

Abstract
The discovery of ketamine as a rapid and robust antidepressant marks the beginning of a new era in the treatment of psychiatric disorders. Ketamine is thought to produce rapid and sustained antidepressant effects through restoration of lost synaptic connections. We investigated this hypothesis in humans for the first time using positron emission tomography (PET) and [11C]UCB-J – a radioligand that binds to the synaptic vesicle protein 2A (SV2A) and provides an index of axon terminal density. Overall, we did not find evidence of a measurable effect on SV2A density 24 hours after a single administration of ketamine in non-human primates, healthy controls (HCs), or individuals with major depressive disorder (MDD) and/or posttraumatic stress disorder (PTSD), despite a robust reduction in symptoms. A post-hoc, exploratory analysis suggests that patients with lower SV2A density at baseline may exhibit increased SV2A density 24 hours after ketamine and that this increase in SV2A was associated with a reduction in depression severity, as well as an increase in dissociative symptoms. These initial findings suggest that a restoration of synaptic connections in patients with lower SV2A at baseline may underlie ketamine’s therapeutic effects, however this needs replication in a larger sample. Further work is needed to build on these initial findings and further establish the nuanced pre- and post-synaptic mechanisms underpinning ketamine’s therapeutic effects.
Introduction
The discovery that ketamine has rapid and robust antidepressant effects is arguably the biggest breakthrough in Psychiatry in recent decades1. Given the pervasive suffering and burden associated with major depressive disorder (MDD) and post-traumatic stress disorder (PTSD)2, along with the limitations of traditional monoaminergic-based treatments in terms of efficacy and speed of action3, the importance of this breakthrough cannot be overstated. It paves the way for a new age of rapidly acting pharmacological treatments for psychiatric disorders. Crucial to this endeavor is understanding the mechanisms underlying the rapid and sustained antidepressant effects of ketamine.
Ketamine is a non-competitive N-methyl-D-aspartate (NMDA) glutamate receptor antagonist. Its discovery as an antidepressant was born out of a shift in focus from the monoamine to the glutamate system, as well as a shift away from simple neurochemical explanations to an emphasis on changes in intrinsic circuitry and plasticity4,5. In a seminal study, the robust, rapid (within 4 hours) and sustained (up to 1 week) antidepressant effects of an intravenous infusion of ketamine were observed for the first time6. Since then, this finding has been replicated multiple times in placebo-controlled studies7 8 and the S-enantiomer of ketamine, in the form of a nasal spray, was FDA approved for treatment-resistant depression in March 20199 and for depression associated with increased suicide risk in August, 2020. Ketamine has also shown promise in treating PTSD10,11 and bipolar disorder12,13.
A decade of research has substantially advanced our understanding of the mechanisms underlying ketamine’s therapeutic effects. A wealth of preclinical research now points to the fundamental process underlying ketamine’s antidepressant effects being an increase in synaptic connections14. This discovery fits with convergent evidence implicating a loss of synaptic connections in mood-related circuitry as a central pathophysiological process underlying depression and other stress-related disorders, such as PTSD15,16. For example, magnetic resonance imaging (MRI) studies demonstrate lower grey matter volume and disrupted functional connectivity in prefrontal cortical (PFC) and limbic regions in both MDD17,18 and PTSD19 20, and post-mortem work indicates a lower number of synapses in the dorsolateral PFC (dlPFC) of depressed individuals, as well as lower density of synaptic signaling proteins21. Further, in the first study of its kind, we used positron emission tomography (PET) to provide the first direct in vivo evidence that lower synaptic density is associated with depression22 (detailed below). Finally, a vast preclinical literature demonstrates that chronic stress leads to synaptic loss in PFC regions and depressive-like behavior14,23–28.
Administration of ketamine is thought to restore these lost synaptic connections. Pioneering preclinical work carried out by Duman et al. indicates that administration of ketamine reverses the synaptic loss and depressive-like behavior induced by chronic stress15,29,30. Specifically, ketamine induces a rapid antidepressant behavioral response that correlates with the number and function of layer V pyramidal dendritic spine synapses in the PFC23,28. Subsequent mechanistic research has shown that ketamine initiates a cascade of events that result in synaptic plasticity31. Specifically, blockade of NMDA receptors and a subsequent disinhibition of glutamatergic neurons triggers a surge in glutamate release32–34, followed by increased activation of the AMPA receptor35. A series of post-synaptic signaling events then occur, including release of brain derived neurotrophic factor (BDNF) and activation of mammalian target of rapamycin complex 1 (mTORC1) signaling, both of which are associated with upregulation of synaptic proteins and, ultimately, synaptogenesis23,28,36,37. Convergent evidence indicates that synaptic deficits observed in stress-related disorders may be precipitated by reduced neurotrophic and mTORC1 signaling38,39. Ketamine therefore appears to be restoring the very mechanisms which underlie synaptic deficits in models of stress, including depression. Indeed, recent in vivo work indicates that ketamine regrows the dendritic spines lost to stress in a targeted manner. Specifically, using two-photon imaging, Moda-Sava et al. showed that stress-induced depressive-like behavior was associated with targeted elimination of postsynaptic dendritic spines on PFC projection neurons and, importantly, that ketamine reversed these effects by selectively rescuing these ‘lost’ spines40. The preclinical evidence that ketamine can restore lost synaptic connections and reverse depressive-like behavior in chronic stress models now appears beyond doubt. The effects of ketamine on synaptic plasticity in humans, however, is less clear, and translating this work into humans is crucial.
In vivo imaging of synaptic density is now possible using PET and [11C]UCB-J - a radioligand that binds to the synaptic vesicle protein 2A (SV2A)41. SV2A is ubiquitously located in synapses throughout the brain42, and can therefore serve as a proxy for the quantification of synaptic density, specifically the density of presynaptic nerve terminals. We previously showed that lower SV2A density is associated with depression severity across individuals with MDD and PTSD, providing the first in vivo confirmation of lower SV2A density in relation to depression in humans22. Importantly, we showed that only those MDD/PTSD individuals with more severe depression exhibited lower SV2A density. Given that ketamine appears to restore eliminated dendritic spines in a homeostatic fashion40, it is possible that the same is true for the presynaptic terminals. Whether ketamine is able to restore ‘lost’ presynaptic terminals, and whether this is associated with an antidepressant response, is unknown. To address this gap, we used [11C]UCB-J PET to determine the effects of ketamine on SV2A density across healthy comparison control (HC), MDD and PTSD individuals. The peak antidepressant effect of ketamine is seen 24 hours post administration, with sustained effects lasting 1–2 weeks43. We therefore imaged participants at baseline and 24 hours post ketamine infusion (0.5mg/kg over 40mins) to determine whether an increase in SV2A density is observable at the time of the peak clinical response. In a parallel [11C]UCB-J PET study in rhesus macaques, we examined the effects of both ketamine and hydroxynorketamine (HNK - a metabolite of ketamine which rapidly induces synaptogenesis and antidepressant-like effects without psychotomimetic effects in animal models)44, on synaptic density at 24 hours, 1 week and 4–6 weeks post-administration. This is the first study to investigate whether ketamine induces an increase in presynaptic nerve terminals (specifically, SV2A) in both healthy and psychiatric human populations, as well as in non-human primates. The findings of this study provide crucial novel provide informative data that are useful in designing future studies attempting to examine ketamine’s effects on synaptogenesis and the potential relationship this may have to clinical measures.
Materials and Methods
Participants
Our primary aim was to explore the effect of a single dose of ketamine on SV2A density 24 hours after administration. A total of n=9 HC HCs (mean age ± SD=35.1 ± 12.7; 6 men, 3 women) and n=12 clinical participants (mean age=39.9 ± 11.2; 8 men, 4 women; 3 MDD, 5 MDD/PTSD, 4 PTSD) were imaged at baseline and 24 hours after ketamine administration. Diagnosis was confirmed at screening (1 week or less prior to PET scanning) using the Structured Interview for DSM-5. Participants with MDD or MDD and comorbid PTSD were in a major depressive episode at screening. Depressive symptoms were also assessed using the Montgomery-Asberg Depression Rating Scale (MADRS), Hamilton Depression Rating Scale (HAMD-17) and the Beck Depression Inventory (BDI) at screening and scan days. PTSD symptoms were assessed using the PTSD checklist for DSM-5 (PCL-5; n=11) or PCL-S (DSM-IV version; n=3). Exclusion criteria were lifetime history of bipolar disorder or schizophrenia, substance use disorder except for tobacco use disorder in the past 12 months, positive urine toxicology or pregnancy test before any scan, significant medical condition, loss of consciousness for more than 5 minutes over lifetime, and contraindications to MRI or PET scanning. Exclusion criteria were the same for the HC group except for the addition of current, history of, or first-degree family history of any DSM-5 disorder, not including tobacco use disorder. The study was approved by Yale University Human Investigation and Radioactive Drug Research Committees. All participants provided written informed consent before inclusion.
Ketamine administration
Racemic ketamine was obtained from the Yale–New Haven Hospital Pharmacy and administered intravenously by constant infusion over 40 minutes at a dose of 0.5mg/kg, which has consistently been shown to have robust and rapid antidepressant effects7,8, and is typically used clinically. The amount of ketamine administered was similar across groups (Table 1). Vital signs (blood pressure and heart rate) were obtained before, during and after ketamine infusion. The effects of ketamine on participants’ mental state were subjectively assessed using the Clinician Administered Dissociative State Scale (CADSS)45 before, 30 minutes after the start of ketamine infusion, and 2 hours after ketamine administration. The HAMD-1746, MADRS47, and BDI48 were used to assess participants’ mood at screening, at baseline scan, immediately after ketamine infusion and 24 hours after ketamine administration (as well as 1 week and 1 month post-ketamine for clinical participants).
Table 1.
Demographic, clinical and radiotracer characteristics for HC and clinical groups imaged before and 24 hours after receiving ketamine
| HC (n=9) | Clinical group (n=12) | |||
|---|---|---|---|---|
| Age (yrs) | 35.11 (12.68) | 39.91 (11.23) | ||
| Sex (m:f) | 6:3 | 8:4 | ||
| Diagnosis | HC | 3 MDD, 4 PTSD, 5 MDD/PTSD | ||
| No. smokers | 2 | 5 | ||
| Age at onset (yrs) | -- | 22.4 (16.6) | ||
| Antidepressant use (n) a | -- | 3 | ||
| Ketamine administered (ml) | 19.23 (2.24) | 21.56 (3.69) | ||
| Baseline | 24hr post-ketamine | Baseline | 24hr post-ketamine | |
| HAMD-17 | 0.88 (0.93) | 0.78 (1.6) | 14.08 (4.42) | 5.92 (4.50) * |
| MADRS | 0.67 (0.87) | 0.78 (1.71) | 18.25 (8.00) | 6.67 (5.07) * |
| BDI | 0.78 (1.30) | 0.89 (1.36) | 21.92 (7.94) | 13.75 (9.40) * |
| PCL-5b | 38.39 (20.33) | 32.71 (17.29) | ||
| PCL-Sc | 30.50 (7.78) | 21.50 (6.36) * | ||
| Injected dose (MBq) | 661.60 (69.83) | 628.75 (89.00) | 490.50 (159.44) | 533.76 (184.64) |
| Injected mass (ng/kg) | 21.83 (9.78) | 16.75 (9.40) * | 14.10 (11.50) | 13.39 (10.42) |
| Free fraction (fp) | 0.28 (0.03) | 0.28 (0.03) | 0.27 (0.02) | 0.28 (0.02) |
| Parent fraction 30mins (%) | 27.35 (5.98) | 26.09 (7.49) | 25.95 (8.19) | 24.33 (6.16) |
| Parent fraction 60mins (%) | 24.19 (3.36) | 24.17 (6.42) | 22.21 (4.95) | 21.39 (4.71) |
| Clearance rate (L/h) | 69.54 (11.00) | 76.82 (16.71) | 81.14 (9.73) | 85.47 (19.74) |
significantly different from baseline at p<0.05, as determined by paired-samples t-tests
HAMD-17: Hamilton depression rating scale (17-item); MADRS: Montgomery-Asberg depression rating scale; BDI: Beck depression inventory
n=1 subject was taking paxil, n=2 subjects were taking wellbutrin
n=6 individuals with PTSD were administered the PCL-5 (DSM-5 version)
n=2 individuals with PTSD were administered the PCL-S (DSM-IV version).
Magnetic Resonance Imaging
T1-weighted MRI scans were acquired on 3-Tesla Siemens Prisma scanner. A high resolution, three-dimensional magnetization prepared rapid acquisition gradient echo (MPRAGE) T1-weighted sequence was used to exclude structural abnormality and for co-registration with PET images (TR=1500ms, TE=2.83ms, FOV=256 × 256mm2, matrix=256 × 256mm2, slice thickness=1.0mm without gap, 160 slices, voxel size 1.0 × 1.0 × 1.0mm3).
Positron Emission Tomography
[11C]UCB-J was synthesized onsite and administered intravenously as a bolus over 1 min using an automated infusion pump (Harvard PHD 22/2000, Harvard Apparatus). Details of radiotracer synthesis are outlined in Supplementary Methods. Participants were scanned on a high-resolution human brain PET camera, the High Resolution Research Tomograph (HRRT). All PET imaging and measurement of the metabolite-corrected arterial input function was performed according to previously described procedures41,49, and outlined in Supplementary Methods. Participants received a baseline [11C]UCB-J scan, after which they were administered ketamine. Participants then returned 24 hours later for their post-ketamine scan.
Image analysis
The primary outcome measure was total volume of distribution (VT), computed parametrically based on 60 minutes of data using the 1 tissue (1T) compartment model and a metabolite-corrected arterial input function, as validated previously49. Distribution volume (VT) is the tissue-to-plasma concentration ratio at equilibrium and reflects total uptake (specific plus nonspecific binding) of the radioligand. The 0–10 minute parametric VT image was registered to each participant’s T1-weighted MRI using a rigid mutual information algorithm, then registered to a template MRI in MNI space using Bioimagesuite (http://bioimagesuite.yale.edu; version 2.5) and nonlinear transformations. Regions of interest were derived from the Automated Anatomical Labeling (AAL) atlas and applied to the parametric images using the combined transformations from template to PET space. A priori ROIs were dlPFC, ACC and hippocampus. PET images were grey matter masked using segmentations derived from the computational anatomy toolbox for SPM12 (Wellcome Department of Cognitive Neurology).
Non-human primate imaging
Four female rhesus monkeys (Macaca mulatta; mean ± SD age=10.8 ± 2.50 yrs; weight=8.50 ± 1.54 kg) were given two sets of [11C]UCB-J PET scans; a) after administration of ketamine and b) after administration of hydroxynorketamine (HNK). Animals were imaged at baseline, then after a 2-week period were given an intramuscular (IM) bolus injection of ketamine at a 0.75mg/kg dose. They were then imaged 24-hours and 1-week after ketamine administration. After a 1–2 month washout period, animals were given a second baseline scan. After a further 2 weeks’ period, they were given an IM injection of HNK at either a 1.5mg/kg dose (first 2 monkeys), or a 3mg/kg dose (second 2 monkeys). The animals were then imaged 24-hours, 1-week and 1-month after HNK administration. Due to a change in sedation regimen after the first baseline scan (to the regimen described above), the second baseline scan was used as comparison data for assessing SV2A density after both ketamine and HNK administration in all animals.
The animals were fasted overnight prior to each PET scan. They were sedated using IM injections of alfaxone (2mg/kg), dexmedetomidine (0.01mg/kg) and midazolam (0.3mg/kg) approximately 2 hours before each PET scan, and anesthesia was subsequently maintained with isoflurane (1.5%−2.5%). A water-jacket heating pad was used to maintain body temperature. Vital signs (heart rate, blood pressure, respirations, EKG, ETCO2 and body temperature) were continuously monitored. Radiotracer synthesis was the same as for the human study, outlined in Supplementary Methods. Dynamic PET scans were performed over 120 mins on a Focus-220 PET scanner (Siemens Preclinical Solutions) and arterial blood samples were collected for generation of the metabolite-corrected arterial input function, as described previously50. Outcome measure was VT, calculated regionally using the 1-tissue compartment model. Regions of interest were derived from a) manual delineation on a single representative anatomic rhesus monkey MR image registered to a template image and b) the INIA19 primate brain template51. Registration parameters were used to apply the ROIs to individual PET scans, as described previously52. The primary ROIs were middle frontal gyrus (INIA19 template), cingulate cortex and hippocampus (manually delineated), corresponding to the primary ROIs in the human study (dlPFC, ACC and hippocampus).
Statistics
Statistical analysis was performed in SPSS v22 (IBM). Shapiro-Wilk tests indicated that the data were normally distributed. Linear mixed-effects models were conducted to examine the effect of ketamine on SV2A density between- and within-groups for the primary ROIs (dlPFC, ACC and hippocampus) in the human study; and the effect of ketamine across scans in the non-human primate study (across middle frontal gyrus, cingulate cortex and hippocampus). In the human study, Group (HC, clinical group), Time (baseline, 24 hours post-ketamine), and Group x Time were entered as fixed factors and subject as a random effect. Correlations between change in symptoms and change in SV2A density were assessed using Pearson’s r. In the non-human primate study, Time (baseline, 24 hours, 1week post-ketamine/HNK) was entered as a fixed factor and subject as a random effect. Findings were considered significant at p<0.05.
Results
Dissociative and antidepressant effects of ketamine
Ketamine administration induced dissociative symptoms, as indicated by significant increases on the depersonalization, derealization and amnesia subscales of the Clinician-Administered Dissociative State Scale (CADSS) in both HC and clinical groups, consistent with the known subjective psychotomimetic effects of ketamine53 (Figure 1A), (HC group - depersonalization: p=0.036, Cohen’s d=0.84; derealization: p=0.006, d=1.25; amnesia: p=0.022, d=0.94; clinical group - depersonalization: p=0.003, d=1.12; derealization: p<0.001, d=1.33; amnesia: p=0.054, d=0.70). These psychotomimetic effects resolved 30 mins after the infusion.
Figure 1. Dissociative and antidepressant effects of ketamine.
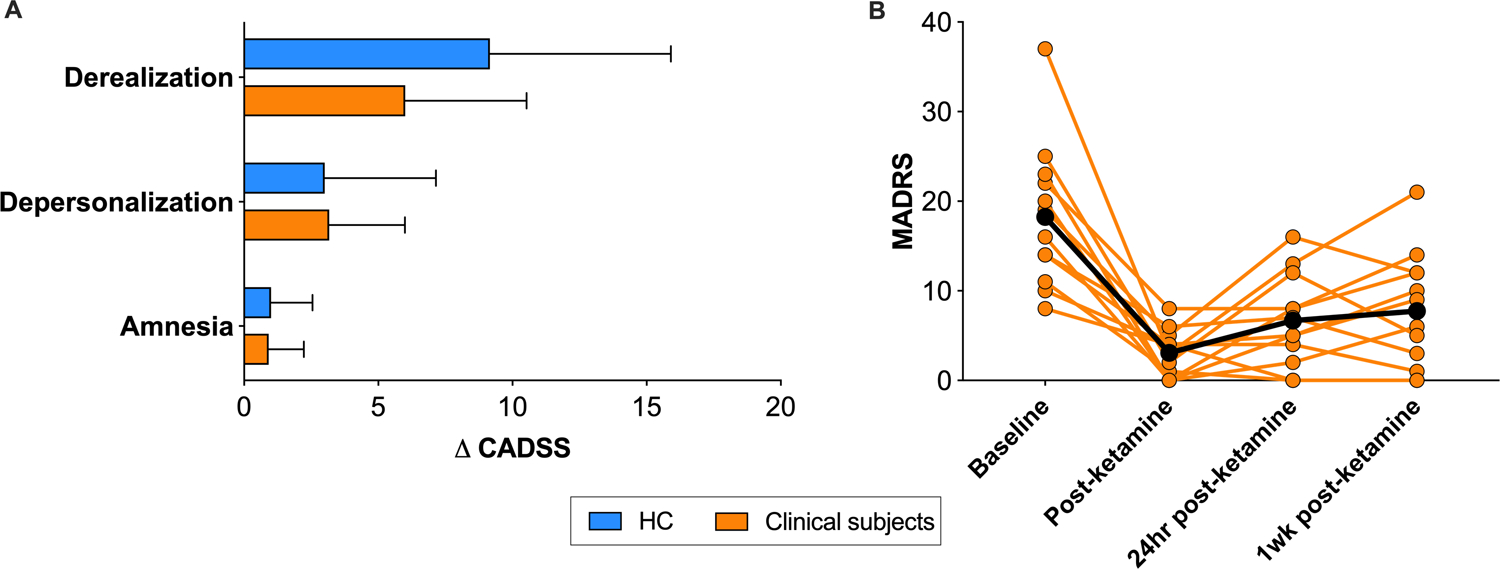
A. Change in CADSS scores during ketamine administration in HCs (blue) and clinical subjects (orange). B. MADRS scores at baseline, immediately, 24 hours and 1 week after ketamine administration in clinical subjects.
Error bars represent standard deviation. CADSS: Clinician administered dissociative scale; MADRS: Montgomery-Asberg Depression Rating Scale.
Ketamine administration also increased heart rate and blood pressure in both HC and clinical groups (p<0.05 at multiple timepoints during infusion), as reported previously54,55. Finally, ketamine produced a robust antidepressant effect in the clinical group (Figure 1B; Table 1), with large reductions in depressive symptoms immediately after ketamine administration (−80±19%, p<0.001, d=−1.74), 24 hours (−66±24%, p<0.001, d=−2.07) and 1-week post-ketamine (−58±28%, p<0.001, d=−2.29).
Effect of ketamine on SV2A density 24 hours after administration
Overall, we did not find evidence of a measurable change in SV2A density ([11C]UCB-J VT) 24 hours after ketamine administration in HC (n=9; mean change 0.5%) or clinical (n=12; mean change 2.4%) groups across primary ROIs [HC group – dlPFC: F1,8=0.09, p=0.78; ACC: F1,8=0.22, p=0.65; hippocampus: F1,8=0.02, p=0.89 (figure 2A); Clinical group – dlPFC: F1,11=0.88, p=0.37; ACC: F1,11=1.72, p=0.22; hippocampus: F1,11=2.70, p=0.13; (Figure 2B)]. Additional regions are shown in Table S1. Further, the effect of ketamine on SV2A density did not differ by group across dlPFC (F1,19=0.65, p=0.43), ACC (F1,19=1.38, p=0.26) or hippocampus (F1,19=0.87, p=0.34). There were no significant correlations found between change in CADSS, depression or PTSD symptoms and change in SV2A density in this small sample. The delivery rate of radiotracer (K1) was significantly increased after ketamine administration across ROIs in clinical participants (all p’s<0.02) but not HCs (Table S2).
Figure 2. Effect of ketamine administration on SV2A density 24 hours after administration in HC and clinical groups.
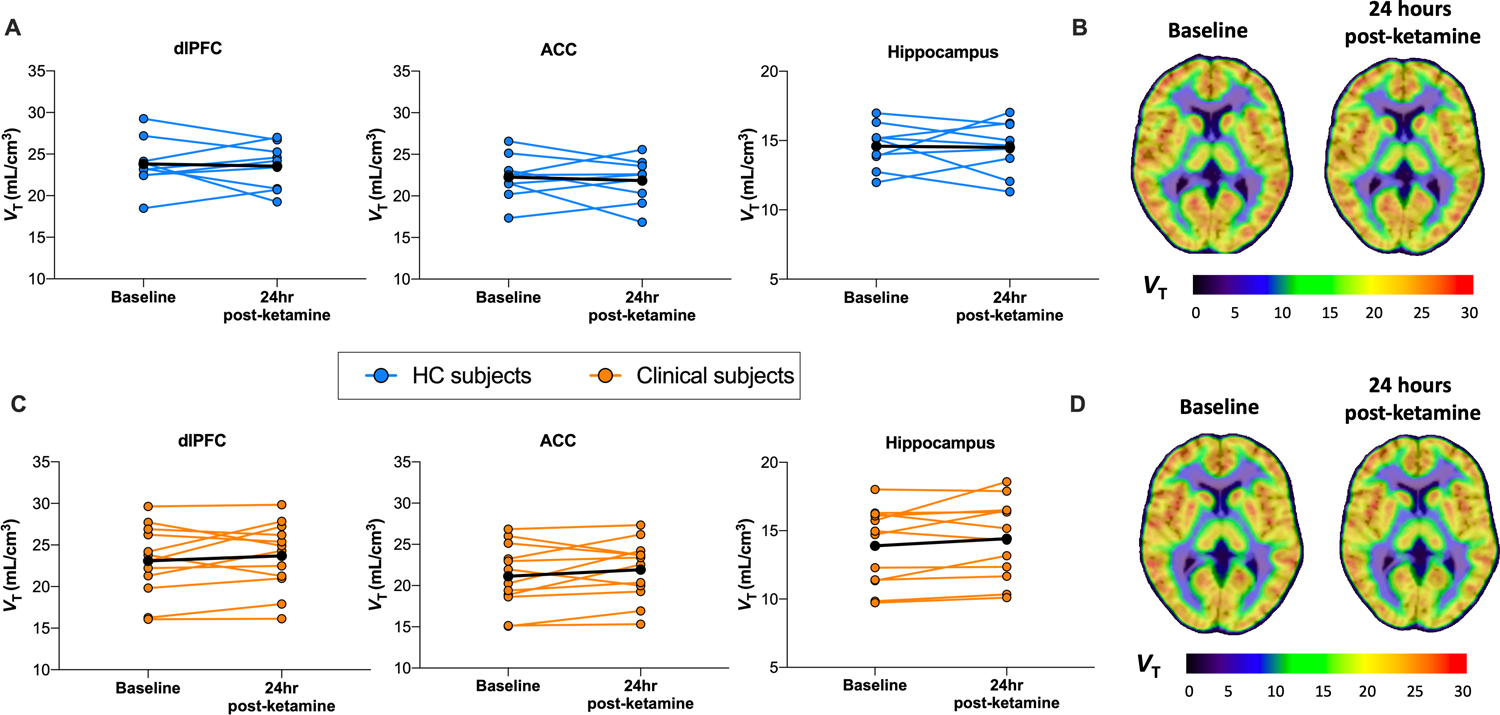
[11C]UCB-J VT (SV2A density) at baseline and 24-hours after ketamine administration in HC subjects across ROIs. B. Mean parametric images of [11C]UCB-J VT overlaid onto MRI for HC group’s baseline (left) and 24-hour post-ketamine scans (right) (in MNI space). C. [11C]UCB-J VT (SV2A density) for HC subjects across ROIs. D. Mean parametric images of [11C]UCB-J VT overlaid onto MRI for clinical group’s baseline (left) and 24-hour post-ketamine scans (right). dlPFC: dorsolateral prefrontal cortex; ACC: anterior cingulate cortex.
Post-hoc analysis based on baseline SV2A stratification
As a post-hoc, exploratory analysis, the clinical group was stratified into subgroups according to their baseline SV2A density, using a median split (n=6 in each group; Table 2). Those with the lowest baseline SV2A density exhibited significantly lower baseline SV2A density compared to a larger cohort of age and sex-matched HCs (n=25; 12 women, 13 men; mean age = 42.0 ± 16.4 yrs) that were imaged with identical methods as part of our previously published work22. In this ‘SV2A deficit’ group, SV2A density was lower across dlPFC (p=0.005), ACC (p=0.001) and hippocampus (p<0.001) (Figure 3A).
Table 2.
Demographic and clinical characteristics of patient subgroups stratified by baseline SV2A (post-hoc analysis)
|
‘No SV2A deficit’
clinical group (n=6) |
‘SV2A deficit’
clinical group (n=6) |
|||
|---|---|---|---|---|
| Age (yrs) | 34.83 (7.49) | 45.00 (12.62) | ||
| Sex (m:f) | 5:1 | 3:3 | ||
| Diagnosis | 3 MDD/PTSD, 3 PTSD | 4 MDD, 2 MDD/PTSD | ||
| No. smokers | 4 | 2 | ||
| Antidepressant use (n)a | 3 | 0 | ||
| Ketamine administered (ml) | 22.02 (3.25) | 21.42 (4.92) | ||
| Baseline | 24hr post-ketamine | Baseline | 24hr post-ketamine | |
| HAMD-17 | 14.00 (4.73) | 6.00 (3.10)* | 14.17 (4.54) | 5.83 (5.91)* |
| MADRS | 17.00 (10.04) | 6.17 (4.36)* | 19.50 (6.02) | 7.17 (6.08)* |
| BDI | 22.33 (8.07) | 13.00 (12.60)* | 21.50 (8.55) | 13.00 (12.60)* |
| PCL-5b | 43.83 (15.41) | 38.00 (11.14) | - | - |
| PCL-Sc | - | - | 30.5 (7.78) | 21.50 (6.36) |
| Injected dose (MBq) | 495.99 (183.63) | 626.97 (116.71) | 485.01 (148.79) | 440.55 (201.31) |
| Injected mass (ng/kg) | 17.34 (12.97) | 16.59 (8.95) | 10.85 (9.88) | 10.18 (11.58) |
| Free fraction (fp) | 0.27 (0.02) | 0.27 (0.02) | 0.26 (0.02) | 0.28 (0.03) |
| Parent fraction 30mins (%) | 24.43 (4.08) | 23.23 (4.22) | 27.47 (11.19) | 25.44 (7.92) |
| Parent fraction 60mins (%) | 21.65 (3.94) | 22.24 (4.71) | 22.78 (6.14) | 20.56 (5.57) |
| Clearance rate (L/h) | 84.60 (6.60) | 82.80 (17.40) | 80.40 (13.20) | 88.20 (23.40) |
HAMD-17: Hamilton depression rating scale (17-item); MADRS: Montgomery-Asberg depression rating scale; BDI: Beck depression inventory
n=1 subject was taking paxil, n=2 subjects were taking wellbutrin
n=6 individuals with PTSD were administered the PCL-5 (DSM-5 version)
n=2 individuals with PTSD were administered the PCL-S (DSM-IV version).
Figure 3. Ketamine-induced increase in SV2A density following ketamine in ‘SV2A deficit’ group (post-hoc analysis).
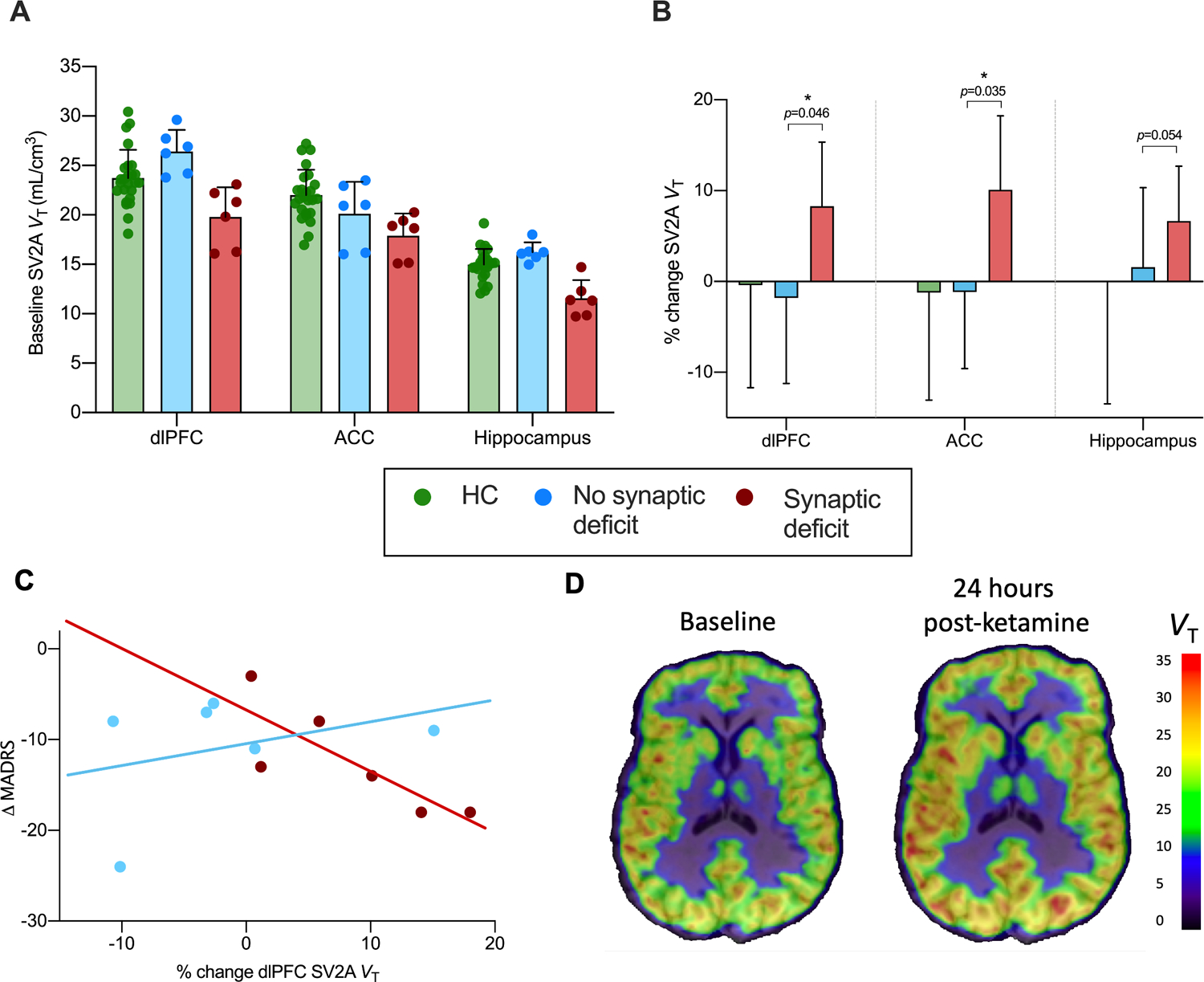
A. Baseline SV2A density for HC (green), ‘SV2A deficit’ (red) and ‘no SV2A deficit’ (blue) groups, indicating significantly lower baseline SV2A density in ‘SV2A deficit’ vs. HC groups. Additional comparison baseline HC data (age-and sex-matched) is included from a separate study. B. Higher % change in SV2A density following ketamine administration in synaptic deficit vs. HC and no synaptic deficit groups. C. Significant negative correlation between reduction in depression severity (MADRS score) and increase in ACC SV2A density following ketamine administration in clinical participants with (red) vs. without (blue) a baseline SV2A deficit. Correlations were also significant in dlPFC and hippocampus. D. Parametric VT images for baseline and 24hr post-ketamine scans for a representative ‘SV2A deficit’ participant with MDD/PTSD. Error bars represent standard deviation. dlPFC: dorsolateral prefrontal cortex; ACC: anterior cingulate cortex.
* significant at p<0.05.
Linear mixed models showed a significant Group x Time interaction across dlPFC (F2,18=5.85, p=0.01), ACC (F2,18=7.79, p=0.004) and hippocampus (F2,18=11.54, p=0.001). Specifically, there was no difference in SV2A density between baseline and 24hr post-ketamine scans in dlPFC (−1.9%, p=0.20, d=−0.22), ACC (−1.2%, p=0.73, d=−0.15) or hippocampus (1.5%, p=0.29, d=0.18) in the ‘no SV2A deficit’ clinical group. However, in the ‘SV2A deficit’ group, SV2A density was significantly higher 24 hours after ketamine administration in dlPFC (8.7% higher, p=0.046, d=1.1) and ACC (10.4% higher, p=0.035, d=1.2), and was just outside significance in the hippocampus (7.0% higher, p=0.054, d=1.0; Figure 3B).
Further, a greater increase in SV2A density was associated with a significantly greater reduction in depressive symptoms in the ‘SV2A deficit’ group, but not in the ‘no SV2A deficit’ group in ACC (SV2A deficit: r=−0.91, p=0.011; no SV2A deficit: r=−0.426, p=0.475; Figure 3C) and dlPFC (SV2A deficit: r=−0.818, p=0.047; no SV2A deficit: r=−0.354, p=0.554). Correlations in the hippocampus were not significant (SV2A deficit: r=−0.678, p=0.139; no SV2A deficit: r=−0.329, p=0.589). Finally, we observed a significant correlation between SV2A density and the derealization scores on the subscale of the CADSS in the ACC (r=0.817, p=0.047; figure S1A), as well as between an increase in derealization symptoms and reduction in depressive symptoms (r=−.882, p=0.020; Figure S1B) in the ‘SV2A deficit’ but not ‘no SV2A deficit’ group. It must be noted that there was a correlation between change in fp (the free fraction of [11C]UCB-J in plasma) and change in VT across primary ROIs in the ‘SV2A deficit’ (dlPFC: r=0.91, p=0.01; ACC: r=0.88, p=0.02; hippocampus: r=0.91, p=0.01) but not ‘no SV2A deficit’ group (all p’s >0.45).
Effect of ketamine and hydroxynorketamine (HNK) on SV2A density in non-human primates
We examined the effect of both ketamine and HNK on [11C]UCB-J VT in healthy rhesus monkeys (n=4) 24 hours, 1 week and 4–6 weeks post administration (Figure 4, Table S3). There were no significant differences in SV2A density between baseline, 24 hour post-ketamine or 1 week post-ketamine scans in middle frontal gyrus (F2,9=0.44, p=0.66), cingulate cortex (F2,9=0.17, p=0.85) or hippocampus (F2,9=0.62, p=0.56). Further, there were no differences in SV2A density 24 hours, 1 week or 4–6 weeks after administration of HNK in middle frontal gyrus (F3,9=0.89, p=0.486), cingulate cortex (F3,9=1.20, p=0.364) or hippocampus (F3,9=0.002, p=1.00). There were no significant differences in injected radioactivity dose, mass, plasma free fraction (Table S4), or in delivery rate of [11C]UCB-J from arterial plasma to brain tissue (Table S5), across timepoints.
Figure 4. No effect of ketamine or HNK administration on SV2A density in non-human primates.
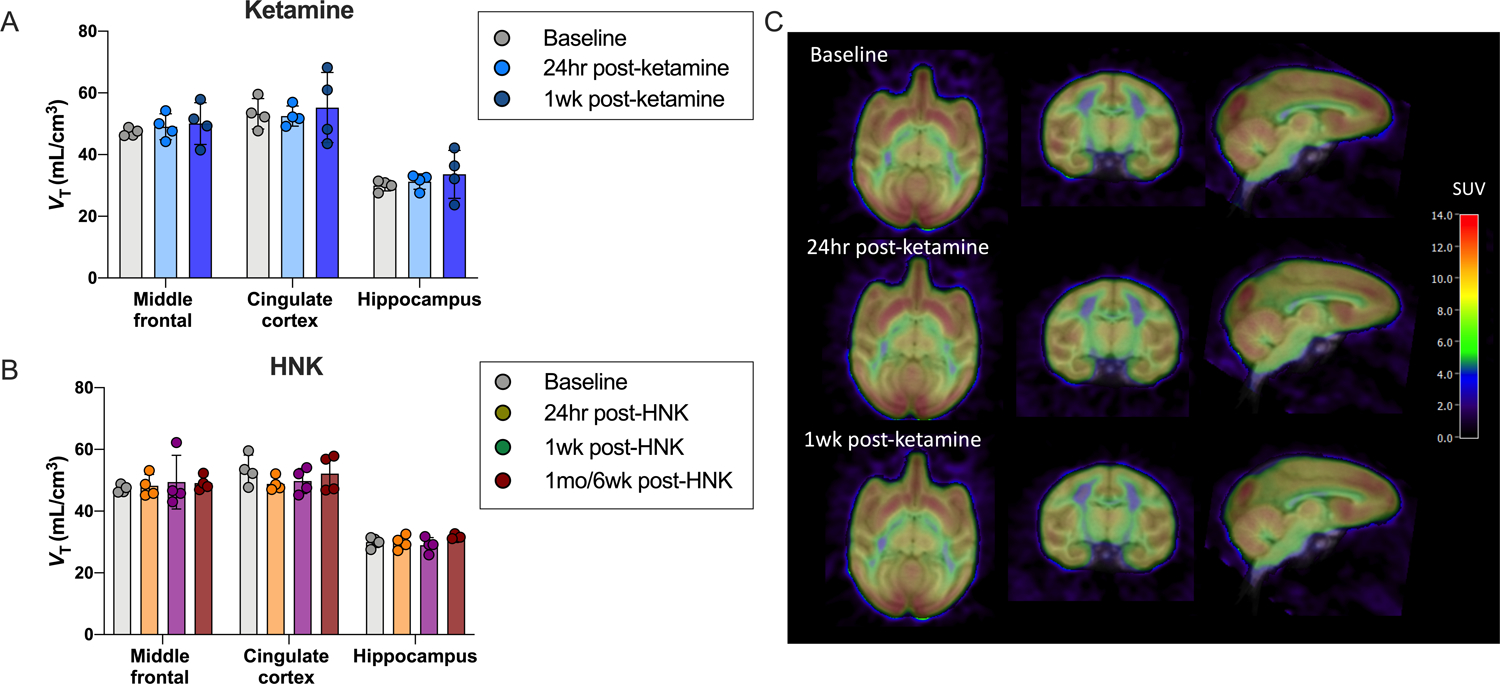
A. [11C]UCB-J VT across primary ROIs from baseline, 24 hour and 1 week post-ketamine scans in non-human primates (n=4) B. [11C]UCB-J VT across primary ROIs from baseline and post-HNK scans (24hr, 1 week and 4–6 weeks) in non-human primates (n=4). C. Representative SUV images overlaid onto MRI (in template space) of baseline, 24hr post-ketamine and 1-week post-ketamine scans from a representative non-human primate.
Discussion
This is the first study to investigate the effect of ketamine on synaptic density (SV2A) in humans. We did not find evidence to suggest ketamine had a detectible effect on SV2A density 24 hours after ketamine administration, as measured with PET, in a sample of n=12 individuals with MDD and/or PTSD or n=9 HCs. Further, we found no evidence of a measurable effect of ketamine or its metabolite HNK in non-human primates 24 hours, 1 week or 4–6 weeks after administration of either drug.
One explanation for our findings is that in stress-related disorders such as MDD and PTSD, synapses are lost predominantly in the postsynaptic compartment. Indeed, ketamine has been shown to restore pruned dendritic spines40. [11C]UCB-J binds solely to SV2A, localized to pre-synaptic nerve terminals, such that we cannot detect changes in the post-synaptic compartment. Our previous data suggest that reductions in the presynaptic compartment are limited to a subgroup of the most severely depressed individuals with MDD/PTSD22. Here, most of the participants were less severely depressed and the extent of presynaptic deficits were variable.
As a post-hoc, exploratory analysis we investigated whether the extent of SV2A deficit at baseline affected the synaptic response to ketamine. Specifically, we stratified the clinical group according to baseline SV2A density and, in this small sample, found that ketamine induced a significant increase in SV2A density in those with the lowest SV2A density at baseline. Further, a greater increase in SV2A density was associated with a greater reduction in depressive symptoms in this group with low baseline SV2A. The increase in SV2A density was also associated with an increase in derealization symptoms (feelings of detachment from oneself and the environment), and the increase in derealization symptoms was in turn associated with a reduction in depression. These associations could reflect target engagement (ketamine-induced glutamate release), leading to dissociative effects, a subsequent elicitation of synaptogenesis and resultant antidepressant response28,56,57. The association between symptoms of derealization and antidepressant response is in line with the evidence base demonstrating a link between the dissociative and therapeutic effects of ketamine58. While one line of thought is that ketamine’s dissociative effects mediate the therapeutic response59, the observation that increasing the dose beyond 0.5mg/kg produces more dissociation but not superior clinical response, suggests that dissociation is more closely associated with adequate dosing and target engagement than therapeutic benefit60. Mechanisms underlying ketamine’s dissociative effects are thought to include distorted sensory information secondary to an imbalance in NMDA and AMPA receptor activation and a surge in glutamate release61. Given the dose-response relationship between the glutamate surge, synaptogenesis and antidepressant response following ketamine administration28,56,57, it is possible that the relationship between derealization and increased SV2A density observed here is mediated by glutamate release. It must be noted that although the observed associations between the antidepressant response, change in SV2A density and symptoms of derealization follow directions as expected based on the literature cited above, the sample size is small and replication of these findings in further studies in humans is crucial. These findings are by no means conclusive, but we report them in order to guide and inform future studies. The association between change in SV2A density (VT) and change in free fraction (fP) of [11C]UCB-J must also be taken into consideration and explored in follow-up studies.
SV2A is ubiquitously and homogenously expressed in synaptic vesicles throughout the brain42. However, using [11C]UCB-J PET, we cannot delineate whether a change in SV2A density reflects a change in synaptic density itself, or a change in either the number of synaptic vesicles or the expression of SV2A. Indeed, one study showed that ketamine induces changes in presynaptic machinery in rats, possibly reflecting a change in the number of synaptic vesicles in the nerve terminals62. Further work is needed to understand ketamine’s effects on pre-synaptic release machinery, and to unravel its effects on pre- vs. post-synaptic mechanisms in association with its antidepressant response.
The formation and elimination of dendritic spines is a highly dynamic process, underscoring the remarkable plasticity of the human brain63. Capturing such a dynamic process at discrete timepoints is therefore a challenging endeavor, made even more so by the likely differential rates of change across individuals. We imaged non-human primates 24-hours, 1-week and 4–6 weeks after administration of ketamine, as well as after administration of HNK, to help elucidate whether an increase in SV2A density was observable beyond the 24-hour timepoint. The lack of change in SV2A density in healthy non-human primates imaged at multiple timepoints post-ketamine/HNK suggests that ketamine may not be inducing a more delayed change in the density of nerve terminals. However, this needs to be examined in humans. It would also be informative to extend our work to examine the synaptic effects of ketamine in models of stress and/or induced synaptic deficits in non-human primates.
Another open question is whether the timing of ketamine’s effects differs between pre- and post-synaptic compartments. Evidence suggests that the pre- and post-synaptic compartments do not respond to ketamine in synchrony62. For example, the increase in glutamate-glutamine cycling, which may reflect changes in presynaptic release machinery, is time-limited, and may even be decreased at the 24-hour timepoint64, such that the major sustained change could be occurring post-synaptically. The effects of ketamine on glutamate release and presynaptic release machinery appears to be much more nuanced than previously thought, with recent evidence showing that ketamine reduces glutamate release and synaptic vesicle recycling in rodents65, in line with earlier work suggesting that ketamine administration reduces the number of vesicles62. If ketamine reduces synaptic vesicle recycling, we might expect to observe lower SV2A density following ketamine administration. However, synaptic vesicle recycling is a highly homeostatic process66, such that the pool of synaptic vesicles may be restored 24 hours later. An important limitation of [11C]UCB-J PET in this context is that we cannot delineate whether a change in SV2A density reflects a change in synaptic density itself, a change in either the number of synaptic vesicles or the expression of SV2A. Further work is needed to understand ketamine’s effects on pre-synaptic release machinery and to unravel its effects on pre- vs. post-synaptic mechanisms in association with its antidepressant response. Another important consideration is sensitivity. While animal studies show that ketamine-induced increases in synaptic proteins and density14 67 40, PET is generally not as sensitive as techniques such as Western blot analysis and two-photon microscopy67, such that human PET imaging may not be capable of capturing the changes detected in animal studies.
Additional limitations include; 1. The clinical subjects in this study had mild to moderate symptom severity. Our previous data suggest that reductions in the presynaptic compartment are limited to a subgroup of the most severely ill individuals with depression/PTSD22. It is possible that we would have observed a detectible signal change in more severely depressed individuals. 2. A single dose may not be enough to induce a signal change in humans and a full treatment course might be needed in order to detect a large enough signal change using PET. 3. Our data indicate that the effect of ketamine on SV2A density was highly variable. Baseline brain measures including glucose metabolism68 and gamma power69, have been associated with differential responses to ketamine raising the possibility of biological subtypes with differential responses to ketamine. Research building on this work should further examine the role of a baseline functional state (e.g., including synaptic density, metabolism, functional network organization) as predictors of the clinical response to ketamine.
In conclusion, we did not find evidence to suggest ketamine has a detectable impact on SV2A density in a sample of MDD and/or PTSD individuals or HCs, 24 hours after administration, as measured by [11C]UCB-J PET. We also observed no change in SV2A density 24 hours, 1-week or 4–6 weeks after a single dose of ketamine in non-human primates. However, an exploratory post-hoc analysis suggests that ketamine may increase SV2A density in those with lower SV2A density at baseline and that the increase in SV2A density was associated with an antidepressant response and symptoms of derealization. This could suggest that a restoration of presynaptic terminals may contribute to ketamine’s antidepressant effects, however this needs to explored in follow-up studies. We add to recent evidence indicating that the mechanisms underlying ketamine’s rapid and sustained antidepressant effects are nuanced and variable. However, we are confident that with continued translational research, these mechanistic underpinnings will become more completely understood and will drive forward the discovery of additional groundbreaking psychiatric treatments.
Supplementary Material
Acknowledgements:
The authors dedicate this work to the late Ronald S. Duman – a colleague, mentor and friend whose pioneering research has made seminal contributions to the neuroscience of depression and PTSD, and to understanding the mechanisms underlying ketamine’s therapeutic effects. We thank the staff at Yale PET Center and the West Haven National Center for PTSD, and all the individuals who took part in this study. Funding support was provided by the Veterans Affairs National Center for PTSD (R.S.D., J.H.K., and I.E.), the National Center for Advancing Translational Science (S.E.H: UL1TR001863), the Nancy Taylor Foundation (I.E.) and the NARSAD Young Investigator Award (S.J.F.).
Conflict of interest:
Dr. Krystal acknowledges the following relevant financial interests. He is a co-sponsor of a patent for the intranasal administration of ketamine for the treatment of depression that was licensed by Janssen Pharmaceuticals, the maker of s-ketamine. He has a patent related to the use of riluzole to treat anxiety disorders that was licensed by Biohaven Pharmaceuticals Medical Sciences. He has stock or stock options in Biohaven Pharmaceuticals Medical Sciences, Sage Pharmaceuticals, Spring Care Inc., EpiVario Inc., Neumora Therapeutics Inc., Terran Biosciences Inc. and Tempero Bio, Inc. He consults broadly to the pharmaceutical industry, but his annual income over the past year did not exceed $5,000 for any organization. He receives over $5,000 in income from the Society of Biological Psychiatry for editing the journal Biological Psychiatry.
Dr. Gerard Sanacora has served as consultant to Allergan, Alkermes, AstraZeneca, Avanier Pharmaceuticals, Axsome Therapeutics, Biogen, Biohaven Pharmaceuticals, Boehringer Ingelheim International GmbH, Bristol-Myers Squibb, Cowen, EMA Wellness, Engrail Therapeutics, Clexio, Denovo Biopharma, Gilgamesh, Hoffman La-Roche, Intra-Cellular Therapies, Janssen, Levo, Lundbeck, Merck, Navitor Pharmaceuticals, Neurocrine, Novartis, Noven Pharmaceuticals, Otsuka, Perception Neuroscience, Praxis Therapeutics, Sage Pharmaceuticals, Servier Pharmaceuticals, Seelos Pharmaceuticals, Taisho Pharmaceuticals, Teva, Valeant, Vistagen Therapeutics, and XW Labs; and received research contracts from AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Johnson & Johnson, Merck, Naurex, and Usona over the past 36 months. Dr. Sanacora holds equity in BioHaven Pharmaceuticals and is a co-inventor on a US patent (#8,778,979) held by Yale University and a co-inventor on US Provisional Patent Application No. 047162-7177P1 (00754) filed on August 20, 2018, by Yale University Office of Cooperative Research. Yale University has a financial relationship with Janssen Pharmaceuticals and may in the future receive financial benefits from this relationship. The University has put multiple measures in place to mitigate this institutional conflict of interest. Questions about the details of these measures should be directed to Yale University’s Conflict of Interest office.
Dr. Sjoerd Finnema is an employee and shareholder of Abbvie. None of the authors declare any conflicts of interest.
Footnotes
Supplementary information is available at Molecular Psychiatry’s website.
References
- 1.Kraus C et al. The influence of ketamine on drug discovery in depression. Drug Discovery Today 24, 2033–2043, doi: 10.1016/j.drudis.2019.07.007 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Organization, W. H. Depression and other common mental disorders: global health estimates. (World Health Organization, 2017). [Google Scholar]
- 3.Gaynes BN et al. What did STAR* D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Psychiatric services 60, 1439–1445 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Sanacora G, Treccani G & Popoli M Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 62, 63–77 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krystal JH, Abdallah CG, Sanacora G, Charney DS & Duman RS Ketamine: a paradigm shift for depression research and treatment. Neuron 101, 774–778 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman RM et al. Antidepressant effects of ketamine in depressed patients. Biological Psychiatry 47, 351–354, doi: 10.1016/S0006-3223(99)00230-9 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Zarate CA et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Archives of general psychiatry 63, 856–864 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Newport DJ et al. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. American Journal of Psychiatry 172, 950–966 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Kaufman MB Pharmaceutical approval update. Pharmacy and Therapeutics 44, 42 (2019). [PMC free article] [PubMed] [Google Scholar]
- 10.Feder A et al. Efficacy of intravenous ketamine for treatment of chronic posttraumatic stress disorder: a randomized clinical trial. JAMA psychiatry 71, 681–688 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Albott CS et al. Efficacy, Safety, and Durability of Repeated Ketamine Infusions for Comorbid Posttraumatic Stress Disorder and Treatment-Resistant Depression. The Journal of clinical psychiatry 79 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Diazgranados N et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Archives of general psychiatry 67, 793–802 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zarate CA Jr et al. Replication of ketamine’s antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biological psychiatry 71, 939–946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duman RS, Aghajanian GK, Sanacora G & Krystal JH Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nature medicine 22, 238–249 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duman RS Neurobiology of stress, depression, and rapid acting antidepressants: remodeling synaptic connections. Depression and anxiety 31, 291–296 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krystal JH et al. Synaptic loss and the pathophysiology of PTSD: implications for ketamine as a prototype novel therapeutic. Current psychiatry reports 19, 74 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Price JL & Drevets WC Neurocircuitry of mood disorders. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 35, 192–216 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaiser RH, Andrews-Hanna JR, Wager TD & Pizzagalli DA Large-scale network dysfunction in major depressive disorder: a meta-analysis of resting-state functional connectivity. JAMA psychiatry 72, 603–611 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kühn S & Gallinat J Gray matter correlates of posttraumatic stress disorder: a quantitative meta-analysis. Biological psychiatry 73, 70–74 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Akiki TJ, Averill CL & Abdallah CG A network-based neurobiological model of PTSD: evidence from structural and functional neuroimaging studies. Current psychiatry reports 19, 81 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang HJ et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nature medicine 18, 1413–1417 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holmes SE et al. Lower synaptic density is associated with depression severity and network alterations. Nature communications 10, 1–10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li N et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biological psychiatry 69, 754–761 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shansky RM & Morrison JH Stress-induced dendritic remodeling in the medial prefrontal cortex: effects of circuit, hormones and rest. Brain research 1293, 108–113 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Radley JJ et al. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cerebral cortex 16, 313–320 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Radley J et al. Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience 125, 1–6 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Banasr M et al. Chronic unpredictable stress decreases cell proliferation in the cerebral cortex of the adult rat. Biological psychiatry 62, 496–504 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Li N et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duman RS, Li N, Liu R-J, Duric V & Aghajanian G Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology 62, 35–41 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdallah CG, Sanacora G, Duman RS & Krystal JH Ketamine and rapid-acting antidepressants: a window into a new neurobiology for mood disorder therapeutics. Annual review of medicine 66, 509–523 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kavalali ET & Monteggia LM Synaptic mechanisms underlying rapid antidepressant action of ketamine. American Journal of Psychiatry 169, 1150–1156 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Ali F et al. Ketamine disinhibits dendrites and enhances calcium signals in prefrontal dendritic spines. Nature Communications 11, 72, doi: 10.1038/s41467-019-13809-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Widman AJ & McMahon LL Disinhibition of CA1 pyramidal cells by low-dose ketamine and other antagonists with rapid antidepressant efficacy. Proceedings of the National Academy of Sciences 115, E3007–E3016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abdallah CG et al. The effects of ketamine on prefrontal glutamate neurotransmission in healthy and depressed subjects. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 43, 2154–2160 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koike H, Iijima M & Chaki S Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behavioural brain research 224, 107–111 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Autry AE et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu R-J et al. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biological psychiatry 71, 996–1005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duman RS & Monteggia LM A neurotrophic model for stress-related mood disorders. Biological psychiatry 59, 1116–1127 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Ota KT et al. REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nature medicine 20, 531–535 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moda-Sava R et al. Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finnema SJ et al. Imaging synaptic density in the living human brain. Science translational medicine 8, 348ra396–348ra396 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Bajjalieh SM, Frantz G, Weimann JM, McConnell SK & Scheller R Differential expression of synaptic vesicle protein 2 (SV2) isoforms. Journal of Neuroscience 14, 5223–5235 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kishimoto T et al. Single-dose infusion ketamine and non-ketamine N-methyl-d-aspartate receptor antagonists for unipolar and bipolar depression: a meta-analysis of efficacy, safety and time trajectories. Psychological medicine 46, 1459–1472 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fukumoto K et al. Activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2R, 6R)-hydroxynorketamine. Proceedings of the National Academy of Sciences 116, 297–302 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bremner JD et al. Measurement of dissociative states with the clinician-administered dissociative states scale (CADSS). Journal of Traumatic Stress: Official Publication of The International Society for Traumatic Stress Studies 11, 125–136 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Hamilton M A rating scale for depression. Journal of neurology, neurosurgery, and psychiatry 23, 56 (1960). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montgomery SA & Åsberg M A new depression scale designed to be sensitive to change. The British journal of psychiatry 134, 382–389 (1979). [DOI] [PubMed] [Google Scholar]
- 48.Beck AT, Steer RA & Carbin MG Psychometric properties of the Beck Depression Inventory: Twenty-five years of evaluation. Clinical psychology review 8, 77–100 (1988). [Google Scholar]
- 49.Finnema SJ et al. Kinetic evaluation and test–retest reproducibility of [11C] UCB-J, a novel radioligand for positron emission tomography imaging of synaptic vesicle glycoprotein 2A in humans. Journal of Cerebral Blood Flow & Metabolism, 0271678X17724947 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nabulsi NB et al. Synthesis and preclinical evaluation of 11C-UCB-J as a PET tracer for imaging the synaptic vesicle glycoprotein 2A in the brain. Journal of Nuclear Medicine 57, 777–784 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Rohlfing T et al. The INIA19 Template and NeuroMaps Atlas for Primate Brain Image Parcellation and Spatial Normalization. Frontiers in Neuroinformatics 6, doi: 10.3389/fninf.2012.00027 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sandiego CM, Weinzimmer D & Carson RE Optimization of PET–MR registrations for nonhuman primates using mutual information measures: A Multi-Transform Method (MTM). Neuroimage 64, 571–581 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krystal JH et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. 51, 199–214 (1994). [DOI] [PubMed] [Google Scholar]
- 54.Esterlis I et al. Ketamine-induced reduction in mGluR5 availability is associated with an antidepressant response: an [11 C] ABP688 and PET imaging study in depression. 23, 824 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holmes SE et al. Measuring the effects of ketamine on mGluR5 using [18F] FPEB and PET. 0271678X19886316 (2019). [DOI] [PMC free article] [PubMed]
- 56.Abdallah CG, Sanacora G, Duman RS & Krystal JH Ketamine and rapid-acting antidepressants: a window into a new neurobiology for mood disorder therapeutics. Annu Rev Med 66, 509–523, doi: 10.1146/annurev-med-053013-062946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Esterlis I et al. Ketamine-induced reduction in mGluR5 availability is associated with an antidepressant response: an [11 C] ABP688 and PET imaging study in depression. Molecular psychiatry 23, 824–832 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luckenbaugh DA et al. Do the dissociative side effects of ketamine mediate its antidepressant effects? Journal of affective disorders 159, 56–61 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yaden DB & Griffiths RR The Subjective Effects of Psychedelics Are Necessary for Their Enduring Therapeutic Effects. ACS Pharmacology & Translational Science, doi: 10.1021/acsptsci.0c00194 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fava M et al. Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD). Molecular psychiatry 25, 1592–1603 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ingram R et al. Some distorted thoughts about ketamine as a psychedelic and a novel hypothesis based on NMDA receptor-mediated synaptic plasticity. Neuropharmacology 142, 30–40, doi: 10.1016/j.neuropharm.2018.06.008 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Müller HK et al. Ketamine regulates the presynaptic release machinery in the hippocampus. Journal of psychiatric research 47, 892–899 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bhatt DH, Zhang S & Gan W-B Dendritic spine dynamics. Annual review of physiology 71, 261–282 (2009). [DOI] [PubMed] [Google Scholar]
- 64.Chowdhury GM et al. Transiently increased glutamate cycling in rat PFC is associated with rapid onset of antidepressant-like effects. Molecular psychiatry 22, 120–126 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lazarevic V, Yang Y, Flais I & Svenningsson P Ketamine decreases neuronally released glutamate via retrograde stimulation of presynaptic adenosine A1 receptors. Molecular psychiatry, 1–11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vitureira N, Letellier M & Goda Y Homeostatic synaptic plasticity: from single synapses to neural circuits. Current Opinion in Neurobiology 22, 516–521, doi: 10.1016/j.conb.2011.09.006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kato T et al. Sestrin modulator NV-5138 produces rapid antidepressant effects via direct mTORC1 activation. The Journal of Clinical Investigation 129, 2542–2554, doi: 10.1172/JCI126859 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li C-T et al. The effects of low-dose ketamine on the prefrontal cortex and amygdala in treatment-resistant depression: A randomized controlled study. Human Brain Mapping 37, 1080–1090, doi: 10.1002/hbm.23085 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nugent AC et al. Ketamine has distinct electrophysiological and behavioral effects in depressed and healthy subjects. Molecular psychiatry 24, 1040–1052, doi: 10.1038/s41380-018-0028-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.