

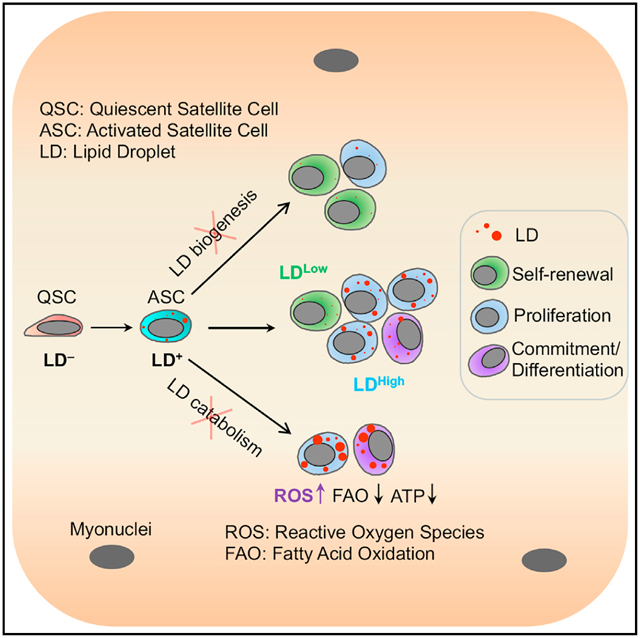
SUMMARY
The lipid droplet (LD) is a central hub for fatty acid metabolism in cells. Here we define the dynamics and explore the role of LDs in skeletal muscle satellite cells (SCs), a stem cell population responsible for muscle regeneration. In newly divided SCs, LDs are unequally distributed in sister cells exhibiting asymmetric cell fates, as the LDLow cell self-renews while the LDHigh cell commits to differentiation. When transplanted into regenerating muscles, LDLow cells outperform LDHigh cells in self-renewal and regeneration in vivo. Pharmacological inhibition of LD biogenesis or genetic inhibition of LD catabolism through knockout of Pnpla2 (encoding ATGL, the rate-limiting enzyme for lipolysis) disrupts cell fate homeostasis and impairs the regenerative capacity of SCs. Dysfunction of Pnpla2-null SCs is associated with energy insufficiency and oxidative stress that can be partially rescued by antioxidant (N-acetylcysteine) treatment. These results establish a direct link between LD dynamics and stem cell fate determination.
Graphical abstract
In brief
Yue et al. identify a role for lipid droplets (LDs) in fate determination and regeneration of satellite cells (SCs). LD distribution in newly divided SCs underlies cell fate, as LDLow cells self-renew while LDHigh cells commit to differentiation. Inhibiting LD biogenesis or catabolism alters SC fate and impairs regeneration.
INTRODUCTION
Lipid droplets (LDs) are dynamic intracellular organelles characterized by a unique architecture with an interface between the dispersed oil phase and the aqueous cytosol (Thiam et al., 2013). LDs are responsible for storing extra cellular fatty acids (FAs) in the forms of triacylglycerol (TAG) as an energy source while minimizing the substantial lipotoxicity (Walther and Farese, 2012). In addition, the degradation of LDs provides reservoirs of lipids for membrane synthesis (Atilla-Gokcumen et al., 2014) and supplies precursors for many signaling mediators (Zechner et al., 2012). Not surprisingly, the dysregulation of LD biogenesis and degradation underlies various pathological conditions and metabolic diseases, such as obesity, neutral lipid storage diseases, and liver and neuron diseases (Krahmer et al., 2013; Liu et al., 2015b; Olzmann and Carvalho, 2019).
Emerging studies have implicated a role for LD metabolism in stem cell behavior. In a mouse model of Alzheimer’s disease, LDs accumulated in adjacent ependymal cells affect the proliferation of forebrain neural stem cells (NSCs) (Hamilton et al., 2015). During Drosophila development, LDs that formed in the glial cell niche preserve the proliferation of NSCs under conditions of oxidative stress by limiting the reactive oxygen species (ROS) (Bailey et al., 2015). These studies highlight a non-cell-autonomous role of LDs in niche supporting cells in regulating stem cell function. Within stem cells, abnormal LD biogenesis was proposed as an effector for the loss of male germline stem cells (GSCs) that caused by dMfn deletion (Demarco et al., 2019). Beyond this study, our knowledge about how LDs function in stem cells is still limited, and a role for LDs in stem cell fate decision has yet to be defined.
Skeletal muscle satellite cells (SCs) are tissue resident stem cells responsible for muscle growth and regeneration. SCs typically reside in a hypoxic microenvironment under the basal lamina and maintain a mitotically quiescent state under homeostatic conditions. In response to muscle injury, they are activated to enter the cell cycle either to self-renew to replenish the stem cell pool or to differentiate to repair the damaged myofiber (Kuang et al., 2008). SCs are self-renewed through symmetric and asymmetric divisions, where symmetric divisions give rise to two daughter cells of identical fate and asymmetric divisions give rise to a stem cell and a committed cell (Kuang et al., 2007). Recent work has elucidated various extrinsic and intrinsic signaling pathways regulating SC fate decisions (Feige et al., 2018; Relaix et al., 2021). Particularly, emerging studies have shed light on the metabolic adaptions of SCs during fate transitions (Pala et al., 2018; Ryall et al., 2015; Yucel et al., 2019), but whether and how cellular metabolism regulates the self-renewal and commitment of SCs remain largely unknown.
In the present study, we report the dynamics of LDs in SCs are linked to cell fates and function. Committed SCs that express Myf5 accumulate abundant LDs during cell division, while self-renewing SCs rarely have LDs. Using pharmacological and genetic tools, we identify that LD homeostasis is required for SC fate decisions. These findings define an indispensable role of LDs in metabolic regulation of stem cell fate.
RESULTS
LDs are highly dynamic at various states of SCs
While studying PAX7+ SC dynamics on cultured single myofibers, we discovered abundant LDs (marked by Bodipy) in the doublets of newly divided SCs but not in quiescent SCs (QSCs) (Figure S1A; Videos S1 and S2). Time-course imaging showed that LDs appeared in 31% of SCs as early as 12 h in culture, shortly after SC activation marked by MyoD expression (Figures 1A and 1B). The frequency of LD-containing SCs continually increased and reached 97% by 48 h, when nearly all SCs have undergone one round of cell division (Figures 1A and 1B). The number of LDs per SC also increased during activation and division, from 1.0 to 4.9 LDs at 20 h and 48 h (Figure 1C).
Figure 1. Dynamics of LDs during homeostasis of SCs.
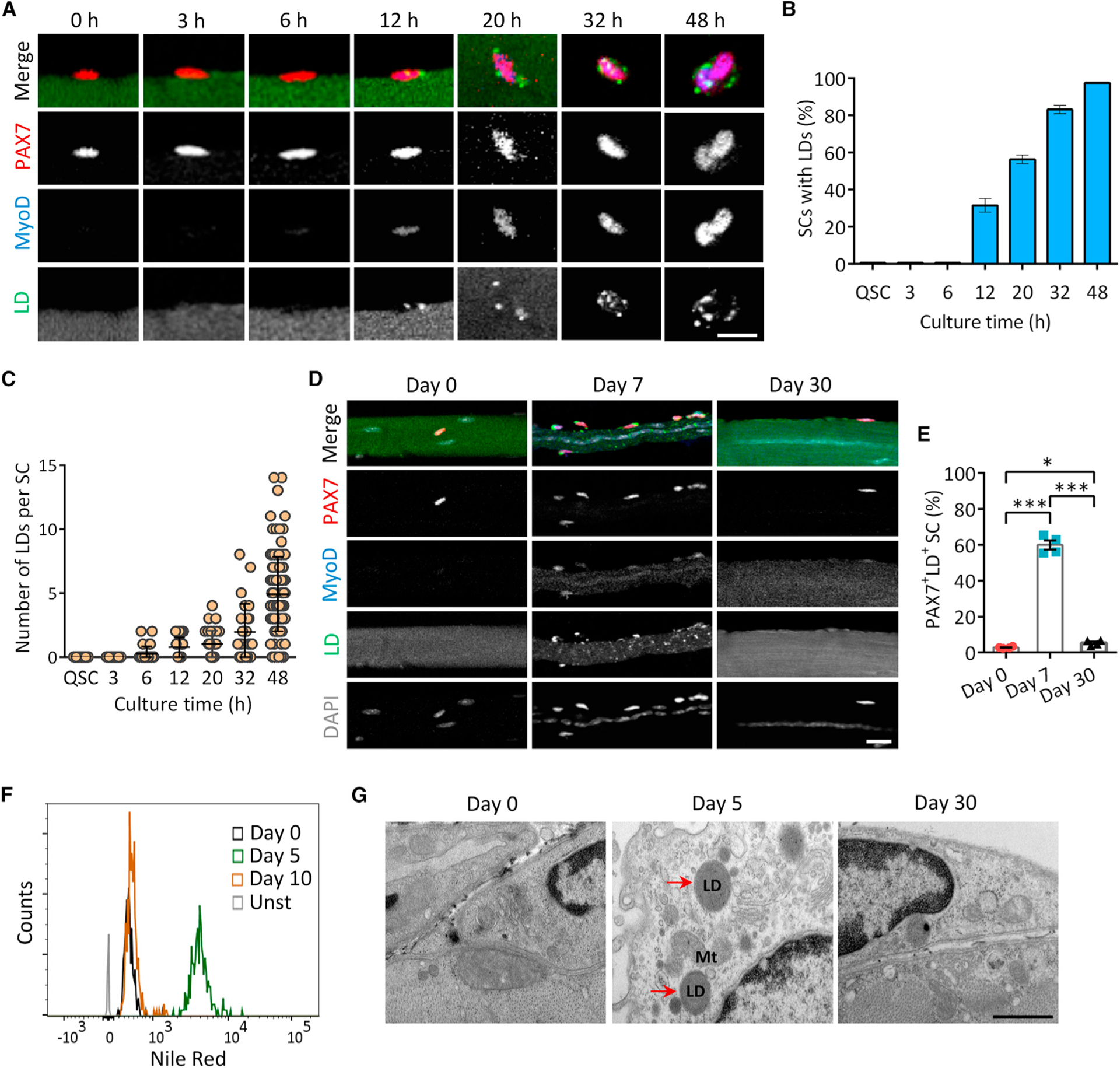
(A) Time course images showing progressive accumulation of LDs (labeled with BODIPY493/503) during activation and division of SCs on cultured myofiber. Scale bar, 10 μm.
(B) Percentage of LD-carrying SCs on myofiber shown in (A) (n = 4 mice). An average of 736 SCs were analyzed at each time point.
(C) Quantification of the number of LDs per SC at various time points (n = 4 mice).
(D) Representative images of LD dynamics in SCs during muscle regeneration in vivo. Scale bar, 10 μm.
(E) Percentage of LD-carrying SCs during muscle regeneration in vivo (n = 4 mice). An average of 402 SCs were analyzed at each time point.
(F) LD contents in ASCs (day 5) compared with QSCs (day 0) and self-renewed SCs (day 10) during muscle regeneration. SCs were isolated from Pax7nGFP mouse and subjected to Nile red staining and flow cytometry analysis (n = 3 mice). Unst, unstained.
(G) Transmission electron microscopy imaging of LDs in SCs resided in injured and non-injured TA muscles. Red arrows indicate LDs. Mt, mitochondria. Scale bar, 500 nm.
Data are mean ± standard deviation; Student’s t test. *p < 0.05, ***p < 0.001. See also Figure S1.
We also examined LDs in vivo before and after cardiotoxin (CTX)-induced activation of SCs, LDs were abundant in activated SCs (ASCs, PAX7+MyoD+, 60%) at 7 days post injury (dpi), but were rare in QSCs (PAX7+MyoD−, 2.8%) before muscle injury (0 dpi) or in self-renewed SCs (SSCs, PAX7+MyoD−, 5.4%) at 30 dpi when regeneration was completed (Figures 1D and 1E). To validate these results, we isolated SCs from Pax7nGFP mice, stained with Nile red to label neutral lipids, and performed flow cytometry analysis. Nile red fluorescence was stronger at 5 dpi compared with 0 and 10 dpi (Figures 1F and S1B). We further examined LDs in SCs by transmission electron microscopy. No LDs were visible in QSCs or SSCs at 0 and 30 dpi, whereas LDs were observed in SCs, often in close proximity to mitochondria, at 5 dpi (Figure 1G). Together, our results reveal previously unappreciated LD dynamics in SCs during myogenesis.
Exogenous FAs contribute to LD biogenesis in SCs
To determine the source of LDs in SCs, we first examined the contribution of protein and fatty acid-rich fetal bovine serum (FBS) to LDs in SC-derived primary myoblasts. LDs were abundant in myoblasts cultured with 10%–20% FBS, but were reduced in 5% FBS cultures and rapidly depleted when FBS was deprived (Figure S1C). In contrast, the abundance of LDs was not affected by media glucose levels (Figure S1D). Live-cell imaging of Pax7nGFP SCs with BODIPY558/568-labeled FAs (C12 Red) on extensor digitorum longus (EDL) myofibers showed that red signal was not detected in QSCs, but appeared as clear droplets in ASCs (12–24 h), and became abundant in proliferating SCs (36–48 h) (Figure S1E). The lack of labeled LDs in SCs before 12 h was not due to slow C12 penetration, as C12 penetrated cells within 10 min of pulse in primary myoblasts and appeared inside LDs by 4 h (Figure S1F). These data point to FAs as the source of LDs and suggest that LD metabolism is highly active in proliferating SCs.
LD abundance specifies SC fates
Using the myofiber explant model, we noted that a fraction of newly divided doublet SCs cultured for 42 h exhibited unequal LD distribution (LDLow/High) between sister cells (Figure 2A), where one cell contained obviously more LDs (average = 5.1, defined as LDHigh) than the other cell (average = 1.2, defined as LDLow) (Figure 2B). We thus investigated whether the abundance of LDs is associated with the fate choice of newly divided SCs. We found that sister cells exhibiting asymmetric LDLow/High distribution were predominantly in apical-basal orientation while sister cells with symmetric LD distribution (LDHigh/High or LDLow/Low) were mostly planarly oriented (Figure 2C). As the orientation of SC division determines cell fates (Feige et al., 2018; Kuang et al., 2007), these observations implicate an association of LDs with SC fate determination. In support of this, self-renewed (PAX7+ MyoD−) SCs had less LD accumulation than proliferating (PAX7+ MyoD+) and differentiating (PAX7−MyoD+) cells at 72 h on cultured myofibers (Figures 2D and 2E). Similarly, on CTX-injured EDL myofibers at 7 dpi, numerous LDs were observed in a majority of proliferating and differentiating SCs in contrast with the few LDs in self-renewing SCs (Figures 2F and 2G). Thus, LD abundance is correlated with the cell fate status of SCs.
Figure 2. LDs in newly divided SCs specify differentiation versus self-renewal cell fate.
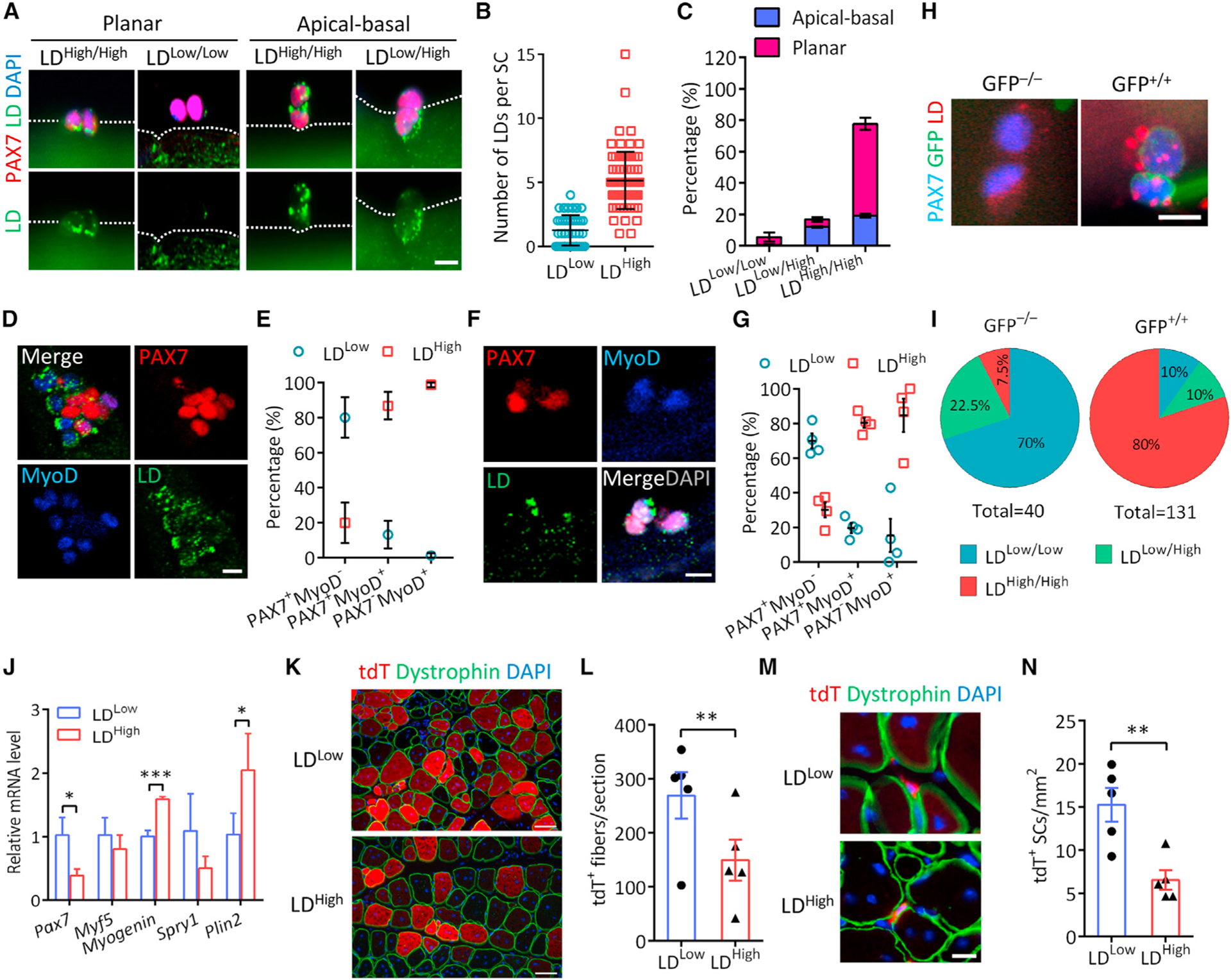
(A and B) LD abundance (A) and the number of LDs per cell (B) in newly divided SC doublets on fiber at 42 h in culture (n = 4 mice). Scale bar, 10 μm.
(C) Percentages of LDLow/Low, LDLow/High, and LDHigh/High SC doublets in apical-basal and planar orientations (n = 3 mice). A total of 271 doublets were analyzed.
(D and E) Images showing LD abundance associated with SC fate (D) and quantification of LDLow and LDHigh cells among SC populations (E) on cultured myofibers at 72 h (n = 3 mice). A total of 227 cells were analyzed. Scale bar, 10 μm.
(F and G) Images showing heterogeneous LD abundance in SCs (F) and quantification of LDLow and LDHigh cells among quiescent, PSCs and CSCs (G) from injured EDL muscle. n = 4 mice. Scale bar, 10 μm.
(H and I) Immunostaining of LDs in SC doublets (H) and percentages of LDLow/Low, LDHigh/Low, and LDHigh/High doublets among self-renewing (GFP−/−) and CSCs(GFP+/+) (I) on myofibers isolated from Myf5Cre;Rosa26sfGFP mice at 48 h. n = 4 mice, 40 GFP−/− and 131 GFP+/+ doublets were analyzed. Scale bar, 10 μm.
(J) Expression of genes related to myogenic commitment and LD biosynthesis in LDLow and LDHigh SCs FACS-isolated from Pax7CreER;Rosa26tdTomato muscles at 5.5 dpi. n = 3 mice.
(K and L) Images showing tdT+ myofibers (K) and quantification of the number of tdT+ myofibers (L) in regenerated muscles of NRG mice transplanted with FACS-isolated LDLow and LDHigh SCs at 14 dpi. n = 5 mice. Scale bar, 50 μm.
(M and N) Images showing tdT+ SCs (M) and quantification of the number of tdT+ SCs (N) in transplanted muscles. n = 5 mice. Scale bar, 10 μm.
Data are mean ± SD; Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S2.
To further confirm this notion, we established Myf5Cre; Rosa26LSL-sfGFP/+ reporter mice to track non-committed (GFP−) and committed (GFP+) SCs in newly divided SC doublets on myofiber culture (Kuang et al., 2007). At 48 h, the vast majority (~80%) of GFP+ doublets was LDHigh/High, whereas most (~70%) GFP− doublets were LDLow/Low (Figures 2H and 2I). Consistent results were observed when individual GFP+ and GFP− SCs were quantified (Figures S2A and S2B). Moreover, the levels of QSC genes (Pax7 and Spry1) were higher in LDLow than in LDHigh SCs, but the levels of myogenic and LD-specific genes were higher in LDHigh than in LDLow SCs (Figure 2J). These data further support the notion that the abundance of LDs specifies the fate status of SCs.
We also evaluated the function of LDHigh and LDLow SCs in vivo through transplantation of fluorescence activated cell sorting (FACS)-isolated tdT+ LDLow and tdT+ LDHigh cells from regenerating muscles of Pax7CreER;Rosa26tdTomato mice based on LipidSpot610 labelling (Figures S2C–S2E). The indelible tdT marks the lineage of transplanted cells. At 14 d post-transplantation, LDLow SCs consistently gave rise to approximately two times more tdT+ myofibers than did LDHigh SCs in both NRG and mdx recipient mice (Figures 2K, 2L, S2F, and S2G). In addition, more dystrophin+ myofibers were observed in mdx muscles transplanted with LDLow SCs (Figures S2F and S2H). Moreover, more tdT+ SCs were found in muscles transplanted with LDLow compared with LDHigh SCs in both NRG and mdx mice (Figures 2M, 2N, S2I, and S2J). These results demonstrate that LDLow SCs are superior to LDHigh SCs in self-renewal and regenerative capacity.
SC fate progression is coupled with changes in neutral lipid metabolism
To explore the molecular mechanisms underlying LD dynamics during SC fate transitions, we performed single-cell RNA-sequencing (scRNA-seq) of Pax7+ SCs at 0 (non-injured), 5, and 10 dpi (Figure S3A). We obtained high-quality transcriptional profiles of 3,995 SCs (250, 2,835, and 910 at 0, 5, and 10 dpi, respectively) (Figure 3A). Unsupervised clustering identified six unique clusters (Figure 3B), labeled as QSCs, SSCs (self-renewal), ASCs, proliferating SCs (PSCs), committed SCs (CSCs), and differentiating SCs (DSCs) based on the top-10 gene signatures of each cluster (Figure S3B). Violin plots highlighted the enrichment of Pax7 in QSCs, SSCs, ASCs, and PSCs, with complementary expression of differentiation markers (Myog, Tnnt2, and Acta2) in CSCs and DSCs (Figures 3C and S3C). Hence, our collection of cells encompass the known heterogeneity and recapitulate the dynamics of SC fate transitions during muscle regeneration (Oprescu et al., 2020).
Figure 3. SC fate progression is coupled with changes in neutral lipid metabolism.
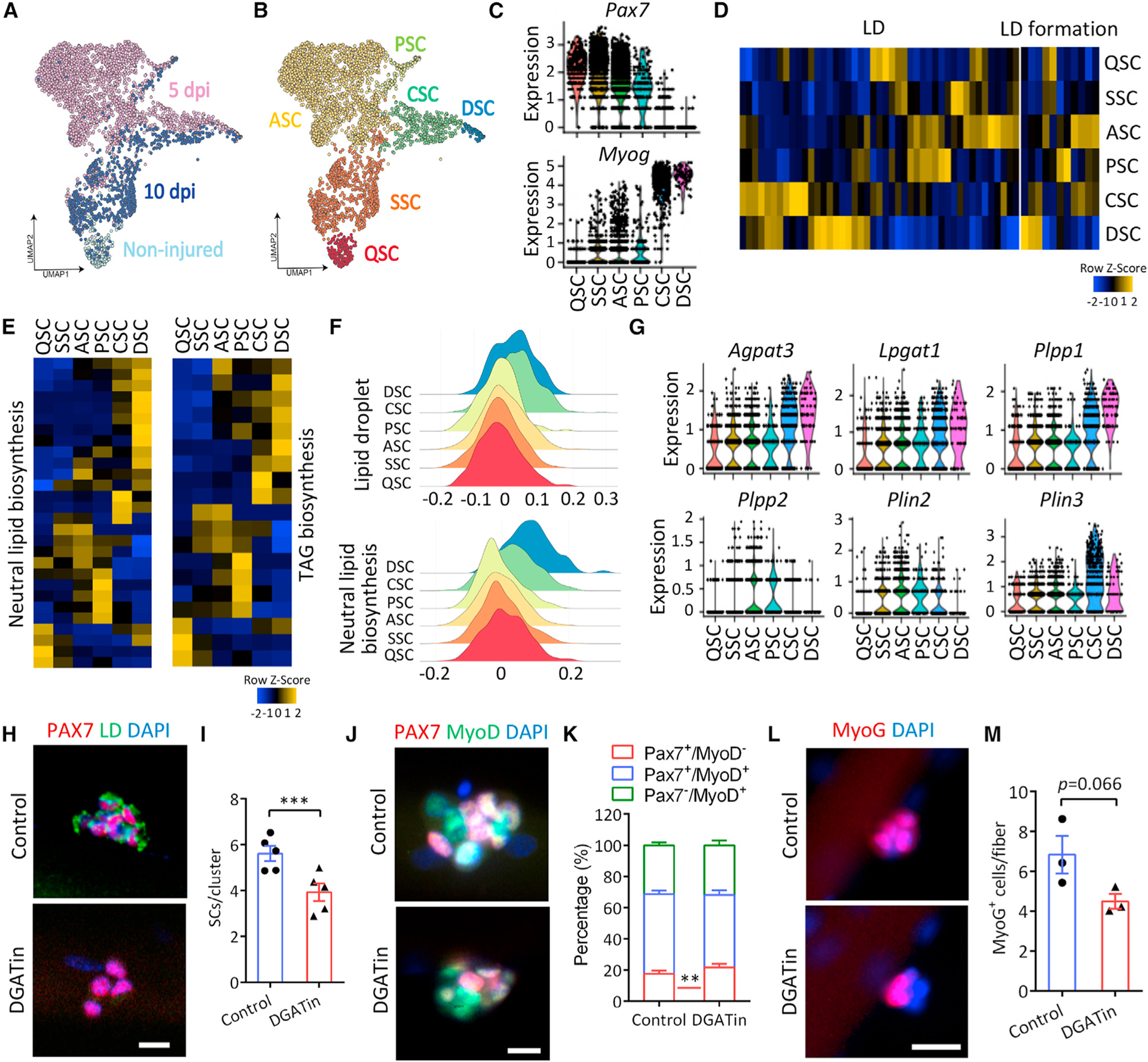
(A) UMAP embedding of scRNA-seq data on SCs isolated from non-injured muscles (n = 250) and injured muscles at 5 dpi (n = 2,835) and 10 dpi (n = 910), colored by isolation time.
(B) UMAP embedding of scRNA-seq data colored by subpopulation identity.
(C) Violin plot showing the expression of SC markers informing cluster annotation.
(D and E) Heatmaps of the genes annotated in gene ontology (GO) term LD (GO: 0005811) and LD formation (GO: 0140042) (D), and neutral lipid (GO: 0046460) and TAG biosynthetic process (GO: 0019432) and (E) in SC clusters.
(F) Density plot visualizing enrichment of genes in GO term LD and neutral lipid biosynthetic process in SC clusters.
(G) Violin plot showing the expression of key LD biosynthesis-related genes.
(H and I) Inhibition of LD biogenesis in SCs with DGAT inhibitors (DGATin) (H) and quantification of PAX7+ SCs per cluster (I) on cultured single myofibers at 72 h (n = 5 mice). Scale bar, 10 μm.
(J and K) Immunostaining PAX7 and MyoD (J) and quantification of SC population on 72 h cultured single myofibers (K) (n = 5 mice). Scale bar, 10 μm.
(L and M) Immunostaining of MyoG (L) and quantification of MyoG+ cells per myofiber (M) with control and DGATin treatment for 72 h (n = 3 mice). Scale bar, 20 μm.
Data are mean ± standard deviation; Student’s t test. **p < 0.01, ***p < 0.001. See also Figure S3.
We then analyzed the lipid metabolic pathways enriched in the six clusters, and found that genes annotated as gene ontology (GO) terms LD, LD formation, neutral lipid, TAG, and glycerolipid biosynthetic processes were sequentially enriched in QSC/SSC, ASCs, PSCs, CSCs, and DSCs (Figures 3D, 3E, and S3D). Functional enrichment density plot further confirmed the clear enrichment of LD and neutral lipid biosynthetic genes in CSCs and DSCs (Figure 3F). A violin plot revealed that the expression of TAG synthesis related genes Agpat3, Lpgat1, and Plpp1 was apparently upregulated in CSCs and DSCs compared with other cell clusters, while Plpp2 and LD membrane related gene Plin2 were expressed in ASCs and PSCs, but rarely detected in QSCs or DSCs (Figure 3G).
In contrast with the lipid biosynthetic process, mitochondrial FA beta-oxidation (FAO, underlying lipid catabolic process) related genes were sequentially enriched in line with myogenic commitment (Figure S3E). In addition, several genes encoding TAG lipolytic enzymes such as Pnpla1, Plb1, and Ddhd2 were upregulated in PSCs and CSCs, respectively (Figure S3F). Notably, the expression of Pnpla2, encoding the initial and rate-limiting enzyme in TAG hydrolysis, was increased in DSCs compared with QSCs and SSCs (Figure S3F), although only detected in a fraction of DSCs (Figure S3G). The scRNA-seq results suggest that both the biosynthesis and catabolism of LD are activated progressively during myogenic commitment of QSCs and deactivated in SSCs upon completion of muscle regeneration.
Inhibition of LD biosynthesis promotes SC quiescence without affecting differentiation
We next examined whether the disruption of LD biosynthesis affects SC fate and function. The pharmacological inhibition of DGAT1 and DGAT2 (hereafter referred to as DGATin), two enzymes catalyzing the last step of TAG synthesis for LD formation, effectively decreased the accumulation of LDs in PSCs on cultured myofibers (Figure 3H). In parallel, the number of PAX7+ cells was significantly decreased by 30.1% in DGATin compared with the control groups (Figure 3I). The decrease in PAX7+ cells was mainly due to an increase in SC quiescence (PAX7+MyoD−) and a slight decrease in proliferation (PAX7+MyoD+) (Figures 3J and 3K). Although DGATin tended to decrease the number of MyoG+ cells (Figures 3L and 3M), it did not directly affect myogenic differentiation when administered to differentiating myoblasts (Figures S3H and S3I). These results indicate that the blockade of LD biogenesis promotes quiescence without affecting differentiation.
ATGL deficiency disrupts LD dynamics in SCs and causes abnormal LD accumulation
As LD dynamics are coordinated by a balance of lipid biosynthesis and catabolism (Olzmann and Carvalho, 2019), we also examined whether blocking LD catabolism affects SC fate and function. We focused on ATGL (encoded by the Pnpla2 gene, upregulated in DSCs), the rate-limiting enzyme for LD catabolism (Zechner et al., 2012). ATGL was undetectable in QSCs, but expressed in ASCs and higher in PSCs (Figures S4A and S4B), while its expression further increased during differentiation of primary myoblasts (Figure S4B), echoing the dynamics of LDs in SCs (Figure 1A). Although the ATGL level was indistinguishable between sister SCs (Figure S4C), we observed a substantial fraction of subcellular ATGL puncta that co-localized with LDs in dividing SCs (Figures S4D and S4E), suggesting that ATGL plays a key role in regulating LD metabolism in PSCs.
To explore the role of ATGL-dependent lipolysis in SCs, we generated tamoxifen (TMX)-inducible SC-specific Pnpla2 knockout mice (Pax7CreERT;Pnpla2flox/flox, referred to as Pnpla2PKO). TMX-induced deletion the Pnpla2 allele and loss of the ATGL protein was verified by immunofluorescence and immunoblot (Figures S4F and S4G). Pnpla2PKO led to the excessive accumulation of LDs in cultured SCs on myofiber and primary myoblasts (Figures 4A and S4H), whereas an overexpression of ATGL eliminated LDs in primary myoblasts (Figure S4I). Consistently, the total cellular TAG content was 2.8-fold in Pnpla2-null compared with wild-type (WT) myoblasts (Figure 4B). TAG catabolism can be induced by serum starvation and monitored by release of glycerol. Pnpla2-null myoblasts failed to breakdown LDs and released significantly less glycerol than did WT (Figures S4J–S4L). Thus, ATGL functions to target LDs in PSCs for lipolysis and loss of ATGL impairs LD mobilization in SCs.
Figure 4. Conditional deletion of ATGL leads to abnormal LD accumulation and perturbs cell fate homeostasis of SCs.
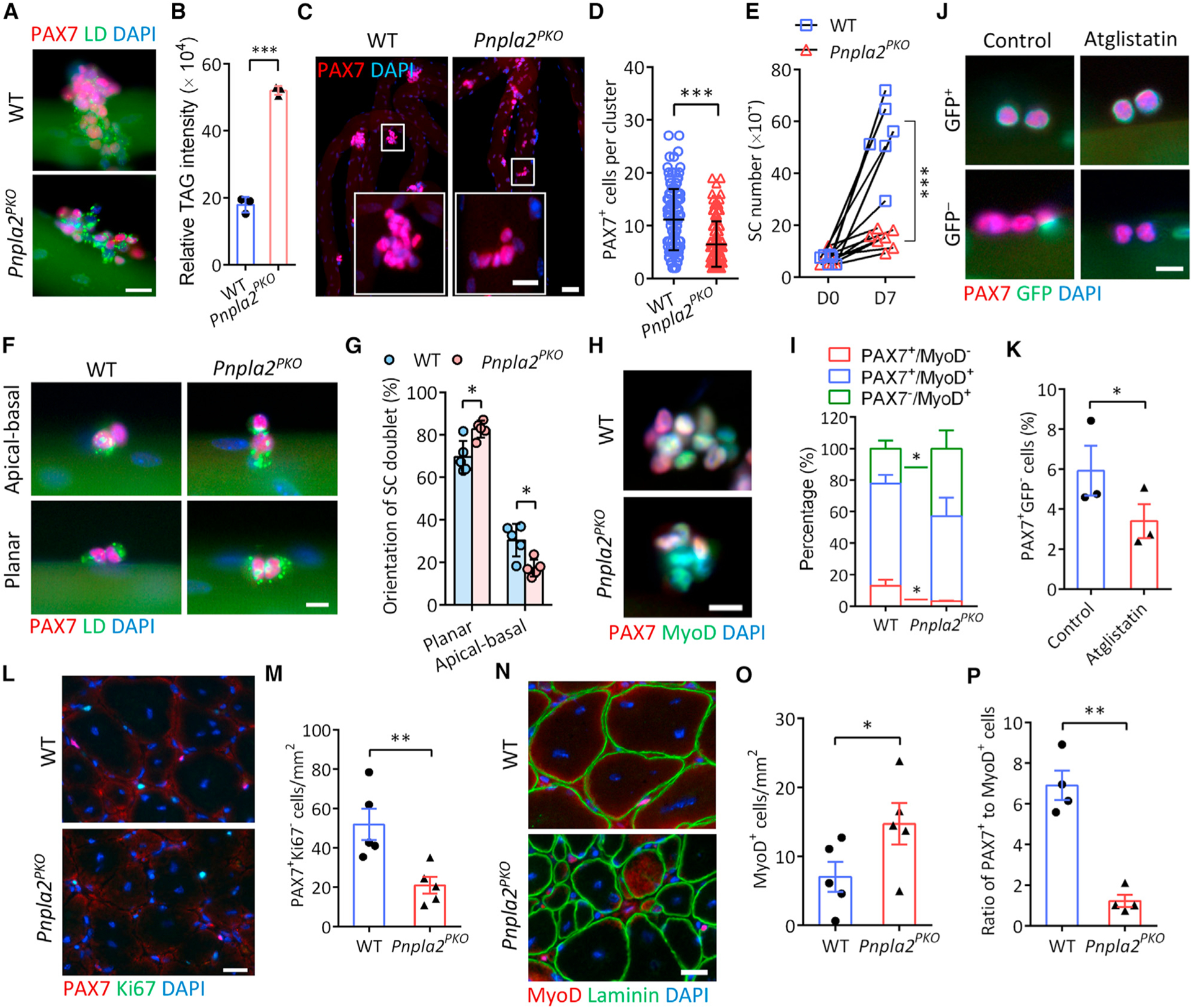
(A) LDs in SCs grown on WT and Pnpla2PKO myofibers cultured for 72 h. Scale bar, 20 μm.
(B) Increased TAG content in Pnpla2PKO SC-derived primary myoblasts measured by lipid profiling (n = 3 mice).
(C and D) Immunostaining of PAX7 (C) and quantification of PAX7+ SCs per cluster (D) on 72 h cultured single myofibers (n = 3 mice). Scale bars, 50 μm and 20 μm for inset.
(E) Growth rate of SCs isolated by FACS from WT and Pnpla2PKO resting muscles. WT: n = 6 mice; Pnpla2PKO: n = 7 mice.
(F and G) Immunostaining of PAX7 and LDs (F) and quantification of planar and apical-basal dividing SC doublets (G) on 48 h cultured single myofibers (n = 5 mice). Scale bar, 10 μm.
(H and I) PAX7 and MyoD immunostaining (H) and quantification of SC population (I) on 72 h cultured single myofibers (n = 4 mice). Scale bar, 10 μm.
(J and K) Representative images of SCs (J) and quantification of the number of GFP− and GFP+ SCs (K) on myofibers isolated from Myf5Cre;Rosa26sfGFP mice cultured 48 h with ATGL inhibitor Atglistatin (50 μM). Scale bar, 10 μm.
(L and M) PAX7 and Ki67 immunostaining (L) and quantification of self-renewed (PAX7+Ki67−) SCs (M) in TA muscles at 14 dpi (n = 5 mice). Scale bar, 20 μm.
(N and O) MyoD and Laminin immunostaining (N) and quantification of MyoD+ SCs (O) in TA muscles at 14 dpi (n = 5 mice). Scale bar, 20 μm.
(P) Ratio of the PAX7+Ki67− (self-renewed) to MyoD+ SCs from (M, O).
Data are mean ± standard deviation; Student’s t test. *p < 0.05, **p < 0.001, ***p < 0.001. See also Figures S4 and S5.
Loss of ATGL perturbs cell fate homeostasis of SCs
We next examined how the disruption of LD metabolism affects SC fate transitions in Pnpla2PKO mice. The loss of ATGL in Pnpla2PKO mice did not alter the number of PAX7+ QSCs in non-injured muscles (Figures S5A and S5B). However, after 72 h in culture, there were significantly fewer PAX7+ cells on Pnpla2PKO myofibers than on WT myofibers (Figures 4C and 4D). We also sorted and cultured SCs from WT and Pnpla2PKO mice and found that the loss of ATGL impaired myoblast proliferation (Figures 4E and S5C). This was further corroborated by a significant decrease in SC numbers in Pnpla2PKO muscles at both 3.5 and 5.5 dpi (Figures S5D and S5E). These data suggest that ATGL is required for SC expansion both in vitro and in vivo.
The impaired proliferation of Pnpla2-null SCs prompted us to ask whether these cells have a propensity for a specific fate. We examined the orientation of newly divided SC doublets on ex vivo myofibers. With the excessive LD accumulation, Pnpla2PKO significantly decreased the percentage of apicalbasally oriented doublets, while increasing planarly oriented doublets (Figures 4F and 4G). Consistently, fewer QSCs (PAX7+ MyoD−) but more DSCs (PAX7−MyoD+) were observed on Pnpla2PKO than on WT myofibers (Figures 4H and 4I). Similar results were observed in FACS-isolated SCs cultured for 7 days (Figure S5F). We treated Myf5Cre;Rosa26sfGFP myofibers with a potent ATGL inhibitor Atglistatin in culture and found that the percentage of PAX7+GFP− SCs was significantly lower in Atglistatin-treated myofibers (3.4%), comparing with controls (5.9%) (Figures 4J and 4K). These data indicate that inhibition of ATGL impairs self-renewal.
We also examined differentiation in vivo, and observed fewer MyoG+ cells in regenerating muscles at 3.5 dpi in Pnpla2PKO compared with WT (Figure S5G), but a higher ratio of MyoG+ to PAX7+ cells (Figure S5H), suggesting that the differentiation per se is accelerated at the expense of self-renewal in Pnpla2PKO mice. This was further supported by the decreased number of SSCs but increased DSCs in Pnpla2PKO transverse abdominis (TA) muscles at 14 dpi in vivo (Figures 4L–4O), which eventually resulted in a significantly lower ratio of PAX7+ to MyoD+ cells (Figure 4P). Thus, the loss of ATGL disrupts SC cell fate homeostasis by diminishing proliferation and self-renewal and promoting differentiation.
We further examined the effect of Pnpla2PKO on differentiation and fusion directly. When FACS-isolated SCs were immediately induced to differentiate without initial expansion, the percentage of MyHC+ myocytes (differentiation index) was slightly higher, whereas the fusion index (quantified by the number of myotubes with ≥2 nuclei) was significantly lower, resulting in fewer and shorter myotubes in Pnpla2-null than in WT cultures (Figures S5I–S5K). Intriguingly, in Pnpla2-null cultures, MyHC+ cells containing abnormal LDs were rarely fused to form myotubes (Figure S5L). Similar results were observed when FACS-isolated SCs were differentiated after cultured for 7 days (Figure S5M–S5O). These observations suggest that, in the absence of ATGL, SCs differentiate prematurely but fail to fuse efficiently. Taken together, these data indicate that ATGL-mediated LD metabolism is essential for SC fate homeostasis by affecting proliferation, self-renewal, and differentiation.
ATGL-dependent lipolysis is necessary for the regenerative function of SCs
To define the physiological role of ATGL-dependent LD mobilization in vivo, we evaluated muscle regeneration using a CTX injury model (Figure 5A). At 5.5 dpi, Pnpla2PKO TA muscles seemed to be smaller, accumulated more Evans blue dye indicative of membrane leakage, and had significantly lower muscle recovery rates compared with WT (Figures 5B and 5C). Histological analyses showed that Pnpla2PKO muscles had smaller regenerated area and a decreased number and size of newly formed myofibers at 3.5, 5.5, and 10 dpi (Figure 5D). Immunofluorescence staining further revealed a delayed regeneration in Pnpla2PKO muscles, as indicated by the reduced nascent (eMyHC+) and mature (dystrophin+) myofiber number at early (3.5 dpi) and late (10 dpi) stages of regeneration, respectively (Figure 5E). Specifically, the regenerated area in Pnpla2PKO muscle was 18.2% in comparison with 63.1% for WT muscle (Figure 5F). The number and cross-sectional area (CSA) of nascent myofibers in Pnpla2PKO muscles were decreased by 74% and 42.3%, respectively, compared with their WT counterparts at 3.5 dpi (Figures 5G and 5H). Similarly, the number of mature myofibers was also decreased by 62% in Pnpla2PKO muscles compared with WT at 5.5 dpi (Figure 5I). The regenerated Pnpla2PKO myofibers remained smaller than WT counterparts at 30 dpi, when regeneration is completed (Figure 5E). Compared with WT at 10 dpi and 30 dpi, the average CSA of Pnpla2PKO myofiber was significantly reduced by 28% and 24.5% in line with a clear left-shift of the CSA distribution (Figures 5J–5M). These observations demonstrate that ATGL-dependent lipolysis is required for the regenerative capacity of SCs.
Figure 5. ATGL-deficient SCs fail to regenerate muscle in response to acute injury.
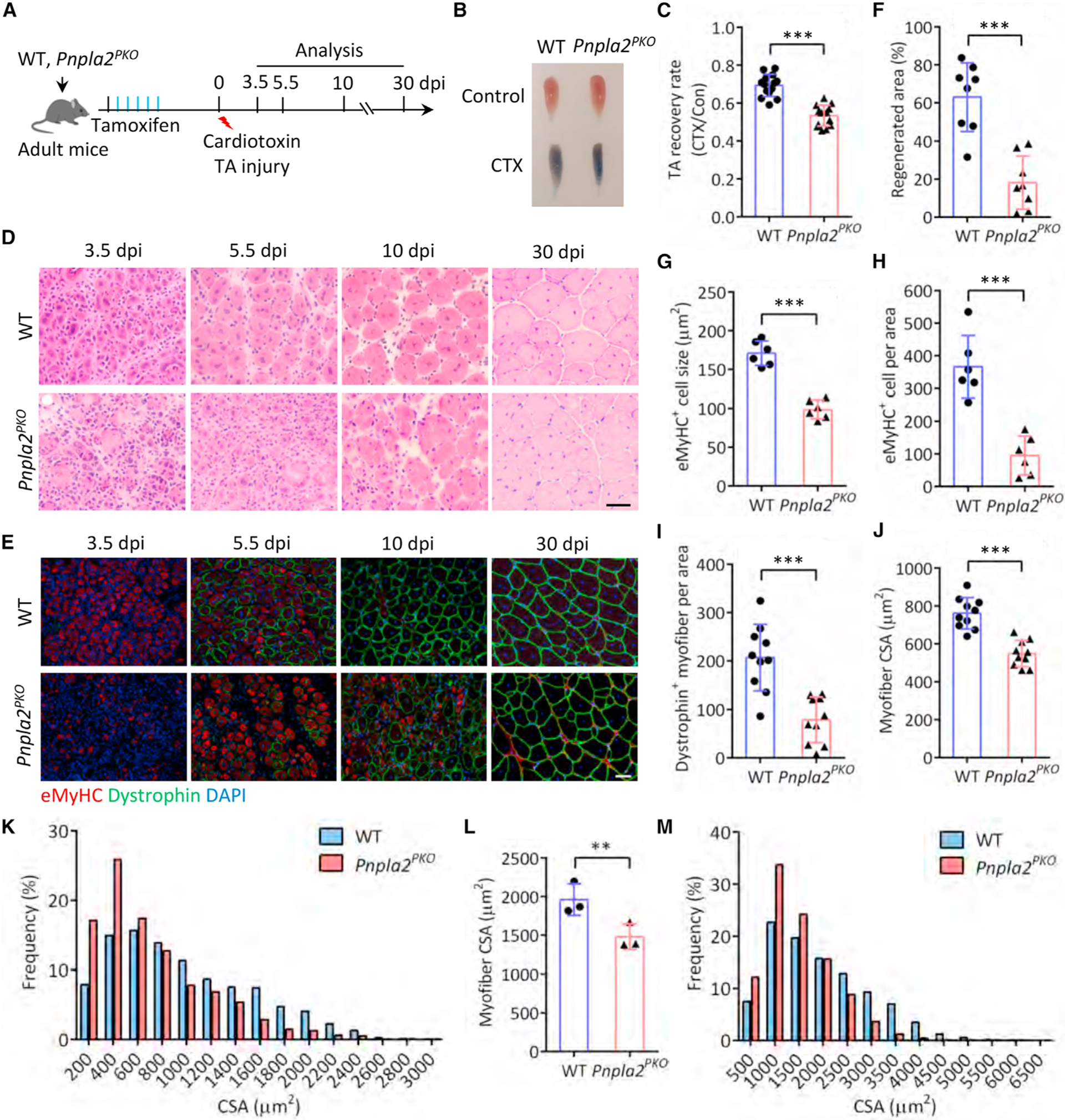
(A) Experimental scheme for WT and conditional knockout Pnpla2PKO mice.
(B) Evans blue dye incorporation in TA muscles at 5.5 dpi.
(C) Recovery rate of TA muscle at 5.5 dpi (n = 14 mice).
(D and E) Hematoxylin and eosin (H&E) staining (D) and immunofluorescence (E) of TA muscle cross-sections. Scale bars, 50 μm.
(F) Regenerated area of TA muscle cross-sections at 5.5 dpi (n = 8).
(G and H) Number (G) and CSA (H) of eMyHC+ myofibers in TA muscle cross-sections at 3.5 dpi (n = 6 mice).
(I) Number of dystrophin+ myofibers in TA muscle cross-sections at 5.5 dpi (n = 10 mice).
(J and K) Average CSA (J) and CSA distribution (K) of dystrophin+ myofibers in TA muscle cross-sections at 10 dpi. For (J) n = 10 mice, for (K) n = 5 mice.
(L and M) Average CSA (L) and CSA distribution (M) of dystrophin+ myofibers in TA muscle cross-sections at 30 dpi (n = 3 mice).
Data are mean ± standard deviation; Student’s t test. **p < 0.01, ***p < 0.001.
ATGL deficiency diminishes FAO flux and leads to cellular oxidative stress in SCs
We next sought to determine the biochemical basis underlying the observed defects in Pnpla2-null SCs. ATGL-dependent lipolysis yields FAs that are released from LDs and subsequently oxidized for energy production in mitochondria (Ahmadian et al., 2011; Zechner et al., 2012). We evaluated the bioenergetics of FACS-isolated Pnpla2-null SCs using Seahorse bioanalyzer. Compared with WT cells, the Pnpla2-null myoblasts had significantly lower oxygen consumption rates (OCR) corresponding to basal respiration, adenosine triphosphate (ATP) production, and non-mitochondrial respiration (Figures 6A and S6A), indicating the decreased overall respiratory activity. To determine the effects on FAO, we treated those myoblasts with Etomoxir, an irreversible pharmacological inhibitor of the rate-limiting enzyme for FAO, carnitine palmitoyltransferase 1. We quantified the endogenous FAO level by subtracting the oxygen consumption rates under Etomoxir treatment from that of vehicle control, and found a remarkable decrease in the basal and maximal FAO capacity in Pnpla2-null myoblasts (Figure 6B), indicative of impaired FAO flux and energy production. In support of this, the levels of cellular acetyl-coenzyme A (CoA) and ATP were both reduced significantly in Pnpla2-null myoblasts (Figures 6C and 6D). Thus, the loss of ATGL diminishes the metabolic flux and energy production of primary myoblasts.
Figure 6. Defective lipolysis impairs energy production and leads to oxidative stress in SCs.
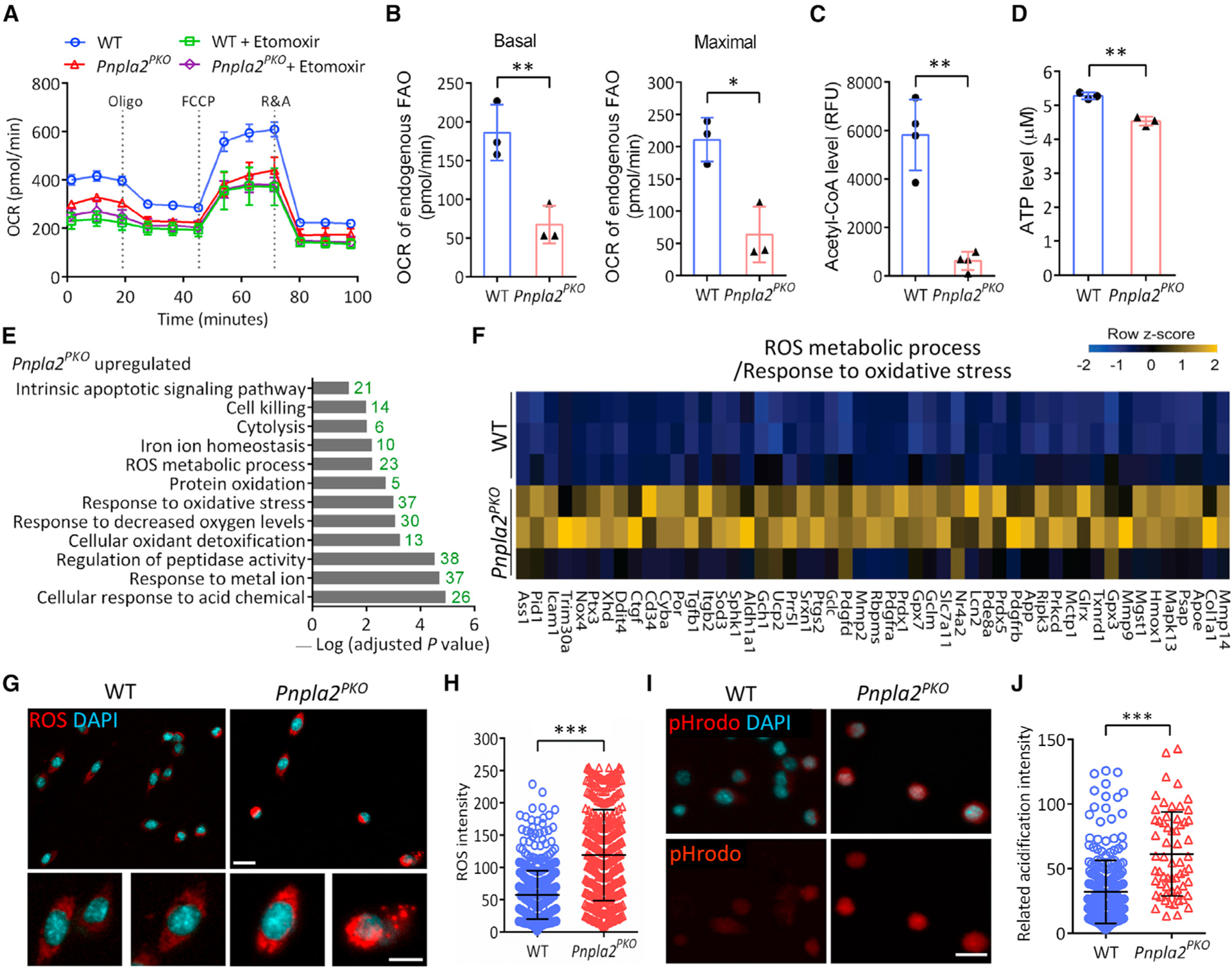
(A) Representative graph showing oxygen consumption rate (OCR) of cultured WT and Pnpla2PKO SC-derived primary myoblasts (n = 3 independent experiments).
(B) Basal and maximal OCR for endogenous mitochondrial FAO calculated by the data obtained in (A) (n = 3 independent experiments, 4 replicates per condition).
(C and D) Cellular acetyl-CoA (C) and ATP (D) levels measured from cultured WT and Pnpla2PKO primary myoblasts (n = 3 mice).
(E) RNA-seq analysis shows enrichment of biological processes related to oxidative stress in Pnpla2PKO SCs (n = 3 mice).
(F) Heatmap showing expression of ROS-related genes in WT and Pnpla2PKO SCs (n = 3 mice).
(G and H) ROS level (G) and quantification (H) in WT and Pnpla2PKO SCs cultured for 7 days after FACS isolation (n = 4 mice). Scale bars, 20 μm and 10 μm for inset.
(I and J) Representative pHrodo staining (I) and quantification (J) showing acidification in Pnpla2PKO SCs. We analyzed 249 cells in WT and 61 cells in Pnpla2PKO mice. Scale bar, 20 μm.
Data are mean ± standard deviation; Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S6.
To gain further insights into how the impaired metabolic flux affects the function of Pnpla2-null primary myoblasts, we performed RNA sequencing on FACS-isolated WT and Pnpla2-null primary myoblasts. Among the 857 differentially expressed genes (adjusted p < 0.05), 152 genes were downregulated and 705 genes were upregulated in Pnpla2-null relative to WT myoblasts (Figure S6B, Table S1), suggesting that Pnpla2PKO mainly activates gene expression in myoblasts. Of note, we found that the expression of Myod1 messenger RNA (mRNA) was significantly upregulated (2.5-fold) in Pnpla2PKO myoblasts, while the expression of SC maker gene Itga7 and Pax7, and Notch1 (a master regulatory signaling for SC self-renewal) was significantly downregulated or tends to decrease (Figure S6C). These transcriptomic results support our in vivo and ex vivo results demonstrating a role for ATGL in SC fate regulation.
A GO analysis unraveled that the expression of genes involved in lipid localization and metabolism was significantly upregulated in Pnpla2-null myoblasts, whereas the expression of genes related to actin cytoskeleton and spindle was significantly downregulated (Figures S6D and S6E), which is consistent with the impaired proliferation and fusion of Pnpla2-null myoblasts. The expression of genes associated with oxidoreductase and electron transfer activities, and nicotinamide adenine dinucleotide binding was increased compared with the WT (Figure S6F), suggesting a potential feedback regulation of cellular respiration to compensate for the insufficient energy production in Pnpla2-null myoblasts. Notably, genes related to biological processes including cellular responses to acid chemical, decreased oxygen levels, and oxidative stress and ROS metabolic process were significantly enriched in Pnpla2-null myoblasts (Figures 6E and 6F), indicating a disruption of cellular redox homeostasis.
To validate the RNA-seq results, we assessed the intracellular ROS level by CellROX staining. While a basal amount of intracellular ROS was detected in WT myoblasts, Pnpla2-null myoblasts exhibited a 2.2-fold increase in ROS signal compared with WT (Figures 6G and 6H), indicating that impaired lipolysis leads to cellular oxidative stress in primary myoblasts. Moreover, a weakly fluorescent signal of pHrodo (a pH indicator) was detected in WT myoblasts, suggesting the neutral pH condition, whereas the signal was markedly elevated in Pnpla2-null myoblasts (Figures 6I and 6J), reflecting the drop of intracellular pH and acidification. Thus, ATGL-dependent LD catabolism is essential for maintaining energy homeostasis and redox balance in SCs.
Reducing ROS by N-acetyl cysteine restores SC homeostasis and regenerative function
Redox balance between ROS generation and scavenging is essential for stem cell homeostasis (Bigarella et al., 2014). We then tested if decreasing ROS levels would decrease oxidative stress and restore the function of Pnpla2-null myoblasts. To this end, FACS-isolated WT and Pnpla2-null SCs were incubated with N-acetyl cysteine (NAC) for 2 days after plating. We found that NAC had no effects on abundance of LDs in WT and Pnpla2PKO cells (Figure S7A). However, compared with vehicle controls, NAC treatment significantly decreased the ROS content in both WT and Pnpla2-null myoblasts (Figures 7A and 7B), and normalized proliferation of Pnpla2-null myoblasts (Figures 7C and 7D).
Figure 7. Reducing oxidative stress rescues cell fate commitment and regenerative deficiency of ATGL-deficient SCs.
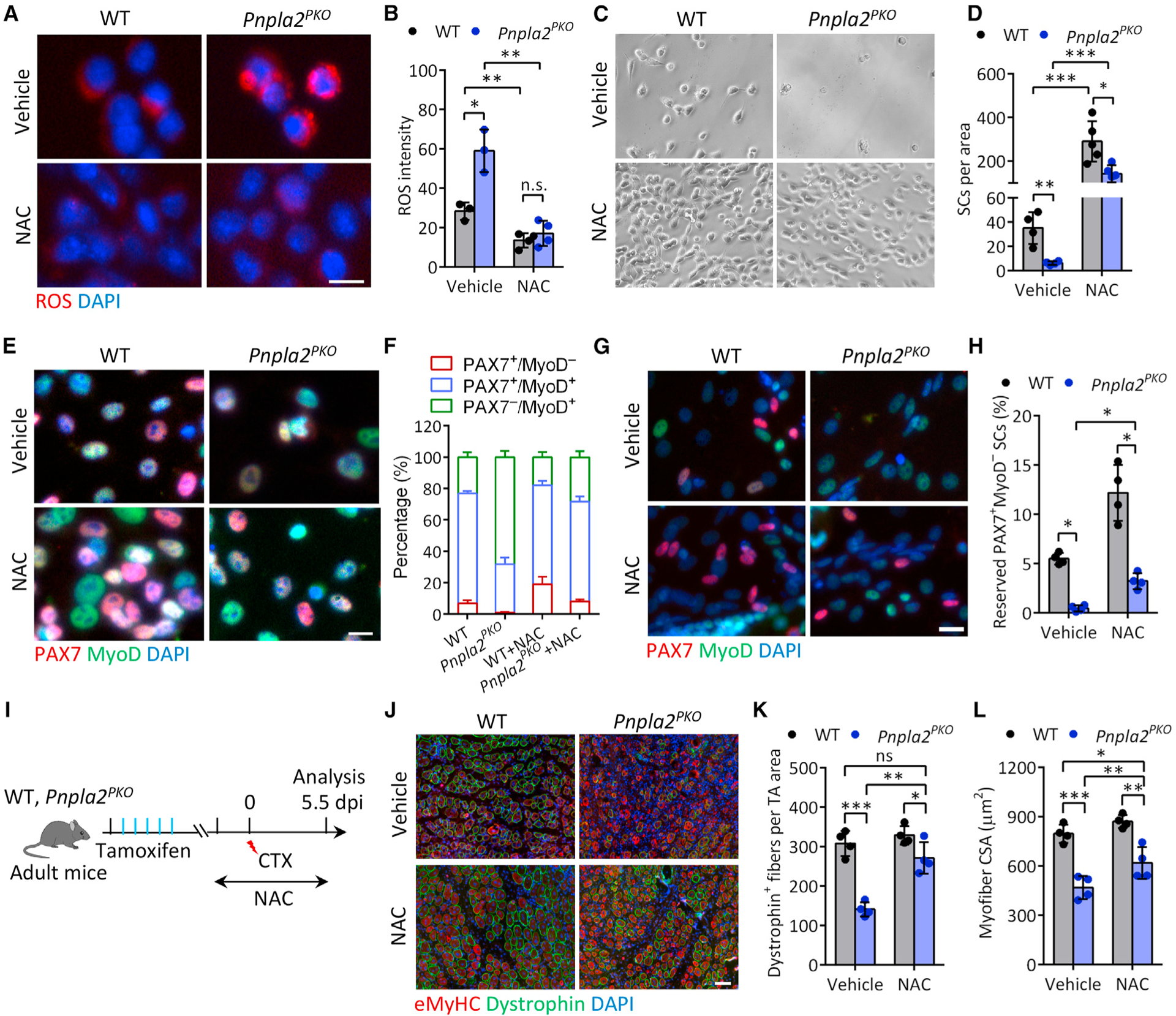
(A and B) Representative ROS staining (A) and quantification of ROS level (B) in FACS-sorted SCs from WT and Pnpla2PKO mice. SCs were cultured for 4 days with or without 5 mM NAC. Vehicle: n = 3 mice, NAC: n = 4 mice. Scale bar, 10 μm.
(C and D) Representative cell growth image (C) and quantification of SC number (D) after NAC treatment. FACS-sorted SCs from WT and Pnpla2PKO mice were cultured for 4 days with or without 5 mM NAC. Vehicle: n = 4 mice, NAC: n = 5 mice.
(E and F) Immunostaining of PAX7 and MyoD (E) and quantification of SC population (F) in WT and Pnpla2PKO cultured muscle cell mixtures treated with 5 mM NAC (n = 3 mice). Scale bar, 10 μm.
(G and H) Immunostaining of PAX7 and MyoD (G) and quantification of quiescent (PAX7+/MyoD−) SCs reserved in cultures (H) upon 2 days differentiation (n = 4 mice). Scale bar, 10 μm.
(I) Experimental scheme for NAC treatment and CTX injury in WT and Pnpla2PKO mice.
(J–L) Immunostaining of eMyHC and dystrophin (J) and quantification of the number (K) and CSA (L) of dystrophin+ myofiber on TA muscle cross-sections at 5.5 dpi after NAC treatment (n = 4 mice). Scale bar, 50 μm.
Data are mean ± standard deviation; Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001. n.s., not significant. See also Figure S7.
Intriguingly, supplementation of NAC promoted self-renewal of both WT and Pnpla2-null myoblasts, as shown by the increase of PAX7+MyoD− cells (Figures 7E and 7F). NAC treatment also increased the percentage of PSCs (PAX7+MyoD+) and decreased the percentage of differentiating (PAX7−MyoD+) cells in Pnpla2-null cultures (Figures 7E and 7F). Examination of the lineage commitment using “reserve” cell assay after induction of differentiation (Shea et al., 2010; Yue et al., 2016) consistently showed that the number of PAX7+MyoD− cells was increased in both WT and Pnpla2-null cultures in the presence of NAC, but the fold increase was more robust in Pnpla2-null cultures (Figures 7G and 7H). Furthermore, NAC treatment significantly increased the fusion index of Pnpla2-null myoblasts compared with vehicle control (FigureS7B). These results suggest that ROS accumulation is at least partially responsible for the defects of Pnpla2-null SCs.
Finally, we assessed whether NAC improves the regenerative defect of Pnpla2PKO muscles in vivo. WT and Pnpla2PKO mice were administrated NAC through drinking water starting at 3 days before CTX injury until sample collection (Figure 7I). Histological analyses showed that regeneration of Pnpla2PKO muscles was improved by NAC (Figure S7C), as indicated by the significant increase in the number and CSA of dystrophin+ myofibers at 5.5 dpi (Figures 7J–7L and S7D). NAC supplementation also restored the number PAX7+ SCs in Pnpla2PKO muscles to the WT level (Figure S7E). In contrast, NAC supplementation had no effects on muscle regeneration in WT mice (Figures 7J–7L, S7C, and S7D). Taken together, these observations indicate that ROS production disrupts SC homeostasis and function in in Pnpla2PKO mice.
DISCUSSION
Emerging evidence has outlined a crucial role for metabolism-associated cellular organelles or vesicles in the regulation of adult stem cell fate, including mitochondria (Zhang et al., 2018), lysosomes (Leeman et al., 2018; Liang et al., 2020), and the autophagosome (Dong et al., 2021; García-Prat et al., 2016). However, despite the presence of LDs in many cell types, the role of LDs in stem cell fate decision is unclear. We provide compelling evidence that LD abundance can distinguish the self-renewing and CSCs during cell division. These findings define an essential role of LDs in regulating SC homeostasis and outline a link between LD metabolism and the fate decision of stem cells.
Our study suggests a sophisticatedly controlled process of LD biogenesis and degradation during SC fate transitions. However, what the upstream factors coordinating these LD dynamics with SC fates are remains unclear. Although various molecules are responsible for the biogenesis of LDs at multiple steps (Walther et al., 2017), the enrichment of lipid biosynthetic genes in CSCs suggest an active transcriptional regulation of LD biogenesis in SCs. Nutrients and growth factors transcriptionally stimulate lipogenic genes through transcription factors downstream of the PI3K/AKT/mTOR pathway (Wang et al., 2015). Because the PI3K/AKT/mTOR pathway is required for SC activation and fate decisions (Rodgers et al., 2014; Wang et al., 2018; Yue et al., 2017), LD biogenesis might be an essential effector downstream of the PI3K/AKT/mTOR pathway in regulating SC fate. This notion is further agreed by the recent finding that the activation of mTOR and downstream Sterol regulatory element-binding protein (SREBP) was responsible for the abnormal LD accumulation caused in Drosophila male GSCs (Demarco et al., 2019). Nevertheless, mechanistic details for the regulation of distinct LD biogenesis and degradation in self-renewal and CSCs require further investigation.
The dynamic changes of LDs in SCs indicate distinct metabolic demands among different SC fate states. It was proposed that FAO is the primary mode of metabolism in QSCs, which is switched to glycolysis in ASCs (Ryall et al., 2015). However, extensive transcriptomic analyses suggest that both FAO and glycolysis are upregulated upon the activation and proliferation of QSCs (Pala et al., 2018). Our scRNA-seq results are more consistent with Pala et al. (2018) and demonstrate that mitochondrial FAO is enriched in activated and CSCs. As FAO can also be mediated peroxisomes, it is imperative to directly test the relative role of peroxisomal and mitochondrial FAO in SCs.
The accumulation of LDs in cells is historically considered as a process that decreases ROS production by sequestering FA from excessive oxidation (Kuramoto et al., 2012; Liu et al., 2015b, 2017). A study in Drosophila found that LD accumulation in the microenvironment protects NSCs from oxidative stress (Bailey et al., 2015). In contrast, our observations showed that an abnormal accumulation of LDs also leads to oxidative stress. Consistent with our observations, it was reported that silencing the coat protein complex I-Arf79F complex, an essential component of trafficking machinery, attenuates the lipolysis pathway in Drosophila intestinal stem cells and leads to the aberrant formation of LDs and increased ROS production (Singh et al., 2016). Thus, LD accumulation resulting from perturbations in cellular metabolism may act as a trigger for ROS generation and oxidative stress.
While the present study outlines a role for LDs in energy metabolism and ROS regulation, functions beyond these warrants future investigation. LD metabolism may provide precursors for bioactive lipid mediators such as phosphatidylinositol 4,5-bisphosphate and prostaglandin E2 (Accioly et al., 2008; Welte and Gould, 2017), both of which are required for the quiescence and maintenance of SCs (Ho et al., 2017; Wang et al., 2018; Yue et al., 2017). It will be interesting to test whether specific signaling lipids determine whether SCs undergo self-renewal or commitment, and whether the asymmetric segregation of signaling lipids governs SC fates. Understanding these mechanisms will be critical for identifying specific pathways that can restore stem cell function for tissue regeneration. In summary, our findings unveil a key role of LDs in regulating SC fate and suggest a potential paradigm for understanding how cellular metabolism regulates asymmetric stem cell divisions.
Limitations of the study
Our study revealed the dynamics of LD biogenesis and degradation during SC fate transitions. However, the upstream factors coordinating the dynamics remain to be elucidated. Our scRNA-seq data have established a foundation for future exploration and validation of the regulatory network that governs LD dynamics during myogenic progression of SCs. We further demonstrated that LD abundance correlates with SC fate and perturbation of LD biogenesis or catabolism disrupts cell fate homeostasis, but it remains to be determined how LDs mechanistically alter cell fate decisions. Although our results point to a role of LD-associated energy and ROS production in SC fate homeostasis and function, several other possibilities remain to be tested. For example, it is possible that LDs-associated bioactive metabolites or phase separation drives SC fate segregation. Even though the current study provided a compelling correlation between LD abundance and SC fate determination, evidence supporting a direct causal relationship between LDs and cell fate of SCs remain to be generated in future studies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Shihuan Kuang, PhD, skuang@purdue.edu.
Materials availability
Materials used in this study are commercially available. Reagents and plasmids generated in this study are available on request.
Data and code availability
RNA-seq data generated from this study are available at the following Gene Expression Omnibus (GEO): GSE150632 (bulk RNA-seq) and GSE150366 (single-cell RNA-seq).
No novel code was written for the analysis of the dataset.
Any additional information required to reanalyze the data reported in this work article is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Mouse lines used in this study were described as follows: Pax7CreERT2 (#012476), Myf5Cre (#007893), Pax7CreERT2(Gaka) (#017763), Rosa26LSL-sfGFP (#021039), Rosa26LSL-tdTomato (#007909), Pnpla2flox/flox (#024278), and mdx (#013141) mouse stocks were purchased from the Jackson Laboratory; Pax7nGFP mouse was provided by Dr. Shahragim Tajbakhsh (Institut Pasteur) (Sambasivan et al., 2013); immunodeficient NOD-Rag1null IL2rgnull (NRG) mice were provided by Purdue University Biological Evaluation Core Facility. Genotyping of the mouse was performed by genomic DNA isolated from ear, and were screened by polymerase chain reaction (PCR) using primers and program following the protocols provided by the supplier. The genotypes of experimental knockout and corresponding control mice are as follows: Pnpla2PKO (Pax7CreERT2;Pnpla2flox/flox) and WT (Pnpla2flox/flox). Mice were housed and maintained in the animal facility with free access to standard rodent chow and water. All procedures involving mice were approved by the Purdue Animal Care and Use Committee. If not stated differently, 2- to 6-month-old adult mice were used for all experiments. Male or female mice were used and always gender matched for each specific experiment.
SC and primary myoblast in vitro culture
FACS-sorted SCs or digested muscle cell mixture were cultured in F-10 Ham’s medium supplemented with 20% FBS (HyClone), 4 ng/mL basic fibroblast growth factor (FGF, Promega), and 1% penicillin-streptomycin (Sigma-Aldrich) on Matrigel-coated culture plates at 37°C, 5% CO2. SC-derived primary myoblasts were cultured on collagen-coated culture plates with the same conditions described above. For myogenic differentiation, SCs or primary myoblasts were seeded on Matrigel-coated culture plates and induced to differentiate with Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco) supplemented with 2% horse serum and 1% penicillin-streptomycin for indicated days.
Single myofiber ex vivo culture
Single EDL myofibers were cultured ex vivo in high glucose DMEM (Gibco) supplemented with 20% FBS, 1% penicillin-streptomycin, and 4 ng/mL basic FGF on horse serum-coated culture plates at 37°C, 5% CO2. Myofibers were collect for further analysis at indicated times.
METHOD DETAILS
TMX, NAC, and Evans blue administration
TMX (Calbiochem, cat#579000) was prepared in corn oil at a concentration of 10 mg/mL. The experimental knockout mice and their corresponding controls were injected intraperitoneally with 2 mg TMX per day per 20 g body weight for 5 consecutive days to induce Cre-mediated deletion, if not stated differently. TMX injections were initiated on adult mice, and experimental mice were used at the time indicated in the Figures and text. NAC (cat#A9165, Sigma-Aldrich) was dissolved in drinking water at a concentration of 1% (Whitehead et al., 2008), adjusted to a pH of 7.4, and filtrated with 0.22-μm filters to sterilize. NAC was administrated to mice uninterruptedly via drinking water 3 days before muscle injury and maintained until the end of experiment. Fresh NAC water is provided thrice weekly. Control animals received normal drinking water. For Evans blue uptake, Evans blue (10 mg/mL in saline) was administered to mice intraperitoneally by 40 μL per 10 g body weight 1 day before being euthanized.
Muscle injury
Muscle injury was induced by CTX injection. Adult mice were anesthetized using a ketamine–xylazine cocktail and CTX (50 μL of 10 μM solution, Sigma-Aldrich) was injected into the tibialis anterior muscles. Muscles were then harvested at the stated time to assess the completion of regeneration and repair.
SC isolation and flow cytometry
SC isolation by flow cytometry was performed as previously described (Liu et al., 2015a). Hind limb muscles from adult mice were minced and digested with 700 U/mL type II collagenase (Worthington) at 37°C for 1 h. Samples were then centrifuged and digested with 100 U/mL type II collagenase and 1 U/mL Dispase II at 37°C for 30 min. Each sample was consecutively filtered through 70-μm cell strainers, and washed with F-10 Ham’s medium containing 10% horse serum. The cell suspension was then centrifuged, treated with red blood cell lysis solution (Promega, cat#Z3141), and washed with phosphate-buffered saline (PBS). For the experimental Pax7CreERT2;Pnpla2flox/flox mice and their corresponding control Pnpla2flox/flox mice, cells were stained with antibody cocktail including CD31-PE (BioLegend, cat#102408), CD45-PE (BioLegend, cat#103106), Sca1-Pacific Blue (BioLegend, cat#108120), and VCAM1-APC (BioLegend, cat#105718) for 20 min at 4°C, prevent from light. SCs were identified and purified by gating with CD31−CD45−Sca1− VCAM1+ using a BD-FACS Aria III FACS system (BD Biosciences).
For single cell RNA-seq analysis, SCs were isolated from hind limb muscles of Pax7CreERT2(Gaka);Rosa26LSL-sfGFP reporter mice with or without injury at the stated time. After digestion as described above, cells were stained with Zombie Violet Live/Dead dye (Biolegend, cat#423113) and live GFP+ SCs were sorted for single cell RNA-seq analysis. For in vivo LD content analysis, SCs were isolated from hind limb muscles of Pax7nGFP mice with or without injury at stated time. SCs were then stained with Nile red (100 ng/mL) and subjected to flow cytometry analysis. For in vivo transplantation assay and gene expression analysis, SCs were isolated from hind limb muscles of Pax7CreERT2(Gaka);Rosa26LSL-tdTomato reporter mice at 5.5 dpi. Cells were stained with LipidSpot610 (T0069, Biotium) for 30 min in PBS at room temperature, washed with PBS, and subjected to FACS. LDLow and LDHigh tdT+ SCs were sorted based on the fluorescent intensity of LipidSpot610. Cells from non-injured muscles with LipidSpot610 staining and injured muscles without staining were set as control for fluorescent compensation.
In vivo SC transplantation
Transplantation experiment was performed following the procedures reported previously with minor modifications (Feige and Rudnicki, 2020; García-Prat et al., 2020; Scaramozza et al., 2019). Briefly, tibialis anterior muscles of transplant recipient immune-deficient NRG and mdx mice were injured with CTX 1 day before transplantation. FACS-isolated tdT+ LDLow and tdT+ LDHigh SCs were centrifuged at 2,500 rpm for 5 min at 4°C. Cells were washed with saline and resuspended in cold saline at the density of 20,000 per 10 μL. In a sterile environment, a previously injured mouse was anesthetized with a ketamine-xylazine cocktail and 10 μL of cell suspension was injected into pre-injured tibialis anterior muscles with a sterile 10-μL Hamilton syringe containing a 28G needle. At 14 days after injury, muscles were harvested and fixed immediately in 4% paraformaldehyde for 30 min at 4°C, protected from light. After washing with PBS and treatment with 100 mM glycine, muscles were dehydrated with 15% and 30% sucrose for 10 h at 4°C. Muscles were then frozen immediately in Optimal cutting temperature (OCT) compound and subjected to further cross-sectional analysis and immunostaining to assess the number of tdT+ myofibers and tdT+ SCs.
SC-derived primary myoblast isolation
SC-derived primary myoblasts were isolated from hind limb muscles of mice at the age of 4–6 weeks. Muscles were minced and digested in type II collagenase and Dispase II mixture for 24 min. The digestions were stopped by adding F-10 Ham’s medium containing 20% FBS. Cells were filtered to remove debris, then centrifuged and cultured in growth medium. For in vitro genetic deletion, SC-derived primary myoblasts isolated from Pax7CreERT2;Pnpla2flox/flox mice were treated with 4-hydroxytamoxifen (4-OHT, 0.4 μM, Sigma-Aldrich) for 2 days to induce Cre-mediated deletion, with methanol vehicle treatment as control.
Single myofiber isolation
Single muscle fibers were isolated from EDL muscles of adult mice and cultured as previously described (Pasut et al., 2013). Whole EDL muscles were dissected and digested with 2 mg/mL type I collagenase (Worthington) in DMEM (Gibco) for 1 h at 37°C, with gentle rotation. Digestion was stopped by transferring EDL muscles to a horse serum coated cell culture dish (60 mm) with 5 mL prewarmed DMEM. Single myofibers were released by gently flushing muscles with large bore glass pipette.
In vitro cell treatments
For serum and glucose deprivation experiments, cultured SC-derived primary myoblasts were switched to the F-10 Ham’s medium containing gradient 20%, 10%, 5%, or 0% FBS, or the DMEM medium containing 25 mM and 5 mM glucose. Cells were washed and fixed for immunofluorescence staining after 24 h culture. For 5-ethynyl-2′-deoxyuridine (EdU) labelling experiments, sorted SCs were cultured for 6 days and incubated with 4 μM EdU in culture medium for 10 h. SC samples with EdU incorporation were fixed in 4% paraformaldehyde in PBS for 10 min. EdU was visualized by the Click-iT method with red fluorescent dye tetramethylrhodamine azide (Invitrogen, cat#T10182). Samples were then subjected to PAX7 staining and imaging. For NAC treatment, SC cultures were treated with 5 mM NAC (pH 7.4, Sigma, cat#A9165) for 2 days and subjected to further experiment and analysis. For the inhibition of LD biogenesis, single myofibers were cultured with 1 μM DGAT1 inhibitor (A922500, Sigma-Aldrich, cat#A1737,) and 2.5 μM DGAT2 inhibitor (PF-06424439, Sigma-Aldrich, cat#PZ0233) for 72 h, immediately after isolation. For inhibition of ATGL, single myofibers were cultured with 50 μM Atglistatin (Cayman Chemical, cat#15284) for 48 h, immediately after isolation.
Fluorescent FA pulse assay
Single myofibers freshly isolated from Pax7nGFP mice were cultured with culture medium (DMEM with 20% FBS, 4 ng/mL basic FGF and 1% penicillin-streptomycin) containing 1 μM BODIPY558/568 C12 Red (Invitrogen, cat#D3835). Myofibers were collected at the indicated time, washed with PBS for three times, and placed on glass slide for live imaging. Hoechst33342 (Invitrogen, cat#H3570) was added 5 min before the imaging for nucleic acid staining. For cultured SC-derived primary myoblasts, 1 μM BODIPY558/568 C12 Red was added in culture medium with Hoechst33342 added 5 min before imaging. Cells were then washed with PBS three times and subjected to live imaging.
Plasmid transfection
SC-derived primary myoblasts were seeded on cover slip in 12-well culture plate and cultured in growth medium. Growth medium was replaced with Opti-MEM media (Gibco) when cells reached to 60% confluence. After incubation for 6 h, pKMyc-ATGL plasmid (gift from Dr. Jun Liu in Mayo Clinic) (Xie et al., 2014) was transfected into cells by lipofectamine 2000 Reagent following the protocol from manufacture. Opti-MEM media was replaced with growth medium after 6 h and cells were cultured for 36 h after transfection and fixed for further analysis.
Transmission electron microscopy
SC-derived primary myoblasts isolated from Pax7CreERT2;Pnpla2flox/flox mice were treated with 4-OHT (0.4 μM, Calbiochem) to induce genetic deletion. Cultured primary myoblasts and isolated tibialis anterior muscles were fixed in a fixative buffer (2.5% glutaraldehyde, 1.5% paraformaldehyde in 0.1M sodium cacodylate buffer) and scraped for collection. Samples were rinsed in deionized water followed by fixation in 2% osmium tetroxide for 1 h. Then, the samples were washed in deionized wat water followed by fixation in 1% uranyl acetate for 15 minutes. After washing with deionized wat water, the samples were dehydrated with series of graded ethanol followed by dehydration in acetonitrile and then were embedded (EMbed 812: DDSA: NMA 5:4:2; 0.22 DMP-30). Ultrathin sections (longitudinal sections) were cut at 70 nm and stained with uranyl acetate and lead citrate. Stained sections were examined under Tecnai T12 transmission electron microscope attached with a Gatan imaging system under 6,000× and 260,00× magnifications.
Hematoxylin and eosin and immunofluorescence staining
Whole muscle tissues were dissected and frozen immediately in an OCT compound. Frozen muscles were cross-sectioned at 10 μm thickness using a Leica CM1850 cryostat. For hematoxylin and eosin staining, the slides were first stained in hematoxylin for 15 min, rinsed in running tap water and then stained in eosin for 1–2 min. Slides were dehydrated in graded ethanol and xylene, and then covered using Permount. For immunofluorescence staining, cross-sections, single myofibers, or cultured cells were fixed in 4% paraformaldehyde in PBS for 10 min, quenched with 100 mM glycine for 10 min, and incubated in blocking buffer (5% goat serum, 2% bovine serum albumin, 0.1% Triton X-100, and 0.1% sodium azide in PBS) for at least 1 h, followed by 1 hour of blocking in a mouse-on-mouse blocking buffer containing mouse IgG blocking reagent (Vector lab, cat#MKB-2213). Samples were then incubated with primary antibodies diluted in blocking buffer overnight at 4°C. After washing with PBS, samples were incubated with secondary antibodies and DAPI for 1 h at room temperature. LDs were stained with Bodipy 493/503 (Invitrogen, cat#D3922) or LipidTOX Deep Red (Invitrogen, cat#H34477) dye as indicated. Antibodies used for immunofluorescence staining are listed in Table S2.
All hematoxylin and eosin staining images were captured using a Nikon D90 digital camera mounted on a microscope with a 20× objective. All immunofluorescent images were captured using a Leica DM 6000B microscope with a 20× objective, Zeiss LSM 700 Confocal with a 63× objective, or Nikon A1R-MP Confocal with a 40× objective. Images for experimental and associated control samples were captured using identical parameters. Confocal Z-stacks were projected as maximum intensity images for each channel and merged into a single image.
Single cell RNA-seq analysis
FACS-sorted SCs from tibias anterior muscles of non-injured, 5.5 and 10 dpi with CTX in Pax7CreERT2(Gaka);Rosa26LSL-sfGFP mice (n = 3 male pooled mice/timepoint) were re-counted and loaded onto the 10× Chromium v3 3′ expression platform for single-cell RNA-sequencing. Samples were processed per the manufacturer’s instructions and sequenced on a NovaSeq to target 50,000 reads per cell. All samples were processed together on the same day.
For data analysis, barcodes and reads were mapped to Grcm38/mm10 (Mus Musculus) using Cell Ranger v3.1 with default settings. The resulting expression/barcode matrix was imported into R Studio (RStudio Team, 2015) for analysis with Seurat v3.1 (Satija et al., 2015; Stuart et al., 2019). Since each timepoint contained one sample and all samples were processed together, all data were merged together before analysis. Cells with more than 15% mitochondrial reads and feature counts greater than 6,000 or fewer than 400 were filtered out. Seurat’s SCTransform was used for data normalization and the top 10 principal components were used for Louvain clustering and Uniform manifold approximation and projection (UMAP) embedding. Clustering results were analyzed to determine if there was any contamination from non-SCs. Cells in Lyz2+ clusters were removed, as these represented contaminating immune cells from both 5.5 and 10 dpi. Data were then re-analyzed using Seurat’s SCTransform, selecting the top 10 PCs for Louvain clustering (resolution of 0.05) and subsequent UMAP embedding. FindAllMarkers function was used to evaluate gene expression enriched in each cluster with a p value of less than 0.05 (Wilcoxon rank-sum test) and ranked by avgLog2Fold-change. The top 10 markers were then visualized via Heatmap to determine specificity and aide with nomenclature for cluster identities. Further mapping of known SC markers at various stages of quiescence, proliferation, activation, and differentiation were also evaluated to aide with cluster annotation. These markers included Pax7, Myog, Mki67, Tnnt2, and Acta2.
For GO term analysis, gene lists for LD (GO: 0005811), LD formation (GO: 0140042), glycerolipid biosynthetic process (GO: 0045017), neutral lipid biosynthetic process (GO: 0046460), triglyceride biosynthetic process (GO: 0019432), triglyceride catabolic process (GO: 0019433), and FAO (GO: 0006635) were downloaded from MGI. Genes annotated with either positive or negative regulatory roles were excluded from the GO term pathway to avoid contradiction. To provide higher confidence or greater specificity of genes for each GO term, GO annotation evidenced by an experimental evidence code was restricted for the analysis, instead of similarity evidence codes. In addition, genes characterized into peroxisomal FAO in GO: 0006635 were analyzed separately from mitochondrial FAO to better compare the two pathways. Density plots were generated to visualize gene sets for GO terms using AddModuleScore function in Seurat, with the control features set as all genes expressed and then plotted using the RidgePlot function.
Bulk cell RNA-seq analysis
FACS-sorted SCs were cultured for 7 days in growth medium and switched to DMEM with 2% horse serum for 16 h. Total RNA was extracted using TRIzol reagent according to the manufacturer’s instructions, and subjected to RNA-seq analysis. Briefly, RNA quality analysis was performed by agarose gel electrophoresis and Agilent 2100. A complementary DNA library was then constructed using poly(A) selected RNA, and sequencing was performed according to the Illumina HiSeq standard protocol. Raw reads from RNA-seq libraries are filtered to remove reads containing adapters or with low quality. Statistics analysis of data production and quality was performed to confirm the sequencing quality. Reference genome and gene annotation files were downloaded from a genome website browser (NCBI/UCSC/Ensembl). TopHat2 (Kim et al., 2013) was used for mapping the filtered reads to the reference genome (Grcm38/mm10, Mus Musculus). For the quantification of gene expression level, HTSeq V0.6.1 (Anders et al., 2015) was used to analyze the read numbers mapped for each gene. The FPKM of each gene was calculated based on the gene read counts mapped to genes or exons. A differential expression analysis was performed using the DESeq2 R/EdgeR R package (Love et al., 2014) with the threshold of significance set as adjusted a p value of less than 0.05.
Real-time PCR
FACS-isolated tdT+LDLow and tdT+LDHigh SCs were centrifuged at 2,500 rpm for 5 min at 4°C. Total RNA was extracted from cells using PicoPure RNA Isolation Kit (Applied Biosystems, cat#KIT0204) according to the manufacturer’s instructions. 300 ng of total RNA was reverse transcribed using random primers with M-MLV reverse transcriptase (Invitrogen, cat#28025021). Real-time PCR was carried out in a Roche Lightcycler 96 Real time PCR system (Roche) with FastStart Essential DNA Green Master (Roche, cat#06402712001) and gene-specific primers that listed in Table S3. The 2−ΔΔCt method was used to analyze the relative changes in each gene’s expression normalized against Gapdh mRNA expression.
Protein extraction and Western blot analysis
Total protein was isolated from cells using RIPA buffer containing 25 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate and, 0.1% sodium dodecyl sulfate (SDS). Protein concentrations were determined using Pierce BCA Protein Assay Reagent (Thermo Scientific, cat#23225). Proteins were separated by SDS-PAGE, transferred to a polyvinylidene fluoride membrane, blocked in 5% fat-free milk for 1 h at room temperature and then incubated with primary antibodies in 5% fat-free milk overnight at 4°C. The membrane was then incubated with secondary antibody for 1 h at room temperature. Antibodies used for Western blot analysis are listed in Table S2. Immunodetection was performed using enhanced Western Blotting Chemiluminescence Luminol Reagent (Santa Cruz Biotechnology, cat#sc-2048) and detected with a FluorChem R System (Proteinsimple).
TAG measurement by lipidomics analysis
SC-derived primary myoblasts isolated from Pax7CreERT2;Pnpla2flox/flox mice were treated with 4-OHT (0.4 μM, Calbiochem) for 2 days to induce genetic deletion. Cultured primary myoblasts were then collected by centrifuge for lipid extraction and liquid chromatography/mass spectrometry. Lipids were extracted using the Bligh and Dyer extraction method (Bligh and Dyer, 1959). Cell pellets were lysed by adding 200 μL H2O and mixed with 250 μL of chloroform and 450 μL of methanol. After incubation at room temperature for 15 min, 250 μL of chloroform and 250 μL of water were added, and the sample was centrifuged for 10 min at 16,000×g. The bottom organic phase containing lipids was transferred to a new tube and dried using a speedvac centrifuge (Savant Speedvac, Thermo Scientific), and dried lipid extracts were subjected to spectrometry analysis.
Targeted lipid profiling was performed using discovery multiple reaction monitoring (MRM) profiling methods and instrumentation as described de Lima et al. (2018). Specifically, for sample preparation, dried lipid extracts were diluted in 500 μL of methanol/chloroform 3:1 (v/v) (stock solution). The stock solution was further diluted 50× in injection solvent (acetonitrile/methanol/ammonium acetate 300 mM 3:6.65:0.35 (v/v)) and 8 μL of this solution was used for the profiling analysis of each lipid class using a micro-autosampler (G1377A) to the ESI source of an Agilent 6410 triple quadrupole mass spectrometer (Agilent Technologies). A capillary pump was connected to the autosampler and operated at a flow rate of 7 μL/min and pressure of 150 bar. Capillary voltage on the instrument was 3.5–5 kV and the gas flow 5.1 L/min at 300°C. For the mass spectrometry analysis, relative amounts of ion abundances were used for statistics. The values of ion intensities for each of the MRMs monitored were normalized by total ion intensity of all MRMs in the method for a given sample. Fold change was calculated by dividing values of ion intensities for each of the MRMs measured in each sample by the ion intensity of the corresponding MRM in the blank.
Lipolysis assay
Lipolytic activity was measured by a PicoProbe Lipolysis Fluorometric Assay Kit (BioVision Inc, cat#K578-100) according to the manufacturer’s instructions with proper optimizations. FACS-sorted SCs were cultured in 48-well plate for 7 days and gently washed with 150 μL of lipolysis wash buffer two times. After adding 200 μL lipolysis assay buffer into each well, cells were treated with isoproterenol (final concentration 100 nM) for 6 h to stimulate lipolysis. After stimulation, 50 μL of medium was collected and incubated with the reaction mix containing a glycerol assay buffer, PicoProbe, glycerol enzyme mix, and glycerol developer for 60 min at room temperature, protected from light. Diluted glycerol standards were included together with the samples to generate a standard curve. The fluorescent intensity was then measured at Ex/Em = 535/587 nm with a Spark 10M multimode microplate reader (TECAN), and the glycerol concentration was calculated based on the standards following the manufacturer’s instructions. The remained cells were lysed and cellular protein contents were measured to normalize the glycerol level in each sample.
Cellular acetyl-CoA measurement
Cellular acetyl-CoA contents in SC-derived primary myoblasts were measured by PicoProbe Acetyl-CoA Fluorometric Assay kit (BioVision Inc, cat#K317) according to the manufacturer’s instructions. In brief, SC-derived primary myoblasts were rinsed with cold PBS and lysed with 1× ice-cold cell lysis buffer (20 mM Tris buffer with pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM Glycerol phosphate, 1 mM Na3VO4) plus 1 mM PMSF. The cell lysate was deproteinized with 1 N perchloric acid and then neutralized with 3 M KHCO3. Samples were quenched for erasing free CoA and cellular acetyl CoA was converted to CoA by adding conversion enzyme. The CoA is reacted to form NADH which interacts with PicoProbe to generate fluorescence. The fluorescence intensity was measured at Ex/Em = 535/587 nm with Spark 10M multimode microplate reader (TECAN), and the acetyl-CoA concentration was calculated based on the standards following the manufacturer’s instructions.
Cellular ATP detection
Cellular ATP levels were measured using an ATP Detection Assay Kit (Cayman Chemical, cat#700410). SC-derived primary myoblasts were rinsed with cold PBS and lysed with ice-cold 1× ATP detection sample buffer. Cell lysates and standards were plated into a 96-well plate. 100 μL of freshly prepared reaction mixture (1× ATP detection sample buffer, d-luciferin, and luciferase) was added to each sample and the plate was incubate at room temperature for 20 min, protected from light. Luminescence intensity was detected by Spark 10M multimode microplate reader (TECAN), and ATP concertation was calculated according to the manufacturer’s instructions.
Seahorse mitochondrial respiration analysis
Mitochondrial respiration was measured with Seahorse XFe24 Analyzer (Agilent Technologies) according to the manual of Seahorse XF Cell Mito Stress Test Kit (Agilent Technologies, cat#103015-100). Briefly, SCs isolated by FACS were plated on Matrigel-coated XF24 cell culture microplate at a density of 10,000 cells per well. Seahorse sensor cartridge was hydrated with calibrant in a non-CO2 incubator at 37°C for overnight 1 day before measurement. On the day of measurements, cells were washed twice and switched to Seahorse XF base medium (pH7.4, Agilent Technologies, cat#103334-100) supplemented with 1 mM sodium pyruvate, 1 mM l-glutamine, and 5 mM glucose. Cells were equilibrated at 37°C in a non-CO2 incubator for 1 hour. For FAO inhibition, Etomoxir (Sigma-Aldrich, cat#E1905) were added to cells at a final concentration of 50 μM 15 min before the measurement. The oxygen consumption rate was monitored at the basal state and after sequential injection of the mitochondrial compounds oligomycin (1.5 μM), FCCP (3 μM), and Rotenone/antimycin A (1 μM) to induce mitochondrial stress. All mitochondrial respiration rates were generated and automatically calculated by the Seahorse Wave software with normalization to the cellular protein contents.
Intracellular ROS detection
The cellular ROS level was detected by CellROX Deep Red Reagent (Invitrogen, cat#C10422). FACS-sorted SCs were cultured in 24-well plate for 4 or 7 days. The culture medium was changed 12 h before the test. CellROX Deep Red reagent was added into the cells at a final concentration of 5 μM, and incubates for 45 min at 37°C, protected from light. Cells were counterstained with Hoechst33342 and replaced with new culture medium after washing, then subjected to the live imaging immediately.
Intracellular pH level detection
The cellular pH level was detected by pHrodo Red AM Intracellular pH Indicator (Invitrogen, cat# P35372). FACS-sorted SCs were cultured in 24-well plate for 4 days. The culture medium was changed 12 h before the test. We mixed 1 μL pHrodo Red with 10 μL PowerLoad concentrate and added it to 1 mL culture medium. Cells were incubated with pHrodo Red for 30 min at 37°C, protected from light. Cells were counterstained with Hoechst33342 and replaced with new culture medium after wash, then subjected to live imaging immediately.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experiments involving mice were performed with a minimum of three biological replicates. All muscle histological analysis including the CSA of myofibers, and the percentage of regenerated area, the number of regenerating, and regenerated myofibers, tdT+ myofiber, myogenic cells, and tdT+ SCs per area were quantified by Fiji-ImageJ software with MuscleJ plugin (Mayeuf-Louchart et al., 2018; Schindelin et al., 2012) or by Adobe PhotoShop software. The differentiation index was quantified as the ratio of the nuclei number in MyHC+ cultures to total nuclei number. The fusion index was quantified as the ratio of the nuclei number in MyHC+ myotubes (≥2 nuclei) to the total nuclei number. The ROS intensity and the fluorescent signal of pHrodo Red in SCs was quantified by Fiji-ImageJ software. Sample size or replicate number (designated as “n”) for each experiment are indicated in the figure legends. Statistical analyses and graphing were performed using GraphPad Prism 6.0 (GraphPad Software). All experimental data are represented as mean ± standard deviation. Statistical significance was determined by the Student t test under two-tailed. The p values are indicated as p < 0.05, p < 0.01, and p < 0.001, and a p value of less than 0.05 was considered as significant, while a p value of greater than 0.05 was considered as not significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-PAX7 | DSHB | Cat# PAX7, RRID:AB_2299243 |
| Mouse monoclonal anti-MyoD | Santa Cruz Biotechnology | Cat# sc-377460, RRID:AB_2813894 |
| Mouse monoclonal anti-MyoG | DSHB | Cat# F5D, RRID:AB_2146602 |
| Mouse monoclonal anti-MyHC | DSHB | Cat# MF 20, RRID:AB_2147781 |
| Mouse monoclonal anti-eMyHC | DSHB | Cat# F1.652, RRID:AB_528358 |
| Rabbit polyclonal anti-Laminin | Sigma | Cat# L9393, RRID:AB_477163 |
| Rabbit polyclonal anti-Dystrophin | Abcam | Cat# ab15277, RRID:AB_301813 |
| Rabbit polyclonal anti-Ki67 | Abcam | Cat# ab15580, RRID:AB_443209 |
| Rabbit polyclonal anti-ATGL | Cell Signaling Technology | Cat# 2439, RRID:AB_2167953 |
| Mouse monoclonal anti-GAPDH | Santa Cruz Biotechnology | Cat# sc-32233, RRID:AB_627679 |
| PE Rat anti-mouse CD31 antibody | BD Biosciences | Cat# 553373, RRID:AB_394819 |
| PE anti-mouse CD45 antibody | eBioscience | Cat# 12-0451-82, RRID:AB_465668 |
| Pacific Blue anti-mouse Ly-6A/E (Sca-1) antibody | BioLegend | Cat# 122520, RRID:AB_2143237 |
| APC anti-mouse CD106 antibody | BioLegend | Cat# 105718, RRID:AB_1877141 |
| Alexa 568 goat anti-mouse IgG1 | Invitrogen | Cat# A-21124, RRID:AB_2535766 |
| Alexa 488 goat anti-mouse IgG1 | Invitrogen | Cat# A-21121, RRID:AB_2535764 |
| Alexa 647 goat anti-mouse IgG2b | Invitrogen | Cat# A-21242, RRID:AB_2535811 |
| Alexa 488 goat anti-rabbit IgG | Invitrogen | Cat# A-11034, RRID:AB_2576217 |
| Alexa 647 goat anti-rabbit IgG | Invitrogen | Cat# A-21244, RRID:AB_2535812 |
| HRP AffiniPure goat anti-mouse IgG | Jackson ImmunoResearch | Cat# 115-035-003, RRID:AB_10015289 |
| HRP AffiniPure goat anti-rabbit IgG | Jackson ImmunoResearch | Cat# 111-035-003, RRID:AB_2313567 |
| Chemicals, peptides, and recombinant proteins | ||
| Tamoxifen (TMX) | Calbiochem | 579000 |
| 4-Hydroxytamoxifen (4-OHT) | Sigma-Aldrich | H6278 |
| N-acetyl cysteine (NAC) | Sigma-Aldrich | A9165 |
| Evans blue | Sigma-Aldrich | E2129 |
| Cardiotoxin | Sigma-Aldrich | 217503 |
| Ketamine HCl | Akron | 59399-114-10 |
| Xylazine | Akron | 59399-110-20 |
| Collagenase, Type I | Worthington | LS004197 |
| Collagenase, Type II | Worthington | LS004179 |
| Dispase II | Sigma-Aldrich | 04942078001 |
| Zombie Violet Live/Dead dye | Biolegend | cat#423113 |
| Red blood cell lysis solution | Promega | Z3141 |
| Ham’s F-10 Nutrient Mix | Gibco | 11550043 |
| Dulbecco’s Modified Eagle Medium | Gibco | 11995065 |
| Fetal bovine serum | HyClone | SH30080.03 |
| Donor Horse Serum | Corning | MT35030CV |
| Opti-MEM | Gibco | 31985070 |
| Penicillin-Streptomycin | Sigma-Aldrich | P4333 |
| Seahorse XF base medium | Agilent Technologies | 103334-100 |
| Phosphate-buffered saline | Gibco | 21600-069 |
| Nile red | Invitrogen | N1142 |
| LipidSpot610 | Biotium | T0069 |
| CellROX Deep Red Reagent | Invitrogen | C10422 |
| pHrodo Red AM Intracellular pH Indicator | Invitrogen | P35372 |
| HCS LipidTOX™ Deep Red Neutral Lipid Stain | Invitrogen | H34477 |
| BODIPY 493/503 | Invitrogen | D3922 |
| BODIPY 558/568 C12 | Invitrogen | D3835 |
| M.O.M. (Mouse on Mouse) Blocking Reagent | Vector lab | MKB-2213 |
| Hoechst33342 | Invitrogen | H3570 |
| DAPI | Invitrogen | D1306 |
| Paraformaldehyde | Sigma-Aldrich | P6148 |
| Glycine | Sigma-Aldrich | 50046 |
| Sucrose | Fisher Scientific | S5-500 |
| Glucose | Sigma-Aldrich | G7021 |
| O.C.T. Compound | Fisher Scientific | 23-730-571 |
| Fibroblast growth factor, basic | Promega | 9PIG507 |
| BD Matrigel Matrix | BD Biosciences | 356235 |
| Collagen from rat tail | Sigma-Aldrich | C7661 |
| 5-Ethynyl-2′-deoxyuridine (EdU) | Cayman Chemical | 20518 |
| Tetramethylrhodamine (TAMRA) azide | Invitrogen | T10182 |
| A922500 | Sigma-Aldrich | A1737 |
| PF-06424439 | Sigma-Aldrich | PZ0233 |
| Atglistatin | Cayman Chemical | 15284 |
| Lipofectamine 2000 Transfection Reagent | Invitrogen | 11668030 |
| Glutardialdehyde | Sigma-Aldrich | 8.20603 |
| Sodium cacodylate | Sigma-Aldrich | 97068 |
| Osmium tetroxide | Sigma-Aldrich | 201030 |
| Uranyl acetate | Fisher Scientific | 18-607-644 |
| Ethanol | Decon Labs, Inc | 2701G |
| Acetonitrile | Fisher Scientific | A998-1 |
| EMbed-812 kit | Fisher Scientific | 50-980-391 |
| Goat serum | MP Biomedicals | 08642921 |
| Bovine serum albumin | GeminiBio | 700-105P |
| TRIzol Reagent | Sigma-Aldrich | T9424 |
| Chloroform | VWR Chemicals | BDH1109 |
| Methanol | Fisher Scientific | A412-20 |
| NP-40 | Thermo Scientific | 85124 |
| Sodium deoxycholate | Sigma-Aldrich | D6750 |
| Sodium Dodecyl Sulfate (SDS) | Fisher Scientific | 02-004-080 |
| Ammonium acetate | Fisher Scientific | A637-500 |
| Perchloric acid | Sigma-Aldrich | 244252 |
| PMSF | Calbiochem | 7110-OP |
| L-Glutamine solution | Sigma-Aldrich | G7513 |
| Sodium pyruvate | Sigma-Aldrich | P5280 |
| Etomoxir | Sigma-Aldrich | E1905 |
| Critical commercial assays | ||
| M-MLV reverse transcriptase | Invitrogen | 28025021 |
| PicoPure RNA Isolation Kit | Applied Biosystems | KIT0204 |
| FastStart Essential DNA Green Master | Roche | 06402712001 |
| Pierce BCA Protein Assay Reagent | Thermo Scientific | 23225 |
| Western Blotting Chemiluminescence Luminol Reagent | Santa Cruz Biotechnology | sc-2048 |
| PicoProbe Lipolysis Fluorometric Assay Kit | BioVision Inc | K578-100 |
| PicoProbe Acetyl-CoA Fluorometric Assay kit | BioVision Inc | K317 |
| ATP Detection Assay Kit | Cayman Chemical | 700410 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent Technologies | 103015-100 |
| Chromium Single Cell 3′ GEM Library & Gel Kit v3 | 10x Genomics | PN-1000075 |
| Chromium Single Cell B Chip Kit | 10x Genomics | PN-1000153 |
| Chromium i7 Multiplex Kit | 10x Genomics | PN-120262 |
| Deposited data | ||
| Bulk RNA-seq data of WT and Pnpla2 KO satellite cells | This paper | GEO: GSE150632 |
| Single cell RNA-seq data of satellite cells during regeneration | This paper | GEO: GSE150366 |
| Experimental models: Organisms/strains | ||
| Mouse: B6;129-Pax7tm2.1(cre/ERT2)Fan/J | The Jackson Laboratory | JAX stock: #012476 |
| Mouse: B6.Cg-Pax7tm1(cre/ERT2)Gaka/J | The Jackson Laboratory | JAX stock: #017763 |
| Mouse: B6.129S4-Myf5tm3(cre)Sor/J | The Jackson Laboratory | JAX stock: #007893 |
| Mouse: B6;129-Gt(ROSA)26Sortm5(CAG-Sun1/sfGFP)Nat/J | The Jackson Laboratory | JAX stock: #021039 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J | The Jackson Laboratory | JAX stock: #007909 |
| Mouse: B6N.129S-Pnpla2tm1Eek/J | The Jackson Laboratory | JAX stock: #024278 |
| Mouse: D2.B10-Dmdmdx/J | The Jackson Laboratory | JAX stock: #013141 |
| Mouse: Pax7nGFP | Sambasivan et al., 2013 | N/A |
| Mouse: NOD-Rag1null IL2rgnull | Purdue University Biological Evaluation Core Facility | N/A |
| Oligonucleotides | ||
| See Table S3 for all primers | This paper | N/A |
| Recombinant DNA | ||
| pKMyc-ATGL | Xie etal., 2014 | N/A |
| Software and algorithms | ||
| Seurat v3.1 | Stuart et al., 2019 | Satija lab: satijalab.org/seurat/ |
| Cell Ranger v3.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/3.0/#cellrangertab |
| RStudio | RStudio Team, 2015 | RRID:SCR_000432 |
| TopHat2 | Kim et al., 2013 | RRID:SCR_013035 |
| HTSeq V0.6.1 | Anders et al., 2015 | RRID:SCR_005514 |
| DESeq2 | Love et al., 2014 | RRID:SCR_015687 |
| BD FACSDiva Software | BD Biosciences | RRID:SCR_001456 |
| FlowJo 10 | FLOWJO, LLC | RRID:SCR_008520 |
| Fiji-ImageJ | Schindelin et al., 2012 | RRID:SCR_002285 |
| MetaMorph Microscopy Automation and Image Analysis Software | Molecular Devices, LLC | RRID:SCR_002368 |
| Agilent Masshunter Quantitative Analysis software | Agilent Technologies | RRID:SCR_015040 |
| Seahorse Wave | Agilent Technologies | RRID:SCR_014526 |
| Adobe Photoshop | Adobe Inc. | RRID:SCR_014199 |
| NIS-Elements | Nikon Instruments | RRID:SCR_014329 |
| ZEN 2010 Microscopy Software | Carl Zeiss Microscopy GmbH | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Prism 6.0 | GraphPad Prism | RRID:SCR_002798 |
Highlights.
LDs are highly dynamic during myogenic progression of satellite cells
Satellite cells with few LDs hold higher self-renewal capacity than those with many LDs
Inhibiting biogenesis or catabolism of LDs disrupts satellite cell fate homeostasis
Excessive accumulation of LDs leads to energy insufficiency and oxidative stress
ACKNOWLEDGMENTS
We thank Dr. Shahragim Tajbakhsh (Institut Pasteur) for providing Pax7nGFP mouse, Dr. Jun Liu (Mayo Clinic) for providing pKMyc-ATGL plasmid, Dr. Jason Cannon and Rachel Foguth for assistance on Seahorse analysis, Dr. Daoguo Zhou for help with confocal microscopy, Di Huang for help with data analysis, and Jun Wu for mouse colony maintenance. We are grateful to Purdue Flow Cytometry and Cell Separation, Metabolite Profiling, Electron Microscopy, and Imaging Facilities. The research was supported by NIH-R01AR071649 (S.K.), NIH-R03AR068108 (M.D.), Muscular Dystrophy Association-MDA516161 (F.Y.), NIH-F31AR077424 (S.O.), NIH-P30CA023168 (Purdue Center for Cancer Research), and NIH Shared Instrumentation Grant-S10DO20029.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110267.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Accioly MT, Pacheco P, Maya-Monteiro CM, Carrossini N, Robbs BK, Oliveira SS, Kaufmann C, Morgado-Diaz JA, Bozza PT, and Viola JP (2008). Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 68, 1732–1740. [DOI] [PubMed] [Google Scholar]
- Ahmadian M, Abbott MJ, Tang T, Hudak CS, Kim Y, Bruss M, Hellerstein MK, Lee H-Y, Samuel VT, and Shulman GI (2011). Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 13, 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atilla-Gokcumen GE, Muro E, Relat-Goberna J, Sasse S, Bedigian A, Coughlin ML, Garcia-Manyes S, and Eggert US (2014). Dividing cells regulate their lipid composition and localization. Cell 156, 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, Lechene CP, Postle AD, and Gould AP (2015). Antioxidant role for lipid droplets in a stem cell niche of Drosophila. Cell 163, 340–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigarella CL, Liang R, and Ghaffari S (2014). Stem cells and the impact of ROS signaling. Development 141, 4206–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, and Dyer WJ (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol 37, 911–917. [DOI] [PubMed] [Google Scholar]
- de Lima CB, Ferreira CR, Milazzotto MP, Sobreira TJP, Vireque AA, and Cooks RG (2018). Comprehensive lipid profiling of early stage oocytes and embryos by MRM profiling. J. Mass Spectrom 53, 1247–1252. [DOI] [PubMed] [Google Scholar]
- Demarco RS, Uyemura BS, D’Alterio C, and Jones DL (2019). Mitochondrial fusion regulates lipid homeostasis and stem cell maintenance in the Drosophila testis. Nat. Cell Biol 21, 710–720. [DOI] [PubMed] [Google Scholar]
- Dong S, Wang Q, Kao Y-R, Diaz A, Tasset I, Kaushik S, Thiruthuvanathan V, Zintiridou A, Nieves E, and Dzieciatkowska M (2021). Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 591, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige P, Brun CE, Ritso M, and Rudnicki MA (2018). Orienting muscle stem cells for regeneration in homeostasis, aging, and disease. Cell Stem Cell 23, 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige P, and Rudnicki MA (2020). Isolation of satellite cells and transplantation into mice for lineage tracing in muscle. Nat. Protoc 15, 1082–1097. [DOI] [PubMed] [Google Scholar]
- García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, and Serrano AL (2016). Autophagy maintains stemness by preventing senescence. Nature 529, 37–42. [DOI] [PubMed] [Google Scholar]
- García-Prat L, Perdiguero E, Alonso-Martín S, Dell’Orso S, Ravichandran S, Brooks SR, Juan AH, Campanario S, Jiang K, and Hong X (2020). FoxO maintains a genuine muscle stem-cell quiescent state until geriatric age. Nat. Cell Biol 22, 1307–1318. [DOI] [PubMed] [Google Scholar]
- Hamilton LK, Dufresne M, Joppé SE, Petryszyn S, Aumont A, Calon F, Barnabé-Heider F, Furtos A, Parent M, and Chaurand P (2015). Aberrant lipid metabolism in the forebrain niche suppresses adult neural stem cell proliferation in an animal model of Alzheimer’s disease. Cell Stem Cell 17, 397–411. [DOI] [PubMed] [Google Scholar]
- Ho AT, Palla AR, Blake MR, Yucel ND, Wang YX, Magnusson KE, Holbrook CA, Kraft PE, Delp SL, and Blau HM (2017). Prostaglandin E2 is essential for efficacious skeletal muscle stem-cell function, augmenting regeneration and strength. Proc. Natl. Acad. Sci. U S A 114, 6675–6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahmer N, Farese RV, and Walther TC (2013). Balancing the fat: lipid droplets and human disease. EMBO Mol. Med 5, 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang S, Gillespie MA, and Rudnicki MA (2008). Niche regulation of muscle satellite cell self-renewal and differentiation. Cell Stem Cell 2, 22–31. [DOI] [PubMed] [Google Scholar]
- Kuang S, Kuroda K, Le Grand F, and Rudnicki MA (2007). Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 129, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto K, Okamura T, Yamaguchi T, Nakamura TY, Wakabayashi S, Morinaga H, Nomura M, Yanase T, Otsu K, and Usuda N (2012). Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J. Biol. Chem 287, 23852–23863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeman DS, Hebestreit K, Ruetz T, Webb AE, McKay A, Pollina EA, Dulken BW, Zhao X, Yeo RW, and Ho TT (2018). Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 359, 1277–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R, Arif T, Kalmykova S, Kasianov A, Lin M, Menon V, Qiu J, Bernitz JM, Moore K, and Lin F (2020). Restraining lysosomal activity preserves hematopoietic stem cell quiescence and potency. Cell Stem Cell 26, 359–376.e357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Cheung TH, Charville GW, and Rando TA (2015a). Isolation of skeletal muscle stem cells by fluorescence-activated cell sorting. Nat. Protoc 10, 1612–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, MacKenzie KR, Putluri N, Maletić-Savatić M, and Bellen HJ (2017). The glia-neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab. 26, 719–737.e716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, and Quintana A (2015b). Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeuf-Louchart A, Hardy D, Thorel Q, Roux P, Gueniot L, Briand D, Mazeraud A, Bouglé A, Shorte SL, and Staels B (2018). MuscleJ: a high-content analysis method to study skeletal muscle with a new Fiji tool. Skelet. Muscle 8, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, and Carvalho P (2019). Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol 20, 137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprescu SN, Yue F, Qiu J, Brito LF, and Kuang S (2020). Temporal dynamics and heterogeneity of cell populations during skeletal muscle regeneration. iScience 23, 100993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pala F, Di Girolamo D, Mella S, Yennek S, Chatre L, Ricchetti M, and Tajbakhsh S (2018). Distinct metabolic states govern skeletal muscle stem cell fates during prenatal and postnatal myogenesis. J. Cell Sci 131, jcs212977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasut A, Jones AE, and Rudnicki MA (2013). Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. J. Vis. Exp, e50074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relaix F, Bencze M, Borok M, Der Vartanian A, Gattazzo F, Mademtzoglou D, Perez-Diaz S, Prola A, Reyes-Fernandez P, and Rotini A (2021). Perspectives on skeletal muscle stem cells. Nat. Commun 12, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, Brunson C, Mastey N, Liu L, and Tsai C-R (2014). mTORC1 controls the adaptive transition of quiescent stem cells from G 0 to G Alert. Nature 510, 393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RStudio Team (2015). RStudio: Integrated Development for R, 42 (RStudio, Inc; ), p. 84. http://www.rstudio.com. [Google Scholar]
- Ryall JG, Dell’Orso S, Derfoul A, Juan A, Zare H, Feng X, Clermont D, Koulnis M, Gutierrez-Cruz G, and Fulco M (2015). The NAD+-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell 16, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambasivan R, Comai G, Le Roux I, Gomès D, Konge J, Dumas G, Cimper C, and Tajbakhsh S (2013). Embryonic founders of adult muscle stem cells are primed by the determination gene Mrf4. Dev. Biol 381, 241–255. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaramozza A, Park D, Kollu S, Beerman I, Sun X, Rossi DJ, Lin CP, Scadden DT, Crist C, and Brack AS (2019). Lineage tracing reveals a subset of reserve muscle stem cells capable of clonal expansion under stress. Cell Stem Cell 24, 944–957.e945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, and Schmid B (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea KL, Xiang W, LaPorta VS, Licht JD, Keller C, Basson MA, and Brack AS (2010). Sprouty1 regulates reversible quiescence of a self-renewing adult muscle stem cell pool during regeneration. Cell Stem Cell 6, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SR, Zeng X, Zhao J, Liu Y, Hou G, Liu H, and Hou SX (2016). The lipolysis pathway sustains normal and transformed stem cells in adult Drosophila. Nature 538, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM III, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiam AR, Farese RV Jr., and Walther TC (2013). The biophysics and cell biology of lipid droplets. Nat. Rev. Mol. Cell Biol 14, 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther TC, Chung J, and Farese RV Jr. (2017). Lipid droplet biogenesis. Annu. Rev. Cell Dev. Biol 33, 491–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther TC, and Farese RV Jr. (2012). Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem 81, 687–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Zhu H, Situ C, Han L, Yu Y, Cheung TH, Liu K, and Wu Z (2018). p110α of PI3K is necessary and sufficient for quiescence exit in adult muscle satellite cells. EMBO J. 37, e98239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Viscarra J, Kim S-J, and Sul HS (2015). Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol 16, 678–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte MA, and Gould AP (2017). Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 1260–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead NP, Pham C, Gervasio OL, and Allen DG (2008). N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J. Physiol 586, 2003–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Langlais P, Zhang X, Heckmann BL, Saarinen AM, Mandarino LJ, and Liu J (2014). Identification of a novel phosphorylation site in adipose triglyceride lipase as a regulator of lipid droplet localization. Am. J. Physiol. Endocrinol. Metab 306, E1449–E1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yucel N, Wang YX, Mai T, Porpiglia E, Lund PJ, Markov G, Garcia BA, Bendall SC, Angelo M, and Blau HM (2019). Glucose metabolism drives histone acetylation landscape transitions that dictate muscle stem cell function. Cell Rep. 27, 3939–3955.e3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Bi P, Wang C, Li J, Liu X, and Kuang S (2016). Conditional loss of Pten in myogenic progenitors leads to postnatal skeletal muscle hypertrophy but age-dependent exhaustion of satellite cells. Cell Rep. 17, 2340–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Bi P, Wang C, Shan T, Nie Y, Ratliff TL, Gavin TP, and Kuang S (2017). Pten is necessary for the quiescence and maintenance of adult muscle stem cells. Nat. Commun 8, 14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, and Madeo F (2012). FAT SIGNALS-lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 15, 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Menzies KJ, and Auwerx J (2018). The role of mitochondria in stem cell fate and aging. Development 145, dev143420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data generated from this study are available at the following Gene Expression Omnibus (GEO): GSE150632 (bulk RNA-seq) and GSE150366 (single-cell RNA-seq).
No novel code was written for the analysis of the dataset.
Any additional information required to reanalyze the data reported in this work article is available from the lead contact upon request.