

Abstract
Molecularly targeted therapeutics have revolutionized the treatment of BRAFV600E-driven malignant melanoma, but the rapid development of resistance to BRAF kinase inhibitors (BRAFi) presents a significant obstacle. The use of clinical antimalarials for the investigational treatment of malignant melanoma has shown only moderate promise, attributed mostly to inhibition of lysosomal-autophagic adaptations of cancer cells, but identification of specific antimalarials displaying single-agent antimelanoma activity has remained elusive. Here, we have screened a focused library of clinically used artemisinin-combination therapeutic (ACT) antimalarials for the apoptotic elimination of cultured malignant melanoma cell lines, also examining feasibility of overcoming BRAFi-resistance comparing isogenic melanoma cells that differ only by NRAS mutational status (BRAFi-sensitive A375-BRAFV600E/NRASQ61 versus BRAFi-resistant A375-BRAFV600E/NRASQ61K). Among ACT antimalarials tested, mefloquine (MQ) was the only apoptogenic agent causing melanoma cell death at low micromolar concentrations. Comparative gene expression-array analysis (A375-BRAFV600E/NRASQ61 versus A375-BRAFV600E/NRASQ61K) revealed that MQ is a dual inducer of endoplasmic reticulum (ER) and redox stress responses that precede MQ-induced loss of viability. ER-tracker™ DPX fluorescence imaging and electron microscopy indicated ER swelling, accompanied by rapid induction of ER stress signaling (phospho-eIF2α, XBP-1s, ATF4). Fluo-4 AM-fluorescence indicated the occurrence of cytosolic calcium overload observable within seconds of MQ exposure. In a bioluminescent murine model employing intracranial injection of A375-Luc2 (BRAFV600E/NRASQ61K) cells, an oral MQ regimen efficiently antagonized brain tumor growth. Taken together, these data suggest that the clinical antimalarial MQ may be a valid candidate for drug repurposing aiming at chemotherapeutic elimination of malignant melanoma cells, even if metastasized to the brain and BRAFi-resistant.
Keywords: malignant melanoma, NRAS-driven BRAFi-resistance, antimalarial chemotherapeutics, mefloquine, ER stress, brain metastases, gene expression array analysis
1. Introduction
Metastatic melanoma is a malignant tumor originating from neural crest-derived melanocytes causing the majority of skin cancer-related deaths.1 Molecularly targeted therapeutics have revolutionized the treatment of BRAFV600E-driven malignant melanoma, but BRAF inhibitor (BRAFi) resistance compromises long-term survivorship of patients.2–4
Importantly, in treatment-naïve melanoma tumors, NRAS mutations rarely occur together with either BRAF or PTEN mutations, a phenomenon referred to as oncogene exclusion.5–8 In contrast, during the course of BRAFV600E-targeted BRAFi-treatment, emergence of resistance often involves NRAS mutations [predominantly in codon 61 (Q61R, Q61K) observable in approximately 20% of patients], a unique co-occurrence of BRAF and NRAS mutations facilitated and selected for by BRAFi-based therapy.3, 9
In addition to BRAFi-resistance compromising clinical outcomes in melanoma patients, an urgent need exists for the identification of molecular therapeutics that can target these drug-resistant cells at metastatic sites including the brain.10–12 Indeed, brain metastases are a frequent complication in patients with metastatic melanoma. Up to 45% of patients develop clinically documented brain tumors during their lifetime, and it has been reported that brain lesions are observable in more than 70 % of melanoma patients at autopsy.10, 12–15
The use of clinical antimalarials for the investigational treatment of malignant melanoma has shown moderate promise, attributed mostly to inhibition of lysosomal-autophagic adaptations of cancer cells.16–18 Artemisinin-combination-therapeutics (ACT) are now standard of care medications used as antimalarials worldwide.19 Beyond clinically used artemisinin-endoperoxides (including dihydroartemisinin acting through induction of oxidative stress), numerous ACT agents of the amino-quinoline class (such as amodiaquine, primaquine, chloroquine, piperaquine, mefloquine, among others) have shown autophagy-directed inhibitory activity that might be harnessed for experimental melanoma therapy, particularly in the context of BRAFi-resistance that might render these cells more sensitive to autophagy inhibition.16–18, 20–24 However, identification of specific antimalarials displaying single agent activity targeting BRAFi-resistant melanoma has remained challenging.
Anti-melanoma activity of the amino-quinoline antimalarial chloroquine has been investigated in preclinical models focusing on therapeutic induction of apoptosis and autophagic blockade but clinical efficacy benefitting melanoma patients remains unsatisfactory.22, 24, 25 Interestingly, the combinatorial use of temozolomide and chloroquine targeting BRAFi-resistant post-treatment melanoma cells (from patients who failed treatment with either vemurafenib or dabrafenib) has recently shown promise in murine models attributed to induction of inflammasome-dependent pyroptotic cell death facilitated by chloroquine-dependent inhibition of autophagy.24
Here, we have screened a focused library of clinical ACT antimalarials [including chloroquine, amodiaquine, piperaquine, lumefantrine, and mefloquine (MQ)] for the apoptotic elimination of cultured malignant melanoma cell lines focusing on BRAFi-resistance. To this end, we have employed a stringent genetic model of BRAFi-resistance using isogenic melanoma cells that differ only by NRAS mutational status (BRAFi-sensitive A375-BRAFV600E/NRASQ61 vs. BRAFi-resistant A375-BRAFV600E/NRASQ61K) as explored and validated by us recently.3, 26 Here, we demonstrate feasibility of using the clinical antimalarial mefloquine for the therapeutic elimination of BRAFi-resistant malignant melanoma cells accompanied by early induction of ER stress and apoptosis targeting brain metastases in a bioluminescent murine model of the human intracranial disease stage.
2. MATERIALS AND METHODS
2.1. Chemicals
All chemicals including clinical antimalarials were purchased from Sigma Chemical Co (St. Louis, MO, USA). Mefloquine was the (±) -racemic mixture (as used in clinical antimalarial formulations).
2.2. Cell culture
Human malignant melanoma cells were purchased as follows: (BRAFV600E/NRAS wildtype) LOX-IMVI (SCC201, Millipore Sigma), G361 (CRL-1424, ATCC), SK-MEL-28 (HTB-72, ATCC).3, 27–32 Moreover, vemurafenib-sensitive BRAFV600E-mutated A375-Luc2 (CRL-1619-LUC2) and vemurafenib-resistant A375-Luc2 (isogenic variant containing both BRAFV600E and NRASQ61K mutations; CRL-1619IG-2-LUC2) were purchased from ATCC.3, 27–32 For generation of CRL-1619IG-2-LUC2, the parental BRAFV600E/NRASQ61 human malignant melanoma cells were CRISPR/Cas9 gene edited, generating an isogenic variant containing both BRAFV600E and mutated NRASQ61K, followed by transduction using a lentiviral vector encoding firefly luciferase (LUC2) under control of the EF-1alpha promoter; differential sensitivity to vemurafenib treatment as a function of NRAS mutational status was confirmed as published before.3 All cells were maintained as published recently.33, 34
2.3. Flow Cytometric Analysis of Cell Viability
After 24 h treatment with clinical antimalarials [15 μM; amodiaquine, chloroquine, lumefantrine, piperaquine and mefloquine (MQ)], viability of melanoma cells was assessed by annexinV-FITC/propidium iodide (PI) dual staining with flow cytometric analysis using an apoptosis detection kit according to manufacturer’s specifications (APOAF, Sigma Aldrich).33, 35
2.4. Caspase-3 activation assay
MQ-induced (15 μM; 24 h) caspase-3 activation was examined by flow cytometric analysis using a cleaved/activated caspase-3 (asp 175) antibody (Alexa Fluor 488 conjugate, Cell Signaling) following our published procedure.33, 35
2.5. Immunoblot analysis
Cellular protein extraction, 4–15% gradient SDS-PAGE gel electrophoresis (Bio-Rad laboratories), transfer to PVDF membrane, and immunoblot development were performed as published recently.3, 33 The following rabbit anti-human antibodies were used (obtained from Cell Signaling): cleaved PARP-1 (5625), p-eIF2α (3398), total eIF2α (5324), SQSTM1 (8025), LAMP1 (9091), LC3-I/II (12741), cytochrome c (11940), ATF-4 (11815), and XBP-1s (40435). Equal protein loading was examined by β-actin detection using a mouse monoclonal antibody (Sigma Aldrich); secondary antibodies: HRP-conjugated goat anti-rabbit or goat anti-mouse (Jackson ImmunoResearch Laboratories). Densitometric image analysis was performed using Image Studio™ Lite quantification software (LI-COR Biosciences). For cytochrome c immunodetection, cell fractionation (mitochondrial versus cytosolic) was performed using the Mitochondria/Cytosol fractionation kit (ab65320, Abcam). Purity of fractions was confirmed according to kit instructions.
2.6. Assessment of mitochondrial transmembrane potential (Δψm)
The potentiometric dye 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1; Sigma, T4069) was used to examine the mitochondrial transmembrane potential [Δψm; JC-1 monomeric green fluorescence (depolarized mitochondria, detector FL-1) vs. JC-1 aggregate red fluorescence (polarized mitochondria, detector FL-2) fluorescence] following our previously published procedure.35
2.7. Human Stress & Toxicity PathwayFinder RT2 Profiler™ gene expression array analysis
After MQ treatment (15 μM; 6 h), total mRNA from cultured A375-BRAFV600E/NRASQ61 and A375-BRAFV600E/NRASQ61K (200,000 in 35 mm dish format) was isolated using the RNeasy Mini kit (Qiagen) following our published standard procedures. Reverse transcription was then performed using the RT2 First Strand kit (Qiagen) from 500 ng total RNA. For gene expression array analysis, the human Stress & Toxicity PathwayFinder RT2 Profiler™ technology (Qiagen), assessing expression of 84 stress response-related genes, was used as published before.35, 36 Quantitative PCR was run using the following conditions: 95 °C (10 min), followed by 40 cycles at 95 °C (15 s) alternating with 60 °C (1 min) (Applied Biosystems). Gene-specific products were normalized to a group of 5 housekeeping genes (ACTB, B2M, GAPDH, HPRT1, RPLP0) and quantified using the comparative ΔΔCt method (ABI Prism 7500 sequence detection system user guide).
2.8. Individual RT-qPCR analysis
Total cellular mRNA was isolated using the Qiagen RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. Human primer probes [DDIT3 (Hs_00358796_g1), HSPA6 (Hs_00275682_s1), HSPA1A (Hs_00359163_s1), EGR1 (Hs_00152928_m1), HMOX1 (Hs_00157965_m1), SOD2 (Hs_00167309_m1), RSP18 (housekeeping gene; Hs_01375212_g1)], were obtained from Thermo Fisher Scientific. After cDNA synthesis, quantitative PCR reactions were performed as follows: 10 min (95 °C) followed by 15 s (95 °C), 1 min (60 °C), 40 cycles, using the ABI7500 Real-Time PCR System (Applied Biosystems). Amplification plots were generated, and Ct values were recorded as published before.34
2.9. Cellular ER and Ca2+ Fluo-4 AM imaging
For ER organelle imaging, photostable ER-Tracker™ Blue-White DPX dye staining (ThermoFisher Scientific) was used. Briefly, cells were seeded in 6-well plate format (100,000/well), drugged accordingly (15 μM MQ; 6 h) and stained with 1 mM ER-tracker probe for 1 h in the dark (37 °C; 5% CO2). DAPI filter settings were used to image cells (EVOS fluorescent microscope; ThermoFisher Scientific). For Calcium imaging, cells were stained with Fluo-4 AM dye (3 μM in HBSS; ThermoFisher Scientific) for 1 h in the dark at room temperature.37 The Fluo-4 loaded cells were washed with HBSS and then exposed to MQ (15 μM) before fluorescent image acquisition (using a FITC filter).
2.10. Transmission electron microscopy
Cells were fixed in situ with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4), post-fixed in 1% osmium tetroxide in cacodylate buffer, washed, scraped and pelleted as described recently.21 Cells were then stained in 2% aqueous uranyl acetate, dehydrated through a graded series (50, 70, 90, and 100%) of ethanol and infiltrated with Spurr’s resin (Sigma, EM0300), then allowed to polymerize overnight at 60 °C. Sections (50 nm) were cut, mounted onto uncoated 150-mesh copper grids, and stained with 2% lead citrate. Sections were examined in a CM12 transmission electron microscope (FEI) operated at 80 kV with digital image collection.
2.11. Detection of intracellular oxidative stress
Levels of MQ-induced intracellular oxidative stress were analyzed by flow cytometry using 2′,7′ - dichlorodihydrofluorescein diacetate (DCFH-DA) as a sensitive nonfluorescent precursor dye according to a published standard procedure.34, 35 Briefly, cells were treated with MQ (15 μM, ≤ 3 h), followed by DCFH-DA loading (5 μg/ml), incubated for 1 h in the dark (37 °C, 5% CO2), then harvested and analyzed immediately by flow cytometry.
2.12. MQ chemotherapeutic intervention in a bioluminescent murine model of human intracranial malignant melanoma
A published standard procedure generating and assessing melanoma brain metastases in live mice was employed.38 To this end, anesthetized SCID mice (male, 10 weeks old, 10 in total) received A375-Luc2 (BRAFV600E/NRASQ61K; CRL-1619IG-2-LUC2) cells (100,000 cells/animal) through stereotaxic methods.39, 40 The cells were implanted unilaterally into the striatum [coordinates: anterior-posterior (AP): +0.74; medial-lateral (ML): +1.2; dorsal-ventral (DV): −3.0] via intracranial injections (50,000 cells/ μl of sterile PBS; 2 μl volumes delivered at 0.5 μl/ min). Control animals received 2 μl PBS alone. One day after injection, bioluminescent imaging (Lago CDC bioluminescent imaging system, Spectral Instruments Imaging) was performed followed by pair matching. Starting on d 7 after cell injection until d 17, mice received MQ [50 mg/kg/d in H2O (10% DMSO; 200 μl via gavage); n=5] or solvent control (n=5). On d 18, mice were imaged for final tumor growth assessment and tissue was harvested for IHC analysis. This study was performed in accordance with the recommendations of the National Institutes of Health (University of Arizona Institutional Animal Care and Use Committee; mouse protocol number: IACUC 17–298)
2.13. Immunohistochemistry
Brain tissue was analyzed for detection of proliferating cell nuclear antigen (PCNA) (2586, Cell Signaling Technology) and MELTF (NBP1-85777, Novus Biologicals) as published.20,22 Nuclear staining was performed using Hematoxylin QS counterstain (H3404; Vector Laboratories, Burlingame, CA). Images were captured using an Olympus BX50 and Spot (Model 2.3.0) camera.
2.14. Statistical analysis
Numerical data were analyzed as published recently.3, 34 Unless stated differently, data sets were analyzed employing analysis of variance (ANOVA) with Tukey’s post-hoc test using the Prism 8.4.3 software (Prism Software Corp., Irvine, CA, USA); in respective bar graphs (analyzing more than two groups), means without a common letter differ (p < 0.05). For bar graphs comparing two groups only, statistical significance was calculated employing the Student’s two-tailed t-test, utilizing Excel (Microsoft™). Experiments involved six individual replicates per data point, except for gene expression array analysis (using three independent biological replicates). Nonparametric data analysis of murine experimentation was performed using the Mann–Whitney test. The level of statistical significance was marked as follows: p * < 0.05; p ** < 0.01; p *** < 0.001.
3. RESULTS
3.1. MQ induces cell death in BRAFi-resistant A375 malignant melanoma cells
First, in search of ACT antimalarials displaying single agent apoptogenicity targeting malignant melanoma cells, we examined a focused library of clinical amino-quinoline ACT-antimalarials (15 μM, 24 h) for the ability to induce A375-BRAFV600E/NRASQ61 melanoma cell death as assessed by annexin V/PI-flow cytometric analysis (Figure 1A). Among the chemical entities tested, only MQ [± mefloquine (R/S, S/R-racemic mixture)] caused pronounced cell death detectable upon 24 h continuous exposure to low micromolar concentrations. Next, the detailed dose-response relationship of MQ-induced cell death was established in a panel of cultured human melanoma cells [BRAFV600E/NRAS wildtype: A375, LOX-IMVI, G361, SK-MEL-28; BRAFV600E/mutated NRAS: A375-BRAFV600E/NRASQ61K; Figure 1B).3, 27–32 All melanoma cell lines contained in our test panel displayed sensitivity to MQ-induced cell death observable in the low micromolar concentration range [Figure 1B; MQ (0–30 μM]. Remarkably, MQ displayed cytotoxic efficacy irrespective of BRAFi-resistance status (A375-BRAFV600E/NRASQ61 versus A375-BRAFV600E/NRASQ61K), and a moderate (up to two-fold) increase in MQ-sensitivity of A375-BRAFV600E/NRASQ61K cells (as compared to their isogenic controls) was observed (Figure 1B,C).
Figure 1. MQ impairs viability of cultured human malignant melanoma cell lines including BRAFi-sensitive and NRAS-based BRAFi-resistant A375 isogenic variants.
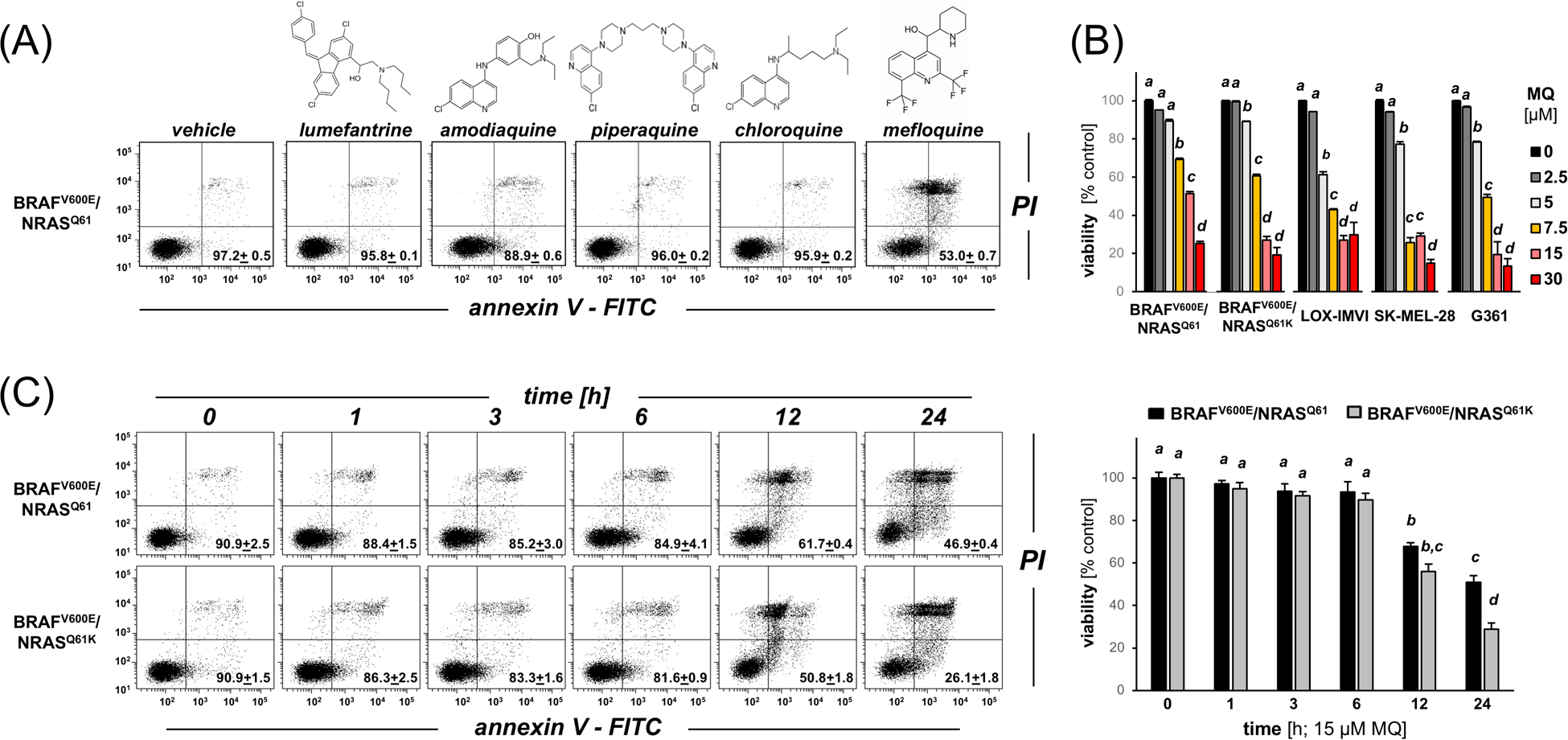
(A) Annexin V/PI-flow cytometric analysis of A375-BRAFV600E/NRASQ61 melanoma cell death induced by exposure to clinical ACT-antimalarials (15 μM, 24 h). The numbers indicate viable (AV−, PI-) in percent of total gated cells (mean ± SD). (B) Dose-response relationship of MQ-induced cell death in A375-BRAFV600E/NRASQ61, A375-BRAFV600E/NRASQ61K, LOX-IMVI, G361, SK-MEL-28 (MQ ≤ 30 μM, 24 h) as assessed in (A). (C) Time course analysis of A375-BRAFV600E/NRASQ61 versus A375-BRAFV600E/NRASQ61K cell death induction [MQ, 15 μM, ≤ 24 h; (left panels: flow cytometric analysis; right bar graph: quantitative analysis)]. For all bar graph depictions, quantitative data analysis employed ANOVA with Tukey’s post hoc test; means without a common letter differ from each other (p<0.05).
Given the remarkable single agent activity of MQ impairing melanoma cell viability (observable even in BRAFi-resistant NRAS isogenic variant), we focused our further studies on comparative examination of MQ-induced cytotoxicity employing our A375 isogenic variants. First, in order to inform the subsequent gene expression array analysis (Figure 2), a detailed time course of cell death induction by MQ was established comparing A375-BRAFV600E/NRASQ61 versus A375-BRAFV600E/NRASQ61K cells (Figure 1C; ≤ 24 h continuous exposure). This analysis indicated that cell viability was fully maintained at 6 h exposure time, followed by loss of viability detectable at 12 h and after. As observed above (Figure 1B), loss of viability induced by MQ exposure (24 h) was almost two-fold more pronounced in A375-BRAFV600E/NRASQ61K cells as compared to the isogenic NRAS wildtype line (Figure 1C).
Figure 2. Comparative array analysis profiles MQ-induced stress response gene expression in isogenic A375-BRAFV600E/NRASQ61 versus A375-BRAFV600E/NRASQ61K cells.
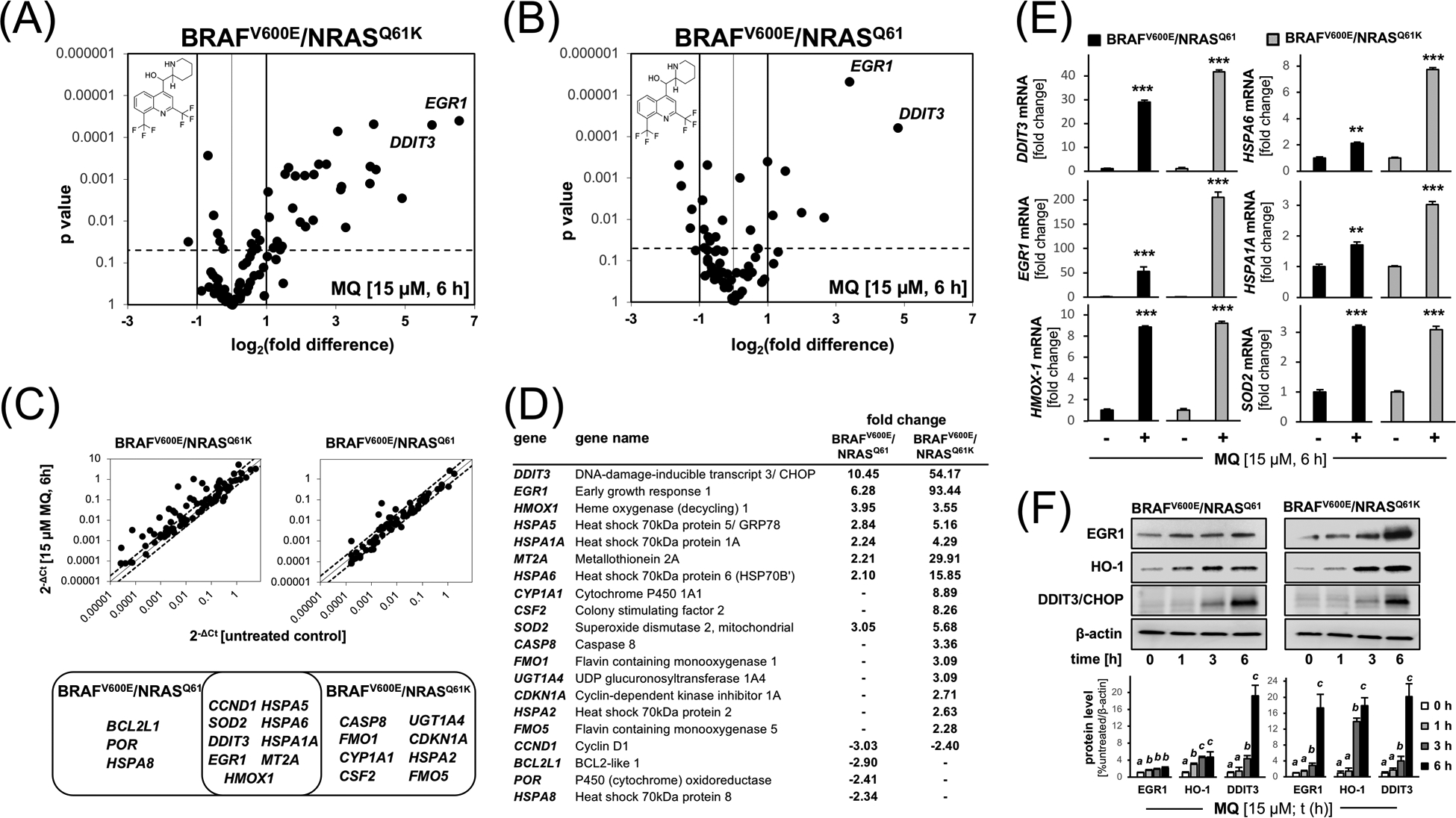
Comparative stress response gene expression analysis in MQ-treated A375-BRAFV600E/NRASQ61K and A375-BRAFV600E/NRASQ61 cells. Volcano blots (panels A and B) depict differential gene expression as detected by the Human Stress and Toxicity Profiler™ RT2 PCR array technology (MQ: 15 μM, 6 h; relative to untreated; cut-off criteria: fold change ≥ 2; p < 0.05). (C) Venn diagram comparing statistically significant MQ-induced expression changes. (Top panels are scatter blot depictions.) (D) Quantitative summary of MQ-induced expression changes comparing isogenic cell lines (cut-off criteria: fold change ≥ 2; p value < 0.05). (E) MQ-induced proteotoxic and oxidative stress response at the single gene expression level confirmed by independent RT-qPCR. Bar graphs depict fold change versus untreated control as a function of genotype. (F) Expression of top three upregulated genes (EGR1, HMOX1, DDIT3; from panel D) examined at the protein level using immunoblot analysis [MQ (15 μM); t ≤ 6 h]; bar graph depiction summarizes densitometric analysis. For bar graph depictions, quantitative data analysis employed ANOVA with Tukey’s post hoc test; means without a common letter differ from each other (p<0.05). For bar graphs comparing two groups only, statistical significance was calculated employing the Student’s two-tailed t-test (**p < 0.01; ***p < 0.001).
3.2. Comparative array analysis: MQ-induced stress response gene expression in isogenic A375-BRAFV600E/NRASQ61 versus A375-BRAFV600E/NRASQ61K melanoma cells
In order to explore the early stress response elicited by short term MQ treatment [15 μM, 6 h; a time point at which viability was not yet impaired (Fig. 1C)], we profiled parental A375-BRAFV600E/NRASQ61 and isogenic BRAFi-resistant A375-BRAFV600E/NRASQ61K cells employing comparative gene expression analysis (Figure 2). Using the Human Stress and Toxicity Profiler™ RT2 PCR array technology, interrogating expression of 84 stress-related genes contained on the array, it was observed that MQ-induced expression changes in isogenic A375 cells affected 20 genes by at least two-fold over untreated control cells (Figure 2A,B). Venn diagram depiction comparing MQ-induced expression changes identified nine genes displaying MQ-induced expression changes shared between the two genotypes including (Figure 2C,D):
proteotoxic stress response genes [i.e. DDIT3 (DNA-damage-inducible transcript 3/ CHOP), HSPA6 (Heat shock 70kDa protein 6), HSPA5 (Heat shock 70kDa protein 5/ GRP78), HSPA1A (Heat shock 70kDa protein 1A);
redox stress-related genes [i.e. EGR1 (early growth response protein 1), HMOX1 (heme oxygenase-1), SOD2 (Superoxide dismutase 2, mitochondrial), MT2A (Metallothionein 2A)].
Remarkably, MQ-induced upregulation of gene expression was generally more pronounced in BRAFi-resistant A375-BRAFV600E/NRASQ61K cells as compared to their isogenic controls (Figure 2D). For example, upregulation of DDIT3 by more than fifty-fold was contrasted by only ten-fold upregulation in the isogenic parental cells. Likewise, upregulation of EGR1 (93-fold) was contrasted by only six-fold upregulation in controls, a trend observed also with other genes including MT2A and HSPA6. Moreover, the MQ-induced proteotoxic and oxidative stress response was also confirmed at the mRNA and protein levels (using independent RT-qPCR and immunoblot analyses), indicating pronounced upregulation of select genes as already observed by array analysis (DDIT3, EGR1, HMOX1, etc.) in both A375 isogenic cell lines (Figure 2E,F).
3.3. MQ elicits ER stress signaling and early calcium dysregulation in isogenic BRAFi-resistant A375-BRAFV600E/NRASQ61K and BRAFi-sensitive A375-BRAFV600E/NRASQ61 melanoma cells
Next, informed by our gene expression data (Figure 2), the early occurrence of MQ-induced ER stress was examined by immunoblot analysis focusing on key effector signaling pathways (phospho-eIF2α, XBP-1s, ATF4; Figure 3A).41, 42 MQ-initiation of ER-related signaling was observed in both cell lines, with A375-BRAFV600E/NRASQ61K cells displaying a more pronounced and rapid MQ-induced upregulation of phospho-eIF2α (detectable within 1 h exposure to MQ); in contrast, parental A375-BRAFV600E/NRASQ61 cells displayed a more pronounced and rapid upregulation of ATF4 (detectable within 1 h exposure to MQ); rapid XBP-1s upregulation was also observed in both isogenic lines detectable within 3–6 h exposure times. Consistent with the early occurrence of ER stress in response to MQ treatment, ER-swelling and enlargement were detectable by quantitative ER-tracker™ DPX fluorescence imaging and electron microscopy (Figure 3B,C).
Figure 3. MQ induces ER stress signaling and early calcium dysregulation in isogenic BRAFi-resistant A375-BRAFV600E/NRASQ61K and BRAFi-sensitive A375-BRAFV600E/NRASQ61 human malignant melanoma cells.
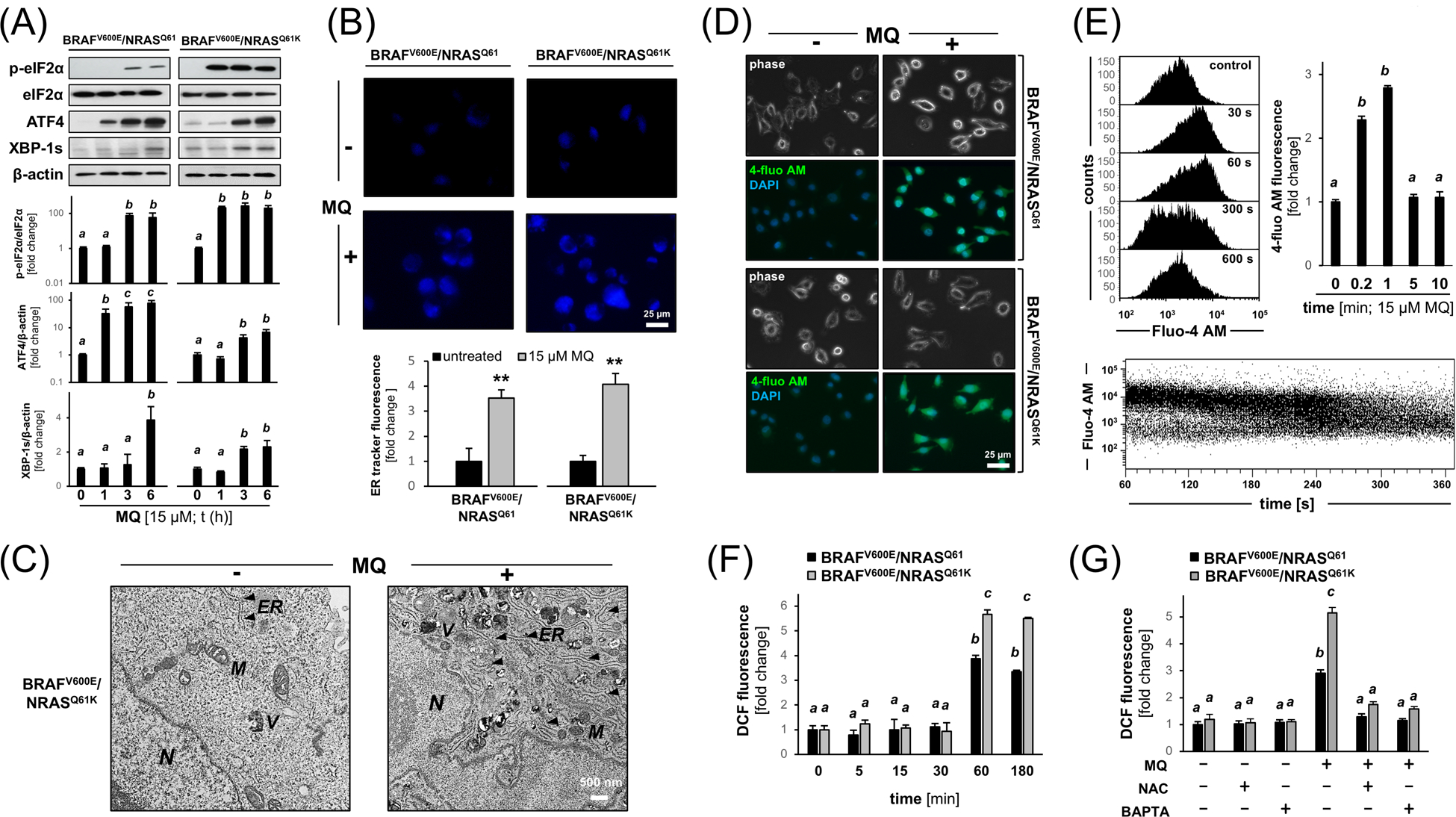
(A) MQ-induced ER stress signaling (phospho-eIF2α, XBP-1s, ATF4) as revealed by immunoblot analysis [MQ (15 μM); t ≤ 6 h]; bar graph depiction summarizes densitometric analysis. (B) ER organelle visualization by ER-tracker™ DPX fluorescence imaging [MQ (15 μM; t ≤ 6 h; 20x magnification)]; bar graph depiction summarizes quantitative analysis. (C) Electron microscopy indicating ER organelle swelling and enlargement observed in untreated versus MQ-exposed (15 μM; 6 h) A375-BRAFV600E/NRASQ61K cells [11,500-fold magnification; scale bar: 500 nm; N; nucleus; M: mitochondria; V: vesicle; ER: endoplasmic reticulum (marked by arrowheads)]. (D) Fluo-4 AM-fluorescence visualization of cytosolic free calcium observable within seconds of exposure to MQ (15 μM; 120 s); phase contrast and DAPI nuclear staining; representative images]. (E) Flow cytometric analysis (histograms) of MQ induced calcium signal (Fluo-4 AM-fluorescence intensity); upper left: representative histograms [0 – 600 s MQ (15 μM) exposure time]; upper right: bar graph depiction with statistical analysis; bottom: time resolved (60 – 360 s) Fluo-4 AM-fluorescence intensity as a function of MQ exposure time. (F) Flow cytometric detection of 2’,7’-dihydrodichlorofluorescein-diacetate (DCFH-DA) oxidation [producing dichlorofluorescein (DCF) fluorescence] as an indicator of cellular oxidative stress in response to MQ exposure (15 μM; ≤ 180 min exposure time) examined in both genotypes; bar graph depiction with statistical analysis. (G) Effect of calcium chelation [BAPTA-AM (20 μM)] or antioxidant intervention [NAC (10 mM)] on MQ-induced cellular oxidative stress (DCF-assay as performed in panel F) examined in both genotypes. For all bar graph depictions, quantitative data analysis employed ANOVA with Tukey’s post hoc test; means without a common letter differ from each other (p<0.05). For bar graphs comparing two groups only, statistical significance was calculated employing the Student’s two-tailed t-test (**p < 0.01).
Next, since calcium dysregulation causing cytosolic overload is an established consequence of ER stress, we employed Fluo-4 AM fluorescence-based calcium visualization (Figure 3D,E).37 Indeed, cytosolic free calcium was observable within seconds of exposure to MQ as detected by fluorescence microscopy, an effect observable in both isogenic A375 melanoma cell variants irrespective of NRAS-genotype, quantified by flow cytometric analysis. Interestingly, calcium homeostasis was restored within 300 s of observation time as revealed by time-resolved (60 – 360 s) continuous monitoring of Fluo-4 AM-fluorescence intensity as a function of MQ exposure time (Figure 3E, bottom panel).37
Next, the occurrence of redox dysregulation as a result of MQ treatment was examined employing flow cytometric detection of 2’,7’-dihydrodichlorofluorescein-diacetate (DCFH-DA) oxidation, an established indicator of cellular oxidative stress known to occur in the context of ER disruption (Figure 3F).41, 43 DCF fluorescence (resulting from indicator dye oxidation) was detectable starting at 60 min of MQ exposure, an early occurrence that places redox dysregulation downstream of earlier calcium dysregulation (observable within seconds of MQ exposure). As observed with calcium release, increase in DCF fluorescence signal was more pronounced in the NRAS-mutated genotype. Furthermore, treatment with the membrane-permeable calcium chelator BAPTA-AM (quenching the consequences of cytosolic overload) prevented MQ-induced increase in DCF fluorescence indicating that early calcium dysregulation is a causative factor upstream of ROS formation, an effect observable in both isogenic variants. (Figure 3G). As a positive control, antioxidant intervention using N-acetyl-L-cysteine (NAC) suppressed ROS formation that occurs as a consequence of MQ exposure.
Taken together these data document that MQ treatment causes early ER stress signaling associated with calcium dysregulation and subsequent induction of oxidative stress, observable in A375-BRAFV600E/NRASQ61 melanoma cells with analogous yet more pronounced effects in the isogenic A375-BRAFV600E/NRASQ61K variant.
3.4. MQ induces apoptotic cell death (with mitochondrial cytochrome C release, caspase activation, and PARP-1 cleavage) targeting BRAFi-resistant A375-BRAFV600E/NRASQ61K melanoma cells, and oral administration blocks tumor growth in a bioluminescent murine model of intracranial BRAFi-resistant malignant melanoma.
Next, in preparation of testing the possibility of therapeutic elimination of BRAFi-resistant melanoma cells (located intracranially in SCID mice) employing systemic administration of MQ, we further explored molecular mechanisms of MQ-induced A375-BRAFV600E/NRASQ61K melanoma cell death in vitro (Figure 4A–E). First, MQ-induced proteolytic activation of caspase-3 activation was confirmed by flow cytometric analysis (Figure 4A). Moreover, cells exposed to the combined action of MQ (15 μM, 24 h) and the pan-caspase inhibitor zVAD-fmk displayed significantly increased survival indicative of an early involvement of caspase-dependent pathways (Figure 4B). Importantly, immunoblot analysis at early time points of MQ exposure confirmed PARP-1 cleavage, an established target of caspase 3 involved in apoptotic execution (Figure 4C). PARP-1 cleavage was observable as early as within 1 h exposure time followed by further increase over 6 h; at the same time, markers of autophagic-lysosomal dysregulation [SQSTM1 (p62), LAMP1] remained largely unchanged during this early observational time frame (with the exception of LC3-I/II displaying mild inhibition indicative of interference with autophagic function), an observation consistent with the rapid occurrence of caspase-dependent pathways preceding disruption of autophagy (Figure 4C).21 Furthermore, mitochondrial release of cytochrome C (accompanied by the corresponding increase in cytosolic cytochrome C level) was detected, an observation consistent with involvement of the mitochondrial pathway of apoptosis (Figure 4D), confirmed further by a concomitant MQ-induced loss of mitochondrial transmembrane potential as detected by JC-1 flow cytometric analysis (Figure 4E).
Figure 4. MQ induces apoptosis in cultured A375-BRAFV600E/NRASQ61K melanoma cells, and oral administration blocks tumor growth in a bioluminescent murine model of intracranial BRAFi-resistant malignant melanoma.
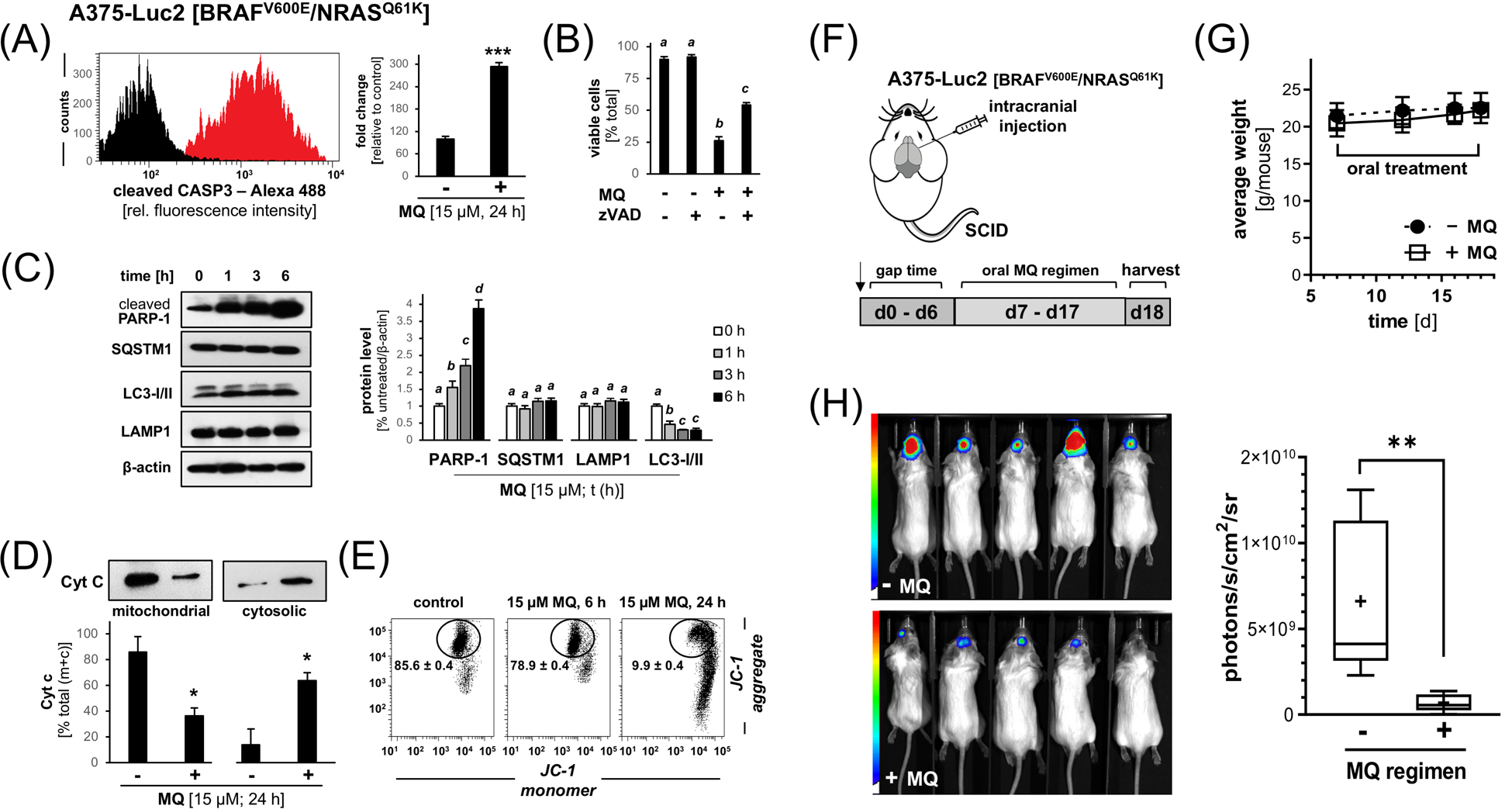
(A) MQ-induced (15 μM, 24 h) caspase-3 activation as examined in cultured A375-BRAFV600E/NRASQ61K cells by flow cytometric detection. (B) Cell death in A375-BRAFV600E/NRASQ61K melanoma cells exposed to the combined action of MQ (15 μM, 24 h) and the pan-caspase inhibitor zVAD-fmk (40 μM; mean ± SD]. (C) PARP-1 cleavage and modulation of autophagic-lysosomal mediators (SQSTM1, LAMP1, LC3-I/II) in response to MQ-exposure (15 μM, ≤ 6 h) in A375-BRAFV600E/NRASQ61K cells as assessed by immunoblot analysis (left panel); bar graph depiction summarizes densitometric analysis (right panel). (D) MQ-induced (15 μM; 24 h) cytochrome C release from cellular fractions (mitochondrial versus cytosolic). (E) Loss of mitochondrial transmembrane potential (Δψm) was assessed by flow cytometric analysis of JC-1 stained cells (15 μM; 6–24 h). Numbers indicate percentage of cells inside the circle displaying intact Δψm [mean ± SD; (p < 0.05)]. (F) Chemotherapeutic MQ-regimen in vivo: A375-Luc2 [BRAFV600E/NRASQ61K] melanoma cells were intracranially injected (n=5 per group) followed by bioluminescent image analysis of brain tumors on d1 (pair matching) and d18 (harvest). Starting on d7 after cell injection until d18, mice received MQ [50 mg/kg/d in H2O (10% DMSO; 200 μL via gavage)] or solvent control. (G) Mouse body weights as a function of treatment group and time (n=5 per group; d7–18). (H) Bioluminescent imaging (d18); bar graph (right panel) depicts quantitative image analysis of bioluminescent intracranial signal. Nonparametric data analysis (Figure 4H) was performed using the Mann–Whitney test (**p < 0.01). For all other bar graph depictions, quantitative data analysis employed ANOVA with Tukey’s post hoc test; means without a common letter differ from each other (p<0.05); for bar graphs comparing two groups only, statistical significance was calculated employing the Student’s two-tailed t-test (*p < 0.05; ***p < 0.001).
Next, based on prior experimental and clinical evidence documenting brain availability of systemic MQ, we tested feasibility of targeting BRAFi-resistant A375-BRAFV600E/NRASQ61K melanoma in a bioluminescent murine model (Figures 4F–H; 5).44, 45 To this end, A375-Luc2 [BRAFV600E/NRASQ61K] melanoma cells were intracranially injected followed by bioluminescent image analysis of brain tumors in SCID mice receiving an oral MQ-regimen as compared to administration of vehicle control only (Figure 4F–H). Indeed, quantitative image analysis at the end of the experiment revealed pronounced tumor growth suppression as substantiated by an almost seven-fold reduction in bioluminescent signal in the MQ-treated group (Figure 4H), a chemotherapeutic effect that occurred without negative impact on average mouse body weight (Figure 4G).
Subsequently, IHC analysis of transverse cuts of brain tissue was performed at the end of the experiment (d 18) in order to confirm suppression of melanoma tumor growth [involving H&E (Figure 5A) and PCNA/ MELTF staining (Figure 5B,C)]. The presence (‘- MQ’ treatment group) or absence (‘+ MQ’ treatment group) of proliferating melanoma cells was substantiated assessing PCNA expression in cells staining positive for the melanocyte-specific antigen melanotransferrin [MELTF (p97); Figures 5B,C].
Figure 5. Immunohistochemical analysis of murine brain tissue confirms MQ-induced attenuation of intracranial BRAFi-resistant malignant melanoma.
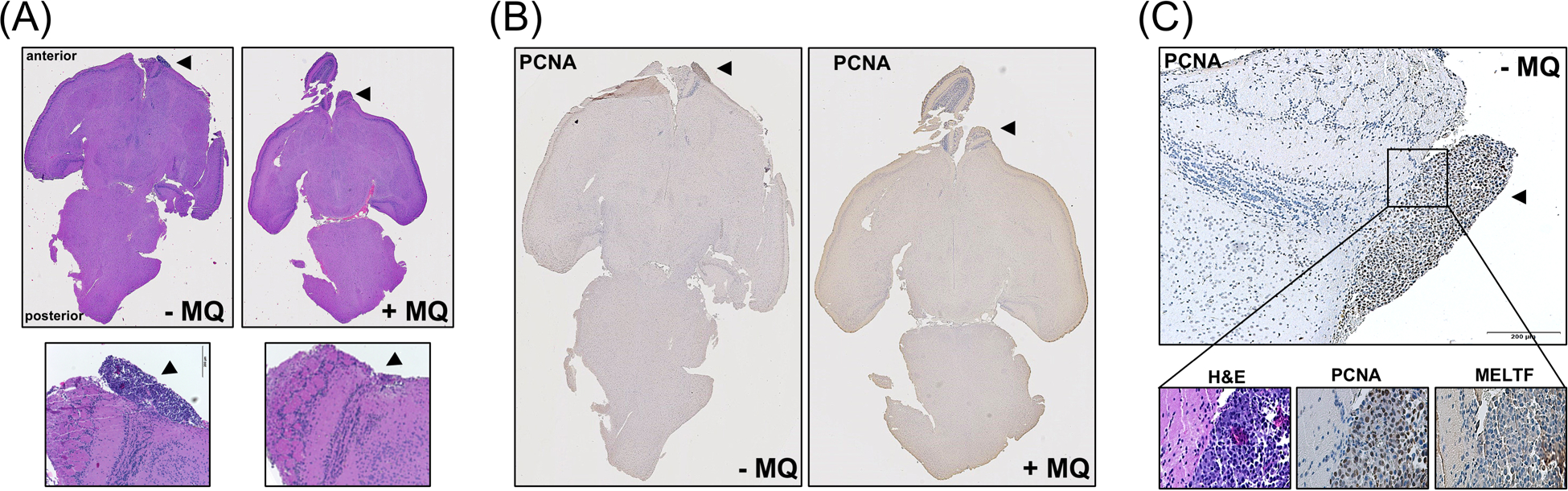
At the end of the experiment (d18), brain tissue was processed for IHC [H&E (panels A, C); PCNA, MELTF (panels B, C). (A) Representative transverse cuts of brain specimens (H&E); top panels: complete specimens (‘- MQ’ versus ‘+ MQ’); bottom panels: enlarged areas (10x magnification; arrows point towards site of cell injection; scale bar: 200 μm). (B) Immunohistochemical staining (PCNA; transverse cuts of brain specimens) from control and MQ-exposed mice. (C) Transverse cut of tumor bearing area [untreated (‘- MQ’) mouse; one representative specimen]; top panel: PCNA (tumor marked by arrow; scale bar: 200 μm); bottom panels (left to right): H&E, PCNA, MELTF.
4. Discussion
Repurposing of clinical antimalarials has shown promise for cancer-directed molecular intervention, and inhibitory modulation of autophagic-lysosomal function has been substantiated as a major molecular mechanism of action underlying cancer cell-directed activity of antimalarials.24 Among various aminoquinoline-antimalarials, cancer cell-directed cytotoxicity of MQ is well documented, involving numerous tentative molecular mechanisms, including autophagic-lysosomal blockade, inhibition of P-glycoprotein-dependent drug efflux, NFκB pathway attenuation through IKK inactivation, and induction of redox dysregulation.46–49
Here we demonstrate for the first time that MQ induces cell death in cultured malignant melanoma cells displaying apoptogenic activity observable even in BRAFi-resistant BRAFV600E/NRASQ61K A375 melanoma cells (Figures 1 and 4), an effect associated with rapid induction of proteotoxic and oxidative stress response gene expression (Figure 2). Moreover, ER stress signaling and rapid onset of calcium dysregulation were detected in response to MQ treatment (Figure 3).
Remarkably, feasibility of targeting intracranial melanoma upon systemic administration of MQ was demonstrated in a bioluminescent murine model, an important preclinical observation given the notoriously unsatisfactory clinical outcomes currently achieved using standard of care interventions in large patient populations afflicted by brain metastases (Figures 4 and 5).10–12 Importantly, in the context of oral MQ targeting brain melanoma in a relevant murine model, it remains to be seen if MQ treatment may also impact molecular pathways involved in invasion and metastasis (such as regulation of EMT-related gene expression) blocking the occurrence of intracranial tumors, a topic of current research activities pursued in our laboratory.34
Interestingly, molecular mechanisms of MQ-associated calcium dysregulation and ER-directed toxicity have been explored before in the context of MQ-induced neurotoxic symptoms observed in malaria patients, representing a well-documented adverse effect of this antimalarial intervention.44, 45, 50–52 This adverse drug action has been attributed to specific molecular interactions including: (i) noncompetitive acetylcholine esterase inhibition, (ii) interaction with voltage-dependent channels, and (iii) ER-associated Ca-ATPase (SERCA) antagonism (mimicking activity of the SERCA-antagonist thapsigargin), all of which might be causing cytosolic overload and dysregulation of calcium homeostasis.44, 52–54 Indeed, ER stress and calcium dysregulation have been identified as critical factors in signal transduction and determination of cancer cell fate, connecting ER, mitochondrial, and lysosomal stress signaling with therapeutic efficacy, a topic with emerging relevance to melanomagenesis and therapy.55–60 However, specific mechanism and molecular targets underlying MQ-induced calcium dysregulation achievable in malignant melanoma cells remain to be identified. It is tempting to speculate that a shared vulnerability to MQ-induced calcium dysregulation might characterize both neurons and melanoma/ melanocytic cells given their common neural crest origin. Also, it should be mentioned that based on the stereochemical diversity of mefloquine [employed here as the clinically-used (±) racemic mixture] either one of the 4 possible stereoisomers might display differential ligand activity modulating a tentative calcium regulatory target, a topic that has been addressed to some extent before comparing mammalian SERCA-directed inhibitory activity of (+)- and (−)- MQ isomers.54
Therapeutic induction of ER stress has shown promise in overcoming BRAFi resistance, and, consistent with our observations (Figure 2D), upregulation of ER stress-related mediators including CHOP (encoded by DDIT3) and GRP78 (encoded by HSPA5) has been observed before in MQ-exposed MDA-MB-231 breast carcinoma cells, but a causative involvement of ER stress in MQ-induced cytotoxicity was not substantiated in that study.61 In our experiments targeting BRAFV600E/NRASQ61K BRAFi-resistant A375 melanoma cells using MQ, ER stress response gene expression (Figure 2), disruption of calcium homeostasis (Figure 3D,E,G), induction of oxidative stress (Figure 3F,G), and mitochondrial dysregulation were observable at early time points (Figure 4D,E), with calcium dysregulation preceding all other changes (upstream of ROS production; Figure 3F,G). However, specific molecular interdependencies and targets relevant to MQ-induced apoptotic elimination of BRAFi-resistant malignant melanoma cells remain to be explored in adequate detail allowing stringent mechanistic conclusions that might inform future clinical decision making. It is also worth mentioning that recent drug discovery efforts have generated MQ-derivatives with improved experimental cancer therapeutic activity, emphasizing the need for more detailed identification of molecular mechanisms specific to MQ that would explain its unique antimelanoma profile distinct from other aminoquinoline antimalarials as described by us here for the first time.62 In addition, for further preclinical and clinical development aiming at repurposing MQ for melanoma intervention, it will be essential to conduct more detailed dose optimization studies identifying clinically relevant MQ regimens and to explore potential MQ synergism with standard of care melanoma therapeutics (including BRAFi and immune checkpoint inhibitors) assessed in treatment-naïve and treatment-resistant tumors.1, 24
Taken together, these novel preclinical data suggest that the clinical antimalarial MQ may be a valid candidate for drug repurposing aiming at chemotherapeutic elimination of malignant melanoma cells, even if BRAFi-resistance and metastasis to the brain have occurred. However, relevance of the cellular BRAFi-resistance model (A375-BRAFV600E/NRASQ61K), employed by us for the first time for drug screening and in vivo testing, might be limited since the specific NRAS mutation examined here is observable only in approximately 20% of patients displaying BRAFi-resistance necessitating a more comprehensive coverage of patient-relevant resistance models to be pursued in the near future.3, 9
ACKNOWLEDGEMENTS
Supported in part by grants from the National Institutes of Health (1R01CA229418, 1P01CA229112, ES007091, ES006694, and UA Cancer Center Support Grant CA023074). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Abbreviations:
- ACT
artemisinin-combination therapeutic
- ANOVA
analysis of variance
- BAPTA-AM
1,2-Bis(2-aminophenoxy) ethane-N,N,N’,N’-tetraacetic acid tetrakis(acetoxymethyl ester)
- BRAFi
BRAF kinase inhibitors
- DAPI
4′,6-diamidino-2-phenylindole
- DCF
dichlorofluorescein
- DCFH-DA
2′,7′ - dichlorodihydrofluorescein diacetate
- ER
endoplasmic reticulum
- JC-1
5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolyl-carbocyanine iodide
- MQ
mefloquine
- NAC
N-acetyl-L-cysteine
- PI
propidium iodide
- ROS
reactive oxygen species
Footnotes
CONFLICT OF INTEREST
All authors declare that there are no conflicts of interest to disclose.
DATA AVAILABILITY STATEMENT
The data sets used or analyzed in the context of the current study are available from the corresponding author by reasonable request.
REFERENCES
- 1.Curti BD, Faries MB. Recent Advances in the Treatment of Melanoma. N Engl J Med. 2021;384(23):2229–2240. [DOI] [PubMed] [Google Scholar]
- 2.Helgadottir H, Rocha Trocoli Drakensjo I, Girnita A. Personalized Medicine in Malignant Melanoma: Towards Patient Tailored Treatment. Front Oncol. 2018;8:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jandova J, Wondrak GT. Vemurafenib Drives Epithelial-to-Mesenchymal Transition Gene Expression in BRAF Inhibitor-Resistant BRAF(V600E)/NRAS(Q61K) Melanoma Enhancing Tumor Growth and Metastasis in a Bioluminescent Murine Model. J Invest Dermatol. 2021. Oct 21:S0022–202X(21)02368-X; online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trojaniello C, Luke JJ, Ascierto PA. Therapeutic Advancements Across Clinical Stages in Melanoma, With a Focus on Targeted Immunotherapy. Front Oncol. 2021;11:670726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petti C, Molla A, Vegetti C, Ferrone S, Anichini A, Sensi M. Coexpression of NRASQ61R and BRAFV600E in human melanoma cells activates senescence and increases susceptibility to cell-mediated cytotoxicity. Cancer Res. 2006;66(13):6503–11. [DOI] [PubMed] [Google Scholar]
- 6.Sensi M, Nicolini G, Petti C, Bersani I, Lozupone F, Molla A, Vegetti C, Nonaka D, Mortarini R, Parmiani G, Fais S, Anichini A. Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma. Oncogene. 2006;25(24):3357–64. [DOI] [PubMed] [Google Scholar]
- 7.Heppt MV, Siepmann T, Engel J, Schubert-Fritschle G, Eckel R, Mirlach L, Kirchner T, Jung A, Gesierich A, Ruzicka T, Flaig MJ, Berking C. Prognostic significance of BRAF and NRAS mutations in melanoma: a German study from routine care. BMC Cancer. 2017;17(1):536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar R, Njauw CN, Reddy BY, Ji Z, Rajadurai A, Klebanov N, Tsao H. Growth suppression by dual BRAF(V600E) and NRAS(Q61) oncogene expression is mediated by SPRY4 in melanoma. Oncogene. 2019;38(18):3504–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao S, Rizos H, Sucker A, Scolyer RA, Gutzmer R, Gogas H, Kefford RF, Thompson JF, Becker JC, Berking C, Egberts F, Loquai C, Goldinger SM, Pupo GM, Hugo W, Kong X, Garraway LA, Sosman JA, Ribas A, Lo RS, Long GV, Schadendorf D. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer. 2015;51(18):2792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen JV, Tawbi H, Margolin KA, Amravadi R, Bosenberg M, Brastianos PK, Chiang VL, de Groot J, Glitza IC, Herlyn M, Holmen SL, Jilaveanu LB, Lassman A, Moschos S, Postow MA, Thomas R, Tsiouris JA, Wen P, White RM, Turnham T, Davies MA, Kluger HM. Melanoma central nervous system metastases: current approaches, challenges, and opportunities. Pigment Cell Melanoma Res. 2016;29(6):627–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vosoughi E, Lee JM, Miller JR, Nosrati M, Minor DR, Abendroth R, Lee JW, Andrews BT, Leng LZ, Wu M, Leong SP, Kashani-Sabet M, Kim KB. Survival and clinical outcomes of patients with melanoma brain metastasis in the era of checkpoint inhibitors and targeted therapies. BMC Cancer. 2018;18(1):490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janavicius M, Lachej N, Anglickiene G, Vincerzevskiene I, Brasiuniene B. Outcomes of Treatment for Melanoma Brain Metastases. J Skin Cancer. 2020;2020:7520924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, Koya RC, Samatar AA, Khanlou N, Braun J, Ruchalski K, Seifert H, Larkin J, Dahlman KB, Johnson DB, Algazi A, Sosman JA, Ribas A, Lo RS. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell. 2015;27(2):240–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardner LJ, Ward M, Andtbacka RHI, Boucher KM, Bowen GM, Bowles TL, Cohen AL, Grossmann K, Hitchcock YJ, Holmen SL, Hyngstrom J, Khong H, McMahon M, Monroe MM, Ross CB, Suneja G, Wada D, Grossman D. Risk factors for development of melanoma brain metastasis and disease progression: a single-center retrospective analysis. Melanoma Res. 2017;27(5):477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rebecca VW, Somasundaram R, Herlyn M. Pre-clinical modeling of cutaneous melanoma. Nat Commun. 2020;11(1):2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, Lazova R, Klump V, Pawelek JM, Xu X, Xu W, Schuchter LM, Davies MA, Herlyn M, Winkler J, Koumenis C, Amaravadi RK. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest. 2014;124(3):1406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ojha R, Leli NM, Onorati A, Piao S, Verginadis II, Tameire F, Rebecca VW, Chude CI, Murugan S, Fennelly C, Noguera-Ortega E, Chu CT, Liu S, Xu X, Krepler C, Xiao M, Xu W, Wei Z, Frederick DT, Boland G, Mitchell TC, Karakousis GC, Schuchter LM, Flaherty KT, Zhang G, Herlyn M, Koumenis C, Amaravadi RK. ER Translocation of the MAPK Pathway Drives Therapy Resistance in BRAF-Mutant Melanoma. Cancer Discov. 2019;9(3):396–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Truong A, Yoo JH, Scherzer MT, Sanchez JMS, Dale KJ, Kinsey CG, Richards JR, Shin D, Ghazi PC, Onken MD, Blumer KJ, Odelberg SJ, McMahon M. Chloroquine Sensitizes GNAQ/11-mutated Melanoma to MEK1/2 Inhibition. Clin Cancer Res. 2020;26(23):6374–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eastman RT, Fidock DA. Artemisinin-based combination therapies: a vital tool in efforts to eliminate malaria. Nat Rev Microbiol. 2009;7(12):864–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cabello CM, Lamore SD, Bair WB 3rd, Qiao S, Azimian S, Lesson JL, Wondrak GT. The redox antimalarial dihydroartemisinin targets human metastatic melanoma cells but not primary melanocytes with induction of NOXA-dependent apoptosis. Invest New Drugs. 2012;30(4):1289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiao S, Tao S, Rojo de la Vega M, Park SL, Vonderfecht AA, Jacobs SL, Zhang DD, Wondrak GT. The antimalarial amodiaquine causes autophagic-lysosomal and proliferative blockade sensitizing human melanoma cells to starvation- and chemotherapy-induced cell death. Autophagy. 2013;9(12):2087–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lakhter AJ, Sahu RP, Sun Y, Kaufmann WK, Androphy EJ, Travers JB, Naidu SR. Chloroquine promotes apoptosis in melanoma cells by inhibiting BH3 domain-mediated PUMA degradation. J Invest Dermatol. 2013;133(9):2247–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodall ML, Wang T, Martin KR, Kortus MG, Kauffman AL, Trent JM, Gately S, MacKeigan JP. Development of potent autophagy inhibitors that sensitize oncogenic BRAF V600E mutant melanoma tumor cells to vemurafenib. Autophagy. 2014;10(6):1120–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmed F, Tseng HY, Ahn A, Gunatilake D, Alavi S, Eccles M, Rizos H, Gallagher SJ, Tiffen JC, Hersey P, Emran AA. Repurposing Melanoma Chemotherapy to Activate Inflammasomes in the Treatment of BRAF/MAPK Inhibitor Resistant Melanoma. J Invest Dermatol. 2021. Oct 22:S0022–202X;(21)02384–8; online ahead of print. [DOI] [PubMed] [Google Scholar]
- 25.Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, Tan KS, Heitjan DF, Rodgers G, Gallagher M, Piao S, Troxel AB, Evans TL, DeMichele AM, Nathanson KL, O’Dwyer PJ, Kaiser J, Pontiggia L, Davis LE, Amaravadi RK. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy. 2014;10(8):1369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chhabra G, Ahmad N. BRAF Inhibitors in Melanoma Management: When Friends Become Foes. J Invest Dermatol. 2021. Dec 3:S0022–202X(21)02485–4; online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O’Meara S, Santarius T, Avis T, Barthorpe S, Brackenbury L, Buck G, Butler A, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Hunter C, Jenkinson A, Jones D, Kosmidou V, Lugg R, Menzies A, Mironenko T, Parker A, Perry J, Raine K, Richardson D, Shepherd R, Small A, Smith R, Solomon H, Stephens P, Teague J, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Reinhold W, Weinstein JN, Stratton MR, Futreal PA, Wooster R. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther. 2006;5(11):2606–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ugurel S, Thirumaran RK, Bloethner S, Gast A, Sucker A, Mueller-Berghaus J, Rittgen W, Hemminki K, Becker JC, Kumar R, Schadendorf D. B-RAF and N-RAS mutations are preserved during short time in vitro propagation and differentially impact prognosis. PLoS One. 2007;2(2):e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schrama D, Keller G, Houben R, Ziegler CG, Vetter-Kauczok CS, Ugurel S, Becker JC. BRAFV600E mutations in malignant melanoma are associated with increased expressions of BAALC. J Carcinog. 2008;7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fattore L, Marra E, Pisanu ME, Noto A, de Vitis C, Belleudi F, Aurisicchio L, Mancini R, Torrisi MR, Ascierto PA, Ciliberto G. Activation of an early feedback survival loop involving phospho-ErbB3 is a general response of melanoma cells to RAF/MEK inhibition and is abrogated by anti-ErbB3 antibodies. J Transl Med. 2013;11:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caenepeel S, Cooke K, Wadsworth S, Huang G, Robert L, Moreno BH, Parisi G, Cajulis E, Kendall R, Beltran P, Ribas A, Coxon A, Hughes PE. MAPK pathway inhibition induces MET and GAB1 levels, priming BRAF mutant melanoma for rescue by hepatocyte growth factor. Oncotarget. 2017;8(11):17795–17809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adam C, Fusi L, Weiss N, Goller SG, Meder K, Frings VG, Kneitz H, Goebeler M, Houben R, Schrama D, Schmidt M. Efficient Suppression of NRAS-Driven Melanoma by Co-Inhibition of ERK1/2 and ERK5 MAPK Pathways. J Invest Dermatol. 2020;140(12):2455–2465 e10. [DOI] [PubMed] [Google Scholar]
- 33.Jandova J, Hua AB, Fimbres J, Wondrak GT. Deuterium Oxide (D2O) Induces Early Stress Response Gene Expression and Impairs Growth and Metastasis of Experimental Malignant Melanoma. Cancers (Basel). 2021;13(4):605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jandova J, Wondrak GT. Genomic GLO1 deletion modulates TXNIP expression, glucose metabolism, and redox homeostasis while accelerating human A375 malignant melanoma tumor growth. Redox Biol. 2021;39:101838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis AL, Qiao S, Lesson JL, Rojo de la Vega M, Park SL, Seanez CM, Gokhale V, Cabello CM, Wondrak GT. The quinone methide aurin is a heat shock response inducer that causes proteotoxic stress and Noxa-dependent apoptosis in malignant melanoma cells. J Biol Chem. 2015;290(3):1623–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perer J, Jandova J, Fimbres J, Jennings EQ, Galligan JJ, Hua A, Wondrak GT. The sunless tanning agent dihydroxyacetone induces stress response gene expression and signaling in cultured human keratinocytes and reconstructed epidermis. Redox Biology. 2020;36:101594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vines A, McBean GJ, Blanco-Fernandez A. A flow-cytometric method for continuous measurement of intracellular Ca(2+) concentration. Cytometry A. 2010;77(11):1091–7. [DOI] [PubMed] [Google Scholar]
- 38.Craft N, Bruhn KW, Nguyen BD, Prins R, Liau LM, Collisson EA, De A, Kolodney MS, Gambhir SS, Miller JF. Bioluminescent imaging of melanoma in live mice. J Invest Dermatol. 2005;125(1):159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Madhavan L, Daley BF, Davidson BL, Boudreau RL, Lipton JW, Cole-Strauss A, Steece-Collier K, Collier TJ. Sonic Hedgehog Controls the Phenotypic Fate and Therapeutic Efficacy of Grafted Neural Precursor Cells in a Model of Nigrostriatal Neurodegeneration. PLoS One. 2015;10(9):e0137136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ray S, Corenblum MJ, Anandhan A, Reed A, Ortiz FO, Zhang DD, Barnes CA, Madhavan L. A Role for Nrf2 Expression in Defining the Aging of Hippocampal Neural Stem Cells. Cell Transplant. 2018;27(4):589–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiao S, Cabello CM, Lamore SD, Lesson JL, Wondrak GT. D-Penicillamine targets metastatic melanoma cells with induction of the unfolded protein response (UPR) and Noxa (PMAIP1)-dependent mitochondrial apoptosis. Apoptosis. 2012;17(10):1079–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sicari D, Delaunay-Moisan A, Combettes L, Chevet E, Igbaria A. A guide to assessing endoplasmic reticulum homeostasis and stress in mammalian systems. FEBS J. 2020;287(1):27–42. [DOI] [PubMed] [Google Scholar]
- 43.Reane DV, Rizzuto R, Raffaello A. The ER-mitochondria tether at the hub of Ca2+ signaling. Current Opinion in Physiology. 2020;17(1):261–268. [Google Scholar]
- 44.Dow GS, Hudson TH, Vahey M, Koenig ML. The acute neurotoxicity of mefloquine may be mediated through a disruption of calcium homeostasis and ER function in vitro. Malar J. 2003;2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toovey S Mefloquine neurotoxicity: a literature review. Travel Med Infect Dis. 2009;7(1):2–6. [DOI] [PubMed] [Google Scholar]
- 46.Sukhai MA, Prabha S, Hurren R, Rutledge AC, Lee AY, Sriskanthadevan S, Sun H, Wang X, Skrtic M, Seneviratne A, Cusimano M, Jhas B, Gronda M, MacLean N, Cho EE, Spagnuolo PA, Sharmeen S, Gebbia M, Urbanus M, Eppert K, Dissanayake D, Jonet A, Dassonville-Klimpt A, Li X, Datti A, Ohashi PS, Wrana J, Rogers I, Sonnet P, Ellis WY, Corey SJ, Eaves C, Minden MD, Wang JC, Dick JE, Nislow C, Giaever G, Schimmer AD. Lysosomal disruption preferentially targets acute myeloid leukemia cells and progenitors. J Clin Invest. 2013;123(1):315–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim JH, Choi AR, Kim YK, Yoon S. Co-treatment with the anti-malarial drugs mefloquine and primaquine highly sensitizes drug-resistant cancer cells by increasing P-gp inhibition. Biochem Biophys Res Commun. 2013;441(3):655–60. [DOI] [PubMed] [Google Scholar]
- 48.Xu X, Wang J, Han K, Li S, Xu F, Yang Y. Antimalarial drug mefloquine inhibits nuclear factor kappa B signaling and induces apoptosis in colorectal cancer cells. Cancer Sci. 2018;109(4):1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mereddy GR, Ronayne CT. Repurposing Antimalarial Drug Mefloquine for Cancer Treatment. Transl Med (Sunnyvale). 2018;8:199. [Google Scholar]
- 50.Dow GS, Caridha D, Goldberg M, Wolf L, Koenig ML, Yourick DL, Wang Z. Transcriptional profiling of mefloquine-induced disruption of calcium homeostasis in neurons in vitro. Genomics. 2005;86(5):539–50. [DOI] [PubMed] [Google Scholar]
- 51.Hood JE, Jenkins JW, Milatovic D, Rongzhu L, Aschner M. Mefloquine induces oxidative stress and neurodegeneration in primary rat cortical neurons. Neurotoxicology. 2010;31(5):518–23. [DOI] [PubMed] [Google Scholar]
- 52.Martins AC, Paoliello MMB, Docea AO, Santamaria A, Tinkov AA, Skalny AV, Aschner M. Review of the mechanism underlying mefloquine-induced neurotoxicity. Crit Rev Toxicol. 2021;51(3):209–216. [DOI] [PubMed] [Google Scholar]
- 53.Caridha D, Yourick D, Cabezas M, Wolf L, Hudson TH, Dow GS. Mefloquine-induced disruption of calcium homeostasis in mammalian cells is similar to that induced by ionomycin. Antimicrob Agents Chemother. 2008;52(2):684–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toovey S, Bustamante LY, Uhlemann AC, East JM, Krishna S. Effect of artemisinins and amino alcohol partner antimalarials on mammalian sarcoendoplasmic reticulum calcium adenosine triphosphatase activity. Basic Clin Pharmacol Toxicol. 2008;103(3):209–13. [DOI] [PubMed] [Google Scholar]
- 55.Deli T, Varga N, Adam A, Kenessey I, Raso E, Puskas LG, Tovari J, Fodor J, Feher M, Szigeti GP, Csernoch L, Timar J. Functional genomics of calcium channels in human melanoma cells. Int J Cancer. 2007;121(1):55–65. [DOI] [PubMed] [Google Scholar]
- 56.Arbabian A, Brouland JP, Gelebart P, Kovacs T, Bobe R, Enouf J, Papp B. Endoplasmic reticulum calcium pumps and cancer. Biofactors. 2011;37(3):139–49. [DOI] [PubMed] [Google Scholar]
- 57.Long T, Su J, Tang W, Luo Z, Liu S, Liu Z, Zhou H, Qi M, Zeng W, Zhang J, Chen X. A novel interaction between calcium-modulating cyclophilin ligand and Basigin regulates calcium signaling and matrix metalloproteinase activities in human melanoma cells. Cancer Lett. 2013;339(1):93–101. [DOI] [PubMed] [Google Scholar]
- 58.Stewart TA, Yapa KT, Monteith GR. Altered calcium signaling in cancer cells. Biochim Biophys Acta. 2015;1848(10 Pt B):2502–11. [DOI] [PubMed] [Google Scholar]
- 59.Raffaello A, Mammucari C, Gherardi G, Rizzuto R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem Sci. 2016;41(12):1035–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chemaly ER, Troncone L, Lebeche D. SERCA control of cell death and survival. Cell Calcium. 2018;69:46–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharma N, Thomas S, Golden EB, Hofman FM, Chen TC, Petasis NA, Schonthal AH, Louie SG. Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer Lett. 2012;326(2):143–54. [DOI] [PubMed] [Google Scholar]
- 62.Rodrigues FA, Bomfim Ida S, Cavalcanti BC, Pessoa C, Goncalves RS, Wardell JL, Wardell SM, de Souza MV. Mefloquine-oxazolidine derivatives: a new class of anticancer agents. Chem Biol Drug Des. 2014;83(1):126–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets used or analyzed in the context of the current study are available from the corresponding author by reasonable request.