

ABSTRACT
Here, we report the molecular engineering of nanobodies that bind with picomolar affinity to both SARS-CoV-1 and SARS-CoV-2 receptor-binding domains (RBD) and are highly neutralizing. We applied deep mutational engineering to VHH72, a nanobody initially specific for SARS-CoV-1 RBD with little cross-reactivity to SARS-CoV-2 antigen. We first identified all the individual VHH substitutions that increase binding to SARS-CoV-2 RBD and then screened highly focused combinatorial libraries to isolate engineered nanobodies with improved properties. The corresponding VHH-Fc molecules show high affinities for SARS-CoV-2 antigens from various emerging variants and SARS-CoV-1, block the interaction between ACE2 and RBD, and neutralize the virus with high efficiency. Its rare specificity across sarbecovirus relies on its peculiar epitope outside the immunodominant regions. The engineered nanobodies share a common motif of three amino acids, which contribute to the broad specificity of recognition. Our results show that deep mutational engineering is a very powerful method, especially to rapidly adapt existing antibodies to new variants of pathogens.
KEYWORDS: Antibody engineering, nanobodies, deep mutational scanning, yeast surface display, SARS-CoV-2
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the cause of the current COVID-19 pandemic, which has had devastating consequences in many countries. To date, vaccination of populations appears to be the most effective approach to control the magnitude of the epidemic and to effectively protect the health of individuals. In high-risk infected patients, passive immunization by infusion of monoclonal antibodies constitutes a very interesting and complementary approach to vaccination. Indeed, several studies demonstrated that early administration of monoclonal antibodies blocks entry of the virus in human cells by targeting the viral Spike protein and prevents progression to severe forms of the disease in patients. 1–3
Since early 2020, numerous studies have been dedicated to the development of monoclonal antibodies targeting the Spike protein and more particularly its receptor-binding domain (RBD). 4 Multiple SARS-CoV-2 antibodies described in the literature bind the receptor-binding motif (RBM) of the Spike protein, i.e., the interaction site between the RBD domain and ACE2. These monoclonal antibodies often exhibit excellent affinities, very good ability to neutralize the SARS-CoV-2 virus, and a protective effect in vivo. Yet, most of these antibodies have a relatively narrow recognition spectrum, with a lack of recognition of RBD domains from other members of the sarbecovirus subgenus, including SARS-CoV-1. 5 Over the past year, new strains have emerged with mutations in the RBM that affect the transmissibility of the virus, but also contribute to the escape from host immunity. 6–9 Expansion of new viral variants in the overall population seems to be favored by host immune pressure from prior infection 10,11 or to vaccination. Indeed, RBM is targeted by a large proportion of neutralizing antibodies and hence appears to be an immunodominant region. Many RBM residues are permissive to mutations, which may preserve binding to ACE2, but might affect the recognition by antibodies from patients 12,13 or by therapeutic monoclonal antibodies and consequently alter their protective efficacy. Antibodies such as bamlanivimab or etesevimab are subject to significant loss of affinity for emerging SARS-CoV-2 variants,12,14 which affects their neutralization potency by a factor up to 1000. 12,15 Most of the antibodies have been produced by animal immunization or B cell cloning from infected patients and hence are derived from the immunodominant B-cell repertoires, which are also involved in the selective pressure on the virus and thus allow viral escape. This limitation illustrates the need for alternative strategies to develop antibodies with high neutralizing activity, broad spectrum of recognition of circulating sarbecoviruses and limited sensitivity to immune escape adaptations.
At the beginning of the current pandemic, the proximity of the Spike protein sequences of the SARS-CoV-1 and SARS-CoV-2 strains led several teams to search for cross-reactive antibodies from SARS-CoV-1 convalescent patients or by screening monoclonal antibody libraries obtained following the 2003 epidemic. 16–18 However, most monoclonal antibodies have restricted specificity for either SARS-CoV-1 or SARS-CoV-2 19 and the ability to cross-neutralize both strains is a relatively uncommon feature. 20,21 Sera of patients infected with either SARS-CoV-1 or SARS-CoV-2 show poor cross-reactivity, suggesting that common epitopes are rare. The few examples of cross-reactive and neutralizing antibodies described in the literature include antibodies S309 17 (from which sotrovimab is derived), antibodies S2H97,5 ADG222 or the bivalent VHH-Fc antibody rimteravimab, derived from the VHH72 nanobody. 18,23
VHH72, a molecule originally resulting from a llama immunization with the SARS-CoV-1 Spike protein, can recognize the corresponding SARS-CoV-2 antigen.18 Monovalent binding affinity of VHH-72 to the SARS-CoV-2 RBD is moderate (~39–60 nM) and markedly weaker than that of the SARS-CoV-1 RBD (1.2 nM). 18,23 Despite relatively fast dissociation rates, this VHH is nonetheless able to neutralize SARS-CoV-2 pseudovirus when expressed as dimer or VHH-Fc.18 A published structure reveals that its epitope is relatively conserved within sarbecoviruses and is located in the RBD core, distant from the ACE2 binding site, probably because of its involvement in the dynamics between “up” and “down” conformations of the Spike protein. 23 This VHH72 epitope therefore has advantages of conservation across sarbecoviruses and limited permissivity to mutations, but only exhibits a moderate affinity, which potentially impedes its full potential for therapeutic applications.
We therefore engineered the VHH72 antibody by combining deep mutational scanning (DMS) and yeast surface display (YSD) to generate antibody candidates, expressed as VHH-Fc, with greatly improved affinity and broad cross-reactivity for SARS-CoV-1 and SARS-CoV-2 antigens and cross-neutralization of SARS-CoV-2 variants Alpha, Beta, Gamma and Delta.
Results
For affinity maturation of the VHH-72 nanobody, two successive mutational approaches were combined. We first used DMS to map permissive positions and identify substitutions demonstrating beneficial effects on antigen binding. We then generated focused combinatorial libraries with relatively modest size (<107 clones) in which diversity is essentially restricted to beneficial or neutral substitutions. These libraries were then screened to identify clones combining different substitutions conferring them a substantial gain in affinity.
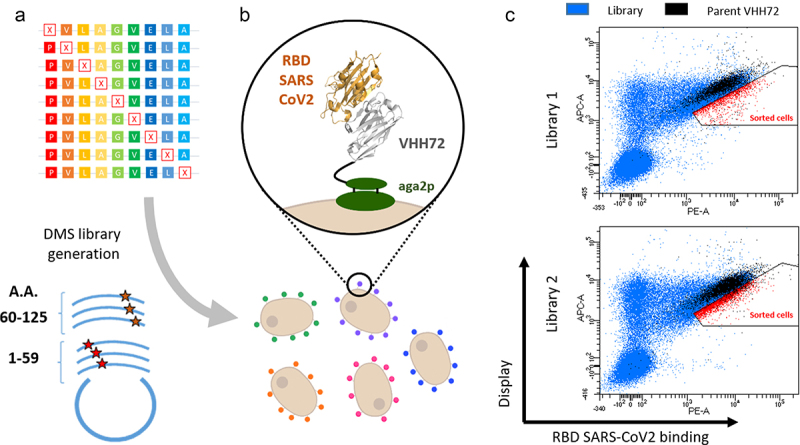
Two libraries encompassing all single amino changes in the VHH72 nanobody were designed with a single NNK degenerate codon for a given cDNA molecule, spanning, respectively, amino acid positions 1–59 and 60–125 (Figure 1a). A YSD system based on the Aga2p/Aga1p anchoring proteins allowed both protein expression and functional discrimination of clones based on their affinity for the SARS-CoV-2 RBD antigen (Figure 1b). The two libraries were cloned in the plasmid pNT VHH72, transformed in S. cerevisiae EBY100 and their expression was induced. To finely compare the RBD binding of clones harboring mutations with VHH72, we introduced a small percentage (typically 5–10%) of cells expressing the parental clone along with intracellular enhanced green fluorescent protein (eGFP), using another plasmid (pSWG VHH72 eGFP). In that manner, parental clones pre-identified by their fluorescence signal corresponding to the eGFP expression (in black) can be readily identified during fluorescence-activated cell sorting (FACS) analysis to precisely define adequate sorting gates (Figure 1c). Upon induction and incubation with biotinylated RBD SARS-CoV-2 antigen, cells displaying variants with strong surface expression levels and augmented RBD binding relative to the parental clone were sorted (in red, Figure 1c). Overall, these sorted populations correspond to a relatively low percentage of cells expressing the VHH construct (5.5% and 11% in libraries L1 and L2, respectively), most mutants having similar or impaired RBD binding levels.
Figure 1.
Deep mutational scanning probing VHH-72 binding to the RBD domain from SARS-CoV2 Spike protein. (a) Two DNA libraries of VHH72 harbouring a single mutation (each corresponding to regions encompassing amino acids 1–59 and 60–125 of the nanobody) were transformed into yeast using gap repair recombination. (b) General principle of functional screening by yeast surface display. Cells are incubated with biotinylated RBD antigen and labelled with secondary reporters before FACS analysis to determine VHH expression and antigen binding. (c) Bivariate flow cytometry analysis of libraries L1 and L2 of yeast cells expressing VHH72 variants on their surface. Cells were double-labelled with biotinylated antigen/Streptavidin–PE (RBD SARS-CoV2 binding) and anti-HA tag antibody coupled to APC (VHH expression). Cells corresponding to clones of the DMS libraries are represented in blue. Libraries were spiked with 10% of clonal cells expressing parental VHH72 along with eGFP protein (represented in black) to discriminate cells with increased antigen binding levels. Selected cells (in red) were sorted and sequenced with illumina deep sequencing.
Alt Text: Deep mutational scanning probing VHH-72 binding to the RBD domain from SARS-CoV2 Spike protein. (a) Scheme illustrating the generation of DMS libraries, their cloning in plasmid and transfection of yeast cells. (b) Scheme representing yeast cells each expressing a mutated VHH72 nanobody. (c) Cytometry dot-plot figures obtained from the analysis of yeast expressing DMS libraries of VHH72.
For each library, plasmid DNA was sequenced for both sorted and unsorted cells to evaluate the respective frequency of clones in these populations. For each position and every substitution, enrichment ratios were calculated and represented as a functionality map (Figure 2 and Figure S1), which reveals uneven distributions of enriched clones (in blue) and depleted clones (in red). Most mutations in complementarity-determining region 2 (CDR2) and CDR3 are associated with a significant depletion in the sorted population, with the exception of a few positions (e.g., A98 or L101). In contrast, several positions in CDR1 are relatively permissive (e.g., T28, E31 or Y32). More intriguingly, a few positions demonstrate contrasted enrichment scores depending on the amino acid substitution involved, such as S57 for which four mutations (alanine, glycine, asparagine or methionine) are enriched, while all charged and most hydrophobic amino acids have strong depletion scores. For positions T103 and V104, all substitutions have strong depletion scores except for valine (T103), and tryptophan or tyrosine residues (V104) for which important enrichment is observed.
Figure 2.
Paratope mapping of the VHH72 nanobody and design of combinatorial libraries for the selection of clones with improved affinity for SARS-COV-2 RBD antigen. NGS-based heatmap representing enrichment values of each VHH72 single mutant after functional sorting in FACS. Enrichment score is a base 2 log function of enrichment between sorted and unsorted VHH72 yeast populations for a given amino acid substitution. Corresponding table is coloured in blue for enriched mutations and in red for depleted mutations. Black squared substitutions were selected for the design of combinatorial libraries to identify VHH72 variants with improved binding properties. Due to the large generated diversity, two separate libraries were designed (with respective theoretical diversities of 4.14e4 and 2.63e7 clones).
Alt Text: Paratope mapping of the VHH72 nanobody and design of combinatorial libraries for the selection of clones with improved affinity for SARS-COV-2 RBD antigen. Table indicating in for each amino acid position (in column) and each substitution (in rows) the enrichment of the corresponding mutation in the DMS setup. Corresponding table is colored in blue for enriched mutations and in red for depleted mutations. Black squared substitutions were selected for the design of combinatorial libraries to identify VHH72 variants with improved binding properties.
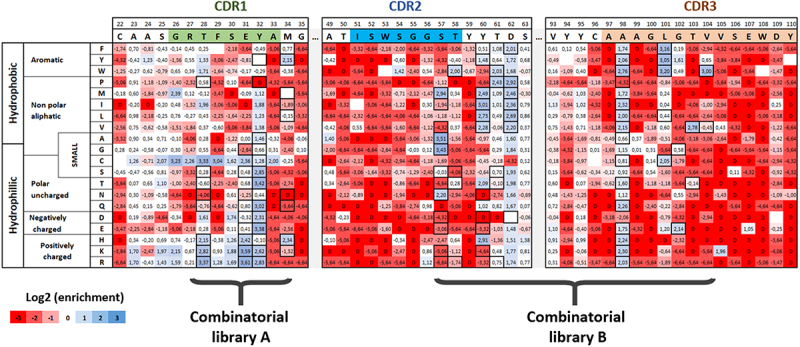
Based on these exhaustive mutational data, we generated optimized combinatorial libraries gathering all mutations with enrichment scores exceeding a threshold of 2 in the DMS experiment (i.e., a four-fold enrichment). We chose this threshold because it represented, in our opinion, a balance between a fairly significant enrichment with respect to the parental clone and the diversity of the combinatorial libraries generated downstream, which seems important to limit to numbers of variants compatible with our screening capacities. Custom degenerate primers were generated using the algorithm Swiftlib 24 and assembled in two libraries of approximately 4.104 and 1.107 clones (Figure S2), corresponding to the CDR1 for library A and CDR2+ CDR3 regions for library B, respectively. Both libraries were generated using splicing by overlap extension PCR (SOE-PCR). The parental amino acid was systematically included for all positions in the design, in contrast to cysteine substitution, which was excluded whenever possible. Because of the limited flexibility of codon degeneracy, a limited number of unwanted mutations were included in the libraries, without compromising their overall quality. The final design of the two libraries is indicated with black bolded squares in Figure 2.
After transformation in yeast cells, combinatorial libraries A and B were sorted to select variants with improved affinity for the SARS-CoV-2 antigen. Selection steps were initiated at 10 nM RBD SARS-CoV-2, a concentration below the KD of the parental antibody, to obtain a good dynamic range in antigen-binding signal. Regardless of their overall good expression on the surface of yeast cells, the best clones of library A demonstrated antigen binding levels similar to the parental clones, without visible affinity enhancement. In sharp contrast, library B comprises a large proportion of clones with improved RBD binding (approximately 30% of induced cells in the first selection round, Figure 3a). The top 2% clones (in the red gate) displayed a very significant increase in antigen binding. The corresponding cells were sorted and submitted to a second round of selection. In addition to the conventional selection of clones based on low antigen concentrations, we also introduced koff-based constraints to drive the selection of clones with long dissociation times. Thus, clones from the first round were incubated with 1 nM RBD biotinylated antigen for two hours, before washing and incubation with an excess amount of 100 nM non-biotinylated antigen to limit possible re-association of the antigen upon dissociation. After two more hours, clones with highest residual binding to biotinylated RBD were sorted (right panel, Figure 3a).
Figure 3.
Yeast Surface Display-based screening of libraries and sorting of VHH72 clones with improved binding for the RBD SARS-CoV-2 antigen. (a) Library A was incubated with 10 nM biotinylated SARS-CoV-2 RBD and analysed in FACS. Few clones with improved antigen binding were detectable. Library B was sorted twice at respective concentrations of 10 nM and 1 nM biotinylated SARS-CoV-2 RBD. In the second round, a selection based on long dissociation times (slow koff) was performed. After equilibrium with 1 nM biotinylated antigen, an excess amount of 100 nM non-biotinylated antigen was introduced to drive dissociation and limit re-association. (b) Clones selected from library B after two rounds of FACS selection. All selected clones display strong binding to both SARS-CoV-1 and SARS-CoV-2 RBD antigens. (c) Sequence logos of clones contained in library B before sorting (in red) and after two rounds of FACS selection for improved affinity (in blue).
Alt Text: Yeast Surface Display-based screening of libraries and sorting of VHH72 clones with improved binding for the RBD SARS-CoV-2 antigen (a) Cytometry dot-plot figures obtained from the analysis of yeast expressing combinatorial libraries of VHH72. (b) Clones selected from library B display strong binding to both SARS-CoV-1 and SARS-CoV-2 RBD antigens. (c) Sequence logos of clones representing the frequency of amino acid substitutions in unsorted library and in sorted library. Positions 56, 103 and 104 demonstrate a substantial shift in frequencies.
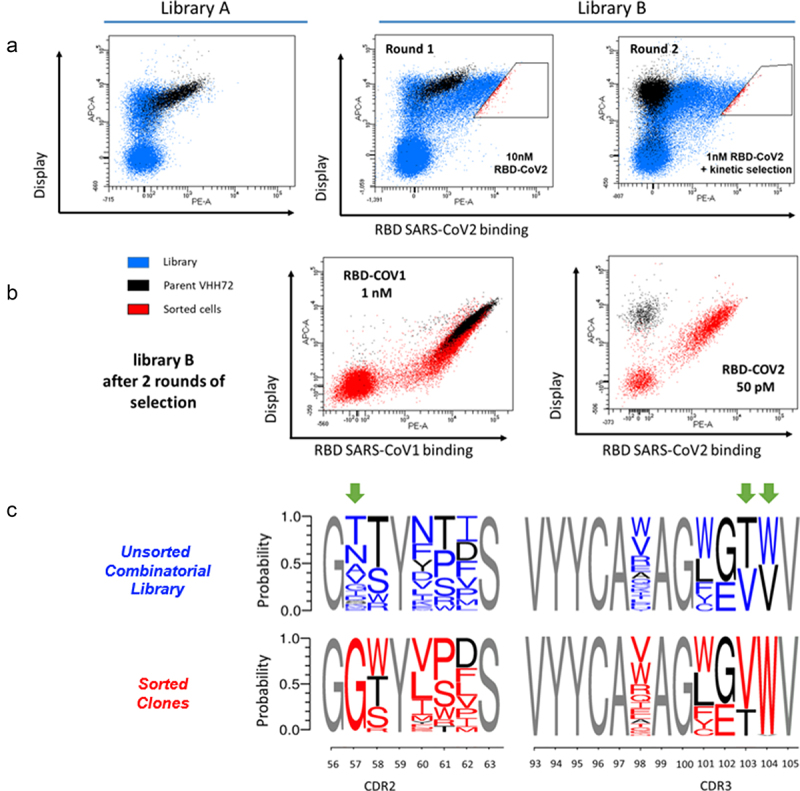
After these selection steps, enriched clones displayed very significant binding levels to SARS-CoV-2 RBD at low concentration of antigen (50 pM, Figure 3b), in contrast to the parental clone that exhibits negligible-binding signal. Notably, the vast majority of enriched clones still bind to SARS-CoV-1 RBD at 1 nM (Figure 3b). A second next-generation sequencing (NGS) campaign was undertaken to measure the evolution of amino acid frequencies in sorted and unsorted populations in the CDR2 and CDR3 regions. The frequency of mutants in the unsorted library is consistent with the theoretical library design (upper panel Figure 3c). After two steps of selection, some mutations (in blue, Figure 3c) are greatly enriched over the parental amino acid (in black), especially S57G, T103V and V104W. Remarkably, S57G substitution is present in more than 97% of clones, replacing the 11 amino acids allowed in the library design. Similarly, tryptophan residue in position 104 (V104W) also has singular enrichment (96%) over valine, the parental amino acid. The amino acid distribution is altered for positions 58, 60, 61 and 62, with visible enrichment of hydrophobic residues over tyrosine at position 60 and threonine 61 being replaced principally by proline or serine residues.
To further investigate the gain of affinity conferred by these substitutions to these VHH molecules, we selected representative clones of highly enriched VHH based on the NGS data. We therefore chose five different VHH (variants VHH72.6, VHH72.65, VHH72.66, VHH72.71 and VHH72.76) each combining five to eight substitutions from the parental antibody (Figure S3). Additionally, we examined the influence of the three principal substitutions, S57G, T103V and V104W, either separately or in combinations. Each mutant was expressed as a VHH-Fc construct. Upon transfection, VHH-Fc molecules were purified, characterized by analytical SEC and SDS-PAGE (Figure S8) and their affinities for the different RBD antigens determined using biolayer interferometry (BLI). Each of the five engineered mutants exhibits strong sub-nanomolar affinities for RBD SARS-CoV-2 in a VHH-Fc format (Figure 4a). The apparent antigen affinity for the Wuhan strain antigen is very high, with double-digit picomolar affinity constants for mutants VHH72.66 and VHH72.71. In addition, binding of these molecules to SARS-CoV-2 RBD is characterized by much longer dissociation rates compared to the parental molecule. For each VHH-Fc, the affinity is only marginally affected by mutations present within the Alpha, Beta, Gamma and Delta strains SARS-CoV-2 strains (Figure 4a and Figure S4a–c). However, binding to the antigen of the recently emerged Omicron strain was greatly affected by the numerous mutations present in its sequence, with affinities in the 200–800 nM range (Figure S9). Interestingly, the five selected VHH-Fc molecules retain very good binding to the SARS-CoV-1 RBD domain. Their affinity constants are in the double-digit picomolar range, with a visible improvement over the parent molecule, whose affinity for the RBD domain of SARS-CoV-1 is in the nanomolar range.
Figure 4.
Affinity of VHH-Fc single-chain antibodies to RBD domains of SARS-CoV and SARS-CoV-2. (a) Bio-Layer Interferometry analysis of VHH-Fc immobilized proteins on anti-human Fc biosensors. Apparent binding kinetics of interaction between the VHH-Fc and the various RBD domains from SARS-CoV variants were evaluated in real time. Binding curves were fitted using a global 1:1 model. (b) Isoaffinity graph representation of kon and koff values for engineered clones and selected single, double and triple mutants, compared to the parental antibody and rimteravimab.
Alt Text: Affinity of VHH-Fc single-chain antibodies to RBD domains of SARS-CoV and SARS-CoV-2. (a) Table indicating the affinities of the VHH72 variants for the tested antigens. (b) graph representation of kon (x-axis) and koff (y-axis) values for engineered clones and selected single, double and triple mutants, compared to the parental antibody and rimteravimab. Engineered clones have much higher affinities, i.e., lower koff.
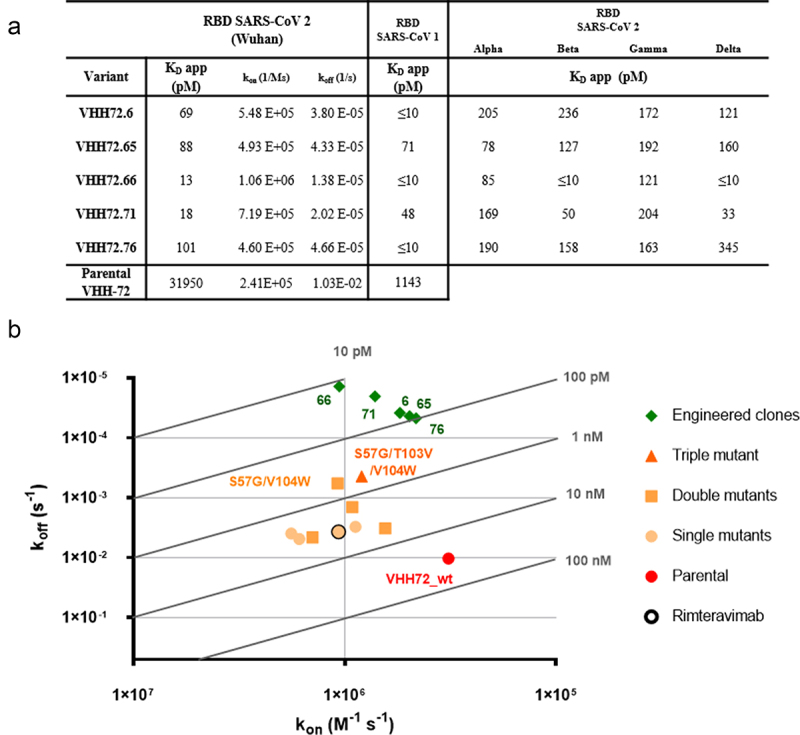
BLI experiments also showed that the S57G, T103V and V104W mutations individually and in combination increased the affinity for the SARS-COV-2 RBD antigen (in yellow, Figure 4b). Higher affinities were observed for the S57G/T103V/V104W triple mutant and for the S57G/V104W double mutation (in orange, Figure 4b). These two combinations confer sub-nanomolar affinities to the VHH-Fc but are nonetheless significantly lower than those observed for the engineered mutants issued from the screening process (in green, Figure 4b). As expected given the constraints introduced during selection, the differences in apparent affinity constants observed are mainly explained by differences in dissociation constants. On the contrary, association constants were not significantly affected by the engineering steps. Overall, these data illustrate that these five engineered mutants not only show excellent affinities with long dissociation rates but also broad-spectrum recognition of known SARS-CoV strains.
Next, we sought to verify that the engineered mutants were all able to antagonize the binding of RBD proteins to the human ACE2 receptor. To this end, we developed a competitive indirect ELISA test. Engineered VHH-Fc showed an increased ability to antagonize the binding of RBD to ACE2, with 50% inhibitory concentration (IC50) values lower than 0.1 µg/mL, up to 220-fold lower than those observed with the parent molecule VHH72-Fc (Figure 5a,b). Our data show very similar IC50 values independently of the antigen tested (RBD of Wuhan, Beta or Delta strains, Figure S5)) for each of the VHH-Fc molecules tested, confirming the large spectrum of recognition of those molecules.
Figure 5.
Protein-based competition ELISA and authentic virus cell-based neutralization assay. (a) Assessment of the ability of the selected VHH-Fc antibodies to block the interaction between ACE2 and the RBD domain of SARS-CoV-2 strain Delta in a competitive ELISA setup. Values represented correspond to three independent experiments. (c) Fifty percent inhibitory concentration (IC50) of the different VHH-Fc for the SARS-CoV2 variants Wuhan, Gamma and Delta by competitive ELISA was calculated. Data points represent mean values ± standard deviation of three independent experiments for each RBD from SARS-CoV2 variant. (b), (d) Neutralization of authentic SARS-CoV-2 virus (Wuhan strain) by the indicated VHH72-Fc constructs.
Alt Text: Protein-based competition ELISA and authentic virus cell-based neutralization assay. (a) Graph curves of the competitive ELISA data demonstrating better ability of engineered clones to antagonize RBD/ACE2 binding. Assessment of the ability of the selected VHH-Fc antibodies to block the interaction between ACE2 and the RBD domain of SARS-CoV-2 strain Delta in a competitive ELISA setup. Values represented correspond to three independent experiments. (b) Neutralization curves of authentic SARS-CoV-2 virus (Wuhan strain) by VHH72-Fc constructs. (c) Table describing the ELISA IC50 of all VHH-Fc construct (d) Table describing the neutralizing titers and the difference of VHH ability to neutralize the virus.
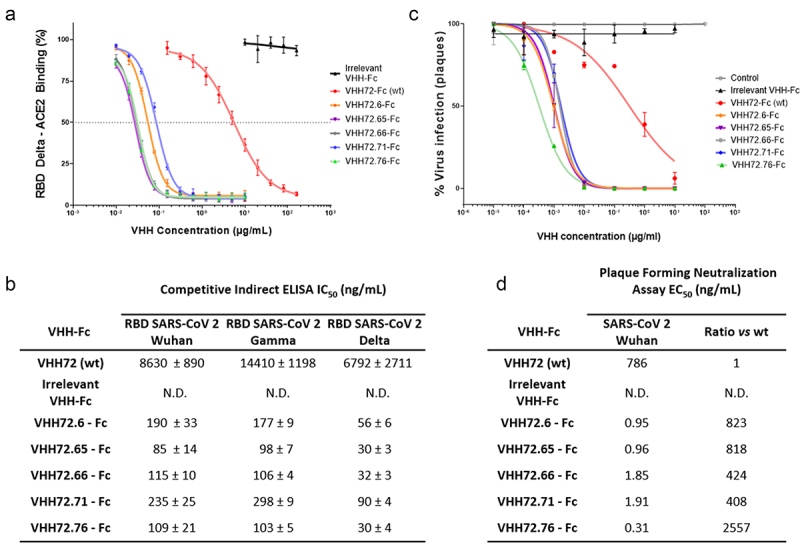
To confirm the ability of these single-chain antibodies to neutralize the SARS-CoV-2 virus and prevent its entry into human cells, we set up a cell-based virus neutralization assay. All VHH-Fc molecules tested demonstrate an excellent ability to neutralize cellular infection by SARS-CoV-2. The reference VHH-Fc (VHH72-Fc) has a neutralization titer of 780 ng/mL (10 nM), and the optimized VHH-Fc all have significantly improved neutralization titers. The improvements over the parental clone range from a factor of 408 for VHH72.71-Fc to 2557 for VHH72.76-Fc. These results showed that engineered molecules exhibited very low neutralizing titers, down to 0.31 ng/mL (3.9 pM) for VHH72.76, objectively demonstrating the impact of sequence optimization (Figure 5c,d).
We next tried to understand the mechanisms by which the introduced mutations in engineered VHH molecules increase the binding to the antigen. Based on the crystallographic structure of the VHH72/RBD SARS-CoV-1 complex, a model of the VHH72- SARS-CoV-2 RBD antigen was built and further refined by molecular dynamics (MD) simulation. Three independent 100 ns MD trajectories were calculated. The initial model suggests that the VHH epitope (in cyan, Figure 6a) does not include any of the substitutions naturally present in the Alpha, Beta, Delta and Gamma SARS-CoV-2 strains, which seems consistent with the observed cross-reactivity.
Figure 6.
Molecular modelling of the interaction between VHH-72 harbouring substitutions S57G/T103V/V104W with SARS-CoV-2 RBD. (a) Homology model of interaction between VHH72 with substitutions S57G/T103V/V104W and SARS-CoV-2 RBD domain. Amino acid substitutions present in the different SARS-CoV-2 variants of concern (VOCs) are shown in red. RBD amino acids within 4.5 Å of any VHH-72 atom were defined as the epitope and stained cyan. Residues included in the receptor binding motif (RBM) are shown in sand color. (b) Location of residues in the epitope of the VHH72 that diverge from SARS-CoV-2 and SARS-CoV-1 (in Orange). Amino acid positions for which substitutions are present in our selected variants are shown in blue. (c), (e) Representative structures extracted from the MD simulation of the SARS-COV-2 RBD – VHH72 complex for state 1 (c) and state 2 (e). The main chains of the proteins are shown in cartoon representation colored in green for SARS CoV-2 RBD and in cyan for VHH72 with the exception of residues G57, V103 and W104 colored in purple. Important residues for the interaction between the two proteins are shown in stick representation coloured by element. In Figure 6c, the movements of residues SARS-CoV-2 RBD Y369 and VHH72 W104 are shown using coloured arrows (green for SARS Cov2 RBD Y369 and purple for VHH72 W104). The χ1 monitored along the MD simulation are shown in (d) for residues W104 (in red) of VHH72 and Y369 (in Orange).
Alt Text: Molecular modeling of the interaction between VHH-72 harboring substitutions S57G/T103V/V104W with SARS-CoV-2 RBD. (a) Three-dimensional model of SARS-CoV-2 RBD domain in interaction with VHH72 with substitutions S57G/T103V/V104W. The epitope is distal from the ACE2 receptor-binding interface. (b) Three-dimensional model indicating that residues mutated in selected variants are in the vicinity of polymorphic positions diverging from SARS-CoV-2 and SARS-CoV-1 within the epitope. (c), (e) Representative three-dimensional structures extracted from the MD simulation of the SARS-COV-2 RBD – VHH72 complex for state 1 and state 2. Residues SARS-CoV-2 RBD Y369 and VHH72 W104 are moving during the course of the modeling trajectory. (d) Graphical representation representing the χ1 angle in degree monitored along the MD simulation for residues W104 (in red) of VHH72 and Y369 (in orange) during the course of the modeling trajectory.
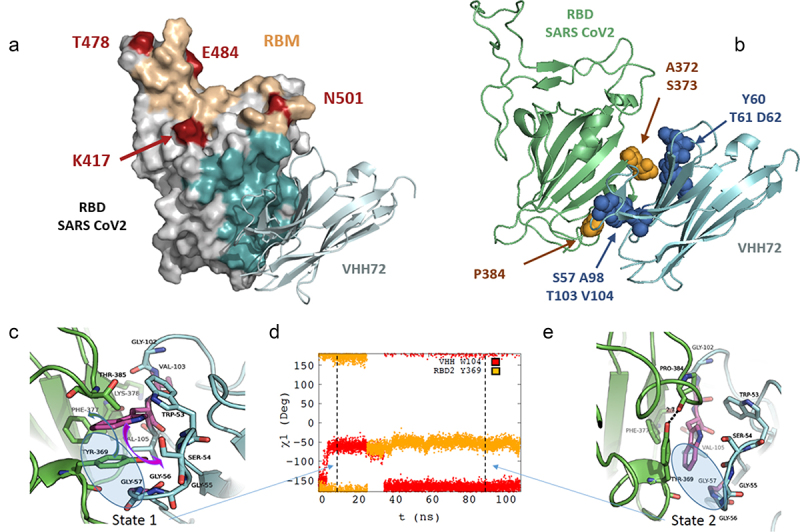
The model shows an immediate proximity of the amino acid at position 57 to positions 103 and 104 of VHH72, which is consistent with the visible coevolution of these three residues in our data set. The model also suggests a direct interaction of the side chains of the amino acids at positions 103 and 104 with the antigen, essentially by creating a network of van der Waals interactions with hydrophobic groups of SARS-CoV-2 RBD. Along the MD simulations, two states characterized by different χ1 values for residues W104 of VHH72 and Y369 of SARS-CoV-2 RBD are systematically found (Figure 6c,e and Figure S6A). In state 1, the side chain of VHH72 W104 is located in the center of a hydrophobic cavity formed by the side chains of F377 and Y369 of SARS-CoV-2 RBD and residue V105 and W53 of VHH72 (Figure 6c). The orientation of W104 is allowed because the side chain of SARS-CoV-2 RBD Y369 has moved from χ1 = −60° in SARS-CoV-1 RBD-VHH72 complex, structure 6WAQ) to χ1 = +180° in the VHH/SARS-CoV-2 model (State 2, Figure 6d and Figure S6A-C). It must be noted that the correlated changes of the χ1 dihedral angles of SARS-CoV-2 RBD Y369 and VHH72 W104 are accompanied by uncorrelated changes of their respective χ2 (Figure S6D-F). The S57G mutation is required to enable this χ1 rotation because the lack of a side chain in position 57 of VHH72 leaves space to accommodate the SARS-CoV-2 RBD Y369 side chain (Figure 6c, blue circle). In this regard, the distance between the CH2 atom of the side chain of VHH72 W104 and the Ca of VHH72 G57 clearly shows that a bulky side chain in position 57 could not accommodate a tryptophan residue in position 104 (Figure S7)). During MD trajectory 1, after about 25 ns, the side chains of SARS-CoV-2 RBD Y369 flip back to a position similar to the one observed in the SARS-CoV-1 RBD-VHH72 complex (χ1 = – 60°, Figure 6e). This motion is accompanied by a rotation of the VHH72 W104 side chain, which also involves evolution of the χ2 of these two residues, which reach an equilibrium state in the last part of the MD trajectories (Figure S6). Overall, our modeling study explains the strong cooperativity between the VHH72 residues located at the interface with RBD antigensin particular, between W104 and G57 and to a lesser extent with V105 that contribute to form the hydrophobic cavity that accommodates the interface with SARS-CoV-2 RBD.
The mutations present in the engineered variants (variants VHH72.6, VHH72.65, VHH72.66 VHH72.71 and VHH72.76) are located in close proximity to the antigen (in blue, Figure 6b) and distributed in two distinct areas (S57, A98, T103, V104 and Y60, T61, D62 respectively). These mutations are present in the vicinity of three polymorphic amino acids that differ within the epitope of the RBD domains of SARS-CoV-1 and SARS-CoV-2 (in orange, Figure 6b). Positions S57, A98, T103, V104 are located in the environment of SARS-CoV-2 Proline 384, while positions Y60, T61 and D62 are in the proximity of SARS-CoV-2 A372 and S373. Engineered variants share common substitutions in the first region (specifically S57G, A98V, T103V and V104W) but incorporate different sets of substitutions in the second region. Hence, the slight differences in affinity and neutralization of the different variants can be attributed to substitutions at positions Y60, T61, and D62.
Discussion
Monoclonal antibodies are becoming a major new class of drugs with very diverse fields of application, including oncology and inflammatory diseases, but also for a number of severe infectious diseases such as those caused by respiratory syncytial virus, 25 Ebola 26 or SARS-CoV-2. Numerous methods are available to obtain new antibodies, whether by immunization of animals, screening of naive or synthetic libraries or by identification within the immune repertoires of patients recovering from infectious diseases. In the context of the COVID-19 pandemic, a wide range of approaches have been used to rapidly generate monoclonal antibodies against SARS-CoV-2 and to accelerate their clinical evaluation.27,28 Over time, some of these antibodies have shown certain limitations, in particular decreased efficacy due to mutations present in emerging strains.15,27 Very recently, most SARS-CoV-2 antibodies approved or in clinical trials were found to exhibit a complete loss of neutralization of the Omicron variant.29,30 Hence, these mutations alter not only their affinity but also their ability to neutralize the virus and to provide effective protection. As a result, a second generation of antibodies might be required to overcome these limitations, and could be obtained by reshaping the initial sequences by conferring them the necessary properties, such as affinity and selectivity, to have optimal therapeutic efficacy for treatment in humans.
From this perspective, many teams have proposed a wide range of methods to generate candidates with the expected properties, mostly increased affinity.31–34 Affinity maturation aims at improving biological activity by adjusting the kinetic parameters of the binding to the target, which in turn may confer greater therapeutic efficacy.25,35 However, the magnitude of this effect depends largely on the epitope recognized by the antibody and the initial affinity along with the format of the antibody and its valence.25 Several recent studies have described affinity maturation of VHH or conventional antibodies to enhance their binding to SARS-CoV-2 antigens, by CDR-swapping approaches, 36 saturation mutagenesis in CDRs 37,38 or light-chain shuffling. 38
In recent years, DMS approaches have emerged as a powerful tool for understanding protein/protein interactions. DMS explores in a selected protein all possible unique substitutions, i.e., all unique mutations for each position. DMS defines the mutational landscape of the protein and aids understanding of the interaction modalities, as recently shown for the RBD/ACE2 and RBD/antibodies interactions.12,13,39,40 In this work, we used these systematic mutagenesis data to engineer new nanobodies with improved affinity and tailored selectivity. We named this process deep mutational engineering (DME) and applied it to VHH72.
The therapeutic potential of VHH72 resides in its ability to neutralize SARS-CoV-2 in spite of its moderate affinity by recognizing an uncommon conserved class IV epitope.41 The marked difference in affinity of the parent molecule for the antigens of the two viruses is probably explained by the presence of three substitutions found in the contact zones with VHH72 (residues A372, S373 and P384 in SARS-CoV-2 epitope are threonine, phenylalanine and alanine residues, respectively, in the SARS-CoV-1 epitope). On this basis, we first sought to improve the affinity of VHH72 by searching all individual favorable mutations conferring enhanced binding to SARS-CoV-2 RBD. After an exhaustive evaluation of the individual role of each amino acid and the influence of all possible substitutions, we demonstrated that it is possible to combine and cumulate individually favorable mutations in the VHH72 sequence. By generating combinatorial libraries and performing two consecutive selection steps, we were able to identify many mutants with very high affinity for the SARS-CoV-2 antigen using DME. After screening of variants for an increased binding to the SARS-CoV-2 antigen, we verified that the affinity for the initial antigen, the RBD domain of SARS-CoV-1 was preserved. Interestingly, in the present case, selection of nanobodies for cross-reactivity was not required, as enriched clones had a preserved binding at low RBD SARS-CoV-1 concentrations. The best clones characterized even showed a significantly improved affinity for the SARS-CoV-1 antigen, below 100 pM. Otherwise, it would have been perfectly conceivable to introduce an additional selection step to select clones based on their SARS-CoV-1 RBD binding in order to retain only clones with the desired cross-reactivity.
The vast majority of our selected clones incorporate a subset of mutations consisting of S57G, T103V and V104W substitutions. This combination alone confers a significant gain in affinity for the SARS-CoV-2 antigen, by approximately 2 logs, resulting in sub-nanomolar binding affinity. The DMS data for these three key positions show very low permissiveness, with very few favorable substitutions. This contrasting pattern seems ultimately more promising in terms of affinity maturation than the positions with many enriched substitutions (e.g., D62 or A98), which could indicate a certain permissiveness or an influence on the expression levels. Interestingly, all the molecules emerging from the screening incorporate other substitutions (between two to five additional changes) that further increase their affinity for the antigen, to subnanomolar affinities. This suggests that beyond this subset of core mutations, additional substitutions contribute to further increase the affinity for the antigen. The accumulation of several favorable and potentially additive changes could be one of the important advantages of this DMS-based affinity maturation method. This could potentially allow for greater affinity gains especially in comparison with random mutagenesis approaches such as error-prone PCR methods.31 Indeed, our method generates libraries with a large proportion of active clones whereas accumulation of several random mutations per clone would most probably result in mostly inactive molecules.
After engineering, our selected VHH-Fc molecules are not only capable of recognizing both SARS-CoV-1 and SARS-CoV-2 antigens but also most emerging and pathogenic variants except omicron with very high affinity and balanced affinity profiles. These results illustrate that it is possible to significantly increase the affinity of antibodies or VHHs by molecular engineering to adapt them to new related antigens. Importantly, the gains in affinity here observed for the engineered molecules translate into an increase in their potential to not only neutralize the interaction between RBD and the human ACE2 but also the entry of the SARS-CoV-2 virus into human cells. Unlike many class IV antibodies, which often are weakly neutralizing,42 our molecules demonstrate a strong capacity to neutralize the SARS-CoV-2 virus, with low EC50 in the ng/mL and picomolar range. Recently, rimteravimab (XVR011), a bivalent nanobody-Fc molecule derived from VHH72 that incorporates a single substitution corresponding to S56A, has shown protection in mice and Syrian hamster animal models.23 Rimteravimab, which was evaluated in a clinical trial of hospitalized patients, has a monovalent binding affinity in the nanomolar range, evaluated at 6 nM,23 which is about 100 to 200 times lower than our best candidates and a significantly lower neutralization potential. Thus, it would be interesting to verify that the gain of affinity acquired by molecular engineering translates into a better protection in animals and for therapeutic applications as a curative treatment. Mutations conferring an increase of the affinity for the SARS-CoV-2 antigen appear to be in close proximity to the epitope. In particular, T103V and V104W are located in the direct vicinity of the polymorphic amino acids, including position P384, thus contributing to a better accommodation of the paratope to the SARS-CoV-2 antigen. Altogether, our results suggest that it is possible to tailor antibodies with known specificity to other related antigens, provided that the original antibody or nanobody has somehow a detectable initial binding to the antigen.
The Omicron (BA.1) variant was discovered just one month before we submitted this study for publication and was therefore rapidly investigated. In contrast to all other variants, our molecules bind to Omicron (BA.1) variant with an affinity close to 20 nM only. An important drop in the affinity is therefore observed for this mutant, but the affinity remains at a comparable level to therapeutic antibodies (sotrovimab) still used to treat patients infected by the BA.1 variant. At variance, many therapeutic antibodies (such bamlanivimab/etesevimab and casirivimab/imdevimab) that were highly active with the alpha to delta variants lost their capacity to neutralize BA.1 variant 29 and have been discontinued for the treatment of patients infected by BA.1 variant. The subnamolar affinity of our engineered molecules limits the impact of mutations at least for BA.1. Further, the DME approach seems very relevant to rapidly adapt existing molecules to generate new antibodies of high affinity and controlled specificity. An immediate perspective of this work could be to adapt VHH72 or one of the many antibodies affected by omicron mutations to this new antigen for the generation of broadly neutralizing antibodies that could be molecules of interest to fight the current pandemic.
Materials and methods
DMS Library design and generation of combinatorial libraries for affinity maturation
Two DMS libraries of VHH72 variants with single amino acid mutations were generated by splicing by overlap extension PCR (SOE-PCR) using degenerate NNK primers. A library was generated for the two targeted regions (positions 1 to 59 and 61 to 125) corresponding to a diversity of 1888 and 2112 DNA variants, respectively.
Design of mutagenic primers containing degenerate codon was performed using SwiftLib (http://rosettadesign.med.unc.edu/SwiftLib/). Two libraries were designed corresponding to the CDRH1 and CDRH2+ CDRH3 regions.
Yeast surface display of VHH
Preparation of competent yeast cells EBY100 (ATCC® MYA-4941) and library transformation were performed according to Benatuil et al. 43 Gap repair transformations were made in plasmid pNT VHH72 between restriction sites NheI and NotI with 1 µg of digested vector and a molar ratio of 12:1 (library/digested vector). After transformation, cells were cultivated in 250 mL of SD-CAA medium. 44 After a passage to an OD600 of 0.25, cells were grown at 30°C until OD600 0.5–1.0 and re-suspended in 50 mL of SG-CAA for induction.44
Flow cytometry
For library sorting, 107 to 2.108 induced cells were washed in PBSF (phosphate-buffered saline (PBS), bovine serum albumin (BSA) 0.1%) and resuspended in PBSF containing the appropriate antigen concentration using an appropriate volume to avoid ligand depletion as performed in Hunter et al. 45 After 3 hours of incubation at 20°C with agitation, cells were washed with ice-cold PBSF to avoid dissociation. Cells were incubated on ice for 15 minutes with anti-HA antibody (Invitrogen HA Tag Mouse anti-Tag, DyLight® 650 conjugate, Clone: 2–2.2.14; 1:100 dilution) and Streptavidin-PE (Thermo Fisher Scientific; catalog number S866; 1:100 dilution). Cells were subsequently washed with ice-cold PBSF and sorted with a BD FACS Aria™ III cytometer using BD FACSdiva™ software.
Deep sequencing and analysis of NGS data
Plasmid DNA of each yeast population was extracted using Zymoprep Yeast Plasmid Miniprep II and prepared for sequencing as described in Medina-Cucurella and Whitehead. 46 Two-step PCR was performed to amplify the region of interest and add Illumina adapters and barcodes for multiplexing. Deep sequencing was performed with an Illumina ISeq 100 device (2x150 bp, 300 cycles) with at least 300,000 reads per population. Reads were demultiplexed, and each sample was processed separately using the Galaxy platform (https://usegalaxy.org/) using the functions described in Blankenberg et al.47 First, paired reads were joined (Fastq Joiner). A trim was then performed (Fastq Trimmer) on reads to keep just the region of interest in the correct frame. A quality filter (Filter FASTQ) was applied to eliminate reads with a minimum quality score under 30. Next, DNA sequences were translated in protein sequences and identical sequences were grouped. Sequences not repeated at least two times were filtered out. Using the software RStudio, single-mutants were selected to allow calculation of enrichment ratios for each single mutation.
Production and purification of the VHH72-Fc ligands
VHH72-Fc and RBD SARS-CoV-2 constructs were obtained by transient transfection of HEK293 FreestyleTM (Thermo). Synthetic genes corresponding to selected VHH72 mutants were ordered to IDT as gblocks and cloned into the mammalian expression vector pcDNA 3.4 VHH72-Fc. Genes coding for the various RBD domains with the corresponding mutations (see below) were cloned in the pCAGGS RBD-SARS-CoV-2 plasmid, which was a kind gift from the Florian Krammer lab. Mutation for Alpha variant: N501Y. Beta variant: K417N, E484K, N501Y. Gamma variant: K417T, E484 K, N501Y. Delta variant: L452R, T478K. Omicron: G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505 H. HEK293 FreestyleTM was transiently transfected at a density of 2.5 106 cells/mL in 100 mL Freestyle medium (Thermo-Fisher) by addition of 150 µg plasmid and 1.8 mL of linear polyethylenimine (PEI, 0.5 mg/ml) (Polysciences). After 24 hours, 100 mL of fresh medium were added. After seven days at 37°C, 120 rpm, 8% CO2, supernatant was purified using HiTrap Protein A for VHH-Fc proteins or HisTrap Excel for RBD proteins, following the manufacturer’s instructions (GE Healthcare). Size-exclusion chromatography was performed (HiPrep Sephacryl-S-200 HR or S100HR) with PBS.
Affinity measurement by BLI
Binding kinetics were determined using an Octet RED96 instrument (ForteBio). Anti-hIgG Fc Capture (AHC) Biosensors (Fortebio) were loaded with VHH-Fc molecules (50 nM) for 60 seconds. After baseline using kinetic buffer (PBS, BSA 0.5% (w/v) and Tween 20 0.05% (v/v)), association of RBD SARS-CoV-2 or RBD SARS-CoV-1 (Sinobiological) was measured at different concentrations (50 nM to 0.78 nM) for 300 seconds before dissociation in kinetic buffer for 1500 seconds. Data of the control without antigen were subtracted from all binding curves, and binding kinetics were fitted using a global 1:1 Langmuir-binding model.
Virus neutralization assay
VERO E6 (ATCC CRL-1586) cells were seeded in DMEM medium (Gibco) + 5% fetal calf serum (FCS) (Hyclone) in sterile 12-well plates (Falcon) at 2.5 105 cells per well and incubated for 24 hours at 37°C – 9% CO2. Purified SARS-CoV-2 virus (strain 2019-nCov/Italy-INMI1, provided by the European Virus Archive goes Global, EVAg) is diluted in DMEM + 2.5% FCS to 400 PFU/mL and mixed 1:1 with each VHH dilution, ranging from 200 µg/mL to 0.2 ng/mL. They were incubated under agitation for one hour at 37°C, 9% CO2. Then, the culture medium of each plate containing the VERO E6 cells was removed and 500 µL of each VHH/virus mixture added to each well in duplicate. The virus suspension was incubated for 45 minutes at 37°C, 9% CO2 and then removed. Two milliliters of carboxymethyl cellulose (Merck) 2% in DMEM + 10% FCS were then added. After 72 hours of incubation at 37°C, 9% CO2, the medium was removed and the cells stained with crystal violet for 20 minutes at room temperature. After removal of the crystal violet and washing in PBS, the lysis plaques in each well were counted.
Molecular modelling of the VHH72-SARS-CoV-2 RBD
We used the structure of SARS-CoV-2 RBD-VHHE as template to build a model of the SARS-CoV-2 RBD-VHH72 complex. The initial structure of VHH72 was built by homology modeling using the MODELLER software 48 and the coordinates of VHH72 in PDB structure 6WAQ as template. 18 The coordinates of the SARS-CoV-2 RBD were taken from the structure 7KN5 49 and the model of VHH72 was positioned by superimposing the model of VHH72 on the VHHU ones. This initial model was refined by a protocol of energy minimization and MD simulation under positional restraints. During this first step, the solvent was treated implicitly. Then, the resulting structure of the SARS-CoV-2 RBD-VHH72 was studied by MD in explicit solvent. All calculations were carried out with NAMD 50 with the CHARMM36 force field. 51 The initial structure was immersed in a TIP3P water box using the solvate tool of VMD. 52 The limit of the box was set such that any solute atom was located at a distance at least equal to 12 Å of the limit in each direction. Then, the system was neutralized using the autoionize plugin of VMD with Na+ ions. The two-step protocol MD simulations (equilibration and production) was achieved in the NPT ensemble using Langevin dynamics. A time step of 2 fs was used in the equilibrium, and the production runs with the SHAKE method to constrain bond vibration involving hydrogens.53 A cutoff of 12 Å was used for non-bonded interactions and a dielectric constant of 1.0 for electrostatic interactions. Multiple timestepping was used with the rRESPA method to calculate long-range interaction forces every 4 fs. We used periodic boundary conditions and Particle Mesh Ewald (PME) method 54 to treat the long-range electrostatic interactions with a real space grid of 1 Å. The temperature was set to 310 K and the pressure at 1.0 atm. The first step started with several 3000 step cycles of energy minimization under positional restraints. From one cycle to the next one, the force constant applied for the positional restraints was gradually decreased to reach 0 kcal. mol−1 A−2 at the end of the minimization step. Then, a 1 ns equilibration step of MD simulation was run after random initialization of the velocities. This step was followed by a 110 ns MD production step. The random initialization of the velocities allows independent trajectories to be run. To ensure reproducibility of the results, three 100 ns MD trajectories of the SARS-CoV-2 RBD-VHHE were calculated starting with different initial velocities. Analysis of the trajectories was achieved using in-house scripts written in the macrolanguage of CHARMM v42b1.55 Figures were produced with PyMol [PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.] and Gnuplot 5.1.
Competitive indirect enzyme-linked immunoassay
Nunc™ MaxiSorp 96-well Immuno-Plates (Thermo Fisher Scientific, Illkirch, France) were coated with 200 µL/well of AffinityPure goat anti-mouse IgG+IgM (H + L) antibody (Catalog number 115–005-044, Jackson ImmunoResearch Laboratories Inc., Pennsylvania, USA) at 10 µg/mL in 50 mM potassium phosphate buffer, and incubated overnight (16 h) at 22 ± 2°C. Plates were then saturated by the addition of 300 µL per well of enzyme immunoassay (EIA) buffer (0.1 M phosphate buffer, pH 7.4, 0.15 M NaCl, 0.1% bovine serum albumin, and 1% sodium azide) and stored at 4°C until use (at least for 24 h). Two-fold serial dilutions of the different VHH-Fc were prepared in EIA buffer ranging from 0.029 µg/mL to 30 µg/mL (for VHH72.6-Fc, VHH72.65-Fc, VHH72.66-Fc, VHH72.71-Fc, VHH72.76-Fc) or from 0.470 to 480 µg/mL (for VHH72-Fc and irrelevant VHH-Fc). Simultaneously, a solution of human ACE2-mouse Fc tag (Catalog number 10,108-H05H, Sino Biological Inc., Eschborn, Germany) at 900 ng/mL in EIA buffer and a solution of biotinylated RBD (recombinant RBD from the SARS-CoV-2 variants Wuhan, Gamma and Delta) at 300 ng/mL in EIA buffer were prepared. Solutions were distributed in the coated plates as follows: 50 µL/well of ACE-2-mouse Fc tagged, 50 µL/well of the different concentrations of each VHH-Fc (or EIA buffer in the control wells to determine the B0 (100%) binding signal), and 50 µL/well of biotinylated RBD. Plates were incubated at 4°C for 18 h, and, after three washes, 150 µL of Pierce™ Streptavidin-PolyHRP (ThermoFisher Scientific, Illkirch, France) diluted 25,000 fold in EIA buffer without sodium azide, were distributed in each well and incubated for 30 minutes at 22 ± 2°C. Finally, plates were extensively washed, 150 µL of TMB (1-Step Ultra TMB-ELISA, ThermoFisher Scientific, Illkirch, France) distributed in each well, and after 30 minutes incubation at 22 ± 2°C, 150 µL of a 2 N H2SO4 solution were added to stop the enzymatic reaction. Absorbances at 450 and 620 nm were determined. Results were expressed as a percentage of B0 (the signal obtained in the absence of VHH-Fc (control wells)).
Size-exclusion chromatography
A Superdex-200 10/300 GL (GE Healthcare) column was used on an AKTA Pure system (Cytiva). Runs were monitored with Unicorn TM 7.6 software (Cytiva). Sample was applied on the column using a flow rate of 0.5 mL/min with PBS as running buffer.
RBD antigen biotinylation
Biotinylation of SARS-CoV2 RBD was performed with EZ-Link sulfo-NHS-LC-Biotin following the manufacturer’s instructions (Thermo Fisher Scientific). A solution containing 23 µM SARS-CoV2 RBD was incubated with a 20-fold molar excess of sulfosuccinimidyl-6-[biotin-amido]hexanoate (Thermo Fisher Scientific) in PBS for 30 min at 4°C. Unreacted biotin was removed using desalting column (Zeba™ Spin desalting column, Thermo Fisher Scientific).
Supplementary Material
Acknowledgments
This research was funded by the French joint ministerial program of R&D against CBRNE threats. This publication was supported by the European Virus Archive goes Global (EVAg) project that has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 653316. Molecular dynamics calculations were carried out at the computing facility of I2BC and at the TGCC HPC computing center [Grant No. AP010313314].
Funding Statement
This work was supported by the French joint ministerial program of R&D against CBRNE threats.
Abbreviations
DMS: Deep mutational scanning
RBD: Receptor binding domain
YSD: Yeast surface display
Disclosure statement
A.L., S.D, B.M, and H.N. are inventors on FR patent application no. 2111541 entitled “AGENT DE LIAISON ayant une affinité améliorée pour la prévention et le traitement des MALADIES liées aux sarbecovirus”.
Supplementary materials
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2076775
References
- 1.Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with covid-19. N Engl J Med. 2021;384:238–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Libster R, Perez Marc G, Wappner D, Coviello S, Bianchi A, Braem V, Esteban I, Caballero MT, Wood C, Berrueta M.. Early high-titer plasma therapy to prevent severe covid-19 in older adults. N Engl J Med. 2021;384(7):610–18. doi: 10.1056/NEJMoa2033700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen P, Nirula A, Heller B, Gottlieb RL, Boscia J, Morris J, Huhn G, Cardona J, Mocherla B, Stosor V.. SARS-CoV-2 neutralizing antibody LY-CoV555 in outpatients with covid-19. N Engl J Med. 2021;384(3):229–37. doi: 10.1056/NEJMoa2029849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raybould MIJ, Kovaltsuk A, Marks C, Deane CM, Wren J. CoV-AbDab: the coronavirus antibody database. Bioinformatics. 2020;37(5):734–35. doi: 10.1093/bioinformatics/btaa739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Starr TN, Czudnochowski N, Liu Z, Zatta F, Park YJ, Addetia A, Pinto D, Beltramello M, Hernandez P, Greaney AJ. SARS-CoV-2 RBD antibodies that maximize breadth and resistance to escape. Nature. 2021;597(7874):97–102. doi: 10.1038/s41586-021-03807-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weisblum Y, Schmidt F, Zhang F, DaSilva J, Poston D, Lorenzi JC, Muecksch F, Rutkowska M, Hoffmann -H-H, Michailidis E. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife. 2020;9. doi: 10.7554/eLife.61312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen RE, Zhang X, Case JB, Winkler ES, Liu Y, VanBlargan LA, Liu J, Errico, JM, Xie X, Suryadevara N, et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat Med. 2021;27(4):717–26. doi: 10.1038/s41591-021-01294-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang P, Nair MS, Liu L, Iketani S, Luo Y, Guo Y, Wang M, Yu J, Zhang B, Kwong PD. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature. 2021;593(7857):130–35. doi: 10.1038/s41586-021-03398-2. [DOI] [PubMed] [Google Scholar]
- 9.Liu Z, VanBlargan LA, Bloyet LM, Rothlauf PW, Chen RE, Stumpf S, Zhao H, Errico JM, Theel ES, Liebeskind MJ, et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe. 2021;29(3):477–88.e4. doi: 10.1016/j.chom.2021.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kistler KE, Bedford T. Evidence for adaptive evolution in the receptor-binding domain of seasonal coronaviruses OC43 and 229e. eLife. 2021;10:e64509. doi: 10.7554/eLife.64509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eguia RT, Crawford KHD, Stevens-Ayers T, Kelnhofer-Millevolte L, Greninger AL, Englund JA, Boeckh MJ, Bloom JD. A human coronavirus evolves antigenically to escape antibody immunity. PLoS Pathog. 2021;17(4):e1009453. doi: 10.1371/journal.ppat.1009453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Starr TN, Greaney AJ, Dingens AS, Bloom JD. Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016. Cell Rep Med. 2021;2(4):100255. doi: 10.1016/j.xcrm.2021.100255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greaney AJ, Loes AN, Crawford KHD, Starr TN, Malone KD, Chu HY, Bloom JD. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe. 2021;29(3):463–76.e6. doi: 10.1016/j.chom.2021.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Widera M, Wilhelm A, Hoehl S, Pallas C, Kohmer N, Wolf T, Rabenau HF, Corman V, Drosten C, Vehreschild M, et al. Bamlanivimab does not neutralize two SARS-CoV-2 variants carrying E484K in vitro. medRxiv. February 24 2021;2021: 21252372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Planas D, Veyer D, Baidaliuk A, Staropoli I, Guivel-Benhassine F, Rajah MM, Planchais C, Porrot F, Robillard N, Puech J. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature. 2021;596(7871):276–80. doi: 10.1038/s41586-021-03777-9. [DOI] [PubMed] [Google Scholar]
- 16.Lv H, Wu NC, Tsang OT, Yuan M, Perera R, Leung WS, So RTY, Chan JMC, Yip GK, Chik TSH, et al. Cross-reactive antibody response between SARS-CoV-2 and SARS-CoV infections. Cell Rep. 2020;31(9):107725. doi: 10.1016/j.celrep.2020.107725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinto D, Park YJ, Beltramello M, Walls AC, Tortorici MA, Bianchi S, Jaconi S, Culap K, Zatta F, De Marco A, et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020;583(7815):290–95. doi: 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- 18.Wrapp D, De Vlieger D, Corbett KS, Torres GM, Wang N, Van Breedam W, Roose K, van Schie L, Hoffmann M, Pöhlmann S, et al. Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell. 2020;181(5):1004–15 e15. doi: 10.1016/j.cell.2020.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ju B, Zhang Q, Ge J, Wang R, Sun J, Ge X, Yu J, Shan S, Zhou B, Song S, et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature. 2020;584(7819):115–19. doi: 10.1038/s41586-020-2380-z. [DOI] [PubMed] [Google Scholar]
- 20.Bates TA, Weinstein JB, Farley S, Leier HC, Messer WB, Tafesse FG. Cross-reactivity of SARS-CoV structural protein antibodies against SARS-CoV-2. Cell Rep. 2021;34(7):108737. doi: 10.1016/j.celrep.2021.108737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang C, Li W, Drabek D, Okba NMA, van Haperen R, Osterhaus A, van Kuppeveld FJM, Haagmans BL, Grosveld F, Bosch B-J. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat Commun. 2020;11(1):2251. doi: 10.1038/s41467-020-16256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rappazzo CG, Tse LV, Kaku CI, Wrapp D, Sakharkar M, Huang D, Deveau LM, Yockachonis TJ, Herbert AS, Battles MB, et al. Broad and potent activity against SARS-like viruses by an engineered human monoclonal antibody. Science. 2021;371(6531):823. doi: 10.1126/science.abf4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schepens B, van Schie L, Nerinckx W, Roose K, Van Breedam W, Fijalkowska D, Devos S, Weyts W, De Cae S, Vanmarcke S. An affinity-enhanced, broadly neutralizing heavy chain-only antibody protects against SARS-CoV-2 infection in animal models. Sci Transl Med. 2021;13(621):eabi7826. doi: 10.1126/scitranslmed.abi7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobs TM, Yumerefendi H, Kuhlman B, Leaver-Fay A. SwiftLib: rapid degenerate-codon-library optimization through dynamic programming. Nucleic Acids Res. 2015;43(5):e34. doi: 10.1093/nar/gku1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu H, Pfarr DS, Tang Y, L-L A, Patel NK, Watkins JD, Huse WD, Kiener PA, Young JF. Ultra-potent antibodies against respiratory syncytial virus: effects of binding kinetics and binding valence on viral neutralization. J Mol Biol. 2005;350(1):126–44. doi: 10.1016/j.jmb.2005.04.049. [DOI] [PubMed] [Google Scholar]
- 26.Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, Wollen S, Ploquin A, Doria-Rose NA, Staupe RP, Bailey M. Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science (New York, N.Y.). 2016;351(6279):1339–42. doi: 10.1126/science.aad5224. [DOI] [PubMed] [Google Scholar]
- 27.Nathan R, Shawa I, De La Torre I, Pustizzi JM, Haustrup N, Patel DR, Huhn G. A narrative review of the clinical practicalities of bamlanivimab and etesevimab antibody therapies for SARS-CoV-2. Infect Dis Ther. 2021;10(4):1933–47. doi: 10.1007/s40121-021-00515-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corti D, LisaPurcell LA, Snell G, Veesler D. Tackling COVID-19 with neutralizing monoclonal antibodies. Cell. 2021;184(12):3086–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Planas D, Saunders N, Maes P, Guivel-Benhassine F, Planchais C, Buchrieser J, Bolland WG, Porrot F, Staropoli I, Lemoine F, et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature. 2021;602(7898): 671–375. [DOI] [PubMed] [Google Scholar]
- 30.Touret F, Baronti C, Bouzidi HS, de Lamballerie X. In vitro evaluation of therapeutic antibodies against a SARS-CoV-2 Omicron B.1.1.529 isolate. Sci Rep. January 1 2022;12(1):4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simons JF, Lim YW, Carter KP, Wagner EK, Wayham N, Adler AS, Johnson DS. Affinity maturation of antibodies by combinatorial codon mutagenesis versus error-prone PCR. mAbs. 2020;12(1):1803646. doi: 10.1080/19420862.2020.1803646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Packer MS, Liu DR. Methods for the directed evolution of proteins. Nat Rev Genet. 2015;16(7):379–94. doi: 10.1038/nrg3927. [DOI] [PubMed] [Google Scholar]
- 33.Rajpal A, Beyaz N, Haber L, Cappuccilli G, Yee H, Bhatt RR, Takeuchi T, Lerner RA, Crea R. A general method for greatly improving the affinity of antibodies by using combinatorial libraries. Proc Natl Acad Sci U S A. 2005;102(24):8466–71. doi: 10.1073/pnas.0503543102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skamaki K, Emond S, Chodorge M, Andrews J, Rees DG, Cannon D, Popovic B, Buchanan A, Minter RR, Hollfelder F. In vitro evolution of antibody affinity via insertional scanning mutagenesis of an entire antibody variable region. Proc Natl Acad Sci U.S.A. 2020;117(44):27307–18. doi: 10.1073/pnas.2002954117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maynard JA, Maassen CBM, Leppla SH, Brasky K, Patterson JL, Iverson BL, Georgiou G. Protection against anthrax toxin by recombinant antibody fragments correlates with antigen affinity. Nat Biotechnol. 2002;20(6):597–601. doi: 10.1038/nbt0602-597. [DOI] [PubMed] [Google Scholar]
- 36.Zupancic JM, Desai AA, Schardt JS, Pornnoppadol G, Makowski EK, Smith MD, Kennedy AA, Garcia de Mattos Barbosa M, Cascalho M, Lanigan TM. Directed evolution of potent neutralizing nanobodies against SARS-CoV-2 using CDR-swapping mutagenesis. Cell Chem Biol. 2021;28(9):1379–1388.e7. doi: 10.1016/j.chembiol.2021.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schoof M, Faust B, Saunders RA, Sangwan S, Rezelj V, Hoppe N, Boone M, Billesbølle CB, Puchades C, Azumaya CM. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive Spike. Science. 2020;370(6523):1473–79. doi: 10.1126/science.abe3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rouet R, Mazigi O, Walker GJ, Langley DB, Sobti M, Schofield P, Lenthall H, Jackson J, Ubiparipovic S, Henry JY. Potent SARS-CoV-2 binding and neutralization through maturation of iconic SARS-CoV-1 antibodies. mAbs. 2021;13(1):1922134. doi: 10.1080/19420862.2021.1922134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KHD, Dingens AS, Navarro MJ, Bowen JE, Tortorici MA, Walls AC. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182(5):1295–310 e20. doi: 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greaney AJ, Starr TN, Barnes CO, Weisblum Y, Schmidt F, Caskey M, Gaebler C, Cho A, Agudelo M, Finkin S, et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat Commun. 2021;12(1):4196. doi: 10.1038/s41467-021-24435-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zupancic JM, Schardt JS, Desai AA, Makowski EK, Smith MD, Pornnoppadol G, Garcia de Mattos Barbosa M, Cascalho M, Lanigan TM, Tessier JM, Engineered multivalent nanobodies potently and broadly neutralize SARS-CoV-2 variants. Adv Ther. 2021;4(8):2100099. doi: 10.1002/adtp.202100099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shrestha LB, Tedla N, Bull RA. Broadly-neutralizing antibodies against emerging SARS-CoV-2 variants. Front Immunol. 2021;12(4025): 752003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benatuil L, Perez JM, Belk J, Hsieh CM. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng Des Sel. 2010;23(4):155–59. doi: 10.1093/protein/gzq002. [DOI] [PubMed] [Google Scholar]
- 44.Sivelle C, Sierocki R, Ferreira-Pinto K, Simon S, Maillere B, Nozach H. Fab is the most efficient format to express functional antibodies by yeast surface display. mAbs. 2018;10(5):720–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hunter SA, Cochran JR. Cell-binding assays for determining the affinity of protein-protein interactions: technologies and considerations. Methods Enzymol. 2016;580:21–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Medina-Cucurella AV, Whitehead TA. Characterizing protein-protein interactions using deep sequencing coupled to yeast surface display. Methods Mol Biol. 2018;1764:101–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blankenberg D, Gordon A, Von Kuster G, Coraor N, Taylor J, Nekrutenko AGalaxy T, et al. Manipulation of FASTQ data with Galaxy. Bioinformatics. 2010;26(14):1783–85. doi: 10.1093/bioinformatics/btq281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Šali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 49.Koenig PA, Das H, Liu H, Kummerer BM, Gohr FN, Jenster LM, Schiffelers LDJ, Tesfamariam YM, Uchima M, Wuerth JD, et al. Structure-guided multivalent nanobodies block SARS-CoV-2 infection and suppress mutational escape. Science. 2021;371(6530). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Phillips JC, Hardy DJ, Maia JDC, Stone JE, Ribeiro JV, Bernardi RC, Buch R, Fiorin G, Hénin J, Jiang W, et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys. 2020;153(4):044130. doi: 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang J, MacKerell AD Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem. 2013;34(25):2135–45. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 53.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-integration of cartesian equations of motion of a system with constraints - molecular-dynamics of N-Alkanes. J Comput Phys. 1977;23(3):327–41. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- 54.Darden T, York D, Pedersen L. Particle mesh Ewald: an N.Log(N) Method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–92. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 55.Brooks BR, Brooks CL 3rd, Mackerell AD Jr., Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30(10):1545–614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.