

Summary
Many insect cells are encapsulated within the exoskeleton and cannot be dissociated intact, making them inaccessible to single-cell transcriptomic profiling. We have used single-nucleus RNA sequencing to extract transcriptomic information from multiple Drosophila tissues. Here, we describe procedures for the (1) dissociation of single nuclei, (2) isolation of single nuclei using two popular cell sorters, and (3) preparation of libraries for Smart-seq2 and 10× Genomics. This protocol enables generation of high-quality transcriptomes from single nuclei and can be applied to other species.
For complete details on the use and execution of this protocol, please refer to McLaughlin et al. (2021) and Li et al. (2022).
Subject areas: Genomics, Model Organisms, RNAseq, Sequencing
Graphical abstract
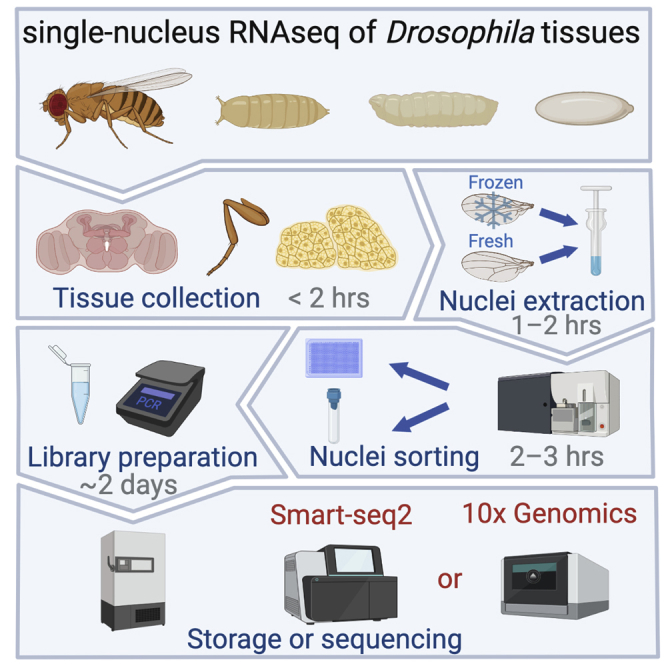
Highlights
-
•
Single-nucleus RNA-seq can profile transcriptomes from many Drosophila tissues
-
•
High-quality nuclei can be extracted from fresh or frozen samples
-
•
Fluorescence-activated cell sorting accurately captures Drosophila nuclei
-
•
Fly nuclei can be profiled using 10× Genomics or Smart-seq2 platforms
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Many insect cells are encapsulated within the exoskeleton and cannot be dissociated intact, making them inaccessible to single-cell transcriptomic profiling. We have used single-nucleus RNA sequencing to extract transcriptomic information from multiple Drosophila tissues. Here, we describe procedures for the (1) dissociation of single nuclei, (2) isolation of single nuclei using two popular cell sorters, and (3) preparation of libraries for Smart-seq2 and 10× Genomics. This protocol enables generation of high-quality transcriptomes from single nuclei and can be applied to other species.
Before you begin
Background
Over the last decade, the study of gene expression has undergone a technical revolution that has enabled high-resolution profiling of individual cells through single-cell RNA sequencing (scRNA-seq). Application of scRNA-seq to multiple tissues and developmental stages in Drosophila (Croset et al., 2018; Li et al., 2017; Karaiskos et al., 2017; Tattikota and Perrimon, 2021) has revealed novel cell types (Brunet Avalos et al., 2019; Fu et al., 2020; Hung et al., 2020; Rust et al., 2020), molecular mechanisms of brain development and aging (Davie et al., 2018; Konstantinides et al., 2018; Kurmangaliyev et al., 2020; Li et al., 2017, 2020; Özel et al., 2021; Tauc et al., 2021; Xie et al., 2021), and pathways underpinning disease (Ariss et al., 2018; Genovese et al., 2019; Ji et al., 2019). Additional studies are summarized in a review by Li (2020). However, most peripheral tissues in adult Drosophila are tightly associated with the hardened cuticle and their cells cannot be isolated using traditional dissociation protocols, making them inaccessible to profiling by scRNA-seq.
Here, we present a single-nucleus RNA sequencing (snRNA-seq) protocol that has been utilized to generate high-quality transcriptomes of cells that reside in tissues that are and are not associated with the cuticle (Li et al., 2022; McLaughlin et al., 2021). Unlike cells, nuclei are more resistant to mechanical assaults and can be isolated from cells in cuticle-associated tissues, such as fly legs, wings, and antennae. snRNA-seq offers the following advantages over scRNA-seq: (1) dissected tissue can be kept in long-term storage at –80°C; (2) reduced cell-type sampling bias; and (3) dissociation procedures and microfluidics devices better capture nuclei than large or fragile cells. Using the protocol below, we have performed both Smart-seq2 and 10× Genomics profiling. Below is a protocol for the dissociation, sorting, and sequencing of Drosophila nuclei which can be adapted to obtain single-nucleus transcriptomes from a wealth of insect species.
Preparation of crosses for tissue isolation
Timing: 1 h
-
1.
Identify a driver line (GAL4, QF2, LexA) that best labels the cell populations of interest. Alternatively, determine the wild-type strain to be used for tissue collection.
-
2.
Set up crosses between driver lines and nuclear GFP lines. We compared nuclear localized (nls) GFP (UAS-GFP.nls; Bloomington Drosophila Stock Center #4775), UAS-unc84-GFP, and lamin-GFP (UAS-lam-GFP) lines and found that the GFP signal in UAS-GFP.nls expressing flies was abolished after nuclear isolation while the other two lines showed strong GFP signal. Therefore, we suggest using UAS-unc84-GFP or UAS-lam-GFP to label nuclei. This is presumably because both GFP tagged Unc-84 and Lamin proteins interact with nuclear membrane proteins, while GFP.nls does not.
Note: Certain GAL4 lines driving UAS-unc84-GFP or UAS-lam-GFP may cause lethality. We suggest testing both GFP reporters in advance.
-
3.
We recommend rearing crosses at 25°C to facilitate suitable GFP expression and transferring files to fresh food every 2–3 days.
Note: We estimate the number of crosses to prepare and amount of tissue required for processing based on three factors: (1) total cell/nucleus number in the tissue; (2) targeted nucleus number for sequencing; and (3) recovery rate of nuclei from fluorescence-activated cell sorting (FACS). Typically, our FACS recovery rate is ∼5%.
Prepare for Smart-seq2 protocol
Prepare oligos, tagmentation stocks, barcoding plates, and program equipment for library preparation steps.
-
4.
Dissolve all primers to requisite concentrations in nuclease-free water or TE buffer. Aliquot the primers and store them at –20°C for a maximum of 2 years. Template switching oligo (TSO) primers should be kept at –80°C in PCR strips for a maximum of 2 years.
CRITICAL: Avoid multiple freeze thaw cycles for TSO primers.
-
5.Prepare TAPS-PEG (TAPS; (3-(Tris(hydroxymethyl) methylamino) propane-1-sulfonic acid); PEG; polyethylene glycol) stocks and constituents:
-
a.Make 40% w/w solution of PEG 8000 and pass it through a 0.22 μm filter unit.
-
b.Make 3 mL of 50× TAPS using the provided calculations and pass it through a 0.22 μm filter unit.
-
c.Make 30 mL 5× TAPS-MgCl2 using the provided calculations and make sure the pH is 8.4.
-
d.Make 60 mL 2.5× TAPS-PEG stock solution using the provided calculations:
-
i.Aliquot it into 1.5 mL tubes.
-
ii.Mix tubes for 1 h at room temperature (∼20°C).
-
iii.Store at 4°C for a maximum of 2 years.
-
i.
-
a.
-
6.
Prepare the barcoding plates containing either 96 or 384 different Nextera XT-compatible barcoding primer combinations according to the manufacturer’s guidelines. Barcoding primers, xGen™ 10nt UDI Primers, can be obtained from Integrated DNA Technologies. These plates can be stored at –20°C for a maximum of 2 years and can be reused multiple times. The specific barcodes in each well will be used to assign reads to different cells following sequencing and de-multiplexing.
-
7.
Pre-program thermal cyclers with each program used in this protocol. For all thermal cycling conditions, the heated lid option should be used (ideally set to 105°C) to avoid evaporation of the contents of each well.
Prepare lysis plates
These are the lysis plates that nuclei will be sorted into during the FACS step. Lysis plates can be made in two formats, 96-well or 384-well, and stored at –80°C for a maximum of 2 years. Below are details for making both 96-well and 384-well lysis plates.
-
8.Create stock of External RNA Controls Consortium (ERCC) RNA Spike-In mixture to be used at a 1:3000 dilution in nuclease-free water. ERCCs function as an external control (Munro et al., 2014) for sources of variability in downstream cDNA generation and amplification steps. Store at –80°C in PCR tube strips for a maximum of 2 years.
-
a.Create 200 μL working solution of ERCCs (this should be enough for >15, 96-well or >30, 384-well plates) using the calculations provided.
-
a.
-
9.
Make lysis buffer using the calculations provided.
-
10.
Using either a multichannel pipet or automated liquid handling robot (such as MANTIS®), dispense 4 μL of lysis buffer into each well of a 96-well plate or 0.5 μL of lysis buffer into each well of a 384-well plate.
-
11.
Briefly centrifuge each plate to ensure that all lysis buffer is in the bottom of the wells.
-
12.
Once each plate is completed, seal it with an aluminum plate seal, and immediately store it on dry ice. Lysis plates should be stored at –80°C if not used immediately.
Prepare for 10× Genomics protocol
-
13.If the 10× Single Cell Reagent Kit (e.g., V3.1) is new, take the TSO tube from −20°C reagent box, centrifuge briefly, and resuspend TSO using 80 μL Low TE Buffer.
-
a.Vortex for 15 s at maximum speed, centrifuge briefly, and leave it at room temperature (∼20°C) for ≥ 30 min.
-
b.Store the resuspended TSO at −80°C.
-
a.
-
14.
If the 10× Reagent Kit is not new, take the resuspended TSO and Single Cell 3′ v3.1 Gel Beads from −80°C and equilibrate them to room temperature (∼20°C) for 30 min before loading the chip.
-
15.
Remove the Single Cell 3′ v3.1 Gel Beads from −80°C and equilibrate them to room temperature (∼20°C) for 30 min before loading the chip.
Note: We suggest removing these two reagents from −80°C just before FACS to make sure they are ready to use after FACS sorting.
Cleaning, buffer preparation, and equipment setup for nuclear dissociation
-
16.Clean work area and tools:
-
a.Clean Dounce homogenizer with 70% ethanol followed by dH2O. Heat sterilize the Dounce homogenizer in aluminum foil at 200°C for a minimum of 2 h. This step can be done in advance and sterile Dounce sets can be stored in aluminum foil in a clean area.
-
b.Clean bench top and dissection area with 70% ethanol followed by RNaseZAP to remove RNases.
-
c.Clean dissection dishes, forceps, and microdissection scissors with 70% ethanol followed by RNAseZAP.
-
a.
-
17.Preparation of buffers (see complete recipes in materials and equipment section):
-
a.Prepare 10 mL of homogenization buffer using the calculations provided.
-
b.Prepare 10 mL of resuspension buffer using the calculations provided.
-
c.Prepare 1.5 mL nuclease-free tubes for collection of dissected tissue containing 100–200 μL of Schneider’s Drosophila Medium.
-
d.Prepare dissection buffer (15 mL conical tubes containing Schneider’s Drosophila medium).
-
a.
-
18.Equipment set up:
-
a.Pre-cool the centrifuge for 1.5 mL tubes to 4°C.
-
b.If using a pestle and pestle motor for homogenization, pre-cool the pestle on ice.
-
a.
Tissue dissection and collection
-
19.
Keep tubes containing dissection and collection buffer on ice for the duration of the dissection procedure.
-
20.
Coat a P20 pipet tip by pipetting extraneous tissues (we typically use fly fat body) up and down into the tip to reduce the static that will cause tissue of interest to stick to the inner wall of the tip.
-
21.
Dissect tissue of interest in ice cold Schneider’s medium.
-
22.Adult cuticle-associated tissues can be difficult to collect because once dissected they will float in Schneider’s medium due to surface tension. For cuticle-associated tissues, we recommend using a three-well spot plate with the following setup for dissections:
-
a.Fill one well with 100% ethanol and fill the other two wells with Schneider’s medium.
-
b.Submerge the flies in 100% ethanol for 10 s in the first well.
-
c.Submerge and leave flies in Schneider’s medium in the second well.
-
d.Dissect flies, one at a time, in the third well.
-
a.
-
23.
Once dissected, move tissues to 1.5 mL tube containing ∼200 μL of Schneider’s medium.
-
24.
Quickly spin samples down using a mini-bench top centrifuge.
Optional: Samples can be flash frozen at this step for long-term storage (several months). Seal the 1.5 mL tube with parafilm and place directly into liquid nitrogen > 30 s and move the sample to a –80°C freezer.
-
25.
Fresh samples can be immediately processed for nuclear extraction.
Note: We suggest dissecting tissue in less than 2 h to preserve RNA quality. If enough samples cannot be obtained within this time frame, multiple batches of collected tissue can be combined during nucleus dissociation. However, we suggest minimizing the number of tubes used for tissue collection, because tissues will be pooled prior to nuclear dissociation and samples can be lost during this step.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Ribonuclease Inhibitor (RNase Inhibitor) | Takara/Clontech | 2313A |
| 10 mM dNTP Mix | Thermo Fisher Scientific | R0193 |
| USB Dithiothreitol (DTT), 100 mM | Thermo Fisher Scientific | 707265ML |
| MgCl2 | MilliporeSigma | 63069 |
| Low TE Buffer (10 mM Tris-HCl pH 8.0, 0.1 mM EDTA) | Thermo Fisher Scientific | 12090-015 |
| 5 M Betaine Solution | MilliporeSigma | B0300 |
| Lambda Exonuclease | New England BioLabs | M0262 |
| AMPure XP Beads | Beckman Coulter | A63881 |
| TAPS (3-(Tris(hydroxymethyl) methylamino) propane-1-sulfonic acid) free acid ≥99%, high purity. | VWR | 97064-208 |
| Tn5 | Picelli et al. (2014) or Nextera XT DNA Sample Preparation Kit | n/a |
| Buffer EB | QIAGEN | 19086 |
| 50× Protease Inhibitor Cocktail | Promega | G6521 |
| RNasin Plus Ribonuclease Inhibitor | Promega | N2615 |
| Critical commercial assays | ||
| KAPA HiFi HotStart ReadyMix | Roche | KK2602 |
| ERCC Spike-In Mixture | Thermo Fisher Scientific | 4456740 |
| Nextera XT DNA Sample Preparation Kit | Illumina | FC-131-1096 |
| Smartscribe Reverse Transcriptase | Clontech/Takara | 639538 |
| Chromium Next GEM Single Cell 3′ GEM, Library & Gel Bead Kit v3.1 | 10× Genomics | 1000128 |
| Chromium Next GEM Chip G Single Cell Kit | 10× Genomics | 1000120 |
| Single Index Kit T Set A, 96 rxn | 10× Genomics | 1000213 |
| KAPA HiFi PCR Kit | Roche | 7958838001 |
| High Sensitivity D5000 Reagents | Agilent Technologies | 5067–5593 |
| Experimental models: Organisms/strains | ||
| UAS-Lam-GFP (D. melanogaster) | Bloomington Drosophila Stock Center | BDSC_97378 |
| UAS-unc84-GFP (D. melanogaster) | Henry et al. (2012) | n/a |
| Oligonucleotides | ||
| IS PCR primer: AAGCAGTGGTATCAACGCAGAGT | Picelli et al. (2014) | n/a |
| Oligo-dT30VN: AAGCAGTGGTATCAACGCAGAG TACTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTVN |
Picelli et al. (2014) | n/a |
| xGen™ 10nt UDI Primers | Integrated DNA Technologies | 10008055 10008056 10008057 |
| TSO: AAGCAGTGGTATCAACGCAGAGTACAT rGrG+G (‘rG’=riboguanosine; ‘+G’= LNA-modified guanosine) |
Exiqon; Picelli et al. (2014) | n/a |
| Other | ||
| Adhesive PCR Plate Foil Seals | Bio-Rad | MSF1001 |
| Adhesive PCR Plate Seals | ThermoFisher | AB0558 |
| DNA LoBind Tubes (1.5 mL) | Eppendorf | 022431021 |
| DynaMag-2 Magnetic Rack | Fisher Scientific | 12-321-D |
| Falcon 5 mL Round Bottom Polystyrene Test Tube, with Cell Strainer Snap Cap | Corning | 352235 |
| FLOWMI 40 μM Cell Strainers | Bel-Art | H13680-0040 |
| Hard-Shell PCR Plates (384-well) | Bio-Rad | HSP3481 |
| Hard-Shell PCR Plates (96-well) | Bio-Rad | HSP9631 |
| Kimble Pellet Pestle | Grainger | 6HAY5 |
| Kimble Pellet Pestle Motor | Grainger | 6HAZ6 |
| Three-well spot plate | Electron Microscopy Sciences | 71574-05 |
| Wheaton Dounce Tissue Grinder (1 mL) | DWK Life Sciences | 357538 |
| High Sensitivity D5000 Ladder | Agilent Technologies | 5067–5594 |
| High Sensitivity D5000 ScreenTape | Agilent Technologies | 5067–5592 |
| Hoechst-33342 | ThermoFisher | H3570 |
| Beckman Coulter SPRIselect | Beckman Coulter | B23318 |
| C-Chip™ Disposable Hemacytometers by SKC, Inc | Fisher Scientific | 22-600-100 |
Materials and equipment
Note: For all Smart-seq2 master mix steps in this protocol, we make more master mix than needed to fill our desired number of plates. The calculations below reflect this excess. If this is not suitable, use the values from the ‘1 well’ column to re-calculate the amount of reagent needed for the desired final volume of master mix.
ERCC working solution
| Reagent | Amount (μL) |
|---|---|
| ERCC stock | 10 |
| Nuclease-free water | 185 |
| RNase Inhibitor | 5 |
50× TAPS (0.5 M)
| Reagent | Amount |
|---|---|
| TAPS powder | 364.9 mg |
| Nuclease-free water | 3 mL |
5× TAPS-MgCl2
| Reagent | Amount (mL) |
|---|---|
| 50× TAPS | 3 |
| 1 M MgCl2 | 0.75 |
| Nuclease-free water | 26.25 |
Note: After making 5× TAPS-MgCl2 solution, it needs to be sterilized using a 0.22 μM filter unit.
2.5× TAPS-PEG
| Reagent | Amount (mL) |
|---|---|
| 5× TAPS-MgCl2 | 30 |
| 40% w/w PEG 8000 | 30 |
Lysis Buffer Master Mix
| Reagent | 96-Well plate |
384-Well plate |
||||
|---|---|---|---|---|---|---|
| 1 well (μL) | 1 plate (μL) | 10 plates (μL) | 1 well (μL) | 1 plate (μL) | 10 plates (μL) | |
| Nuclease-free water | 2.68 | 294.8 | 2948 | 0.335 | 134 | 1340 |
| RNase Inhibitor | 0.1 | 11 | 110 | 0.0125 | 5 | 50 |
| ERCC working solution | 0.1 | 11 | 110 | 0.0125 | 5 | 50 |
| 10% Triton | 0.02 | 2.2 | 22 | 0.0025 | 1 | 10 |
| 10 mM dNTP | 1 | 110 | 1100 | 0.125 | 50 | 500 |
| 100 μM oligo-dT30VN primer | 0.1 | 11 | 110 | 0.0125 | 5 | 50 |
Homogenization Buffer
| Reagent | Amount | Final concentration | Storage |
|---|---|---|---|
| Nuclease Free water | 9.2 mL | RT | |
| Sucrose (nuclease free) | 0.856 g | 250 mM | RT |
| 1 M Tris (pH 8.0) | 100 μL | 10 mM | 4°C |
| 1 M KCl | 250 μL | 25 mM | 4°C |
| 1 M MgCl2 | 50 μL | 5 mM | 4°C |
| 10% Triton-x 100 | 100 μL | 0.1% | 4°C |
| RNasin Plus | 50 μL | 0.5% | −20°C |
| 50× protease inhibitor | 200 μL | 1× | −20°C aliquots |
| 20 mM DTT | 50 μL | 0.1 mM | −20°C |
Resuspension buffer
| Reagent | Amount | Final concentration |
|---|---|---|
| 1× PBS | 9.4 mL | |
| 10% BSA | 500 μL | 0.5% |
| RNasin Plus | 100 μL | 0.5% |
Reverse Transcription Master Mix
| Reagent | 96-Well plate |
384-Well plate |
||||
|---|---|---|---|---|---|---|
| 1 well (μL) | 1 plate (μL) | 4 plates (μL) | 1 well (μL) | 1 plate (μL) | 4 plates (μL) | |
| Nuclease-free water | 0.14 | 15.4 | 61.6 | 0.0175 | 8.75 | 35 |
| 5× first strand buffer (from Takara kit) | 2 | 220 | 880 | 0.25 | 125 | 500 |
| RNase inhibitor | 0.25 | 27.5 | 110 | 0.031 | 15.5 | 62 |
| 100 mM DTT (Invitrogen) | 0.5 | 55 | 220 | 0.063 | 31.5 | 126 |
| 1 M MgCl2 | 0.06 | 6.6 | 26.4 | 0.0075 | 3.75 | 15 |
| 5 M Betaine | 2 | 220 | 880 | 0.25 | 125 | 500 |
| 100 μM TSO | 0.1 | 11 | 44 | 0.0125 | 6.25 | 25 |
| SmartScribe II Reverse Transcriptase | 0.95 | 104.5 | 418 | 0.12 | 60 | 240 |
cDNA Amplification Master Mix
| Reagent | 96-Well plate |
384-Well plate |
||||
|---|---|---|---|---|---|---|
| 1 well (μL) | 1 plate (μL) | 4 plates (μL) | 1 well (μL) | 1 plate (μL) | 4 plates (μL) | |
| Nuclease-free water | 2.14 | 235.4 | 941.6 | 0.27 | 135 | 540 |
| 10 μM IS PCR primer | 0.25 | 27.5 | 110 | 0.031 | 15.6 | 62.4 |
| 2× Kapa HiFi HotStart ReadyMix | 12.5 | 1375 | 5500 | 1.56 | 780 | 3120 |
| Lambda Exonuclease | 0.11 | 12.1 | 48.4 | 0.014 | 7 | 28 |
cDNA Tagmentation Master Mix
| Reagent | 96-Well plate |
384-Well plate |
||||
|---|---|---|---|---|---|---|
| 1 well (μL) | 1 plate (μL) | 4 plates (μL) | 1 well (μL) | 1 plate (μL) | 4 plates (μL) | |
| Tn5 | 0.128 | 14.08 | 56.32 | 0.016 | 8 | 32 |
| Nuclease-free water | 4.352 | 478.72 | 1914.88 | 0.544 | 272 | 1088 |
| TAPS-PEG (2.5×) | 5.12 | 563.2 | 2252.8 | 0.64 | 320 | 1280 |
Note: Tn5 reagent can be either synthesized or can be taken from the Nextera XT DNA Sample Preparation Kit.
Indexing PCR Master Mix
| Reagent | 96-well plate |
384-well plate |
||||
|---|---|---|---|---|---|---|
| 1 well (μL) | 1 plate (μL) | 4 plates (μL) | 1 well (μL) | 1 plate (μL) | 4 plates (μL) | |
| KAPA HiFi DNA Polymerase | 0.64 | 70.4 | 281.6 | 0.08 | 40 | 160 |
| 5× KAPA HiFi Fidelity Buffer | 6.4 | 704 | 2816 | 0.8 | 400 | 1600 |
| 10 mM KAPA dNTP Mix | 0.96 | 105.6 | 422.4 | 0.12 | 60 | 240 |
| Nuclease-free water | 1.6 | 176 | 704 | 0.2 | 100 | 400 |
Step-by-step method details
Nuclei dissociation
During the following steps, nuclei are extracted from Drosophila tissues (Figure 1). This protocol is compatible with fresh or frozen tissues. If performing this dissociation protocol on frozen tissue, allow the tissue to thaw on ice for ∼5 min and quickly spin down samples (∼30 s) in Schneider’s medium using a mini-benchtop centrifuge prior to beginning.
Optional: If using large tissues, such as intact heads, use a pestle motor and disposable plastic pestles to grind the samples for 60 s on ice. Make sure pestles are pre-cooled on ice before beginning.
-
1.
Discard Schneider’s medium without disrupting pellet.
-
2.
Add 1000 μL of homogenization buffer and transfer sample to the 1 mL Dounce.
-
3.
Release nuclei on ice by 20 strokes of the loose Dounce pestle and 40 strokes of the tight Dounce pestle. Avoid inducing bubbles.
-
4.
Filter 1000 μL sample through a 35 μm cell strainer into a 5 mL round bottom polystyrene FACS tube.
-
5.
Filter samples again using a 40 μm Flowmi cell strainer into a 1.5 mL tube.
-
6.
Centrifuge for 10 min at 1,000 × g at 4°C.
Note: The slow centrifugation speed is necessary to avoid damaging the nuclei.
-
7.
Discard supernatant and do not disturb the pellet.
-
8.
Re-suspend the nuclei using the desired amount of resuspension buffer. Pipet > 20 times to completely re-suspend the nuclei in 300–600 μL of resuspension buffer. Typically, we use 500 μL for resuspension.
-
9.
Use a 40 μm Flowmi cell strainer to filter the sample into a new 5 mL FACS tube. Keep the tube on ice.
Note: In our experience, nuclei are much stickier than whole cells. For researchers making the single-nucleus suspension for the first time, we suggest confirming that the sample contains mostly single nuclei. To do this, take 10 μL of sample and stain it with Hoechst for more than 5 min, place it on a hemocytometer, and visualize it under a 20× objective on an epifluorescence microscope.
Figure 1.

Workflow detailing the dissection and dissociation of Drosophila tissues for single-nucleus RNA-seq
FACS of nuclei
In this step, individual nuclei are collected into either one tube for 10× Genomics (steps 10–18) or 96 or 384-well plates for Smart-seq2 (steps 19–24). This protocol has been tested with the Sony SH800 sorter and the BD FACS Aria III using the 100 μm nozzle.
Note: Prior to sorting and after nuclear extraction, add 20 μL of unstained nuclei to 200 μL of resuspension buffer as an unstained control for setting up FACS gates. Add Hoechst-33342 (at 1:1000) to the remaining nuclei and incubate for more than 5 min. When we sort wild-type tissues, we collect all Hoechst+ nuclei. When sorting GFP+ nuclei, first gate for Hoechst+ events and then gate the GFP+ population.
Note: The following steps are for 10× genomics.
-
10.
Set up the sorter prior to nuclear extraction to avoid leaving the nuclei on ice for too long. This will take roughly 30 min. After setting up the sorter, pre-cool the collection chamber to 4°C.
-
11.
Load the unstained nuclei to set up the negative gate.
Note: Always run the unstained control first.
-
12.
Prepare the collection tube by adding 400 μL of resuspension buffer to a 1.5 mL nuclease-free tube, and vortex the tube to coat it.
-
13.
Load Hoechst-stained nuclei and the collection tube. The nucleus events and the debris events should be separated in the Hoechst channel. Sorting gate should be set tightly to the cluster of nuclei to exclude debris. The overall Hoechst signal for the debris will be slightly increased from the stained nuclei compared with the unstained sample (Figure 2).
-
14.
Sort nuclei into the collection tube depending on the number of desired 10× runs. 100k nuclei are typically collected for one 10× run.
-
15.
After sorting, centrifuge the collection tube for 10 min at 1,000 × g at 4°C. We use a swinging-bucket rotor for this in a refrigerated centrifuge, such as an Eppendorf® Centrifuge 5430 R with Eppendorf® Swing-bucket rotor S-24-11-AT.
-
16.
Carefully discard the supernatant without touching the bottom of the tube. The pellet is often too small to be seen. Alternatively, the supernatant can be temporarily saved in a new nuclease-free tube on ice and discarded only after re-suspended nuclei are verified in the next step.
-
17.Resuspend the pellet with 45.2 μL resuspension buffer.
-
a.Take 2 μL of nuclei and mix with 8 μL resuspension buffer.
-
b.Load 10 μL diluted nuclei into a hemocytometer and count the number, according to the manufacturer’s guidelines, under a 20× objective of an epifluorescence microscope based on Hoechst signal (Figure 3). Calculate the concentration of nuclei.
-
a.
-
18.
Now the nuclei are ready to be loaded to the 10× Chromium Controller.
Note: The following steps are for Smart-seq.
-
19.
Follow steps 10 and 11 from the above 10× Genomics FACS section to prepare the sorter and set up the gate with unstained and stained nuclei.
-
20.Adjust the plate position to ensure that the FACS collection solution drops in the center of each well.
-
a.Place a new aluminum seal on an empty plate and put the plate in the collection chamber.
-
b.Test the accuracy of the sorter by dropping 25 events onto the 4 corner wells. Adjust the position of 4 corner wells one-by-one to ensure that the drop is in the center of each well.
-
a.
-
21.Prepare collection plates, 96-well or 384-well.
-
a.Thaw a lysis plate for about 1 min at room temperature (∼20°C).
-
b.Quickly spin the plate down using a mini-benchtop centrifuge for 30 s to ensure that the lysis buffer is in the bottom of each well. Thaw one plate at a time.
-
a.
-
22.
Load Hoechst stained nuclei and a fresh collection plate. Sort single nuclei into individual wells.
-
23.
After collection is finished, immediately seal the plate with an aluminum seal and spin the plate using a mini-benchtop centrifuge at room temperature (∼20°C) for 1 min, and immediately put the plate onto dry ice.
-
24.
The plates now can be stored at –80°C for several months or can be immediately processed to make sequencing libraries.
Note: Since polyploidy is common for many fly tissues, different nucleus populations may be observed during FACS according to the Hoechst signal, which reflects DNA content or nuclear size. For example, we observed 6 different populations with different Hoechst signal intensities for the fly testis and confirmed that the Hoechst signal intensities correlated well with nuclear size (Li et al., 2022). For the fly head, we observed 2 different populations with different Hoechst signals (Figure 2). We recommend collecting nuclei of each size to ensure that all cell populations are adequately represented.
Figure 2.

Representative FACS plots of nuclei from adult fly heads
Left: plot of unstained nuclei. Right: plot of nuclei stained with Hoechst. The two populations of nuclei in the polygon are sorted for snRNA-seq.
Figure 3.

Example image of nuclei following FACS for 10× Genomics platform
Scale bar, 25 um.
10× Genomics: Library preparation and sequencing
In this step, the Chromium Next GEM Single Cell 3′ Reagent Kits v3.1 are used. Polyadenylated mRNAs from each individual nucleus are labeled with an individual 10× barcode during cDNA synthesis, followed by cDNA amplification and 3′ Gene expression library construction. The Single Cell 3′ Gene Expression libraries comprise standard Illumina paired-end constructs which begin with P5 and end with P7. The library is compatible with multiple Illumina sequencers.
-
25.
Capture nuclei using the 10× Chromium Controller. When loading nuclei to the 10× controller, aim to recover 10k nuclei for each channel. The 10× Genomics User Guide suggests loading 16k nuclei in order to recover 10k. However, we observed that loading 1.5-fold more (24k nuclei total) than recommended allowed us to recover about 10k cells after sequencing. This is likely because fly nuclei are smaller than mammalian nuclei.
Note: We load more nuclei than recommended because: (1) FACS can capture cell debris that are the same size as nuclei as ‘events’; (2) FACS cell counts can be overestimated; and (3) centrifugation following FACS can cause nuclei loss.
-
26.
Make sequencing libraries. Follow the 10× Chromium Next GEM Single Cell 3′ Reagent Kits v3.1 User Guide to perform GEM Generation & Barcoding, post GEM-RT cleanup, cDNA amplification, and 3ʹ gene expression library construction. Representative cDNA and library traces are shown in Figure 4.
-
27.
Sequencing. Single-cell sequencing libraries can be sequenced in most Illumina sequencing systems. We sequenced most of our libraries using Illumina NovaSeq 6000 sequencer with minimum sequencing depth of 50,000 reads/cell using the read lengths 28 bp Read1, 8 bp i7 Index, 91 bp Read2.
Figure 4.

Representative electropherograms showing the DNA distribution of cDNA and the sequencing library from 10× Genomics using the Agilent Bioanalyzer system
Smart-seq2: First-strand synthesis, cDNA amplification, and library preparation
This portion of the protocol is modified specifically for fly nuclei from Picelli et al. (2014), Li et al. (2017), and Jaeger et al. (2020).
-
28.
Primer Annealing ∼5 min.
Anneal oligo-dT30VN primer to RNA.-
a.Place frozen 96 or 384-well plate(s) into pre-warmed thermal cycler(s) to anneal primers using the following program:Primer Annealing Program
Steps Temperature Time Cycles Primer annealing 72°C 3 min 1 Hold 4°C ∞ -
b.Make reverse transcription (RT) master mix, on ice, while the plates are in the thermal cyclers for annealing, using the provided calculations.
-
a.
-
29.
Reverse Transcription (RT) ∼ 1.75 h.
Perform RT reaction to obtain cDNA from single nuclei.-
a.Remove one plate at a time from the thermal cycler and immediately store it on ice.
-
b.Using either a multichannel pipet or automated liquid handling robot (such as MANTIS®), dispense 6 μL of RT master mix into each well of a 96-well plate or 0.75 μL of RT master mix into each well of a 384-well plate.
-
c.Cover plate with an adhesive PCR plate seal.
-
d.Briefly vortex and then centrifuge each plate to ensure that all solution is in the bottom of the wells.
-
e.Place plate back into pre-warmed thermal cycler and run the RT reaction using the following program:Reverse Transcription Program
Steps Temperature Time Cycles RT and template switching 42°C 90 min 1 Enzyme inactivation 70°C 5 min 1 Hold 4°C ∞  Pause point: Following RT reaction, plates can be stored at –20°C for up to two weeks.
Pause point: Following RT reaction, plates can be stored at –20°C for up to two weeks. -
f.Once plates have < 30 min left of the RT reaction, begin making the cDNA amplification master mix, on ice, using the provided calculations.
-
a.
-
30.
cDNA Amplification ∼ 3 h.
Perform PCR to amplify the cDNA generated from the RT reaction.-
a.Remove plates from thermal cyclers and store them on ice.
-
b.Using either a multichannel pipet or automated liquid handling robot (such as MANTIS®), dispense 15 μL of RT master mix into each well of a 96-well plate or 1.9 μL of RT master mix into each well of a 384-well plate.
-
c.Cover plate(s) with adhesive PCR plate seal(s).
-
d.Briefly vortex and then centrifuge each plate to ensure that all solution is in the bottom of the wells.
-
e.Place plate(s) back into pre-warmed thermal cycler(s) and run the cDNA amplification reaction using the following program:cDNA Amplification Program
Steps Temperature Time Cycles Incubation 37°C 30 min 1 Initial Denaturation 95°C 3 min 1 Denaturation 98°C 20 s 28 cycles Annealing 67°C 15 s Extension 72°C 4 min Final extension 72°C 5 min 1 Hold 4°C ∞ Note: This is largely the same protocol that we use with single-cells; however, for single-cells we repeat the cDNA amplification for 25 cycles instead of 28.Pause point: Plates can be stored at –20°C for several months.
-
a.
-
31.
Dilution of cDNA 30 min.
Dilute cDNA for downstream processing.-
a.By this step, the cDNA will be very concentrated and must be diluted using elution buffer. For 96-well plates, add 20 μL of elution buffer to each well. For 384-well plates, add 10 μL of elution buffer to each well.Note: cDNA concentration can be quantitated, using Quant-iT™ PicoGreen™ dsDNA Assay Kit or similar assay, and diluted to 0.32 ng/μL per well following the manufacturer’s instructions. However, for 384-well plates, we find that diluting each sample with the same amount of elution buffer works equivalently well. Further, custom dilutions for each well of 384-well plates are difficult to achieve if researchers do not have access to a liquid handling robot.
-
b.If using 96-well plates, merge cDNA from 4 different 96-well plates into a single 384-well plate. This step can be done manually using multichannel pipet or automated liquid handling robot.Pause point: Plates can be stored at –20°C for several months.
-
a.
-
32.
Tagmentation 30 min.
Fragment the cDNA for the library preparation.-
a.Make the cDNA tagmentation master mix on ice using the provided calculations.
-
b.Using either a multichannel pipet or automated liquid handling robot (such as MANTIS®) distribute 1.2 μL of tagmentation master mix into each well of a new 384-well plate.
-
c.Using either a multichannel pipet or automated liquid handling robot (such as Mosquito® LV) distribute 0.4 μL of diluted cDNA to the 384-well plate containing tagmentation mixture.
-
d.Cover plate(s) with adhesive PCR plate seal(s).
-
e.Briefly vortex and then centrifuge each plate to ensure that all solution is in the bottom of the wells.
-
f.Place the plate(s) into pre-warmed thermal cycler(s) and run the following tagmentation reaction:cDNA Tagmentation Program
Steps Temperature Time Cycles Tagmentation 55°C 5 min 1 Hold 4°C ∞ -
g.Once the tagmentation reaction is complete, add 0.4 μL of 0.1% SDS into each well of the 384-well plate. This will prevent over-tagmentation by neutralizing the reaction.
-
h.Briefly vortex and then centrifuge each plate to ensure that all solution is in the bottom of the wells.
-
a.
-
33.
Addition of Indexes and Amplification of Libraries 1 h.
Barcode and amplify the libraries.-
a.Make the indexing PCR master mix using the provided calculations.
-
b.Using either a multichannel pipet or automated liquid handling robot (such as MANTIS®) distribute 1.2 μL of the indexing PCR master mix into each well of the 384-well plate containing the tagmented cDNA.
-
c.Using either a multichannel pipet or automated liquid handling robot (such as Mosquito® LV) distribute 1 μL from each well of a barcoding plate into the corresponding wells of the 384-well plate containing the tagmented cDNA and indexing PCR master mix.
-
d.Cover the plate(s) with adhesive PCR plate seal(s).
-
e.Briefly vortex and then centrifuge each plate to ensure that all solution is in the bottom of the wells.
-
f.Place the plate(s) into pre-warmed thermal cycler(s) and run the following indexing PCR reaction:Indexing PCR Program
Steps Temperature Time Cycles Initial Extension 72°C 3 min 1 Initial Denaturation 95°C 30 s 1 Denaturation 95°C 10 s 10 cycles Annealing 55°C 30 s Extension 72°C 1 min Final extension 72°C 5 min 1 Hold 4°C ∞ -
g.Once the indexing reaction is complete, use either a multichannel pipet or automated liquid handling robot (such as Mosquito® LV) to pool 1 μL of each well from the entire plate into the first columns of an empty 384-well plate.
-
h.Manually pipet the contents of the pooled wells into 1.5 mL tubes.Pause point: libraries can be stored at –20°C for several months.
-
a.
-
34.
Library Purification (Bead Clean-Up) 1.5 h.
Clean-up and size selection of final libraries.-
a.Prepare an 80% (vol/vol) ethanol solution in nuclease-free water.
-
b.Equilibrate AMPure XP beads to room temperature (∼20°C) for 20 min. Vortex the beads to ensure that they are mixed with the buffer.
-
c.Aliquot 400 μL of each library into fresh 1.5 mL tubes and add AMPure XP beads at a 0.8:1 bead:cDNA ratio (320 μL) to each tube.
-
d.Vortex briefly and incubate bead–cDNA mixture at room temperature (∼20°C) for 6 min.
-
e.Put bead–cDNA mixture on compatible magnetic rack and wait until the solution completely clears (< 3 min).
-
f.Remove and discard supernatant.
-
g.Wash beads in 600 μL of 80% ethanol.
-
h.Remove and discard all residual 80% ethanol from tubes.
-
i.Repeat steps g and h for a total of two washes.
-
j.Remove tubes from magnetic rack and add 100 μL of elution buffer. Pipet the mixture to ensure it is thoroughly mixed.
-
k.Incubate at room temperature (∼20°C) for 6 min.
-
l.Place tubes back on magnetic rack and incubate until the solution completely clears.
-
m.Collect supernatant (100 μL) and transfer to a new 1.5 mL tube.
-
n.Add AMPure beads at a 0.7:1 bead:cDNA ratio (70 μL) to the supernatant, pipet to mix well.
-
o.Incubate beads in cDNA for 6 min at room temperature (∼20°C).
-
p.Put bead–cDNA mixture on magnetic rack and wait until the solution is completely clear.
-
q.Remove and discard supernatant.
-
r.Wash beads with 800 μL of 80% ethanol.
-
s.Remove and discard all residual 80% ethanol from tubes.
-
t.Repeat steps r and s for a total of two washes.
-
u.Allow magnetic beads to dry on the rack for ∼5 min.
-
v.Carefully remove the tube from the magnet and add 30 μL of elution buffer.
-
w.Pipet the bead–elution buffer mixture and ensure that it is thoroughly mixed.
-
x.Incubate a room temperature (∼20°C) for 6 min.
-
y.Place tubes back on magnetic stand and incubate until the solution completely clears.
-
z.Collect and keep supernatant (30 μL) in new 1.5 mL tube.Pause point: libraries can be stored at –20°C for several months.
-
a.
-
35.
Final Quality Control of Libraries 30 min.
Evaluate the quality and concentration of undiluted libraries using Bioanalyzer (Agilent 2200) or TapeStation (Agilent 2200) before sequencing. A good library should have the DNA peak at about 500 bp with no small DNA peaks around 100 bp which mostly consist of primer dimers (Figure 5).
-
36.
Sequencing.
Figure 5.

Representative electropherograms showing the DNA distribution of the sequencing library from Smart-seq2 using the Agilent Bioanalyzer system
By this stage, samples are ready to be sequenced using most Illumina platforms. We have used the NovaSeq6000 system (Illumina) with 100 paired end reads with 2 × 8 bp index reads. Samples should be sequenced to a depth of ∼1 million reads/nucleus.
Expected outcomes
These stepwise protocols will equip researchers with the tools necessary to extract transcriptomic information from single nuclei in Drosophila tissues. At the conclusion of these protocols, single-nucleus libraries will be compatible for sequencing. After sequencing, alignment, and quality filtration, transcriptional content of individual nuclei will be accessible through bioinformatic pipelines like Scanpy (Wolf et al., 2018) or Seurat (Stuart et al., 2019). Further, these protocols can be easily adapted for use in other species; thus, providing transcriptomic access, at the single-cell level, to a range of arthropods and non-insect species.
Limitations
Performing snRNA-seq on Drosophila tissues can be a complex and resource-intensive process, regardless of the sequencing platform. Therefore, it is important to consider the limits of these technologies. We will discuss two limitations in this section. Additional discussions of the limitations and comparisons of Smart-seq2 and 10× Genomics platforms can be found elsewhere (Baran-Gale et al., 2018; Wang et al., 2021; Jaeger et al., 2020).
One possible limitation is that nuclei contain only a fraction of the RNA content present within the entire cell, and snRNA-seq may not detect enough transcripts to characterize or distinguish cell types. We directly compared cells and nuclei of two different neuronal types profiled using Smart-seq2 and found that when we included intronic reads in our nuclear transcriptomes we detected 70%–90% of the genes detected using scRNA-seq, indicating that snRNA-seq has a higher dropout rate when compared to scRNA-seq (McLaughlin et al., 2021). Yet, we were able to use our nuclear dataset, obtained from either Smart-seq2 or 10× Genomics platforms, to differentiate closely related cell types (McLaughlin et al., 2021; Li et al., 2022).
Second, nuclear isolation causes the rupture of cells which can lead to ambient RNA from these cells adhering to the nuclei in suspension. This ambient RNA can be incorporated into droplets during FACS, barcoded, sequenced with a nucleus’ native RNA, and appear during data analysis. Unfortunately, this is a common phenomenon for single-cell and single-nucleus RNAseq and cannot be completely avoided from the experimental side. There are bioinformatic tools, like DecontX (Yang et al., 2020), that can subtract ambient RNA from the gene expression values.
Troubleshooting
Problem 1
Few nuclei or large clumps of tissue are present on slide when visualizing dissociated nuclei prior to sorting.
Potential solution
Increase the number of strokes with the tight pestle when releasing nuclei. We suggest increasing the stroke number by increments of 10 and observing the quality of the dissociation using Hoechst–33342 staining under a microscope. Increasing the stroke number too drastically can impair nuclear quality.
Problem 2
Following Smart-seq2 thermal cycling steps, the volume of each well in the 96 or 384 well plate is variable or inconsistent.
Potential solution
Vigorously run a film sealing roller or paddle over adhesive PCR film once applied to the plate. Pay close attention to the wells around the edges of the plate as they are most susceptible to evaporation during thermal cycling steps.
Problem 3
cDNA isolated from the Smart-seq2 protocol is of poor quality.
Potential solution
If the ERCC RNA spike-in included in the lysis buffer appears to be degraded, make new lysis plates. If ERCC spike-in appears to be amplified as expected, it is likely that the sample quality was poor. Ensure that: 1) samples are kept on ice from time of dissection through sorting; 2) sorting time does not exceed 20 min/plate; 3) once sorted, plates are quickly spun down and stored on ample dry ice; and 4) plates are kept on dry or wet (depending on the step) ice prior to RT reaction.
Problem 4
Not enough nuclei are collected after FACS.
Potential solution
Multiple sets of tissues can be dissected, flash-frozen and stored at –80°C. These samples can be later combined during the nuclei extraction step to increase total nucleus number. We have not observed detrimental effects from isolating nuclei from frozen tissue. Also, note that centrifugation steps will result in the loss of some nuclei.
Problem 5
There is no clear nucleus population during FACS setup.
Potential solution
First, take 10 μL of the nucleus suspension (before FACS), put it on a hemocytometer, and check the Hoechst staining for the presence of nuclei under a fluorescent microscope. If enough nuclei are in the suspension but cannot be visualized in the FACS, adjust the FACS settings, like the forward scatter detectors. Drosophila nuclei are small and may not be easily separated from debris based on size. To ensure that you are collecting nuclei, collect each sub-population of Hoechst-positive nuclei in the FACS plot and verify that they are indeed nuclei by visualizing them on a slide.
Problem 6
Nuclei stick together or form aggregates.
Potential solution
Make sure BSA is added in the nucleus resuspension buffer.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hongjie Li (hongjie.li@bcm.edu).
Materials availability
This study did not generate new or unique resources.
Acknowledgments
BioRender software was used to create the Graphical Abstract and Figure 1. We thank Yunming Wu for comments on the protocol. C.N.M. is a Howard Hughes Medical Institute Fellow of the Damon Runyon Cancer Research Foundation (DR2390-20). L.L. is supported by an NIH grant (DC-005982) and the Howard Hughes Medical Institute. L.L. and S.Q.R. are supported by Wu Tsai Neuroscience Institute (Neuro-omics program). H.L. is a CPRIT Scholar in Cancer Research (RR200063) and supported by National Institutes of Health (R00AG062746).
Author contributions
C.N.M. and Y.Q. designed, implemented, and wrote the protocol. H.L. conceived and designed the study. H.L., L.L., and S.R.Q. reviewed and edited the protocol.
Declaration of interests
The authors declare no competing financial interests.
Contributor Information
Stephen R. Quake, Email: steve@quake-lab.org.
Liqun Luo, Email: lluo@stanford.edu.
Hongjie Li, Email: hongjie.li@bcm.edu.
Data and code availability
This study did not generate new or unique code.
References
- Ariss M.M., Islam A.B.M.M.K., Critcher M., Zappia M.P., Frolov M.V. Single cell RNA-sequencing identifies a metabolic aspect of apoptosis in Rbf mutant. Nat. Commun. 2018;9:5024. doi: 10.1038/s41467-018-07540-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet Avalos C., Brugmann R., Sprecher S.G. Single cell transcriptome atlas of the drosophila larval brain. Elife. 2019;8:e50354. doi: 10.7554/eLife.50354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baran-Gale J., Chandra T., Kirschner K. Experimental design for single-cell RNA sequencing. Brief. Funct. Genom. 2018;17:233–239. doi: 10.1093/bfgp/elx035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croset V., Treiber C.D., Waddell S. Cellular diversity in the drosophila midbrain revealed by single-cell transcriptomics. Elife. 2018;7:e34550. doi: 10.7554/eLife.34550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie K., Janssens J., Koldere D., De Waegeneer M., Pech U., Kreft Ł., Aibar S., Makhzami S., Christiaens V., Bravo González-Blas C., et al. A single-cell transcriptome atlas of the aging Drosophila brain. Cell. 2018;174:982–998. doi: 10.1016/j.cell.2018.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Huang X., Zhang P., van de Leemput J., Han Z. Single-cell RNA sequencing identifies novel cell types in Drosophila blood. J. Genet. Genom. 2020;47:175–186. doi: 10.1016/j.jgg.2020.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese S., Clément R., Gaultier C., Besse F., Narbonne-Reveau K., Daian F., Foppolo S., Luis N.M., Maurange C. Coopted temporal patterning governs cellular hierarchy, heterogeneity and metabolism in drosophila Neuroblast tumors. Elife. 2019;8:e50375. doi: 10.7554/elife.50375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry G.L., Davis F.P., Picard S., Eddy S.R. Cell type-specific genomics of Drosophila neurons. Nucleic Acids Res. 2012;40:9691–9704. doi: 10.1093/nar/gks671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung R.J., Hu Y., Kirchner R., Liu Y., Xu C., Comjean A., Tattikota S.G., Li F., Song W., Ho Sui S., Perrimon N. A cell atlas of the adult Drosophila midgut. Proc. Natl. Acad. Sci. U.S.A. 2020;117:1514–1523. doi: 10.1073/pnas.1916820117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger B.N., Yángüez E., Gesuita L., Denoth-Lippuner A., Kruse M., Karayannis T., Jessberger S. Miniaturization of Smart-seq2 for single-cell and single-nucleus RNA sequencing. STAR Protoc. 2020;1:100081. doi: 10.1016/j.xpro.2020.100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji T., Zhang L., Deng M., Huang S., Wang Y., Pham T.T., Smith A.A., Sridhar V., Cabernard C., Wang J., Yan Y. Dynamic MAPK signaling activity underlies a transition from growth arrest to proliferation in Drosophila scribble mutant tumors. Dis. Model. Mech. 2019;1:dmm040147. doi: 10.1242/dmm.040147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaiskos N., Wahle P., Alles J., Boltengagen A., Ayoub S., Kipar C., et al. The Drosophila embryo at single-cell transcriptome resolution. Science. 2017;358:194–199. doi: 10.1126/science.aan3235. [DOI] [PubMed] [Google Scholar]
- Konstantinides N., Kapuralin K., Fadil C., Barboza L., Satija R., Desplan C. Phenotypic convergence: distinct transcription factors regulate common terminal features. Cell. 2018;174:622–635.e13. doi: 10.1016/j.cell.2018.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurmangaliyev Y.Z., Yoo J., Valdes-Aleman J., Sanfilippo P., Zipursky S.L. Transcriptional programs of circuit assembly in the Drosophila visual system. Neuron. 2020;108:1045–1057.e6. doi: 10.1016/j.neuron.2020.10.006. [DOI] [PubMed] [Google Scholar]
- Li H., Li T., Horns F., Li J., Xie Q., Xu C., Wu B., Kebschull J.M., McLaughlin C.N., Kolluru S.S., et al. Single-cell transcriptomes reveal diverse regulatory strategies for olfactory receptor expression and axon targeting. Curr. Biol. 2020;30:1189–1198.e5. doi: 10.1016/j.cub.2020.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Single-cell RNA sequencing in Drosophila: Technologies and applications. Wiley Dev Biol. 2020;e396 doi: 10.1002/wdev.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Horns F., Wu B., Xie Q., Li J., Li T., Luginbuhl D.J., Quake S.R., Luo L. Classifying Drosophila olfactory projection neuron subtypes by single-cell RNA sequencing. Cell. 2017;171:1206–1220.e22. doi: 10.1016/j.cell.2017.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Janssens J., De Waegeneer M., Kolluru S.S., Davie K., Gardeux V., Saelens W., David F.P.A., Brbić M., Spanier K., et al. Fly cell atlas: a single-nucleus transcriptomic atlas of the adult fruit fly. Science. 2022;375:eabk2432. doi: 10.1126/science.abk2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin C.N., Brbić M., Xie Q., Li T., Horns F., Kolluru S.S., Kebschull J.M., Vacek D., Xie A., Li J., et al. Single-cell transcriptomes of developing and adult olfactory receptor neurons in Drosophila. Elife. 2021;10:e63856. doi: 10.7554/elife.63856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S., Lund S., Pine P., et al. Assessing technical performance in differential gene expression experiments with external spike-in RNA control ratio mixtures. Nat Commun. 2014;5:5125. doi: 10.1038/ncomms6125. [DOI] [PubMed] [Google Scholar]
- Özel M.N., Simon F., Jafari S., Holguera I., Chen Y.C., Benhra N., El-Danaf R.N., Kapuralin K., Malin J.A., Konstantinides N., Desplan C. Neuronal diversity and convergence in a visual system developmental atlas. Nature. 2021;589:88–95. doi: 10.1038/s41586-020-2879-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S., Faridani O.R., Björklund Å.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Rust K., Byrnes L.E., Yu K.S., Park J.S., Sneddon J.B., Tward A.D., Nystul T.G. A single-cell atlas and lineage analysis of the adult Drosophila ovary. Nat. Commun. 2020;11:5628. doi: 10.1038/s41467-020-19361-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauc H.M., Rodriguez-Fernandez I.A., Hackney J.A., Pawlak M., Ronnen Oron T., Korzelius J., Moussa H.F., Chaudhuri S., Modrusan Z., Edgar B.A., Jasper H. Age-related changes in polycomb gene regulation disrupt lineage fidelity in intestinal stem cells. Elife. 2021;10:e62250. doi: 10.7554/elife.62250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattikota S.G., Perrimon N. Preparation of Drosophila larval blood cells for single-cell RNA sequencing. Bio Protoc. 2021;11:e4127. doi: 10.21769/BioProtoc.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., He Y., Zhang Q., Ren X., Zhang Z. Direct comparative analyses of 10X genomics Chromium and smart-seq2. Genom. Proteomics Bioinform. 2021;19:253–266. doi: 10.1016/j.gpb.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q., Brbic M., Horns F., Kolluru S.S., Jones R.C., Li J., Reddy A.R., Xie A., Kohani S., Li Z., et al. Temporal evolution of single-cell transcriptomes of drosophila olfactory projection neurons. Elife. 2021;10:e63450. doi: 10.7554/elife.63450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S., Corbett S.E., Koga Y., Wang Z., Johnson W.E., Yajima M., Campbell J.D. Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biol. 2020;21:57. doi: 10.1186/s13059-020-1950-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate new or unique code.