

Abstract
APOE4 is the first identified genetic risk factor and remains as the strongest predictor for late-onset Alzheimer’s disease (AD). Studies of AD patients, AD patient-specific induced pluripotent stem cell-derived neurons and cerebral organoids, and human apoE4-expressing and apoE-deficient mouse models clearly demonstrate that apoE4 provokes neuroinflammation, impairs cerebrovasculature, and exacerbates amyloid and tau pathologies. ApoE expression is greatly upregulated in disease-associated microglia in mouse models of amyloidosis and in human microglia from AD brains. Importantly, genetic knock-down or depletion of apoE in mice greatly attenuates neuroinflammation and alleviates amyloid and tau pathologies. Similar beneficial effects can be achieved when apoE reduction is induced by the overexpression of apoE metabolic receptor LDLR. Towards therapeutic implications, administration of apoE antisense oligonucleotides or apoE siRNAs leads to significant pharmacological effects, i.e., significant alleviation of AD pathologies in mouse models. Therefore, apoE reduction represents a promising therapeutic strategy for the treatment of AD patients carrying the APOE ε4 allele. In this review, we summarize evidence and rationale on why and how we target apoE4 reduction for AD therapy.
Keywords: apoE4, LDLR, Alzheimer’s disease, therapeutic approach
1. Introduction
Alzheimer’s disease (AD) is the most common form of age-related neurodegenerative disease characterized by detrimental cognitive impairments with the pathological amyloid-β (Aβ) plaques and tau-containing neurofibrillary tangles (NFTs) as the two major hallmarks of this incurable disease (Guo et al 2020, Kloske & Wilcock 2020, Koutsodendris et al 2021, Shi & Holtzman 2018b, Yamazaki et al 2019). The current AD therapies are dominated by symptomatic treatments with three inhibitors of cholinesterase and one blocker of N-methyl-D-aspartate (NMDA) receptor, which partially ameliorate cognitive and behavioral symptoms in AD patients for a short period (Cummings 2021, Ferreira-Vieira et al 2016). Aducanmab is the first potential disease-modifying therapy approved by FDA in 2021, but its clinical benefits need further investigation (Gandy et al 2021, Musiek et al 2021). Therefore, there is an urgent need to develop new disease-modifying therapies for AD treatment.
The human apolipoprotein E (APOE) gene has three common polymorphic alleles (ε2, ε3 and ε4) that result from two single-nucleotide polymorphisms in the coding sequence where the ε4 allele is the strongest genetic risk factor for late-onset AD (Corder et al 1993, Saunders et al 1993, Sims et al 2020). The risk for AD is 3–4-fold higher in individuals carrying one APOE ε4 allele and about 10–15-fold higher in those carrying two APOE ε4 alleles (Corder et al 1993, Saunders et al 1993, Sims et al 2020). Although there is only about 20% of the population carrying the APOE ε4, this isoform is vastly enriched in AD patients with over 60% of symptomatic AD patients carrying at least one APOE ε4 allele (Corder et al 1993, Farrer et al 1997).
ApoE is highly expressed in the liver hepatocytes populating the majority of plasma apoE protein with its main role being transporting lipoproteins by binding to its receptors (Goldstein & Brown 2015). Brain is second to liver with regards to apoE expression levels, and brain apoE is mainly produced by astrocytes and to a lesser extent by microglia, oligodendrocytes, vascular mural cells and neurons (Flowers & Rebeck 2020). ApoE functions as a transporter of cholesterol and other lipids between cells and is a modulator of the inflammatory response in normal brains (Flowers & Rebeck 2020). In AD pathogenesis, apoE4 exerts its detrimental effects mainly via a ‘toxic’ gain of function mechanism to provoke neuroinflammation, impair cerebrovasculature, and exacerbate amyloid and tau pathologies (Kloske & Wilcock 2020, Koutsodendris et al 2021, Shi & Holtzman 2018b, Yamazaki et al 2019). In addition, apoE4 proteins are poorly lipidated (Gong et al 2002, Hu et al 2015), thus loss-of-physiological function of apoE4 also contributes to AD pathogenesis (Kloske & Wilcock 2020, Koutsodendris et al 2021, Shi & Holtzman 2018b, Yamazaki et al 2019). As an emerging therapeutic target for AD, there are various approaches targeting apoE4, including the use of the CRISPR/Cas9 system to convert APOE4 to APOE2 or APOE3 for AD treatment (McDade et al 2021, Safieh et al 2019, Williams et al 2020, Yamazaki et al 2019). In this review, we summarize evidence and rationale highlighting that apoE4 reduction represents a promising therapeutic strategy for AD.
2. Rationale for apoE4 reduction in AD therapy
Studies of AD patients, AD patient-specific induced pluripotent stem cell (iPSC)-derived neurons and cerebral organoids, human apoE4-targeted replacement mice and apoE-deficient mice clearly demonstrate that apoE4 is associated with detrimental effects in diverse pathological contexts and thereby provide a strong rationale for apoE4 reduction in AD therapy (Fig. 1).
Fig. 1.
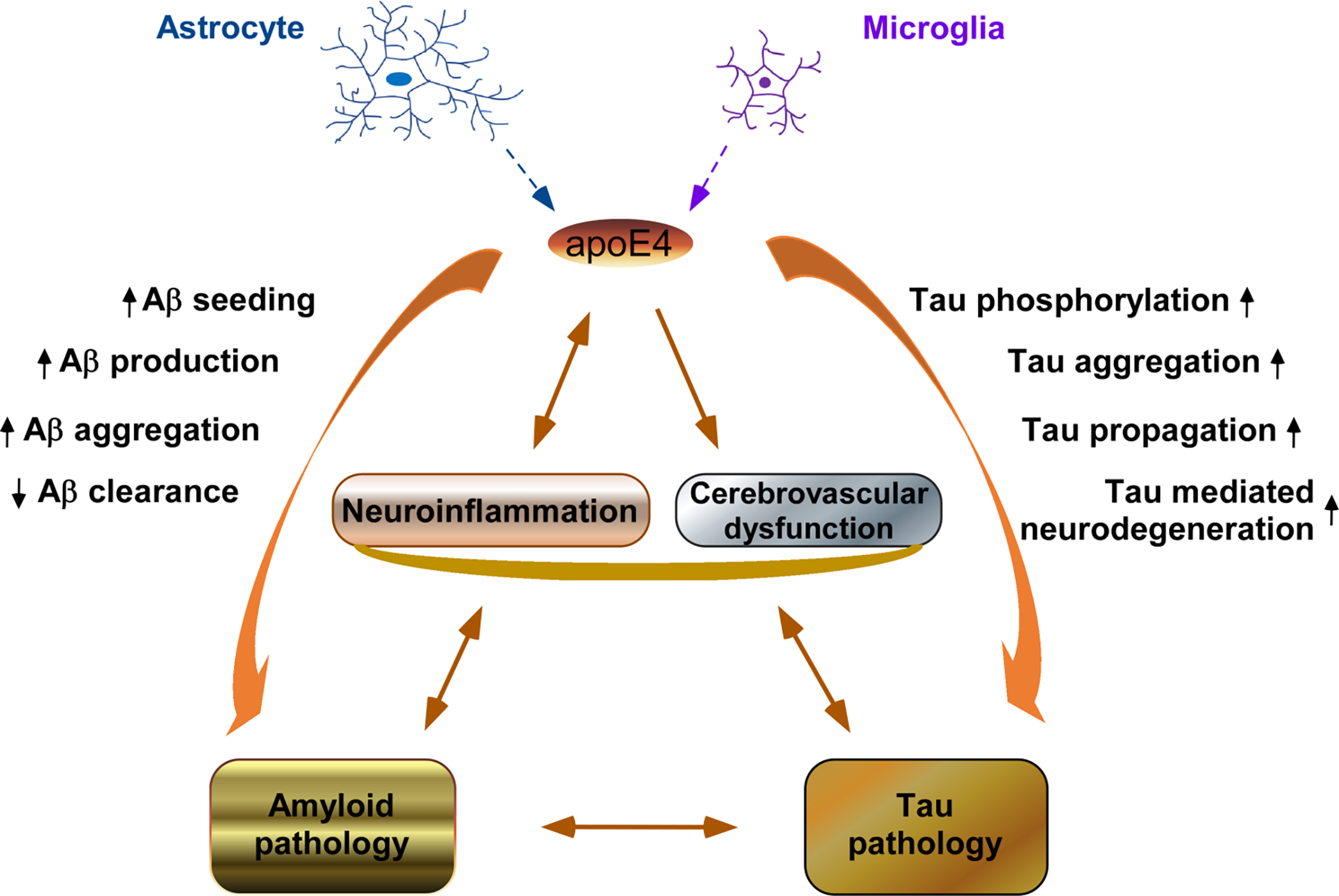
ApoE4 provokes neuroinflammation, impairs cerebrovasculature, and exacerbates amyloid and tau pathologies in the AD brain. ApoE4 is mainly produced by astrocytes and microglia in the AD brain and displays a central role in AD pathogenesis. Therefore, apoE4 reduction represents a promising therapeutic strategy for the treatment of AD.
2.1. ApoE4 exacerbates and apoE depletion alleviates amyloid pathology
Aβ, the main component of amyloid plaques in AD brain, is generated by sequential proteolytic cleavage of the amyloid precursor protein (APP). It is well established that there is a strong positive correlation between the APOE ε4 allele and Aβ pathology in human AD brains with the highest Aβ burden found in homozygous APOE ε4 carriers (Bertram et al 2007, Mishra et al 2018, Polvikoski et al 1995, Premkumar et al 1996, Reiman et al 2009, Schmechel et al 1993). Studies with human iPSC-derived neurons and organoids confirm that apoE4 increases Aβ production and aggregation as well as decreases Aβ clearance (Lin et al 2018, Wang et al 2018, Zhao et al 2020). Experimentally, expression of human apoE4 in APPV717F transgenic mice leads to more than 10-fold of fibrillar deposits when compared to apoE3-expressing mice (Holtzman et al 2000a). Further studies also indicate that there are apoE isoform-dependent effects (E4>E3>E2) on promoting brain Aβ deposition in PDAPP transgenic mice (Bales et al 2009). Mechanistically, apoE4 exacerbates amyloid pathology by promoting Aβ production and aggregation, and inhibiting Aβ clearance and degradation (Koutsodendris et al 2021, Yamazaki et al 2019).
Critically, APP transgenic mice lacking endogenous mouse apoE display a significant decrease of amyloid pathology (Bales et al 1999, Bales et al 1997, Bien-Ly et al 2012, Holtzman et al 2000a, Holtzman et al 2000b). While amyloid deposition in APP transgenic mice occurs in an APOE gene dose-dependent manner in which mice containing two copies of human APOE gene exhibit more amyloid deposition than those with only one copy (Bales et al 1999, Bien-Ly et al 2012, Holtzman et al 2000a, Holtzman et al 2000b), apoE-deficient APP transgenic mice display virtually no fibrillar Aβ deposits nor neuritic degeneration (Bales et al 1999, Bales et al 1997, Holtzman et al 2000a, Holtzman et al 2000b). Additionally, astrogliosis and microgliosis, typical in mice with amyloidosis, are also reduced in the absence of apoE (Bales et al 1999). This evidence provides a strong rationale that reduction of apoE is an attractive strategy for alleviating amyloid pathology in AD patients carrying the APOE ε4 allele.
2.2. ApoE4 exacerbates and apoE depletion attenuates tau pathology and tau-mediated neurodegeneration
Intracellular neurofibrillary tangles with hyperphosphorylated tau protein in neurons is another hallmark of AD pathology. It is generally believed that the detrimental effects of apoE4 is mainly associated with the induction of earlier and more abundant amyloid pathology in the AD patients carrying the APOE ε4 allele. Importantly, increasing evidence demonstrates that apoE4 not only exacerbates amyloid pathology but might also independently aggravate tau pathology and tau-mediated neurodegeneration (Koutsodendris et al 2021, Yamazaki et al 2019). In clinical studies, APOE ε4 is associated with more severe medial temporal tau pathology, neurodegeneration, and memory impairment (Filippini et al 2009, Therriault et al 2020, Wang et al 2021b, Weigand et al 2021). Moreover, an APOE3 Christchurch (R136S) homozygote carrying a causal AD mutation (E280A) in the presenilin1 (PSEN1) gene displays minimal tau pathology and neurodegeneration and is resistant to autosomal dominant AD (Arboleda-Velasquez et al 2019), highlighting that apoE could play a critical role in the onset and progression of tau pathology in AD patients. Studies with postmortem human brains with sporadic primary tauopathy also reveal that the presence of an ε4 allele is corelated with more severe regional neurodegeneration (Shi et al 2017a). Studies with human iPSC-derived neurons and cerebral organoids demonstrate that apoE4 enhances tau phosphorylation and exacerbates synapse loss and neurodegeneration (Lin et al 2018, Wang et al 2018, Zhao et al 2020). Mechanistically, apoE4 aggravates the tau pathology via promoting tau phosphorylation, aggregation, propagation, and tau-mediated neurodegeneration (Koutsodendris et al 2021, Yamazaki et al 2019).
Interestingly, apoE4 knock-in (KI) mice exhibit an age-dependent loss of hilar GABAergic interneurons and deficits in learning and memory, which is more profound when the mice express neurotoxic apoE fragments (Andrews-Zwilling et al 2010). By creating the PS19 tau transgenic mice on a human apoE2/3/4 knock-in (KI) background, Shi et al. have demonstrated that PS19-apoE4 KI mice display more extensive tau pathology and tau-mediated neurodegeneration when compared to PS19-apoE2 KI or PS19-apoE3 KI mice (Shi et al 2017a). Importantly, PS19 mice with apoE knockout (KO) are strongly protected from tau-mediated neurodegeneration when compared to PS19-apoE2/3/4 KI mice and non-transgenic (wild-type) mice (Shi et al 2017a). In addition, selective depletion of astrocytic apoE4 markedly reduces tau pathology and tau-mediated neurodegeneration in PS19-apoE4 KI mice (Wang et al 2021a). Together, these studies provide a strong rationale that reduction of apoE4 level is an attractive strategy for alleviating tau pathology and tau-mediated neurodegeneration in AD therapy.
2.3. ApoE4 provokes and apoE depletion attenuates neuroinflammation
Neuroinflammation is another major pathological feature that is strongly corelated with the Aβ plaque and NFT pathologies and modulates AD progression (Kloske & Wilcock 2020, Shi & Holtzman 2018a). Glial cells, particularly microglia, are the main cells responsible for the neuroinflammation in the brain. While astrocytes are the primary source of brain apoE, microglia are the second major type of cells producing abundant apoE in the brain in particular when these immune cells are activated. Variants of triggering receptor expressed on myeloid cells 2 (TREM2) is another major risk factor of late-onset AD (Guerreiro et al 2013, Jonsson et al 2013, Magno et al 2021). TREM2 is a cell surface receptor mainly expressed in microglia within CNS. Importantly, apoE is a ligand of TREM2 and can trigger TREM2 signaling, thereby regulating microglia function and neuroinflammation (Atagi et al 2015, Bailey et al 2015, Jendresen et al 2017, Yao et al 2019, Yeh et al 2016). In AD mouse models, there is a subset of microglia exhibiting a neurodegenerative/disease-associated phenotype characterized by upregulation of pro-inflammatory genes (Keren-Shaul et al 2017, Krasemann et al 2017). The phenotype of disease-associated microglia (DAM) is dependent on the TREM2-apoE pathway, and deletion of apoE might prevent DAM development (Krasemann et al 2017). However, it remains to be clarified whether there is differential expression of these DAM genes in human AD brains (van Wageningen et al 2021).
In clinical studies, APOE ε4 carriers have higher levels of activated microglia and pro-inflammatory cytokines than non-ε4 carriers (Egensperger et al 1998, Olgiati et al 2010, Schram et al 2007), and the increase of the pro-inflammatory cytokines is associated with greater cognitive decline in old age (Schram et al 2007). Experimentally, apoE4-expressing mice exhibit higher levels of glial activation, cytokine production and synaptic marker loss after the inflammatory challenge with lipopolysaccharide (LPS) when compared to apoE2- and apoE3-expressing mice (Zhu et al 2012). In addition, apoE4-expressing 5xFAD mice display higher levels of glial activation and the proinflammatory cytokine interleukin-1β (IL-1β) as well as larger and more intensely stained Aβ plaques than apoE3-expressing 5xFAD mice (Rodriguez et al 2014). Therefore, apoE4 accelerates AD pathogenesis by inducing dysregulation of microglia and astrocytes and provoking neuroinflammation (Shi et al 2021, Shi et al 2017b). Importantly, several studies have demonstrated that apoE reduction or depletion attenuates neuroinflammation and alleviates pathological changes in various mouse models of amyloid and tau pathology (Kim et al 2011, Krasemann et al 2017, Shi et al 2017a, Ulrich et al 2018, Wang et al 2021a), providing additional evidence that apoE4 reduction is an attractive therapeutic strategy for AD patients carrying the APOE ε4 allele.
2.4. ApoE4 impairs cerebrovasculature
Cerebrovascular dysfunction has been recognized as a major pathological process of AD (Montagne et al 2017, Yamazaki et al 2019). Breakdown of the blood-brain barrier (BBB) is an early biomarker of human cognitive decline (Montagne et al 2020, Nation et al 2019, Nguyen et al 2021). Although both Aβ and tau can exert vasoactive and vasculotoxic effects, leading to neurovascular dysfunction and BBB breakdown in AD brains (Bennett et al 2018, Montagne et al 2017), apoE4 exhibits direct toxic effects on cerebrovasculature independently of amyloid and tau pathologies (Montagne et al 2020, Montagne et al 2017). Notably, strictly lobar microbleeds are found more often in APOE ε4 carriers than APOE ɛ4 non-carriers (Knol et al 2020, Vernooij et al 2008). Experimentally, vascular defects have been detected in human apoE4-targeted replacement (apoE4-TR) mice, but not apoE3-TR mice, at 2 weeks of age (Bell et al 2012). In addition, specific expression of apoE4, but not apoE3, in vascular mural cells impairs cerebrovascular function and spatial learning in mice (Yamazaki et al 2021). Therefore, apoE4 reduction could ameliorate the cerebrovascular dysfunction in the AD brain.
3. ApoE reduction via its receptor LDLR attenuates AD pathology
The low-density lipoprotein receptor (LDLR) is an important metabolic receptor for regulating apoE levels in the brain. Recent studies have demonstrated that LDLR up-expression or induction is an effective approach for brain apoE reduction to mitigate pathological changes in AD brains.
3.1. Overexpression of LDLR for apoE reduction
ApoE primarily binds to LDLR and LDLR-related protein 1 (LRP1) in the brain, and both LDLR and LRP1 belong to the LDLR family and are metabolic receptors that mediate the endocytosis and clearance of apoE lipoproteins (Holtzman et al 2012). While deletion of either LDLR or LRP1 significantly increases apoE levels in brain (Cao et al 2006, Fryer et al 2005, Liu et al 2007), the effects of LDLR overexpression on amyloid and tau pathologies are distinctly different from that of LRP1 overexpression. Although overexpression of a functional LRP1 minireceptor resulted in a 25% reduction of brain apoE levels, there was an age-dependent increase of soluble Aβ in the brain of PDAPP transgenic mice (Zerbinatti et al 2004). In addition, LRP1 mediates tau endocytosis and promotes tau propagation between neurons (Rauch et al 2020). In contrast, overexpression of LDLR is an effective approach to significantly induce apoE reduction and consequent alleviation of pathological changes in AD mouse models (Castellano et al 2012, Kim et al 2009, Shi et al 2021). Specifically, overexpression of LDLR in the brain of APP/PS1 transgenic mice result in a 50–90% reduction of brain apoE levels, which is correlated with marked attenuation of Aβ aggregation, induction of Aβ clearance and suppression of plaque-associated neuroinflammatory responses (Kim et al 2009). Importantly, 2-fold LDLR protein overexpression is sufficient to reduce more than 50% of brain apoE levels and Aβ accumulation, suggesting that modulation of LDLR expression is an applicable approach to reduce brain apoE levels for AD therapy (Kim et al 2009). Similarly, overexpression of LDLR in PS19 tauopathy mice also results in a significant reduction of brain apoE levels and inhibition of tau phosphorylation, which strongly correlate with one another, and consequent amelioration of tau pathology and neurodegeneration (Shi et al 2021). Together, these studies demonstrate that up-regulation of LDLR can lead to reduction of apoE and alleviation of AD pathologies.
ApoE receptor 2 (ApoER2) and VLDL receptor (VLDLR), which are the two closest family members of LDLR, regulate adult synaptic plasticity via mediation of Reelin signaling in CNS (Lane-Donovan & Herz 2017). While LDLR is a metabolic receptor of apoE, apoE4 can greatly inhibit the recycling of apoER2 to the cell surface and thereby severely impair Reelin signaling in neurons (Chen et al 2010, Feng et al 2020, Xian et al 2018). Therefore, it could be interesting to determine whether LDLR up-regulation attenuates the inhibition of apoE4 on apoER2 recycling and restores Reelin signaling in neurons.
3.2. Inhibition of IDOL for apoE reduction
Inducible degrader of the LDLR (IDOL) is an E3 ubiquitin ligase that binds to LDLR cytoplasmic tail and thereby triggers LDLR ubiquitination and degradation (Zhang et al 2011). IDOL plays a primary role in regulating brain LDLR protein and has a limited effect on hepatic LDLR levels in mice (Choi et al 2015, Hong et al 2014), although the IDOL-LDLR axis effectively regulates hepatic lipoprotein metabolism in primates (Hong et al 2014). Interestingly, IDOL deficiency resulted in a significant reduction of apoE levels, induction of Aβ clearance, decrease of amyloid plaque burden, and amelioration of neuroinflammation in APP/PS1 mice (Choi et al 2015). Using an antisense oligonucleotide (ASO) to reduce IDOL expression in the brains of APP/PS1 male mice, it was demonstrated that IDOL reduction led to a significant increase of brain LDLR levels, decrease of apoE levels, and alleviation of amyloid pathology and cognitive decline (Gao et al 2020). Together, these findings provide further evidence that apoE reduction is a promising therapeutic strategy for AD patients carrying the APOE ε4 allele.
3.3. Inhibition of PCSK9 for apoE reduction
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is another negative regulator of LDLR. PCSK9 binds to the extracellular domain of LDLR and thereby induces LDLR degradation in endosomes/lysosomes (O’Connell & Lohoff 2020). PCSK9 plays a crucial role in regulating LDLR metabolism, and loss-of-function (LOF) mutations of PCSK9 are associated with a marked increase of hepatic LDLR levels and a significant decrease of plasma LDL cholesterol levels (Cohen et al 2005, Mayne et al 2011). PCSK9 is expressed at a low level in adult brain but its expression is greatly upregulated in neurological disorders (O’Connell & Lohoff 2020). Studies with post-mortem human brains reveal that both mRNA and protein levels of PCSK9 are upregulated in frontal cortices of AD patients compared to controls (Picard et al 2019). Notably, PCSK9 levels in cerebrospinal fluid (CSF) are higher in AD APOE ɛ4 carriers than AD APOE ɛ4 non-carriers (Zimetti et al 2017). In addition, CSF PCSK9 levels are strongly correlated with CSF apoE levels in cognitively normal subjects with a parental history of late-onset AD (Picard et al 2019), suggesting that PCSK9 could regulate apoE metabolism via LDLR in the brain. Interestingly, it has been reported that pretreatment with PCSK9 inhibitor Pep2–8 (a small peptide) reduces Aβ42 levels and attenuates neuroinflammation and brain damage induced by cardiac ischemia/reperfusion injury in rats (Apaijai et al 2019), and that PCSK9 inhibitor alirocumab (a monoclonal antibody) significantly alleviates cognitive impairment and amyloidosis induced by high fat cholesterol diet in rats (Abuelezz & Hendawy 2021). However, it is unclear whether brain apoE is associated with the therapeutic effects of PCSK9 inhibitors in these experimental rat models. Nevertheless, these findings suggest that PCSK9 inhibition represents a therapeutic approach for AD therapy.
4. Upregulation of microglia apoE expression in AD brains
While some studies have shown that apoE levels are upregulated in human AD brains and CSF when compared to controls (Akram et al 2012, Lee et al 2020, Sihlbom et al 2008, Toledo et al 2014), others have reported that apoE levels are not changed (Martinez-Morillo et al 2014, Pirttila et al 1996, Schmidt et al 2014) or decreased (Bertrand et al 1995, Cruchaga et al 2012, Talwar et al 2016). Although studies on apoE mRNA or protein levels in human AD brains have yielded contradictory results, recent studies with single-cell RNA-sequencing and transcriptomic analyses of isolated microglia have revealed that Apoe is one of the most upregulated genes in a subset of microglia with neurodegenerative phenotype in amyloid mouse models (Keren-Shaul et al 2017, Krasemann et al 2017). In addition, apoE is enriched in amyloid plaques as a major constituent and promotes Aβ aggregation (Huynh et al 2017, Liu et al 2017, Ma et al 1994). Proteomic analyses have also demonstrated that apoE protein levels are greatly increased in plaque-associated microglia in amyloid mice (Parhizkar et al 2019). Importantly, it has been demonstrated that APOE is greatly upregulated in human AD microglia (HAM) from human AD brains (Mathys et al 2019, Srinivasan et al 2020, Zhou et al 2020). Together, significant upregulation of apoE in DAM from AD mouse models and HAM from human AD brains provide additional rationale that apoE reduction can be an attractive therapeutic strategy for AD patients carrying the APOE ε4 allele. It is important to mention that if the increased expression of apoE in DAM is a critical response to amyloid development, this process might be protective in the early amyloid phase of the disease but could be detrimental in the late phase when neurodegenerative can be accelerated by microglia overactivation. As such, in regard to apoE function in microglia, reduction of apoE as a therapeutic strategy might depend on the stage of the disease although further investigations are needed in this critically important research field.
5. Pharmacological approaches to directly induce apoE4 reduction for AD therapy
Genetic knockdown or deficiency of apoE leads to a marked attenuation of disease severity in APP and tau transgenic mouse models, thereby providing a strong rationale for developing pharmacological invention strategies to directly induce apoE4 reduction for AD therapy (Fig. 2).
Fig. 2.

Therapeutic approaches of apoE4 reduction for AD therapies. ApoE4 reduction leads to attenuation of neuroinflammation and alleviation of amyloid and tau pathologies in the AD brain. Specific apoE ASOs and siRNAs are promising therapeutic approaches to directly reduce apoE levels for the treatment of AD patients carrying APOE ε4 allele. In addition, the IDOL/PCSK9-LDLR-apoE axis provides an alternative approach to indirectly reduce apoE4 levels for AD therapies. Specific IDOL ASOs, and PCSK9 inhibitors Pep2–8 and Alirocumab (Aliro) display significant therapeutic effects on alleviation of pathological changes in experimental AD models.
5.1. Reduction of apoE4 level by apoE-specific ASOs for AD therapy
ASOs, a short single-strand of chemically modified nucleic acid polymers generally consisting of 16–25 nucleotides, are designed to alter the expression of their specific target genes through a variety of mechanisms. The FDA has approved eight ASO drugs since 2016, and the approval of the ASO-based drug nusinersen provides a strong basis for using this technology to develop novel and effective therapies for neurodegenerative diseases (Bennett et al 2021, Leavitt & Tabrizi 2020). Currently, ASO targeting MAPT (Tau) for AD treatment is under Phase 1/2 clinical trial (Bennett et al 2021).
Using ASOs to reduce human apoE expression in the brains of APP/PS1–21 mice homozygous for human APOE ε2, ε3 or ε4 allele, Huynh et al. demonstrated that apoE4 reduction prior to plaque deposition led to a significant decrease in Aβ pathology, and that lowering apoE4 after Aβ seeding resulted in a reduction in plaque-associated neuritic dystrophy (Huynh et al 2017). Using ASO against human APOE, Litvinchuk et al. further demonstrated that reduction of apoE4 protein levels by ~50% in the P301S/ApoE4 mouse model resulted in significant protection against tau pathology and associated neurodegeneration as well as inhibition of neuroinflammation (Litvinchuk et al 2021). Therefore, ASO-mediated apoE4 reduction can alleviate not only amyloid pathology but also tau pathology and tau-mediated neurodegeneration.
5.2. Reduction of apoE4 levels by apoE-specific siRNAs for AD therapy
Double-stranded small-interfering RNA (siRNA) is another major type of oligonucleotide-based therapeutics. The FDA has approved four siRNA-based therapeutics including the recently approved drug inclisiran which targets PCSK9 for the treatment of hypercholesterolaemia (FDA 2021, Zhang et al 2021). With the advances in siRNA chemical modifications for potent and persistent inhibition of gene expression throughout the brain, this technology has great potential in developing novel and effective therapies for neurological disorders (Alterman et al 2019). Encouragingly, it was recently reported that tissue-specific apoE siRNAs completely silenced apoE expression in the brain, had no effects on systemic cholesterol and greatly inhibited amyloid plaque formation in APP/PS1 transgenic mice (Ferguson 2021, Khvorova et al 2020). These findings demonstrate that siRNA-mediated apoE reduction is a promising strategy for AD therapy.
5.3. Reduction of apoE4 levels using apoE antibodies
Anti-apoE immunotherapy is one of the therapeutic approaches that specifically target detrimental apoE4 (Safieh et al 2019). ApoE antibodies can suppress amyloidosis progression, alleviate amyloid pathology and improve cognitive function through multiple mechanisms including modification of glia responses (Kim et al 2012, Liao et al 2014, Liao et al 2018, Xiong et al 2021), regulation of proinflammatory cytokines (Kim et al 2012), amendment of the apoE4-induced apoER2 reduction (Luz et al 2016), and improvement of cerebrovascular function (Xiong et al 2021). Interestingly, long-term treatment (weekly intraperitoneal injections for 21 weeks) with the anti-apoE antibody HJ6.3 leads to the downregulation of brain apoE levels in APP/PS1 mice (Liao et al 2014), suggesting that apoE reduction might contribute to the therapeutic effects of HJ6.3 in amyloid pathology.
6. Perspectives and conclusion
Brain apoE is primarily produced by glial cells and plays a critical role in lipid transport for neuronal maintenance and repair (Lanfranco et al 2020), although apoE4 proteins are poorly lipidated and display an impaired capacity in lipid transport when compared to apoE3 proteins (Gong et al 2002, Hu et al 2015). There is a long debate about the benefits of apoE induction or reduction during the pathological changes of AD (Belloy et al 2019, Liao et al 2018). While expression of apoE2 and apoE3 might be beneficial (Li et al 2020, Robert et al 2020), the increased expression of apoE4 is evidently detrimental for AD progression as discussed in this review. Nuclear receptor liver X receptors (LXRs) and retinoid X receptor (RXR) are obligate heterodimers and play an important role in apoE expression/secretion and cholesterol homeostasis in the brain (Liang et al 2004). Both the LXR agonists such as TO901317 and RXR agonists such as bexarotene upregulate apoE expression, but these agonists have showed inconsistent results on Aβ deposition and cognitive function in AD mouse models (Li et al 2020, Williams et al 2020). Bexarotene is an FDA-approved drug for the treatment of cutaneous T-cell lymphoma. In AD clinical trials, bexarotene failed to produce any cognitive benefits (Cummings et al 2016). In addition, as a direct transcriptional target of LXR, IDOL expression is induced by LXR ligand and reduced by LXR knockout (Zelcer et al 2009). Therefore, LXR agonists could stimulate IDOL expression, which results in downregulation of LDLR, and subsequent aggravation of the pathological changes of AD. ApoE lipidation is controlled by ATP binding cassette A1 (ABCA1), which is also transcriptionally regulated by LXR and RXR (Koldamova et al 2014). As the LXR/RXR system has many targets, selective activation of ABCA1 is an attractive therapeutic strategy to increase apoE lipidation for the treatment of AD (Boehm-Cagan et al 2016, Lanfranco et al 2020, Moulton et al 2021). Nevertheless, the evidence presented here highlights that apoE reduction is a promising therapeutic strategy for treatment of AD patients carrying the APOE ε4 allele. Importantly, apoE4 can drive amyloid pathology during the seeding stage (Liu et al 2017); thus, apoE4 reduction could have both prophylactic and therapeutic applications.
There is always a concern that complete depletion of apoE expression could be harmful for neuronal maintenance, repair, and brain homeostasis (Belloy et al 2019, Liao et al 2018). Particularly, apoE mediates the transport of cholesterol and other lipids, which is essential for synaptogenesis and neuronal growth/survival in normal brains (Flowers & Rebeck 2020, Li et al 2021, Li et al 2020, Martin et al 2014, Yu et al 2021). In addition, peripheral apoE, which is produced primarily from the liver, is a key player in cholesterol metabolism and has prominent effects on cardiovascular disease (Belloy et al 2019). Therefore, apoE depletion might result in unintended adverse effects in APOE3/APOE4 heterozygotes. Interestingly, a patient carrying a mutation associated with complete absence of apoE exhibited normal cognitive, neurological, and retinal function, and had no significant symptoms of cardiovascular disease, suggesting that there might be alternative mechanisms to fulfill the apoE function in the brain (Mak et al 2014). Nevertheless, the therapeutic application of apoE4 reduction should aim to reduce apoE levels, but not completely deplete apoE, in AD patients. Indeed, apoE reduction at about 50% by apoE ASO treatment or LDLR overexpression is sufficient to alleviate the pathological changes in AD mouse models (Kim et al 2009, Litvinchuk et al 2021).
While specific apoE ASOs and siRNAs are attractive therapeutic approaches to directly reduce apoE levels for AD patients carrying APOE ε4 allele, developing novel and CNS-specific small molecules to reduce apoE4 levels is another promising therapeutic strategy. Unfortunately, there are currently no such small molecule apoE4 inhibitors reported in the literature. Thereby, it is desirable to develop high-throughput screening approaches to identify compounds that reduce apoE4 levels (Krishnan et al 2020). Small molecules that reduce apoE4 levels in brain, particularly those targeting apoE and LDLR, could represent novel therapeutic tools in AD.
The IDOL/PCSK9-LDLR-apoE axis provides an alternative approach to indirectly reduce apoE4 levels for AD therapy (Fig. 2). Besides LDLR, both IDOL and PCSK9 also promote the degradation of ApoER2 and VLDLR (Hong et al 2010, Poirier et al 2008). While targeting IDOL is an effective way to reduce apoE brain levels (Choi et al 2015, Gao et al 2020), complete inactivation of IDOL induces constitutive overexpression of ApoER2 and consequent impairment of learning and memory in mice (Gao et al 2017). In addition, IDOL plays a critical role in the regulation of neuronal VLDLR expression and systemic energy balance (Lee et al 2019). Currently, it is unclear whether IDOL is dysregulated in human AD brains. Further studies are required to verify that targeting IDOL or PCSK9 is an applicable approach to modulate LDLR-apoE axis in the brain for AD therapy. The siRNA-based PCSK9 inhibitor inclisiran, which increases LDLR in hepatocytes, has been recently approved for the treatment of hypercholesterolemia (FDA 2021). It will be interesting to examine whether a similar siRNA strategy targeting brain PCSK9 is able to enhance brain LDLR levels and thereby reduce apoE4 levels for AD treatment.
In conclusion, APOE4 is the strongest genetic risk factor for AD with high prevalence (Saunders et al 1993, Sims et al 2020). Mounting evidence indicates that apoE4 provokes multiple AD pathogenic pathways and pathologies, supporting the notion that apoE reduction is a promising therapeutic approach in the treatment of AD patients carrying the APOE ε4 allele. With the recent advances in technologies for increasing bioavailability and stability and minimizing toxicity of ASOs and siRNAs, apoE4 reduction therapies are likely to be evaluated on safety and efficacy in clinical trials in the near future.
Highlights.
ApoE4 provokes neuroinflammation and exacerbates amyloid and tau pathologies
Genetic knock-down or depletion of apoE alleviates amyloid and tau pathologies
ApoE reduction via its receptor LDLR attenuates AD pathologies
ApoE4 reduction by apoE specific ASOs and siRNAs alleviates AD pathologies
Acknowledgements
We thank Hongmei Li for the critical reading of the manuscript. This work was supported in part by grants from the National Institutes of Health R21AG065653 to Y.L. and RF1AG062110 to C.-C. Liu and the Florida Department of Health Ed and Ethel Moore Alzheimer’s Disease Research Program Pilot Grant 9AZ09 to Y.L., and multiple grants from the NIH and Cure Alzheimer’s Fund to G.B.
Footnotes
Competing interests
GB consults for SciNeuro and Lexeo, is on the scientific advisory board for Kisbee, has consulted for AbbVie, E-Scape, Eisai, and Vida Ventures. Other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abuelezz SA, Hendawy N. 2021. HMGB1/RAGE/TLR4 axis and glutamate as novel targets for PCSK9 inhibitor in high fat cholesterol diet induced cognitive impairment and amyloidosis. Life Sci 273: 119310. [DOI] [PubMed] [Google Scholar]
- Akram A, Schmeidler J, Katsel P, Hof PR, Haroutunian V. 2012. Association of ApoE and LRP mRNA levels with dementia and AD neuropathology. Neurobiol Aging 33: 628 e1–28 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alterman JF, Godinho B, Hassler MR, Ferguson CM, Echeverria D, et al. 2019. A divalent siRNA chemical scaffold for potent and sustained modulation of gene expression throughout the central nervous system. Nat Biotechnol 37: 884–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews-Zwilling Y, Bien-Ly N, Xu Q, Li G, Bernardo A, et al. 2010. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci 30: 13707–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apaijai N, Moisescu DM, Palee S, McSweeney CM, Saiyasit N, et al. 2019. Pretreatment With PCSK9 Inhibitor Protects the Brain Against Cardiac Ischemia/Reperfusion Injury Through a Reduction of Neuronal Inflammation and Amyloid Beta Aggregation. J Am Heart Assoc 8: e010838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arboleda-Velasquez JF, Lopera F, O’Hare M, Delgado-Tirado S, Marino C, et al. 2019. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat Med 25: 1680–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, et al. 2015. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J Biol Chem 290: 26043–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CC, DeVaux LB, Farzan M. 2015. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J Biol Chem 290: 26033–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Liu F, Wu S, Lin S, Koger D, et al. 2009. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J Neurosci 29: 6771–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Verina T, Cummins DJ, Du YS, Dodel TC, et al. 1999. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. P Natl Acad Sci USA 96: 15233–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, et al. 1997. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet 17: 263–4 [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, et al. 2012. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485: 512–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloy ME, Napolioni V, Greicius MD. 2019. A Quarter Century of APOE and Alzheimer’s Disease: Progress to Date and the Path Forward. Neuron 101: 820–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CF, Kordasiewicz HB, Cleveland DW. 2021. Antisense Drugs Make Sense for Neurological Diseases. Annu Rev Pharmacol 61: 831–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RE, Robbins AB, Hu M, Cao X, Betensky RA, et al. 2018. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. P Natl Acad Sci USA 115: E1289–E98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. 2007. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 39: 17–23 [DOI] [PubMed] [Google Scholar]
- Bertrand P, Poirier J, Oda T, Finch CE, Pasinetti GM. 1995. Association of apolipoprotein E genotype with brain levels of apolipoprotein E and apolipoprotein J (clusterin) in Alzheimer disease. Brain Res Mol Brain Res 33: 174–8 [DOI] [PubMed] [Google Scholar]
- Bien-Ly N, Gillespie AK, Walker D, Yoon SY, Huang Y. 2012. Reducing human apolipoprotein E levels attenuates age-dependent Abeta accumulation in mutant human amyloid precursor protein transgenic mice. J Neurosci 32: 4803–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm-Cagan A, Bar R, Liraz O, Bielicki JK, Johansson JO, Michaelson DM. 2016. ABCA1 Agonist Reverses the ApoE4-Driven Cognitive and Brain Pathologies. J Alzheimers Dis 54: 1219–33 [DOI] [PubMed] [Google Scholar]
- Cao D, Fukuchi K, Wan H, Kim H, Li L. 2006. Lack of LDL receptor aggravates learning deficits and amyloid deposits in Alzheimer transgenic mice. Neurobiol Aging 27: 1632–43 [DOI] [PubMed] [Google Scholar]
- Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, et al. 2012. Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Abeta clearance in a mouse model of beta-amyloidosis. Proc Natl Acad Sci U S A 109: 15502–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Durakoglugil MS, Xian X, Herz J. 2010. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A 107: 12011–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Gao J, Kim J, Hong C, Kim J, Tontonoz P. 2015. The E3 ubiquitin ligase Idol controls brain LDL receptor expression, ApoE clearance, and Abeta amyloidosis. Sci Transl Med 7: 314ra184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. 2005. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet 37: 161–5 [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, et al. 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261: 921–3 [DOI] [PubMed] [Google Scholar]
- Cruchaga C, Kauwe JS, Nowotny P, Bales K, Pickering EH, et al. 2012. Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer’s disease. Hum Mol Genet 21: 4558–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings J 2021. New approaches to symptomatic treatments for Alzheimer’s disease. Mol Neurodegener 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JL, Zhong K, Kinney JW, Heaney C, Moll-Tudla J, et al. 2016. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene Xin moderate Alzheimer’s disease. Alzheimers Res Ther 8: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egensperger R, Kosel S, von Eitzen U, Graeber MB. 1998. Microglial activation in Alzheimer disease: Association with APOE genotype. Brain Pathol 8: 439–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, et al. 1997. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. Jama 278: 1349–56 [PubMed] [Google Scholar]
- FDA. 2021. Drugs@FDA: FDA-Approved Drugs. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&varApplNo=214012.
- Feng M, Cui D, Li Y, Shi J, Xiang L, et al. 2020. Carnosic Acid Reverses the Inhibition of ApoE4 on Cell Surface Level of ApoER2 and Reelin Signaling Pathway. J Alzheimers Dis 73: 517–28 [DOI] [PubMed] [Google Scholar]
- Ferguson CM. 2021. Modulating ApoE with Tissue Specific siRNAs in Alzheimer’s Disease. GSBS Dissertations and Theses. 10.13028/qk23-zg81. Retrieved from https://escholarship.umassmed.edu/gsbs_diss/1132. [DOI] [Google Scholar]
- Ferreira-Vieira TH, Guimaraes IM, Silva FR, Ribeiro FM. 2016. Alzheimer’s disease: Targeting the Cholinergic System. Curr Neuropharmacol 14: 101–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippini N, Rao A, Wetten S, Gibson RA, Borrie M, et al. 2009. Anatomically-distinct genetic associations of APOE epsilon4 allele load with regional cortical atrophy in Alzheimer’s disease. Neuroimage 44: 724–8 [DOI] [PubMed] [Google Scholar]
- Flowers SA, Rebeck GW. 2020. APOE in the normal brain. Neurobiol Dis 136: 104724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer JD, Demattos RB, McCormick LM, O’Dell MA, Spinner ML, et al. 2005. The low density lipoprotein receptor regulates the level of central nervous system human and murine apolipoprotein E but does not modify amyloid plaque pathology in PDAPP mice. J Biol Chem 280: 25754–9 [DOI] [PubMed] [Google Scholar]
- Gandy S, Knopman DS, Sano M. 2021. Talking points for physicians, patients and caregivers considering Aduhelm(R) infusion and the accelerated pathway for its approval by the FDA. Mol Neurodegener 16: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Littman R, Diamante G, Xiao X, Ahn IS, et al. 2020. Therapeutic IDOL Reduction Ameliorates Amyloidosis and Improves Cognitive Function in APP/PS1 Mice. Mol Cell Biol 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Marosi M, Choi J, Achiro JM, Kim S, et al. 2017. The E3 ubiquitin ligase IDOL regulates synaptic ApoER2 levels and is important for plasticity and learning. Elife 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. 2015. A century of cholesterol and coronaries: from plaques to genes to statins. Cell 161: 161–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong JS, Kobayashi M, Hayashi H, Zou K, Sawamura N, et al. 2002. Apolipoprotein E (ApoE) isoform-dependent lipid release from astrocytes prepared from human ApoE3 and ApoE4 knock-in mice. J Biol Chem 277: 29919–26 [DOI] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, et al. 2013. TREM2 variants in Alzheimer’s disease. N Engl J Med 368: 117–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Zhang D, Zeng Y, Huang TY, Xu H, Zhao Y. 2020. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol Neurodegener 15: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, et al. 2000a. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 97: 2892–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Fagan AM, Mackey B, Tenkova T, Sartorius L, et al. 2000b. Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann Neurol 47: 739–47 [PubMed] [Google Scholar]
- Holtzman DM, Herz J, Bu G. 2012. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med 2: a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong C, Duit S, Jalonen P, Out R, Scheer L, et al. 2010. The E3 ubiquitin ligase IDOL induces the degradation of the low density lipoprotein receptor family members VLDLR and ApoER2. J Biol Chem 285: 19720–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong C, Marshall SM, McDaniel AL, Graham M, Layne JD, et al. 2014. The LXR-Idol axis differentially regulates plasma LDL levels in primates and mice. Cell Metab 20: 910–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Liu CC, Chen XF, Zhang YW, Xu H, Bu G. 2015. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Abeta metabolism in apoE4-targeted replacement mice. Mol Neurodegener 10: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh TV, Liao F, Francis CM, Robinson GO, Serrano JR, et al. 2017. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of beta-amyloidosis. Neuron 96: 1013–23 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendresen C, Arskog V, Daws MR, Nilsson LNG. 2017. The Alzheimer’s disease risk factors apolipoprotein E and TREM2 are linked in a receptor signaling pathway. J Neuroinflamm 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, et al. 2013. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368: 107–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, et al. 2017. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169: 1276–90 e17 [DOI] [PubMed] [Google Scholar]
- Khvorova A, Ferguson F, Rogaev E. 2020. OLIGONUCLEOTIDES FOR TISSUE SPECIFIC APOE MODULATION. https://patents.justia.com/patent/20200362341. [Google Scholar]
- Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, et al. 2009. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron 64: 632–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Eltorai AEM, Jiang H, Liao F, Verghese PB, et al. 2012. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of A beta amyloidosis. Journal of Experimental Medicine 209: 2149–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, et al. 2011. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J Neurosci 31: 18007–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloske CM, Wilcock DM. 2020. The Important Interface Between Apolipoprotein E and Neuroinflammation in Alzheimer’s Disease. Front Immunol 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knol MJ, Lu D, Traylor M, Adams HHH, Romero JRJ, et al. 2020. Association of common genetic variants with brain microbleeds: A genome-wide association study. Neurology 95: e3331–e43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova R, Fitz NF, Lefterov I. 2014. ATP-binding cassette transporter A1: from metabolism to neurodegeneration. Neurobiol Dis 72 Pt A: 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsodendris N, Nelson MR, Rao A, Huang Y. 2021. Apolipoprotein E and Alzheimer’s Disease: Findings, Hypotheses, and Potential Mechanisms. Annu Rev Pathol [DOI] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, et al. 2017. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47: 566–81 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan N, Chen X, Donnelly-Roberts D, Mohler EG, Holtzman DM, Gopalakrishnan SM. 2020. Small Molecule Phenotypic Screen Identifies Novel Regulators of LDLR Expression. ACS Chem Biol 15: 3262–74 [DOI] [PubMed] [Google Scholar]
- Lane-Donovan C, Herz J. 2017. The ApoE receptors Vldlr and Apoer2 in central nervous system function and disease. J Lipid Res 58: 1036–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfranco MF, Ng CA, Rebeck GW. 2020. ApoE Lipidation as a Therapeutic Target in Alzheimer’s Disease. Int J Mol Sci 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavitt BR, Tabrizi SJ. 2020. Antisense oligonucleotides for neurodegeneration. Science 367: 1428–29 [DOI] [PubMed] [Google Scholar]
- Lee EG, Tulloch J, Chen S, Leong L, Saxton AD, et al. 2020. Redefining transcriptional regulation of the APOE gene and its association with Alzheimer’s disease. PLoS One 15: e0227667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SD, Priest C, Bjursell M, Gao J, Arneson DV, et al. 2019. IDOL regulates systemic energy balance through control of neuronal VLDLR expression. Nat Metab 1: 1089–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang J, Li D, He C, He K, et al. 2021. Astrocytic ApoE reprograms neuronal cholesterol metabolism and histone-acetylation-mediated memory. Neuron 109: 957–70 e8 [DOI] [PubMed] [Google Scholar]
- Li Z, Shue F, Zhao N, Shinohara M, Bu G. 2020. APOE2: protective mechanism and therapeutic implications for Alzheimer’s disease. Mol Neurodegener 15: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Lin S, Beyer TP, Zhang Y, Wu X, et al. 2004. A liver X receptor and retinoid X receptor heterodimer mediates apolipoprotein E expression, secretion and cholesterol homeostasis in astrocytes. J Neurochem 88: 623–34 [DOI] [PubMed] [Google Scholar]
- Liao F, Hori Y, Hudry E, Bauer AQ, Jiang H, et al. 2014. Anti-ApoE Antibody Given after Plaque Onset Decreases A beta Accumulation and Improves Brain Function in a Mouse Model of A beta Amyloidosis. Journal of Neuroscience 34: 7281–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Li A, Xiong M, Bien-Ly N, Jiang H, et al. 2018. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J Clin Invest 128: 2144–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YT, Seo J, Gao F, Feldman HM, Wen HL, et al. 2018. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 98: 1141–54 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvinchuk A, Huynh TV, Shi Y, Jackson RJ, Finn MB, et al. 2021. Apolipoprotein E4 Reduction with Antisense Oligonucleotides Decreases Neurodegeneration in a Tauopathy Model. Ann Neurol 89: 952–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Zhao N, Fu Y, Wang N, Linares C, et al. 2017. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 96: 1024–32 e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zerbinatti CV, Zhang J, Hoe HS, Wang B, et al. 2007. Amyloid precursor protein regulates brain apolipoprotein e and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 56: 66–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz I, Liraz O, Michaelson DM. 2016. An Anti-apoE4 Specific Monoclonal Antibody Counteracts the Pathological Effects of apoE4 In Vivo. Curr Alzheimer Res 13: 918–29 [DOI] [PubMed] [Google Scholar]
- Ma J, Yee A, Brewer HB Jr., Das S, Potter H. 1994. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature 372: 92–4 [DOI] [PubMed] [Google Scholar]
- Magno L, Bunney TD, Mead E, Svensson F, Bictash MN. 2021. TREM2/PLCgamma2 signalling in immune cells: function, structural insight, and potential therapeutic modulation. Mol Neurodegener 16: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak AC, Pullinger CR, Tang LF, Wong JS, Deo RC, et al. 2014. Effects of the absence of apolipoprotein e on lipoproteins, neurocognitive function, and retinal function. JAMA Neurol 71: 1228–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MG, Pfrieger F, Dotti CG. 2014. Cholesterol in brain disease: sometimes determinant and frequently implicated. EMBO Rep 15: 1036–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Morillo E, Hansson O, Atagi Y, Bu G, Minthon L, et al. 2014. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathol 127: 633–43 [DOI] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, et al. 2019. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570: 332–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayne J, Dewpura T, Raymond A, Bernier L, Cousins M, et al. 2011. Novel loss-of-function PCSK9 variant is associated with low plasma LDL cholesterol in a French-Canadian family and with impaired processing and secretion in cell culture. Clin Chem 57: 1415–23 [DOI] [PubMed] [Google Scholar]
- McDade E, Llibre-Guerra JJ, Holtzman DM, Morris JC, Bateman RJ. 2021. The informed road map to prevention of Alzheimer Disease: A call to arms. Mol Neurodegener 16: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Blazey TM, Holtzman DM, Cruchaga C, Su Y, et al. 2018. Longitudinal brain imaging in preclinical Alzheimer disease: impact of APOE epsilon4 genotype. Brain 141: 1828–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, et al. 2020. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 581: 70–+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Zhao Z, Zlokovic BV. 2017. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? Journal of Experimental Medicine 214: 3151–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulton MJ, Barish S, Ralhan I, Chang J, Goodman LD, et al. 2021. Neuronal ROS-induced glial lipid droplet formation is altered by loss of Alzheimer’s disease-associated genes. Proc Natl Acad Sci U S A 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, Gomez-Isla T, Holtzman DM. 2021. Aducanumab for Alzheimer disease: the amyloid hypothesis moves from bench to bedside. J Clin Invest 131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nation DA, Sweeney MD, Montagne A, Sagare AP, D’Orazio LM, et al. 2019. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nature Medicine 25: 270–+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen B, Bix G, Yao Y. 2021. Basal lamina changes in neurodegenerative disorders. Mol Neurodegener 16: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell EM, Lohoff FW. 2020. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) in the Brain and Relevance for Neuropsychiatric Disorders. Front Neurosci 14: 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olgiati P, Politis A, Malitas P, Albani D, Dusi S, et al. 2010. APOE epsilon-4 allele and cytokine production in Alzheimer’s disease. Int J Geriatr Psychiatry 25: 338–44 [DOI] [PubMed] [Google Scholar]
- Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, et al. 2019. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci 22: 191–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard C, Poirier A, Belanger S, Labonte A, Auld D, et al. 2019. Proprotein convertase subtilisin/kexin type 9 (PCSK9) in Alzheimer’s disease: A genetic and proteomic multi-cohort study. PLoS One 14: e0220254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirttila T, Soininen H, Heinonen O, Lehtimaki T, Bogdanovic N, et al. 1996. Apolipoprotein E (apoE) levels in brains from Alzheimer disease patients and controls. Brain Res 722: 71–7 [DOI] [PubMed] [Google Scholar]
- Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, et al. 2008. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem 283: 2363–72 [DOI] [PubMed] [Google Scholar]
- Polvikoski T, Sulkava R, Haltia M, Kainulainen K, Vuorio A, et al. 1995. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med 333: 1242–7 [DOI] [PubMed] [Google Scholar]
- Premkumar DR, Cohen DL, Hedera P, Friedland RP, Kalaria RN. 1996. Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer’s disease. Am J Pathol 148: 2083–95 [PMC free article] [PubMed] [Google Scholar]
- Rauch JN, Luna G, Guzman E, Audouard M, Challis C, et al. 2020. LRP1 is a master regulator of tau uptake and spread. Nature 580: 381–+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Chen KW, Liu XF, Bandy D, Yu MX, et al. 2009. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. P Natl Acad Sci USA 106: 6820–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert J, Button EB, Martin EM, McAlary L, Gidden Z, et al. 2020. Cerebrovascular amyloid Angiopathy in bioengineered vessels is reduced by high-density lipoprotein particles enriched in Apolipoprotein E. Mol Neurodegener 15: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GA, Tai LM, LaDu MJ, Rebeck GW. 2014. Human APOE4 increases microglia reactivity at A beta plaques in a mouse model of A beta deposition. J Neuroinflamm 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safieh M, Korczyn AD, Michaelson DM. 2019. ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC Med 17: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, et al. 1993. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43: 1467–72 [DOI] [PubMed] [Google Scholar]
- Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, et al. 1993. Increased Amyloid Beta-Peptide Deposition in Cerebral-Cortex as a Consequence of Apolipoprotein-E Genotype in Late-Onset Alzheimer-Disease. P Natl Acad Sci USA 90: 9649–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Becker H, Zerr I. 2014. Cerebrospinal fluid apolipoprotein E concentration and severity of cognitive impairment in patients with newly diagnosed Alzheimer’s disease. Am J Alzheimers Dis Other Demen 29: 54–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schram MT, Euser SM, de Craen AJ, Witteman JC, Frolich M, et al. 2007. Systemic markers of inflammation and cognitive decline in old age. J Am Geriatr Soc 55: 708–16 [DOI] [PubMed] [Google Scholar]
- Shi Y, Andhey PS, Ising C, Wang K, Snipes LL, et al. 2021. Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms. Neuron 109: 2413–26 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Holtzman DM. 2018a. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol 18: 759–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Holtzman DM. 2018b. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol 18: 759–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao LZ, et al. 2017a. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549: 523–+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao LZ, et al. 2017b. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549: 523–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sihlbom C, Davidsson P, Sjogren M, Wahlund LO, Nilsson CL. 2008. Structural and quantitative comparison of cerebrospinal fluid glycoproteins in Alzheimer’s disease patients and healthy individuals. Neurochem Res 33: 133-–240. [DOI] [PubMed] [Google Scholar]
- Sims R, Hill M, Williams J. 2020. The multiplex model of the genetics of Alzheimer’s disease. Nat Neurosci 23: 311–22 [DOI] [PubMed] [Google Scholar]
- Srinivasan K, Friedman BA, Etxeberria A, Huntley MA, van Der Brug MP, et al. 2020. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talwar P, Sinha J, Grover S, Agarwal R, Kushwaha S, et al. 2016. Meta-analysis of apolipoprotein E levels in the cerebrospinal fluid of patients with Alzheimer’s disease. J Neurol Sci 360: 179–87 [DOI] [PubMed] [Google Scholar]
- Therriault J, Benedet AL, Pascoal TA, Mathotaarachchi S, Chamoun M, et al. 2020. Association of Apolipoprotein E epsilon4 With Medial Temporal Tau Independent of Amyloid-beta. JAMA Neurol 77: 470–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo JB, Da X, Weiner MW, Wolk DA, Xie SX, et al. 2014. CSF Apo-E levels associate with cognitive decline and MRI changes. Acta Neuropathol 127: 621–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Ulland TK, Mahan TE, Nystrom S, Nilsson KP, et al. 2018. ApoE facilitates the microglial response to amyloid plaque pathology. J Exp Med 215: 1047–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wageningen TA, Gerrits E, Palacin IBS, Huitinga I, Eggen BJL, van Dam AM. 2021. Exploring reported genes of microglia RNA-sequencing data: Uses and considerations. Glia 69: 2933–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernooij MW, van der Lugt A, Ikram MA, Wielopolski PA, Niessen WJ, et al. 2008. Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology 70: 1208–14 [DOI] [PubMed] [Google Scholar]
- Wang C, Najm R, Xu Q, Jeong DE, Walker D, et al. 2018. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med 24: 647–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Xiong M, Gratuze M, Bao X, Shi Y, et al. 2021a. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 109: 1657–74 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YT, Pascoal TA, Therriault J, Kang MS, Benedet AL, et al. 2021b. Interactive rather than independent effect of APOE and sex potentiates tau deposition in women. Brain Commun 3: fcab126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigand AJ, Thomas KR, Bangen KJ, Eglit GML, Delano-Wood L, et al. 2021. APOE interacts with tau PET to influence memory independently of amyloid PET in older adults without dementia. Alzheimers Dement 17: 61–69 [DOI] [PubMed] [Google Scholar]
- Williams T, Borchelt DR, Chakrabarty P. 2020. Therapeutic approaches targeting Apolipoprotein E function in Alzheimer’s disease. Mol Neurodegener 15: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xian X, Pohlkamp T, Durakoglugil MS, Wong CH, Beck JK, et al. 2018. Reversal of ApoE4-induced recycling block as a novel prevention approach for Alzheimer’s disease. Elife 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong M, Jiang H, Serrano JR, Gonzales ER, Wang C, et al. 2021. APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Science Translational Medicine 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Liu CC, Yamazaki A, Shue F, Martens YA, et al. 2021. Vascular ApoE4 Impairs Behavior by Modulating Gliovascular Function. Neuron 109: 438–47 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu GJ. 2019. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nature Reviews Neurology 15: 501–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Coppola K, Schweig JE, Crawford F, Mullan M, Paris D. 2019. Distinct Signaling Pathways Regulate TREM2 Phagocytic and NFkappaB Antagonistic Activities. Front Cell Neurosci 13: 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. 2016. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 91: 328–40 [DOI] [PubMed] [Google Scholar]
- Yu TS, Tensaouti Y, Stephanz EP, Chintamen S, Rafikian EE, et al. 2021. Astrocytic ApoE underlies maturation of hippocampal neurons and cognitive recovery after traumatic brain injury in mice. Commun Biol 4: 1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelcer N, Hong C, Boyadjian R, Tontonoz P. 2009. LXR Regulates Cholesterol Uptake Through Idol-Dependent Ubiquitination of the LDL Receptor. Science 325: 100–04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbinatti CV, Wozniak DF, Cirrito J, Cam JA, Osaka H, et al. 2004. Increased soluble amyloid-beta peptide and memory deficits in amyloid model mice overexpressing the low-density lipoprotein receptor-related protein. Proc Natl Acad Sci U S A 101: 1075–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Fairall L, Goult BT, Calkin AC, Hong C, et al. 2011. The IDOL-UBE2D complex mediates sterol-dependent degradation of the LDL receptor. Genes Dev 25: 1262–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MM, Bahal R, Rasmussen TP, Manautou JE, Zhong XB. 2021. The growth of siRNA-based therapeutics: Updated clinical studies. Biochem Pharmacol 189: 114432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Fu Y, Yamazaki Y, Ren Y, Davis MD, et al. 2020. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat Commun 11: 5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Song WM, Andhey PS, Swain A, Levy T, et al. 2020. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26: 131–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Nwabuisi-Heath E, Dumanis SB, Tai LM, Yu C, et al. 2012. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia 60: 559–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimetti F, Caffarra P, Ronda N, Favari E, Adorni MP, et al. 2017. Increased PCSK9 Cerebrospinal Fluid Concentrations in Alzheimer’s Disease. J Alzheimers Dis 55: 315–20 [DOI] [PubMed] [Google Scholar]