

Summary
mTORC1 controls cellular metabolic processes in response to nutrient availability. Amino acid signals are transmitted to mTORC1 through the Rag GTPases, which are localized on the lysosomal surface by the Ragulator complex. The Rag GTPases receive amino acid signals from multiple upstream regulators. One negative regulator, GATOR1, is a GTPase activating protein (GAP) for RagA. GATOR1 binds to the Rag GTPases via two modes: an inhibitory mode and a GAP mode. How these two binding interactions coordinate to process amino acid signals is unknown. Here, we resolved three cryo-EM structural models of the GATOR1-Rag-Ragulator complex, with the Rag-Ragulator subcomplex occupying the inhibitory site, the GAP site, and both binding sites simultaneously. When the Rag GTPases bind to GATOR1 at the GAP site, both Rag subunits contact GATOR1 to coordinate their nucleotide loading states. These results reveal a potential GAP mechanism of GATOR1 during the mTORC1 inactivation process.
Keywords: mTOR complex 1 (mTORC1), Rag GTPase, enzyme mechanism, GATOR1, GAP, nutrient sensing
Graphical Abstract
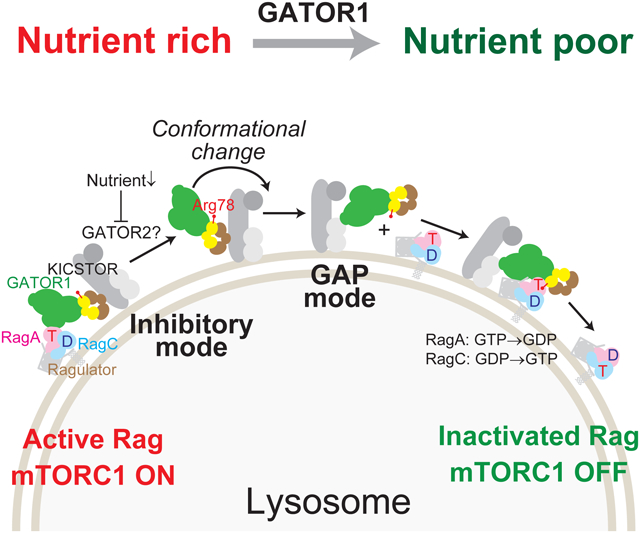
eTOC blurb:
How GATOR1, a negative regulator of mTORC1, suppresses signaling on a mechanistic level has remained poorly understood. Here, Egri et al. describe three structural models of the GATOR1-Rag-Ragulator complex. They reveal that GATOR1 modulates the nucleotide status of both Rag subunits to regulate mTORC1 signaling.
Introduction
Mechanistic target of Rapamycin complex 1 (mTORC1) is a central regulator of cellular metabolism that coordinates nutrient resources with cellular growth (Ben-Sahra and Manning, 2017; Dibble and Cantley, 2015; Kim and Guan, 2019; Liu and Sabatini, 2020; Mossmann et al., 2018). Activation of mTORC1 occurs under abundant nutrient and growth factor conditions and requires two small GTPase units: the Rag GTPase heterodimer which in the presence of abundant amino acids, recruits mTORC1 to the lysosomal surface (Kim et al., 2008; Sancak et al., 2008), and the Rheb GTPase that responds to growth factor signals and stimulates the kinase activity of mTORC1 (Inoki et al., 2003; Li et al., 2004; Menon et al., 2014). This coincidence detector ensures that cells will activate the proliferation program only under favorable nutrient conditions.
In human cells, a network of proteins senses and processes amino acid signals. Central to the network is the heterodimeric Rag GTPase (Figure 1A) (Nicastro et al., 2017). In contrast to monomeric signaling GTPases such as Ras, Rab, or Rho, each functional unit of the Rag GTPases contains two subunits, with RagA or RagB tightly bound to RagC or RagD, forming a stable, obligate heterodimer (Schürmann et al., 1995; Sekiguchi et al., 2001). Each Rag subunit consists of two domains: a P-loop nucleotide binding domain (NBD) and a C-terminal roadblock domain (CRD) that mediates dimerization (Gong et al., 2011; Jeong et al., 2012). Nucleotide loading on the Rag GTPase heterodimer is unique compared to canonical GTPases. When RagA binds GTP and RagC binds GDP, the Rag GTPases adopt an active conformation that interacts with the Raptor subunit of mTORC1, facilitating the recruitment of mTORC1 to the lysosomal surface (Anandapadamanaban et al., 2019; Rogala et al., 2019; Sancak et al., 2008). In contrast, when RagA binds GDP and RagC binds GTP, the corresponding conformation rejects mTORC1 and releases it to the cytosol, thus inactivating the pathway (Sancak et al., 2008). Currently, only these two out of the four theoretical nucleotide loading states have been associated with biological functions. The dual GTP-loaded or GDP-loaded states are considered to be unstable and their roles in amino acid sensing are unclear (Shen et al., 2017).
Figure 1. Ragulator modulates the biochemical properties of GATOR1.
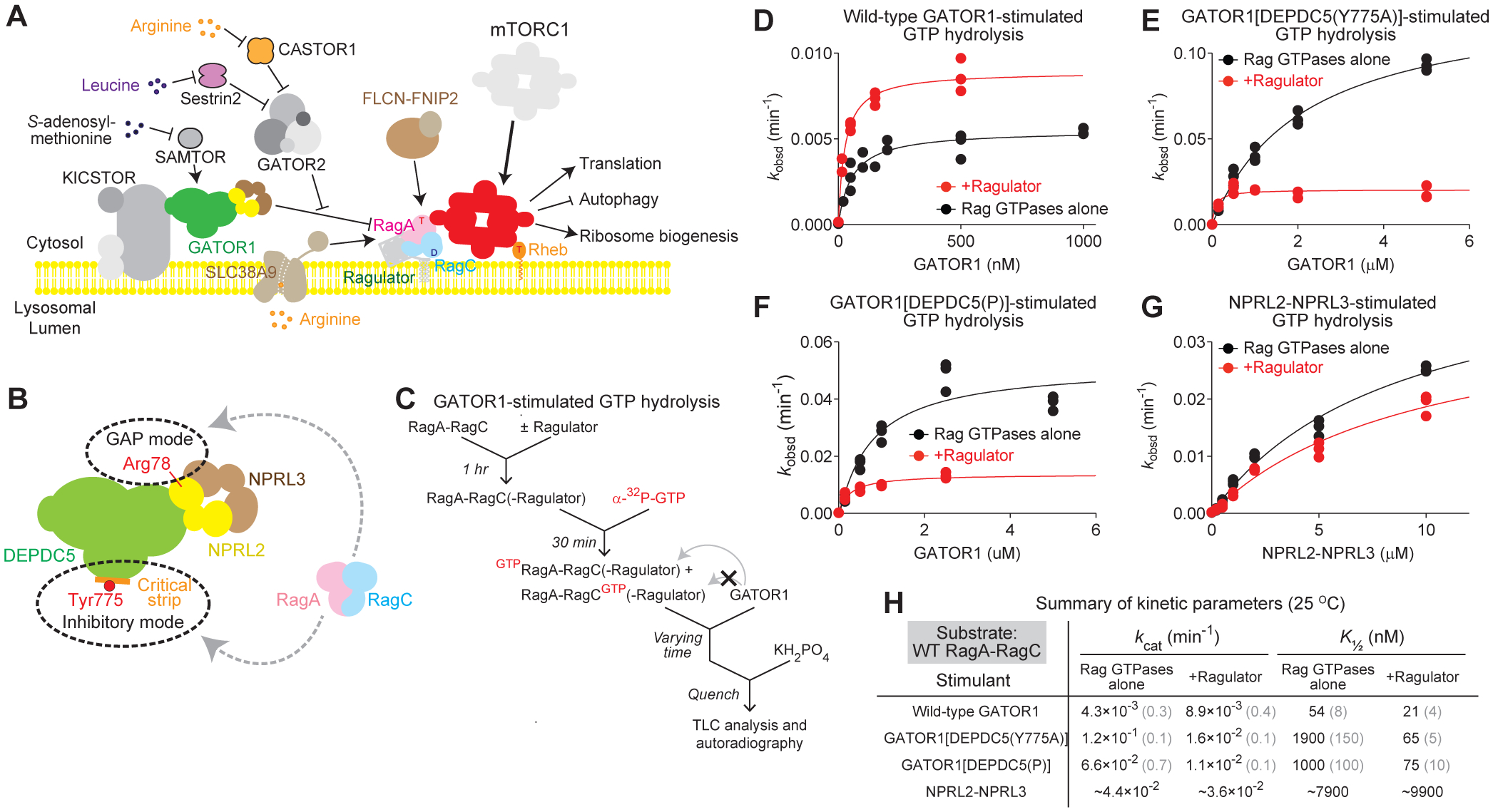
A. Protein components of the amino acid sensing branch of the mTORC1 pathway.
B. GATOR1 binds to the Rag GTPase heterodimer via two modes: an inhibitory mode and a GAP mode. A point mutation on the critical strip, DEPDC5(Y775A), blocks binding in the inhibitory mode.
C. Scheme for GATOR1-stimulated GTP hydrolysis assay (GAP assay)..
D–G. Stimulated GTP hydrolysis by wild-type Rag GTPases in the presence or absence of Ragulator, with wild-type GATOR1 (D), GATOR1[DEPDC5(Y775A)] (E), GATOR1[DEPDC5(P)] (F), and NPRL2-NPRL3 (G) as stimulant..
H. Summary of kinetic parameters from panels D–G. Gray numbers in parentheses denote the SEMs of the reported values calculated from three independent experiments.
The Rag GTPase heterodimer does not contain any lipidation motif, and its lysosomal localization is dictated by Ragulator (Figure 1A) (Bar-Peled et al., 2012; Sancak et al., 2010). Ragulator is a heteropentameric protein complex consisting of two pairs of heterodimerized subunits (LAMTOR2 and LAMTOR3; LAMTOR4 and LAMTOR5) and a lipidated scaffold protein (LAMTOR1) that encloses the dimerized-dimer. In addition to its function to tether the Rag GTPases on the lysosomal membrane, Ragulator also carries out non-canonical guanine nucleotide-exchange factor (GEF) activity on the Rag GTPases, as it specifically accelerates GTP release, but not GDP release, from a single Rag subunit, RagC (Shen and Sabatini, 2018).
The dimerized architecture of the Rag GTPases endows them with unique biochemical properties. Intrinsically, the two nucleotide binding domains of the Rag heterodimer coordinate their nucleotide loading states through extensive crosstalk between the subunits (Shen et al., 2017). When one subunit binds GTP, it becomes dominant and controls the behavior of the other subunit by (1) inhibiting the second GTP from binding, and (2) in the case of spontaneous binding, stimulating the hydrolysis of the second GTP. These mechanisms ensure that only one subunit is loaded with GTP, and therefore, only defined signals are directed towards mTORC1. Compared with other GTPases, the intrinsic GTP off-rate and GTP hydrolysis rate for the Rag GTPases are both extremely slow. As a consequence, once the Rag heterodimer is “locked” into either the active or the inactive state, it can generate a stable signal towards mTORC1. However, the slow intrinsic rates could pose a problem when the nutrient status from the environment changes, so upstream regulators are necessary to facilitate rapid interconversion between the functional states.
An important negative regulator of the pathway is GATOR1 (Figure 1A) (Bar-Peled et al., 2013; Panchaud et al., 2013). GATOR1 is a heterotrimeric complex consisting of DEPDC5 (DEP Domain-Containing protein 5), NPRL2 (Nitrogen permease regulator 2-like protein), and NPRL3 (Nitrogen permease regulator 3-like protein). GATOR1 is a GTPase activating protein (GAP) for the RagA subunit, and its lysosomal localization is dictated by KICSTOR (Figure 1A) (Wolfson et al., 2017). Upon nutrient deprivation, GATOR1 is activated and it stimulates GTP hydrolysis of RagA, which converts the Rag GTPase heterodimer from the active into the inactive state. Distinct from canonical GAPs, GATOR1 binds to the Rag GTPases via two distinct modes - an inhibitory mode and a GAP mode (Figure 1B) (Shen et al., 2018, 2019a). A previous cryo-EM structural model of the GATOR1-Rag complex captured the inhibitory mode, in which a β-strand of DEPDC5, termed the ‘critical strip’, directly binds to the nucleotide binding pocket of RagA in its GTP-bound conformation (Shen et al., 2018). This binding has nano-molar affinity and is therefore predominant with wild-type GATOR1. However, the stimulatory effect on GTP hydrolysis by RagA at the inhibitory site is very mild, and it was proposed that this mode of binding exists to prevent hyper-activation of GATOR1. In contrast, the GAP mode was identified using genetic approaches (Shen et al., 2019a). An arginine residue on the Longin domain of NPRL2, Arg78, was suggested to be the “arginine finger” that carries out the catalytic activity of GATOR1. The interaction between GATOR1 and the Rag GTPases in the GAP mode is ~40-fold weaker than in the inhibitory mode, but the catalytic efficiency is ~30-fold higher. Because of the relatively weak binding affinity, there is no structural information about the GAP mode, nor is it understood whether or how GATOR1 regulates its GAP activity and coordinates the nucleotide loading states of both Rag subunits.
In this study, we used cryo-EM to resolve three structural models of the GATOR1-Rag-Ragulator complex. We captured the GATOR1-Rag-Ragulator complex in (1) the GAP mode only, (2) the inhibitory mode only, and (3) both modes simultaneously. Specifically, in the GAP mode, all three subunits of GATOR1 make extensive contacts with both Rag subunits, forming an intricate network of interactions that hold the Rag GTPase heterodimer in an open conformation. Our structural models reveal a unique GAP mechanism that allows GATOR1 to regulate the nucleotide loading states of the Rag GTPase heterodimer.
Results
Ragulator modulates the enzymatic activity of GATOR1
As a non-canonical GEF for RagC, Ragulator directly modulates the nucleotide loading states of the Rag GTPases (Shen and Sabatini, 2018). Therefore, it is conceivable that Ragulator could also influence the biochemical properties of other upstream regulators of the Rag GTPases. We assessed whether Ragulator modulates the stimulatory effect of GATOR1 (Figure 1C). When we used wild-type GATOR1 as the stimulant (Figure 1D), the kcat of GATOR1-stimulated hydrolysis by the Rag GTPases increases by two-fold in the presence of Ragulator, and the K½ value decreases to half (Figure 1H). This suggests that Ragulator likely modulates the preferred binding site for the Rag GTPases on GATOR1, resulting in an increase in the population of Rag GTPases bound in the GAP mode.
To probe how Ragulator impacts the Rag GTPases bound specifically in the GAP mode, we utilized previously identified GATOR1 mutants that impair Rag GTPase binding in the inhibitory mode. These mutants either contain a point mutation, Y775A, or a combination of mutations localized at the center of the critical strip, named mutant P [YDLLP(775–779)GSGSG], which disrupts the β-strand conformation (Shen et al., 2018). We observed striking effects with these two mutants (Figure 1E and 1F). In the absence of Ragulator, the K½ of GATOR1[DEPDC5(Y775A)] increases by ~40-fold in comparison to wild-type GATOR1 (1900 nM vs. 54 nM, Figure 1D vs. 1E, summarized in 1H). In the presence of Ragulator, the K½ value returns to the nano-molar range (65nM, Figure 1E and 1H), suggesting that Ragulator significantly stabilizes the binding of the Rag GTPases in the GAP mode. In the absence of Ragulator, the stimulatory effect (kcat) of the complex in the GAP mode is ~30-fold higher than that of the inhibitory mode (0.12 min−1 vs. 0.0042 min−1, Figure 1E vs. 1F, summarized in 1H). In the presence of Ragulator, however, the stimulatory effects for wild-type GATOR1 and GATOR1[DEPDC5(Y775A)] are within two-fold of one another (0.0089 min−1 vs. 0.016 min−1, Figure 1D vs. 1E, summarized in 1H). We observed similar behaviors with mutant P (Figure 1F and 1H). Taken together, we concluded that Ragulator enhances the binding of the Rag GTPases to GATOR1 in the GAP mode and imposes regulation on the GAP reaction by decreasing the catalytic efficiency.
To probe the effect of Ragulator on the catalytic step of the GAP activity, we used NPRL2-NPRL3 heterodimer as a stimulant. Ragulator does not cause any changes in either kcat or K½ values (Figure 1G and 1H), suggesting that DEPDC5 plays a major role in mediating the Ragulator-induced effect on the GAP activity. Moreover, K½ for NPRL2-NPRL3-stimulated GTP hydrolysis is much higher than that for either wild-type GATOR1 or GATOR1[DEPDC5(Y775A)] (Figure 1H), indicating the binding between NPRL2-NPRL3 and the Rag GTPases is weaker than the binding between the full GATOR1 complex and the Rag GTPases. Therefore, DEPDC5 must directly contact the Rag GTPases in the GAP mode.
RagA-RagC(F92A) traps the Rag GTPases in the GAP mode
The dramatic effect of Ragulator on GATOR1-stimulated GTP hydrolysis enables us to search for Rag mutants that preferentially bind at the GAP site. We screened for a series of point mutants of the Rag GTPases, and found RagA-RagC(F92A) (Figure 2A–2C). When RagA-RagC(F92A) serves as the substrate for GATOR1[DEPDC5(Y775A)], the K½ value remains in the low-nanomolar range (Figure 2B and 2C, cf. Figure 1H), suggesting that RagA-RagC(F92A) preferentially occupies the GAP site. Furthermore, when we included Ragulator in the reaction, we observed no further modulation of the GAP activity, suggesting RagA-RagC(F92A) is likely prepositioned in a conformation that Ragulator induces in wild-type Rag GTPases (Figure 2B and 2C).
Figure 2. RagA-RagC(F92A) preferentially binds GATOR1 in the GAP mode.
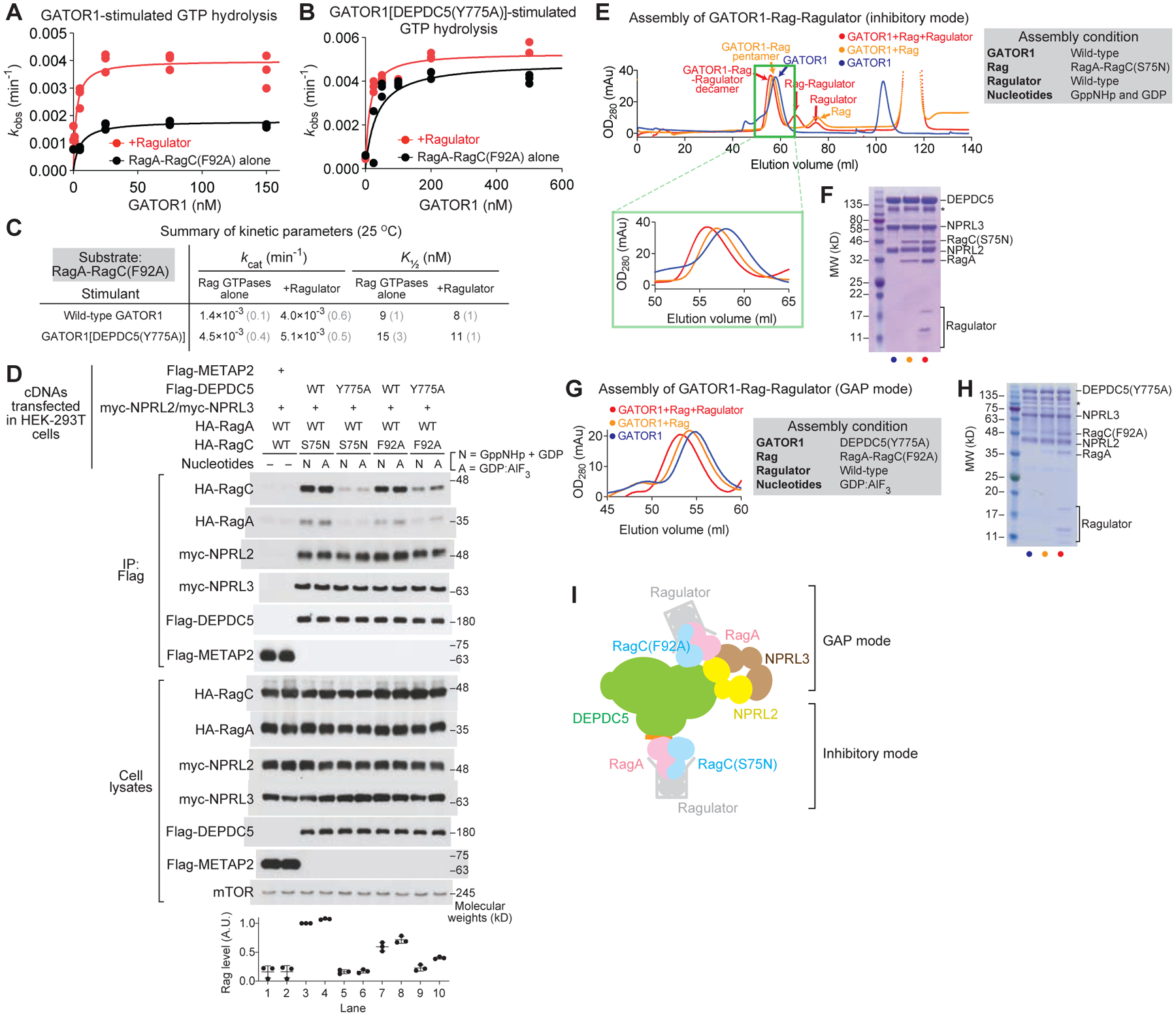
A & B. Stimulated GTP hydrolysis by RagA-RagC(F92A) in the presence or absence of Ragulator, with wild-type GATOR1 (A) or GATOR1[DEPDC5(Y775A)] (B) as stimulant.
C. Summary of kinetic parameters from panels A and B. Gray numbers in parentheses denote the SEMs of the reported values calculated from three independent experiments.
D. In vivo binding assay between GATOR1 and the Rag GTPases.
E. Gel-filtration profiles for the assembly of the GATOR1-Rag-Ragulator complex in the inhibitory mode.
F. Coomassie staining gel for the eluate at high molecular weight region. Subunits of GATOR1, RagA-RagC(S75N), and Ragulator can be visualized. Asterisk denotes a non-specific band commonly observed in GATOR1 purification.
G. Gel-filtration profiles for the assembly of the GATOR1-Rag-Ragulator complex in the GAP mode.
H. Coomassie staining gel for the eluate at high molecular weight region. Subunits of GATOR1[DEPDC5(Y775A)], RagA-RagC(F92A), and Ragulator can be visualized. Asterisk denotes non-specific bands commonly observed in GATOR1 purification.
I. Scheme for the binding between GATOR1 and Rag-Ragulator.
We tested the interaction between the Rag GTPases and GATOR1 in cells using a co-immunoprecipitation (co-IP) assay (Figure 2D). Compared with a control protein, METAP2 (Figure 2D, lanes 1 & 2), wild-type DEPDC5 strongly interacts with RagA-RagC(S75N) in the presence of either GppNHp or GDP:AlF3 (Figure 2D, lanes 3 & 4). This interaction is weakened by introducing the Y775A mutation on DEPDC5 (Figure 2D, lanes 3 & 4 vs. lanes 5 & 6). Moreover, binding between RagA-RagC(S75N) and GATOR1 in the GAP mode is insensitive to the nucleotide identity (Figure 2D, lane 5 vs. lane 6). In contrast, when we probed the binding between the RagA-RagC(F92A) mutant and GATOR1 in the GAP mode, we observed enhanced binding, specifically in the condition where GDP:AlF3 serves as the nucleotides (Figure 2D, lane 9 vs. lane 10).
Based on the reaction conditions identified above, we prepared the GATOR1-Rag-Ragulator complex in different binding modes as follows. To enrich the inhibitory mode, we incubated RagA-RagC(S75N) with wild-type GATOR1 and Ragulator in the presence of GppNHp and GDP. As RagC(S75N) is defective in binding GTP (or its analog GppNHp), the nucleotide loading state naturally becomes GppNHpRagA-RagC(S75N)GDP. Indeed, all ten subunits eluted in a single peak from a HiLoad 16/600 gel filtration column (Figure 2E and 2F, denoted as I-mode). To form the complex in the GAP mode, we incubated RagA-RagC(F92A) with GATOR1[DEPDC5(Y775A)] and Ragulator in the presence of GDP:AlF3 (Figure 2G). This complex also eluted out in a single peak (Figure 2H, denoted as G-mode). In an attempt to capture both binding interactions simultaneously, we mixed the two Rag mutants with Ragulator and wild-type GATOR1, and collected the eluate at the high molecular weight region. As shown below, we observed both binding sites on GATOR1 occupied (Figure 2I, denoted as D-mode).
Structural determination of the GATOR1-Rag-Ragulator complexes
We carried out cryo-EM single particle analysis for the three complexes assembled above (Figure S1A, S2A, and S3A). Well-defined particles can be clearly visualized in micrographs taken by a 300 kV cryo-EM (Figure S1B, S2B, and S3B). Reference-free two-dimensional (2D) classification revealed fine structural details of the complex with views from different orientations (Figure S1C, S1G, S2C, S2G, S3C, and S3I). For the I-mode and G-mode datasets, three-dimensional (3D) refinements from a homogeneous subset of 3D classification generated the final envelopes at 4.0 Å (I-mode) and 3.9 Å (G-mode) resolution, respectively (Gold-standard criteria, Figure S1D, S2D). For the D-mode dataset, we obtained two populations of particles after 3D classification (Figure S3A). Within 34.4% of the particles, we observed Coulomb potential densities corresponding to two copies of the Rag-Ragulator complex, which occupy the two binding modes simultaneously, while the other 65.6% of the particles only contains density corresponding to the GAP mode. The resolutions for the two populations are 4.1 Å and 3.5 Å, respectively (Figure S3D and S3E). Taken together, these maps reveal the molecular architecture of the GATOR1-Rag-Ragulator complex in different conformations (Figure 3A–3C).
Figure 3. Cryo-EM structural models of the GATOR1-Rag-Ragulator complex.
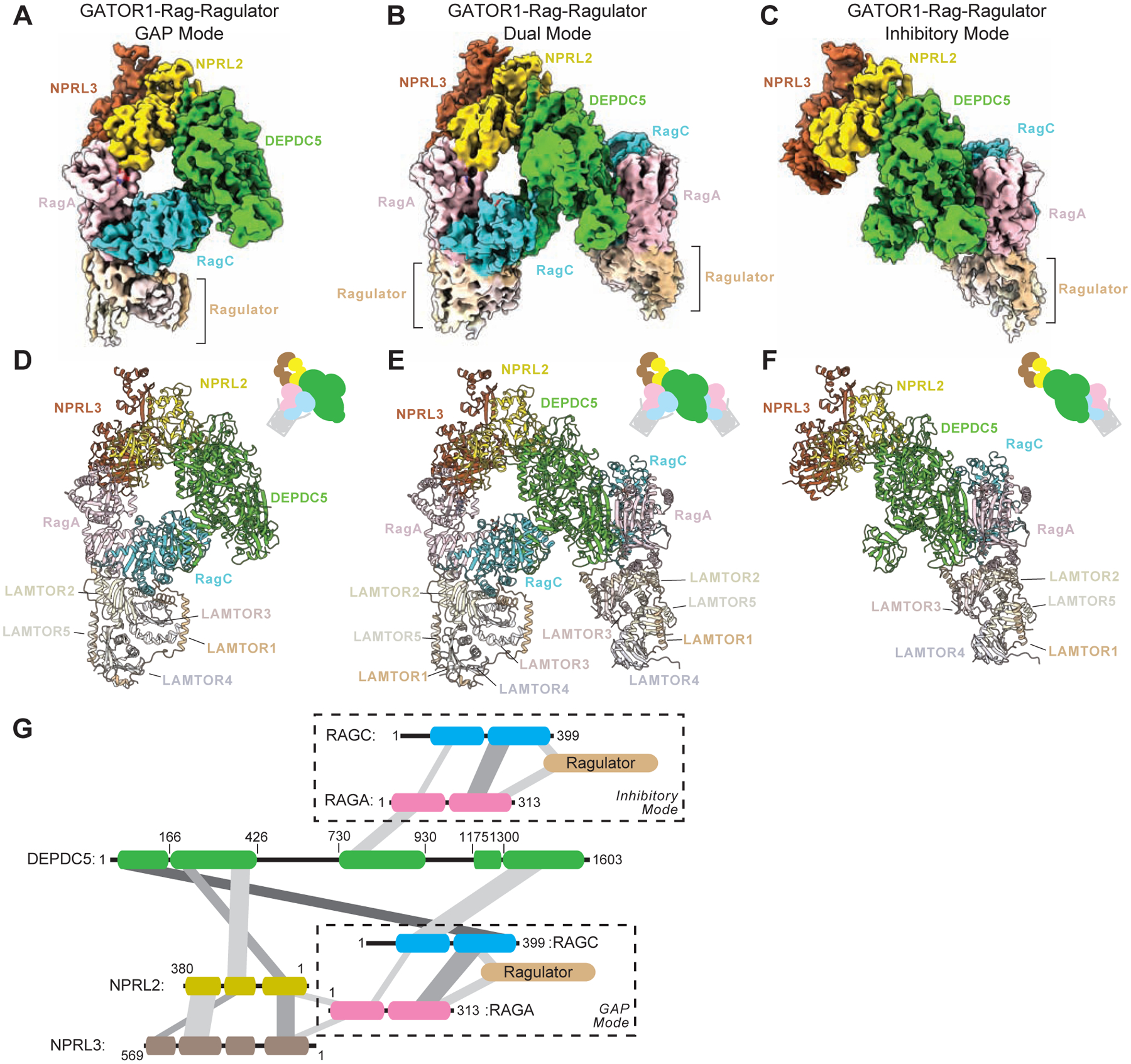
A–C. Cryo-EM density maps for the GATOR1-Rag-Ragulator complex in G-mode (A, EMD-25653), D-mode (B, EMD-25654), and I-mode (C, EMD-25652). Subunits were colored as following: DEPDC5 in green, NPRL2 in yellow, NPRL3 in brown, RagA in pink, RagC in cyan, LAMTOR1 (p18) in light brown, LAMTOR2 (p14) in light yellow, LAMTOR3 (MP1) in light pink, LAMTOR4 (c7orf59) in light purple, and LAMTOR5 (HBXIP) in beige.
D–F. Atomic models for the GATOR1-Rag-Ragulator complex in G-mode (D, PDB:7T3B), D-mode (E, PDB:7T3C), and I-mode (F, PDB:7T3A). Subunits were colored the same as above.
G. Interaction graph within the GATOR1-Rag-Ragulator complex.
We built structural models based on the maps resolved above (Figure 3D–3F, S1E, S2E, S3F, S3G, see Methods for details). According to local resolution estimates (Figure S1F, S2F, and S3H), the core region of the maps exhibited better resolution, and thus allows for de novo model building by tracing the backbone and registering bulky residues. For the peripheral region, especially the C-terminal domain of NPRL3, the cryo-EM density becomes scattered, preventing direct model building. We therefore harnessed the AlphaFold algorithm and used the predicted 3D structural model for NPRL3 as a reference (Jumper et al., 2021). We divided the predicted model into globular domains that contain ~80 residues each, and fit these individual domains into our cryo-EM density map (Figure S4A–S4E). Satisfactorily, the precise fit of the backbone as well as the matching of bulky residues and cryo-EM density generated a complete model for the C-terminal domain of NPRL3 (Figure S4F–S4H).
In all three complexes, the Rag-Ragulator complex was resolved at two positions, in which the Rag GTPase heterodimer directly contacts GATOR1, and Ragulator binds to the CRD of the Rag GTPase at the distal end. For the I-mode, RagA binds to the critical strip of DEPDC5, consistent with the previously resolved GATOR1-Rag complex (Figure 3F). For the G-mode, RagA-RagC was located near the Longin domain heterodimer of NPRL2-NPRL3, also consistent with the previous genetic prediction (Figure 3D). For the D-mode, two copies of Rag-Ragulator subcomplexes sandwich the GATOR1 complex in the middle (Figure 3E), suggesting the two binding modes are not mutually exclusive in vitro. We further mapped out the interactions among the subunits, and depicted a complicated network of contacts (Figure 3G).
Architecture of the GATOR1-Rag-Ragulator complex in two modes
The Rag GTPase heterodimer differentially interacts with GATOR1 at the two binding sites (Figure 4A and 4B). In the inhibitory mode, the critical strip on DEPDC5 contacts the NBD of RagA and no contact between GATOR1 and RagC(S75N) is observed (Figure 4A). Here, RagA adopts the GTP-bound conformation, in which Switch I forms a short α-helix (αGSI) and swings to the top of the nucleotide binding pocket, covering the bound nucleotide (GppNHp in I-mode, GDP:AlF3 in D-mode). This helical conformation is consistent with the previously resolved crystal structure of Gtr1p-Gtr2p and the Rag GTPases (Anandapadamanaban et al., 2019; Jeong et al., 2012), and allows for interaction with the critical strip on DEPDC5. As αGSI only forms when RagA binds GTP or its analogue, but not GDP, the inhibitory interaction can only be secured when the Rag GTPases are in the GTPRagA-RagCGDP configuration, a state into which RagA-RagC(S75N) locks.
Figure 4. Architecture of the GATOR1-Rag-Ragulator complex.
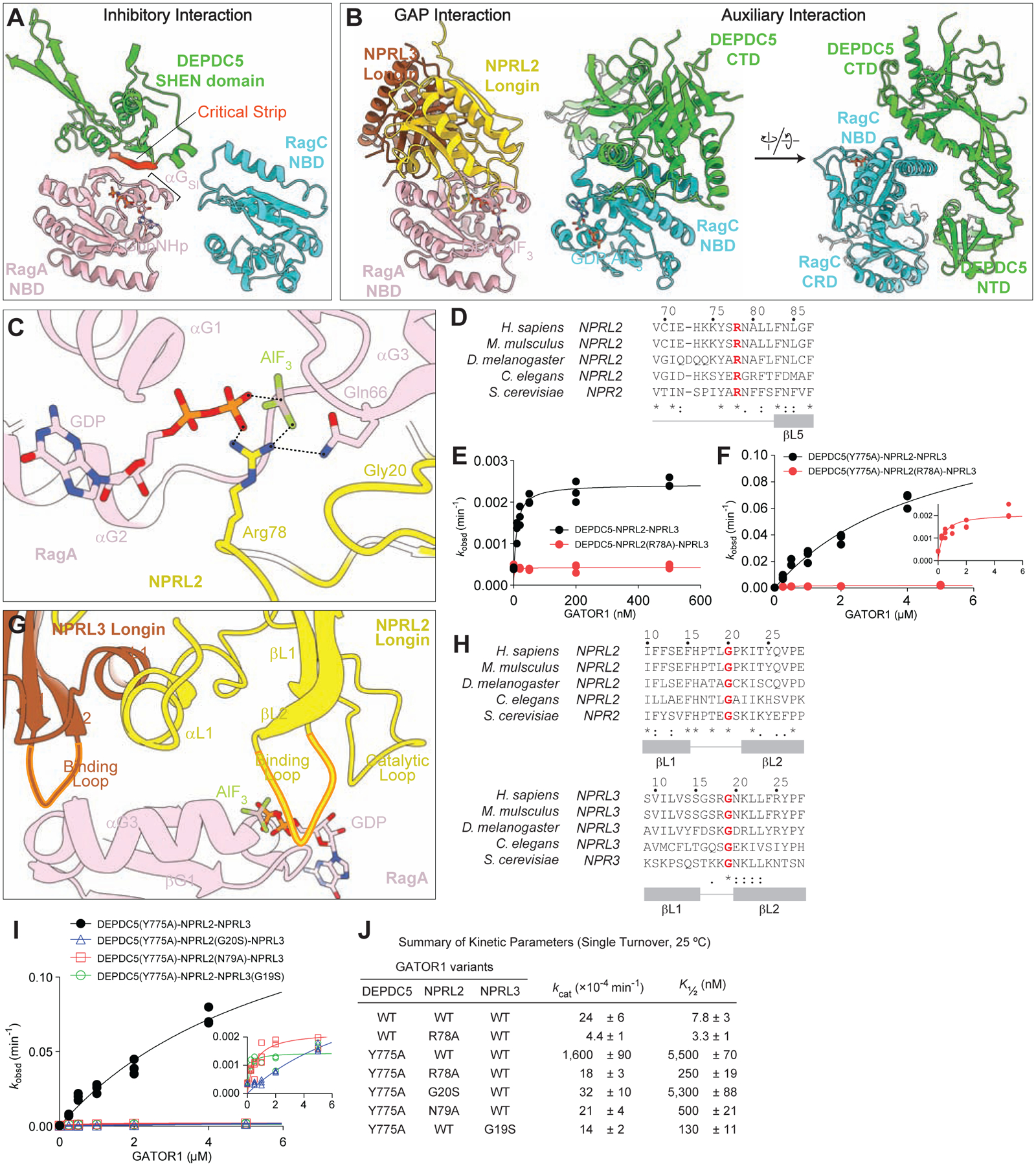
A. Interaction between the Rag GTPase heterodimer and GATOR1 in the inhibitory mode.
B. Interaction between the Rag GTPase heterodimer and GATOR1 in the GAP mode.
C. Arg78 of NPRL2 points at the nucleotide bound to RagA, serving as an arginine finger to catalyze GTP hydrolysis by RagA.
D. Sequence conservation of Arg78 across species.
E. Stimulated GTP hydrolysis assay by GATOR1 containing NPRL2(R78A) mutation.
F. Stimulated GTP hydrolysis assay by GATOR1 containing DEPDC5(Y775A) and NPRL2(R78A) mutations.
G. Two loops on NPRL2 and NPRL3 coordinate the binding to RagA-NBD.
H. Sequence conservation of the two loops on NPRL2 and NPRL3.
I. Stimulated GTP hydrolysis assay by GATOR1 containing mutations on the loop of NPRL2 and NPRL3 (inset shows zoom-in of NPRL2 and NPRL3 mutants).
J. Summary of kinetic parameters from panels 4E, 4F, and 4I. Mean ± SEMs from three independent experiments are reported.
In the GAP mode, we observed extensive contacts between both Rag subunits and all three subunits of GATOR1 (Figure 4B). Here, both Rag subunits bind GDP:AlF3, and their Switch I regions adopt the GTP-bound conformation (Figure S4I–S4L). Such nucleotide binding configuration is in contrast to previous structures, in which the two subunits were almost always loaded with ‘opposite’ nucleotides (GTP vs. GDP) (Anandapadamanaban et al., 2019; Fromm et al., 2020; Lawrence et al., 2019; Rogala et al., 2019; Shen et al., 2018, 2019b). Combined with the biochemical evidence below, we defined the two interfaces as the GAP interface and the auxiliary interface, respectively. The GAP interface is formed between the heterodimerized Longin domains of NPRL2-NPRL3 and the NBD of RagA (Figure 4B), in which an arginine residue, Arg78 of NPRL2, inserts into the nucleotide binding pocket of RagA and points directly at the GDP:AlF3 molecule (Figure 4C and Figure S4M). The auxiliary interface is formed between RagC and the N-terminal domain (NTD) and C-terminal domain (CTD) of DEPDC5 (Figure 4B). As GATOR1 has been shown to stimulate GTP hydrolysis on RagA, this additional contact with RagC was unexpected and as demonstrated below, secures the nucleotide loading state of the released product.
NPRL2-NPRL3 interacts with RagA to execute the catalytic reaction
To probe the biological function of the contacts between NPRL2-NPRL3 and RagA, we characterized a series of point mutations at the GAP interface. We first assessed the Arg78 residue of NPRL2 (Figure 4C). This arginine residue localizes in a loop between the fourth and fifth β-strand of the Longin domain, and is evolutionarily conserved (Figure 4D). Upon mutation to an alanine residue, we observed severe defects in stimulating GTP hydrolysis by the Rag GTPases, either in the context of wild-type GATOR1 (Figure 4E) or GATOR1 containing the DEPDC5(Y775A) mutation (Figure 4F, summarized in Figure 4J). Together with the structural evidence, we assigned Arg78 of NPRL2 as the catalytic arginine finger.
In addition to the catalytic loop that contains Arg78, two additional loops on NPRL2 and NPRL3 directly contact RagA-NBD in our structural model (Figure 4G). These two loops localize between the first and second β-strand of the Longin domain, and are evolutionarily conserved (Figure 4H). To probe the physiological role of these loops, we harnessed HEK-293T cell lines in which individual GATOR1 subunits were knocked out using CRISPR-Cas9. In these cell lines, mTORC1 becomes hyperactive regardless of the nutrient condition, as the phosphorylation of its downstream target (pT389-S6K1) remains high in the presence and absence of amino acids (Figure S5A). We can then introduce subunit mutants back into these cell lines to test their function. As a positive control, addition of wild-type NPRL2 restored the response to amino acids in NPRL2-knockout cells (Figure S5A). Notably, when we mutated the conserved Gly20 residue to a serine, it failed to sensitize this cell line to amino acids (Figure S5A and S5B), suggesting its necessity in mediating amino acid signals. To further evaluate its GAP activity in vitro, we recombinantly purified GATOR1 that contains NPRL2(G20S) and measured the ability to stimulate GTP hydrolysis (Figure 4I and 4J). We observed that NPRL2(G20S) failed to stimulate GTP hydrolysis by the Rag GTPases, suggesting a glycine residue is necessary to maintain the correct conformation of the binding loop on NPRL2. We obtained analogous results for Gly19 on NPRL3 (Figure S5C and S5D, Figure 4I and 4J). These results support the functional importance of the two loops in mediating the binding between NPRL2-NPRL3 and RagA, and we therefore defined them as the binding loops.
DEPDC5 interacts with RagC to secure the nucleotide loading state of the GAP product
GATOR1 is a GAP for RagA (Bar-Peled et al., 2013; Panchaud et al., 2013; Shen et al., 2018), and thus far there is no evidence to associate its function with RagC. We observed extensive interactions between DEPDC5 and RagC in our structural model (Figure 5A). Specifically, DEPDC5-NTD contacts RagC-CRD, and DEPDC5-CTD contacts RagC-NBD. The residues at the interface are evolutionarily conserved (Figure 5B), suggesting their functional importance. To formally test this hypothesis, we utilized an HEK-293T cell line in which DEPDC5 was knocked out (Figure 5C and Figure S5E). We generated a series of constructs carrying DEPDC5 mutation at the auxiliary interface and expressed them individually in DEPDC5-KO cells. Although single point mutants behaved like wild-type DEPDC5 and were still capable of sensitizing cells to external amino acids (Figure S5E), a mutant with a truncation of the NTD of DEPDC5 [Δ165, DEPDC5(166–1603)] completely failed to do so (Figure 5C), as well as a double mutant in which the two contacting interfaces are simultaneously disrupted [DEPDC5(Y82A, R1407A)] (Figure S5F). Importantly, this defect is not caused by structural alteration of GATOR1, as (1) the DEPDC5(Δ165) mutant co-immunoprecipitates a similar amount of NPRL2, NPRL3, and the Rag GTPases in cells (Figure 5D), and (2) a Coomassie staining gel after gel-filtration chromatography confirms the co-elution of all three GATOR1 subunits for the DEPDC5(Δ165) mutant (Figure S5G). These results suggested that the interactions on the auxiliary interface between DEPDC5 and RagC are likely redundant and are necessary to maintain normal function of GATOR1 in cells.
Figure 5. DEPDC5 coordinates the nucleotide loading states of RagA and RagC.
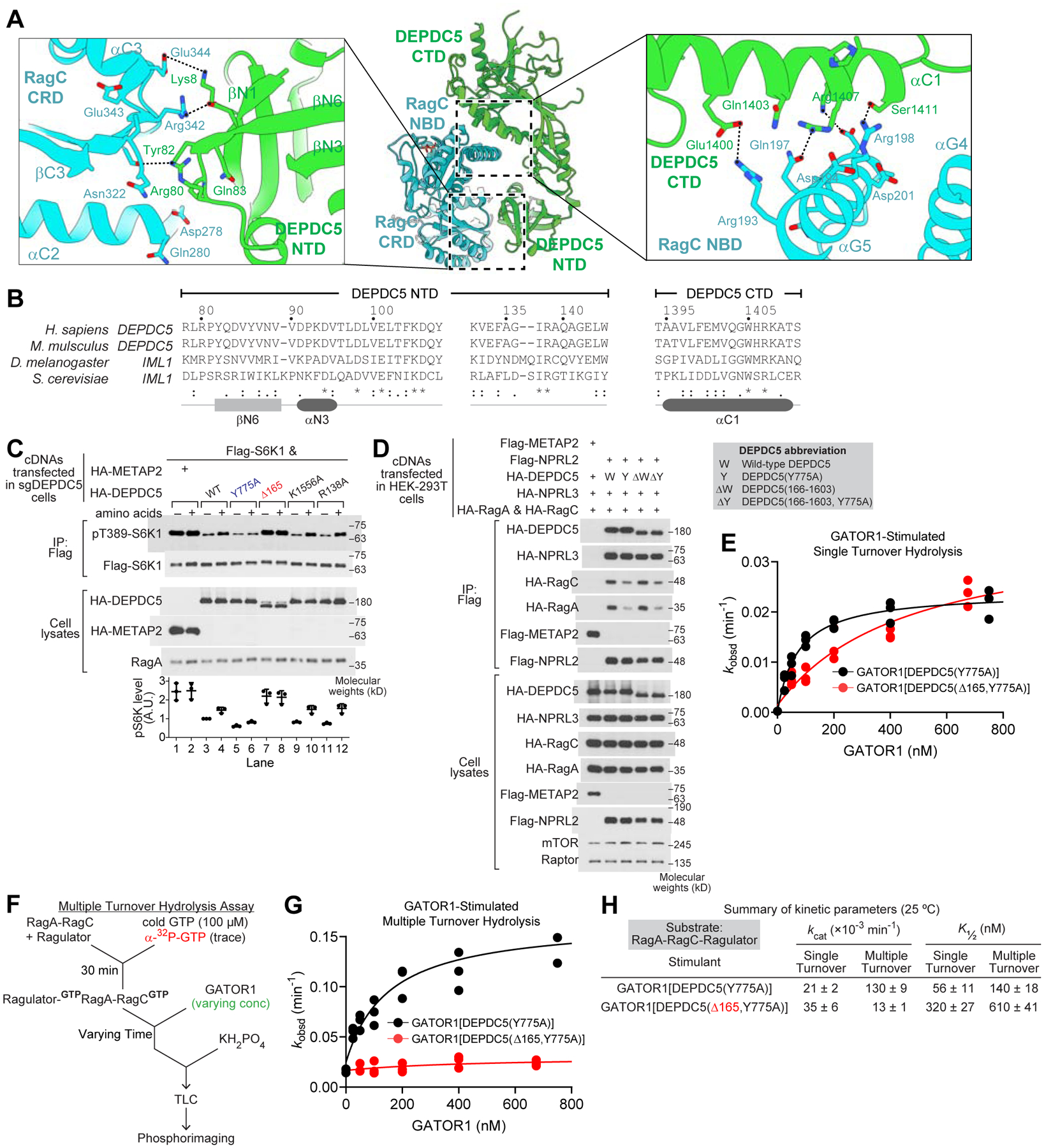
A. Interaction between DEPDC5 and the RagC subunit is mediated by two contact surfaces: the RagC C-terminal roadblock domain (CRD) and DEPDC5 N-terminal domain (NTD, left), as well as the RagC nucleotide binding domain (NBD) and DEPDC5 C-terminal domain (CTD, right).
B. Sequence conservation of DEPDC5-NTD and DEPDC5-CTD.
C. Effect of expression of various DEPDC5 mutants on the ability of sgDEPDC5 cells to restore mTORC1 signaling in the presence and absence of amino acids.
D. Co-immunoprecipitation of NPRL2, NPRL3, and the Rag GTPase heterodimer with the DEPDC5 variants.
E. Single turnover GTP hydrolysis assay by GATOR1 containing DEPDC5(Δ165) mutation.
F. Experimental setup for the multiple turnover GTP hydrolysis assay.
G. Multiple turnover GTP hydrolysis assay stimulated by GATOR1 containing DEPDC5(Δ165, Y775A) mutation.
H. Summary of GTP hydrolysis kinetics for Rag-Ragulator under single- and multiple-turnover conditions at 25 °C. Mean ± SEMs from three independent experiments are reported.
To investigate the biochemical properties of the auxiliary interface, we included the recombinantly purified Δ165 mutant in our GAP assay and compared it with full length GATOR1. Under the single turnover condition, GATOR1[DEPDC5(Δ165,Y775A)] behaves similarly to GATOR1[DEPDC5(Y775A)], with an increase in K½ for the Δ165 mutant (Figure 5E and 5H). The kcat value remains similar, suggesting the catalytic interface is still functional. However, when we included the mutant in a multiple turnover GTP hydrolysis assay (Figure 5F), in which both Rag subunits were forced to bind GTP simultaneously, we observed a striking difference: the Δ165 mutant failed to stimulate GTP hydrolysis by the Rag GTPase heterodimer (Figure 5G and 5H). This result indicates that the auxiliary interface is functionally active only under the condition when both Rag subunits are GTP-bound.
Conformation of the Rag GTPases in the GAP mode
Based on the structural models and biochemical characterization, we were curious to find that the two Rag mutants preferentially occupy different binding sites. To investigate the probable cause, we first compared their conformations. We overlaid the CRDs of the two heterodimers, which show minimal difference, and employed two parameters to compare the relative orientation of the two NBDs (Figure 6A). First, d measures the distance between the N-terminal tips of the two αG5 helices on the Rag GTPases, and represents the relative distance between the NBDs. Second, θ measures the angle formed by three points: the C- and N-terminal tips of the αG5 helix of RagA, and the N-terminal tip of the αG5 helix of RagC, thus reflecting the relative rotation. Both RagA-RagC(F92A) and RagA-RagC(S75N) show an open conformation, similar to the open conformation of Gtr1p-Gtr2p, in which both Gtr subunits are bound to GppNHp (Gong et al., 2011). However, they are significantly different from the closed conformation of Gtr1p-Gtr2p, in which Gtr1p binds GTP and Gtr2p binds GDP (Jeong et al., 2012) (Figure 6A and 6B). Interestingly, RagA-RagC(F92A) at the GAP site opens even wider than RagA-RagC(S75N), as d and θ are larger for RagA-RagC(F92A). This is likely caused by the induced-fit mediated by GATOR1 and the Rag GTPases in the GAP mode in order to form both the GAP interface and the auxiliary interface.
Figure 6. Conformation of the Rag GTPases in the inhibitory mode and in the GAP mode.

A. The Rag GTPase heterodimer adopts different conformations upon binding to upstream regulators or downstream effectors..
B. Summary of the conformational parameters for Rag GTPases at different states in panel A.
C. Multiple turnover GTP hydrolysis assay stimulated by GATOR1[DEPDC5(Y775A)].
D. Thermodynamic cycle for GATOR1 and nucleotide binding to the Rag GTPase heterodimer.
E. Hypothetical in vivo binding of two copies of the Rag-Ragulator complex on the lysosomal membrane to GATOR1.
F. Relative orientation of GATOR1 to the lysosomal membrane dictates its GAP function. In the inhibitory mode, Arg78 of NPRL2 localizes away from the lysosomal surface.
To probe the effect of nucleotide loading states on the global conformation of the Rag GTPase heterodimer, we utilized a previously developed crosslinking assay, in which radioactively labeled guanine nucleotide was incubated with the Rag GTPases, followed by ultraviolet (UV) irradiation to induce crosslinking between the bound nucleotide and the corresponding subunit (Shen et al., 2017). We can then resolve the binding to individual subunits by SDS-PAGE (Figure S6A). For RagA-RagC(F92A), both subunits bind to guanine nucleotides at a similar affinity to wild-type Rag GTPases, suggesting that the F92A mutation does not alter the nucleotide binding preference (Figure S6B–S6I). For RagA-RagC(S75N), the mutation on RagC specifically abolishes its binding to GTP, but not GDP (Figure S6B–S6I). This difference in binding preference matches the structural model, and correlates the dual GTP-loaded state of GTPRagA-RagC(F92A)GTP with the open conformation in the GAP mode.
Coordination of the nucleotide binding states of the Rag subunits
Prior to this study, two out of the four nucleotide loading states of the Rag GTPase heterodimer have been correlated with its biological functions: GTPRagA-RagCGDP being the active state and GDPRagA-RagCGTP being the inactivated state (Kim et al., 2008; Sancak et al., 2008). The dual GTP-loaded state has previously been considered improbable for apo Rag GTPases due to the locking mechanism that actively prevents two GTP molecules from binding simultaneously (Shen et al., 2017). To probe this question, we forced both Rag subunits to bind GTP simultaneously by including a saturating concentration of GTP at 100 μM, which is much higher than the KM (~10 μM) (Shen et al., 2017), and measured the stimulatory effect of GATOR1 on the GTP hydrolysis rate (Figure 6C). The kcat/KM value here is over five-fold higher than when the Rag GTPase heterodimer is singly loaded (0.31 vs. 0.06 μM−1 min−1, cf. Figure 1E), suggesting the dual GTP-loaded Rag heterodimer is a more favorable substrate for the GAP mode.
Summarizing the results above, we obtained a first glimpse of how GATOR1 controls the nucleotide loading states of the Rag GTPases (Figure 6D). As a negative regulator, the substrate for GATOR1 is the active form of the Rag GTPases, GTPRagA-RagCGDP (Figure 6D, top left). Because the dual GTP-loaded Rag GTPase heterodimer is a better substrate for GATOR1 (cf. Figure 6C), K2 is greater than K3 (Figure 6D, vertical equilibria). To maintain the balance around the thermodynamic cycle, K4 then must be greater than K1 (Figure 6D, horizontal equilibria, K1K2 = K3K4 so that ΔG = 0). In other words, upon GATOR1 binding to the singly GTP-loaded Rag GTPase heterodimer (GTPRagA-RagCGDP), the energy barrier for GTP binding to the RagC subunit is lowered, resulting in the dual GTP-loaded state. Reaching this state is particularly useful here, as after GATOR1 stimulates GTP hydrolysis on the RagA subunit, the released product will naturally become the inactivated form (GDPRagA-RagCGTP). If GATOR1 does not assist RagC to bind GTP and simply catalyzes GTP hydrolysis on RagA (K3), the released product will be in a dual GDP-loaded state, which has a potential risk of returning to the active configuration, as previous study has shown that RagA and RagC have similar association rates to GTP (Shen et al., 2017). Taken together, we concluded that GATOR1 coordinates the nucleotide loading states of both Rag subunits, and ensures that only the inactivated form of Rag GTPases is released once the GAP reaction finishes. Moreover, we could tentatively define the dual GTP-loaded state (GTPRagA-RagCGTP) for the Rag GTPase heterodimer as an on-pathway intermediate for GATOR1-simulated GTP hydrolysis of RagA.
Relative orientation to the lysosomal surface
The Rag GTPase heterodimer is anchored on the lysosomal membrane through the N-terminal lipid tail on the LAMTOR1 subunit of Ragulator (Shigeyki et al., 2009). When the Rag GTPase heterodimer is activated, recruitment of mTORC1 also happens on the lysosomal membrane (Sancak et al., 2008). Therefore, in order to switch off the activated Rag GTPases, GATOR1, as a constitutively lysosomal-anchored complex (Wolfson et al., 2017), has to follow additional spatial constraints along the membrane. In our structural model, the first 45 residues at the N-terminus of the LAMTOR1 subunit cannot be resolved, likely due to the flexibility caused by the proline- and serine-rich sequence, but we can clearly visualize the first α-helix on LAMTOR1 that connects to this flexible linker (Figure S7E and S7F). We noticed that, when the two binding sites are simultaneously occupied, as in D-mode, the flexible linkers on the two copies of LAMTOR1 reside on opposite sides of the complex, and thus likely extend in opposite directions (red circle, Figure 6E). Taking the spatial constraints along the lysosomal membrane into account, we calculated that the minimum distance between the visible tips of the two lipid anchors (Ala46 of LAMTOR1 in the inhibitory mode and Asp49 of LAMTOR1 in the GAP mode) is 100 – 120 Å (see Methods for details). Using an estimation of 3.5 Å/amino acid, the maximum length of the flexible linker is 150 Å. Therefore, if the D-mode were to form on the lysosomal surface, two boundary conditions must be satisfied: (1) Anchor-I should be flattened along the membrane to minimize the distance of the arginine finger to the lysosome, and (2) Anchor-G must be fully extended, in order for RagA to reach the arginine finger, which points away from the lysosome (Figure 6E). Considering the enthalpy cost in a fully extended conformation for Anchor-G and the entropy cost in excluding solvent molecules for Anchor-I, we believe that formation of the D-mode in vivo is likely improbable.
Summarizing the results above, we speculate that the two binding sites on GATOR1 may be mutually exclusive to one another for the Rag-Ragulator complexes that localize on the lysosomal membrane. GATOR1 may bind to the Rag-Ragulator complex either in the inhibitory mode, or in the GAP mode, but not both simultaneously (Figure 6F). Therefore, the GAP activity of GATOR1 is likely restricted by its orientation relative to the lysosomal membrane, and we speculate that upstream regulators such as KICSTOR, GATOR2, and/or amino acid sensors contribute to controlling this unique feature in order to coordinate mTORC1 activity with nutrients.
Discussion
As an essential negative regulator of the mTORC1 pathway, GATOR1 converts the Rag GTPase heterodimer from the active state to the inactivated state upon amino acid deprivation. Here, we presented three structural models for the GATOR1-Rag-Ragulator complex to reveal the molecular mechanism of GATOR1. In the inhibitory mode, the GAP activity of GATOR1 is restricted by spatial constraints imposed by the lysosomal membrane. We proposed that this mode serves as an off-pathway trap in vivo that prevents hyperactivation of GATOR1. In the GAP mode, GATOR1 mediates extensive interactions with the Rag GTPase heterodimer using the GAP interface and the auxiliary interface. These interactions coordinate the nucleotide loading states of both Rag subunits. Our models identify a unique GAP mechanism and provide a structural basis for further investigation into the regulation of the activity of GATOR1.
Due to the heterodimerized architecture of the Rag GTPases, their regulators need to coordinate the nucleotide loading states of both subunits. This is especially difficult considering that the two subunits are always bound to opposite nucleotides (GTP vs. GDP) in the two stable functional states. Therefore, to convert the Rag GTPase heterodimer from the active to the inactive configuration, or vise versa, requires: (1) conversion of the GTP-bound subunit to GDP-bound, and (2) conversion of the GDP-bound subunit to GTP-bound. GATOR1 and FLCN-FNIP2 have been identified as GAPs for RagA and RagC, respectively. If no other regulation is applied, the product for the GAP reaction will be dual GDP-loaded Rag GTPases. This is promiscuous as RagA and RagC have similar association rates to GTP, and therefore have a 50% chance of loading a GTP molecule to the undesired subunit. Here, we proposed a mechanism for GATOR1 to coordinate the nucleotide loading states for both Rag subunits, thus resolving the inefficiency outlined above. Using the auxiliary interaction formed between DEPDC5 and RagC, GATOR1 promotes RagC into its GTP-bound state while stimulating GTP hydrolysis on RagA with the arginine finger on NPRL2. These coordinated actions ensure that only the product, GDPRagA-RagCGTP, is released to inactivate mTORC1 signaling.
To compare the molecular architecture of the two GAPs in the mTORC1 pathway, we overlaid the Rag GTPases in the two structural models (Figure S7A and S7B) (Lawrence et al., 2019; Shen et al., 2019b). GATOR1 and FLCN-FNIP2 interact with the NBDs of both RagA and RagC. GATOR1 forms two interfaces on the lateral side of the Rag GTPase heterodimer, and pulls the NBDs away from each other (Figure S7A). In contrast, FLCN-FNIP2 wedges between the NBDs, and pushes them apart (Figure S7B). These two GAPs cause dramatically different conformations of the Rag GTPase heterodimer (Figure S7C and S7D). However, one common feature is that despite serving as the GAP for a specific subunit, they contact both Rag subunits. We showed here that GATOR1 can manipulate the nucleotide loading states for both Rag subunits to control the functional state of the heterodimer. Accordingly, we suspect a similar mechanism may also be present for FLCN-FNIP2.
The Rag GTPase heterodimer functions through a unique locking mechanism that maintains the singly GTP-loaded state. Here, we found that GATOR1 disrupted this intersubunit communication by pulling the two subunits away from each other, and thus allows for the second GTP to bind to RagC. Such an interruption is observed in structural studies for other upstream regulators such as SLC38A9 (Fromm et al., 2020), FLCN-FNIP2 (Lawrence et al., 2019; Shen et al., 2019b), and downstream effectors, Raptor/mTORC1 (Anandapadamanaban et al., 2019; Rogala et al., 2019). Accordingly, we suspect that interruption of the intersubunit communication may be a necessary step before commitment to a specific functional state. In general, these structural models suggest that the Rag GTPase heterodimer functions through a complex molecular mechanism, which is yet to be explored to fully understand how it transmits amino acid signals to mTORC1.
Limitations of the study
Based on the current models, we do not know what triggers the release of GATOR1 from the inhibitory mode and activates its GAP activity. As GATOR2 is the main receiver of the amino acid signals, it is conceivable that (1) in a nutrient deprived condition, GATOR2 resolves the GATOR1-Rag interaction in the inhibitory mode, and liberates the GAP activity of GATOR1 so as to turn off the mTORC1 pathway, or (2) in a nutrient rich condition, GATOR2 confines GATOR1 to the inhibitory mode, and switches off its GAP activity to turn on the mTORC1 pathway. Further structural and biochemical characterization is required to clarify this critical step of amino acid sensing.
STAR Methods
RESOURCE AVAILABILITY
LEAD CONTACT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kuang Shen (Kuang.Shen@umassmed.edu).
MATERIALS AVAILABILITY
Plasmids generated in this study are derived from the ones deposited to Addgene (Key Resource Table) and are available with no restrictions.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Raptor | EMD Millipore | 09-217 |
| Rabbit anti-HA | Cell Signaling Technology | 3724 |
| Rabbit anti-Flag | Cell Signaling Technology | 14793 |
| Rabbit anti-S6K1 | Cell Signaling Technology | 9202 |
| Rabbit anti-pT389-S6K1 | Cell Signaling Technology | 9205 |
| Rabbit anti-mTOR | Cell Signaling Technology | 2983 |
| Anti-rabbit, HRP-linked antibody | Cell Signaling Technology | 7074 |
| Bacterial Strains | ||
| BL21(DE3) | NEB | C2527 |
| LOBSTR | Kerafast | EC1002 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| HEPES | Sigma-Aldrich | H3375 |
| Potassium acetate | Sigma-Aldrich | P1190 |
| Magnesium chloride | Sigma-Aldrich | M8266 |
| Sodium chloride | Sigma-Aldrich | S7563 |
| DTT | Sigma-Aldrich | D5545 |
| Chaps | Sigma-Aldrich | C5070 |
| Triton | Sigma-Aldrich | T9284 |
| Imidazole | Sigma-Aldrich | I5513 |
| GTP | Sigma-Aldrich | G8877 |
| GDP | Sigma-Aldrich | G7127 |
| TCEP | Sigma-Aldrich | C4706 |
| ATP | Sigma-Aldrich | A7699 |
| α-32P-GTP | PerkinElmer | BLU006H250UC |
| DMEM | Thermal Fisher Scientific | 10564-011 |
| SMM 293-TII | Sinobiological | M293TII |
| Deposited data | ||
| Atomic model for GATOR1-Rag-Ragulator (Inhibitory mode) | Protein Data Bank | 7T3A |
| Atomic model for GATOR1-Rag-Ragulator (GAP mode) | Protein Data Bank | 7T3B |
| Atomic model for GATOR1-Rag-Ragulator (Dual mode) | Protein Data Bank | 7T3C |
| EM density map for GATOR1-Rag-Ragulator (Inhibitory mode) | EM Data Bank | 25652 |
| EM density map for GATOR1-Rag-Ragulator (GAP mode) | EM Data Bank | 25653 |
| EM density map for GATOR1-Rag-Ragulator (Dual mode) | EM Data Bank | 25654 |
| Raw images for western blots and Coomassie staining gel | Mendeley | DOI: 10.17632/4vnfng4w4b.1 |
| Experimental Models: Cell Lines | ||
| Human: HEK-293T | ATCC | CRL-3216 |
| Human: FreeStyle 293-F | Thermal Fisher Scientific | R79007 |
| Recombinant DNA | ||
| [pSK109] pETDuet-1 RagA-RagC | Addgene | 99641 |
| [pSK235] pK RagA-RagC | Addgene | 99649 |
| [pSK315] pRK5 Flag-DEPDC5 | Addgene | 46327 |
| [pSK163] pRK5 HA-NPRL2 | Addgene | 99709 |
| [pSK164] pRK5 HA-NPRL3 | Addgene | 46330 |
| [pSK66] pRK5 Flag-S6K1(rat) | Addgene | 100509 |
| Software and Algorithms | ||
| Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Relion | (Zivanov et al., 2018) | https://www3.mrc-lmb.cam.ac.uk/relion/index.php?title=Main_Page |
| SerialEM | (Mastronarde, 2005) | http://bio3d.colorado.edu/SerialEM/ |
| cryoSPARC | (Punjani et al., 2017) | https://guide.cryosparc.com/ |
| Coot | (Emsley et al., 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix | (Liebschner et al., 2019) | http://www.phenix-online.org/ |
| Other | ||
| Flag-M2 affinity resin | Sigma-Aldrich | A2220 |
| Ni-NTA affinity resin | EMD Millipore | 30230 |
| Cellulose 300 PEI thin layer chromatography plates | Sorbtech | 1122126 |
DATA AND CODE AVAILABILITY
Atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under the accession number 7T3A (Inhibitory mode), 7T3B (GAP mode), and 7T3C (Dual mode). Electron density maps have been deposited in EM Data Bank under the accession number EMD-25652 (Inhibitory mode), EMD-25653 (GAP mode), and EMD-25654 (Dual mode). These coordinates and maps are publicly available as of the date of publication. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. The access numbers are listed in the key resources table. Raw cryo-EM movies reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK-293T cells were obtained from American Type Culture Collection (ATCC) and were maintained in an incubator set at 37 °C and 5% CO2. They were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% inactivated fetal bovine serum (Gibco, Life Technologies, Thermo Fisher), 20 mM HEPES, 2 mM glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin.
HEK-293 FreeStyle cells were obtained from ThermalFisher Scientific and were maintained in a New Brunswick S41i shaker (Eppendorf) set at 37 °C, 135 rpm, 8% CO2, and 100% humidity. They were cultured in SMM 293-TII media (Sinobiological), supplemented with 100 IU/ml penicillin and 100 μg/ml streptomycin.
BL21(DE3) and LOBSTR E. Coli strains were grown at 37 °C in a New Brunswick S44i shaker (Eppendorf) in LB media. To induce protein expression, they were transformed by the corresponding plasmids (see below), propagated at 37 °C, and induced with IPTG at 18 °C.
METHOD DETAILS
Protein purification
The Rag GTPase heterodimer was purified as previously described (Shen et al., 2017). In brief, co-expression of C-terminally His-tagged RagA and tagless RagC was induced by IPTG in BL21(DE3) cells. The heterodimer was then purified through Q Sepharose (Cytiva), Ni-NTA (Qiagen), MonoQ (Cytiva), and Superdex 200 (Cytiva) columns sequentially. For Rag mutants, single point mutation was introduced using the QuikChange protocol (Agilent), and the corresponding protein was purified following a similar protocol.
The pentameric Ragulator complex was purified as previously described (Shen and Sabatini, 2018). In brief, the five subunits were co-transformed and expressed in BL21(DE3) strain. The pentameric complex was purified through glutathione (Pierce, Thermo Fisher), Ni-NTA, MonoQ, and Superdex 200 columns sequentially.
Human GATOR1 was expressed and purified as previously described (Shen et al., 2018). In brief, HA-tagged NPRL2 and NPRL3 were co-expressed with Flag-tagged DEPDC5 in FreeStyle 293 cells. The complex was isolated using Flag-M2 beads (Sigma) in Triton lysis buffer (40 mM NaHEPES, pH 7.4; 5 mM MgCl2; 100 mM NaCl; 10 mM Na4P2O7; 10 mM sodium β-glycerol phosphate; 1% Triton X-100; and one tablet of EDTA-free protease inhibitor (Roche) per 25 ml of buffer) and further purified by a Superdex 200 gel-filtration column. For GATOR1 mutants, mutations were introduced using the QuikChange protocol, and the corresponding protein complex was purified following a similar protocol.
The GATOR1-Rag-Ragulator complex was assembled as follows. For the complex in the inhibitory mode, 200 μg of wild-type GATOR1 was incubated with 200 μg of RagA-RagC(S75N) and 200 μg of wild-type Ragulator in a total volume of 500 μl for ten hours at 4 °C. The assembly solution contains 50 mM NaHEPES (pH 7.4), 100 mM NaCl, 2 mM MgCl2, 2 mM DTT, 0.1% CHAPS, 100 μM GppNHp, and 100 μM GDP. The decameric complex was separated from excess Rag GTPases and Ragulator on a Superdex 200 column.
For the complex in the GAP mode, 200 μg of GATOR1[DEPDC5(Y775A)] was incubated with 200 μg of RagA-RagC(F92A) and 200 μg of wild-type Ragulator in a total volume of 500 μl for ten hours at 4 °C. The assembly solution contains 50 mM NaHEPES (pH 7.4), 100 mM NaCl, 2 mM MgCl2, 2 mM DTT, 0.1% CHAPS, 100 μM GDP, 1 mM AlCl3, and 20 mM NaF. The decameric complex was separated from excess Rag GTPases and Ragulator on a Superdex 200 column.
For the complex in which both binding sites were occupied, 200 μg of wild-type GATOR1 was incubated with 100 μg of RagA-RagC(F92A), 100 μg of RagA-RagC(S75N), and 200 μg of wild-type Ragulator in a total volume of 500 μl for ten hours at 4 °C. The assembly solution contains 50 mM NaHEPES (pH 7.4), 100 mM NaCl, 2 mM MgCl2, 2 mM DTT, 0.1% CHAPS, 100 μM GDP, 1 mM AlCl3, and 20 mM NaF. The 17-mer complex was separate from excess Rag GTPases and Ragulator on a Superdex 200 column.
All GATOR1-Rag-Ragulator complexes were concentrated down to ~5 μg/μl in a 100kDa molecular weight cut-off concentrator (Amicon) and ultracentrifuged at 100,000×g for 30 minutes prior to plunge freezing grids.
Cryo grids preparation
Cryo grids were prepared immediately after protein purification. For the I-mode complex, 3 μL of sample was applied to a glow-discharged 400-mesh quantifoil 1.2/1.3 Au grid (Quantifoil, Großlöbichau Germany) on a Mark IV Vitrobot (FEI/ThermoFisher). The sample chamber on the Vitrobot was kept at 5 °C with a targeted relative humidity level at 95%. The grid was blotted from both sides for 3 seconds with blot force set at 3. After blotting the grid was rapidly plunge-frozen into a liquid ethane bath cooled by liquid nitrogen.
For the G-mode and D-mode complex, 300-mesh Quantifoil 1.2/1.3 Au grids (Quantifoil, Großlöbichau Germany) were made hydrophilic by glow discharging for 30 seconds with a current of −20 mA in a Pelco EasiGlow system. Cryo grids were prepared using a Mark IV Vitrobot. The chamber of the Vitrobot was kept at 5 °C and 95% relative humidity. The blotting time was 3 seconds with the blotting force set at 3. 3 μL of sample was applied to the glow-discharged grid and then rapidly plunge-frozen into a liquid ethane bath.
Image collection and processing
For the I-mode, cryo grids were imaged on the Janelia Krios #2, an FEI Titan Krios cryo electron microscope (FEI/ThermoFisher), operated at 300 kV. The microscope was equipped with a Gatan K2 Summit direct electron detector. Images were taken on the K2 camera in dose fractionation (movie) mode at a calibrated magnification of 38167.9, corresponding to 1.31 Å per physical pixel (0.655 Å per super-resolution pixel). The dose rate on the specimen was set to be 5.83 electrons per Å2 per second and total exposure time was 10 s, resulting in a total dose of 58.3 electrons per Å2. With dose fractionation set at 0.25 s per frame, each movie series contained 40 frames and each frame received a dose of 1.46 electrons per Å2. Automated data collection was carried out using SerialEM (Mastronarde, 2005) with a nominal defocus range set from −1.5 to −3 μm. Camera gain reference map was taken at the start of the data collection session but not applied to each movie series to limit file size. To further save disk space, each movie series was saved in tiff format with LZW compression.
For the G-mode grids, two datasets were collected at the University of Massachusetts Chan Medical School on a 300 kV FEI Titan Krios cryo electron microscope equipped with a Gatan K3 camera and Gatan Bioquantum Imaging Filter set to a slit width of 20 eV. The final exposure was collected in dose fractionation mode on the K3 camera at a nominal magnification of 105,000, corresponding to 0.83 Å per physical pixel in the image (0.415 Å per super-resolution pixel). The dose rate on the specimen was set to be 25.06 electron per unbinned pixel and total exposure time was 1.47 s, resulting in a total dose of 53.26 electrons per Å2. With dose fractionation set at 0.047 s per frame, each movie series contained 31 frames and each frame received a dose of 1.71 electrons per Å2. Fully automated data collection was carried out using SerialEM with a nominal defocus range set from −1.5 to −3.1 μm. For the first data set, 18 images were taken per image-shift group, with 2 images per hole for a total of 8273 dose fractionated movies. For the second data set, 9 images were taken per image-shift group, with 1 image per hole for a total of 2504 dose fractionated movies. A combined total of 10777 dose fractionation movies were collected in this session.
For D-mode grids, data was collected at the University of Massachusetts Chan Medical school on a 300 kV FEI Titan Krios cryo electron microscope equipped with a Gatan K3 camera and Gatan Bioquantum Imaging Filter set to a slit width of 20 eV. The final exposure was collected in dose fractionation mode on the K3 camera at a nominal magnification of 105,000, corresponding to 0.83 Å per physical pixel in the image (0.415 Å per super-resolution pixel). The dose rate on the specimen was set to be 24.26 electron per unbinned pixel and total exposure time was 1.47 s, resulting in a total dose of 52.63 electrons per Å2. With dose fractionation set at 0.046 s per frame, each movie series contained 32 frames and each frame received a dose of 1.64 electrons per Å2. Fully automated data collection was carried out using SerialEM with a nominal defocus range set from −1.5 to −3.1 μm and 9 images take per image-shift group. A total of 2884 dose fractionation movies were collected in this session.
Data processing was first initiated in Relion (Zivanov et al., 2018). The motionCorr2 implementation was used to correct for beam-induced motion (Zheng et al., 2017). The aligned movies were then imported into cryoSPARC (v3.2) (Punjani et al., 2017) for patch-CTF estimation. Following, 2,000 particles were manually picked and further classified into 10 distinct groups, which were used as templates for automated particle picking across the dataset. From the automated particle picking, roughly 40–50% of picked particles were selected and extracted from micrographs. These extracted particles were subjected to numerous, sequential rounds of reference-free 2D classification, followed by ab initio reconstruction. Further particle curation was carried out by an additional round of 3D classification (4 classes), and finally homogenous refinement was performed to generate a final 3D reconstruction density map. The final map obtained from each dataset was sharpened with a B-factor determined from the Guinier plot generated as output from the previous homogeneous refinement step. Local resolution was estimated using cryoSPARC.
Model Building and Refinement
GATOR1-Rag-Ragulator atomic models were generated using Coot (Emsley et al., 2010). We first took a rigid-body approach to dock previously generated structural models into our cryo-EM density map, including the crystal structure of RagC-NBD bound to GppNHp (PDB: 3LLU), Ragulator-Rag(CRD) complex (PDB: 6EHR), and DEPDC5-NPRL2-NPRL3-RagA-RagC (PDB: 6CES). Despite repetitive trials using focused refinement and Baeysian polishing, two regions on the cryo-EM maps remained scattered, likely due to local movement of the protein subunits. The first region is where the C-terminal domain of NPRL3 localizes, specifically residues 192–567. This region was poorly resolved in the previous maps, and the model was tentatively built (Shen et al., 2018). To resolve this problem, we harnessed the newly-developed AlphaFold algorithm to dock structural predictions NPRL3 into our cryo-EM density maps. We divided the predicted model into globular domains that contain ~80 residues each, and fit these individual domains into our experimentally determined cryo-EM density map. The precise fit of the backbone as well as the matching of bulky residues and cryo-EM density generated a complete model for the C-terminal domain of NPRL3. Where necessary, some small regions of the model were built de novo by tracing the peptide backbone, and registering the location of bulky residues (e.g. Phe or Trp). Compared with the previous model, the current model fits density better. The second poorly resolved region is where the LAMTOR4-LAMTOR5 heterodimer resides. For the completeness of the model, we simply docked the crystal structure into the density without drawing any conclusion regarding their conformation (de Araujo et al., 2017; Mu et al., 2017; Su et al., 2017; Yonehara et al., 2017; Zhang et al., 2017).
To build the nucleotides, we followed the extra densities in the nucleotide binding pocket of the Rag GTPases and utilized previously resolved GDP-AlF3 molecule in crystal structures as a template. We noticed that the density for the AlF3 moiety is weaker than that for the GDP moiety, so a slightly lower contour is required to visualize it. We speculated that as the AlF3 mimics the gamma-phosphate at the transition state, it may exist in multiple conformations, vibrating around the central position, thus leading to weakened density than the bound GDP, which is in a stable binding conformation. In addition to the structural density, the presence of AlF3 is also supported by two pieces of biochemical evidence. First, the GAP-mode complex cannot form without AlF3. If we include only GDP, but no AlF3, in the complex formation buffer, no GATOR1-Rag-Ragulator complex can be assembled as determined by size exclusion chromatography. This suggests that AlF3 must bridge the GDP molecule to mimic a GTP molecule, so that the Rag GTPases can be loaded at the correct nucleotide loading state for complex formation with GATOR1. Second, Switch I regions of RagA and RagC are both in the upward conformation. As a feature of the Arf-family GTPases, including the Rags, the upward conformation of Switch I can be seen primarily when they bind GTP or its analogue, but not GDP. This has been observed in previous structural studies (Anandapadamanaban et al., 2019; Rogala et al., 2019; Shen et al., 2018). In this case, only GDP-AlF3 can satisfy this requirement.
To differentiate the Rag GTPase mutants in the D-mode model, we utilized the following structural and biochemical evidence. First, for the Rag GTPase mutant localized at the GAP binding site, the Switch I region of RagC is in the upward conformation, suggesting it is binding to a GTP analogue. This is consistent with the biochemical behavior of RagC(F92A), but not RagC(S75N) (cf. Supplementary Figure 6). Furthermore, the sidechain density for residue 92 is small, suggesting it is not a bulky phenylalanine residue. In addition, RagA-RagC(S75N) has been previously shown incapable of binding in the GAP mode (Shen et al., 2018). Second, for the Rag GTPase mutant localized at the inhibitory binding site, we observed that the Switch I region of RagC is in the downward conformation. This is consistent with the biochemical behavior of RagC(S75N), but not RagC(F92A), as well as with the previously resolved GATOR1-RagA-RagC(S75N) model (Shen et al., 2018). The above evidence allows us to differentiate the identity of the two bound Rag GTPase mutants.
PHENIX was used to perform real-space refinements of our constructed models (Liebschner et al., 2019). Furthermore, to assess the geometries of our models we used MolProbity (Williams et al., 2018). Lastly, corrected Fourier shell correlation curves were calculated to compare our constructed atomic models with their corresponding cryo-EM density maps.
Estimation of the distance between the two lipid anchors
We wrote a Matlab script to exhaustively search the distance relative to the lysosomal membrane. First, we anchored the tip of the visible part of Anchor-I (Ala46 of LAMTOR1 in the inhibitory mode) to the origin, mimicking the lysosomal anchor in the Inhibitory mode. Then we rotated the Dual mode complex around at 1° increment, and ruled out any orientation where the complex clashes with the membrane. With all the permitted orientations, the distance between the tip of Anchor-G (Asp49 of LAMTOR1 in the GAP mode) to the membrane was measured to be at least 100 Å. This calculation was based on the assumption that the first 45 amino acids of Anchor-I take up no space, while in reality, with the spacing between Ala46 of Anchor-I and the lysosomal membrane, the actual distance between Anchor-G and the lysosomal membrane will only be longer, making the D-mode less likely to exist under physiological conditions.
Preparation of Cell Lysates and Immunoprecipitates
CRISPR-knockout cell lines were generated in previous studies (Chantranupong et al., 2014; Wolfson et al., 2017). Cell lysates and immunoprecipitates were prepared as previously established (Shen et al., 2017). In brief, two million HEK-293T cells or cells with a GATOR1 subunit knocked out were first plated onto a 10 cm dish. Twenty-four hours later, the cells were transfected with the plasmids indicated in the figure panels. Thirty-six hours later, cells were rinsed once with PBS and lysed in Triton Lysis Buffer [40 mM NaHEPES, pH 7.4; 5 mM MgCl2; 10 mM Na4P2O7; 10 mM sodium β-glycerol phosphate; 1% Triton X-100; and one tablet of EDTA-free protease inhibitor (Roche) per 25 ml of buffer]. The lysates were cleared and immunoprecipitated with Flag-M2 affinity gel. Following immunoprecipitation, the gel was washed once with Triton Lysis Buffer and three times with Triton Lysis Buffer supplemented with 500 mM sodium chloride. Immunoprecipitated proteins were denatured by 2xSDS buffer, resolved by SDS-PAGE gels, and analyzed by immunoblotting.
Kinetic Measurements
To determine the stimulatory effect of the various GATOR1 mutants on the hydrolysis rate of the Rag GTPases we used previously established kinetic assays (Shen et al., 2017). Briefly, under single turnover conditions, increasing concentrations of GATOR1 (10–500 nM for wild-type, and 0.2–5 μM for DEPDC5(Y775A)-containing protein) was incubated with 50 nM Rag GTPase heterodimer. The Rag GTPases were preloaded with ~ 0.1 nM of α-32P-GTP. To measure the reaction progress, small aliquots of the reaction were aliquoted at various time points and quenched using 0.75 M KH2PO4 (pH 3.3). The time points were analyzed by thin layer chromatography (TLC) plates, and imaged and quantified with phosphorimaging screens. Linear regression was used to fit the fraction of hydrolyzed GTP against time, to generate the observed rate constants (kobsd) corresponding to each concentration. The kobsds were fit to a single binding equation.
In a multiple turnover condition, increasing concentrations of GATOR1 (0.2–5 μM) were incubated with 2 μM Rag GTPase heterodimer. The Rag GTPases are preloaded with a vast excess of cold GTP (100 μM), doped with ~1 nM of α-32P-GTP. This setup ensures that both subunits of the Rag GTPase are dual-loaded with GTP. The reaction progress is measured and analyzed in the same manner as for the single turnover conditions.
QUANTIFICATION AND STATISTICAL ANALYSIS
All the kinetic assays were measured three times, and the results were reported with Mean ± SEM.
Supplementary Material
Table 1.
Summary of cryo-EM data collection, 3D reconstruction, and model refinement
| GATOR1-Rag-Ragulator (I-mode) | GATOR1-Rag-Ragulator (G-mode) | GATOR1-Rag-Ragulator (D-mode) | |
|---|---|---|---|
| Imaging parameters and 3D reconstruction | |||
| Microscope | Janelia Farm Titan Krios #2 | UMass cryo-EM facility | UMass cryo-EM facility |
| Grids | Quantfoil R 1.2/1.3 Au 400 mesh | Quantfoil R 1.2/1.3 Au 300 mesh | Quantfoil R 1.2/1.3 Au 300 mesh |
| Calibrated magnification | 38168 | 105100 | 105100 |
| Acceleration voltage [kV] | 300 | 300 | 300 |
| Pixel size [Å] | 1.31 | 0.83 | 0.83 |
| Total dose [e−/Å2] | 58.3 | 54.2 | 52.6 |
| Exposure time [s] | 10 | 1.7 | 1.5 |
| Defocus range [μm] | −1.0 to −3.5 | −1.5 to −3.0 | −1.5 to −3.0 |
| Particles in final 3D reconstruction | 251,583 | 139,231 | 56,117 |
| Resolution (‘Gold-standard’ at FSC 0.143) [Å] | 4.02 | 3.9 | 4.08 |
| Model refinement | |||
| Resolution in phenix.real_space_refine [Å] | 4 | 3.5 | 4 |
| No. atoms: protein | 23,310 | 23,278 | 32,464 |
| Ligands / ions | GppNHp, GDP | GDP, AlF3 | GDP, AlF3 |
| r.m.s. deviations: bond lengths [Å] | 0.006 | 0.004 | 0.006 |
| r.m.s. deviations: bond angles [°] | 1.172 | 1.044 | 1.232 |
| MolProbity score | 1.75 | 1.86 | 0.44 |
| EMRinger score | 1.53 | 1.4 | 0.43 |
| Clash score | 7.26 | 6.53 | 10.62 |
| Rotamer outliers [%] | 0.04 | 0.04 | 0.03 |
| Ramachandran angles: favored [%] | 90.71 | 93.03 | 89.29 |
| Ramachandran angles: allowed [%] | 9.26 | 6.93 | 10.68 |
| Ramachandran angles: outliers [%] | 0.04 | 0.04 | 0.03 |
| PDB | 7T3A | 7T3B | 7T3C |
| EMDB | 25652 | 25653 | 25654 |
Three cryo-EM structural models of the GATOR1-Rag-Ragulator complex resolved
GATOR1 secures nucleotide status of both subunits of the Rag GTPase heterodimer
Rag GTPase binding modes are predicted to be mutually exclusive in vivo
Acknowledgements
We thank all members of the Shen Laboratory for helpful insights. We thank D. Lambright (UMass Chan Medical School) and X. Wang (FEI/Thermo Fisher) for helpful discussions, and R. Davis, J. Richter, C. Peterson, and G. Liu for critical reading of the manuscript. This work was supported by a grant from the NIH (K22CA241362) to Ku.S., and the 2020 Mello Scholarship from the Program in Molecular Medicine (UMass Chan Medical School) to S.B.E.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
Ku.S. is a consultant for Casma Therapeutics.
References
- Anandapadamanaban M, Masson GR, Perisic O, Berndt A, Kaufman J, Johnson CM, Santhanam B, Rogala KB, Sabatini DM, and Williams RL (2019). Architecture of human Rag GTPase heterodimers and their complex with mTORC1. Science 366, 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Araujo MEG, Naschberger A, Fürnrohr BG, Stasyk T, Dunzendorfer-Matt T, Lechner S, Welti S, Kremser L, Shivalingaiah G, Offterdinger M, et al. (2017). Crystal structure of the human lysosomal mTORC1 scaffold complex and its impact on signaling. Science 358, 377–381. [DOI] [PubMed] [Google Scholar]
- Bar-Peled L, Schweitzer LD, Zoncu R, and Sabatini DM (2012). Ragulator Is a GEF for the Rag GTPases that Signal Amino Acid Levels to mTORC1. Cell 150, 1196–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, and Sabatini DM (2013). A Tumor Suppressor Complex with GAP Activity for the Rag GTPases That Signal Amino Acid Sufficiency to mTORC1. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, and Manning BD (2017). mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol 45, 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, Spooner E, Isasa M, Gygi SP, and Sabatini DM (2014). The Sestrins Interact with GATOR2 to Negatively Regulate the Amino-Acid-Sensing Pathway Upstream of mTORC1. Cell Rep. 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble CC, and Cantley LC (2015). Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 25, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm SA, Lawrence RE, and Hurley JH (2020). Structural mechanism for amino acid-dependent Rag GTPase nucleotide state switching by SLC38A9. Nat. Struct. Mol. Biol 27, 1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong R, Li L, Liu Y, Wang P, Yang H, Wang L, Cheng J, Guan K-L, and Xu Y (2011). Crystal structure of the Gtr1p–Gtr2p complex reveals new insights into the amino acid-induced TORC1 activation. Genes Dev. 25, 1668–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, and Guan K-L (2003). Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J-H, Lee K-H, Kim Y-M, Kim D-H, Oh B-H, and Kim Y-G (2012). Crystal Structure of the Gtr1p GTP -Gtr2p GDP Protein Complex Reveals Large Structural Rearrangements Triggered by GTP-to-GDP Conversion. J. Biol. Chem 287, 29648–29653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, and Guan K-L (2019). mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol 21, 63–71. [DOI] [PubMed] [Google Scholar]
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, and Guan K-L (2008). Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol 10, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence RE, Fromm SA, Fu Y, Yokom AL, Kim DJ, Thelen AM, Young LN, Lim C-Y, Samelson AJ, Hurley JH, et al. (2019). Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Inoki K, and Guan K-L (2004). Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity. Mol. Cell. Biol 24, 7965–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung L-W, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. Sect. Struct. Biol 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GY, and Sabatini DM (2020). mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol 21, 183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, and Manning BD (2014). Spatial Control of the TSC Complex Integrates Insulin and Nutrient Regulation of mTORC1 at the Lysosome. Cell 156, 771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossmann D, Park S, and Hall MN (2018). mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 18, 744–757. [DOI] [PubMed] [Google Scholar]
- Mu Z, Wang L, Deng W, Wang J, and Wu G (2017). Structural insight into the Ragulator complex which anchors mTORC1 to the lysosomal membrane. Cell Discov. 3, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicastro R, Sardu A, Panchaud N, and De Virgilio C (2017). The Architecture of the Rag GTPase Signaling Network. Biomolecules 7, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchaud N, Péli-Gulli M-P, and Virgilio CD (2013). Amino Acid Deprivation Inhibits TORC1 Through a GTPase-Activating Protein Complex for the Rag Family GTPase Gtr1. Sci. Signal [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Rogala KB, Gu X, Kedir JF, Abu-Remaileh M, Bianchi LF, Bottino AMS, Dueholm R, Niehaus A, Overwijn D, Fils A-CP, et al. (2019). Structural basis for the docking of mTORC1 on the lysosomal surface. Science 366, 468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, and Sabatini DM (2008). The Rag GTPases Bind Raptor and Mediate Amino Acid Signaling to mTORC1. Science 320, 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, and Sabatini DM (2010). Ragulator-Rag Complex Targets mTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell 141, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schürmann A, Brauers A, Maßmann S, Becker W, and Joost H-G (1995). Cloning of a Novel Family of Mammalian GTP-binding Proteins (RagA, RagBs, RagB1) with Remote Similarity to the Ras-related GTPases *. J. Biol. Chem 270, 28982–28988. [DOI] [PubMed] [Google Scholar]
- Sekiguchi T, Hirose E, Nakashima N, Ii M, and Nishimoto T (2001). Novel G Proteins, Rag C and Rag D, Interact with GTP-binding Proteins, Rag A and Rag B*. J. Biol. Chem 276, 7246–7257. [DOI] [PubMed] [Google Scholar]
- Shen K, and Sabatini DM (2018). Ragulator and SLC38A9 activate the Rag GTPases through noncanonical GEF mechanisms. Proc. Natl. Acad. Sci 115, 9545–9550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Choe A, and Sabatini DM (2017). Intersubunit Crosstalk in the Rag GTPase Heterodimer Enables mTORC1 to Respond Rapidly to Amino Acid Availability. Mol. Cell 68, 552–565.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Huang RK, Brignole EJ, Condon KJ, Valenstein ML, Chantranupong L, Bomaliyamu A, Choe A, Hong C, Yu Z, et al. (2018). Architecture of the human GATOR1 and GATOR1–Rag GTPases complexes. Nature 556, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Valenstein ML, Gu X, and Sabatini DM (2019a). Arg-78 of Nprl2 catalyzes GATOR1-stimulated GTP hydrolysis by the Rag GTPases. J. Biol. Chem 294, 2970–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Rogala KB, Chou H-T, Huang RK, Yu Z, and Sabatini DM (2019b). Cryo-EM Structure of the Human FLCN-FNIP2-Rag-Ragulator Complex. Cell 179, 1319–1329.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeyki N, Hondo A, Kasai A, Koike M, Saito K, Uchiyama Y, and Okada M (2009). The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK–ERK pathway to late endosomes. EMBO J. 28, 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M-Y, Morris KL, Kim DJ, Fu Y, Lawrence R, Stjepanovic G, Zoncu R, and Hurley JH (2017). Hybrid Structure of the RagA/C-Ragulator mTORC1 Activation Complex. Mol. Cell 68, 835–846.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, et al. (2018). MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson RL, Chantranupong L, Wyant GA, Gu X, Orozco JM, Shen K, Condon KJ, Petri S, Kedir J, Scaria SM, et al. (2017). KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 543, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonehara R, Nada S, Nakai T, Nakai M, Kitamura A, Ogawa A, Nakatsumi H, Nakayama KI, Li S, Standley DM, et al. (2017). Structural basis for the assembly of the Ragulator-Rag GTPase complex. Nat. Commun 8, 1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Wang R, Wang Z, Wang X, Wang F, and Ding J (2017). Structural basis for Ragulator functioning as a scaffold in membrane-anchoring of Rag GTPases and mTORC1. Nat. Commun 8, 1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. ELife 7, e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under the accession number 7T3A (Inhibitory mode), 7T3B (GAP mode), and 7T3C (Dual mode). Electron density maps have been deposited in EM Data Bank under the accession number EMD-25652 (Inhibitory mode), EMD-25653 (GAP mode), and EMD-25654 (Dual mode). These coordinates and maps are publicly available as of the date of publication. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. The access numbers are listed in the key resources table. Raw cryo-EM movies reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.