

Abstract
A major goal of cancer research is to understand how mutations distributed across diverse genes affect common cellular systems, including multi-protein complexes and assemblies. Two challenges— how to comprehensively map such systems and how to identify which are under mutational selection— have hindered this understanding. Here, we create a comprehensive map of cancer protein systems integrating new and published multi-omic interaction data at multiple scales of analysis. We then develop a unified statistical model that pinpoints 395 specific systems under mutational selection across 13 cancer types. This map, called NeST (Nested Systems in Tumors), incorporates canonical processes and new discoveries, including a PIK3CA-actomyosin complex that inhibits PI3K signaling and recurrent mutations in collagen complexes that promote tumor proliferation. These systems can be used as clinical biomarkers and implicate a total of 548 genes in cancer evolution and progression. This work shows how disparate tumor mutations converge on protein assemblies at different scales.
One Sentence Summary:
Mutations in diverse genes converge on common protein complexes and larger assemblies defined by systematic network mapping.
Substantial progress in cataloging the molecular basis of cancer has come from genomic, transcriptomic, and proteomic profiling of thousands of patients by consortia such as The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC). However, very different patterns of mutations are observed across different tumors and across different cells within the same tumor (1), hindering the interpretation of cancer genomes. Although some known cancer driver genes are mutated frequently in a statistically significant number of samples, the importance of the much greater number of low-frequency genetic events has remained largely unclear (2) (Fig. 1A).
Fig. 1. Rationale for a multiscale map of cancer systems and overview of its assembly.
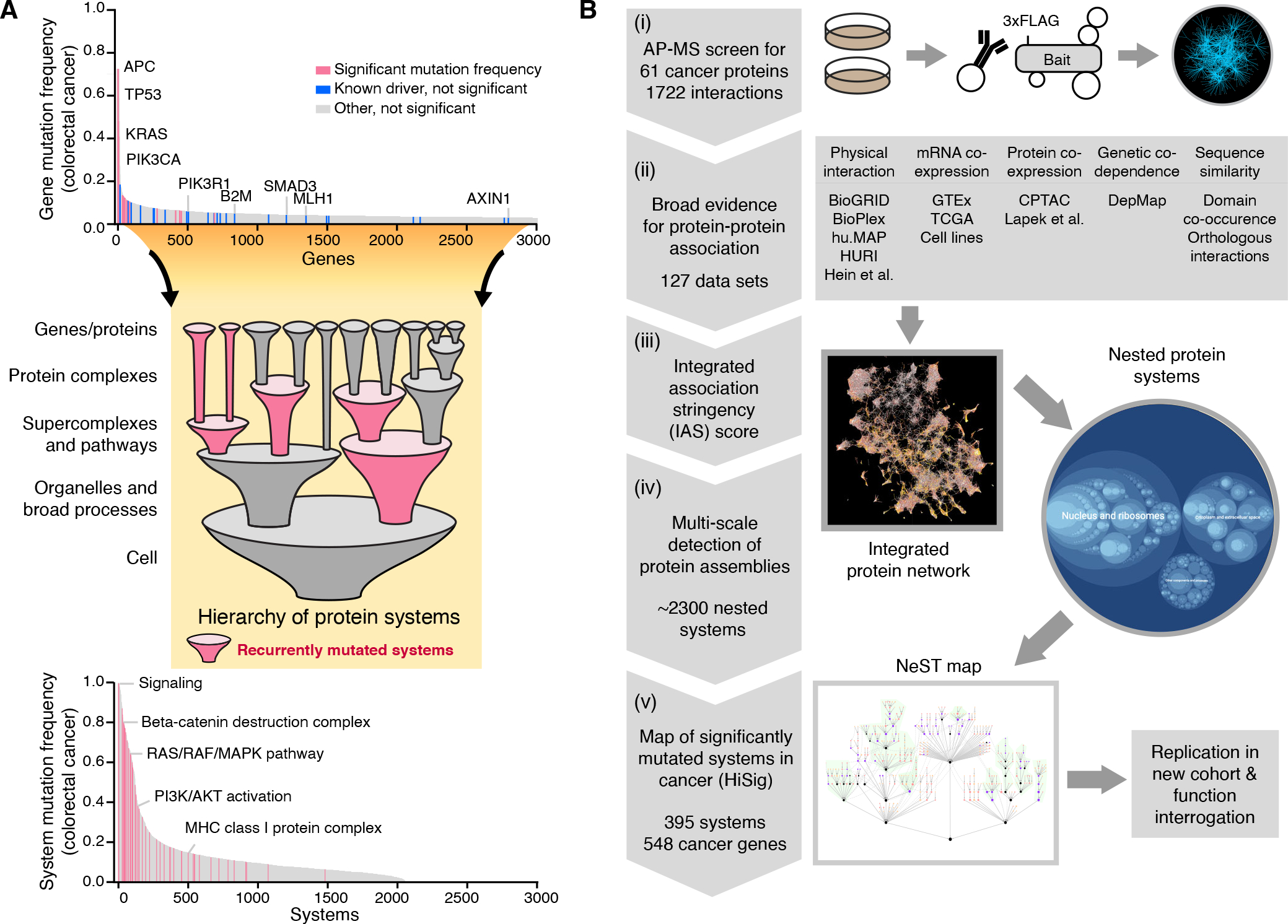
(A) Cancer genes missed by single-gene mutation analysis can be recovered by identification of significantly mutated systems. In the distribution of gene mutation frequencies in colorectal adenocarcinoma (COAD, top), analysis of significantly mutated genes (pink, TCGA pan-cancer atlas) (37) misses a number of known colorectal cancer driver genes (blue, COSMIC Cancer Gene Census) (72). Representative genes from both categories are labeled. When evaluating mutation significance in a hierarchy of protein systems (middle), driver genes missed in the single-gene analysis can be recovered within significantly mutated systems (pink, bottom). (B) Pipeline for assembly of the cancer systems map. (i) Generate cancer protein interactions using affinity purification mass spectrometry (AP-MS). (ii) Collect previous protein association evidence of five major data types. (iii) Integrate all evidence to derive an integrated association stringency (IAS) score network for all pairs of 19,035 human proteins, (iv) Identify a hierarchy of protein systems by multiscale community detection, (v) Identify recurrently mutated systems in the hierarchy by HiSig, defining a cancer systems map, which is validated in independent cohorts and functional studies.
A powerful way to interpret the many rare mutations is to organize them into networks of genes that participate in commonly dysregulated cellular components or processes (3–5). Alterations may be observed infrequently at the nucleotide or gene level but can be substantially more common when considering impacts in a larger biological process. For instance, the TCGA analysis of head and neck squamous cell carcinoma (6) reported genetic changes to a “cell differentiation pathway” in 64% of HPV-negative tumors, combining mutations across four genes that were each altered more rarely (AJUBA, TP63, NOTCH1 and FAT1, each individually 7% - 32% of patients). Systematic aggregation of mutated genes in the form of gene sets (7–10) or molecular networks connecting pairs of functionally related genes (11–20) has been very useful in identifying higher-order systems of genes under mutational selection in cancer, many of which would otherwise be missed.
While this strategy is promising, realizing a complete systems-level description of cancer mutations will require overcoming two challenges in particular. First is to assemble accurate and comprehensive knowledge maps of dysregulated cellular components and processes (henceforth called cellular “systems” for generality). Unlike whole-genome sequencing, which has provided complete information on tumor genomes, systematic efforts to map cancer cellular systems are just beginning (21–28). In this respect, proteomics efforts have used techniques such as affinity purification, proximity labeling, co-elution mass spectrometry, and yeast-two-hybrid assays to create catalogs of human protein interactions, which have been useful for defining protein complexes and larger cellular components (29–33). Thus far, however, large-scale protein interaction surveys have not focused on cancer proteins in particular, and experiments have been typically conducted in model organisms or cell lines chosen for experimental tractability rather than cancer relevance. A second challenge is to identify mutational selection on biological systems larger than those encoded by single genes. Cellular components functioning in cancer can occupy a range of biophysical scales— from individual residues and domains in proteins (34–37) to impacts on multi-protein complexes (38–41), signaling networks (42, 43), and classical and membraneless organelles (44, 45). Analyzing the incidence of cancer mutations at only one of these scales misses components under mutational selection at all others. Accordingly, it remains largely unclear which multi-gene systems and scales represent the key focal points on which mutations converge.
To address these challenges, we integrated existing data resources with a collection of systematic protein interaction networks centered on cancer proteins in cancer-relevant conditions, with an emphasis on breast and head-and-neck cancers as described in two companion papers (46, 47). Using these data, we constructed a structured map of protein systems, not restricted to one scale but organized across a hierarchy of cellular components and processes. We then developed a unified statistical model to identify systems under mutational selection considering all scales simultaneously. Together, these analyses define a compendium of protein complexes, signaling pathways, and larger assemblies with evidence for recurrent mutation in cancer.
Results
Interaction Mapping and Integrative Analysis Yield a Hierarchy of Protein systems
We amassed a large compendium of cancer protein-protein interactions (PPIs) based on affinity purification mass spectrometry (AP-MS) of 61 proteins with established roles in cancer, combining the separate screens in breast and head-and-neck tissues described in our two companion papers (citations to be updated). In these companion studies, proteins were epitope tagged (3xFLAG), expressed, then purified from a panel of cell lines representing malignant and non-malignant breast tissues (tumor: MDA-MB-231, MCF-7; normal: MCF-10A; 40 tagged proteins) and/or head-and-neck tissues (tumor: CAL-33, SCC-25; normal: HET-1A; 30 tagged proteins, of which 9 were also investigated in breast, Table S1). Copurified proteins were then identified by mass spectrometry using PPI confidence scoring algorithms (Materials and Methods). Here, we combined data across all proteins and cell lines, yielding a total of 1722 distinct interactions. Approximately 85% of PPIs had not been reported in previous AP-MS datasets or in curated databases (30, 48–50), showing that these experiments had substantially enlarged the known interactomes of many cancer proteins (Table S1).
We integrated these experiments with a broad collection of human PPI data from previous studies (29, 30, 49, 51, 52) alongside four additional types of evidence previously shown to inform protein physical associations (Fig. 1B). These additional types were: (a) correlated levels of protein abundance over many cell lines or tissues (53–55), (b) correlated mRNA transcript levels over many cell lines or tissues (54), (c) genetic co-dependencies, as revealed by correlated cell growth outcomes for CRISPR knockdowns performed over many cell lines (57), and (d) sequence-based associations, including protein pairs with sequence domains that frequently interact and protein pairs with orthologs that interact in model species (58). For each evidence type, we performed a broad survey of studies relevant to tumor samples, tumor cell lines, and human tissues, resulting in a compendium of 127 datasets in total (Fig. 1B; Table S2). We employed an established method of biological network integration (59), based on supervised machine learning, to quantitatively weigh and combine all evidence to create a single integrated association stringency (IAS) score for each pair of human proteins (Fig. 2A; Fig. S1A; 19,035 proteins and 1.8 × 108 scored protein pairs; Materials and Methods). This integration system was trained for the ability to interconnect proteins in the same cellular component or biological process recorded in the Gene Ontology reference database (60). PPIs were the most informative evidence type for this task, followed by sequence similarity and protein co-expression (Fig. S1B, C). The resulting IAS network (Fig. 2B) has been made available for download, browsing and query (http://ccmi.org/nest; Data S1).
Fig. 2. Integration and multiscale organization of protein networks.
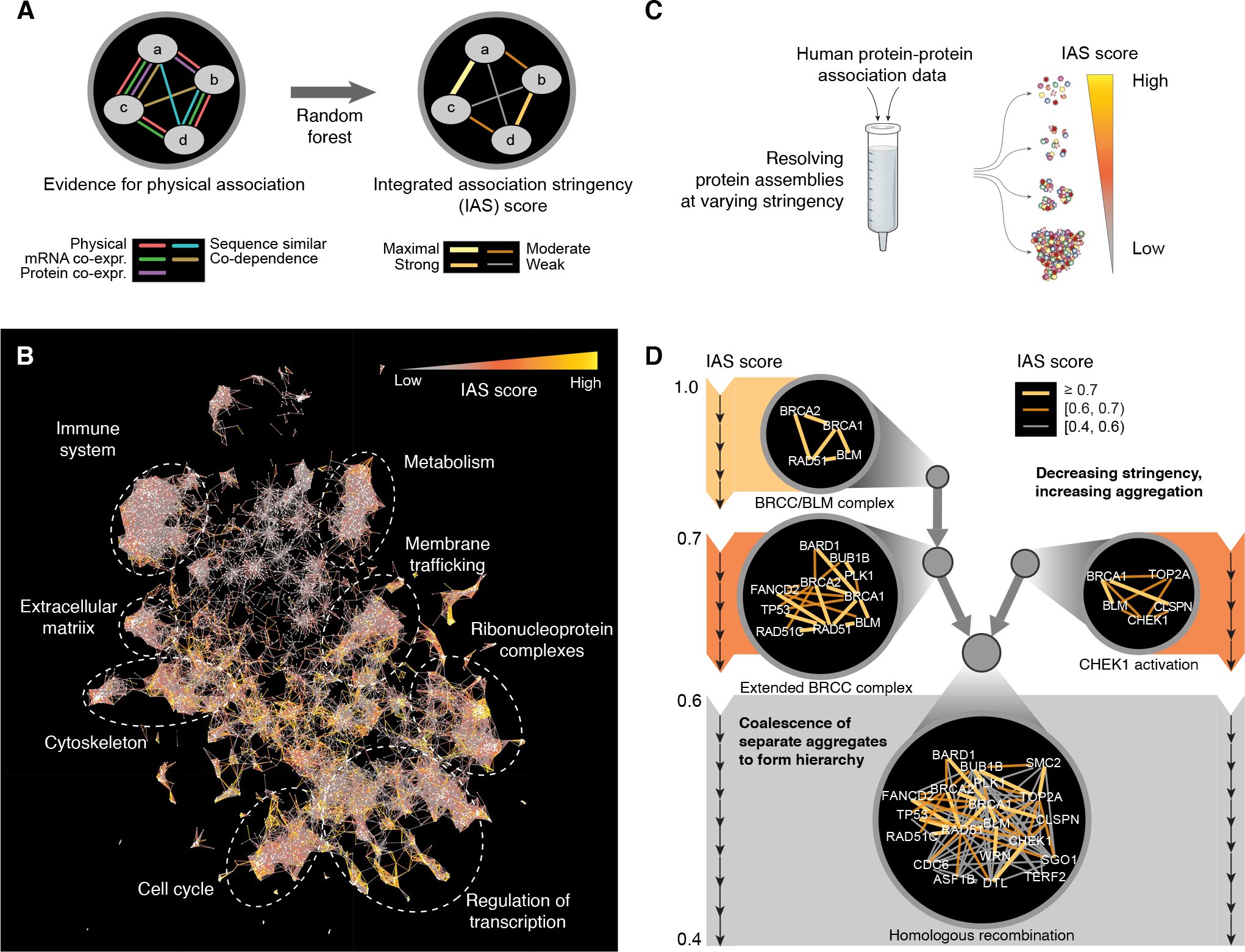
(A) Types of network evidence are combined in a machine learning framework (random forest) to determine an integrated association stringency (IAS) score among protein pairs. (B) Visualization of overall IAS network structure highlighting major large-scale systems (text labels and dashed circles). (C) Process of multiscale community detection, whereby protein systems of increasing size are discovered as the threshold IAS score is progressively lowered. (D) As the IAS threshold decreases (top to bottom), strongly associated protein systems are detected then subsequently expanded to include proteins with moderate-to-weak associations. Each circle indicates a protein system, and edge colors (yellow, orange, grey) indicate decreasing stringencies of association.
Because the IAS score forms a continuous metric of physical association, robust assemblies of interacting proteins can be identified at specific IAS thresholds (Fig. 2C). To catalog these assemblies over the entire range, we performed a hierarchical community detection analysis (Materials and Methods). By this procedure, the IAS threshold was initialized to its maximum (most stringent) value and then iteratively relaxed to a lower (more permissive) value; at each step, progressively larger and less stringently associated protein assemblies were detected. Small assemblies of strongly associating proteins were subsumed within larger assemblies as the IAS threshold decreased, creating a hierarchy (Fig. 2C). For example, four homologous recombination (HR) proteins BRCA1, BRCA2, RAD51, and BLM associated under a stringent IAS threshold, reconstituting a variant of the BRCC complex (61) (which we called the “BRCC/BLM complex”). As the IAS threshold decreased, this complex expanded to encompass a larger group incorporating BARD1, BUB1B, FANCD2, PLK1, RAD51C, and TP53 (“Extended BRCC complex”) which then consolidated with a distinct assembly containing BRCA1, BLM, TOP2A, CHEK1, and CLSPN (“CHEK1 activation”) and other proteins to form a supercluster of 20 proteins broadly involved in HR (Fig. 2D). Rather than merely supply an inventory of HR factors, however, the hierarchical analysis reveals how larger protein assemblies are organized from smaller ones.
When applied to the entire IAS network, hierarchical community detection resulted in identification of a total of 2338 protein assemblies (Data S2). Notably in this analysis, proteins were allowed to cluster in multiple distinct assemblies when such affiliation was supported by the interaction data. For example, β-catenin (CTNNB1) has well-established pleiotropic functions, with separate roles in the β-catenin destruction complex and adherens junction (62); accordingly, its interaction patterns placed it in distinct assemblies corresponding to each of these two aspects (Fig. S1D, E). Since small assemblies tended to correspond to protein complexes and signaling scaffolds, while larger groups more closely represented broad cellular processes, we adopted the general name “protein systems” to describe entities at any scale.
Identifying Recurrently Mutated Systems at Multiple Scales
We next sought to identify which systems were under pressure for recurrent mutations in cancer. Given a list of systems, each consisting of a set of proteins, a straightforward analysis might be to test for statistical enrichment of mutations within each system individually (63). However, for overlapping or nested systems, such tests are confounded because mutations impacting one system create correlated enrichments in other systems with overlapping components. Our goal was to determine whether the gene mutation frequencies in a cancer cohort were best explained by separate selection pressures on individual genes, or more parsimoniously by pressure on a small set of systems. For this purpose, we developed a unified statistical model of mutation, called HiSig, to determine a set of systems that optimally explains the observed mutation pattern for all genes while seeking to minimize the number of systems required for such explanation (Figs. 3A, S2). The HiSig model accounts for overall mutation burden, protein length, and other factors when analyzing mutation patterns to arrive at an expected mutation frequency but does not attempt to model phenotypic impacts of mutation (Materials and Methods). The significance of mutational enrichment was evaluated using permutation testing at a fixed false discovery rate (FDR, Materials and Methods).
Fig. 3. Convergence of mutations within protein systems.
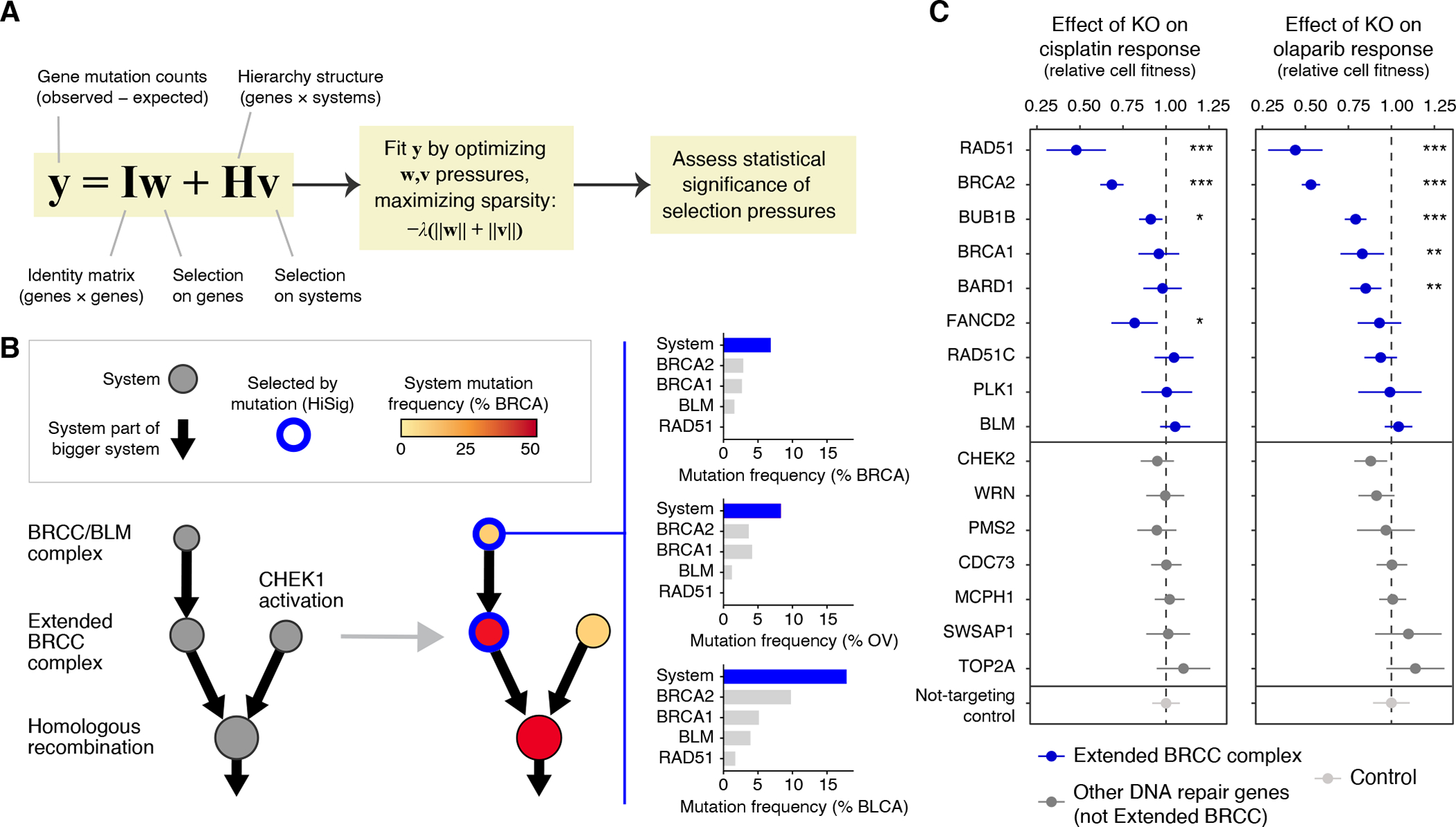
(A) HiSig model for identification of significantly mutated systems. A regularized lasso regression model is fit to the observed mutation counts (y) of genes in a cohort, by adjusting mutation pressures (w, v) on genes and systems, respectively. The λ term penalizes models that lack sparsity, i.e., placing pressures on many genes or systems. Systems harboring frequent mutations that are not well-explained by pressures on any single gene will have positive pressures v after solving the penalized regression. (B) Hierarchy of systems related to homologous recombination, extracted from the IAS network and analyzed for mutation frequency in breast cancer (BRCA, red color gradient on nodes). Blue node borders indicate significantly mutated systems selected by HiSig. Bar charts at right show system- and gene-level mutation frequencies for the “BRCC/BLM complex” in breast, ovarian and bladder cancers (BRCA, OV, BLCA). (C) Effects of CRISPR-Cas9 gene knockouts (rows) on the response to cisplatin (left) or olaparib (right) in the SW1710 bladder cancer cell line. Error bars: mean ± 95% CI. The cell fitness resulting from each knockout is compared to that of non-targeting controls via t-test with Benjamini-Hochberg multiple-testing correction, with significant differences indicated: *: P < 0.05; **: P < 0.01; ***: P < 0.001.
As an example of mutational analysis at the systems level, we again turned to homologous recombination (HR) and its subsystems as discussed in the previous section. HR defects can be caused by driving loss-of-function mutations in BRCA1, BRCA2 and related proteins in an effect that has been called “BRCAness” (64, 65). Consistent with this expectation, HiSig identified significant mutational pressure on the “BRCC/BLM complex” and “Extended BRCC complex” in breast and ovarian carcinomas, two tumor types for which BRCAness had been well-studied (64, 65) (Fig. 3B). While mutations in individual proteins in the BRCC/BLM complex were rare and failed to achieve strong statistical significance, with < 3% for each gene in breast tumors (66), mutations in the four BRCC/BLM genes converged for an aggregate mutation frequency of 7%, exceeding random expectation (95% of confidence interval: 0.4%−3.6%; P = 0.0008, log-normal distribution). Thus, the systems-level analysis was able to recognize evidence of a well-known systems-level effect, BRCAness, despite the lack of strong signals from individual genes.
HiSig also identified mutational selection for the same HR systems in bladder urothelial carcinoma (Fig. 3B, 18%; versus 95% of confidence interval of random expectation: 1.2%−11.1%; P = 0.0029, log-normal distribution), a tumor type for which significant mutation rates of individual BRCAness proteins had not previously been identified (67) perhaps due to a higher background mutation rate in this cancer. To corroborate this finding, we performed CRISPR-Cas9 disruptions to genes encoding the “Extended BRCC complex” in bladder cancer cells and scored each for sensitivity to olaparib or cisplatin, which are phenotypes indicative of HR deficiency and thus BRCAness (68). Six gene disruptions to this system caused sensitization to one or both drugs, as compared to a control set of gene disruptions none of which produced significant sensitization (Fig. 3C).
A Cancer Systems Map Integrating 13 Tumor Types
We applied HiSig to analyze somatic mutation patterns from 6,251 exomes, representing 13 tumor types with sufficient sample numbers and mutation burdens (37): Bladder Urothelial Carcinoma (BLCA), Breast Invasive Carcinoma (BRCA), Colorectal Adenocarcinoma (COAD), Glioblastoma (GBM), Head and Neck Squamous Cell Carcinoma (HNSC), Kidney Renal Clear Cell Carcinoma (KIRC), Liver Hepatocellular Carcinoma (LIHC), Lung Adenocarcinoma (LUAD), Lung Squamous Cell Carcinoma (LUSC), Ovarian Serous Cystadenocarcinoma (OV), Skin Cutaneous Melanoma (SKCM), Stomach Adenocarcinoma (STAD), and Uterine Corpus Endometrial Carcinoma (UCEC). HiSig analysis identified 319 systems with evidence for mutational selection in one or more cancer types. To unify these systems into a pan-cancer hierarchy, we added sufficient higher-order systems to link them, yielding a final map of 395 protein systems which we called NeST (Nested Systems in Tumors, http://ccmi.org/nest/; Fig. 4A, Data S3). Systems were named by a team of in-house curators using a combination of expert knowledge, literature analysis, and gene set enrichment (Materials and Methods). In general, NeST organizes numerous small systems with tissue-specific mutation patterns within a few large systems relevant to greater numbers of tumor types (e.g. cell cycle progression, immune systems, and transcription, Fig. 4A), providing a systematic reconstruction of hallmark processes altered in cancer (69).
Fig. 4. Pan-cancer map of mutated systems (NeST).
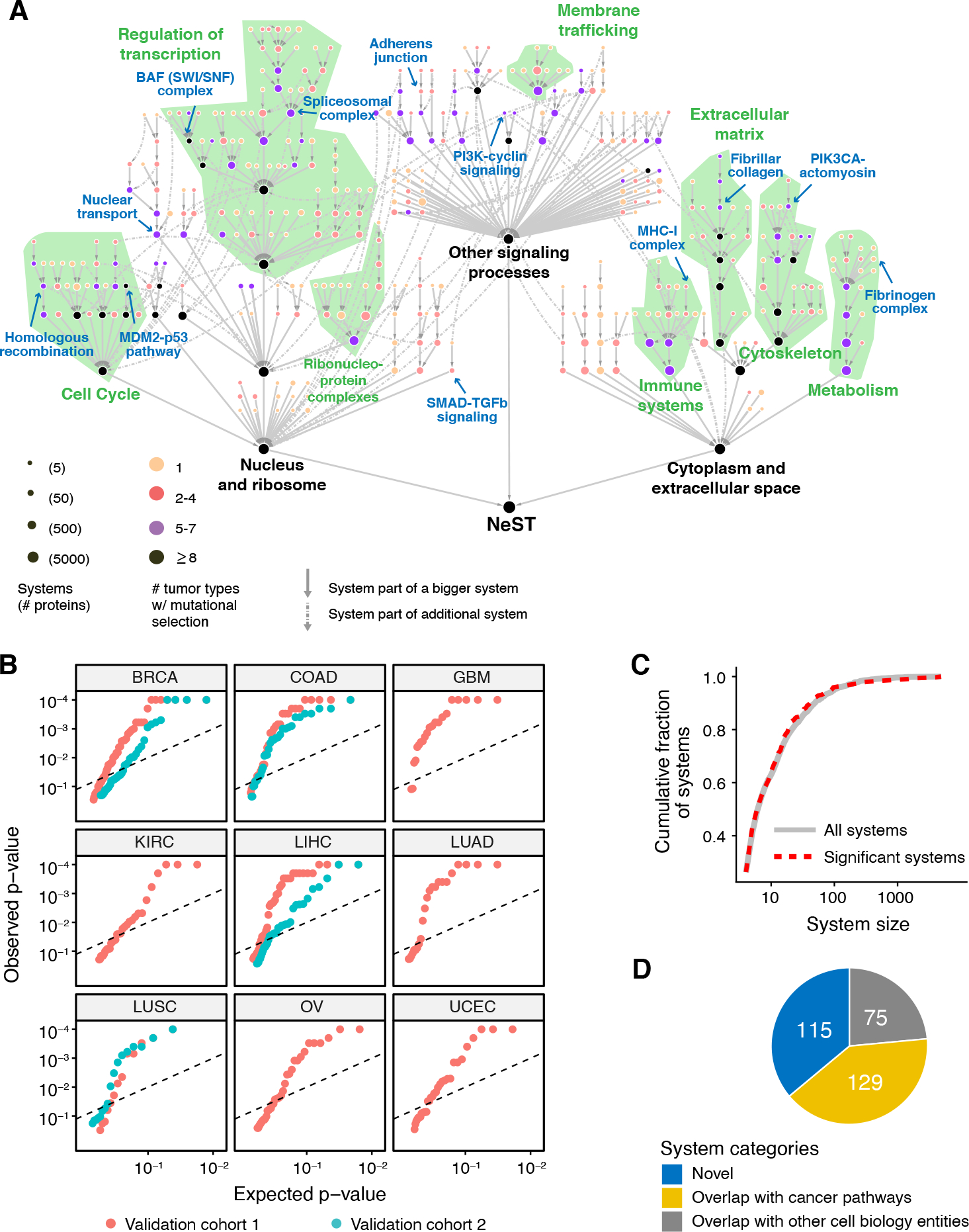
(A) NeST map assembled from the union of significantly mutated systems identified by HiSig in each of 13 tumor types. Nodes represent protein systems, with color indicating number of cancer types for which a system is mutated. Gray arrows between systems (s→t) show hierarchical containment of the first system (s) in the second (t). For systems contained by two or more larger ones (representing pleiotropy), these additional containment relations are shown as dashed arrows. Green text and regions show correspondence to the large-scale components of the IAS network in Fig. 2B. Blue text and arrows refer to examples highlighted in text or figures. (B) Recapitulation of significantly mutated systems in independent validation cohorts. For each system (red points), the qq-plots show HiSig p-values of recurrent mutation as determined in the validation cohort versus the values expected by chance. The 3×3 plot grid shows validation in nine tissue types. When multiple validation cohorts are available for a tissue type, values for the second validation cohort are shown in cyan. Dashed line shows a 10% FDR cutoff. (C) Numbers of proteins per system for significantly mutated systems (red dashes) versus all systems (black) extracted from the IAS network. (D) Composition of NeST systems by relationships to prior knowledge of cell biology and cancer pathways.
To systematically validate the collection of recurrently mutated NeST systems, we examined whole-exome sequencing data from independent patient cohorts representing nine tumor tissue types, for a total validation set of 4077 tumor exomes (Materials and Methods). Per tissue type, we observed that 37%−66% of NeST systems were also recurrently mutated in the tissue-matched validation cohorts (Benjamini-Hochberg FDR < 0.1 by HiSig; Figs. 4B, S3A; Table S3). This range of validation percentages was comparable to that of validating individual mutated genes and was significantly better than achieved if the tissue types used for discovery and validation were decoupled and randomized (e.g., pairing a BRCA validation cohort with the mutated systems discovered for LUSC; Fig. S3A).
The number of recurrently mutated systems in each cancer type ranged from 11 (SKCM) to 84 (BLCA) and was anticorrelated with the genome-wide mutation burden of each type (Spearman ρ = −0.59; P = 0.03; Fig. S3B), reflecting the difficulty of demonstrating significant mutation rates in very highly mutated cancers (70). Notably, the size distribution of recurrently mutated systems (as determined by HiSig) was similar to that of all systems (Fig. 4C), suggesting that selective pressures operate at all scales of cell biology rather than at one resolution only, such as protein complexes or narrowly defined pathways. Application of HiSig to alternative hierarchies of human protein systems curated from literature (Gene Ontology, Fig. S3C) identified significantly fewer mutated systems, demonstrating the specific value and relevance of NeST as a resource for the study of cancer. We also repeated HiSig analysis on the IAS-derived hierarchy, considering transcription-altering copy number alterations (CNAs) rather than point mutations and indels (see Supplementary Text). This analysis found 20 additional systems enriched for CNAs (Table S4), including a cyclin-containing system containing CDKN2A (p16/ARF) and a transcriptional regulator complex containing EP300 and CREBBP (Fig. S4).
Comparing NeST to our recent comprehensive literature survey of published cancer pathways (71), we saw that 40% of significantly mutated systems (129/319) recapitulated an established cancer mechanism (Fig. 4D; Table S5). Examples of these 129 established systems included “PIK3-cyclin signaling,” which integrated mutations in subunits of the PI3K holoenzyme (PIK3CA, PIK3R1, PIK3R2) with mutations in the downstream oncoprotein cyclin D1 (CCND1), as well as “SMAD-TGFβ signaling,” a system encompassing mutations to SMAD transcription factors, the TGFβ receptor TGFBR1, and functionally related proteins. While both of these complexes contained individual cancer proteins with high mutation rates (PIK3CA, SMAD4), mutations in other proteins in these complexes (PIK3R1, SMAD3) had been too rare to meet the significance thresholds of previous analyses (37) despite having functionally validated roles in cancer (72).
Among the remaining 60% of systems (190/319), 75 recapitulated well-known cellular components that had not been previously associated with recurrent cancer mutations, whereas 115 were best described as novel protein assemblies (Materials and Methods). Below, we investigate several of the discoveries in greater detail with biophysical and functional assays, exemplifying the use of NeST to generate biological hypotheses and inform their investigation.
A PIK3CA-Actomyosin Assembly Regulating the PI3K/AKT Pathway
HiSig identified recurrent mutations in a novel variant of the actomyosin complex, a cytoskeletal component regulating cell shape, motility, and membrane organization (73, 74). Multiple direct physical interactions from the AP-MS data newly linked actomyosin proteins with the p110α subunit (PIK3CA) of phosphatidylinositol 3-kinase (PI3K), forming a system we named “PIK3CA-actomyosin complex” (Fig. 5A). This system was under significant mutational pressure in five cancer types (BLCA: 36%, BRCA: 38%, COAD: 42%, HNSC: 29%, STAD: 32%), integrating frequent mutations in PIK3CA with less common mutations in numerous actomyosin genes including non-muscle type II myosins (NM2 proteins MYH9 and MYH10; Fig. 5B). This mutation pressure was seen to validate in BRCA and COAD independent cohorts (Fig. S5A; Table S3; large secondary cohorts were unavailable for BLCA, HNSC, STAD). While cytoskeletal remodeling is a downstream effect of PI3K/AKT signaling (75, 76), actomyosin proteins had not been previously shown to physically associate with PIK3CA, nor had their mutations in cancer been widely studied.
Fig. 5. A PIK3CA-actomyosin assembly regulating the PI3K/AKT pathway.
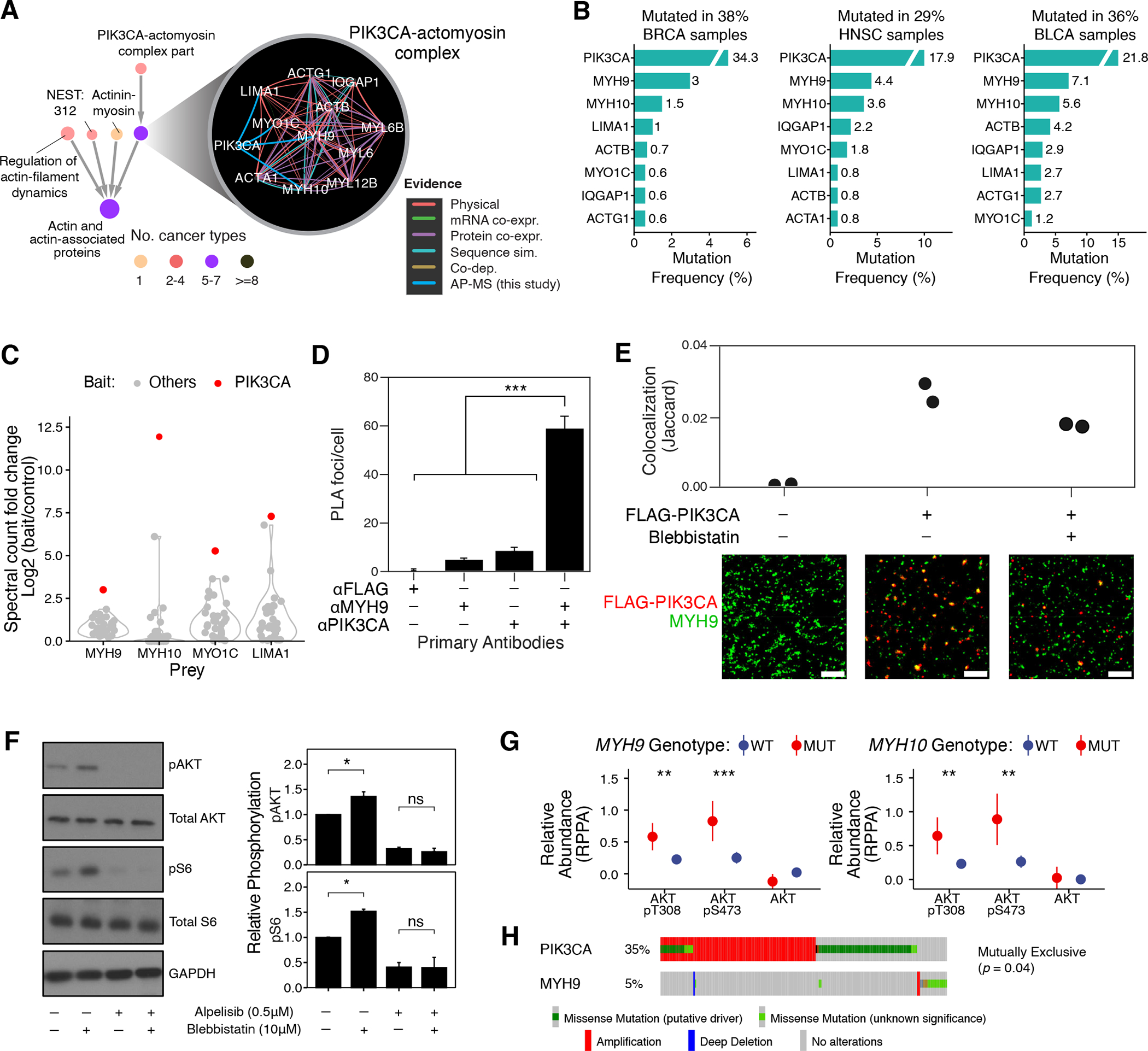
(A) Interactions defining the PIK3CA-actomyosin complex and the context of this system in the NeST hierarchy. (B) Mutation frequencies of genes in this system (shown top seven frequently mutated genes in each cohort). (C) Signal (spectral count fold change versus control) of actomyosin proteins (MYH9, MYH10, MYO1C, LIMA1) in AP-MS experiments with cancer protein baits. Red dots represent signal as interactors of PIK3CA in the CAL-33 cell line; gray dots show corresponding data for other cancer proteins. (D) Per-cell counts of proximity ligation foci when probing for PIK3CA and MYH9 in CAL-33 cells; significantly more foci are observed when probing for both proteins. ***: P <0.001 by one-tailed Student’s T test. (E) CAL-33 cell lines expressing FLAG-PIK3CA (as used in AP/MS experiments) were probed with anti-FLAG and anti-MYH9 antibodies and imaged by DNA-PAINT. Substantial colocalization is observed in small, membrane-proximal puncta. Representative subfields are shown below; scale bar is 500nm. (F) CAL-33 cells were treated with DMSO, blebbistatin (10 μM) and/or alpelisib (0.5 μM) and harvested with indicated antibodies for immunoblotting (N = 3; a representative image is displayed). Signals were normalized relative to total protein and loading control. Error bars indicate mean ± standard error; *: P < 0.05 by one-way ANOVA; images are representative of 3 independent experiments; N = 6 for all measurements. (G) Comparison of the RPPA-measured abundance of indicated phosphorylated/total proteins between cancer cell lines (78) (N = 899) with MYH9 (N = 108) or MYH10 (N = 82) mutations and the wild-type lines. (H) Genomic alterations of PIK3CA and MYH9 in the TCGA-HNSC cohort show a pattern of mutual exclusivity. Wilcoxon rank sum test was used for G and H. *: P < 0.05; **: P < 0.01; ***: P < 0.001; ****: P < 0.0001.
In support of the physical association, we found that actomyosins bind specifically to PIK3CA and generally not to other cancer protein baits assayed by our AP-MS experiments (Fig. 5C). We were also able to validate the association by proximity ligation assay, demonstrating that PIK3CA and MYH9 colocalize in CAL-33 cells (Fig. 5D). To determine more precisely where this colocalization occurs, we performed super-resolution microscopy and found both molecules associated in small, membrane-proximal puncta (Figs. 5E, S5C).
We next investigated the functional consequences of PIK3CA-actomyosin physical interaction by assaying canonical readouts of PI3K signaling, phosphorylation of AKT (p-AKT) and of ribosomal protein S6 (p-S6), in response to NM2 inhibition by blebbistatin (77). Blebbistatin treatment increased the level of p-AKT and p-S6 in CAL-33 cells, but this effect was suppressed by additional treatment with alpelisib, an FDA-approved PIK3CA inhibitor (Fig. 5F; Materials and Methods). Conversely, in SCC-25 cells, where PIK3CA-actoymosin interactions were not observed, blebbistatin treatment did not affect either readout (Fig. S5D). PIK3CA inhibition by NM2 was further supported by Reverse Phase Protein Array (RPPA) data for 899 cell lines from the Cancer Cell Line Encyclopedia (CCLE) (78), which showed that cells harboring MYH9 or MYH10 mutations have significantly elevated p-AKT in comparison to cells lacking such mutations (Fig. 5G). Furthermore, genomic alterations in MYH9 and PIK3CA were mutually exclusive, a sign of functional dependency (P < 0.05; Fig. 5H). Together, these results suggest that the actomyosin complex directly inhibits PIK3CA signaling.
Beyond the example of the PIK3CA-actomyosin complex, we noted that many of the systems in NeST were driven by new protein interactions from our AP-MS screens (112 systems; Fig. S6A; Table S6). For example, new AP/MS experiments showed MAP kinase proteins copurifying with HLA-A to form a new protein system (Fig. S6B), suggesting that known crosstalk between the MAPK pathway and major histocompatibility complex internalization may be mediated by direct MAP kinase-HLA interaction (79). Enrichment for the new interactions was greatest in systems mutated in BRCA (31 systems) and substantial in HNSC tumors (25 systems), the two tumor types used for AP-MS data generation; such enrichment was also observed in other tumor types (Fig. S6C).
Destabilizing Mutations in Collagen Systems Promote Tumor Progression
HiSig detected selection pressure on a system consisting of 11 fibrillar collagen proteins in 7 cancer types, with particularly high mutation frequencies for SKCM (71%), LUSC (49%), and LUAD (49%) (Fig. 6A–C). Recurrent mutations in this system were validated in independent cohorts of lung cancers (Fig. S7B; Table S3). Fibrillar collagens are the most abundant proteins of the extracellular matrix (80, 81), the structure and composition of which can promote a tumor microenvironment conducive to invasive growth (77,82,83). Although differential expression of collagen proteins between tumor and normal tissues had been reported (84), thus far somatic mutations in collagens had not been associated with cancer, save for very rare tumor types (85, 86).
Fig. 6. Destabilizing mutations in collagen systems promote tumor progression.
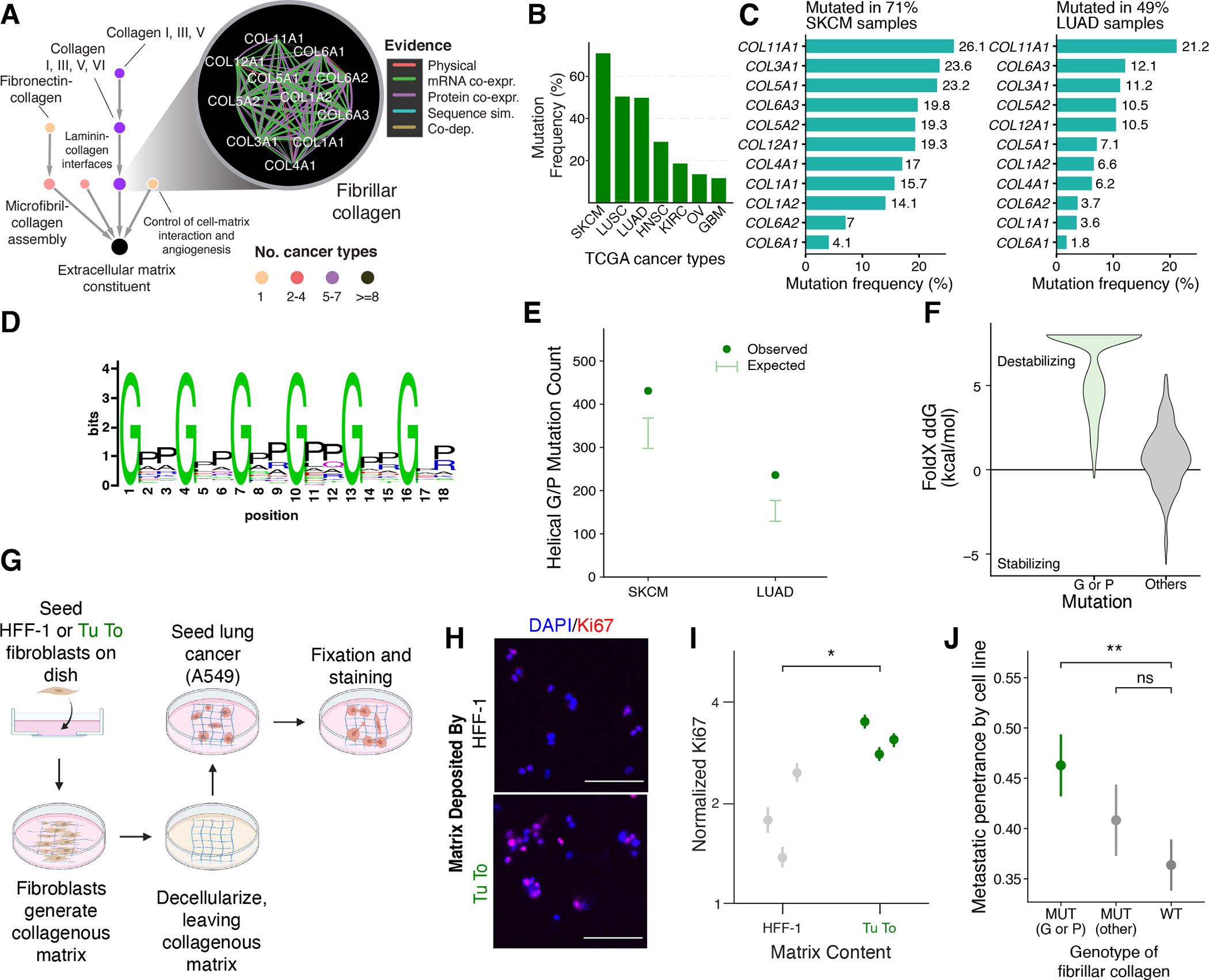
(A) Interactions defining the fibrillar collagen system and the context of this system in the NeST hierarchy. (B) Mutation frequencies of this system across TCGA tumor cohorts in which the system was identified by HiSig. (C) Mutation frequencies per collagen protein in TCGA melanoma (SKCM) and lung adenocarcinoma (LUAD) cohorts. (D) Amino-acid sequence tendency of the collagen triple-helix repeats. Image created based on the PFAM domain PF01391. (E) dN/dS analysis of collagen mutations in SKCM and LUAD cohorts. Error bars represent 95% confidence intervals based on a binomial distribution according to the background mutation rates of collagen genes. (F) Protein stability change upon point mutations on the Glycine/Proline positions (G/P) versus other positions in the triple-helix repeats, predicted by FoldX 5.0 (Materials and Methods) (88). (G) Schematic of matrix-deposition assay. (H) Immunofluorescence images of A549 cells on matrix deposited by the indicated cell lines. Scale bars: 100μm. (I) Quantification of immunofluorescence across three biological replicates (points) for each condition. Error bars indicate 95% confidence intervals. For HFF-1, N = 445, 860, 774; for Tu To, N = 2316, 2114, 1810. *: P < 0.05 by one-tailed Student’s T Test. (J) Associations between fibrillar collagen mutation status and the metastatic penetrance of cancer cell lines (87) (N = 146, 123, 219), assessed by Wilcoxon rank sum test *: P < 0.05; **: P < 0.01; ns: not significant.
Collagen proteins contain helical repeat regions rich in glycine and proline residues (Fig. 6D), and these domains are critical for proper folding of collagen into trimeric bundles (80). In SKCM and LUAD, we found that non-silent mutations in collagens significantly tend to modify these glycine or proline codons, even when correcting for the abundance of C or G transversions in these cancers (87) (Fig. 6E). Computational modeling (88) predicted that the specific mutations observed in tumor samples strongly destabilize collagen proteins (Fig. 6F).
We therefore hypothesized that mutations of collagens may promote tumor progression by disrupting protein folding and thus organization of the extracellular matrix. To test our hypothesis, we allowed fibroblasts with or without G/S mutations in collagen genes (Tu-To or HFF-1 cell lines, respectively, see Supplementary Text) to deposit matrix in culture vessels. After decellularizing this matrix, we seeded it with A549 LUAD cells and observed expression of Ki67, a standard marker of cell proliferation (Fig. 6G; Materials and Methods). We found greatly increased cell proliferation specifically in the collagen-mutant matrix, suggesting that collagen mutations can perturb the cellular environment to favor tumor growth (Fig. 6H, I). We also compared matrix deposited by HFF-1 fibroblasts that were engineered to overexpress COL1A1 with and without the G281S mutation, and we similarly observed increased proliferation of A549 cells on the mutant matrix (Fig. S7B, C). In yet further supporting analysis, we found that collagen mutations are associated with increased metastasis in mouse xenograft models (89) (Figs. 6J, S7D). Together, these data support a role for collagen mutations in disrupting the tumor microenvironment to favor cancer progression.
Protein Systems as Clinical Biomarkers
We examined the extent to which NeST systems could serve as prognostic biomarkers, nominating those systems in which several criteria were met: 1) HiSig identifies the system as under mutational selection in a given tumor type; 2) significant differences in survival are observed between patients with mutations in the system and those without; and 3) the survival association cannot be trivially explained by mutations in any single gene in the system. By these criteria, and with additional correction for mutational burden (Materials and Methods), we identified a total of 25 prognostic associations (FDR < 0.3, Fig. 7A; Table S7). Among the prognostic systems, we observed poor prognoses in GBM tumors associated with mutations in a system integrating PI3K components (PIK3CA, PIK3R1, PIK3CB) with GRB2 and its binding partner GAB1 (“Proximal receptor tyrosine kinase (RTK) signaling”, Fig. 7B, C). While mutations in PIK3CA and PIK3R1 were each weakly associated with the progression-free survival time, integrating these mutations resulted in a stronger prognostic effect (Fig. 7C). Another prognostic system, representing histone functions in the DNA damage response, brought together 15 proteins which individually had low mutation frequencies (< 3.5%) and no prognostic association. However, integrating all of these mutations led to an unexpectedly high rate of mutation (14.3%) with poor prognosis in COAD tumors (Fig. 7D, E).
Fig. 7. Protein systems inform clinical markers and lists of cancer genes.
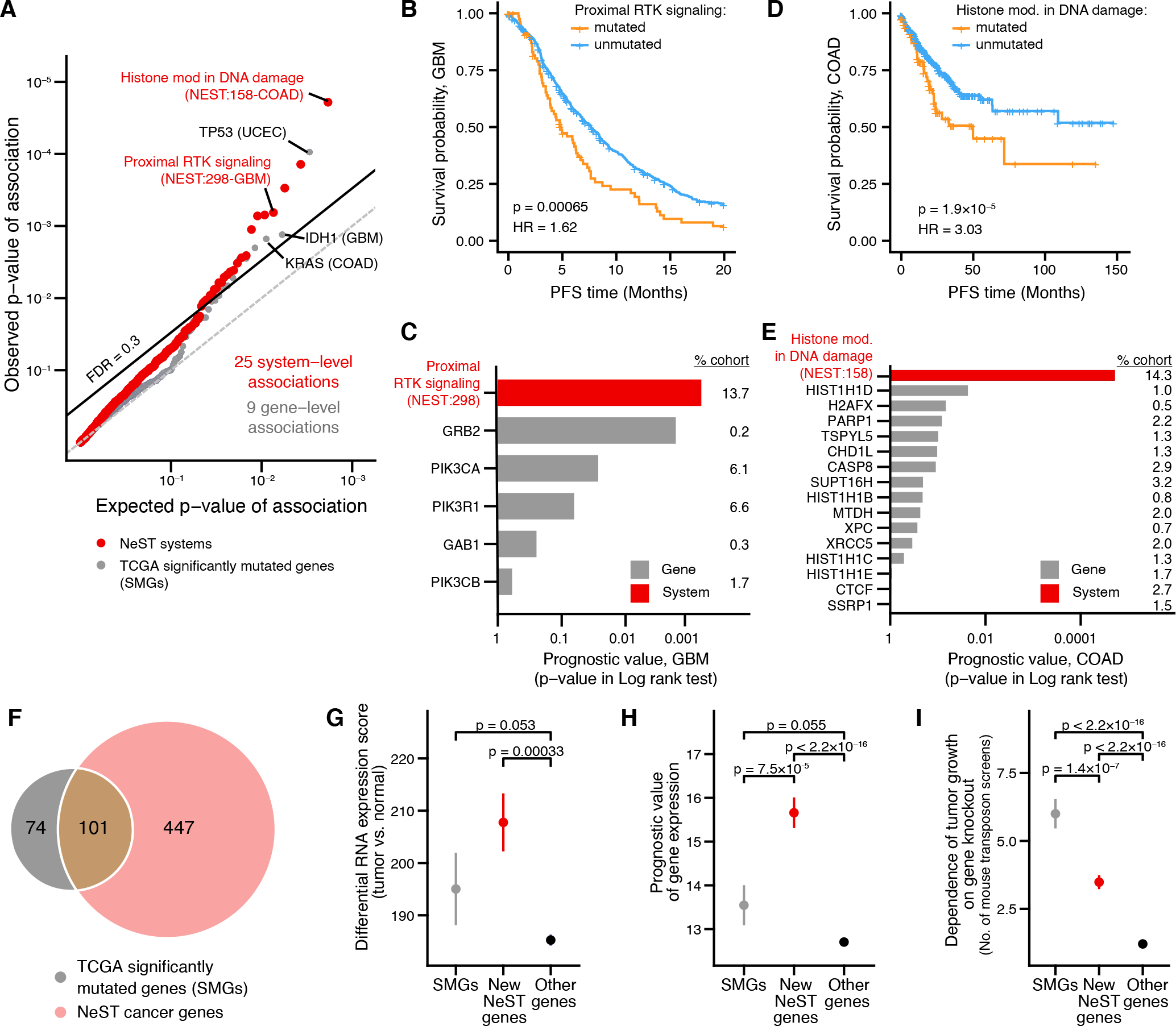
(A) Significance of prognostic association between biomarker status (mutated/unmutated) and progression free survival (PFS). Red points represent systems; gray points represent genes. Observed P-values are plotted versus those expected when performing the same number of tests at random. The dashed line indicates a cutoff of FDR = 0.3. Relevant tissue type for each association shown in parentheses. (B-E) Selected NeST systems associated with prognosis: (B, C) NeST:298, “Proximal RTK signaling.” (D, E) NeST:158, “Histone modification in DNA damage response.” Kaplan-Meier curves (B, D) stratify tumors based on mutation status of each system. Lower bar charts (C, E) indicate significance of association (log-rank test) between mutation status and PFS for systems (black bars) versus individual genes in those systems (gray bars) Numbers on the right indicate the percentage of patients in that cohort with mutations in the indicated gene. (F) Venn diagram showing the common and distinct genes identified by significantly mutated systems in NeST (pink) versus TCGA PanCancer analysis (gray) (37). (G-I) Functional support for NeST cancer genes: (G) RNAseq tumor-normal differential expression in TCGA (126). (H) Prognostic value of mRNA expression in TCGA (127). (I) Number of times a gene has been identified in independent cancer genetic screens in mice (128). Whisker plots show mean ± stderr. Asterisk (*) indicates significant difference by one-sided Wilcoxon rank sum test, P < 0.05.
While the preceding analyses demonstrate advantages of interpreting cancer genomes through a map of protein systems, lists of cancer genes remain very useful for clinical applications, such as the design of diagnostic gene panels. In this respect, we considered that the recurrently mutated systems identified in NeST might include many genes not implicated in current cancer gene panels or databases (37, 72, 90, 91). The designation of a NeST system requires mutations in multiple constituent genes that cannot be better explained by any other system (HiSig approach, Materials and Methods). Thus, we conservatively nominated two genes per system having the highest relative mutation frequencies, yielding a non-redundant catalog of 548 NeST cancer genes (Table S8). This NeST systems gene catalog covered 101 of 179 significantly mutated genes (SMGs) reported in previous TCGA cancer-driver analyses for the 13 tissues studied (37); the remaining 78 SMGs were not components of a larger system under selection. Moreover, our analysis nominated 447 genes not previously associated with cancer by their somatic mutations (Fig. 7F). As an ensemble, mutations in NeST cancer genes were predicted as less deleterious than mutations in TCGA SMGs, while significantly more deleterious than mutations in background genes not in either category (Fig. S8). Conversely, NeST cancer genes were differentially expressed between tumor and normal samples to a greater extent than previously reported SMGs (Fig. 7G) and, individually, their expression levels offered greater prognostic value (Fig. 7H). Additionally, orthologs of both NeST genes and SMGs were significantly enriched for tumor growth effects in transposon-based forward genetic screens in mice (86) (Fig. 7I; Materials and Methods).
Discussion
In their original description of the “Hallmarks of Cancer”, Hanahan and Weinberg (92) predicted that “complexities of cancer, described in the laboratory and clinic, will become understandable in terms of a small number of underlying principles.” Independently, in his seminal vision regarding the future of molecular biology (93), Bruce Alberts described the cell as a “collection of protein machines” where “each of these protein assemblies interacts with other large complexes of proteins in an elaborate network.” Our joint study, which includes the accompanying manuscripts (Swaney et al. and Kim et al.), represents a systematic effort to combine these two visions– organizing the diverse mutation patterns of cancer into underlying principles represented by a multi-scale map of complexes and larger protein machines. This map, NeST, derives from an end-to-end data generation and analysis pipeline consisting of protein network collection, multi-omics integration, structural and predictive modeling, and model visualization (Fig. 1B). It serves not only as a useful abstraction for understanding cancer cell biology, but as a tool to discover new protein systems and their association with cancer.
Defining a collection of biological systems requires recognizing physical and functional boundaries between these entities, a process with inherent ambiguities. Even when assigning mutations to genes, the gene boundary might or might not include introns, alternatively spliced exons, promoters, or enhancers. Likewise, the systems in NeST are defined based on evidence that the proteins are densely connected by interactions of a certain stringency. One source of ambiguity concerns the parameters used for community detection. In constructing NeST, we optimized these parameters with respect to the ability of systems to explain mutation rates, although other means of defining systems boundaries could be explored. Nonetheless, we found that most systems could be validated by mutation patterns in second cohorts (Fig. 4B) and many of the associated genes had independent functional evidence (Fig. 7F–I), supporting the relevance of the current pipeline.
Comprehensive interaction screens, directed to a panel of 61 cancer proteins in multiple cell lines (citations to be updated), contributed greatly to the discovery of protein systems in this study. In general, physical protein interaction strongly informed IAS scores (Fig. S1A, C), and the specific baits chosen for AP/MS were known cancer proteins in cancer cells. While new interaction data from BRCA and HNSC cell lines were critical to defining protein systems mutated in those diseases (Kim et al., Swaney et al., accompanying manuscripts), they also contributed to protein systems mutated in other cancer types (Fig. S6). Similar observations have emerged from projects such as ENCODE, in which analysis of chromatin factors in selected cell lines has defined transcriptional regulatory networks with broad relevance across tissues and diseases (94, 95). These findings suggest that additional interaction screens in a modest number of cellular contexts might achieve reasonable coverage of protein complexes driving most cancer types.
Beyond recapitulating known cancer mechanisms, the NeST map identifies a number of recurrently mutated systems that do not overlap significantly with previously published cancer pathways (71) (Fig. 4D; Table S5). Such systems might be newly identified for one or more of the following distinct aspects of the present study: (i) the expanded content and scope of protein-protein associations in the input network, which integrates new AP-MS experiments targeting cancer protein interactions with a compendium of diverse multi-omics data; (ii) the explicit identification of distinct protein assemblies in the network (systems) over a continuum of scales; and (iii) the scoring of mutation significance at the level of systems rather than genes, in a manner that is distinct from approaches such as network propagation or gene set enrichment.
NeST serves as a resource for tumor genome interpretation and prognosis. The search for a well-defined and interpretable set of causative mutations and prognostic markers has been hindered by the “long tail” of cancer mutations (1) (Fig. 1A). This distribution of mutations across genes simultaneously makes candidate mutations numerous and statistical tests to assert their significance underpowered. By rationally consolidating mutations into fewer classes that maintain functional association, fewer statistical tests are required and each class contains more samples. Indeed, we are able to identify more prognostic systems than prognostic gene-level mutations (Fig. 7A). As interactome-mapping and patient-sequencing efforts continue over the next years, we expect to improve our identification of protein systems and their associations with cancers.
Finally, while this study has focused on the analysis of the cancer genome, cancer is by no means the only disease in which diverse mutations converge on a narrower set of common processes and phenotypes. The concept of a hierarchy of protein systems, likewise, is not specific to cancer cells or their cell types of origin. The analysis presented here thus serves as a model to understand the large array of other diseases influenced by complex genetic alterations to somatic cells or to the germline.
Materials and Methods
Targeted Interrogation of Cancer Protein Networks by AP-MS
Experimental acquisition of AP-MS data for 61 cancer proteins (baits) is described in the two companion papers (citations to be updated). Briefly, protein spectral counts were determined by MaxQuant (96) and used for PPI confidence scoring by two complementary algorithms, SAINTexpress (97) and CompPASS (98, 99). Outputs of these two algorithms were combined into a unified metric called the PPI score. We retained interactions above a PPI score cutoff (0.8 for breast cell lines, 0.9 for head and neck cell lines), resulting in 1722 non-redundant interactions discovered in one or more of six cell lines (Table S1).
Acquisition of Network Data in Major Categories of Experimental Features
In addition to the new AP-MS data, we considered major types of protein pairwise associations recorded in public databases: physical protein-protein interaction, mRNA co-expression, protein co-expression, co-dependence, and sequence-based relationships. Data obtained under each of these feature categories resulted in a total of 127 individual features on human protein pairs (Table S2), described below. As the namespace for human proteins, we used the symbols of protein-coding genes in the HUGO Gene Nomenclature Committee database (HGNC) (100), yielding 19,035 distinct proteins. We converted gene and protein names from other namespaces (Ensembl, Entrez, Uniprot) to HUGO nomenclature during the preprocessing of networks.
(1). Physical protein-protein interaction (PPI) networks.
PPI networks were drawn from five recent data-driven studies: (1) BioPlex 2.0, based on immunoprecipitation of tagged proteins followed by detection of interactors by mass spectrometry (MS) (30, 98); (2) A network based on MS that considers interaction stoichiometry and cellular abundances of interactors (50); (3) hu.Map, based on protein co-fractionation followed by MS (29); (4) Human Reference Interactome (the “HI-II-14” dataset), a comprehensive yeast-two-hybrid network (51); (5) BioGRID, a literature-curated PPI database (49), restricted to high-confidence PPIs (the “multi-validated” category) and excluding entries based on high-throughput techniques to reduce redundancy with the other four networks. Each PPI network was sparse, with direct interaction relationships for a minority of protein pairs. However, two proteins that fall in the same network neighborhood may be associated with one another through network proximity, even if they are not directly connected by physical interaction. Thus, each of the five networks mentioned above were processed using a network embedding algorithm, node2vec (101), which represents the network neighborhood of each node (i.e. protein) as a high-dimensional vector and calculates the Euclidean distance between these vectors for each node pair. We specified 32 node2vec dimensions with all other parameters set to default. Each PPI network thus contributed one sparse feature (the original network) and one dense feature (the embedded pairwise distances), for ten features in this category in total.
(2). mRNA co-expression networks.
Since the amount of available mRNA co-expression data is currently higher and more diverse than for other types, we split these datasets into three categories: co-expression in cell line collections (36 features); co-expression in human tumor samples (28 features); and co-expression in healthy human tissues (28 features). RNA expression levels in cell lines were obtained from the CCLE (Cancer Cell Line Encyclopedia) and GDSC (Genomics of Drug Sensitivity in Cancer) projects (102, 103). RNA expression levels for human tumor samples were obtained from TCGA (The Cancer Genome Atlas) studies documented in the cBioPortal repository (104). RNA expression levels for healthy human tissue samples were obtained from the GTEx (Genotype-Tissue Expression) data portal (105). Genes with very low expression levels (median TPM <= 1) in these data sets were excluded. The Pearson correlation coefficients of expression were calculated for all remaining gene pairs across all samples in a collection, and also across subsets of samples from the same tissue of origin if the subset size was larger than 40 (104, 105).
(3). Protein co-expression networks.
In parallel to RNA expression, protein expression levels were obtained from CPTAC studies of breast and ovarian tumor samples (54, 55) and a study of breast cancer cell lines (51). The Pearson correlation coefficients were calculated for all pairs of characterized proteins.
(4). Co-dependency networks.
For each human gene, we accessed its gene dependency profile, the vector of fitness scores resulting from CRISPR knockdown of that gene across a panel of 485 cell lines. These data were obtained from the DepMap project release 18Q3 (https://depmap.org/portal/) (106). We used these data to compute a co-dependency score for each gene pair as the Pearson correlation of the two corresponding gene dependency profiles. A global correlation profile over all cell-line tissue types as well as correlation profiles per tissue of origin were generated. We focused on 7 tissues with more than 20 cell lines, resulting in a total of 8 features.
(5). Sequence-based relationships.
For features based on sequence relationships, such as protein-domain co-occurrence and interactions of orthologous genes in other organisms, we used the log-likelihood score for each evidence code documented in HumanNet 1.0 (58) including CE-CX, CE-GT, CE-LC, CE-YH, DM-PI, HS-DC, HS-GN, HS-PG, SC-CX, SC-GT, SC-LC, SC-MS, SC-TS, and SC-YH (defined in http://www.functionalnet.org/humannet/HumanNet.v1.evidence_code.txt).
Using Network Features to Formulate the Integrated Association Stringency (IAS) score
AP-MS interaction data and the 127 additional network features were used as inputs to a two-stage random forest regression model, trained to best recover the proximity of protein pairs in the Gene Ontology (GO). To compute this proximity we used the GossTo tool (107) to calculate the Resnik semantic similarity score (108) for all protein pairs, based on the GO Biological Process (BP) and Cellular Component (CC) branches as of May 2017. Only evidence codes related to experimental support were used (evidence codes IDA, IPI, IMP, IGI, IEP, TAS, IC) considering “is_a” and “part_of” relationships. This procedure resulted in approximately 9.5×107 protein pairs with semantic similarity values, which were used as training data. In the first model stage, seven random forest predictors were trained, each representing one category of network features (see above), each with 640 decision trees. In the second model stage, the predictions of the seven regression models were used as input features and trained again against semantic similarity values, to produce a final model output. Outputs of both stages used out-of-bag (OOB) predictions, a standard approach for random forest models (109). We also explored standard five-fold cross-validation and did not observe a significant difference in prediction performance versus the OOB procedure. New AP-MS interactions identified for protein pairs (see above) were incorporated by setting the physical interaction PPI feature of that pair to the maximum in the input of the second stage random forest.
The output of this model was taken as the unified IAS score for each protein pair (1.8×108 pairs, which also included protein pairs not used in the training). Random forests were implemented using the Scikit-learn Python package (110) with “max_tree_depth” set to 20. This setting was determined to be near-optimal for most feature categories when sweeping over max_tree_depths of 5, 10, 15 … 35, 40. Comparison between the OOB accuracy and training accuracy showed the OOB procedure did not cause overfitting (Fig. S1A). The parameter “max_features” was set to 1 for categories with fewer than 10 features (protein co-expression and co-dependence) and to 0.5 otherwise. Other parameters were set to default. Network features were set to 8-bit signed integer arrays (NumPy data type “int8”) during the training to reduce running time. The two-stage random forest model performed similarly to an alternative procedure of training on all 127 features simultaneously in terms of correlation to GO proximity score (Fig. S1B). However, the two-stage approach enabled an analysis of the contribution of features by their broad categories (Fig. S1C).
Multi-Scale Protein Network Community Detection
Hierarchical protein community detection was based on the CliXO clique detection algorithm, which was revised for bug fixes, redesigned parameters, and code optimization(111). CliXO inputs a weighted network and outputs a hierarchy of communities detected in that network, as the threshold of edge weight (here, IAS score) is lowered (Fig. 2C, D). The new version of CliXO (v1.0) had better performance than the previous version (v0.3) in a benchmark task, in which algorithms were used to recover a ground-truth GO hierarchy from an input weighted network reflecting the Resnik semantic similarity scores defined by the same hierarchy (Fig. S2A; see also Supplementary Text).
The algorithm consists of three parameters, α, β, and m. The parameter α reflects the step size by which network stringency (threshold IAS score) is lowered for progressive cycles of clique detection; a smaller α tends to generate a deeper hierarchy, in which the differences between parent and child communities tend to be smaller. β reflects the stringency of merging overlapping cliques; a higher β tends to merge cliques less frequently, resulting in a broader hierarchy with more sibling systems with larger overlaps. Finally, each community is assigned a score adapted from the Newman-Girvan modularity (112), and those with a modularity score less than m (i.e. communities that are more likely to emerge by chance) are rejected from the hierarchy (Fig. S2B).
This CliXO algorithm was applied to identify protein communities at moderate-to-high interaction stringency thresholds (IAS ≥ 0.3), which captured the vast majority of protein associations driven by physical interaction (Fig. S1C). Below this threshold, the integrated network had a much higher edge density, leading to impractical run-time requirements. Instead, we used HiDeF, a scalable community detection method we recently developed (113), to efficiently extract large-scale protein systems at low stringency (IAS < 0.3, see Supplementary Text).
HiSig: Identification of Recurrently Mutated Cancer Systems
Given a set of partially overlapping and/or nested systems, each consisting of a set of proteins, we developed a unified statistical model of mutation to precisely pinpoint the systems with strong evidence for mutational selection. This model, HiSig, was inspired by the technique of overlapping group lasso regression (114) albeit with a different mathematical formulation. HiSig code is available online(115).
A comprehensive list of exome-wide somatic mutations identified in the 13 TCGA tumor cohorts considered in this study (BLCA, BRCA, COAD, GBM, HNSC, KIRC, LIHC, LUAD, LUSC, OV, STAD, SKCM, UCEC) was obtained from the NCI Genomic Data Commons as a MAF (Mutation Annotation Format) file (116) (https://gdc.cancer.gov/about-data/publications/mc3-2017).We considered the following types of somatic mutation events: “Missense_Mutation”, “Nonsense_Mutation”, “Frame_Shift_del”, “Frame_Shift_Ins”, “Splice_Site”, “Splice_Region”, and “Nonstop_Mutation” and removed others, such as silent mutations. We did not opt to incorporate any model of phenotypic impact from mutations into HiSig, given the model’s complexity and the general lack of consensus about which method of phenotypic prediction is optimal. For each cohort and gene, we recorded the number of tumors in that cohort in which that gene was observed to have at least one somatic mutation event (Ng_obs). This observed number was compared to the expected number of mutations for that gene and cohort (Ng_exp), computed using MutSigCV 1.4 with default settings (https://software.broadinstitute.org/cancer/cga/mutsig_download) (36). The expected mutation value accounts for covariates of mutation tendency, including gene length, mRNA expression level, replication timing, and trinucleotide context of the mutation, which are integral parts of the MutSigCV statistical model (36). Note that Ng_exp is an internal variable not included in the MutSigCV output, requiring a trivial code modification to access its value. We then defined the corrected mutation count of each gene g as:
with ε = 5 as a pseudocount to avoid taking the logarithm of zero. The vector of corrected mutation counts for all m genes, denoted y, was fit using the following model of mutation pressure, illustrated graphically in Fig. 3A:
In this model, H is an m × n matrix representing the assignment of μ genes to n systems, in which Hij = 1 if gene i is a member of system j and 0 otherwise. I is an m × m identity matrix. The vectors w and v model the positive selection pressures on genes and systems, respectively, that have given rise to the mutation counts in y. Values for these vectors were solved by linear regression with L1 lasso regularization using the R package glmnet (117) with parameters lambda.min = 10−4, nlambda = 500, standardize = False, and lower.limit = 0. These settings produce a family of optimal solutions to w and v under different strengths of regularization λ. Large λ tends to select a few large systems (zero w, sparse v), whereas small λ tends to select every gene as a model of its own mutation counts (dense w, zero v). Each system t, among the set of systems T, was assigned a selective pressure S over all regularization penalties:
For each λ this equation calculates the fraction of the weight of a system t among all weights in the linear equation; the maximum fraction attained is returned as S(t). An empirical p-value was calculated by comparing S(t) of the actual hierarchy against 10,000 random hierarchies in which the hierarchy structure H is permuted with respect to gene labels (i.e., permuting the rows in H). The false discovery rate (FDR) is calculated using the Benjamini-Hochberg procedure with the Python package statsmodels (v0.9). In this study, we defined systems with FDR < 0.25 as recurrently mutated.
To optimize the hierarchical structure for HiSig analysis, we scanned the CliXO parameters (α, β, m, see above), yielding an ensemble of systems hierarchies of varying size and complexity (Fig. S2F, G). The HiSig procedure was applied to each of these hierarchies to determine recurrently mutated systems in each tumor type. Notably, a higher overall number of systems in a hierarchy did not necessarily imply that more of these systems would be scored as under mutation pressure, since larger hierarchies also must test more systems, adversely affecting the FDR. Among this ensemble, we chose the parameters α=0.07, β=0.5, m=0.005, as the hierarchy generated under these parameters was an optimal tradeoff between the model parsimony and the power of detecting more significantly mutated systems (Fig. S5C, D). We used this parameter set for all subsequent analyses.
Naming of NeST Systems
NeST systems were named by a team of in-house curators, based on expert knowledge, literature analysis, and GSEA analysis. Where possible, names were chosen to agree with existing literature about the functional relationship between systems’ substituent genes. The naming process had no influence on the composition of these systems or their inclusion in NeST, which remains a truly data-driven construct.
Independent Cancer Cohorts for Validation
For validation of NeST systems in independent tumor cohorts, we selected 13 tumor cohorts with sufficient whole exome sequencing data (>100 samples per cohort) that were independent of the TCGA cohorts used to define NeST. The validation cohorts include a total of 4077 tumor samples from three ICGC (International Cancer Genome Consortium) datasets (breast, liver, lung; samples from TCGA studies were removed), seven CPTAC (Clinical Proteomic Tumor Analysis Consortium) datasets (brain, breast, colon, lung, kidney, ovary, uterus), and two datasets from focused studies (colon, liver). MAF files recording the somatic mutations of these studies were obtained from the ICGC data portal release 27 (https://dcc.icgc.org/releases/release_27), CPTAC data portal at the Genomic Data Commons (https://portal.gdc.cancer.gov/), and the cBioPortal (104), respectively. The MAF files of ICGC samples were processed by the icgcSimpleMutationToMAF function in the maftools package (118). MAF files were then used as input to MutSigCV 1.4 and followed by HiSig analysis, similar to the procedure described above for analysis of TCGA data.
Each validation cohort was paired with the TCGA discovery cohort of matching tissue type (Fig. 4C). For each of these pairwise comparisons, systems identified by HiSig as recurrently mutated in the discovery cohort (see above) were examined for significance in the validation cohort – if HiSig FDR < 0.1 the system was marked as “validated”. FDR was computed from the HiSig p-value using the Benjamini-Hochberg procedure, with number of multiple tests equal to the number of recurrently mutated systems in the discovery cohort. To provide a reference for this analysis, we performed similar analyses for individual genes identified by MutSigCV, and for systems across cohorts of mismatched cancer types (Fig. S3A).
Evaluation of the Novelty of NeST Systems
Overlap between NeST and known cancer pathways was determined based on 241 literature curated pathways collected in our previous survey (71). NeST systems with significant overlap with any of these pathways (at least 2 overlapping genes; p < 0.05; hypergeometric test adjusted by Bonferroni correction) were marked as having “overlap with cancer pathways”. Remaining systems were tested for significant overlap with 16,064 gene sets in Gene Ontology (including terms in Cellular Component, Biological Process, and Molecular Function), and those with significant overlaps (same statistical definition as above) were marked as “overlap with other cell biology entities” (Fig. 4D; Table S5).
CRISPR/Cas9 Screen for Determination of BRCAness
The protocol for arrayed CRISPR-Cas9 screening was adapted from previous work (119). To obtain reliable cell counts for cell proliferation, SW1710 bladder cancer cells were RFP-labeled using the Incucyte NucLight Red Lentivirus Reagent (cat. no. 4476), and transfected cells were selected using puromycin (3 μg/ml). Three different crRNAs were obtained for each gene from Dharmacon. crRNA:tracrRNA duplexes were formed by initially incubating 4 μl of crRNA (160 μM) with 4 μl of tracrRNA (160 μM) for 45 min at 37°C. Duplexes were incubated with 8 μl of Cas9-NLS protein (40 μM) at 37°C for 15 min. These crRNPs were then aliquoted (4 μl) into 96-well V-bottom plates in arrayed format in a random order to mitigate potential positional effects. Nucleofection of crRNPs into cells were conducted using the SE cell line 4D-nucleofector kit (Lonza, cat. no. V4SC-1960). 200,000 SW1710 RFP-labeled cells per well were resuspended in 20 μl supplemented SE buffer and mixed with crRNPs. Cells were nucleofected on the Amaxa 4D-Nucleofector System, using program CM-137.
Following nucleofection, 80 μl of prewarmed media was added and the cells were recovered at 37°C. Nucleofected cells from 96-well plates were then transferred to a 384-well plate such that each well has 4 technical replicates of about 500 cells. After 24 h of recovery, the initial cell counts were measured using an ArrayScan (Thermo Fisher Scientific). Immediately after obtaining the initial cell counts, cells were treated with olaparib (10 μM) or cisplatin (600 nM). The selected doses were those resulting in 80% cell death compared to no treatment in wild-type cells based on dose-response curves determined in prescreens. Final cell counts were measured after ~6 d, when the wells nucleofected with non-targeting control were confluent.
As a control, BRCA1 (cat. no. CM-003461–02, Dharmacon) was tested to have a gene-editing efficiency of 87.6% under these conditions through TIDE analysis (120). Phenotypic analysis was conducted by normalizing the cell proliferation in drug-treated conditions to the cell proliferation in the no-treatment condition. This approach eliminated potential relative cell growth defects in the no-treatment condition as a result of individual gene knockouts. Differential drug sensitivity was determined by comparison of the mean cell proliferation of 24 total replicates per gene to the mean value obtained from non-targeting control. The statistical significance of the phenotypic effect (Fig. 3C) was determined by a paired sample t-test.
Analysis of the PIK3CA-Actomyosin system
Proximity Ligation Assay
CAL-33 cell lines were fixed in 4% paraformaldehyde (in PBS) for 15 min at room temperature, and then washed in PBS. Cells were permeabilized with 0.25% Triton X-100 (in PBS) for 10 min at room temperature. Unspecific binding sites were blocked by incubating cells in blocking solution included in the Duolink® In Situ Red Starter Kit Mouse/Rabbit (Sigma-Aldrich, Cat# DUO92101). Primary antibody incubation was performed at 4°C overnight. The different combinations of primary antibodies used were: anti-PIK3CA (Invitrogen, Cat# MA5–17149) diluted at 1/400; anti-FLAG (Cell Signaling Technologies, Cat# 14793S) diluted at 1/800, and anti-MYH9 (ThermoFisher, Cat#PA-29673), diluted at 1/500. Primary antibodies were diluted in Duolink® Antibody Diluent. Detection was performed according to the manufacturer’s protocol. Briefly, after probe incubation for an hour at 37°C, a ligation-ligase solution was added to each sample for 30 minutes at 37 °C, and then washed twice for 2 minutes with 1x Duolink® In Situ Wash Buffer A. An Amplification-Polymerase solution was added for 100 min at 37 °C, and then washed twice for 10 minutes with 1x Duolink In Situ Wash Buffer B. The slides were mounted using Duolink® In Situ Mounting Medium with DAPI. Protein-protein interactions appear as red dots.
Samples were imaged on a Nikon Ti2-E (Nikon) microscope is equipped with a CREST X-Light spinning disk confocal (Crest Optics), Celeste Light Engine (Lumencor), Piezo stage (Mad City Labs), and a Prime 95B 25mm CMOS camera (Photometrics) using a Plan Apo VC 100x/1.4 Oil (Nikon). The red PLA dye was measured by exciting with 561nm laser and capturing with 607/36m filter. Nuclei/DAPI were excited with a 405nm laser and captured with 450/50m filter. Z stacks were set to capture the height of all cells in the field of view and images were taken to capture > 150 cells per condition. PLA spots in cells were segmented in 3D and counted using GA3 analysis in NIS Elements (v. 5.30.01 build 1541, Nikon).
Cell culture and immunostaining.
CAL-33 cells were maintained in Gibco DMEM (ThermoFisher, 11995073) supplemented with 10% fetal bovine serum (Fisher Scientific, 26–140-079) and 1% Penicillin-Streptomycin (Corning, Cat# 30–002-CI), and incubated at 37°C with 5% CO2. For superresolution imaging experiments, cells were cultured in a poly-L-lysine-coated (Sigma-Aldrich, P9155) Lab-Tek® II 8-well chambered coverglass (ThermoFisher, 155360). Doxycycline (Clontech, 631311) was added to each well at 1 μg/mL at the time of cell plating to induce 3xFLAG-PIK3CA expression. After 36 to 48 hrs, the cells reached 60–70% confluency and were fixed and stained using the same protocols as described (121). Briefly, fixation was done with 3.7% paraformaldehyde (PFA) in 1x PHEM buffer at room temperature for 20 min. After three quick PBS (ThermoFisher, 14190144) washes, the sample was quenched with fresh 0.1% sodium borohydride in PBS for 7 minutes, washed with PBS (3x), and permeabilized with 0.3% saponin (Sigma-Aldrich,47036) in PBS for 30 minutes. The sample was then rinsed with PBS and blocked with Image-iT® FX signal enhancer (ThermoFisher, I36933) at room temperature for 30 min. Following three quick rinses with PBS, the sample underwent a second blocking in PBS with 3% goat serum (Abcam, ab7481) and 5% salmon sperm DNA (ThermoFisher, AM9680) for 30 minutes with gentle rocking. The sample was then incubated with a mouse monoclonal anti-FLAG® M2 antibody (Sigma-Aldrich, F1804, dilution ratio 1:1000) and a rabbit anti-Myosin IIA antibody (Sigma-Aldrich, M8064, dilution ratio 1:150) in the same BSA and Salmon DNA blocking buffer at room temperature on a rocker for 90 min. The sample was then rinsed with PBS (3x, 5 min each) before incubating with DNA-conjugated donkey anti-rabbit IgG (H+L) (Jackson Immuno Research, 711–005-152, dilution ratio 1:100) and donkey anti-mouse IgG (H+L) (Jackson Immuno Research, 715–005-150, dilution ratio 1:200) in blocking buffer. Here, the anti-mouse and anti-rabbit secondary antibodies were pre-conjugated with different DNA oligos, namely DS1 (sequence: 5’-TATACATCTAAATACATCTAAT) and DS2 (sequence: 5’-TTATCTACATATATCT ACATAT), respectively, using a procedure as previously reported (121). The incubation also took place on a rocker at room temperature for 1 hour, followed by thorough PBS rinses. The sample was then post-fixed with 3.7% PFA and 0.1% glutaraldehyde in a PHEM buffer at room temperature for 10 min. Of note, this post-fixation step is critical to subsequent DNA-PAINT imaging.
DNA-PAINT imaging.
All superresolution fluorescence data were taken on a custom-built single-molecule imaging system used in our previous work(121). For multicolor imaging, the targets were imaged sequentially with exchange-PAINT using our modified protocol (121, 122). Gold nanorods (50 nm, BBI Solutions, EM. GC50/4) added to the samples were used as fiduciary markers for both stage drift correction and image registration. All imaging was performed using Atto643 (Atto-tec, AD643) labeled imager strands IS1 (sequence: 5’-TTAGATGTAT) or IS2 (sequence: 5’ ATATGTAGAT) in imaging buffer C (1x PBS with 500 mM NaCl) containing 10–15% ethylene carbonate (EC) (Sigma-Aldrich, 676802). For each target, typically 40,000 frames of raw DNA-PAINT image data were acquired at 50 ms exposure time using micromanager(123). Image analysis, including single-molecule localization and subsequent coordinate filtering, sorting, and rendering was performed using in-house Matlab scripts as previously described(124). For localization sorting, events that appeared within a defined number of frames (typically 5) and distance (typically 85 nm or 0.5 pixel on our setup) were combined into a single event with averaged coordinates. The assorted localizations were then used for final image rendering, and the rendered images were exported as TIF files for further analysis and annotations in Fiji(125).
Signaling assay.
Serum-starved cells from head-and-neck cancer cell lines (CAL-33, SCC-25) were treated with a combination of 0.5μM Alpelisib (MedChemExpress HY-15244) and/or 10μM (−)-Blebbistatin (Selleckchem S7099) with the appropriate vehicle control for 30 minutes. Cells were lysed and subsequently cleared by centrifugation. After normalization of protein concentrations, samples were boiled in Laemmli sample buffer (BioRad) and subjected to immunoblot analysis. Antibodies (Cell Signaling Technologies) were used at the indicated dilutions: p-AKT Ser473 (cat. no. 4060, 1:5000), AKT (cat. no. 9272, 1:1000), p-S6 Ser235/236 (cat. no. 2211, 1:5000), S6 (cat. no. 2317, 1:1000), GAPDH (cat. no. 2118, 1:10,000). Results were quantified using ImageJ using the ratio of phosphorylated/total signal normalized to the vehicle control of each replicate (N=3, Fig. 5F, S5D).
Association of mutation status with RPPA data.
Reverse-phase protein array (RPPA) quantifications of phosphorylated/total proteins in each of 899 cancer cell lines, (CCLE_RPPA_20181003.csv), along with the mutation profiles of these cell lines, (CCLE_DepMap_18q3_maf_20180718.txt), were downloaded from the CCLE data portal (https://portals.broadinstitute.org/ccle/data) (75). Synonymous mutations and mutations outside of protein coding regions were excluded from this analysis.
Analysis of Collagen Mutations
dN/dS analysis.
To analyze mutational selection on collagen complexes, we considered the most prominent collagen mutation types for SKCM and LUAD, i.e., C->T or G->A transitions for SKCM, and transitions and transversions on C/G nucleotides for LUAD. Background mutation rates were calculated by examining the silent positions of the collagen genes in the analyzed systems. Triple-helical regions of collagen proteins were defined using annotations in the Uniprot database. Statistical significance and 95% confidence intervals (Fig. 6E) were calculated based on a one-tailed binomial test.
Structure-based stability analysis.
All point mutations in the trimeric helix region were considered. A crystal structure of collagen I trimeric helix region (PDB ID: 1BKV) was used as a template, which contains three identical polypeptide chains, each with 10 trimeric repeats. For each point mutation, nine residues surrounding the mutated position were threaded onto the middle of the template (fourth to sixth repeats). Two structure models representing the wild-type and the mutant were created (differing by one residue at the mutated position) and scored by the FoldX 5.0 suite (86) with the “BuildModel” command. The change of protein stability upon point mutation (ΔΔG) was defined as the “total energy” of the mutant model subtracted by that of the wild-type model. Since the magnitude of destabilization is often non-physiological, because of the fixed backbone structures in this type of analysis, we set the maximum of ΔΔG to 8.0 kcal/mol when displaying results (Fig. 6F).
Association of mutation status with metastatic phenotypes.
Metastatic potential and penetrance of 488 cancer cell lines were obtained from the MetMap 500 dataset (https://depmap.org/metmap/data) (89). Mutation profiles of these cell lines (CCLE_DepMap_18q3_maf_20180718.txt) were downloaded from the CCLE data portal (https://portals.broadinstitute.org/ccle/data) (78). Synonymous mutations and mutations outside of protein coding regions were excluded from this analysis.
Cell lines and plasmids.
HFF-1 (ATCC® SCRC-1041™), Tu To (ATCC® CRL-1298™), and A549 cells (ATCC® CCL-185™) were obtained from the American Type Culture Collection (ATCC). pEGFP-N2-COL1A1 was a gift from David Stephens (Addgene plasmid # 66602; http://n2t.net/addgene:66602; RRID:Addgene_66602). pMD2.G was a gift from Didier Trono (Addgene plasmid # 12259; http://n2t.net/addgene:12259; RRID:Addgene_12259) pCMV-dR8.2 dvpr was a gift from Bob Weinberg (Addgene plasmid # 8455; http://n2t.net/addgene:8455; RRID:Addgene_8455). The COL1A1 ORF was introduced into pLenti CMV Blast DEST (706–1), which was a gift from Eric Campeau & Paul Kaufman (Addgene plasmid # 17451; http://n2t.net/addgene:17451; RRID:Addgene_17451), and, with pCMV-dR8.2 dvpr and pMD2.G, was used to stably introduce CMV-COL1A1 into HFF-1 cells via standard lentiviral packaging and infection protocols as described (28). Site-directed mutagenesis was used to introduce the G281S mutation into COL1A1 as described (28).
Whole-exome Sequencing of Tu To.
All cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum (fbs, Corning, Corning, NY) and 0.1% gentamicin (Gibco Thermofisher, Waltham, MA). These cells were harvested in order to isolate their genomic DNA using a Wizard® Genomic DNA Purification Kit (Promega, Madison, WI, USA), following the manufacturer’s instructions (Wizard® Genomic DNA Purification Kit ). The isolated genomic DNA was sequenced utilizing an Illumina NovaSeq 6000. We used Terra (https://terra.bio/), a cloud computing platform for genomics to perform germline mutation calling of Tu To. Paired-end FASTQ files of Tu To whole-exome sequencing were used as the input, and we used the “Sequence-Format-Conversion/Paired-FASTQ-to-Unmapped-BAM” and “Exome-Analysis-Pipeline/ExomeGermlineSingleSample” workflows to identify the germline variants.
Fibroblast-derived matrix experiments.
WT and mutant fibroblast-derived matrices were generated following the basic protocol outlined in (124) with adaptations. Media was supplemented with Dextran Sulfate sodium salt from Leuconostoc spp., MW ~ 500,000 g/mole (D8906, Sigma-Aldrich, St. Louis, MO, USA) as described in (127) with a concentration of 100 ug/mL instead of the addition of ascorbic acid. Thickness of each matrix was measured using a Nikon TiE microscope with a 10x objective as the z-distance of the cell layer focal plane from the dish bottom focal plane. Once the fibroblast-derived matrix thickness reached 20–30 microns, the matrices were decellularized following the protocol in (126) and included the addition of DNase I (Thermo Fisher Scientific Baltics, Vilnius, Lithuania). After decellularization, A549 cells were seeded on top of the matrices at a density of 30,000 cells/mL and incubated for 25 hours. The cells were then fixed using 4% paraformaldehyde (15710-s, Electron Microscopy Sciences, Hatfield, PA, USA) for 30 minutes at room temperature. The fixed samples were stained with primary antibody against Ki67 (1:400 dilution, 8D5, Cell Signaling Technology) and secondary antibody Alexa FluorTM 546 Goat anti-Mouse (1:2000 dilution, A11003, Life Technologies, Eugene, OR, USA). DAPI (1:1000 dilution, 4083, Cell Signaling, Danvers, MA, USA) was utilized to counterstain the nuclei. Fluorescent images were obtained using a Nikon TiE inverted microscope (Nikon Instruments Inc., Melville, NY) with a 10x objective lens. The images were observed and taken in NIS Elements Software (Nikon Instruments Inc.).
Clinical Analysis of Protein Systems
Each NeST system was tested for association between the mutation status of the system and the progression-free patient survival (PFS) time. Associations were tested only in the tumor type(s) for which the system was recurrently mutated (HiSig). Clinical data from TCGA patients (Pancancer Atlas), including PFS, were downloaded from the cBioPortal (104). Statistical significance was evaluated by log rank test, with p-values adjusted by the Benjamini-Hochberg procedure. We noted that PFS time is significantly associated with the overall mutation burden in several tumor types (BLCA, OV, STAD, UCEC); thus, we used a multivariable Cox proportional-hazards model for all analysis, using the mutation status and the total number of mutations in the sample (Log10 transformed) as independent variables. By this procedure, the p-values assigned to mutation status by this model considered the statistical association after removing the confounding effects of mutation burden. This process identified a total of 38 associations (FDR < 0.3) between a system and PFS. Next, we tested the association of PFS with the mutation status of individual genes defined as cancer drivers by the TCGA Pancancer Atlas (37) for each of the 13 analyzed tumor types. From all combinations of single genes and tumor types, 9 significant prognostic associations (i.e., gene/PFS) were detected (FDR < 0.3; Table S7). We then excluded any PFS-associated system containing a gene which was (i) determined to be prognostic in that same tumor type and (ii) accounted for more than 80% of tumor cases with mutations in that system. After subtracting these systems, a total of 25 (system, tumor type) associations were found remaining, i.e., associations which could not be attributed to mutations within only a single gene.
Survey of Cancer Genes Based on Systems-Level Analysis
To create a catalog of cancer genes based on systems in the NeST map (Table S8), we conservatively selected the top two genes with the highest mutation rates per system, adjusted for pleiotropic genes included in multiple systems. Raw gene mutation frequency is not a suitable metric to rank the importance of a gene to a system, since a gene with high mutation frequency (e.g., TP53) would be ranked first for every system in which it participates, despite contributing to many different systems. Rather, we distributed the total mutation count of a gene across the systems in which it participates, proportioned according to the weights assigned to each system by HiSig (v vector, see HiSig method above). In particular, we translated Mg, the total mutation counts for gene g, to Mgs, the mutation counts for gene g ascribed to system s, according to the following function:
where wg and vs are the weights given to gene g and system s in the regression model by HiSig. Thus, for each system 𝑠𝑠, the genes were ranked by their values of Mgs, which penalized genes with high pleiotropy. The resulting NeST gene catalog was analyzed against three types of validation as follows:
Differential expression in tumors.
For each gene, we examined differential expression between tumor and normal samples in nine TCGA cohorts which each had >40 normal samples (BRCA, HNSC, KIRC, LIHC, LUAD, LUSC, PRAD, THCA, COAD). Differential expression was evaluated using a Kolmogorov-Smirnov (KS) test, by comparing the percentile rankings of gene RSEM values downloaded from the UCSC Xena platform (128) (https://xenabrowser.net/datapages/). A summary statistic (Fig. 7G) was derived by summing up the negative log of the p-values across these cohorts.
Prognostic value of gene expression.
For each gene and tissue type, we examined log-rank P values scoring the significance of association between mRNA expression level and patient survival, using Kaplan-Meier analysis. These P values were obtained from the Human Protein Atlas (HPA) (129) for each of the 13 TCGA cohorts analyzed in our study (https://www.proteinatlas.org/about/download). A summary statistic (Fig. 7H) was derived by summing up the negative log of the p-values across these cohorts.
Mouse genetic screens.
We used the Candidate Cancer Gene Database (CCGD) (130) (http://ccgd-starrlab.oit.umn.edu, downloaded on 4 June 2019), which contains literature-curated evidence from transposon-based forward genetic screens in mice. The number of studies in which a gene was disrupted by transposon insertional mutagenesis in mice tumors was used as the summary statistic (Fig. 7I).
Supplementary Material
Acknowledgments
We gratefully acknowledge the very helpful comments and suggestions of Drs. Cherie Ng and Jianzhu Ma.
Funding:
This work was supported by the Cancer Cell Map Initiative (NCI U54 CA209891), the National Resource for Network Biology (P41 GM103504) and the Cytoscape Project (R01 HG009979). B.T. was also supported by funding from the NIH (F32 CA239336). E.S. was also supported by funding from the NCI (5F30CA236404-02). J.F.K. was also supported by funding from the NIH (R50 CA243885). S.I.F. and M.H. were supported by a National Science Foundation CAREER award MCB-1651855 (to S.I.F.) and an American Cancer Society Research Scholar Grant RSG-21-033-01-CSM (to S.I.F.). K.T. and X.N. were supported by the Cancer Systems Biology Consortium (NCI U54 CA209988) and funding from the NIGMS (R01 GM132322). B.K. was funded under an NIH K00 grant (CA212456). M.K. was also supported by the Martha and Bruce Atwater Breast Cancer Research Program, UCSF Prostate Cancer Program, and Benioff Initiative for Prostate Cancer Research. J.S.G. was supported by NIH award #R01DE026870. This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a National Institutes of Health SIG grant (#S10 OD026929).
Footnotes
Competing Interests: T.I. is co-founder of Data4Cure, Inc., is on the Scientific Advisory Board, and has an equity interest. T.I. is on the Scientific Advisory Board of Ideaya BioSciences, Inc., has an equity interest, and receives sponsored research funding. The terms of these arrangements have been reviewed and approved by the University of California San Diego in accordance with its conflict-of-interest policies. J.S.G. is the Member of the Board of Scientific Advisors for Vividion, Oncoceutics Pharmaceuticals, and Domain Pharmaceuticals (not directly relevant to this study). The laboratory of N.J.K. has received research support from Vir Biotechnology and F. Hoffmann-La Roche. N.J.K. is a shareholder of Tenaya Therapeutics and has received stocks from Maze Therapeutics and Interline Therapeutics and has consulting agreements with the Icahn School of Medicine at Mount Sinai, New York, Maze Therapeutics and Interline Therapeutics.
Data and Materials Availability: All data are available in the main text and supplementary materials. The software used in this work are archived on Zenodo(109, 129, 130).
References and Notes
- 1.Garraway LA, Lander ES, Lessons from the cancer genome. Cell 153, 17–37 (2013). doi: 10.1016/j.cell.2013.03.002 [DOI] [PubMed] [Google Scholar]
- 2.Burrell RA, McGranahan N, Bartek J, Swanton C, The causes and consequences of genetic heterogeneity in cancer evolution. Nature 501, 338–345 (2013). doi: 10.1038/nature12625 [DOI] [PubMed] [Google Scholar]
- 3.Mutation Consequences and Pathway Analysis Working Group of the International Cancer Genome Consortium, Pathway and network analysis of cancer genomes. Nat. Methods 12, 615–621 (2015). doi: 10.1038/nmeth.3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim Y-A, Cho D-Y, Przytycka TM, Understanding Genotype-Phenotype Effects in Cancer via Network Approaches. PLOS Comput. Biol. 12, e1004747 (2016). doi: 10.1371/journal.pcbi.1004747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laddach A, Ng JC-F, Chung SS, Fraternali F, Genetic variants and protein-protein interactions: A multidimensional network-centric view. Curr. Opin. Struct. Biol. 50, 82–90 (2018). doi: 10.1016/j.sbi.2017.12.006 [DOI] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Network, Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582 (2015). doi: 10.1038/nature14129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P, The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015). doi: 10.1016/j.cels.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M, KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016). doi: 10.1093/nar/gkv1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, Chakravarty D, Daian F, Gao Q, Bailey MH, Liang W-W, Foltz SM, Shmulevich I, Ding L, Heins Z, Ochoa A, Gross B, Gao J, Zhang H, Kundra R, Kandoth C, Bahceci I, Dervishi L, Dogrusoz U, Zhou W, Shen H, Laird PW, Way GP, Greene CS, Liang H, Xiao Y, Wang C, Iavarone A, Berger AH, Bivona TG, Lazar AJ, Hammer GD, Giordano T, Kwong LN, McArthur G, Huang C, Tward AD, Frederick MJ, McCormick F, Meyerson M, Van Allen EM, Cherniack AD, Ciriello G, Sander C, Schultz N, Cancer Genome Atlas Research Network, Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10 (2018). doi: 10.1016/j.cell.2018.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paczkowska M, Barenboim J, Sintupisut N, Fox NS, Zhu H, Abd-Rabbo D, Mee MW, Boutros PC, Reimand J, PCAWG Drivers and Functional Interpretation Working Group, PCAW Consortium, Integrative pathway enrichment analysis of multivariate omics data. Nat. Commun. 11, 735 (2020). doi: 10.1038/s41467-019-13983-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chuang H-Y, Lee E, Liu Y-T, Lee D, Ideker T, Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 3, 140 (2007). doi: 10.1038/msb4100180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vandin F, Upfal E, Raphael BJ, Algorithms for detecting significantly mutated pathways in cancer. J. Comput. Biol. 18, 507–522 (2011). doi: 10.1089/cmb.2010.0265 [DOI] [PubMed] [Google Scholar]
- 13.Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, Zhu J, Haussler D, Stuart JM, Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 26, i237–i245 (2010). doi: 10.1093/bioinformatics/btq182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hofree M, Shen JP, Carter H, Gross A, Ideker T, Network-based stratification of tumor mutations. Nat. Methods 10, 1108–1115 (2013). doi: 10.1038/nmeth.2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leiserson MDM, Vandin F, Wu H-T, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, Lawrence MS, Gonzalez-Perez A, Tamborero D, Cheng Y, Ryslik GA, Lopez-Bigas N, Getz G, Ding L, Raphael BJ, Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 47, 106–114 (2015). doi: 10.1038/ng.3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho A, Shim JE, Kim E, Supek F, Lehner B, Lee I, MUFFINN: Cancer gene discovery via network analysis of somatic mutation data. Genome Biol. 17, 129 (2016). doi: 10.1186/s13059-016-0989-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horn H, Lawrence MS, Chouinard CR, Shrestha Y, Hu JX, Worstell E, Shea E, Ilic N, Kim E, Kamburov A, Kashani A, Hahn WC, Campbell JD, Boehm JS, Getz G, Lage K, NetSig: Network-based discovery from cancer genomes. Nat. Methods 15, 61–66 (2018). doi: 10.1038/nmeth.4514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dimitrakopoulos C, Hindupur SK, Häfliger L, Behr J, Montazeri H, Hall MN, Beerenwinkel N, Network-based integration of multi-omics data for prioritizing cancer genes. Bioinformatics 34, 2441–2448 (2018). doi: 10.1093/bioinformatics/bty148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reyna MA, Leiserson MDM, Raphael BJ, Hierarchical HotNet: Identifying hierarchies of altered subnetworks. Bioinformatics 34, i972–i980 (2018). doi: 10.1093/bioinformatics/bty613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reyna MA, Haan D, Paczkowska M, Verbeke LPC, Vazquez M, Kahraman A, Pulido-Tamayo S, Barenboim J, Wadi L, Dhingra P, Shrestha R, Getz G, Lawrence MS, Pedersen JS, Rubin MA, Wheeler DA, Brunak S, Izarzugaza JMG, Khurana E, Marchal K, von Mering C, Sahinalp SC, Valencia A, Reimand J, Stuart JM, Raphael BJ, PCAWG Drivers and Functional Interpretation Working Group, PCAWG Consortium, Pathway and network analysis of more than 2500 whole cancer genomes. Nat. Commun. 11, 729 (2020). doi: 10.1038/s41467-020-14367-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pujana MA, Han J-DJ, Starita LM, Stevens KN, Tewari M, Ahn JS, Rennert G, Moreno V, Kirchhoff T, Gold B, Assmann V, Elshamy WM, Rual J-F, Levine D, Rozek LS, Gelman RS, Gunsalus KC, Greenberg RA, Sobhian B, Bertin N, Venkatesan K, Ayivi-Guedehoussou N, Solé X, Hernández P, Lázaro C, Nathanson KL, Weber BL, Cusick ME, Hill DE, Offit K, Livingston DM, Gruber SB, Parvin JD, Vidal M, Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat. Genet. 39, 1338–1349 (2007). doi: 10.1038/ng.2007.2 [DOI] [PubMed] [Google Scholar]
- 22.Rozenblatt-Rosen O, Deo RC, Padi M, Adelmant G, Calderwood MA, Rolland T, Grace M, Dricot A, Askenazi M, Tavares M, Pevzner SJ, Abderazzaq F, Byrdsong D, Carvunis A-R, Chen AA, Cheng J, Correll M, Duarte M, Fan C, Feltkamp MC, Ficarro SB, Franchi R, Garg BK, Gulbahce N, Hao T, Holthaus AM, James R, Korkhin A, Litovchick L, Mar JC, Pak TR, Rabello S, Rubio R, Shen Y, Singh S, Spangle JM, Tasan M, Wanamaker S, Webber JT, Roecklein-Canfield J, Johannsen E, Barabási A-L, Beroukhim R, Kieff E, Cusick ME, Hill DE, Münger K, Marto JA, Quackenbush J, Roth FP, DeCaprio JA, Vidal M, Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 487, 491–495 (2012). doi: 10.1038/nature11288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, Schmitz R, Mitala J, Whiting A, Xiao W, Ceribelli M, Wright GW, Zhao H, Yang Y, Xu W, Rosenwald A, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Wiestner A, Kruhlak MJ, Iwai K, Bernal F, Staudt LM, Essential role of the linear ubiquitin chain assembly complex in lymphoma revealed by rare germline polymorphisms. Cancer Discov. 4, 480–493 (2014). doi: 10.1158/2159-8290.CD-13-0915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krogan NJ, Lippman S, Agard DA, Ashworth A, Ideker T, The cancer cell map initiative: Defining the hallmark networks of cancer. Mol. Cell 58, 690–698 (2015). doi: 10.1016/j.molcel.2015.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowler EH, Wang Z, Ewing RM, How do oncoprotein mutations rewire protein-protein interaction networks? Expert Rev. Proteomics 12, 449–455 (2015). doi: 10.1586/14789450.2015.1084875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eckhardt M, Zhang W, Gross AM, Von Dollen J, Johnson JR, Franks-Skiba KE, Swaney DL, Johnson TL, Jang GM, Shah PS, Brand TM, Archambault J, Kreisberg JF, Grandis JR, Ideker T, Krogan NJ, Multiple Routes to Oncogenesis Are Promoted by the Human Papillomavirus-Host Protein Network. Cancer Discov. 8, 1474–1489 (2018). doi: 10.1158/2159-8290.CD-17-1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kovalski JR, Bhaduri A, Zehnder AM, Neela PH, Che Y, Wozniak GG, Khavari PA, The Functional Proximal Proteome of Oncogenic Ras Includes mTORC2. Mol. Cell 73, 830–844.e12 (2019). doi: 10.1016/j.molcel.2018.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelly MR, Kostyrko K, Han K, Mooney NA, Jeng EE, Spees K, Dinh PT, Abbott KL, Gwinn DM, Sweet-Cordero EA, Bassik MC, Jackson PK, Combined Proteomic and Genetic Interaction Mapping Reveals New RAS Effector Pathways and Susceptibilities. Cancer Discov. 10, 1950–1967 (2020). doi: 10.1158/2159-8290.CD-19-1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drew K, Lee C, Huizar RL, Tu F, Borgeson B, McWhite CD, Ma Y, Wallingford JB, Marcotte EM, Integration of over 9,000 mass spectrometry experiments builds a global map of human protein complexes. Mol. Syst. Biol. 13, 932 (2017). doi: 10.15252/msb.20167490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, Szpyt J, Tam S, Zarraga G, Pontano-Vaites L, Swarup S, White AE, Schweppe DK, Rad R, Erickson BK, Obar RA, Guruharsha KG, Li K, Artavanis-Tsakonas S, Gygi SP, Harper JW, Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509 (2017). doi: 10.1038/nature22366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roux KJ, Kim DI, Burke B, May DG, BioID: A Screen for Protein-Protein Interactions. Curr. Protoc. Protein Sci. 91, 19.23.11–19.23.15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luck K, Kim D-K, Lambourne L, Spirohn K, Begg BE, Bian W, Brignall R, Cafarelli T, Campos-Laborie FJ, Charloteaux B, Choi D, Coté AG, Daley M, Deimling S, Desbuleux A, Dricot A, Gebbia M, Hardy MF, Kishore N, Knapp JJ, Kovács IA, Lemmens I, Mee MW, Mellor JC, Pollis C, Pons C, Richardson AD,Schlabach S, Teeking B, Yadav A, Babor M, Balcha D, Basha O, Bowman-Colin C, Chin S-F, Choi SG, Colabella C, Coppin G, D’Amata C, De Ridder D, De Rouck S, Duran-Frigola M, Ennajdaoui H, Goebels F, Goehring L, Gopal A, Haddad G, Hatchi E, Helmy M, Jacob Y, Kassa Y, Landini S, Li R, van Lieshout N, MacWilliams A, Markey D, Paulson JN, Rangarajan S, Rasla J, Rayhan A, Rolland T, San-Miguel A, Shen Y, Sheykhkarimli D, Sheynkman GM, Simonovsky E, Taşan M, Tejeda A, Tropepe V, Twizere J-C, Wang Y, Weatheritt RJ, Weile J, Xia Y, Yang X, Yeger-Lotem E, Zhong Q, Aloy P, Bader GD, De Las Rivas J, Gaudet S, Hao T, Rak J, Tavernier J, Hill DE, Vidal M, Roth FP, Calderwood MA, A reference map of the human binary protein interactome. Nature 580, 402–408 (2020). doi: 10.1038/s41586-020-2188-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Go CD, Knight JDR, Rajasekharan A, Rathod B, Hesketh GG, Abe KT, Youn J-Y, Samavarchi-Tehrani P, Zhang H, Zhu LY, Popiel E, Lambert J-P, Coyaud E, Cheung SWT, Rajendran D, Wong CJ, Antonicka H, Pelletier L, Raught B, Palazzo AF, Shoubridge EA, Gingras A-C, A proximity biotinylation map of a human cell. Nature 595, 120–124 (2021). doi: 10.1038/s41586-021-03592-2 [DOI] [PubMed] [Google Scholar]
- 34.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE, High frequency of mutations of the PIK3CA gene in human cancers. Science 304, 554 (2004). doi: 10.1126/science.1096502 [DOI] [PubMed] [Google Scholar]
- 35.Muller PAJ, Vousden KH, p53 mutations in cancer. Nat. Cell Biol. 15, 2–8 (2013). doi: 10.1038/ncb2641 [DOI] [PubMed] [Google Scholar]
- 36.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, Pugh TJ, Stransky N, Helman E, Kim J, Sougnez C, Ambrogio L, Nickerson E, Shefler E, Cortés ML, Auclair D, Saksena G, Voet D, Noble M, DiCara D, Lin P, Lichtenstein L, Heiman DI, Fennell T, Imielinski M, Hernandez B, Hodis E, Baca S, Dulak AM, Lohr J, Landau D-A, Wu CJ, Melendez-Zajgla J, Hidalgo-Miranda A, Koren A, McCarroll SA, Mora J, Crompton B, Onofrio R, Parkin M, Winckler W, Ardlie K, Gabriel SB, Roberts CWM, Biegel JA, Stegmaier K, Bass AJ, Garraway LA, Meyerson M, Golub TR, Gordenin DA, Sunyaev S, Lander ES, Getz G, Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013). doi: 10.1038/nature12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, Ng PK-S, Jeong KJ, Cao S, Wang Z, Gao J, Gao Q, Wang F, Liu EM, Mularoni L, Rubio-Perez C, Nagarajan N, Cortés-Ciriano I, Zhou DC, Liang W-W, Hess JM, Yellapantula VD, Tamborero D, Gonzalez-Perez A, Suphavilai C, Ko JY, Khurana E, Park PJ, Van Allen EM, Liang H, Lawrence MS, Godzik A, Lopez-Bigas N, Stuart J, Wheeler D, Getz G, Chen K, Lazar AJ, Mills GB, Karchin R, Ding L, MC3 Working Group, Cancer Genome Atlas Research Network, Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385.e18 (2018). doi: 10.1016/j.cell.2018.02.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niu B, Scott AD, Sengupta S, Bailey MH, Batra P, Ning J, Wyczalkowski MA, Liang W-W, Zhang Q, McLellan MD, Sun SQ, Tripathi P, Lou C, Ye K, Mashl RJ, Wallis J, Wendl MC, Chen F, Ding L, Protein-structure-guided discovery of functional mutations across 19 cancer types. Nat. Genet. 48, 827–837 (2016). doi: 10.1038/ng.3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tokheim C, Bhattacharya R, Niknafs N, Gygax DM, Kim R, Ryan M, Masica DL, Karchin R, Exome-Scale Discovery of Hotspot Mutation Regions in Human Cancer Using 3D Protein Structure. Cancer Res. 76, 3719–3731 (2016). doi: 10.1158/0008-5472.CAN-15-3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao J, Chang MT, Johnsen HC, Gao SP, Sylvester BE, Sumer SO, Zhang H, Solit DB, Taylor BS, Schultz N, Sander C, 3D clusters of somatic mutations in cancer reveal numerous rare mutations as functional targets. Genome Med. 9, 4 (2017). doi: 10.1186/s13073-016-0393-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng F, Zhao J, Wang Y, Lu W, Liu Z, Zhou Y, Martin WR, Wang R, Huang J, Hao T, Yue H, Ma J, Hou Y, Castrillon JA, Fang J, Lathia JD, Keri RA, Lightstone FC, Antman EM, Rabadan R, Hill DE, Eng C, Vidal M, Loscalzo J, Comprehensive characterization of protein-protein interactions perturbed by disease mutations. Nat. Genet. 53, 342–353 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Creixell P, Schoof EM, Simpson CD, Longden J, Miller CJ, Lou HJ, Perryman L, Cox TR, Zivanovic N, Palmeri A, Wesolowska-Andersen A, Helmer-Citterich M, Ferkinghoff-Borg J, Itamochi H, Bodenmiller B, Erler JT, Turk BE, Linding R, Kinome-wide decoding of network-attacking mutations rewiring cancer signaling. Cell 163, 202–217 (2015). doi: 10.1016/j.cell.2015.08.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reimand J, Bader GD, Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol. Syst. Biol. 9, 637 (2013). doi: 10.1038/msb.2012.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bouchard JJ, Otero JH, Scott DC, Szulc E, Martin EW, Sabri N, Granata D, Marzahn MR, Lindorff-Larsen K, Salvatella X, Schulman BA, Mittag T, Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments. Mol. Cell 72, 19–36.e8 (2018). doi: 10.1016/j.molcel.2018.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vyas S, Zaganjor E, Haigis MC, Mitochondria and Cancer. Cell 166, 555–566 (2016). doi: 10.1016/j.cell.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swaney DL, Ramms DJ, Wang Z, Park J, Goto Y, Soucheray M, Bhola N, Kim K, Zheng F, Zeng Y, McGregor M, Herrington KA, O’Keefe R, Jin N, VanLandingham NK, Foussard H, Von Dollen J, Bouhaddou M, Jimenez-Morales D, Obernier K, Kreisberg JF, Kim M, Johnson DE, Jura N, Grandis JR, Gutkind JS, Ideker T, Krogan NJ, A protein network map of head and neck cancer reveals PIK3CA mutant drug sensitivity. Science 374, eabf2911 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim M, Park J, Bouhaddou M, Kim K, Rojc A, Modak M, Soucheray M, McGregor MJ, O’Leary P, Wolf D, Stevenson E, Foo TK, Mitchell D, Herrington KA, Muñoz DP, Tutuncuoglu B, Chen K-H, Zheng F, Kreisberg JF, Diolaiti ME, Gordan JD, Coppé J-P, Swaney DL, Xia B, van ‘t Veer L, Ashworth A, Ideker T, Krogan N, A protein interaction landscape of breast cancer. Science 374, eabf3066 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giurgiu M, Reinhard J, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, Ruepp A, CORUM: The comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res. 47, D559–D563 (2019). doi: 10.1093/nar/gky973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chatr-aryamontri A, Oughtred R, Boucher L, Rust J, Chang C, Kolas NK, O’Donnell L, Oster S, Theesfeld C, Sellam A, Stark C, Breitkreutz B-J, Dolinski K, Tyers M, The BioGRID interaction database: 2017 update. Nucleic Acids Res. 45, D369–D379 (2017). doi: 10.1093/nar/gkw1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orchard S, Kerrien S, Abbani S, Aranda B, Bhate J, Bidwell S, Bridge A, Briganti L, Brinkman FSL, Cesareni G, Chatr-aryamontri A, Chautard E, Chen C, Dumousseau M, Goll J, Hancock REW, Hannick LI, Jurisica I, Khadake J, Lynn DJ, Mahadevan U, Perfetto L, Raghunath A, Ricard-Blum S, Roechert B, Salwinski L, Stümpflen V, Tyers M, Uetz P, Xenarios I, Hermjakob H, Protein interaction data curation: The International Molecular Exchange (IMEx) consortium. Nat. Methods 9, 345–350 (2012). doi: 10.1038/nmeth.1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rolland T, Taşan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, Kamburov A, Ghiassian SD, Yang X, Ghamsari L, Balcha D, Begg BE, Braun P, Brehme M, Broly MP, Carvunis A-R, Convery- Zupan D, Corominas R, Coulombe-Huntington J, Dann E, Dreze M, Dricot A, Fan C, Franzosa E, Gebreab F, Gutierrez BJ, Hardy MF, Jin M, Kang S, Kiros R, Lin GN, Luck K, MacWilliams A, Menche J, Murray RR, Palagi A, Poulin MM, Rambout X, Rasla J, Reichert P, Romero V, Ruyssinck E, Sahalie JM, Scholz A, Shah AA, Sharma A, Shen Y, Spirohn K, Tam S, Tejeda AO, Trigg SA, Twizere J-C, Vega K, Walsh J, Cusick ME, Xia Y, Barabási A-L, Iakoucheva LM, Aloy P, De Las Rivas J, Tavernier J, Calderwood MA, Hill DE, Hao T, Roth FP, Vidal M, A proteome-scale map of the human interactome network. Cell 159, 1212–1226 (2014). doi: 10.1016/j.cell.2014.10.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, Hyman AA, Mann M, A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163, 712–723 (2015). doi: 10.1016/j.cell.2015.09.053 [DOI] [PubMed] [Google Scholar]
- 53.Lapek JD Jr., Greninger P, Morris R, Amzallag A, Pruteanu-Malinici I, Benes CH, Haas W, Detection of dysregulated protein-association networks by high-throughput proteomics predicts cancer vulnerabilities. Nat. Biotechnol. 35, 983–989 (2017). doi: 10.1038/nbt.3955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, Kawaler E, Mundt F, Krug K, Tu Z, Lei JT, Gatza ML, Wilkerson M, Perou CM, Yellapantula V, Huang KL, Lin C, McLellan MD, Yan P, Davies SR, Townsend RR, Skates SJ, Wang J, Zhang B, Kinsinger CR, Mesri M, Rodriguez H, Ding L, Paulovich AG, Fenyö D, Ellis MJ, Carr SA, NCI CPTAC, Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62 (2016). doi: 10.1038/nature18003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou J-Y, Petyuk VA, Chen L, Ray D, Sun S, Yang F, Chen L, Wang J, Shah P, Cha SW, Aiyetan P, Woo S, Tian Y, Gritsenko MA, Clauss TR, Choi C, Monroe ME, Thomas S, Nie S, Wu C, Moore RJ, Yu K-H, Tabb DL, Fenyö D, Bafna V, Wang Y, Rodriguez H, Boja ES, Hiltke T, Rivers RC, Sokoll L, Zhu H, Shih I-M, Cope L, Pandey A, Zhang B, Snyder MP, Levine DA, Smith RD, Chan DW, Rodland KD, CPTAC Investigators, Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 166, 755–765 (2016). doi: 10.1016/j.cell.2016.05.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramani AK, Li Z, Hart GT, Carlson MW, Boutz DR, Marcotte EM, A map of human protein interactions derived from co-expression of human mRNAs and their orthologs. Mol. Syst. Biol. 4, 180 (2008). doi: 10.1038/msb.2008.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boyle EA, Pritchard JK, Greenleaf WJ, High-resolution mapping of cancer cell networks using co-functional interactions. Mol. Syst. Biol. 14, e8594 (2018). doi: 10.15252/msb.20188594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee I, Blom UM, Wang PI, Shim JE, Marcotte EM, Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 21, 1109–1121 (2011). doi: 10.1101/gr.118992.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kramer MH, Farré J-C, Mitra K, Yu MK, Ono K, Demchak B, Licon K, Flagg M, Balakrishnan R, Cherry JM, Subramani S, Ideker T, Active Interaction Mapping Reveals the Hierarchical Organization of Autophagy. Mol. Cell 65, 761–774.e5 (2017). doi: 10.1016/j.molcel.2016.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G, Gene Ontology Consortium, Gene ontology: Tool for the unification of biology. Nat. Genet. 25, 25–29 (2000). doi: 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dong Y, Hakimi M-A, Chen X, Kumaraswamy E, Cooch NS, Godwin AK, Shiekhattar R, Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Mol. Cell 12, 1087–1099 (2003). doi: 10.1016/S1097-2765(03)00424-6 [DOI] [PubMed] [Google Scholar]
- 62.Valenta T, Hausmann G, Basler K, The many faces and functions of β-catenin. EMBO J. 31, 2714–2736 (2012). doi: 10.1038/emboj.2012.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lord CJ, Ashworth A, BRCAness revisited. Nat. Rev. Cancer 16, 110–120 (2016). doi: 10.1038/nrc.2015.21 [DOI] [PubMed] [Google Scholar]
- 65.Turner N, Tutt A, Ashworth A, Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 4, 814–819 (2004). doi: 10.1038/nrc1457 [DOI] [PubMed] [Google Scholar]
- 66.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, Bowlby R, Shen H, Hayat S, Fieldhouse R, Lester SC, Tse GMK, Factor RE, Collins LC, Allison KH, Chen Y-Y, Jensen K, Johnson NB, Oesterreich S, Mills GB, Cherniack AD, Robertson G, Benz C, Sander C, Laird PW, Hoadley KA, King TA, Perou CM, TCGA Research Network, Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 163, 506–519 (2015). doi: 10.1016/j.cell.2015.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, Hinoue T, Laird PW, Hoadley KA, Akbani R, Castro MAA, Gibb EA, Kanchi RS, Gordenin DA, Shukla SA, Sanchez-Vega F, Hansel DE, Czerniak BA, Reuter VE, Su X, de Sa Carvalho B, Chagas VS, Mungall KL, Sadeghi S, Pedamallu CS, Lu Y, Klimczak LJ, Zhang J, Choo C, Ojesina AI, Bullman S, Leraas KM, Lichtenberg TM, Wu CJ, Schultz N, Getz G, Meyerson M, Mills GB, McConkey DJ, Weinstein JN, Kwiatkowski DJ, Lerner SP, TCGA Research Network, Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 171, 540–556.e25 (2017). doi: 10.1016/j.cell.2017.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lord CJ, Ashworth A, PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158 (2017). doi: 10.1126/science.aam7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hanahan D, Weinberg RA, Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). doi: 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 70.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G, Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014). doi: 10.1038/nature12912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kuenzi BM, Ideker T, A census of pathway maps in cancer systems biology. Nat. Rev. Cancer 20, 233–246 (2020). doi: 10.1038/s41568-020-0240-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR, A census of human cancer genes. Nat. Rev. Cancer 4, 177–183 (2004). doi: 10.1038/nrc1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hall A, The cytoskeleton and cancer. Cancer Metastasis Rev. 28, 5–14 (2009). doi: 10.1007/s10555-008-9166-3 [DOI] [PubMed] [Google Scholar]
- 74.Köster DV, Husain K, Iljazi E, Bhat A, Bieling P, Mullins RD, Rao M, Mayor S, Actomyosin dynamics drive local membrane component organization in an in vitro active composite layer. Proc. Natl. Acad. Sci. U.S.A. 113, E1645–E1654 (2016). doi: 10.1073/pnas.1514030113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xue G, Hemmings BA, PKB/Akt-dependent regulation of cell motility. J. Natl. Cancer Inst. 105, 393–404 (2013). doi: 10.1093/jnci/djs648 [DOI] [PubMed] [Google Scholar]
- 76.Wu X, Renuse S, Sahasrabuddhe NA, Zahari MS, Chaerkady R, Kim M-S, Nirujogi RS, Mohseni M, Kumar P, Raju R, Zhong J, Yang J, Neiswinger J, Jeong J-S, Newman R, Powers MA, Somani BL, Gabrielson E, Sukumar S, Stearns V, Qian J, Zhu H, Vogelstein B, Park BH, Pandey A, Activation of diverse signalling pathways by oncogenic PIK3CA mutations. Nat. Commun. 5, 4961 (2014). doi: 10.1038/ncomms5961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kuczek DE, Larsen AMH, Thorseth M-L, Carretta M, Kalvisa A, Siersbæk MS, Simões AMC, Roslind A, Engelholm LH, Noessner E, Donia M, Svane IM, Straten PT, Grøntved L, Madsen DH, Collagen density regulates the activity of tumor- infiltrating T cells. J. Immunother. Cancer 7, 68 (2019). doi: 10.1186/s40425-019-0556-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, Hu K, Andreev-Drakhlin AY, Kim J, Hess JM, Haas BJ, Aguet F, Weir BA, Rothberg MV, Paolella BR, Lawrence MS, Akbani R, Lu Y, Tiv HL, Gokhale PC, de Weck A, Mansour AA, Oh C, Shih J, Hadi K, Rosen Y, Bistline J, Venkatesan K, Reddy A, Sonkin D, Liu M, Lehar J, Korn JM, Porter DA, Jones MD, Golji J, Caponigro G, Taylor JE, Dunning CM, Creech AL, Warren AC, McFarland JM, Zamanighomi M, Kauffmann A, Stransky N, Imielinski M, Maruvka YE, Cherniack AD, Tsherniak A, Vazquez F, Jaffe JD, Lane AA, Weinstock DM, Johannessen CM, Morrissey MP, Stegmeier F, Schlegel R, Hahn WC, Getz G, Mills GB, Boehm JS, Golub TR, Garraway LA, Sellers WR, Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019). doi: 10.1038/s41586-019-1186-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bradley SD, Chen Z, Melendez B, Talukder A, Khalili JS, Rodriguez-Cruz T, Liu S,Whittington M, Deng W, Li F, Bernatchez C, Radvanyi LG, Davies MA, Hwu P, Lizée G, BRAFV600E Co-opts a Conserved MHC Class I Internalization Pathway to Diminish Antigen Presentation and CD8+ T-cell Recognition of Melanoma. Cancer Immunol. Res. 3, 602–609 (2015). doi: 10.1158/2326-6066.CIR-15-0030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shoulders MD, Raines RT, Collagen structure and stability. Annu. Rev. Biochem. 78, 929–958 (2009). doi: 10.1146/annurev.biochem.77.032207.120833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fang M, Yuan J, Peng C, Li Y, Collagen as a double-edged sword in tumor progression. Tumour Biol. 35, 2871–2882 (2014). doi: 10.1007/s13277-013-1511-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Velez DO, Tsui B, Goshia T, Chute CL, Han A, Carter H, Fraley SI, 3D collagen architecture induces a conserved migratory and transcriptional response linked to vasculogenic mimicry. Nat. Commun. 8, 1651 (2017). doi: 10.1038/s41467-017-01556-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Riegler J, Labyed Y, Rosenzweig S, Javinal V, Castiglioni A, Dominguez CX, Long JE, Li Q, Sandoval W, Junttila MR, Turley SJ, Schartner J, Carano RAD, Tumor Elastography and Its Association with Collagen and the Tumor Microenvironment. Clin. Cancer Res. 24, 4455–4467 (2018). doi: 10.1158/1078-0432.CCR-17-3262 [DOI] [PubMed] [Google Scholar]
- 84.Bell SE, Mavila A, Salazar R, Bayless KJ, Kanagala S, Maxwell SA, Davis GE, Differential gene expression during capillary morphogenesis in 3D collagen matrices: Regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J. Cell Sci. 114, 2755–2773 (2001). doi: 10.1242/jcs.114.15.2755 [DOI] [PubMed] [Google Scholar]
- 85.Simon M-P, Pedeutour F, Sirvent N, Grosgeorge J, Minoletti F, Coindre J-M, Terrier-Lacombe M-J, Mandahl N, Craver RD, Blin N, Sozzi G, Turc-Carel C, O’Brien KP, Kedra D, Fransson I, Guilbaud C, Dumanski JP, Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nat. Genet. 15, 95–98 (1997). doi: 10.1038/ng0197-95 [DOI] [PubMed] [Google Scholar]
- 86.Tarpey PS, Behjati S, Cooke SL, Van Loo P, Wedge DC, Pillay N, Marshall J, O’Meara S, Davies H, Nik-Zainal S, Beare D, Butler A, Gamble J, Hardy C, Hinton J, Jia MM, Jayakumar A, Jones D, Latimer C, Maddison M, Martin S, McLaren S, Menzies A, Mudie L, Raine K, Teague JW, Tubio JMC, Halai D, Tirabosco R, Amary F, Campbell PJ, Stratton MR, Flanagan AM, Futreal PA, Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat. Genet. 45, 923–926 (2013). doi: 10.1038/ng.2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, Islam SMA, Lopez-Bigas N, Klimczak LJ, McPherson JR, Morganella S, Sabarinathan R, Wheeler DA, Mustonen V, Getz G, Rozen SG, Stratton MR, PCAWG Mutational Signatures Working Group, PCAWG Consortium, The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020). doi: 10.1038/s41586-020-1943-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Delgado J, Radusky LG, Cianferoni D, Serrano L, FoldX 5.0: Working with RNA, small molecules and a new graphical interface. Bioinformatics 35, 4168–4169 (2019). doi: 10.1093/bioinformatics/btz184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jin X, Demere Z, Nair K, Ali A, Ferraro GB, Natoli T, Deik A, Petronio L, Tang AA, Zhu C, Wang L, Rosenberg D, Mangena V, Roth J, Chung K, Jain RK, Clish CB, Vander Heiden MG, Golub TR, A metastasis map of human cancer cell lines. Nature 588, 331–336 (2020). doi: 10.1038/s41586-020-2969-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Beaubier N, Tell R, Huether R, Bontrager M, Bush S, Parsons J, Shah K, Baker T, Selkov G, Taxter T, Thomas A, Bettis S, Khan A, Lau D, Lee C, Barber M, Cieslik M, Frankenberger C, Franzen A, Weiner A, Palmer G, Lonigro R, Robinson D, Wu Y-M, Cao X, Lefkofsky E, Chinnaiyan A, White KP, Clinical validation of the Tempus xO assay. Oncotarget 9, 25826–25832 (2018). doi: 10.18632/oncotarget.25381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sharaf R, Pavlick DC, Frampton GM, Cooper M, Jenkins J, Danziger N, Haberberger J, Alexander BM, Cloughesy T, Yong WH, Liau LM, Nghiemphu PL, Ji M, Lai A, Ramkissoon SH, Albacker LA, FoundationOne CDx testing accurately determines whole arm 1p19q codeletion status in gliomas. Neurooncol. Adv. 3, vdab017 (2021). doi: 10.1093/noajnl/vdab017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hanahan D, Weinberg RA, The hallmarks of cancer. Cell 100, 57–70 (2000). doi: 10.1016/S0092-8674(00)81683-9 [DOI] [PubMed] [Google Scholar]
- 93.Alberts B, The cell as a collection of protein machines: Preparing the next generation of molecular biologists. Cell 92, 291–294 (1998). doi: 10.1016/s0092-8674(00)80922-8 [DOI] [PubMed] [Google Scholar]
- 94.Cheng Y, Ma Z, Kim B-H, Wu W, Cayting P, Boyle AP, Sundaram V, Xing X, Dogan N, Li J, Euskirchen G, Lin S, Lin Y, Visel A, Kawli T, Yang X, Patacsil D, Keller C, Giardine B, Kundaje A, Wang T, Pennacchio LA, Weng Z, Hardison RC, Snyder MP, Mouse ENCODE Consortium, Principles of regulatory information conservation between mouse and human. Nature 515, 371–375 (2014). doi: 10.1038/nature13985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ding H, Douglass EF Jr., Sonabend AM, Mela A, Bose S, Gonzalez C, Canoll PD, Sims P, Alvarez MJ, Califano A, Quantitative assessment of protein activity in orphan tissues and single cells using the metaVIPER algorithm. Nat. Commun. 9, 1471 (2018). doi: 10.1038/s41467-018-03843-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cox J, Mann M, MaxQuant enables high peptide identification rates, individualized p.p.b.- range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008). doi: 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- 97.Teo G, Liu G, Zhang J, Nesvizhskii AI, Gingras A-C, Choi H, SAINTexpress: Improvements and additional features in Significance Analysis of INTeractome software. J. Proteomics 100, 37–43 (2014). doi: 10.1016/j.jprot.2013.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, Vaites LP, Ordureau A, Rad R, Erickson BK, Wühr M, Chick J, Zhai B, Kolippakkam D, Mintseris J, Obar RA, Harris T, Artavanis-Tsakonas S, Sowa ME, De Camilli P, Paulo JA, Harper JW, Gygi SP, The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 162, 425–440 (2015). doi: 10.1016/j.cell.2015.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sowa ME, Bennett EJ, Gygi SP, Harper JW, Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 (2009). doi: 10.1016/j.cell.2009.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yates B, Braschi B, Gray KA, Seal RL, Tweedie S, Bruford EA, Genenames.org: The HGNC and VGNC resources in 2017. Nucleic Acids Res. 45, D619–D625 (2017). doi: 10.1093/nar/gkw1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Grover A, Leskovec J, node2vec: Scalable Feature Learning for Networks. KDD 2016, 855–864 (2016). doi: 10.1145/2939672.2939754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P Jr., de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L,Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V,Myer VE, Weber BL, Porter J, Warmuth M, Finan, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA, The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). doi: 10.1038/nature11003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H, Cokelaer T, Greninger P, van Dyk E, Chang H, de Silva H, Heyn H, Deng X, Egan RK, Liu Q, Mironenko T, Mitropoulos X, Richardson L, Wang J, Zhang T, Moran S, Sayols S, Soleimani M, Tamborero D, Lopez-Bigas N, Ross-Macdonald P, Esteller M, Gray NS, Haber DA, Stratton MR, Benes CH, Wessels LFA, Saez-Rodriguez J, McDermott U, Garnett MJ, A Landscape of Pharmacogenomic Interactions in Cancer. Cell 166, 740–754 (2016). doi: 10.1016/j.cell.2016.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N, Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013). doi: 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.GTEx Consortium, The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348, 648–660 (2015). doi: 10.1126/science.1262110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, Goodale A, Lee Y, Ali LD, Jiang G, Lubonja R, Harrington WF, Strickland M, Wu T, Hawes DC, Zhivich VA, Wyatt MR, Kalani Z, Chang JJ, Okamoto M, Stegmaier K, Golub TR, Boehm JS, Vazquez F, Root DE, Hahn WC, Tsherniak A, Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784 (2017). doi: 10.1038/ng.3984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Caniza H, Romero AE, Heron S, Yang H, Devoto A, Frasca M, Mesiti M, Valentini G, Paccanaro A, GOssTo: A stand-alone application and a web tool for calculating semantic similarities on the Gene Ontology. Bioinformatics 30, 2235–2236 (2014). doi: 10.1093/bioinformatics/btu144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Resnik P, Others, Semantic similarity in a taxonomy: An information-based measure and its application to problems of ambiguity in natural language. J. Artif. Intell. Res. 11, 95–130 (1999). doi: 10.1613/jair.514 [DOI] [Google Scholar]
- 109.Breiman L, Random forests. Mach. Learn. 45, 5–32 (2001). doi: 10.1023/A:1010933404324 [DOI] [Google Scholar]
- 110.Pedregosa F et al. , Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011). [Google Scholar]
- 111.Zheng F, CliXO-1.0: v1.0 beta (2021); 10.5281/zenodo.4717237. [DOI]
- 112.Newman MEJ, Modularity and community structure in networks. Proc. Natl. Acad. Sci. U.S.A. 103, 8577–8582 (2006). doi: 10.1073/pnas.0601602103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zheng F, Zhang S, Churas C, Pratt D, Bahar I, Ideker T, HiDeF: Identifying persistent structures in multiscale ‘omics data. Genome Biol. 22, 21 (2021). doi: 10.1186/s13059-020-02228-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jacob L, Obozinski G, Vert J-P, paper presented at the Proceedings of the 26th Annual International Conference on Machine Learning (2009. ). [Google Scholar]
- 115.Zheng F, HiSig (2021); 10.5281/zenodo.4722465. [DOI]
- 116.Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, Hess J, Ma S, Chiotti KE, McLellan M, Sofia HJ, Hutter C, Getz G, Wheeler D, Ding L, MC3 Working Group, Cancer Genome Atlas Research Network, Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 6, 271–281.e7 (2018). doi: 10.1016/j.cels.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Friedman J, Hastie T, Tibshirani R, Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010). doi: 10.18637/jss.v033.i01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mayakonda A, Lin D-C, Assenov Y, Plass C, Koeffler HP, Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756 (2018). doi: 10.1101/gr.239244.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hultquist JF, Schumann K, Woo JM, Manganaro L, McGregor MJ, Doudna J, Simon V, Krogan NJ, Marson A, A Cas9 Ribonucleoprotein Platform for Functional Genetic Studies of HIV-Host Interactions in Primary Human T Cells. Cell Rep. 17, 1438–1452 (2016). doi: 10.1016/j.celrep.2016.09.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brinkman EK, Chen T, Amendola M, van Steensel B, Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 (2014). doi: 10.1093/nar/gku936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Civitci F, Shangguan J, Zheng T, Tao K, Rames M, Kenison J, Zhang Y, Wu L, Phelps C, Esener S, Nan X, Fast and multiplexed superresolution imaging with DNA-PAINT-ERS. Nat. Commun. 11, 4339 (2020). doi: 10.1038/s41467-020-18181-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jungmann R, Avendaño MS, Woehrstein JB, Dai M, Shih WM, Yin P, Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat. Methods 11, 313–318 (2014). doi: 10.1038/nmeth.2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, Stuurman N, Advanced methods of microscope control using μManager software. J. Biol. Methods 1, e10 (2014). doi: 10.14440/jbm.2014.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nan X, Collisson EA, Lewis S, Huang J, Tamgüney TM, Liphardt JT, McCormick F, Gray JW, Chu S, Single-molecule superresolution imaging allows quantitative analysis of RAF multimer formation and signaling. Proc. Natl. Acad. Sci. U.S.A. 110, 18519–18524 (2013). doi: 10.1073/pnas.1318188110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). doi: 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Beacham DA, Amatangelo MD, Cukierman E, Preparation of extracellular matrices produced by cultured and primary fibroblasts. Curr. Protoc. Cell Biol. 33, 10.19.11–10.19.21 (2006). [DOI] [PubMed] [Google Scholar]
- 127.Lareu RR, Subramhanya KH, Peng Y, Benny P, Chen C, Wang Z, Rajagopalan R, Raghunath M, Collagen matrix deposition is dramatically enhanced in vitro when crowded with charged macromolecules: The biological relevance of the excluded volume effect. FEBS Lett. 581, 2709–2714 (2007). doi: 10.1016/j.febslet.2007.05.020 [DOI] [PubMed] [Google Scholar]
- 128.Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, Zhu J, Haussler D, Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 38, 675–678 (2020). doi: 10.1038/s41587-020-0546-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, von Feilitzen K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnström H, Glimelius B, Sjöblom T, Edqvist P-H, Djureinovic D, Micke P, Lindskog C, Mardinoglu A, Ponten F, A pathology atlas of the human cancer transcriptome. Science 357, eaan2507 (2017). doi: 10.1126/science.aan2507 [DOI] [PubMed] [Google Scholar]
- 130.Abbott KL, Nyre ET, Abrahante J, Ho Y-Y, Isaksson Vogel R, Starr TK, The Candidate Cancer Gene Database: A database of cancer driver genes from forward genetic screens in mice. Nucleic Acids Res. 43, D844–D848 (2015). doi: 10.1093/nar/gku770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zheng F, Zhang S, Churas C, HiDeF v1.0.0 (2020); 10.5281/zenodo.4059074. [DOI] [Google Scholar]
- 132.Yu MK, Ma J, Ono K, Zheng F, Fong SH, Gary A, Chen J, Demchak B, Pratt D, Ideker T, DDOT: A Swiss Army Knife for Investigating Data-Driven Biological Ontologies. Cell Syst. 8, 267–273.e3 (2019). doi: 10.1016/j.cels.2019.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kramer M, Dutkowski J, Yu M, Bafna V, Ideker T, Inferring gene ontologies from pairwise similarity data. Bioinformatics 30, i34–i42 (2014). doi: 10.1093/bioinformatics/btu282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yang J, Leskovec J, in Proceedings of the 6th ACM International Conference on Web Search and Data Mining (2013), pp. 587–596. [Google Scholar]
- 135.Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E, Fast unfolding of communities in large networks. J. Stat. Mech. 2008, P10008 (2008). doi: 10.1088/1742-5468/2008/10/P10008 [DOI] [Google Scholar]
- 136.Tibshirani R, Regression shrinkage and selection via the lasso. J. R. Stat. Soc. B 58, 267–288 (1996). doi: 10.1111/j.2517-6161.1996.tb02080.x [DOI] [Google Scholar]
- 137.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G, GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011). doi: 10.1186/gb-2011-12-4-r41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pratt D, Chen J, Welker D, Rivas R, Pillich R, Rynkov V, Ono K, Miello C, Hicks L, Szalma S, Stojmirovic A, Dobrin R, Braxenthaler M, Kuentzer J, Demchak B, Ideker T, NDEx, the Network Data Exchange. Cell Syst. 1, 302–305 (2015). doi: 10.1016/j.cels.2015.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pratt D, Chen J, Pillich R, Rynkov V, Gary A, Demchak B, Ideker T, NDEx 2.0: A Clearinghouse for Research on Cancer Pathways. Cancer Res. 77, e58–e61 (2017). doi: 10.1158/0008-5472.CAN-17-0606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shannon P et al. , Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). doi: 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zheng F, Integrated association stringency (IAS) network (2021); 10.5281/zenodo.4516939. [DOI]
- 142.Zheng F, IAS catalog of multi-scale protein systems (2021); 10.5281/zenodo.4654236. [DOI]
- 143.Zheng F, NeST (Nested Systems in Tumor) Cytoscape session (2021); 10.5281/zenodo.4726267. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.