

Abstract
The effect of the presence of ammonia on [1-13C]glucose metabolism in the rumen fibrolytic bacterium Fibrobacter succinogenes S85 was studied by 13C and 1H nuclear magnetic resonance (NMR). Ammonia halved the level of glycogen storage and increased the rate of glucose conversion into acetate and succinate 2.2-fold and 1.4-fold, respectively, reducing the succinate-to-acetate ratio. The 13C enrichment of succinate and acetate was precisely quantified by 13C-filtered spin-echo difference 1H-NMR spectroscopy. The presence of ammonia did not modify the 13C enrichment of succinate C-2 (without ammonia, 20.8%, and with ammonia, 21.6%), indicating that the isotopic dilution of metabolites due to utilization of endogenous glycogen was not affected. In contrast, the presence of ammonia markedly decreased the 13C enrichment of acetate C-2 (from 40 to 31%), reflecting enhanced reversal of the succinate synthesis pathway. The reversal of glycolysis was unaffected by the presence of ammonia as shown by 13C-NMR analysis. Study of cell extracts showed that the main pathways of ammonia assimilation in F. succinogenes were glutamate dehydrogenase and alanine dehydrogenase. Glutamine synthetase activity was not detected. Glutamate dehydrogenase was active with both NAD and NADP as cofactors and was not repressed under ammonia limitation in the culture. Glutamate-pyruvate and glutamate-oxaloacetate transaminase activities were evidenced by spectrophotometry and 1H NMR. When cells were incubated in vivo with [1-13C]glucose, only 13C-labeled aspartate, glutamate, alanine, and valine were detected. Their labelings were consistent with the proposed amino acid synthesis pathway and with the reversal of the succinate synthesis pathway.
Fibrobacter succinogenes is a major rumen fibrolytic bacterium. It uses cellulose, glucose, and cellobiose as carbon and energy sources and ammonia as its sole nitrogen source (21). Ammonia is the prime source for protein synthesis in the rumen, accounting for the synthesis of 50 to 70% of bacterial nitrogen (13). Most of the studies of ammonia assimilation in the rumen have dealt with total ruminal content, and only a few papers have given detailed information on individual species (18, 23). In particular, the pathways of ammonia assimilation and amino acid synthesis in F. succinogenes are still unknown, although its carbon metabolism has been extensively studied (14, 15, 17). Glucose is metabolized through glycolysis into succinate, acetate, and a little formate. A part of the carbohydrates metabolized is stored as glycogen. Analysis by 13C and 1H nuclear magnetic resonance (NMR) was previously used to monitor in vivo the storage and degradation of glycogen in resting cells of different strains of F. succinogenes (8, 16), and a futile cycling of glycogen was shown: glycogen was simultaneously stored and degraded when bacteria were supplied with an exogenous carbon source. Furthermore, F. succinogenes was shown to accumulate glycogen throughout the exponential growth phase even when ammonia was not limiting (8, 16), whereas bacteria usually accumulate glycogen when ammonia (or another factor) limits growth (19). In addition to the futile glycogen cycle, a reversal of glycolysis and of the succinate synthesis pathway was found in F. succinogenes (16). These unusual features prompted us to investigate the relationship between ammonia assimilation and glycogen storage and also the effect of the presence of ammonia on the futile cycles. The enzymes responsible for ammonia assimilation and amino acid synthesis from intermediates of glucose metabolism were also sought.
The main pathways of ammonia assimilation were glutamate dehydrogenase (GLDH) and alanine dehydrogenase (ADH). The results obtained by 13C-NMR and 13C-filtered spin-echo difference (13C-FSED) 1H-NMR spectroscopy suggest a modulation of carbon metabolism by ammonia in F. succinogenes S85.
MATERIALS AND METHODS
Culture conditions.
F. succinogenes S85 (ATCC 19169) was grown for 15 h on a chemically defined medium (8) with 3 g of cellobiose per liter.
Preparation and incubation of cells.
For in vivo NMR experiments, cells were prepared as described by Matheron et al. (16). The cells harvested in the late log phase were spun (6,000 × g, 10 min, 4°C) and resuspended in a reduced 50 mM potassium phosphate–0.4% Na2CO3–0.05% cysteine buffer (pH 7.1). The cells at a final concentration of 5 mg of protein · ml−1 were incubated with 32 mM [1-13C]glucose, with or without 13 mM (NH4)2SO4, depending on the experiment. The cells were incubated at 37°C either in the spectrometer (in vivo NMR) or in a water bath and then sampled.
Each experiment was carried out at least three times with three different cultures.
Preparation of cell extracts.
For 13C-FSED 1H-NMR and 13C-NMR experiments, samples taken at the end of the incubation were frozen in liquid nitrogen and thawed three times. After being spun (15,000 × g, 10 min, 4°C) to remove the cell debris, the supernatants were analyzed by NMR.
For determination of enzyme activities (spectrophotometry or 1H NMR), cells in late log phase were collected by spinning (4,500 × g, 12 min, 4°C) and washed anaerobically in a sterile reduced buffer (50 mM potassium phosphate, 0.4% Na2CO3, 0.05% cysteine-HCl, 5 mM MgCl2 [pH 7.1]). The cells were disrupted anaerobically by sonication with a Branson Sonifier cell disrupter, model B15 (three treatments of 10 s each, 30 s apart), in an ice bath. After being spun (82,000 × g, 30 min, 5°C) in CO2-filled centrifuge tubes, supernatants were immediately used as cell extracts.
NMR spectroscopy.
NMR spectra were recorded on a Bruker MSL 300 spectrometer operating at 300.13 MHz for 1H NMR and 75.4 MHz for 13C NMR. The 2H resonance of D2O (10%) was used to lock the field and for shimming.
In vivo 13C-NMR experiments were performed at 37°C as previously described, with a 10-mm-diameter probe (14). In vitro 13C-NMR experiments were performed with a 5-mm-diameter probe (60° pulse, 2-s relaxation delay, 8 kilobytes of data, 1,000 scans). The low-power Waltz-16 proton-decoupling program was used to avoid sample heating. A pure benzene capillary, centered in the NMR tube, was used as an external reference resonating at 128.5 ppm from tetramethylsilane and to normalize the integrals of 13C-NMR spectra.
1H-NMR spectra, unless otherwise stated, were acquired with a 13C-FSED pulse sequence and a 5-mm-diameter inverse probe (1H-13C-15N) (16) by the following formula: preparation − (90°)H − τ/2 − (180°)H/(α)X − τ/2 − (90°)X − FID(1H), where the subscripts denote the nucleus experiencing the pulse (H, proton; X, 13C), τ is the evolution interval, α is pulse angle, and FID is free induction decay. The last carbon pulse is a purging pulse. Spectra with an α of 0 or 180° were acquired in subsequent scans and were stored independently in two blocks of memory. Extensive phase cycling was used to compensate for quadrature detection artifacts (CYCLOPS) and 180° pulse imperfections (EXORCYCLE). In a preparation period the solvent resonance was saturated by the DANTE pulse train [(γ/2π)B1 = 5,500 Hz; pulse duration, 15 μs; pulse repetition, 250 μs] for a period of 2 s. τ was adjusted according to the one-bond C,H constant for succinate and acetate [τ = 1/1J(C,H) = 7.2 ms]. After 8 dummy scans, 256 scans were accumulated in 8 kilobytes of memory. The acquisition time was 1.024 s, the spectral width was 4,000 Hz, and the scan repetition time was 3.03 s.
The determination of percentages of 13C enrichment of succinate and acetate is fully described in the work of Matheron et al. (16).
Enzyme activities.
The experimental conditions for the enzymatic assays are reported in Table 1. The assays for GLDH (reaction I), glutamine synthetase (GS) (reaction II), glutamate-pyruvate transaminase (reaction V), and glutamate-oxaloacetate (OAA) transaminase (reaction VI) activities were adapted from the work of Erfle et al. (7), except that the products of reactions II, V, and VI were detected by 1H NMR. Activity of ADH was assayed in a system similar to that in reaction I except that pyruvate was used in place of α-ketoglutarate.
TABLE 1.
Assay conditions for determination of enzyme activities
| Testa | Enzyme | Reactionb | Test conditionsc |
|---|---|---|---|
| Spectro | l-GLDH | I 1 | 8 mM αKG, 0.25 mM NAD(P)H, 90 mM AcNH4, 0.2 mg of protein/ml |
| I 2 | 8 mM glutamate, 0.25 mM NAD(P), 0.2 mg of protein/ml | ||
| NMR | GS | II 1 | 20 mM glutamate, 11 mM ATP or GTP, 80 mM AcNH4, 1.8 mg of protein/ml ± PLP |
| Spectro | l-ADH (0.2) | III 1 | 8 mM pyruvate, 0.25 mM NAD(P)H, 90 mM AcNH4, 0.2 mg of protein/ml |
| III 2 | 8 mM alanine, 0.25 mM NAD(P), 0.2 mg of protein/ml | ||
| NMR | Aspartase or ammonia lyase | IV 1 | 20 mM fumarate, 20 mM AcNH4, 1.8 mg of protein/ml ± 0.2 mM PLP |
| NMR | Glutamate-pyruvate transaminase | V 1 | 20 mM pyruvate, 20 mM glutamate, 1.8 mg of protein/ml ± 0.2 mM PLP |
| V 2 | 20 mM alanine, 20 mM αKG ± 0.12 mM PLP | ||
| Spectro | V 2 | 12 mM alanine, 12 mM αKG, 0.18 mM NADH, 12 U of lactate dehydrogenase, 0.5 mg of protein/ml ± 0.12 mM PLP | |
| NMR | Glutamate-OAA transaminase | VI 1 | 20 mM OAA, 20 mM glutamate, 0.9 mg of protein/ml ± 0.2 mM PLP |
| VI 2 | 20 mM aspartate, 20 mM αKG, 0.9 mg of protein/ml ± 0.12 mM PLP | ||
| Spectro | VI 2 | 12 mM aspartate, 12 mM αKG, 0.18 mM NADH, 12 U of malate dehydrogenase, 0.5 mg of protein/ml ± 0.12 mM PLP |
Spectro, reactions were monitored at 365 nm by spectrophotometry; NMR, reactions were monitored by 1H NMR at 300.13 MHz (5-mm-diameter 1H-13C-15N probe, 90° pulse, 3-s relaxation delay, 300 scans, 16 kilobytes of data, water suppression with a DANTE sequence).
1, forward reaction; 2, reverse reaction.
All the reactions were performed with 50 mM KH2PO4 (pH 7.5) at 37°C. Controls were carried out systematically without substrate or cofactor. AcNH4, ammonium acetate; αKG, α-ketoglutarate; PLP, pyridoxal phosphate.
Metabolite assays.
Protein concentration was determined by the Bradford method (3), with bovine serum albumin as the standard.
Succinate, acetate, and glucose were assayed with a Boehringer kit.
Ammonia was assayed by the phenol-hypochlorite reaction described by Weatherburn (24).
Glycogen was quantified by a glucose assay after hydrolysis with amyloglucosidase as previously described (16).
Chemicals.
[1-13C]glucose (99% labeled) was purchased from Eurisotop (Saint Aubin, France). All enzymes and chemicals were purchased from Sigma or Boehringer.
RESULTS
Effect of ammonia on glucose metabolism in F. succinogenes S85 cells.
Resting cells of F. succinogenes S85 (5 mg of protein · ml−1) were incubated with 32 mM [1-13C]glucose at 37°C in the absence of ammonia or in the presence of 26 mM ammonia. Kinetics of [1-13C]glucose utilization were monitored in vivo by 13C-NMR spectroscopy. Figure 1A presents a spectrum recorded after 27 min of incubation in the presence of ammonia. As in the absence of ammonia, disappearance of the two glucose anomer signals, β-[1-13C]glucose (96.4 ppm) and α-[1-13C]glucose (92.6 ppm), was associated with detection of signals of [2-13C]succinate (34.5 ppm), [2-13C]acetate (23.7 ppm), [6-13C]glycogen (61.0 ppm), and [1-13C]glycogen (100.1 ppm); the labeling at the C-6 position of glycogen results from glycolysis reversal after isomerization at the triose-phosphate level (8, 16). The ratio of the integrals of [1-13C] and [6-13C]glycogen (about 3) was not significantly affected by the presence of ammonia, indicating that ammonia did not modulate this reversal.
FIG. 1.

Proton-decoupled 13C-NMR spectra of 32 mM [1-13C]glucose utilization by resting cells of F. succinogenes (5 mg of protein · ml−1) incubated in the presence of 26 mM ammonia. (A) In vivo 13C-NMR spectrum registered after 27 min of incubation. (B) 13C-NMR spectrum of extracts of cells incubated for 35 min. Peaks: 1, [1-13C]glycogen; 2, β-[1-13C]glucose; 3, α-[1-13C]glucose; 4, [6-13C]glycogen; 5, [2-13C]aspartate; 6, [3-13C]aspartate; 7, [2-13C]succinate; 8, [2-13C]acetate; 9, [4-13C]valine; 10, [3-13C]alanine; 11, [2-13C]glutamate; 12, [2-13C]alanine, 13, [3-13C]glutamate.
Additional signals were observed both in the in vivo spectrum (Fig. 1A) and in the 13C-NMR spectrum (Fig. 1B) of extracts; they will be discussed later.
The time course of [1-13C]glucose utilization and metabolite production is presented in Fig. 2. In the presence of ammonia, glucose consumption was slowed by 34% (Fig. 2A) and [1-13C]glycogen storage decreased by 50% (Fig. 2B), with storage stopping after 15 min. The same was observed for [6-13C]glycogen (not shown). The rate of production of [2-13C]succinate was unchanged (Fig. 2C), while that of [2-13C]acetate was increased by 30% (Fig. 2D).
FIG. 2.

Time-dependent changes in signal integrals of β-[1-13C]glucose and α-[1-13C]glucose (A), [1-13C]glycogen (B), [2-13C]succinate (C), and [2-13C]acetate (D) during utilization of 32 mM [1-13C]glucose in the presence (○) or in the absence (●) of 26 mM ammonia, measured from in vivo 13C-NMR spectra.
In parallel, glucose and ammonia utilization, succinate and acetate production, and glycogen content were enzymatically or chemically assayed in supernatants or frozen and thawed extracts of cells incubated under similar conditions in a water bath at 37°C (Table 2). Glucose consumption calculated after 10 min of incubation (0.30 μmol · mg of protein−1 · min−1) was reduced by 33% (0.20 μmol · mg of protein−1 · min−1) in the presence of 26 mM ammonia. The ammonia consumption was 60 nmol · mg of protein−1 · min−1. The presence of ammonia had no effect on the kinetics of succinate synthesis, while the synthesis of acetate was increased 1.5-fold. The accumulation of glycogen was decreased twofold during the first 10 min and threefold by the end of the incubation.
TABLE 2.
Concentration of metabolites produced or substrates consumed by cells of F. succinogenes in the presence or absence of ammonia
| Time (min) | Consumption (mM) of:
|
Production (mM) of:
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Glucose
|
NH4+ | Acetate
|
Succinate
|
Glycogen
|
|||||
| Ca | NH4+b | C | NH4+ | C | NH4+ | C | NH4+ | ||
| 1 | 1.0 | 0.2 | 0.7 | 0.0 | 0.5 | 0.6 | 0.6 | 0.6 | 0.3 |
| 5 | 7.6 | 4.0 | 1.5 | 0.8 | 1.3 | 2.9 | 2.7 | 1.7 | 0.7 |
| 12 | 16.8 | 10.7 | 3.8 | 2.0 | 2.9 | 6.8 | 6.3 | 2.9 | 1.3 |
| 19 | 23.4 | 16.8 | 5.6 | 3.1 | 4.3 | 10.2 | 9.7 | 4.7 | 1.9 |
| 26 | 31.2 | 21.2 | 7.6 | 4.2 | 6.1 | 14.6 | 14.5 | 6.5 | 2.1 |
C, control experiment. Cells were incubated at 37°C with 32 mM glucose in the absence of ammonia. Metabolites and substrates were assayed in the supernatant.
Conditions were the same as those described under footnote a except that cells were incubated in the presence of 26 mM ammonia.
The results were analyzed in terms of the yields of metabolites relative to glucose consumption (Fig. 3). The addition of ammonia increased the rates of glucose conversion into succinate and acetate 1.4-fold and 2.2-fold, respectively (Fig. 3A and B), decreasing the ratio of succinate to acetate from 3.5 to 2.1. Storage of glycogen was halved (Fig. 3C). For example, the utilization of 20 mM glucose led to the synthesis of 8.3 mM succinate, 2.6 mM acetate, and 4.0 mM glycogen (glucose equivalent) in the absence of ammonia versus, respectively, 11.9, 5.5, and 1.95 mM in the presence of ammonia. Thus, the addition of ammonia modifies the relative proportions of the metabolites.
FIG. 3.

Production of succinate (A), acetate (B), and glycogen (C) relative to glucose consumption by resting cells incubated in the presence (○) or in the absence (●) of 26 mM ammonia. Metabolite concentrations were measured by enzymatic assays. Glycogen concentrations are given as glucose equivalents.
To determine whether the presence of ammonia influenced the futile glycogen cycling or the reversal of the succinate synthesis pathway previously observed (16), the succinate and acetate 13C enrichments were quantified at the end of the incubations by 1H NMR. We used a 13C-FSED pulse sequence previously developed by us (16) to improve the precision of the quantification (less than 1% error). The percentage of enrichment of succinate C-2 was not significantly changed by the presence of 26 mM ammonia (without ammonia, 20.8%, and with ammonia, 21.6% [Table 3]). This is consistent with the equivalent amounts of total succinate measured with and without ammonia (Table 2) and of [2-13C]succinate measured with and without ammonia (Fig. 2). We previously showed that the difference between the theoretical and the measured percentages of enrichment of succinate C-2 reflects the contribution of intracellular unlabeled glycogen to succinate production (16). With or without ammonia, this difference was 3.6 or 4.2%, respectively (Table 3), indicating that about 16% of the glucose entering glycolysis comes from prestored glycogen (see Fig. 6 in the work of Matheron et al. [16]).
TABLE 3.
Percentages of 13C enrichment of the CH3 of acetate and the CH2 of succinate
| Metabolite | Test conditionsa | % of labeling of C-2
|
Contribution (%) to the deficitb
|
|||
|---|---|---|---|---|---|---|
| Theoretical | Measured | Deficitb | Glycogen degradation | Reversal of succinate synthesis pathway | ||
| Succinate | C | 25 | 20.8 | 4.2 | 4.2 | |
| NH4+ | 25 | 21.6 | 3.6 | 3.6 | ||
| Acetate | C | 50 | 40 | 10 | 8.4c | 1.6 |
| NH4+ | 50 | 31 | 19 | 7.2c | 11.8 | |
C, control experiment. Cells were incubated for 35 min at 37°C with 32 mM [1-13C]glucose in the absence of NH4+. NH4+, ammonia experiment. Conditions were the same as those described for the control experiment but 26 mM NH4+ was present.
The deficit of labeling corresponds to the difference between the theoretical and experimental values. It reflects the contribution of glycogen degradation and of the reversal of the succinate synthesis pathways to the production of metabolites.
Deduced from the succinate value.
The percentage of enrichment of acetate was decreased from 40 to 31% by the presence of ammonia (Table 3), while the total amount of acetate increased in the enzymatic assays (Table 2). With acetate, both intracellular unlabeled glycogen degradation and the reversal of the succinate synthesis pathway contribute to the isotopic dilution of C-2 (16). The contribution due to the unlabeled glycogen can be determined from dilution of the enrichment of C-2 of succinate. Unlabeled glycogen contributes in the same proportion to succinate and acetate synthesis, leading to 8.4% of the 10% deficit of labeling of acetate C-2 in the incubation carried out in the absence of ammonia. Thus, 1.6% of this deficit is due to the reversal of the succinate synthesis pathway. In the presence of ammonia, unlabeled glycogen contributes 7.2% to the deficit of labeling and the reversal of the succinate synthesis pathway accounts for 11.8% of the deficit of labeling (Table 3). These results indicate that the presence of ammonia increases this reversal sevenfold.
13C-NMR experiments were performed with extracts from cells incubated with [1-13C]glucose with or without ammonia under conditions allowing detection of carboxylates (not shown). The signal of acetate C-1 (δ = 181.8 ppm) was increased in the presence of ammonia. This result is consistent with the increase in the reversal of the succinate pathway. We previously explained that because of scrambling at the fumarate level, the reversal gives rise to both [1-13C]acetate and [2-13C]acetate (16).
Ammonia assimilation and amino acid synthesis. (i) Enzyme activities.
Enzymes involved in the synthesis of amino acids from intermediates of carbon metabolism (α-ketoglutarate, pyruvate, OAA, and fumarate), either by direct amination from ammonia or by transamination, were sought in F. succinogenes sonicated extracts. Activities of dehydrogenases or transaminases were monitored by either spectrophotometry or 1H NMR (Table 4).
TABLE 4.
Investigation of enzymes involved in amino acid synthesis in F. succinogenes
| Reaction no. | Tested enzyme | Reactions | Sp act of enzyme with NH4+ concn (mM)a of:
|
NMRb | |
|---|---|---|---|---|---|
| 13 | 0.5 | ||||
| I | l-GLDH | α-Ketoglutarate + NH3 + NAD(P)H → l-glutamate + NAD(P)+ + H2O | 80 | 120 | |
| Reverse reaction | 7 | ||||
| II | GS | l-Glutamate + ATP or GTP + NH3 → l-glutamine + ADP or GDP + H2O + Pi | − | ||
| III | l-ADH | Pyruvate + NADPH + NH3 → l-alanine + NADP+ + H2O | 15 | ||
| Reverse reaction | 0 | ||||
| IV | Aspartase or ammonia lyase | Fumarate + NH3 → l-aspartate | − | ||
| V | Glutamate-pyruvate transaminase | Pyruvate + l-glutamate → l-alanine + α-ketoglutarate | + | ||
| Reverse reaction | 17.3 | + | |||
| VI | Glutamate-OAA transaminase | OAA + l-glutamate → l-aspartate + α-ketoglutarate | + | ||
| Reverse reaction | 36 | + | |||
Specific activities are expressed in nanomoles of synthesized products per minute and per milligram of protein. They are the averages of at least three values. The NH4+ concentration is that in the culture medium of the bacterium.
NMR means that the results were obtained from the analysis of 1H-NMR spectra.
The two main pathways of ammonia assimilation in bacteria are the amination of α-ketoglutarate by GLDH (reaction I) and the synthesis of glutamine from glutamate by GS (reaction II). The ammonia concentration in the culture medium may affect the respective contributions of the two enzymes to ammonia assimilation (18, 23). We therefore assayed the activities of the two enzymes in F. succinogenes cells cultured with two different ammonia concentrations: 0.5 and 13 mM. The specific activities of NAD-GLDH were 80 nmol · mg of protein−1 · min−1 in extracts of cells grown with 13 mM ammonia and 120 nmol · mg of protein−1 · min−1 in extracts of cells grown with 0.5 mM ammonia (Table 4). GLDH activities were equivalent with NADH or NADPH as the cofactor in extracts of cells grown with 13 mM ammonia. Under this condition, the reverse reaction was 10 times less efficient and occurred only with NADP as the cofactor (Table 4). Our results extend the previous conclusions of Joyner and Baldwin (12), who found only an NADP-GLDH activity in extracts of F. succinogenes S85.
The GS (reaction II) activities in extracts incubated with ammonia and ATP or GTP were investigated by 1H NMR. No resonance corresponding to the signals of glutamine was detected on the spectrum (not shown).
Activity of ADH (reaction III) was detected in the presence of NADPH (15 nmol · mg of protein−1 · min−1) (Table 4). The reverse reaction was not found.
Aspartase (reaction IV) activity in extracts incubated with fumarate and ammonia was investigated by 1H NMR. Under these conditions, only malate, resulting from the activity of fumarase, was detected on the spectrum (not shown).
Activities of the reverse reactions of glutamate-pyruvate transaminase (reaction V) and glutamate-OAA transaminase (reaction VI), measured by spectrophotometry, were 17.3 and 36 nmol · mg of protein−1 · min−1, respectively (Table 4). Both direct and reverse reactions were shown by 1H NMR (Table 4).
The spectra of incubations performed to show transaminations V and VI in the forward direction are presented in Fig. 4. On the spectrum in Fig. 4A, obtained at the end of an incubation with pyruvate and glutamate (reaction V), a singlet at 2.37 ppm, a multiplet at 2.09 ppm, and a triplet at 2.34 ppm were assigned to the protons bound to pyruvate C-3 and glutamate C-3 and C-4. Resonances centered at 1.46 ppm (doublet), 2.44 ppm (triplet), and at 3.01 ppm (triplet) were assigned, respectively, to protons borne by alanine C-3 and α-ketoglutarate C-3 and C-4, the transamination products. The doublet observed at 1.46 ppm is not symmetric, because it overlaps the singlet signal of a proton borne by C-3 of hydrated pyruvate at 1.48 ppm. The resonances of protons borne by the glutamate C-2 overlapped those of sugars (not shown).
FIG. 4.

1H-NMR spectra of extracts incubated for 30 min with 20 mM glutamate and 20 mM pyruvate (A) or 20 mM oxaloacetate (B). Peaks: 1, α-ketoglutarate; 2, pyruvate; 2′, hydrated form of pyruvate; 3, glutamate; 4, alanine; 5, aspartate.
At the end of the incubation (reaction VI) with OAA and glutamate (spectrum in Fig. 4B), resonances were assigned to protons borne by the carbon atoms of pyruvate, glutamate, and α-ketoglutarate and also by aspartate C-3 (doublets centered at 2.79 and 2.68 ppm, corresponding to the AB part of the ABX system). The resonances corresponding to OAA (initial substrate) were not observed on the spectrum in Fig. 4B owing to the very fast exchange between a proton and deuterium from D2O (enol form of OAA). An unexpected doublet centered at 1.46 ppm was assigned to the protons bound to alanine C-3. This signal was observed in a control experiment in which extracts were incubated only with aspartate (not shown). Alanine may result from decarboxylation of aspartate by an aspartate decarboxylase. Finally, the signal at 2.37 ppm, assigned to pyruvate, can be explained by transamination (reaction V) of the alanine coming from decarboxylation of aspartate and reacting with the α-ketoglutarate present in the incubation mixture.
(ii) In vivo observations.
In vivo 13C-NMR spectra of cells incubated with [1-13C]glucose in the presence of ammonia (Fig. 1A) showed signals that were identified as [3-13C]aspartate (37.0 ppm), [2-13C]aspartate (52.8 ppm), [3-13C]alanine (17.0 ppm), and [4-13C]valine (17.2 and 18.5 ppm). These resonances increased for the first 10 min and then remained at a constant level. No other amino acids were detected on the in vivo 13C-NMR spectra, suggesting that their intracellular concentrations were very low owing to their rapid incorporation into proteins. Additional signals were detected on spectra of extracts of the cells at the end of the incubation, recorded under conditions allowing higher sensitivity (Fig. 1B). They were assigned to [2-13C] and [3-13C]glutamate (55.08 and 27.35 ppm, respectively) and to [2-13C]alanine (51.4 ppm).
Modifications of the ammonia concentration in the incubation mixture and addition of essential volatile fatty acids (isobutyrate and valerate [5]) did not promote amino acid accumulation in the cells. Preincubation of cells for 20 min in the presence of tetracycline or chloramphenicol (1 mg/ml) did not change the observed amino acid pattern. No modification was induced even when cells were incubated in culture medium instead of buffer.
The labeling of C-3 of alanine and aspartate and of C-2 of glutamate was as expected from the metabolism of [1-13C]glucose via reactions III and V, reaction VI, and reaction I, respectively (Fig. 5). The labeling of C-4 of valine is consistent with the classical pathway of valine synthesis from two [3-13C]pyruvates as building blocks (9) (Fig. 5). Labeling of C-4 of glutamate was expected, resulting from the incorporation of [2-13C]acetyl coenzyme A and leading to the formation of α-ketoglutarate. The signal of the [4-13C]glutamate probably overlapped the signal of [2-13C]succinate.
FIG. 5.

Pathway of formation of succinate, acetate, and amino acids from [1-13C]glucose. EMP, Embden Meyerhof Parnas pathway. Reaction I, GLDH; reaction III, ADH; reaction V, glutamate-pyruvate transaminase; reaction VI, glutamate-OAA transaminase. Symbols: 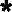 , 13C enrichment obtained via forward pathway; ▴, 13C enrichment obtained after reversion of the succinate synthesis pathway.
, 13C enrichment obtained via forward pathway; ▴, 13C enrichment obtained after reversion of the succinate synthesis pathway.
The unexpected labelings of C-3 of glutamate and of C-2 of aspartate and alanine are explained by the reversal of the succinate synthesis pathway through scrambling at the fumarate level leading to [2-13C] and [3-13C]OAA and to [2-13C] and [3-13C]pyruvate. [2-13C]OAA gives rise to [2-13C]aspartate (reaction VI). [2-13C]alanine can result from either [2-13C]pyruvate (Fig. 5) or decarboxylation of [2-13C]aspartate (see above). The labeling of valine C-2 and C-3, also expected from the reversal of the succinate pathway (Fig. 5), was not detected on the 13C-NMR spectrum (Fig. 4). However, given the low intensities of the C-4 signals of valine, the signal-to-noise ratio would be too low to allow detection of C-2 and C-3 signals.
DISCUSSION
Our results show the activity of a reversible GLDH in F. succinogenes S85. In the reaction leading to glutamate synthesis, GLDH activity was measured with NADH or NADPH as the cofactor in sonicated extracts of cells grown in a medium containing 0.5 or 13 mM ammonia.
We found no GS activity in F. succinogenes S85. The enzyme may be present in the cells, but its activity may be very low and therefore undetected by 1H NMR. It is also possible that the synthesis of glutamine in F. succinogenes occurs via asparagine synthetase, replacing GS in a coupled system similar to that of GS-glutamine:α-oxoglutarate aminotransferase (GOGAT), as shown previously for other rumen bacteria (7).
We showed the synthesis of alanine from pyruvate and ammonia (ADH activity [reaction III]), with NADPH as the cofactor. The reverse reaction was not detected.
GLDH and ADH thus seem to be the main pathways of direct assimilation of ammonia in F. succinogenes. These systems are generally considered low-affinity systems for the integration of ammonia, while the GS-GOGAT couple is the highest-affinity enzyme system (23). In the rumen, GS is thought to play a major role when the ammonia concentration is low while GLDH is thought to be predominant when the rumen ammonia concentration is high (18, 23). The rumen ammonia concentration varies with diet, from 1 mM with a low-protein diet to 40 mM, transiently, with a nitrogen-rich diet (23). However, enzyme activities measured in individual rumen bacterial species do not always fit this general assumption and suggest that GLDH may be the main pathway of ammonia assimilation. For example, in Selenomonas ruminantium cultured with low concentrations of ammonia, GS activity was predominant but GLDH was still active and never repressed and GOGAT was not overexpressed (20). In Ruminobacter amylophilus, although ammonia limitation resulted in repression of GLDH and stimulation of GS, GOGAT was not detected (11). In Ruminococcus flavefaciens, GLDH was induced in ammonia-limited cultures (6), and in Streptococcus bovis, GLDH activity was much higher than GS activity (10).
In the nonruminal species Bacteroides fragilis, NAD- and NADP-dependent GLDH activities with high affinities for ammonia (Km = 0.8 mM) and a negligible GS activity were found. GLDH activity was not repressed under ammonia limitation (25). These results recall those obtained with F. succinogenes.
The role of ADH in ammonia assimilation in the rumen is not yet fully understood, although enriched alanine was the main labeled amino acid when rumen microbial cells were incubated with 15NH4+ (2). However, results obtained with pure cultures may differ significantly from results obtained with rumen content. Assimilation of 15NH4+ by pure culture of the predominant noncellulolytic bacteria Prevotella bryantii B14, Selenomonas ruminantium HD4, and Streptococcus bovis ES1 was shown to be affected by the addition of amino acids or peptides to the culture medium, but glutamate and aspartate were always formed de novo to a greater extent than the other amino acids, including alanine (1).
The two transaminases most commonly found in rumen microorganisms (23), i.e., glutamate-pyruvate and glutamate-OAA transaminases, were found in F. succinogenes by both enzymatic assays and 1H NMR. This last approach is very easy and straightforward, allowing unambiguous identification of the reaction products.
In in vivo incubations in the presence of ammonia and [1-13C]glucose, only a few amino acids were detected by 13C NMR, suggesting a very efficient incorporation of amino acids into proteins in F. succinogenes cells. The detected amino acids were aspartate, glutamate, alanine, and valine. Aspartate results from the activity of glutamate-OAA transaminase (reaction VI), glutamate results from GLDH activity (reaction I) or from transaminations (reactions V and VI), alanine can be synthesized from ADH (reaction III) or glutamate-pyruvate transaminase (reaction V) activity or from decarboxylation of aspartate, and valine can be synthesized, as in many bacteria, from the condensation of two pyruvates (9) via α-acetolactate. This intermediate was previously identified in F. succinogenes extracts (17).
The most important result in this work concerns the modification of fluxes of carbon metabolism induced by the presence of ammonia. When ammonia was added to resting cells metabolizing glucose, the flux to glycogen synthesis was decreased and those to acetate and succinate syntheses were increased. We showed that, under these conditions, NH4+ was consumed and that amino acids and macromolecules were synthesized, probably in place of glycogen. The presence of ammonia also modified the ratio of succinate to acetate in favor of acetate. The increase in the rate of conversion of glucose into acetate may be explained by the need of ATP for the synthesis of amino acids, ATP being directly provided by the acetate synthesis pathway. This increase in acetate production at the expense of succinate may result from the reversal of the succinate synthesis pathway (see below).
Although glycogen synthesis was decreased by 50%, the reversal of glycolysis from triose-phosphate (16) was not modified by the presence of ammonia, as the ratio of the integrals of [1-13C] and [6-13C]glycogen (about 3) was not affected by the presence of ammonia. The storage of [1-13C]glycogen results from incorporation of [1-13C]glucose-6-phosphate (G 6P) synthesized either directly from [1-13C] glucose or after reversal of glycolysis, whereas the storage of [6-13C]glycogen results only from reversal of glycolysis. The constancy of the ratio of [1-13C]glycogen to [6-13C]glycogen indicates that the flux of G 6P entering the pathway of glycogen synthesis with or without reversal of glycolysis is not modulated by ammonia, suggesting a mechanism of regulation of this flux.
The analysis of the percentage of 13C enrichment of succinate C-2, measured by 13C-FSED 1H NMR, indicated that the contribution of glycogen to the synthesis of the metabolites was not modified by the presence of ammonia and therefore that futile glycogen cycling was still effective under this condition. With or without ammonia, about 16% of the glucose molecules entering glycolysis come from prestored glycogen. However, the percentage of 13C enrichment of C-2 of acetate was decreased from 40 to 31%. This result was explained by an increase (sevenfold) in the flux through the reverse succinate pathway. This reversal was confirmed (i) by the labeling of the carboxylate of acetate and (ii) by the labeling of the amino acids. The labeling of aspartate C-2 reflects the labeling of OAA C-2 and thus proves the reversal from fumarate to OAA (Fig. 5). The labeling of acetate C-1, previously observed in the absence of ammonia (16), reflects the labeling of pyruvate C-2 (Fig. 5). Pyruvate can be synthesized from OAA via two routes: either directly through the activity of OAA-decarboxylase or indirectly through the combined activities of phosphoenolpyruvate-carboxykinase and pyruvate kinase. OAA-decarboxylase and phosphoenolpyruvate-carboxykinase activities could not be measured in F. succinogenes extracts owing to the very high activity of malate dehydrogenase (unpublished results). Consequently, we do not yet know which of these two enzymes is involved in this step.
Such a reversal from OAA to pyruvate was also observed in rabbit kidney tubule (4). It was due to OAA-decarboxylase activity and was greatly stimulated by the addition of ammonia to the cells. In these cells, the addition of NH4Cl also increased glucose conversion into metabolites (4). These results recall those obtained with F. succinogenes cells and reflect the regulation of carbon metabolism by ammonia.
Until now, no information concerning nitrogen metabolism in F. succinogenes has been available. Our work shows some of the enzymes involved in ammonia assimilation and amino acid synthesis in this bacterium. Furthermore, the use of 13C and 1H NMR enabled us to quantify the modulation of carbon fluxes by ammonia. Also, we showed that the futile cycles previously found in F. succinogenes, i.e., reversal of glycolysis, simultaneous synthesis and degradation of glycogen, and reversal from fumarate to pyruvate, were still effective in the presence of ammonia, under conditions closer to physiological conditions. It would now be interesting to determine whether these futile cycles are present under conditions where succinate does not accumulate, such as in the rumen. For this purpose, we could apply a similar approach to coincubations of cells of F. succinogenes and of a succinate-utilizing strain such as Selenomonas ruminantium (22).
ACKNOWLEDGMENTS
This work was supported by a grant to C.M. from the Centre National de la Recherche Scientifique and the Région Auvergne. T.L. was an invited professor at the Université Blaise Pascal of Clermont-Ferrand.
REFERENCES
- 1.Atasoglu C, Valdés C, Walker N D, Newbold C J, Wallace R J. De novo synthesis of amino acids by the ruminal bacteria Prevotella bryantii B14, Selenomonas ruminantium HD4, and Streptococcus bovis ES1. Appl Environ Microbiol. 1998;64:2836–2843. doi: 10.1128/aem.64.8.2836-2843.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blake J S, Salter D N, Smith R H. Incorporation of nitrogen into rumen bacterial fractions of steers given protein- and urea-containing diets. Ammonia assimilation into intracellular bacterial amino acids. Br J Nutr. 1983;50:769–782. doi: 10.1079/bjn19830148. [DOI] [PubMed] [Google Scholar]
- 3.Bradford M M. A rapid sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 4.Chauvin M-F, Mégnin-Chanet F, Martin G, Lhoste J-M, Baverel G. The rabbit kidney tubule utilizes glucose for glutamine synthesis. A 13C NMR study. J Biol Chem. 1994;269:26025–26033. [PubMed] [Google Scholar]
- 5.Dehority B A, Scott H W, Kowaluk P. Volatile fatty acid requirements of cellulolytic rumen bacteria. J Bacteriol. 1967;94:537–543. doi: 10.1128/jb.94.3.537-543.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duncan P A, White B A, Mackie R I. Purification and properties of NADP-dependent glutamate dehydrogenase from Ruminococcus flavefaciens FD-1. Appl Environ Microbiol. 1992;39:4032–4037. doi: 10.1128/aem.58.12.4032-4037.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erfle J D, Sauer F D, Mahadevan S. Effect of ammonia concentration on activity of enzymes of ammonia assimilation and on synthesis of amino acids by mixed rumen bacteria in continuous culture. J Dairy Sci. 1977;60:1064–1072. doi: 10.3168/jds.s0022-0302(77)83989-1. [DOI] [PubMed] [Google Scholar]
- 8.Gaudet G, Forano E, Dauphin G, Delort A-M. Futile cycling of glycogen in Fibrobacter succinogenes as shown by in situ 1H NMR and 13C NMR investigation. Eur J Biochem. 1992;207:155–162. doi: 10.1111/j.1432-1033.1992.tb17032.x. [DOI] [PubMed] [Google Scholar]
- 9.Gottschalk G. Bacterial metabolism. New York, N.Y: Springer-Verlag; 1985. [Google Scholar]
- 10.Griffith C J, Carlsson J. Mechanism of ammonia assimilation in streptococci. J Gen Microbiol. 1974;82:253–260. doi: 10.1099/00221287-82-2-253. [DOI] [PubMed] [Google Scholar]
- 11.Jenkinson H F, Buttery P J, Lewis D. Assimilation of ammonia by Bacteroides amylophilus in chemostat cultures. J Gen Microbiol. 1979;113:305–313. [Google Scholar]
- 12.Joyner A E, Baldwin R L. Enzymatic studies of pure cultures of rumen microorganisms. J Bacteriol. 1966;92:1321–1330. doi: 10.1128/jb.92.5.1321-1330.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mackie R L, Kistner A. Some frontiers of research in basic ruminant nutrition. S Afr J Anim Sci. 1985;15:72–75. [Google Scholar]
- 14.Matheron C, Delort A-M, Gaudet G, Forano E. Simultaneous but differential metabolism of glucose and cellobiose in Fibrobacter succinogenes cells, studied by in vivo13C NMR. Can J Microbiol. 1996;42:1091–1099. doi: 10.1139/m96-140. [DOI] [PubMed] [Google Scholar]
- 15.Matheron C, Delort A-M, Gaudet G, Forano E. Re-investigation of glucose metabolism in Fibrobacter succinogenes, using NMR spectroscopy and enzymatic assays. Evidence for pentose phosphate phosphoketolase and pyruvate formate lyase activities. Biochim Biophys Acta. 1997;1355:50–60. doi: 10.1016/s0167-4889(96)00118-8. [DOI] [PubMed] [Google Scholar]
- 16.Matheron C, Delort A-M, Gaudet G, Forano E, Liptaj T. 13C and 1H nuclear magnetic resonance study of glycogen futile cycling in strains of the genus Fibrobacter. Appl Environ Microbiol. 1998;64:74–81. doi: 10.1128/aem.64.1.74-81.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller T L. The pathway of formation of acetate and succinate from pyruvate by Bacteroides succinogenes. Arch Microbiol. 1978;117:145–152. doi: 10.1007/BF00402302. [DOI] [PubMed] [Google Scholar]
- 18.Morrison M, Mackie R I. Nitrogen metabolism by ruminal microorganism: current understanding and future perspective. Aust J Agric Res. 1996;47:227–246. [Google Scholar]
- 19.Preiss J. Bacterial glycogen synthesis and its regulation. Annu Rev Microbiol. 1984;38:419–458. doi: 10.1146/annurev.mi.38.100184.002223. [DOI] [PubMed] [Google Scholar]
- 20.Smith C J, Hespell R B, Bryant M P. Regulation of urease and ammonia assimilatory enzymes in Selenomonas ruminantium. Appl Environ Microbiol. 1981;42:89–96. doi: 10.1128/aem.42.1.89-96.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stewart C S, Flint H J. Bacteroides (Fibrobacter) succinogenes, a cellulolytic anaerobic bacterium from the gastrointestinal tract. Appl Microbiol Biotechnol. 1989;30:433–439. [Google Scholar]
- 22.Strobel H J, Russel J B. Succinate transport by a ruminal selenomonad and its regulation by carbohydrate availability and osmotic strength. Appl Environ Microbiol. 1991;57:248–254. doi: 10.1128/aem.57.1.248-254.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wallace R J, Onodera R, Cotta M A. Metabolism of nitrogen-containing compounds. In: Hobson P N, Stewart C S, editors. The rumen microbiol ecosystem. London, United Kingdom: Blackie Academic and Professional; 1997. pp. 661–684. [Google Scholar]
- 24.Weatherburn M W. Phenol-hypochlorite reaction for determination of ammonia. Anal Chem. 1967;39:971–974. [Google Scholar]
- 25.Yamamoto L A, Sabe A, Saito H, Ishimoto M. The pathway of ammonia assimilation in Bacteroides fragilis. J Gen Microbiol. 1984;30:499–508. [Google Scholar]