

Abstract
Linker histone H1.2 (H1.2), encoded by HIST1H1C (H1C), is a major H1 variant in somatic cells. Among five histone H1 somatic variants, upregulated H1.2 was found in human hepatocellular carcinoma (HCC) samples and in a diethylnitrosamine (DEN)‐induced HCC mouse model. In vitro, H1.2 overexpression accelerated proliferation of HCC cell lines, whereas H1.2 knockdown (KD) had the opposite effect. In vivo, H1.2 insufficiency or deficiency (H1c KD or H1c KO) alleviated inflammatory response and HCC development in DEN‐treated mice. Mechanistically, H1.2 regulated the activation of signal transducer and activator of transcription 3 (STAT3), which in turn positively regulated H1.2 expression by binding to its promoter. Moreover, upregulation of the H1.2/STAT3 axis was observed in human HCC samples, and was confirmed in mouse models of methionine‐choline‐deficient diet induced nonalcoholic steatohepatitis or lipopolysaccharide induced acute inflammatory liver injury. Disrupting this feed‐forward loop by KD of STAT3 or treatment with STAT3 inhibitors rescued H1.2 overexpression‐induced proliferation. Moreover, STAT3 inhibitor treatment‐ameliorated H1.2 overexpression promoted xenograft tumor growth. Therefore, H1.2 plays a novel role in inflammatory response by regulating STAT3 activation in HCC, thus, blockade of the H1.2/STAT3 loop is a potential strategy against HCC.
Keywords: feed‐forward loop, hepatocellular carcinoma, histone H1.2, inflammation, STAT3
This study showed that histone H1.2 was upregulated in hepatocellular carcinoma samples and promoted hepatocarcinogenesis by regulating signal transducer and activator of transcription 3 (STAT3) activation. STAT3 in turn upregulated H1.2 by binding to its promoter to form a feed‐forward H1.2/STAT3 loop. The H1.2/STAT3 loop was further identified in inflammatory liver injury models.

Abbreviations
- BP
BP‐1‐102
- CT
control
- DEN
diethylnitrosamine
- FOS
Fos proto‐oncogene, AP‐1 transcription factor subunit
- H1C
HIST1H1C
- HCC
hepatocellular carcinoma
- HFD1D
male offspring under high‐fat diet plus DEN stress
- HFD2D
male offspring from maternal obesity under high‐fat diet plus DEN stress
- HFD3D
male offspring from maternal and grandmaternal obesity under high‐fat diet plus DEN stress
- IHC
immunohistochemistry
- IL‐6
interleukin‐6
- KD
knockdown
- KO
knockout
- KPNA2
karyopherin subunit alpha 2
- LPS
lipopolysaccharide
- luc.
luciferase
- MCD
methionine‐choline‐deficient
- NC
normal chow
- NCD
normal chow plus DEN
- NS
not significant
- OD
optical density
- qPCR
quantitative real‐time PCR
- S3I
S3I‐201
- SOCS3
suppressor of cytokine signaling 3
- STAT3
signal transducer and activator of transcription 3
- TC‐PTP
T‐cell protein tyrosine phosphatase
- TIM23
translocase of inner mitochondrial membrane 23
- TSS
transcription start site;Vec, vector
- Vec
vector
1. INTRODUCTION
Liver cancer is the fourth leading cause of death worldwide, with HCC the most dominant type. 1 Over the past decade, neither surgery nor chemotherapy has proven appreciably effective in improving HCC outcomes. 2 Hence, understanding the mechanisms behind HCC and identifying effective therapeutic targets are urgently required.
Genetic and epigenetic alterations all contribute to the development and progression of HCC. 3 Compared to epigenetic alterations, such as DNA methylation and histone modifications, linker histone H1 alterations are less investigated in HCC. 4 The linker histone H1 family, well known as chromatin structural proteins, consists of five somatic variants (H1.1–H1.5) in mammals. 5 In most somatic cells, histone H1.2 coded by H1C is one of the predominant variants. 5 Although KO of H1.2 in mouse shows no anatomic or histological abnormality, 6 its roles in apoptosis, 7 cell cycle progression, 8 autophagy, 9 and DNA damage repair 10 have been reported, mostly in cultured cells. Recently, H1.2/H1.4 double‐KO has been found to accelerate lymphomas in mouse 11 ; conditional H1.2/H1.3/H1.4 triple‐KO in hematopoietic cells decreases lymphocyte proliferation in mouse. 12 Nevertheless, the role of histone H1 variants, including H1.2 per se, in the development of HCC remains unclear in vivo.
Signal transducer and activator of transcription 3, a pivotal transcription factor that regulates inflammation and innate immunity, plays critical roles in multiple physiological processes, including cell growth, survival, metastasis and angiogenesis. 13 Activation of STAT3 requires phosphorylation on tyrosine 705 (Y705), which mediates its homodimerization, nuclear translocation, and transcription of downstream target genes, including IL‐6, FOS, and SOCS3. 14 Nuclear trafficking of STAT3 is essential to its function, and its nuclear translocation has been reported to be regulated by various importins, including KPNA2. 15 A Y640F mutation that causes constitutive Y705 phosphorylation by stabilizing STAT3 homodimerization (constitutive activation) is found in multiple cancers, especially in inflammation‐associated tumorigenesis, such as HCC. 16 , 17 Therefore, elucidation of new factors that regulate STAT3 activation will reveal insights on HCC development.
Here, we reported a H1.2/STAT3 feed‐forward loop in the development of HCC. H1.2 was upregulated in HCC mouse or human samples that promoted cell proliferation. Knockout or KD of H1.2 markedly alleviated DEN‐induced liver carcinogenesis and proliferation of HCC cell lines by downregulating STAT3 activation. Moreover, disrupting this H1.2/STAT3 axis attenuated the proliferation of HCC cells in vitro and in vivo.
2. MATERIALS AND METHODS
2.1. Mice and experimental design
The Hist1h1c (H1c) KO mice were generated by Biocytogen using a CRISPR/Cas9 based system. Two single guide RNAs were designed to target the upstream of the 5′‐UTR and 3′‐UTR of Hist1h1c exon 1, respectively, using the CRISPR design tool (http://www.sanger.ac.uk/htgt/wge/) (Figure S1). Genotyping was undertaken by PCR as we previously described 18 , 19 with the primers listed in Table S1. Representative genotyping results are shown in Figure S1B. Male BALB/c nude mice were obtained from Hunan SJA Laboratory Animal Co. Ltd. Mice were maintained in a specific pathogen‐free, temperature controlled (22 ± 1°C) animal facility with a 12/12‐h light/dark cycle, and free access to water and food. Animals were handled according to the Guidelines of China Animal Welfare Legislation, and approved by the Committee on Ethics in the Care and Use of Laboratory Animals of the College of Life Sciences, Wuhan University.
For the DEN‐induced HCC mouse model, male mice were intraperitoneally injected with 25 mg/kg DEN (Sigma) at 2 weeks old and killed at 40 weeks old. 20 , 21 Tumors with diameter of 3 mm or more were counted and measured, and liver and serum were collected. Liver samples of mice treated with LPS, and HCC samples of high fat diet plus DEN‐treated male offspring from a multigenerational maternal obesity model were collected as we previously reported. 18 , 21 For the nonalcoholic steatohepatitis model, mice were fed an MCD diet (HFK Bioscience) for 4 weeks.
2.2. Histological, immunohistochemical and immunofluorescent studies
Paraffin‐embedded mouse liver samples were sectioned and stained with H&E. 22 For immunohistochemical studies, mouse liver sections were incubated overnight at 4°C with primary Abs for H1.2, Ki‐67, F4/80, CD3, or Ly6G; a human HCC tissue microarray comprising 15 pairs of tumor and paratumor tissues (Outdo Biotech), was incubated overnight at room temperature with Ab for H1.2 or p‐STAT3Y705. Detailed information regarding the Abs used is provided in Table S2. Positive staining was visualized by 3,3’‐diaminobenzidine substrate following the ABC kit (both from Vector Laboratories). For mouse liver sections, positively stained areas or cells were quantified using ImagePro Plus software (Media Cybernetics) based on four to six different randomly selected fields per sample. For human HCC tissue microarray, a semiquantitative analysis was carried out to evaluate H1.2 or p‐STAT3Y705 levels in a double‐blinded fashion and scored from 0 to 5, which correspond to the percentage of positively stained cells per field (0%, 1%–20%, 20%–40%, 40%–60%, 60%–80%. and 80%–100%, respectively). Pearson’s correlation coefficient analysis between H1.2 and p‐STAT3Y705 levels was carried out using GraphPad Prism version 8.0.
Immunofluorescent staining was undertaken as we previously reported. 23 Mouse liver sections or HepG2 cells were incubated with primary Abs for H1.2, or TIM23 (a protein located in the inner mitochondrial membrane 24 ), or p‐STAT3Y705, with images taken by a TCS SP8 confocal microscope (Leica). Detailed information on Abs used is provided in Table S2.
2.3. RNA sequencing
Total RNA of tumor samples from different groups was isolated and prepared for RNA sequencing as we previously described. 19 , 21 Sequencing was undertaken by Genewiz, with details provided in the supporting information.
2.4. Cell culture, primary hepatocyte isolation, plasmids and treatments
HepG2, Huh7, Hep3B, HCC‐LM3 (LM3), and 293T cells were obtained from China Center for Type Culture Collection. HepG2, Huh7, Hep3B, and 293T cells were cultured in DMEM (Hyclone), and LM3 cells were cultured in MEM (Hyclone), supplemented with 10% FBS (Lonsera) and 1% penicillin–streptomycin (Hyclone). To knock down H1.2, HepG2 or Hep3B cells were transiently transfected with either scrambled shRNA or two different shRNAs targeting H1C (Table S1). To overexpress H1.2, Huh7 or LM3 cells were transiently transfected with pRK‐(h)H1.2‐Flag or pLVX‐(h)H1.2, respectively. To knock down or overexpress STAT3, cells were transiently transfected with two different shRNAs targeting STAT3 (Table S1), WT STAT3 (STAT3‐Flag), a constitutive activated STAT3 (STAT3Y640F‐Flag), or a transactivation‐deficient STAT3 (STAT3Y705F‐Flag), respectively. Primary hepatocytes were isolated from male mice as we described previously. 25 For IL‐6 treatment, cells were starved overnight in medium without FBS, then treated with 50 ng/ml recombinant human IL‐6 (Peprotech) for the indicated times.
2.5. Luciferase reporter assay
Human H1C‐WT or H1C‐mut (mutant, three putative STAT3 binding sites confirmed by ChIP assays (−1423/−1413, −701/−691, and −162/−152) and three sites with the highest predictive scores (−876/866, −963/953, and −329/319) were mutated to A) promoter (−2000/0 relative to the TSS) was cloned into the pGL3‐Basic vector (Promega). Luciferase assays were carried out using a dual‐specific luciferase assay kit (Promega). To evaluate the effect of H1.2 on the activity of STAT3 promoter, cells were transfected with a STAT3 firefly luciferase reporter plasmid together with STAT3Y640F, and shScram or shH1C; to evaluate the effect of STAT3 on the activity of H1C promoter, cells were transfected with H1C‐WT or H1C‐mut firefly luciferase reporter plasmid, together with shScram or shSTAT3. All cells were cotransfected with pRL‐TK (Renilla luciferase reporter plasmid) for normalization. Luciferase assays were undertaken at 48 h after transfection. 19
2.6. Colony formation and MTT assays
Cells were transiently transfected with indicated plasmids; 48 h later, cells were stimulated with or without 50 μM S3I (Selleck) or 10 μM BP (TargetMol) for another 72 h. Standard MTT and colony formation assays were carried out as we described previously. 21 , 26
2.7. Tumor xenograft
Both flanks of nude mice (6 weeks old) were subcutaneously injected with 1 × 107 LM3 cells transfected with empty vector or pLVX‐H1.2, and counted as day 0. S3I (5 mg/kg body weight) was injected intraperitoneally daily from day 9 after cell injection. Tumor sizes were measured every other day. Mice were killed at day 17, tumors were removed and weighed, and volumes were calculated as described. 21 , 27
2.8. Western blot analyses and qPCR
Freshly isolated liver or cultured cells were sonicated in ice‐cold RIPA buffer (Beyotime) and protein concentrations were determined. Western blots were probed with respective primary Abs (Table S2), visualized by enhanced chemiluminescence (Advansta), and quantitated using Quantity One software (Bio‐Rad). The relative levels of targeted proteins were quantitated to the respective loading control in the same sample.
Total RNA was extracted using RNAiso Plus (TaKaRa Biotech.). RNA from each sample was reverse transcribed into cDNA using an M‐MLV First Strand Kit (Invitrogen). Actb for human cell lines or Rn18s for mouse samples was used as internal control. Primers used are provided in Table S1.
2.9. Coimmunoprecipitation and ChIP
Coimmunoprecipitation and ChIP assays were carried out as we previously described. 18 , 28 , 29 Primers and Abs used are provided in Tables S1 and S2, and three different regions of human H1C or mouse H1c promoter ranging from −2000 bp to the TSS were chosen.
2.10. Database analysis
The Cancer Genome Atlas (https://cancergenome.nih.gov/) was used to evaluate the expression of H1.1–H1.5 in HCC patients. Bioinformatic software JASPAR (http://jaspar.genereg.net/cgi‐bin/jaspar_db.pl) was used to analyze STAT3 binding sites within the promoters of human H1C or mouse H1c.
2.11. Statistical analysis
Results were expressed as the mean ± SD. All the cell experiments were repeated at least three times. Data were analyzed using the Kruskal–Wallis test followed by the Mann–Whitney test for more than two‐group comparison, and the Mann–Whitney test was used for two‐group comparison. Differences were considered statistically significant at p < 0.05.
3. RESULTS
3.1. Histone H1.2 is upregulated in liver of HCC patients and mice
H1.2 has the highest transcriptional level among somatic H1 variants in human or mouse liver (Figure S2A,B). To investigate whether H1.2 plays a role in HCC development, the expression profiles of histone H1 variants in Th Cancer Genome Atlas database were analyzed. Significantly upregulated mRNA level of H1C, but not other somatic H1 variants, was found in HCC patients (Figures 1A and S2C). Furthermore, compared to paratumor tissues, significantly higher nuclear H1.2 staining was found in human HCC samples (Figure 1B). Similarly, significantly increased H1.2 levels were found in the tumors of DEN‐treated mice, compared to that in the liver of age‐ and sex‐matched controls; most of H1.2 staining was found in the nuclei but not in the mitochondria (suggested by costaining with DAPI or TIM23, respectively) (Figures 1C and S2D,E). Furthermore, in DEN‐treated mice, significantly higher H1.2 levels were observed in tumors compared with the paratumor tissues (Figure 1D).
FIGURE 1.

Histone H1.2 is elevated in hepatocellular carcinoma (HCC) tumor samples. (A) HIST1H1C (H1C) levels in human HCC tumor (T, red; n = 369) or normal liver tissue (N, gray; n = 160). TPM, transcripts per kilobase of exon model per million mapped reads. (B) Representative images of H1.2 staining on human HCC tumor (T, n = 15) and paratumor (P, n = 15) samples (left panels), with corresponding scores (right panel). Scale bar, 100 μm. (C) Representative western blots (left panel) and quantification results (right panel) of H1.2 in normal mouse livers (N, n = 5) and tumors (T, n = 6) from diethylnitrosamine (DEN)‐treated mice. (D) Representative western blots and quantification results of H1.2 in tumors (T, n = 6) and matched paratumor samples (P, n = 6) from DEN‐treated mice. Results were expressed as mean ± SD. *p < 0.05, **p < 0.01
3.2. Downregulation of H1.2 mitigates HCC development in DEN‐treated mice
To explore whether H1.2 is an oncogenic factor for HCC, we challenged the H1c knockout (H1c KO) or insufficient (H1c KD) mice, as well as their WT littermates, with DEN (Figure 2A,B). In DEN‐stressed mice, KD or KO of H1c caused a significant reduction in tumor number and maximum tumor size compared to their WT littermates (Figure 2C–F). Consistently, the number of Ki‐67 (a cell proliferation marker 30 ) positive cells and the mRNA level of Ccna2 (a cell cycle regulator 31 ) were significantly reduced in the liver of H1c KD and H1c KO mice under DEN stress (Figure 2G–I).
FIGURE 2.

Loss of histone H1.2 alleviates hepatocellular carcinoma tumorigenesis in mice. (A, B) H1c knockdown (KD) or knockout (KO) efficiency at mRNA (A) and protein (B) levels in the livers of H1c+/+ (WT), H1c+/− (H1c KD), and H1c−/− (H1c KO) mice. Bands were from the same gel (or blot); black dotted lines delineate the splicing boundary. (C–F) Representative images of dissected livers (C) and H&E stained liver sections (D), and quantification results of tumor number (E) and maximal (Max) tumor size (F) of the indicated groups. Arrows indicate tumors, dotted lines indicate the outline of tumors. Scale bar, 100 μm. (G, H) Representative images of Ki‐67 staining (G) with quantification results (H) in mouse liver tumors. Scale bar, 50 μm. (I) Ccna2 mRNA level. WT group, n = 6; H1c KD group, n = 12; H1c KO group, n = 10. Results are expressed as mean ± SD. *p < 0.05, **p < 0.01. P, paratumor; T, tumor
The role of H1.2 in HCC progression was next verified in vitro. Among four HCC cell lines we examined, HepG2 and Hep3B cells have higher, whereas LM3 and Huh7 cells have lower, H1.2 protein levels (Figure S3A). Thus, H1.2 was successfully knocked down in HepG2 or Hep3B cells, but overexpressed in LM3 or Huh7 cells (Figure S3B–E). Significantly decreased cell proliferation was indicated by colony formation and MTT assays in H1.2 KD HepG2 and Hep3B cells (Figure S3F–I). Consistently, increased cell proliferation was observed in H1.2 overexpressed LM3 and Huh7 cells (Figure S3J–M). These results suggested that H1.2 promoted both hepatocarcinogenesis in vivo and cell proliferation of HCC cell lines in vitro.
3.3. Downregulation of H1.2 suppresses hepatic inflammation and STAT3 signaling in DEN‐treated mice
To investigate how H1.2 promotes HCC development, RNA sequencing was carried out using tumor samples. Compared to WT mice, 4973 and 2342 differentially expressed genes (>1.5‐fold change) were identified in H1c KD and H1c KO mice, respectively (Figure 3A, Tables S3 and S4); among 1789 overlapped genes, most genes were associated with inflammatory response (Figures 3A,B and S4A, Table S5). Therefore, two classic inflammation‐related signaling pathways, STAT3 and nuclear factor‐κB, were further examined. Knockout or KD of H1c significantly inhibited phosphorylation of STAT3Y705 but not total STAT3 level in tumor and paratumor tissues (Figure 3C,D), and showed no effect on the level of phosphorylated p65 or IκBα (Figure S4B,C). Furthermore, the transcriptional levels of several STAT3 targeted genes, such as Il‐6, Socs3, Fos, and Mcl‐1 (MCL1 apoptosis regulator), were decreased in H1c KD or H1c KO mice compared to those of WT mice (Figure 3E). Consistently, the cell number of positively stained F4/80 (a macrophage marker 32 ), or CD3 (a T cell marker 33 ), or Ly6G (a neutrophil marker 34 ), was significantly lower in the liver of H1c KD or H1c KO mice compared to that of the WT mice following DEN stress (Figure 3F). Collectively, these results indicated that downregulated STAT3 activation and associated inflammation could contribute to tumor suppression in H1.2 insufficient and deficient mice.
FIGURE 3.
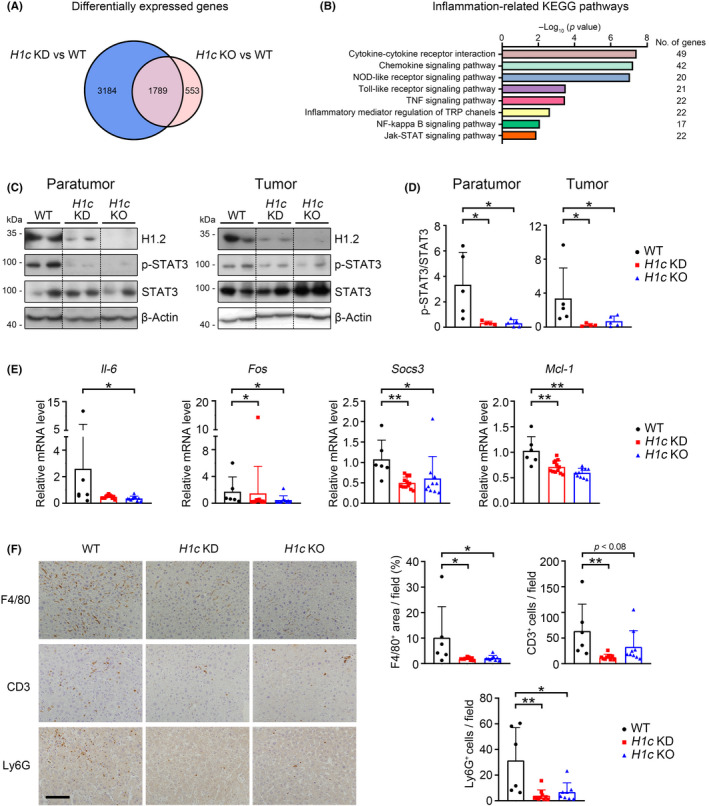
Loss of histone H1.2 suppresses hepatic inflammation by downregulating signal transducer and activator of transcription 3 (STAT3) signaling in diethylnitrosamine‐treated mice. (A) Venn diagram showing differentially expressed genes. (B) Top eight inflammation‐related Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways commonly enriched in H1c knockdown (KD) and H1c knockout (KO) groups. (C, D) Western blot results (C) of H1.2, p‐STAT3, and STAT3 with quantification results (D) in liver paratumors and tumors. Bands were from the same gel (or blot); black dotted lines delineate the splicing boundary. E, mRNA levels of Il‐6, Fos, Socs3, and Mcl‐1. F, Representative images of F4/80, CD3, and Ly6G staining (left panels) with quantification results (right panels). Scale bar, 100 μm. Results expressed as mean ± SD. *p < 0.05, **p < 0.01
3.4. H1.2 regulates activation of STAT3
We next examined whether H1.2 regulates STAT3 activation. In H1.2 KD HepG2 cells or H1.2 KO mouse primary hepatocytes, IL‐6‐induced upregulation of p‐STAT3Y705 was inhibited (Figure 4A,B), and IL‐6‐induced transcriptions of STAT3 targeted genes were downregulated (Figure 4C,D). Luciferase reporter assay confirmed STAT3Y640F as a constitutive active form of STAT3, suggested by the dramatically elevated STAT3 transcriptional activity, as previously reported, 16 which was attenuated by KD of H1.2 (Figure 4E). Furthermore, KD of H1.2 inhibited IL‐6‐induced nuclear translocation of p‐STAT3Y705 (Figure 4F). Overexpression of H1.2 not only significantly promoted STAT3Y705 phosphorylation, but also upregulated STAT3 targeted genes, such as IL‐6, FOS, or SOCS3, with or without IL‐6 stimulation (Figure 4G,H).
FIGURE 4.

Histone H1.2 regulates signal transducer and activator of transcription 3 (STAT3) activity. (A, B) Western blots (left panel) with quantification results (right panel) of H1.2 and p‐STAT3 in HepG2 cells (A) and mouse primary hepatocytes (B) treated with or without 50 ng/ml interleukin‐6 (IL‐6) for 90 min (n = 3). (C) mRNA levels of FOS and IL‐6 in HepG2 cells (n = 3). D, mRNA levels of Fos and Socs3 in mouse primary hepatocytes (n = 3). (E) Effect of H1.2 knockdown on STAT3 activation in 293T cells with or without transfection of STAT3Y640F (n = 3–6). (F) Representative images of p‐STAT3 in shScram‐ and shH1.2‐transfected Hep3B cells treated with or without IL‐6. Scale bar, 50 μm. (G) Western blots of H1.2 and p‐STAT3 (top panel) with quantification results of p‐STAT3 (lower panel) in LM3 cells overexpressing H1.2 treated with or without IL‐6 (n = 3). (H) Quantitative PCR results of HIST1H1C, IL‐6, FOS, and SOCS3 in Huh7 cells overexpressing H1.2 treated with or without IL‐6 (n = 3). I, Western blots of H1.2 and STAT3 in the nucleus and cytoplasm of 293T cells. (J) Immunoblot (IB) analysis of H1.2 and Flag‐tagged karyopherin subunit alpha 2 (KPNA2) in 293T cells transfected with vector or KPNA2‐Flag. Results expressed as mean ± SD from three independent experiments. *p < 0.05, **p < 0.01. CT, control; KO, knockout; luc., luciferase; n.s., not significant; Vec, vector
To explore how H1.2 regulates STAT3, we first investigated whether H1.2 might regulate some secretory factor(s) that affect STAT3 phosphorylation. Medium from shScram or shH1.2 transfected HepG2 cells, with or without IL‐6 treatment, were collected and used to treat HepG2 cells (Figure S5A). Compared with the observation that H1.2 KD downregulated p‐STAT3Y705 level (Figure 4A), no obvious effect on p‐STAT3Y705 level was found in cells treated with medium from H1.2 KD cells (Figure S5B). Next, we found the effects of H1.2 KD on the p‐STAT3Y705 level were retained after inhibiting protein translation by cycloheximide (Figure S5C). Coimmunoprecipitation studies indicated no interaction between H1.2 and TC‐PTP, which regulates STAT3 dephosphorylation in the nucleus 35 (data not shown), while no significant alteration in total TC‐PTP level was found following H1.2 KD, with or without IL‐6 treatment (Figure S5D). As nuclear localization of STAT3 has been reported to protect p‐STAT3 from dephosphorylation, 36 we then investigated whether H1.2 might regulate STAT3 by affecting its nuclear translocation. H1.2 KD increased STAT3 level in cytoplasm and reduced nuclear STAT3 level (Figure 4I), and coimmunoprecipitation showed interaction between H1.2 and KPNA2, an importin mediating nuclear translocation of STAT3 (Figure 4J). 15 Together, these results suggested that H1.2 could regulate STAT3 activation by affecting its nuclear translocation, but not by a noncell‐autonomous manner, or by regulating new protein synthesis or TC‐PTP level.
3.5. HIST1H1C is a target of activated STAT3
We also noticed that IL‐6 treatment significantly upregulated the H1.2 protein levels in HepG2 cells or mouse primary hepatocytes (Figure 4A,B), which made us wonder how IL‐6 affects H1.2 levels. In addition to STAT3 activation, a time‐dependent upregulation of H1.2 was also observed in IL‐6‐treated primary mouse hepatocytes (Figure 5A). Significantly increased protein level of H1.2 was also observed after IL‐6 treatment in HepG2 cells, and most of H1.2 was found in the nuclei but not in the mitochondria (Figures 5B and S6A). As STAT3 is a transcription factor regulating multiple genes, 14 we hypothesized that H1C might also be regulated by STAT3. By analyzing the promoter region (from −2000 bp to the TSS) using JASPAR software (Figure 5C), 40 potential STAT3 binding sites were suggested. A ChIP assay further confirmed accumulation of STAT3 on the promoters of human H1C and mouse H1c, while IL‐6 stimulation increased bindings (Figures 5D–G and S6B,C). Luciferase reporter assays showed that STAT3 KD greatly reduced human H1C promoter activity by 63%–71%, while mutating six putative STAT3‐binding sites similarly reduced the activity of H1C promoter (Figure 5H). Further STAT3 KD only showed mild effects on mutant H1C promoter activity (16%–24%) (Figure 5H). These results suggested H1C as a direct target of STAT3.
FIGURE 5.
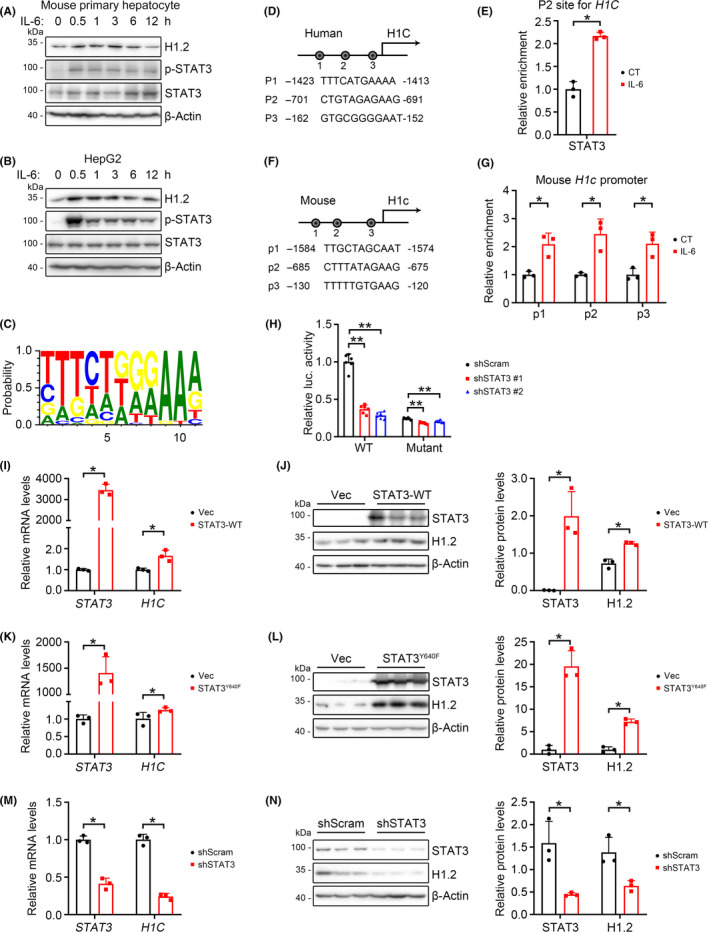
Histone H1.2 is regulated by signal transducer and activator of transcription 3 (STAT3). (A, B) Western blots of H1.2, p‐STAT3, and STAT3 in mouse primary hepatocytes (A) and HepG2 cells (B) treated with interleukin‐6 (IL‐6) for indicated times. (C) Potential STAT3 binding sites on H1C promoter. (D, E) Representative potential STAT3‐binding sites within the 2.0‐kb region upstream of the transcription start site (D) and STAT3 enrichment (E) on HIST1H1C promoter in 293T cells. (F, G) Representative potential STAT3‐binding sites within the 2.0‐kb region upstream of the transcription start site (F) and STAT3 enrichment (G) on Hist1h1c promoter in mouse primary hepatocytes. (H) Relative luciferase (luc.) activity of H1C in 293T cells transfected with indicated plasmids. (I, J) Quantitative PCR (qPCR) (I) and western blot with quantification (J) of H1.2 and STAT3 in 293T cells following transfection of WT STAT3. (K, L) qPCR (K) and western blot with quantification (L) of H1.2 and STAT3 in 293T cells transfected with STAT3Y640F. (M, N) qPCR (M) and western blot with quantification (N) of H1.2 and STAT3 in shScram‐ or shSTAT3‐transfected 293T cells. n = 3 for each group. Results expressed as mean ± SD from three independent experiments. *p < 0.05, **p < 0.01. CT, control; Vec, vector
The mRNA and protein levels of H1.2 were upregulated in 293T cells (a cell line with high transfection efficiency) overexpressing WT STAT3 or STAT3Y640F (Figure 5I–L). Furthermore, either mRNA or protein levels of H1.2 were significantly downregulated in the STAT3 KD 293T cells (Figure 5M,N). Consistently, similar results were found in HepG2 cells when overexpressing STAT3Y640F or knocking down STAT3 (Figure S5D–G).
3.6. H1.2 is positively correlated with p‐STAT3 level in hepatic inflammation
To confirm upregulation of a H1.2/STAT3 axis in hepatic inflammatory stresses, we further examined the correlation between H1.2 and p‐STAT3 in MCD diet‐induced nonalcoholic steatohepatitis or LPS‐induced acute inflammatory liver injury. Significantly upregulated nuclear H1.2 levels, indicated by immunohistochemistry, and significantly upregulated H1.2 and p‐STAT3Y705 levels, indicated by western blots, were found in the liver of MCD‐ or LPS‐treated mice (Figure 6A–D). Moreover, we investigated H1.2 and p‐STAT3Y705 levels in tumor samples from DEN‐induced male offspring from a multigenerational maternal obesity model, which has shown gradually increased hepatic inflammation over generations, as we previously reported. 21 , 37 , 38 Under DEN stress, compared with normal chow or high‐fat diet treated male offspring from lean mothers (NCD or HFD1D), the H1.2 and p‐STAT3Y705 levels of high‐fat diet treated male offspring from obese mothers or obese grandmothers (HFD2D or HFD3D) were both significantly upregulated (Figure 6E). Importantly, in human HCC samples, a positive correlation between the levels of nuclear H1.2 and p‐STAT3 was found (Figure 6F).
FIGURE 6.

H1.2 level is correlated with signal transducer and activator of transcription 3 (STAT3) phosphorylation following hepatic inflammation. (A, B) Representative nuclear H1.2 staining with quantification (A) and western blots with quantification of H1.2, p‐STAT3, and STAT3 (B) in mice injected with lipopolysaccharide (LPS) (n = 3). (C, D) Representative images of H1.2 and H&E stained liver sections, with quantification of nuclear H1.2 levels (C) and western blots with quantification of H1.2, p‐STAT3, and STAT3 (D) in methionine‐choline‐deficient diet (MCD) fed mice (n = 3). (E) Western blots with quantification of hepatic H1.2, p‐STAT3, and STAT3 in a multigenerational maternal obesity mice model. n = 3 for each group. HFD1/2/3D, first/second/third generations of male offspring under high‐fat diet + diethylnitrosamine (DEN); NCD, normal chow + DEN. F, Representative images of hepatic p‐STAT3 staining in tumor (T) and paratumor (P) sections in human hepatocellular carcinoma tissue microarray (top panels), and correlation between H1.2 and p‐STAT3 levels based on staining scores analyzed by Pearson correlation analysis; n = 30 for each group. Results expressed as mean ± SD. *p < 0.05. CT, control; IHC, immunohistochemistry; NC, normal chow
3.7. H1.2 promotes cell proliferation in a STAT3 activity‐dependent manner
To verify whether H1.2 promotes HCC development by regulating STAT3 activity, H1.2 was overexpressed in LM3 cells transfected with transactivation‐deficient STAT3Y705F (Figure S7A,B). The MTT and colony formation assays showed that abrogation of STAT3 transactivation significantly inhibited the H1.2 overexpression‐promoted cell proliferation (Figure 7A,B). Similarly, STAT3 deficiency also prevented H1.2 overexpression‐induced cell proliferation (Figures 7C,D and S7C). Furthermore, two STAT3 inhibitors, 39 , 40 S3I and BP, effectively rescued cells from H1.2 overexpression‐induced proliferation (Figures 7E,F and S7D–F). We further investigated whether S3I, an inhibitor with low in vivo toxicity, 41 restrains the HCC promoting effect of H1.2 in nude mice. Consistent with in vitro results, S3I treatment significantly decreased the H1.2 overexpressing‐induced tumor volume and weight in nude mice (Figure 7G–I). Collectively, these results indicated that H1.2 promoted development of HCC by regulating STAT3 activity.
FIGURE 7.
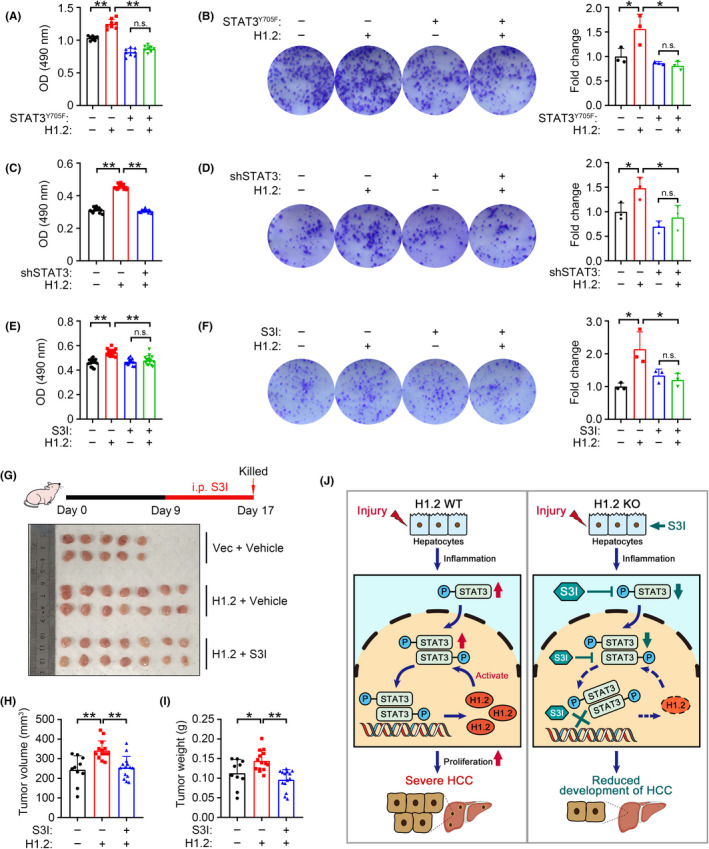
Tumor‐promoting effect of histone H1.2 depends on signal transducer and activator of transcription 3 (STAT3) activity. (A, B) MTT assay (A, n = 15) and representative images with quantification results (B, n = 3) of colony formation in LM3 cells transfected with or without H1.2 and STAT3Y705F. (C, D) MTT assay (C, n = 15) and representative images with quantification (D, n = 3) of colony formation in LM3 cells transfected with or without H1.2 and shSTAT3. (E, F) MTT assay (E, n = 15) and representative images with quantification results (F, n = 3) of colony formation in H1.2‐overexpressing LM3 cells under S3I‐201 (S3I) treatment. (G) Experimental design flowchart (top panel) and dissected xenograft tumors (bottom panel). (H, I) Tumor volume (H) and weight (I) of xenograft tumors from indicated groups. (J) A proposed model for the role of H1.2/STAT3 loop in regulating hepatocellular carcinoma (HCC). Results expressed as mean ± SD from three independent experiments. *p < 0.05, **p < 0.01. KO, knockout; n.s., not significant; OD, optical density; Vec, vector
4. DISCUSSION
In this study, we identified that H1.2 was the most abundant linker histone among five somatic H1 variants in human and mouse liver (Figure S2). H1.2, rather than other variants, upregulated and aggravated hepatocarcinogenesis (Figures 1 and 2). As a replication‐dependent linker histone variant, 5 increased levels of H1.2 in HCC samples are reasonable due to greatly increased numbers of dividing cells in liver cancer; it is interesting that the other four replication‐dependent variants were not upregulated in HCC samples (Figure S2). The physiological/pathological impacts of such H1 variant‐specific alterations in HCC and the underlying mechanisms are worthy of further investigation.
To investigate the physiological roles of H1 variants, multiple lines of single/double/triple H1 variant KO mice have been constructed. Interestingly, single or double KO of H1.2, H1.3, or H1.4 in mouse do not cause overt pathological phenotypes, 6 whereas triple KO of them in mouse leads to embryonic lethality. 42 In our study, although single H1.2 KO did not cause obvious phenotypical changes under normal conditions, carcinogenesis stress revealed its role in HCC (Figure 2). Therefore, an experimental strategy that combines variant KO with a disease model could be applicable to explore the specific physiological/pathological role of individual H1 variants in vivo.
Recently, double KO of H1.2/H1.4 mouse, which shows no obvious pathological phenotype under normal conditions, 42 has been reported to initiate the expression of stem cell genes in germinal center B cells and cause aggressive lymphomas in a diffuse large B‐cell lymphoma model. 11 In contrast, conditional H1.2/H1.3/H1.4 triple KO in hematopoietic cells upregulates genes that control apoptosis and negatively regulate cell proliferation, thus limiting lymphocyte proliferation in mouse. 12 These seemingly opposite phenotypes after H1.2 KO could implicate cell type‐specific or genetic background‐dependent effects of H1.2 on tumorigenesis.
Here, we identified that H1.2 suppressed inflammatory response in the liver by regulating the level of p‐STAT3Y705 and its downstream genes (Figure 3). Previously, we have reported that upregulation of H1.2 is associated with increased expression of inflammatory factors in diabetic retinopathy 9 ; however, we were unclear whether H1.2 directly regulated inflammation. In this study, we showed that H1.2 directly affected inflammatory response in hepatocytes by regulating the protein level and nuclear distribution of activated form of STAT3 (p‐STAT3Y705), subsequently influencing its transcriptional activity (Figure 4). Similar pro‐inflammatory roles of hepatic STAT3 have also been found in other liver diseases, such as viral hepatitis, nonalcoholic steatohepatitis, and chemical‐induced liver injury. 43 , 44 , 45 Consistently, upregulation of the H1.2/STAT3 axis was also found in MCD‐induced nonalcoholic steatohepatitis and LPS‐induced acute inflammatory liver injury (Figure 6). Considering the commonly found constitutive STAT3 activation in many cancers, 46 the existence and impacts of the H1.2/STAT3 axis in other types of cancer will be interesting for further exploration.
As H1.2 locates and is upregulated in the nuclei of HCC tissues and cell lines examined (Figures 2C,D and S6A), it is plausible that H1.2 might regulate STAT3 activity by affecting some nuclear events. Our results showed that H1.2 interacts with KPNA2 (Figure 4J), an importin regulating the nuclear translocation of STAT3. 47 However, H1.2 did not interact with TC‐PTP, a nuclear phosphatase regulating STAT3 dephosphorylation 35 (data not shown). Consistently, H1.2 KD increased STAT3 levels in cytoplasm and reduced nuclear STAT3 levels (Figure 4I). Thus, it would be interesting to investigate how H1.2 mediates STAT3 nuclear translocation through KPNA2.
In conclusion, we found that, following DEN induction, upregulated H1.2 drives HCC development by regulating STAT3 activation to promote inflammatory response and cell proliferation. Activated STAT3 in turn transcriptionally regulates H1.2 to form a feed‐forward H1.2/STAT3 loop, and disrupting the H1.2/STAT3 axis attenuated HCC cell proliferation, which suggests a potential therapeutic target for HCC (Figure 7J).
DISCLOSURE
The authors declare that they have no conflict of interest.
Supporting information
Supplementary Material
Table S3
Table S4
Table S5
ACKNOWLEDGMENTS
The STAT3‐related plasmids were kind gifts from Dr YY Wang, Chinese Academy of Sciences. This work was supported by the Natural Science Foundation of China (91957114, 31871381, and 32021003), the National Key R&D Program of China (2018YFA0800700 and 2019YFA0802701), and the Natural Science Foundation of Hubei Province (2021CFA004).
Wang Q, Chen Y, Xie Y, et al. Histone H1.2 promotes hepatocarcinogenesis by regulating signal transducer and activator of transcription 3 signaling. Cancer Sci. 2022;113:1679–1692. doi: 10.1111/cas.15336
Contributor Information
Kun Huang, Email: kunhuang@hust.edu.cn.
Ling Zheng, Email: lzheng@whu.edu.cn.
REFERENCES
- 1. Likhitsup A, Parikh ND. Economic implications of hepatocellular carcinoma surveillance and treatment: a guide for clinicians. Pharmacoeconomics. 2020;38(1):5‐24. doi: 10.1007/s40273-019-00839-9 [DOI] [PubMed] [Google Scholar]
- 2. Anwanwan D, Singh SK, Singh S, Saikam V, Singh R. Challenges in liver cancer and possible treatment approaches. Biochim Biophys Acta Rev Cancer. 2020;1873(1):188314. doi: 10.1016/j.bbcan.2019.188314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pogribny IP, Rusyn I. Role of epigenetic aberrations in the development and progression of human hepatocellular carcinoma. Cancer Lett. 2014;342(2):223‐230. doi: 10.1016/j.canlet.2012.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Scaffidi P. Histone H1 alterations in cancer. Biochim Biophys Acta. 2016;1859(3):533‐539. doi: 10.1016/j.bbagrm.2015.09.008 [DOI] [PubMed] [Google Scholar]
- 5. Happel N, Warneboldt J, Hanecke K, Haller F, Doenecke D. H1 subtype expression during cell proliferation and growth arrest. Cell Cycle. 2009;8(14):2226‐2232. doi: 10.4161/cc.8.14.8982 [DOI] [PubMed] [Google Scholar]
- 6. Fan Y, Sirotkin A, Russell RG, Ayala J, Skoultchi AI. Individual somatic H1 subtypes are dispensable for mouse development even in mice lacking the H1(0) replacement subtype. Mol Cell Biol. 2001;21(23):7933‐7943. doi: 10.1128/MCB.21.23.7933-7943.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Konishi A, Shimizu S, Hirota J, et al. Involvement of histone H1.2 in apoptosis induced by DNA double‐strand breaks. Cell. 2003;114(6):673‐688. [DOI] [PubMed] [Google Scholar]
- 8. Munro S, Hookway ES, Floderer M, et al. Linker histone H1.2 directs genome‐wide chromatin association of the retinoblastoma tumor suppressor protein and facilitates its function. Cell Rep. 2017;19(11):2193‐2201. doi: 10.1016/j.celrep.2017.05.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang W, Wang Q, Wan D, et al. Histone HIST1H1C/H1.2 regulates autophagy in the development of diabetic retinopathy. Autophagy. 2017;13(5):941‐954. doi: 10.1080/15548627.2017.1293768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Z, Li Y, Tang M, et al. Destabilization of linker histone H1.2 is essential for ATM activation and DNA damage repair. Cell Res. 2018;28(7):756‐770. doi: 10.1038/s41422-018-0048-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yusufova N, Kloetgen A, Teater M, et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature. 2020;589:299‐305. doi: 10.1038/s41586-020-3017-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Willcockson MA, Healton SE, Weiss CN, et al. H1 histones control the epigenetic landscape by local chromatin compaction. Nature. 2020;589:293‐298. doi: 10.1038/s41586-020-3032-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao B, Wang H, Lafdil F, Feng D. STAT proteins ‐ key regulators of anti‐viral responses, inflammation, and tumorigenesis in the liver. J Hepatol. 2012;57(2):430‐441. doi: 10.1016/j.jhep.2012.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu Z, Han Y, Liu Y, et al. CREBZF as a key regulator of STAT3 pathway in the control of liver regeneration in mice. Hepatology. 2019;71(4):1421‐1436. doi: 10.1002/hep.30919 [DOI] [PubMed] [Google Scholar]
- 15. Zhou KX, Huang S, Hu LP, et al. Increased nuclear transporter KPNA2 contributes to tumor immune evasion by enhancing PD‐L1 expression in PDAC. J Immunol Res. 2021;2021:6694392. doi: 10.1155/2021/6694392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pilati C, Amessou M, Bihl MP, et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J Exp Med. 2011;208(7):1359‐1366. doi: 10.1084/jem.20110283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jerez A, Clemente MJ, Makishima H, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T‐cell large granular lymphocyte leukemia. Blood. 2012;120(15):3048‐3057. doi: 10.1182/blood-2012-06-435297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang YU, Xue W, Zhang W, et al. Histone methyltransferase G9a protects against acute liver injury through gstp1. Cell Death Differ. 2020;27(4):1243‐1258. doi: 10.1038/s41418-019-0412-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu C, Wang J, Wei Y, et al. Fat‐specific knockout of Mecp2 upregulates Slpi to reduce obesity by enhancing browning. Diabetes. 2020;69(1):35‐47. doi: 10.2337/db19-0502 [DOI] [PubMed] [Google Scholar]
- 20. Liu S, Sun Y, Jiang M, et al. Glyceraldehyde‐3‐phosphate dehydrogenase promotes liver tumorigenesis by modulating phosphoglycerate dehydrogenase. Hepatology. 2017;66(2):631‐645. doi: 10.1002/hep.29202 [DOI] [PubMed] [Google Scholar]
- 21. Sun Y, Wang Q, Zhang Y, et al. Multigenerational maternal obesity increases the incidence of HCC in offspring via mir‐27a‐3p. J Hepatol. 2020;73(3):603‐615. doi: 10.1016/j.jhep.2020.03.050 [DOI] [PubMed] [Google Scholar]
- 22. Wang C, Xiong M, Yang C, et al. PEGylated and Acylated Elabela analogues show enhanced receptor binding, prolonged stability, and remedy of acute kidney injury. J Med Chem. 2020;63(24):16028‐16042. doi: 10.1021/acs.jmedchem.0c01913 [DOI] [PubMed] [Google Scholar]
- 23. Chen H, Huang Y, Zhu X, et al. Histone demethylase UTX is a therapeutic target for diabetic kidney disease. J Physiol. 2019;597(6):1643‐1660. doi: 10.1113/JP277367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Demishtein‐Zohary K, Azem A. The TIM23 mitochondrial protein import complex: function and dysfunction. Cell Tissue Res. 2017;367(1):33‐41. doi: 10.1007/s00441-016-2486-7 [DOI] [PubMed] [Google Scholar]
- 25. Sun Y, Geng M, Yuan Y, et al. Lmo4‐resistin signaling contributes to adipose tissue‐liver crosstalk upon weight cycling. FASEB J. 2020;34(3):4732‐4748. doi: 10.1096/fj.201902708R [DOI] [PubMed] [Google Scholar]
- 26. Liu X, Zhou Y, Liu X, et al. MPHOSPH1: a potential therapeutic target for hepatocellular carcinoma. Cancer Res. 2014;74(22):6623‐6634. doi: 10.1158/0008-5472.CAN-14-1279 [DOI] [PubMed] [Google Scholar]
- 27. Xu W, Zhang X, Wu J‐L, et al. O‐GlcNAc transferase promotes fatty liver‐associated liver cancer through inducing palmitic acid and activating endoplasmic reticulum stress. J Hepatol. 2017;67(2):310‐320. doi: 10.1016/j.jhep.2017.03.017 [DOI] [PubMed] [Google Scholar]
- 28. Wan D, Liu C, Sun Y, Wang W, Huang K, Zheng L. MacroH2A1.1 cooperates with EZH2 to promote adipogenesis by regulating wnt signaling. J Mol Cell Biol. 2017;9(4):325‐337. doi: 10.1093/jmcb/mjx027 [DOI] [PubMed] [Google Scholar]
- 29. Zhang W, Yang D, Yuan Y, et al. Muscular G9a regulates muscle‐liver‐fat axis by musclin under overnutrition in female mice. Diabetes. 2020;69(12):2642‐2654. doi: 10.2337/db20-0437 [DOI] [PubMed] [Google Scholar]
- 30. Sobecki M, Mrouj K, Colinge J, et al. Cell‐cycle regulation accounts for variability in Ki‐67 expression levels. Cancer Res. 2017;77(10):2722‐2734. doi: 10.1158/0008-5472.CAN-16-0707 [DOI] [PubMed] [Google Scholar]
- 31. Krasnov GS, Puzanov GA, Kudryavtseva AV, et al. Differential expression of an ensemble of the key genes involved in cell‐cycle regulation in lung cancer. Mol Biol (Mosk). 2017;51(5):849‐856. doi: 10.7868/S0026898417050135 [DOI] [PubMed] [Google Scholar]
- 32. Leenen PJ, de Bruijn MF, Voerman JS, Campbell PA, van Ewijk W. Markers of mouse macrophage development detected by monoclonal antibodies. J Immunol Methods. 1994;174(1–2):5‐19. doi: 10.1016/0022-1759(94)90005-1 [DOI] [PubMed] [Google Scholar]
- 33. Mason DY, Cordell J, Brown M, et al. Detection of T cells in paraffin wax embedded tissue using antibodies against a peptide sequence from the CD3 antigen. J Clin Pathol. 1989;42(11):1194‐1200. doi: 10.1136/jcp.42.11.1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fleming TJ, Fleming ML, Malek TR. Selective expression of Ly‐6G on myeloid lineage cells in mouse bone marrow. RB6‐8C5 mab to granulocyte‐differentiation antigen (Gr‐1) detects members of the Ly‐6 family. J Immunol. 1993;151(5):2399‐2408. [PubMed] [Google Scholar]
- 35. Yamamoto T, Sekine Y, Kashima K, et al. The nuclear isoform of protein‐tyrosine phosphatase TC‐PTP regulates interleukin‐6‐mediated signaling pathway through STAT3 dephosphorylation. Biochem Biophys Res Commun. 2002;297(4):811‐817. doi: 10.1016/s0006-291x(02)02291-x [DOI] [PubMed] [Google Scholar]
- 36. Muromoto R, Sekine Y, Imoto S, et al. BART is essential for nuclear retention of STAT3. Int Immunol. 2008;20(3):395‐403. doi: 10.1093/intimm/dxm154 [DOI] [PubMed] [Google Scholar]
- 37. Li J, Huang J, Li JS, Chen H, Huang K, Zheng L. Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J Hepatol. 2012;56(4):900‐907. doi: 10.1016/j.jhep.2011.10.018 [DOI] [PubMed] [Google Scholar]
- 38. Ding Y, Li J, Liu S, et al. DNA hypomethylation of inflammation‐associated genes in adipose tissue of female mice after multigenerational high fat diet feeding. Int J Obes (Lond). 2014;38(2):198‐204. doi: 10.1038/ijo.2013.98 [DOI] [PubMed] [Google Scholar]
- 39. Siddiquee K, Zhang S, Guida WC, et al. Selective chemical probe inhibitor of STAT3, identified through structure‐based virtual screening, induces antitumor activity. Proc Natl Acad Sci USA. 2007;104(18):7391‐7396. doi: 10.1073/pnas.0609757104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Page BDG, Fletcher S, Yue P, et al. Identification of a non‐phosphorylated, cell permeable, small molecule ligand for the STAT3 SH2 domain. Bioorg Med Chem Lett. 2011;21(18):5605‐5609. doi: 10.1016/j.bmcl.2011.06.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chakraborty D, Šumová B, Mallano T, et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat Commun. 2017;8(1):1130. doi: 10.1038/s41467-017-01236-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fan Y, Nikitina T, Morin‐Kensicki EM, et al. H1 linker histones are essential for mouse development and affect nucleosome spacing in vivo. Mol Cell Biol. 2003;23(13):4559‐4572. doi: 10.1128/MCB.23.13.4559-4572.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xiong Q, Wu S, Wang J, et al. Hepatitis B virus promotes cancer cell migration by downregulating mir‐340‐5p expression to induce STAT3 overexpression. Cell Biosci. 2017;7:16. doi: 10.1186/s13578-017-0144-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li Z, Feng PP, Zhao ZB, Zhu W, Gong JP, Du HM. Liraglutide protects against inflammatory stress in non‐alcoholic fatty liver by modulating kupffer cells M2 polarization via cAMP‐PKA‐STAT3 signaling pathway. Biochem Biophys Res Commun. 2019;510(1):20‐26. doi: 10.1016/j.bbrc.2018.12.149 [DOI] [PubMed] [Google Scholar]
- 45. Fan J, Chen Q, Wei L, Zhou X, Wang R, Zhang H. Asiatic acid ameliorates CCL4‐induced liver fibrosis in rats: Involvement of Nrf2/ARE, nf‐kappab/IkappaBalpha, and JAK1/STAT3 signaling pathways. Drug Des Devel Ther. 2018;12:3595‐3605. doi: 10.2147/DDDT.S179876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat Rev Cancer. 2009;9(11):798‐809. doi: 10.1038/nrc2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yoneda Y. Nucleocytoplasmic protein traffic and its significance to cell function. Genes Cells. 2000;5(10):777‐787. doi: 10.1046/j.1365-2443.2000.00366.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Table S3
Table S4
Table S5