated at 37 °C for 10 min. Next, unbound NPs were washed twice by PBS and cells pelleted by centrifugation at 270 g for 5 min. Cells were fixed with 4% PFA (Sigma-Aldrich, St Louis, MO), and capture of NPs by cells determined by flow cytometry using a FACSCalibur instrument (BD Biosciences, San Jose, CA) and analyzed through Flowing software 2.
Determination of IC50 in TZM-bl Cells.
Serial 5-fold dilutions of soluble drugs or polymer NPs (over a concentration range of 0.0002 – 60 nM) were added to TZM-Bl cells (104 cells/well) in a 96 well plate for 1 h prior to infection with VSV-G pseudotyped HIV-1 (LaiΔenv/VSV-G, MOI 0.1). Cells were spinoculated at 2300 rpm at 25°C for 1 h, and returned to culture for an additional 3 days. Cells were lysed with Glo lysis buffer (Promega), and luciferase expression in cell lysates was determined using the Bright-Glo luciferase expression system (Promega) as described previously.67 Nonlinear regression was used to fit the data with a constant Hill coefficient to determine the half-maximal inhibitory concentrations (IC50).
Cell Viability 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenylterazolium bromide (MTT) Assays.
MDMs were plated in a 12-well plate at a concentration of 5 × 105 cells per well. CD169+ MDMs were incubated with ARV-loaded GM3 PLA, DOPS PLA, and GM3 PLGA NPs at a concentration of 1 × 1012 NPs/mL, corresponding to approximately 14 μM RPV and 148 μM CAB for 4 h. After removing the NPs, cells were maintained in the full medium for 5 days. Then cells were washed with 1 × PBS and incubated with RPMI containing 10% MTT solution (5mg/mL) for 1 h at 37°C. After removal of the MTT solution, DMSO and ethanol solution (1:1 ratio) was added and mixed thoroughly. Absorbance was measured at 570 nm using a SpectraMax M5 plate reader (WVR, Radnor Corporate Center, Radnor, PA).
Inhibition of HIV-1 Infection in CD169+ MDMs.
CD169+ MDMs were seeded in a 96-well plate at 5 × 104 cells per well. Soluble drugs or ARV-loaded polymer NPs were added to cells at 1μM RPV equivalent concentration (CAB concentration varied between 6 to 33 μM) and incubated at 37°C for 3 h. Cells were washed extensively, and cultured in fresh medium (RPMI/10% FBS) prior to infection with VSV-G pseudotyped luciferase expressing single cycle HIV-1 (HIVΔenv/Luc VSV-G, 100ng p24gag per well) on days 1, 7, 14, 21, 28 and 35 post drug treatment. Cells were spinoculated as described above, washed three times to remove unbound virus, followed by culture for 3 days. Cells were lysed with Glo lysis buffer (Promega), and luciferase expression in cell lysates was determined using the Bright-Glo luciferase expression system (Promega) as described previously.67 Results were analyzed by one-way ANOVA with Tukey multiple comparisons test using Graphpad Prism. *P ≤ 0.05 **P ≤ 0.01 ***P ≤ 0.001 ****P ≤ 0.0001.
RPV and CAB Quantification in CD169+ MDMs and the Medium.
ARV-loaded GM3 PLA, DOPS PLA, and GM3 PLGA NPs at a concentration of 10 μM RPV in RPMI/10% FBS were added to CD169+ MDMs in a 12-well plate at a concentration of 5 × 105 cells per well, and incubated for 3 h. For these experiments, the NP concentration was adjusted to have the same RPV concentrations (10 μM) for all the conditions, therefore CAB concentration varied between 50 to 165 μM. Next, NPs were removed and cells were washed with RPMI and maintained in RPMI/10% FBS for the duration of the experiment. Half of the medium was exchanged every other day.9 RPV and CAB concentrations in CD169+ MDMs were determined on day 0, 1, 3, 5, 10, 15, 20, and 28. Cells were lysed using 1% Triton-X 100 (Sigma-Aldrich, St Louis, MO) in 1 × PBS, following addition of acetonitrile to extract the drugs. The cell-lysate mixtures were centrifuged at 14,000 rpm for 10 min at 4 °C. RPV and CAB concentrations in the supernatant were determined using HPLC, as described above. ATZ was added as an IS to all the samples. The total protein concentration was monitored in the cell-lysates by a micro bicinchoninic acid (BCA) assay using a Micro BCA™ Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA). To quantify the amount of drugs released from CD169+ MDMs into the culture medium, 300 to 500 μL of medium was added to 1 mL of HPLC-grade acetonitrile and the mixture was centrifuged at 14,000 rpm for 10 min. Supernatant was dried using a SPD Speed Vacuum (SpeedVac, Thermo Fisher Scientific, Waltham, MA) following established procedures.9, 74 Extracted drugs were resuspended in acetonitrile and RPV and CAB concentrations were determined via HPLC analysis following the same method described above.
Confocal Imaging in CD169+ MDMs.
1 × 106 MDMs in 3 mL medium were seeded in culture dishes (Cellvis, Mountain View, CA) and incubated with IFN-λ contained medium at a concentration of 5 ng/ml for 48 h. CD169+ MDMs were incubated with ARV-loaded polymer NPs (5 × 1012 NPs/mL in RPMI/10% FBS) for 4 h at 37 °C, subsequently washed with RPMI to remove the unbound NPs, and were incubated in the complete growth medium for the duration of experiment, up to 25 days. The medium was exchanged every 2 days. On the day of staining, nucleus stain Hoechst was added to cells for 15 min at 37 °C, 5% CO2. Next, cells were fixed in 4% PFA, permeabilized with 0.2% TWEEN 20, and blocked with 1% BSA in 1 × PBS. Antihuman CD169 (Siglec-1, Clone: 7–239), antihuman CD9 (Clone: HI9a), and anti-human CD107a (LAMP-1, Clone: H4A3) mAbs (BioLegend, San Diego, CA) were used to stain for CD169, CD9, and LAMP-1, respectively. Staining was visualized upon addition of Alexa Fluor 647 (Goat anti-mouse, Clone: Poly4053) conjugated secondary antibody. Note that concentrations of primary and the secondary antibodies were 2 μg/mL and 1 μg/mL, respectively. Samples were imaged via Olympus FV3000 scanning confocal microscope. Lasers with excitation wavelength of (405,561,640 nm) and a 60 × (oil) objective were used. Z-stack images were collected at 3 μm steps. The recorded images were processed by ImageJ, and Coloc 2 analysis plugin (ImageJ) was used to calculate the MCC (M1 and M2) values.
Statistical Analysis.
Unless otherwise noted, analysis of statistical significance of data was performed using one-way ANOVA following a subsequent Tukey post-hoc test as implemented in MATLAB. Significant differences at p ≤ 0.05 are demonstrated using one asterisk (*), for p ≤ 0.01 with two asterisks (**), for p ≤ 0.001 with three asterisks (***), and for p ≤ 0.0001 with four asterisks (****).
Ethics Statement.
All human PBMCs used in this article were acquired in an anonymous fashion from NY Biologics, and no identifiable private information was collected, therefore this research does not meet the definition of human subjects research and is determined to be exempt by the Institutional Review Board of the Boston University Medical Center.
Data Availability.
All the data that support the findings of this study are available from the authors upon reasonable request.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Maliheh Aghanasiri for rendering the scheme of molecular structure of the lipid-coated polymer NP (Figure 1A), Zhongkun Zhang for assistance with running prepared HPLC samples (for drug release kinetics experiments), Jacob Berrigan for assistance with protocols for human MDM differentiation, and Serge Zdanovich and Dr. Norman Lee for technical support of HPLC experiments.
Funding
B.M.R. and S.G. acknowledge support from the National Institutes of Health through Grant Nos. R01AI132111 (S.G. and B.M.R.), R01AI064099 (S.G.), P30AI042853 (S.G.) and R01CA138509 (B.M.R.). J.F. was supported by the Immunology Training Program (T32 AI-007309). Research reported in this publication was supported by Boston University Micro and Nano Imaging Facility and the Office of The Director, National Institutes of Health of the National Institutes of Health under Award Number S10OD024993. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing Interests
S.G. and B.M.R. hold patents for GM3-functionalized nanoparticles.
ASSOCIATED CONTENT
Supporting Information.
Supporting information contains an additional methods section for release kinetics determination, 12 supplemental figures (Figure S1 – S12) featuring cell viability assays, isobologram analysis, hydrodynamic DLS data, RPV and CAB release kinetics, CD169 induction determination, RPV and CAB quantification in cell culture medium, confocal localization of NPs, as well as 8 supplemental tables (tables S1 – S8) summarizing encapsulation efficiencies and drug loadings, IC50 values, percentages of RPV/CAB released, remaining RPV/CAB concentrations, zero- and first-order fits, Higuchi fits, and Manders’ colocalization coefficients (MCCs).
REFERENCES
- (1).Spreen WR; Margolis DA; Pottage JC Jr Long-acting Injectable Antiretrovirals for HIV Treatment and Prevention. Cur. Opin. HIV AIDS 2013, 8 (6), 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Soriano V; Barreiro P; de Mendoza C Long-acting Antiretroviral Therapy. Nat. Mater. 2020, 19 (8), 826–827. [DOI] [PubMed] [Google Scholar]
- (3).Mandal S; Kang G; Prathipati PK; Zhou Y; Fan W; Li Q; Destache CJ Nanoencapsulation Introduces Long-acting Phenomenon to Tenofovir Alafenamide and Emtricitabine Drug Combination: A Comparative Pre-exposure Prophylaxis Efficacy Study against HIV-1 Vaginal Transmission. J. Control. Release 2019, 294, 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Mandal S; Kang G; Prathipati PK; Fan W; Li Q; Destache CJ Long-acting Parenteral Combination Antiretroviral Loaded Nano-drug Delivery System to Treat Chronic HIV-1 Infection: A Humanized Mouse Model Study. Antiviral Res. 2018, 156, 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Nowacek AS; Miller RL; McMillan J; Kanmogne G; Kanmogne M; Mosley RL; Ma Z; Graham S; Chaubal M; Werling J NanoART Synthesis, Characterization, Uptake, Release and Toxicology for Human Monocyte–Macrophage Drug Delivery. Nanomedicine 2009, 4 (8), 903–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kirtane AR; Abouzid O; Minahan D; Bensel T; Hill AL; Selinger C; Bershteyn A; Craig M; Mo SS; Mazdiyasni H Development of an Oral Once-weekly Drug Delivery System for HIV Antiretroviral Therapy. Nat. Commun. 2018, 9 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kulkarni TA; Bade AN; Sillman B; Shetty BLD; Wojtkiewicz MS; Gautam N; Hilaire JR; Sravanam S; Szlachetka A; Lamberty BG A Year-long Extended Release Nanoformulated Cabotegravir Prodrug. Nat. Mater. 2020, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hilaire JR; Bade AN; Sillman B; Gautam N; Herskovitz J; Shetty BLD; Wojtkiewicz MS; Szlachetka A; Lamberty BG; Sravanam S Creation of a Long-acting Rilpivirine Prodrug Nanoformulation. J. Control. Release 2019, 311, 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sillman B; Bade AN; Dash PK; Bhargavan B; Kocher T; Mathews S; Su H; Kanmogne GD; Poluektova LY; Gorantla S Creation of a Long-acting Nanoformulated Dolutegravir. Nat. Commun. 2018, 9 (1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pantaleo G; Koup RA Correlates of Immune Protection in HIV-1 Infection: What we know, What We Don’t Know, What We Should Know. Nat. Med. 2004, 10 (8), 806–810. [DOI] [PubMed] [Google Scholar]
- (11).van Kooyk Y; Appelmelk B; Geijtenbeek TB A fatal attraction: Mycobacterium Tuberculosis and HIV-1 Target DC-SIGN to Escape Immune Surveillance. Trends Mol. Med. 2003, 9 (4), 153–159. [DOI] [PubMed] [Google Scholar]
- (12).Visser T Mechanisms of HIV-1 to Escape from the Host Immune Surveillance. Eur. J Clin. Investig. 2000, 30 (8), 740–746. [DOI] [PubMed] [Google Scholar]
- (13).Hill AL; Rosenbloom DI; Nowak MA; Siliciano RF Insight into Treatment of HIV Infection from Viral Dynamics Models. Immunol. Rev. 2018, 285 (1), 9–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chen S; Kumar S; Espada CE; Tirumuru N; Cahill MP; Hu L; He C; Wu L N6-methyladenosine Modification of HIV-1 RNA Suppresses Type-I Interferon Induction in Differentiated Monocytic Cells and Primary Macrophages. PLoS Pathog. 2021, 17 (3), e1009421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sewald X; Ladinsky MS; Uchil PD; Beloor J; Pi R; Herrmann C; Motamedi N; Murooka TT; Brehm MA; Greiner DL Retroviruses use CD169-mediated Trans-infection of Permissive Lymphocytes to Establish Infection. Science 2015, 350 (6260), 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hammonds JE; Beeman N; Ding L; Takushi S; Francis AC; Wang J-J; Melikyan GB; Spearman P Siglec-1 Initiates Formation of the Virus-containing Compartment and Enhances Macrophage-to-T Cell Transmission of HIV-1. PLoS Pathog. 2017, 13 (1), e1006181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gupta P; Lai SM; Sheng J; Tetlak P; Balachander A; Claser C; Renia L; Karjalainen K; Ruedl C Tissue-resident CD169+ Macrophages Form a Crucial Front Line against Plasmodium Infection. Cell Rep. 2016, 16 (6), 1749–1761. [DOI] [PubMed] [Google Scholar]
- (18).Grabowska J; Lopez-Venegas MA; Affandi AJ; Den Haan JM CD169+ Macrophages Capture and Dendritic Cells Instruct: the Interplay of the Gatekeeper and the General of the Immune System. Front. Immunol. 2018, 9, 2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Martinez-Pomares L; Gordon S CD169+ Macrophages at the Crossroads of Antigen Presentation. Trends Immunol. 2012, 33 (2), 66–70. [DOI] [PubMed] [Google Scholar]
- (20).van Dinther D; Veninga H; Iborra S; Borg EG; Hoogterp L; Olesek K; Beijer MR; Schetters ST; Kalay H; Garcia-Vallejo JJ Functional CD169 on Macrophages Mediates Interaction with Dendritic cells for CD8+ T Cell Cross-priming. Cell Rep. 2018, 22 (6), 1484–1495. [DOI] [PubMed] [Google Scholar]
- (21).Gummuluru S; Ramirez N-GP; Akiyama H CD169-dependent Cell-associated HIV-1 Transmission: a Driver of Virus Dissemination. J. Infect. Dis. 2014, 210 (suppl_3), S641–S647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Puryear WB; Akiyama H; Geer SD; Ramirez NP; Yu X; Reinhard BM; Gummuluru S Interferon-inducible Mechanism of Dendritic Cell-mediated HIV-1 Dissemination is Dependent on Siglec-1/CD169. PLoS Pathog. 2013, 9 (4), e1003291. DOI: 10.1371/journal.ppat.1003291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hatch SC; Archer J; Gummuluru S Glycosphingolipid Composition of Human Immunodeficiency Virus Type 1 (HIV-1) Particles is a Crucial Determinant for Dendritic Cell-mediated HIV-1 Trans-infection. J. Virol. 2009, 83 (8), 3496–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Izquierdo-Useros N; Lorizate M; Contreras; Rodriguez-Plata M; Glass B; Erkizia I; Prado J; Casas J; Fabriàs G; Kräusslich H-G; et al. Sialyllactose in Viral Membrane Gangliosides Is a Novel Molecular Recognition Pattern for Mature Dendritic Cell Capture of HIV-1. PLoS Biol. 2012, 10 (4), e1001315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Puryear WB; Yu X; Ramirez NP; Reinhard BM; Gummuluru S HIV-1 Incorporation of Host-cell–derived Glycosphingolipid GM3 Allows for Capture by Mature Dendritic Cells. Proc. Natl. Acad. Sci. U.S.A 2012, 109 (19), 7475–7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Izquierdo-Useros N; Lorizate M; McLaren PJ; Telenti A; Kräusslich H-G; Martinez-Picado J HIV-1 Capture and Transmission by Dendritic Cells: The Role of Viral Glycolipids and the Cellular Receptor Siglec-1. PLoS Pathog. 2014, 10 (7), e1004146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Eshaghi B; Alsharif N; An X; Akiyama H; Brown KA; Gummuluru S; Reinhard BM Stiffness of HIV-1 Mimicking Polymer Nanoparticles Modulates Ganglioside-Mediated Cellular Uptake and Trafficking. Adv. Sci. 2020, 7 (18), 2000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yu X; Feizpour A; Ramirez N-GP; Wu L; Akiyama H; Xu F; Gummuluru S; Reinhard BM Glycosphingolipid-functionalized nanoparticles recapitulate CD169-dependent HIV-1 Uptake and Trafficking in Dendritic Cells. Nat. Commun. 2014, 5 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yu X; Xu F; Ramirez N-GP; Kijewski SD; Akiyama H; Gummuluru S; Reinhard B. r. M. Dressing up Nanoparticles: A Membrane Wrap to Induce Formation of the Virological Synapse. ACS Nano 2015, 9 (4), 4182–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Xu F; Reiser M; Yu X; Gummuluru S; Wetzler L; Reinhard BM Lipid-mediated Targeting with Membrane-wrapped Nanoparticles in the Presence of Corona Formation. ACS Nano 2016, 10 (1), 1189–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Xu F; Bandara A; Akiyama H; Eshaghi B; Stelter D; Keyes T; Straub JE; Gummuluru S; Reinhard BM Membrane-wrapped Nanoparticles Probe Divergent Roles of GM3 and Phosphatidylserine in Lipid-mediated Viral Entry Pathways. Proc. Natl. Acad. Sci. U.S.A. 2018, 115 (39), E9041–E9050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Schafer JJ; Short WR Rilpivirine, a Novel Non-nucleoside Reverse Transcriptase Inhibitor for the Management of HIV-1 Infection: a Systematic Review. Antivir. Ther. 2012, 17 (8), 1495–1502. [DOI] [PubMed] [Google Scholar]
- (33).Trezza C; Ford SL; Spreen W; Pan R; Piscitelli S Formulation and Pharmacology of Long-acting Cabotegravir. Cur. Opin. HIV AIDS 2015, 10 (4), 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Margolis DA; Gonzalez-Garcia J; Stellbrink H-J; Eron JJ; Yazdanpanah Y; Podzamczer D; Lutz T; Angel JB; Richmond GJ; Clotet B Long-acting Intramuscular Cabotegravir and Rilpivirine in Adults with HIV-1 Infection (LATTE-2): 96-Week Results of a Randomised, Open-label, Phase 2b, Non-inferiority Trial. The Lancet 2017, 390 (10101), 1499–1510. [DOI] [PubMed] [Google Scholar]
- (35).Taylor BS; Tieu H-V; Jones J; Wilkin TJ CROI 2019: Advances in Antiretroviral Therapy. Top. Antivir. Med. 2019, 27 (1), 50. [PMC free article] [PubMed] [Google Scholar]
- (36).Zhang L; Chan JM; Gu FX; Rhee J-W; Wang AZ; Radovic-Moreno AF; Alexis F; Langer R; Farokhzad OC Self-assembled Lipid− polymer Hybrid Nanoparticles: a Robust Drug Delivery Platform. ACS Nano 2008, 2 (8), 1696–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Fang RH; Aryal S; Hu C-MJ; Zhang L Quick Synthesis of Lipid−polymer Hybrid Nanoparticles with Low Polydispersity using a Single-step Sonication Method. Langmuir 2010, 26 (22), 16958–16962. [DOI] [PubMed] [Google Scholar]
- (38).Lemke G; Rothlin CV Immunobiology of the TAM Receptors. Nat. Rev. Immunol. 2008, 8 (5), 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Freeman GJ; Casasnovas JM; Umetsu DT; DeKruyff RH TIM Genes: a Family of Cell Surface Phosphatidylserine Receptors that Regulate Innate and Adaptive Immunity. Immunol. Rev. 2010, 235 (1), 172–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Date AA; Shibata A; Bruck P; Destache CJ Development and Validation of a Simple and Isocratic Reversed-phase HPLC Method for the Determination of Rilpivirine from Tablets, Nanoparticles and HeLa Cell Lysates. Biomed. Chromatogr. 2015, 29 (5), 709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Cheow WS; Hadinoto K Factors Affecting Drug Encapsulation and Stability of Lipid–polymer Hybrid Nanoparticles. Colloids Surf. B 2011, 85 (2), 214–220. [DOI] [PubMed] [Google Scholar]
- (42).Albisa A; Piacentini E; Sebastian V; Arruebo M; Santamaria J; Giorno L Preparation of Drug-loaded PLGA-PEG Nanoparticles by Membrane-assisted Nanoprecipitation. Pharm. Res. 2017, 34 (6), 1296–1308. [DOI] [PubMed] [Google Scholar]
- (43).Rajoli RK; Curley P; Chiong J; Back D; Flexner C; Owen A; Siccardi M Predicting Drug–Drug Interactions between Rifampicin and Long-acting Cabotegravir and Rilpivirine using Physiologically Based Pharmacokinetic Modeling. J. Infect. Dis. 2019, 219 (11), 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Maruthapillai A; Palanisamy K; Sunkara M Preparation and Characterization of Rilpivirine Solid Dispersions with the Application of Enhanced Solubility and Dissolution Rate. Beni-Suef Univ. J. of Basic Appl. Sci. 2015, 4 (1), 71–79. [Google Scholar]
- (45).Zhou T; Su H; Dash P; Lin Z; Shetty BLD; Kocher T; Szlachetka A; Lamberty B; Fox HS; Poluektova L Creation of a Nanoformulated Cabotegravir Prodrug with Improved Antiretroviral Profiles. Biomaterials 2018, 151, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Dash S; Murthy PN; Nath L; Chowdhury P Kinetic Modeling on Drug Release from Controlled Drug Delivery Systems. Acta. Pol. Pharm. 2010, 67 (3), 217–223. [PubMed] [Google Scholar]
- (47).Costa P; Lobo JMS Modeling and Comparison of Dissolution Profiles. Eur. J. Pharm. Sci. 2001, 13 (2), 123–133. [DOI] [PubMed] [Google Scholar]
- (48).Fredenberg S; Wahlgren M; Reslow M; Axelsson A The mechanisms of Drug Release in Poly (lactic-co-glycolic acid)-based Drug Delivery systems—a Review. Int. J. Pharm. 2011, 415 (1–2), 34–52. [DOI] [PubMed] [Google Scholar]
- (49).Korsmeyer RW; Gurny R; Doelker E; Buri P; Peppas NA Mechanisms of Solute Release from Porous Hydrophilic Polymers. Int. J. Pharm. 1983, 15 (1), 25–35. [DOI] [PubMed] [Google Scholar]
- (50).Ritger PL; Peppas NA A Simple Equation for Description of Solute Release I. Fickian and Non-fickian Release from Non-swellable Devices in the Form of Slabs, Spheres, Cylinders or Discs. J. Control. Release 1987, 5 (1), 23–36. [PubMed] [Google Scholar]
- (51).Ritger PL; Peppas NA A Simple Equation for Description of Solute Release II. Fickian and Anomalous Release from Swellable Devices. J. Control. Release 1987, 5 (1), 37–42. [PubMed] [Google Scholar]
- (52).Alsharif N; Eshaghi B; Reinhard BM; Brown KA Physiologically Relevant Mechanics of Biodegradable Polyester Nanoparticles. Nano Lett. 2020, 20 (10), 7536–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Makadia HK; Siegel SJ Poly lactic-co-glycolic acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3 (3), 1377–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Gentile P; Chiono V; Carmagnola I; Hatton PV An overview of Poly (lactic-co-glycolic) acid (PLGA)-based Biomaterials for Bone Tissue Engineering. Int. J Mol. Sci. 2014, 15 (3), 3640–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Mohammad AK; Reineke JJ Quantitative Detection of PLGA Nanoparticle Degradation in Tissues Following Intravenous Administration. Mol. Pharm. 2013, 10 (6), 2183–2189. [DOI] [PubMed] [Google Scholar]
- (56).Hartnell A; Steel J; Turley H; Jones M; Jackson DG; Crocker PR Characterization of Human Sialoadhesin, a Sialic Acid Binding Receptor Expressed by Resident and Inflammatory Macrophage Populations. Blood 2001, 97 (1), 288–296. [DOI] [PubMed] [Google Scholar]
- (57).Sun J; Zhang L; Wang J; Feng Q; Liu D; Yin Q; Xu D; Wei Y; Ding B; Shi X Tunable Rigidity of (Polymeric Core)–(Lipid Shell) Nanoparticles for Regulated Cellular Uptake. Adv. Mater. 2015, 27 (8), 1402–1407. [DOI] [PubMed] [Google Scholar]
- (58).Hui Y; Wibowo D; Liu Y; Ran R; Wang H-F; Seth A; Middelberg AP; Zhao C-X Understanding the Effects of Nanocapsular Mechanical Property on Passive and Active Tumor Targeting. ACS Nano 2018, 12 (3), 2846–2857. [DOI] [PubMed] [Google Scholar]
- (59).Anselmo AC; Zhang M; Kumar S; Vogus DR; Menegatti S; Helgeson ME; Mitragotri S Elasticity of Nanoparticles Influences Their Nlood Virculation, Phagocytosis, Endocytosis, and Targeting. ACS Nano 2015, 9 (3), 3169–3177. [DOI] [PubMed] [Google Scholar]
- (60).Hui Y; Yi X; Wibowo D; Yang G; Middelberg AP; Gao H; Zhao C-X Nanoparticle Elasticity Regulates Phagocytosis and Cancer Cell Uptake. Sci. Adv. 2020, 6 (16), eaaz4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kundrotas G; Karabanovas V; Pleckaitis M; Juraleviciute M; Steponkiene S; Gudleviciene Z; Rotomskis R Uptake and Distribution of Carboxylated Quantum Dots in Human Mesenchymal Stem Cells: Cell Growing Density Matters. J. Nanobiotechnology 2019, 17 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Welsch S; Keppler OT; Habermann A; Allespach I; Krijnse-Locker J; Kräusslich H-G HIV-1 Buds Predominantly at the Plasma Membrane of Primary Human Macrophages. PLoS Pathog 2007, 3 (3), e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Tan J; Sattentau QJ The HIV-1-containing Macrophage Compartment: a Perfect Cellular Niche? Trends Microbiol. 2013, 21 (8), 405–412. [DOI] [PubMed] [Google Scholar]
- (64).Deneka M; Pelchen-Matthews A; Byland R; Ruiz-Mateos E; Marsh M In Macrophages, HIV-1 Assembles into an Intracellular Plasma Membrane Domain Containing the Tetraspanins CD81, CD9, and CD53. J. Cell Biol. 2007, 177 (2), 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Dunn KW; Kamocka MM; McDonald JH A Practical Guide to Evaluating Colocalization in Biological Microscopy. Am. J. Phys. Cell Physiol. 2011, 300 (4), C723–C742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Bragagni M; Beneitez C; Martín C; de la Ossa DHP; Mura PA; Gil-Alegre ME Selection of PLA Polymers for the Development of Injectable Prilocaine Controlled Release Microparticles: Usefulness of Thermal Analysis. Int. J. Pharm. 2013, 441 (1–2), 468–475. [DOI] [PubMed] [Google Scholar]
- (67).Miller CM; Akiyama H; Agosto LM; Emery A; Ettinger CR; Swamstrom RI; Henderson AJ; Gummuluru S Virion Associated Vpr Alleviates a Post-integration Block to HIV-1 Infection of Dendritic Cells. J. Virol. 2017, 91 (13) e00051–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Akiyama H; Miller C; Patel HV; Hatch SC; Archer J; Ramirez NG; Gummuluru S Virus Particle Release from Glycosphingolipid-enriched Microdomains is Essential for Dendritic Cell-mediated Capture and Transfer of HIV-1 and Henipavirus. J. Virol. 2014, 88 (16), 8813–8825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Akiyama H; Ramirez NP; Gibson G; Kline C; Watkins S; Ambrose Z; Gummuluru S Interferon-Inducible CD169/Siglec1 Attenuates Anti-HIV-1 Effects of Alpha Interferon. J. Virol. 2017, 91 (21), e00972–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Derdeyn CA; Decker JM; Sfakianos JN; Wu X; O’Brien WA; Ratner L; Kappes JC; Shaw GM; Hunter E Sensitivity of Human Immunodeficiency Virus Type 1 to the Fusion Inhibitor T-20 is Modulated by Coreceptor Specificity Defined by the V3 Loop of gp120. J. Virol. 2000, 74 (18), 8358–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Wehrly K; Chesebro B p24 Antigen Capture Assay for Quantification of Human Immunodeficiency Virus Using Readily Available Inexpensive Reagents. Methods 1997, 12 (4), 288–293. [DOI] [PubMed] [Google Scholar]
- (72).Wiley RD; Gummuluru S Immature Dendritic Cell-derived Exosomes can Mediate HIV-1 Trans infection. Proc. Natl. Acad. Sci. U.S.A. 2006, 103 (3), 738–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Derdeyn CA; Decker JM; Sfakianos JN; Wu X; O’Brien WA; Ratner L; Kappes JC; Shaw GM; Hunter E Sensitivity of Human Immunodeficiency Virus Type 1 to the Fusion Inhibitor T-20 is Modulated by Coreceptor Specificity Defined by the V3 Loop of gp120. J. Virol. 2000, 74 (18), 8358–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Date AA; Shibata A; Goede M; Sanford B; La Bruzzo K; Belshan M; Destache CJ Development and Evaluation of a Thermosensitive Vaginal Gel Containing Raltegravir + Efavirenz Loaded Nanoparticles for HIV Prophylaxis. Ant. Res. 2012, 96 (3), 430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data that support the findings of this study are available from the authors upon reasonable request.