

Abstract
Effective screening modalities are currently available for only a small subset of cancers, and they generally have suboptimal performance with complicated procedures. Therefore, there is an urgent need to develop simple, accurate, and non-invasive methods for early detection of cancers. Genetic and epigenetic alterations in plasma circulating cell-free DNA (cfDNA) have shown the potential to revolutionize methods of early detection of cancers and facilitate subsequent diagnosis to improve survival of patients. The medical interest in cfDNA assays has been inspired by emerging single- and multi-early detection of cancers studies. This review summarizes current technological and clinical advances, in the hopes of providing insights into the development and applications of cfDNA assays in various cancers and clinical scenarios. The key phases of clinical development of biomarkers are highlighted, and the future developments of cfDNA-based liquid biopsies in early detection of cancers are outlined. It is hoped that this study can boost the potential integration of cfDNA-based early detection of cancers into the current clinical workflow.
Keywords: cancer early detection, liquid biopsy, circulating cell-free DNA, multi-cancer early detection, methylation
Graphical abstract
Public summary
-
•
Liquid biopsy, characterized by minimal invasiveness and user friendliness, can identify multiple cancers at the early stage and localize the tissue of origin
-
•
The state-of-the-art technology facilitates the application of circulating cell-free DNA (cfDNA) assays in the early detection of cancers
-
•
cfDNA assays are expected to be integrated into the clinical workflow after technological refinement and clinical trial validation
-
•
The development and application strategies of cfDNA assays in various cancers and clinical scenarios can vary, and the harm-and-benefit should be balanced carefully
Introduction
Cancer morbidity and mortality are growing rapidly worldwide, with 19.29 million new cases of cancer and 9.96 million cancer deaths annually, according to the global cancer statistics in 2021.1 The stage at which cancer is diagnosed is one of the most important predictors of survival. The 5-year survival rate decreases more sharply in patients with cancer at the advanced stage than those with localized disease.2 As for lung cancer, the most prevalent cancer worldwide, the 5-year survival rate was 59%, 32%, and 6% for localized, regional, and distant stages, respectively.2 Thus, the identification of patients at earlier stages when cures are more accessible is critical to the overall cancer survival.
Currently, low-dose computed tomography (LDCT),3 colonoscopy,4 mammography,5 and cervical cytology combined with human papillomavirus testing6 are recommended for lung, colorectal, breast, and cervical cancer screening, respectively, by the National Comprehensive Cancer Network guidelines. However, the clinical applicability of these screening tools is limited by the suboptimal sensitivity and specificity, high invasiveness,7 and poor compliance.8 Moreover, for many deadly cancers, such as ovarian and pancreatic cancer, no screening methods are recommended, and the majority of patients with those cancers are diagnosed at advanced stages with a poor prognosis.9 Thus, it is of great significance to develop simple, accurate, and non-invasive methods for early detection of cancers.
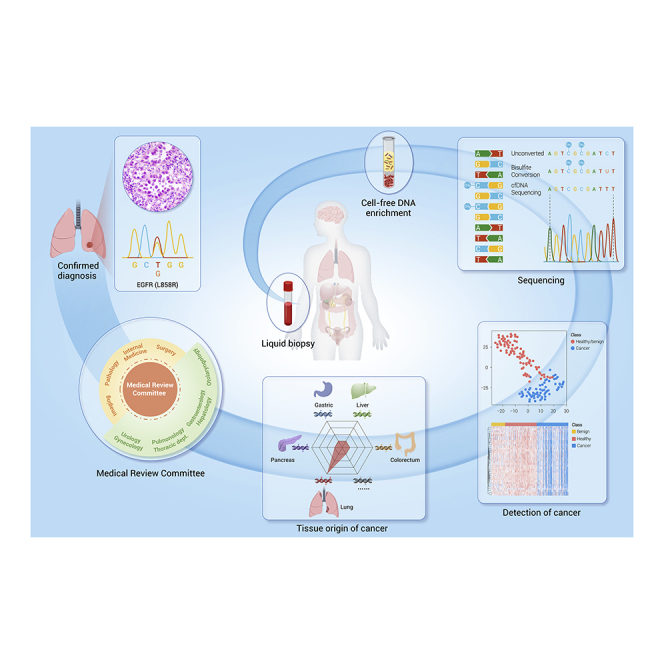
Liquid biopsy, well known for its non-invasiveness or minimal invasiveness, has shown good potential in detecting cancers at the early stage or pre-cancerous lesions, which is also capable of detecting cancers through detecting circulating tumor cells, exosomes, cfDNA, mRNA, microRNAs (miRNAs), proteins, or metabolites etc.10,11 Among them, cfDNA, which is extracellular nucleic acid fragments shed by cancer cells during apoptosis, necrosis, or secretion,12 is the most promising for detecting early stage cancers (Figure 1). cfDNA exhibits the genetic and epigenetic alterations of cancers, including mutations, copy number alterations, chromosomal rearrangements, hypermethylation, and hypomethylation.11 Although cfDNA can be detected in different body fluids, we focus on studies of cfDNA in the blood in particular.
Figure 1.

Comparisons between liquid biopsy and traditional screening approaches
Plasma-based biomarkers generally exhibit aberration early during tumorigenesis and provide abundant signals for analysis, e.g., circulating tumor cells (CTCs), exosomes, cfDNA, mRNA, miRNAs, and proteins. Several molecular alterations also carry tissue-specific patterns, which may help to locate the tissue origin of cancers and facilitate the follow-up diagnostic workup. Plasma-based techniques for early detection may provide solutions for the cancers with or without any recommended screening tools for now (marked by purple).
cfDNA testing shows several advantages over traditional cancer screening tools, making it an ideal approach to the early detection of cancers. First, theoretically, the presence of abnormal cfDNA in the blood can be identified at an ultra-early stage of cancer, before the threshold of conventional clinical measurements, expanding the time window for early intervention. Second, cfDNA-based tests make possible simultaneous detection of multiple cancers by providing information on the tissue of origin (TOO), while the traditional method usually screens a specific cancer at one time. Third, cfDNA testing requires nothing more than a blood draw, which is safer, easier, and less invasive than traditional screening methods, thus increasing compliance. Finally, cfDNA has the potential to depict the entire molecular and (epi)genetic landscape, irrespective of intratumor heterogeneity, aiding in precision treatment and monitoring relapse after early detection.
We have herein comprehensively reviewed the recent advances in plasma cfDNA testing and its applications in early detection of cancers. We begin with the biology of cfDNA and an overview of cfDNA biomarkers and current cfDNA sequencing techniques. We then outline the recent breakthroughs in cfDNA-based liquid biopsy for the early detection of single- and multi-cancer tests. We also discuss the key clinical phases of biomarker development for the early detection of cancers and end with open questions and prospects of development in this rapidly evolving field.
cfDNA biology
cfDNA is believed to be mainly released from cells via apoptosis and necrosis, and also possibly from active secretion.13, 14, 15 The half-life of cfDNA ranges from 16 min to 2.5 h.16, 17, 18 In vivo, cfDNA is either cleared by nuclease action19,20 before being excreted into urine by the kidney21,22 or absorbed by the liver and spleen followed by macrophage degradation.23,24 In vitro, isolated cfDNA needs to be stored at less than −20 °C and go through no more than one freeze-thaw cycle to prevent cfDNA degradation.25 The stability of cfDNA fragments may be increased by their binding with the cell membrane, extracellular vesicles, or proteins.26 The concentration of cfDNA is usually low (within 100 ng/mL) under normal conditions, but significantly increases under certain physiological and pathological conditions, including exercise, inflammation, diabetes,27,28 and cancer.29 Lymphocytes contribute to cfDNA contents primarily in healthy individuals,30 while in patients with cancer, the cfDNA from cancer tissues is significantly increased.28,31
The peak at 166 bp is prominent when cfDNA is measured by sequencing-based approaches, while tumor-derived cfDNA (approximately 144 bp) is shorter than the non-mutated cfDNA.32,33 This difference in length suggests the existence of a nucleosome footprint34 and the fragments of cfDNA are cleaved by apoptotic caspase-dependent endonuclease and released into blood.35 There are 146 bp of DNA twisted around the histone octamer. An additional 20 bp are needed to fix histone H1 outside the nucleosomes at the exit of DNA. Then, an additional 20‒50 bp linker DNA connects neighboring nucleosomes. The caspase-induced DNases periodically cleave DNA within the internucleosomal linker region, while the DNA wrapped around the histone octamer protects DNA from enzymatic cleavage.35 There is growing evidence that DNA is more fragmented in patients with cancer than in healthy individuals, with a significant proportion of fragments shorter than 145 bp.36, 37, 38 Given the difference in length between cfDNA derived from normal and cancerous tissues, some DNA extraction procedures and fragment enrichment technologies can be used to enrich cancerous cfDNA. Along with the development of sequencing techniques, single-stranded DNA library preparation can recover DNA fragments with damaged ends and greatly facilitate the identification of fragments shorter than 100 bp.39
Current sequencing approaches to cfDNA in the early detection of cancers
Strategies for early detection of cancers are generally based on the detection of cancer-related alterations in the cfDNA released from cancer cells, known as circulating tumor DNA (ctDNA). The concentration of ctDNA in plasma is relatively low and accounts for less than 0.01% of the total cfDNA concentration,10,40 especially in early-stage cancers,41 posing a big challenge to early detection of cancers. Currently, mutation, methylation, and fragment patterns are the main sequenced biomarkers of cfDNA for the early detection of cancers. Thus, we focus on current sequencing approaches to cfDNA mutation, methylation, and fragment patterns in the early detection of cancers.
Mutation-based sequencing approach and strategy
An ideal application of mutation-based sequencing approach in the early-stage cancer detection requires a low limit of detection (LOD) to differentiate scarce cfDNA mutation signals from background noise that (e.g., false positivity) may be introduced during DNA extraction, library preparation, target enrichment, the hybridization process, and sequencing itself,42, 43, 44 making the detection of true rare mutations harder. Some of the background noise can be mitigated, such as sequence alterations caused by oxidative stress damage during the extraction, fragmentation, and hybridization processes.42 To increase the analytical sensitivity of targeted sequencing, unique molecular identifiers (UMIs), that is, unique molecular barcodes ligated directly at both ends of library DNA fragments, have been applied to facilitate the bioinformatic alignment of sequences from the same DNA fragment. This UMI strategy can help to minimize artificial errors in subsequent analyses.45, 46, 47 When combined with duplex-UMI, the cancer personalized profiling by deep sequencing (CAPP-Seq) method can achieve a LOD of 0.02% in cfDNA.47
Since prior knowledge of mutations carried by a particular patient with cancer is not available in the early detection, panels including the most frequently mutated genes are usually used in mutation-based sequencing approaches. Using the CAPP-Seq method, the assessment of mutations in 139 genes that are frequently mutated in non-small cell lung cancer (NSCLC) achieved a sensitivity of 85.0% in differentiating early-stage NSCLC from healthy controls.47 The CAPP-Seq method combined with a computational error correction approach and UMI strategy can further reduce selector-wide error rates by 15-fold and improve detection performance. This approach detected EGFR kinase domain mutations with a 92% sensitivity and a 96% specificity and the detection of cfDNA was down to 4 in 105 cfDNA molecules.47 Targeted-error correction sequencing (TEC-seq)48 is another UMI-based technique that measures 58 genes frequently mutated in colorectal, lung, ovarian, and breast cancers. It can detect 50.0%–70.0% of stage I or II cancers across those different cancer types.
However, there are several limitations to cfDNA mutation-based analyses. Targeted sequencing panels are designed mainly based on the existing knowledge of the genetic alterations in cancers. False-negative results are not only inevitable when cancerous mutations are out of the scope of the customized panel, but also made possible by the insufficiency of the plasma ctDNA concentration.29 The concentration of ctDNA is higher in patients with late-stage and metastatic cancers than in patients with early-stage cancers and varies with cancer types.49,50 It is also higher in liver, biliary, esophagus, and ovarian cancers, but relatively lower in prostate, breast, and colon cancers.49 Furthermore, several factors may contribute to the variation of cfDNA concentrations, such as age,50 body mass index,51 and physiological parameters.52 Mutation-based analysis is also limited by the number of mutations initially present in the tumor mass, which varies according to cancers. Melanoma and lung cancer have an average of more than 8.9 mutations/Mbp, while low-grade gliomas, breast, pancreatic, and prostate cancers have an average of less than 2.2 mutations/Mbp.49 Moreover, the tumor mutational burden is lower in patients with early-stage cancer than patients with late-stage cancers,49,50 making it more difficult to detect early-stage cancers with mutation-based technologies. In contrast, false-positive results may occur in a substantial proportion of individuals without cancer,53,54 owing to clonal hematopoiesis (chromatin immunoprecipitation [CHIP])-related mutations. Of the variants detected in cfDNA from NSCLC and healthy controls, 58.0% and 90.0% were also detected in the matched white blood cells (WBCs),55 highlighting the importance of equivalent sequencing depth applied to the matched WBC DNA to exclude the interference of CHIP.
Methylation-based sequencing approach
Epigenetic alteration-based cfDNA sequencing methods have been considered promising alternatives for the early detection of cancers. Methylation of CpG sites is an important epigenetic regulatory mechanism of gene expression, tissue differentiation, organ development, aging, and tumorigenesis.56, 57, 58 The changes of cancer-specific DNA methylation occur early in tumorigenesis, sometimes even before the occurrence of gene mutations.58 A previous study has shown that the changes of methylation were detected in plasma four years before a clinical cancer diagnosis.59 Furthermore, in contrast with typical cancer mutations that only occur in a handful of genomic locations, nearly 30 million methylation sites are scattered across the human genome,60 making them ubiquitous and rich signals for cancer detection. Notably, methylation patterns are usually tissue specific, which makes TOO analysis possible.58
Whole genomic bisulfite sequencing (WGBS) is the gold standard to capture the genome-wide DNA methylation profile.61 However, the prohibitively high cost, low recovery of input DNA, and limited depth of sequencing make it infeasible for clinical application.61 In contrast, owing to the lower cost and higher sequencing depth, next-generation sequencing (NGS)-based methylation sequencing methods have attracted increasing attention. NGS-based methods include bisulfite pretreatment (reduced representation bisulfite sequencing [RRBS]62,63), enzyme digestion (methylation-sensitive restriction enzyme sequencing,64 etc.), and affinity enrichment (methylated DNA immunoprecipitation and high-throughput sequencing [MeDIP-Seq], etc.65). The low recovery rate of DNA after bisulfite conversion is also a concern for NGS-based methods. Therefore, new methods have been developed to enrich methylated DNA fragments or decrease the amount of DNA input.66 The cell-free MeDIP-Seq (cfMeDIP-Seq) is an immunoprecipitation-based protocol adapted from traditional MeDIP-Seq, allowing a genome-wide methylome profiling with low input of DNA in a bisulfite-free procedure.67 In contrast with MeDIP-Seq, RRBS, and WGBS, which require 100‒2,000 ng DNA input, cfMeDIP-Seq generates a nearly perfect linear association between observed and expected numbers of differential methylation regions (r2 = 0.99, p < 0.0001) with only 1‒10 ng DNA input.68 Recent technical advances, such as methylBEAMing,69 single-cell RRBS,70,71 and enhanced linear-splinter amplification sequencing (ELSA-Seq),72 can help to facilitate the application of cfDNA methylation sequencing by decreasing the required amount of DNA input and increasing the analytical sensitivity. For example, ELSA-Seq constructs a single-stranded library and demonstrates a deep methylome coverage, little amplification bias, and ultra-high reproducibility with the inputs down to 500 pg.72 In addition, adjacent CpG sites usually represent co-methylation status, and integrating multiple CpG sites with close genomic distance and high correlation into methylation haplotype blocks can further improve the detection accuracy.70
There are also limitations to the cfDNA-methylation analysis. The epigenetic alterations present in cancer may also exist in other non-cancer tissues, which may lead to false positivity.73 For example, several similar methylation alterations are shared by esophagus cancer and Barrett’s esophagus.74 In addition, methylation alterations accumulate with age, and approximately 5% of the CpG sites exhibit significant changes shared by aging and tumorigenesis.59,75 Meanwhile, false-negative results may occur when the detection signaling is lower than the LOD. Similarly, the detection rate intactly correlates with the cfDNA concentration, which is also influenced by the pathological types of cancers.49 Plus, these methylation-based sequencing approaches potentially fail to detect cancers that are mainly driven by gene mutations, somatic copy number aberrations, or fusion events,58 such as several subtypes of lung cancer mainly driven by EGFRL858R mutation, EML4-ALK fusion, or ERBB2 amplification.76
cfDNA fragmentation-based sequencing approach
Genome-wide fragmentation of cfDNA has recently opened a new area of early detection and TOO of cancer.38,77,78 Whole-genome sequencing (WGS) has revealed that the lengths of cancer-derived cfDNA fragments are more variable than those of non-cancer origin. Differences in cfDNA fragment patterns reflect changes in chromatin structures, as well as other genomic and epigenomic abnormalities in cancer.79,80 Recently, a machine learning model incorporating genome-wide fragmentation can distinguish multiple patients with cancer from healthy controls with sensitivities ranging from 57% to more than 99% at 98% specificity and identify the TOO of cancers in 75% of the cases.77 In addition to the fragment length, the break sites of cfDNA fragments in the genome also reveal a genome-wide map of nucleosome occupancy, providing abundant signaling.34 Despite the ability of WGS to analyze tens to hundreds of tumor-specific abnormalities simultaneously from a minute amount of cfDNA, its application is compromised by low coverage depth and high cost.81 In addition, the cfDNA fragmentation-based approach shares limitations with cfDNA-methylation analysis, such as false positivity caused by physiological and other pathological conditions and false negativity induced by technical limitations. Comparisons between cfDNA mutation, methylation, and fragmentation patterns are summarized in Table 1.
Table 1.
The advantages and limitations relying on cfDNA-based sequencing methods
| Detected objects | Technique | Approach | Advantages |
Disadvantages |
|||
|---|---|---|---|---|---|---|---|
| Common | Specific | Common | Specific | ||||
| Somatic mutations | PCR-based | ddPCR BEAMing |
High signal intensity Low background noise | Ultra-low input | Limited markers Non-tissue-specific markers Susceptible to clonal hematopoiesis |
Low through-put Limited sites |
|
| Next generation sequencing (NGS)-based | Target enriched by amplification | Safe-seqS/Safer-seqS44 Tagged amplicon deep sequencing (TAm-seq)43 |
Low input | Limited sites | |||
| Target enriched by hybrid capture | CAPP-seq46,47 Targeted error correction sequencing (TEC-seq)48 | More sites detected High through-put |
High input High cost |
||||
| Methylation patterns | Restriction enzymes-based | Methylation-sensitive restriction enzymes-PCR (MRE-PCR)64 | Abundant markers Tissue specific High signal intensity |
Low cost | Biological variation (eg, age, cell type composition) | Limited sites | |
| Affinity enrichment-based | cfMeDIP-seq67 | Low cost Genome-wide CpG enriched |
Antibody depended Low resolution |
||||
| Bisulfite conversion-based | MethylBEAMing69 Single-cell RRBS71 ELSA-seq72 |
High resolution More sites detected |
High input Bisulfite conversion noise | ||||
| Fragmentation patterns | Paired-end, low-coverage WGS61 | Abundant markers Tissue-specific |
High cost Low signal intensity |
||||
Advance in early detection of cancers by cfDNA profiling
The development of cfDNA sequencing technology steers the clinical use of cfDNA profiling for early detection of cancers. Currently, clinical trials of cfDNA-centered early detection of single and multi-cancers are both being carried out, with tens to thousands of participants enrolled. Their detailed performances are illustrated in Table 2 and Figure 2.
Table 2.
Quantitative performance of cfDNA-based liquid biopsy in the detection and TOO of cancer
| Classifier | No. of participants | Cancer | Sequencing approach | Sensitivity | Specificity | AUC | TOO |
|---|---|---|---|---|---|---|---|
| Cd-score82 | HCC (n = 1,098), healthy controls (n = 835) | HCC | Targeted bisulfite sequencing | Training, 85.7%; validation, 83.3% |
Training, 94.3%; validation, 90.5% |
Training, 0.966; validation, 0.944 |
NA |
| Wd-score83 | HCC (n = 1,204), CHB or liver cirrhosis (n = 392), benign liver lesions (n = 388), healthy controls (n = 570) | HCC | Genome-wide 5-hmC profiles | Training, 89.6%; vlidation-1, 82.7% |
Training, 78.9%; validation-1, 76.4% |
Training, 0.923; validation-1, 0.884; validation-2, 0.887 |
NA |
| HCC screen score84 | HCC (n = 65), CHB (n = 70); AFP/US-negative CHB (n = 331) | HCC | PCR | Training, 85.0%; validation, 100.0% |
Training, 93.5%; validation 94.0% |
Training, 0.928 validation, NA |
NA |
| HIFI score (5-hmC/motIf/Fragmentation/nucleosome footprInt)85 | HCC (n = 508), liver cirrhosis (n = 2,250), healthy controls (n = 476) | HCC | Low-pass WGS | Training, NA; test, 95.4%; validation, 95.8% |
Training, NA; test 97.8%; validation 95.0% |
Training, NA; test, 0.996; validation, 0.995 |
NA |
| cfDNA fragmentation86 | HCC (n = 159), ICC (n = 26), cHCC-ICC (n = 7); CHB or liver cirrhosis (n = 53), healthy controls (n = 117) | HCC, ICC, cHCC-ICC | WGS | Training, NA; test, 96.8% |
Training, NA; test, 98.8% |
Training, NA; test, 0.995 |
NA |
| Cd-score (9 methylation markers)87 | CRC (n = 801); healthy controls (n = 1,021) | CRC | Targeted methylation sequencing | Training, 87.5%; validation, 87.9% |
Training, 89.9%; validation, 89.6% |
Training, 0.960; validation, 0.960 |
NA |
| Multi-target stool DNA test composite score88 | Asymptomatic persons who were at average risk for CRC, aged 50–84 years (n = 9,989) | CRC | PCR | Total, 92.3%: stage I, 88.0%; stage II, 100%; stage III, 90.0%; stage IV, 78.0% | 86.6% | 0.940 | NA |
| PulmoSeek (Blood-based DNA methylation model)89 | Patients with pulmonary nodules (n = 529) | Lung cancer | Targeted methylation sequencing | Training, NA; Test, 93.3%; validation, 99.0% |
Training, NA; test, 60.0%; validation, 32.5% |
Training, NA; test, 0.829; validation, 0.843 |
NA |
| Lung-CLiP score55 | Early-stage NSCLC (n = 46), risk-matched healthy controls (n = 48) | Lung cancer | CAPP-seq | Training: stage I/II/III sensitivity: 41.0%, 54.0%, 67.0%; validation: stage I/II/III sensitivity: 63.0%, 69.0%, 75.0% | Training, 98.0%; validation, 80.0% | NA | |
| Methylation score90 | Plasma samples: RCC (n = 69), UBC (n = 21), healthy controls (n = 13); Urine samples: RCC (n = 30), healthy controls (n = 15) |
RCC, UBC | cfMeDIP-seq | NA | NA | Plasma, 0.990 (RCC vs healthy); plasma, 0.979 (RCC vs UBC); urine, 0.858 (RCC vs healthy) |
NA |
| LR score59 | Cancers (n = 414): post-diagnosis (n = 223), pre-diagnosis (n = 191) Healthy controls (n = 414) |
Stomach, esophageal, colorectal, lung, and liver cancers | Targeted methylation sequencing | Training: post-diagnosis, 88.2%; pre-diagnosis, 91.4%; test: post-diagnosis, 87.6%; pre-diagnosis, 94.9% | Training, 94.7%; test, 96.1% |
Training: NA; test: post-diagnosis, 0.970; pre-diagnosis 0.990 |
NA |
| Screen positive or negative91 | Chinese men aged 40–62 years (n = 20,174) | NPC | PCR | 97.1% | 98.6% | NA | |
| Circulating proteins and gene mutations (CancerSEEK)28 | Stages I‒III cancers (n = 1,005), healthy controls (n = 812) | Ovarian, liver, stomach, pancreatic, colorectal, lung, and breast, esophageal cancers | PCR | Median, 70.0% (range, 33.0% in breast cancer to 88.0% in ovarian cancer) | 99.1% | 0.910 | Top prediction: 39.0–84.0%; top 2 predictions: 63.0–100.0% |
| Circulating proteins and gene mutations + PET-CT/imaging92 | Women aged 65–75 years (n = 10,006) | Pan-cancer | PCR | 27.1% | 99.6% | NA | |
| Targeted methylation classifier93 | Cancers (n = 2,482), non-cancer (n = 4,207) | Multiple cancers (more than 50 types of cancer) | Targeted methylation assay | Training, 55.2%; validation, 54.9%; stage I, 18%; stage II, 43.0%; stage III, 81.0%; stage IV, 93.0% | Training, 99.8%; validation, 99.3% |
93.0% | |
| Targeted methylation classifier94 | Cancers (n = 2,823), non-cancer (n = 1,254) | Multiple cancers (6 types of cancer) | Targeted methylation assay | 51.5% | 99.5% | 88.7% | |
| cfDNA fragmentation patterns77 | Cancers (n = 236) and healthy controls (n = 245). | Breast, colorectal, stomach, lung, ovarian, pancreatic and biliary tract cancers | WGS | Stage I, 68.0%; stage II, 72.0%; stage III, 79.0%; stage IV, 77.0% | 98.0% | 0.940 | Top prediction: 61.0%; top two predictions: 75.0% |
Abbreviations: AUC, Area under curve; CHB, Chronic HBV infection; cHCC-ICC, Combined HCC and intrahepatic cholangiocarcinoma; ICC, Intrahepatic cholangiocarcinoma; Lung-CLiP, lung cancer likelihood in plasma; NPC, Nasopharyngeal carcinoma; NA, not available; PET-CT, Positron emission tomography/computed tomography; UBC, Urothelial bladder cancer; WGBS, Whole genome bisulfite sequencing.
Figure 2.

The advances of cfDNA-based biomarkers for early detection and tissue-of-origin of cancer
AFP, alpha fetoprotein; AXIN1, axis inhibition protein 1; bCa, bladder cancer; BMP3, bone morphogenetic protein 3; CIN2, cervical intraepithelial neoplasia grade 2; CDO1, cysteine dioxygenase type I; CNV, copy number variation; CTNNB1, catenin beta 1; DCP, des-gamma-carboxy prothrombin; EBV, Epstein-Barr virus; HOXA7, homeobox A7; HOXA9, homeobox A9; Hgb, hemoglobin; IDH, isocitrate dehydrogenase; KRAS, Kirsten rat sarcoma virus; LRG1, leucine-rich alpha-2-glycoprotein 1; NDRG4, N-Myc downstream-regulated gene 4; RASSF1A, Ras association domain family member 1; VIM, vimentin; SNV, single nucleotide variant; SFRP2, Secreted Frizzled Related Protein 2; SOX17, SRY-Box Transcription Factor 17; SEPT9, Septin 9; TIMP1, tissue inhibitor matrix metalloproteinase 1; TAC1, tachykinin precursor 1; TERT, telomerase reverse transcriptase; VHL, Von Hippel-Lindau syndrome; ZFP42, zinc finger protein 42.
Lung cancer
Lung cancer is the most lethal malignancy worldwide, with an estimated 2.2 million new cases and 1.8 million deaths globally in 2020.95 The 5-year survival rate is less than 20%, largely owing to advanced stages at diagnosis, when treatments are less effective than at early stages. LDCT is recommended for screening lung cancer in high-risk populations.96 However, its implementation is hindered by the high false-positive rate,97 low compliance,98 and potential radiation exposure. Thus, efforts have been made to explore cfDNA-based liquid biopsy as a potential complement for LDCT.
Clinically, the adoption of a screening test with relatively high sensitivity and acceptable specificity before LDCT can do much to prevent unnecessary radiation exposure and increase compliance with LDCT. A genome-wide cfDNA fragmentation profiling approach (DNA evaluation of fragments for early interception [DELFI]) detected 91% of stage I‒II and 94% of stage III‒IV lung cancers at 80% specificity.99 Therefore, for individuals at high risk for lung cancer, an annual blood test with DELFI may be recommended and, for those with a positive result, LDCT should be taken subsequently. Similarly, a cfDNA assay containing three methylation markers yielded a sensitivity of 93.0%, but a specificity of 62.0%,100 which may be a pre-screen alternative before LDCT. However, these extremely high sensitivities were only achieved in case-control studies, whose performance in prospective screening studies remains to be explored.
It is not unusual that individuals at high risk for lung cancer refuse LDCT for various reasons, such as limited accessibility and concerns about false positivity. In such scenarios, screening tests with a high specificity and acceptable sensitivity might be conducted as complements. The lung cancer likelihood in plasma integrating genomic profiles, including single nucleotide variants (SNVs) and genome-wide CNVs of cfDNA using machine learning, could discriminate early-stage lung cancer from risk-matched controls with sensitivities of 41.0% in stage I, 54.0% in stage II, and 67.0% in stage III, at 98.0% specificity.55 Analysis of methylation markers in cfDNA could also help detect lung cancer with high specificity, showing a potential to complement LDCT screening. Promoter methylation status of six genes (SOX17, TAC1, HOXA7, CDO1, HOXA9, and ZFP42) had sensitivities ranging from 65.0% to 76.0% and specificities ranging from 74.0% to 84.0%, respectively.100 Additionally, a cfDNA methylation-based early-detection model in a prospective case-control study yielded sensitivities of 23.0% in stage I and 47.0% in stage II at a specificity of 99.0% in lung cancer.93 Another cfDNA methylation early-detection model established in lung cancer showed sensitivities of 37% in stage I and 75.0% in stage II at a specificity of 99.0%.94 These tests are less sensitive than LDCT in some subtypes of lung cancer, especially in lung adenocarcinoma,41 which poses a great challenge to its clinical usefulness. However, by complementing LDCT, cfDNA tests may potentially increase the total number of individuals screened so that more early-stage lung cancer can be detected to save more lives.
The cfDNA tests are also applied to accurate differentiation of malignant pulmonary nodules from benign ones, which are expected to alleviate the subsequent diagnostic burden.101 A 100-feature cfDNA methylation model named PulmoSeek identified the malignant pulmonary nodules from benign ones with a sensitivity of 100.0% at 30.0% specificity, suggesting that the blood test after LDCT screening can decrease unnecessary over-diagnosis.89
Currently, there are no cfDNA-based screening methods that can replace LDCT. One obstacle to cfDNA-based screening in lung cancer is the lack of cfDNA released into the circulation, especially in early-stage lung adenocarcinoma.41 It causes a natural ceiling effect in cfDNA-based screening methods, even though strenuous efforts have been made to increase the analytical sensitivity in early-stage lung cancer. A possible solution is to identify new lung cancer screening biomarkers independent of the quantity of ctDNA in circulation.
Colorectal cancer
Colorectal cancer (CRC) ranks the third in terms of incidence but the second in terms of mortality worldwide, with an estimated 1.9 million new cases and 0.94 million deaths in 2020.81 Most patients with CRC diagnosed with metastatic diseases have a 5-year survival rate of 14%, but this rate is as high as 90% for patients with localized disease.2 The detection of pre-cancerous lesions and early-stage CRC is critical to curative treatment and optimal prognosis.102 Currently, fecal occult blood testing (FOBT),103,104 fecal immunochemical testing (FIT),105, 106, 107 and colonoscopy108 are used for CRC screening. With a specificity more than 95%, the sensitivities of FOBT and FIT are 12.9% and 73.8%, respectively.88,109 These relatively low sensitivities would cause high false-negative results. Although colonoscopy is an effective method for CRC screening, its application is limited by the uncomfortable, invasive, and time-consuming process.7 Thus, new non-invasive methods with sensitivities high enough to prevent unnecessary colonoscopy promise an ideal clinical development roadmap to CRC screening.
A stool-based multi-target test consisting of sequencing the methylation of NDRG4, BMP3, and seven mutation sites of KRAS, together with an immunohistochemical assay for hemoglobin, has been evaluated in a large prospective study of 9,989 participants at average risk for CRC. The sensitivity is higher than FIT (92.3% vs. 73.8%) at the cost of lower specificity (89.8% vs. 96.4%).88 Based on these results, a FDA-approved stool multi-target panel has been commercialized for CRC screening,110 known as Cologuard. However, the compliance of stool-based tests is also affected by the inconvenient procedure.111 Thus, alternatives to stool-based CRC screening are under active exploration, and plasma-based tests are on the radar of further investigation.
Plasma cfDNA methylation in specific genes has been extensively explored for CRC screening. SEPT9, encoding GTP-binding protein Septin 9, is one of the most thoroughly evaluated biomarkers for CRC screening, with sensitivities ranging from 48.0% to 90.0% and specificities ranging from 73.0% to 97.0%.112, 113, 114 A large prospective trial has assessed the efficiency of circulating methylated SEPT9 test for detecting CRC in 7,941 participants, yielding a sensitivity of 48.2% and a specificity of 91.5%, respectively.115 However, the sensitivity of SEPT9 methylation was only 7.9%–38.7% for advanced adenomas (AAs), a pre-cancerous lesion that requires surgical intervention to prevent cancerigenesis.115 Another two well-studied methylation biomarkers under clinical development are VIM, encoding the intermediate filament protein vimentin,69 and SFRP2, encoding secreted frizzled-related protein 2.116 Plasma VIM methylation yields a sensitivity of 59.0% and a specificity of 93.0%, while plasma SFRP2 methylation has a sensitivity of 89.2% and a specificity of 73.0%.
Tests combining several methylation markers have also been developed to improve the accuracy in CRC screening. A diagnostic score integrating 9 methylation markers has been constructed by machine learning, which discriminated patients with CRC from normal controls with an area under the curve (AUC) of 0.96.87 Another two models including multi-methylation locus had similar performances with sensitivities of 84%–86% in CRCs and 42% in AAs at a specificity of 92%.117,118
So far, no head-to-head comparisons have been made to evaluate the effectiveness of various screening modalities. In general, plasma-based CRC screening tests have higher specificities but lower sensitivities than stool-based CRC screening tests. In addition, the low sensitivity in AAs also limits the clinical applications of current plasma-based CRC screening tests.
Hepatocellular carcinoma
Hepatocellular carcinoma (HCC) ranks the sixth in incidence and the third leading cause of cancer-related deaths globally.2 Patients with HCC are generally diagnosed at late stages in the absence of symptoms.119 HCC mostly develop progressively from chronic liver diseases, including hepatitis B virus (HBV)/hepatitis C virus (HCV) infection, obesity-driven non-alcoholic fatty liver disease, and liver cirrhosis.120 Surveillance by ultrasound examination plus alpha-fetoprotein (AFP) every 6 months is recommended as HCC screening for individuals with cirrhosis from any cause or carriers of HBV.121,122 However, ultrasound examination has a low sensitivity of 63.0% (23.0%–91.0%) for early-stage HCC,123 particularly for those within the Milan criteria. AFP alone lacks adequate performance with sensitivities of 60%–80% and specificities of 70%–90%.119 Detection of early-stage HCC in asymptomatic individuals would allow more patients to undergo potentially curative treatments, including hepatic resection and liver transplantation.
The liver is known to be a major contributor of cfDNA owing to its generous blood supply.124 Unsurprisingly, the concentration of cfDNA shedding into blood is the highest in HCC compared with other cancers, as demonstrated in a pan-cancer early detection study,49 making plasma cfDNA an ideal alternative for the early detection of HCC. Currently, cfDNA-based biomarkers for HCC early detection are mainly evaluated by case-control studies. Given that patients with liver diseases, especially liver cirrhosis and HBV/HCV infection, are the target screening population under current guidelines,125 it is essential that a cfDNA-based test be reliable enough to distinguish states of these chronic diseases from HCC. Results from studies only focusing on HCC and healthy controls may not be sufficient in the real-world screening population.
Recently, the feasibility of methylation markers for differentiating HCC from non-HCC has been reported. Hypermethylation in the promoters of RASSF1A, CDKN2A, and CDKN2B was detected in 44 serum samples from HCC, which were collected 1‒9 years before a clinical HCC diagnosis, suggesting that the hypermethylation of these genes is an early event in the development of HCC.126 A six-marker MDM panel had a sensitivity of 95.0% at a specificity of 92.0% to differentiate HCC from both cirrhosis and healthy controls in a validation cohort of 95 patients with HCC and 149 non-HCC controls.127 The panel detected 91.3% of patients (42 of 46) with Barcelona Clinic Liver Cancer (BCLC) stages of 0/A.127 A similar performance was also obtained in another study, where the methylation model had a sensitivity of 83.6% at a specificity of 96.0% in the validation cohort including 67 patients with HCC and 353 patients with liver cirrhosis and healthy controls.128
Genome-wide 5-hydroxymethylcytosines (5-hmC) markers, generated from the oxidation of 5-methylcytosine by the translocation enzymes,129 have also been evaluated in HCC early detection.83 The 5-hmC marker model was constructed in early-stage HCC and non-HCC, and accurately distinguished early HCC (BCLC stage 0/A) from non-HCC (chronic hepatitis B, liver cirrhosis, and healthy controls) with a sensitivity of 82.7% and a specificity of 76.4%.83 The sensitivity and specificity of differentiating HCC from liver cirrhosis were improved to 95.4 and 97.8%, respectively, by combining 5-hmC with nucleosome footprint, 5′ end motif, and fragmentation profiles (HIFI score).85 In this study, the HIFI score outperformed any of these techniques alone and the performance of 5-hmC.83 Notably, the cfDNA fragmentation profile sequenced by WGS was also leveraged to detect early-stage HCC.86 This model showed excellent sensitivities of 95.9% and 97.9% in distinguishing stage I and stage II liver cancer from non-cancers including chronic hepatitis B, liver cirrhosis, and healthy controls at a 98.8% specificity,86 showing the potential of cfDNA fragmentation in the early detection of HCC.
Multi-omics hinting the existence of HCC have also been evaluated. Mutations in TP53, CTNNB1, AXIN1, and the promoter region of TERT, HBV sequence and protein markers including AFP and des-γ-carboxy-prothrombin (DCP), as well as clinical covariates (age and sex)84 were integrated and first trained in a case-control setting and then validated in a prospective cohort of HBV surface antigen positive asymptomatic individuals. The integrated multi-analytics had a sensitivity of 85% and a specificity of 93% in the training cohort. In the prospective cohort, 4 of the 24 test-positive individuals were diagnosed with HCC by subsequent CT scan or magnetic resonance imaging (MRI), or by pathological review 6‒8 months after baseline test, all of which was less than 3 cm in diameter. However, the final specificity and sensitivity need to be evaluated considering the limited time window of follow-up. Despite the recent progress, prospective studies in asymptomatic individuals at high risk of HCC with large sample size are still lacking. Whether those new cfDNA markers could accurately identify patients with early-stage HCC who may benefit from liver resection or transplantation needs to be further evaluated. False positivity of those cfDNA tests in patients with cirrhosis or HBV/HCV infection is another concern, since those liver diseases share similar genetic or epigenetic alterations with HCC.130,131
Renal cancer
Renal cell carcinoma (RCC) is the most prevalent subtype of renal cancer, accounting for approximately 90% of renal cancer.132 So far, no standard screening tests have ever been recommended for RCC, and approximately 30.0% of patients with RCC are diagnosed at late stages.133 Imaging examination exposes patients to unnecessary radiation and does not always distinguish benign renal tumors from RCC.134 PCR sequencing the methylation of VHL and RASSF1A in plasma had relatively low sensitivities of 0%–50.3% and 45.9%–62.9% at specificities of 90.7%–100.0% and 93.0%–93.3%, respectively, for RCC early detection.135,136 A diagnostic classifier based on the cfMeDIP-seq sequenced cfDNA methylation data of 120 patients with RCC and 28 healthy controls separated patients with RCC from healthy controls with an AUC of 0.99.90 However, RCC sheds the least amount of cfDNA into the bloodstream among all the extracranial cancers.137 Even in advanced RCC, ctDNA genomic alterations were only detected in less than 80% of the patients.29 The application of cfDNA in RCC early detection might have limited practicability. Instead, urinary cfDNA is a promising alternative for RCC early detection, although the reported discriminating accuracy of urinary cfDNA is lower than that of plasma cfDNA (AUC, 0.858 vs. 0.99).90 Future studies are warranted to evaluate cfDNA-based tests in RCC early detection.
Intracranial cancers
The current cfDNA-based early detection studies focused on extracranial cancers, and the early detection of intracranial tumors such as glioma remains challenging,29 due to the blood-brain barrier to cfDNA transmission into peripheral plasma.49,138 As demonstrated in pan-cancer studies, glioma shed the least cfDNA into circulation compared with other cancers, even in patients with advanced disease. Only 20%–50% of the patients with glioma harbored detectable ctDNA.29,137 Despite the low ctDNA abundance, plasma cfDNA is attracting attention in the area of intracranial cancer screening given its convenience.
Plasma cfDNA methylation profiles were reported to differentiate glioma from both other extracranial cancers and healthy controls with AUC of 0.99 based on 447 samples.139 Excellent performance was also observed in IDH mutated glioma (AUC = 0.99) and IDH wild-type glioma (AUC = 0.99), as well as in both lower-grade (AUC = 0.99) and higher-grade (AUC = 0.98) glioma, highlighting the potential of cfDNA methylation as an efficient biomarker of glioma.
To accurately identify lesions on imaging is another major challenge to the diagnosis of intracranial cancers owing to the broad differential diagnosis ranging from indolent low-grade tumors to aggressive cancers. To obtain a definite diagnosis, invasive neurosurgery is necessary, but it may introduce neurological morbidities and, probably, anxiety to the patients.140 The cfDNA methylation profiles in plasma showed a potential to discriminate tumors with similar cell-of-origin, including solitary extra-axial tumors (meningioma [AUC = 0.89], hemangiopericytoma [AUC = 0.95]), intra-axial tumors (low-grade glial-neuronal tumor [AUC = 0.93], IDH mutant glioma [AUC = 0.82], IDH wild-type glioma [AUC = 0.71]) and brain metastases from systemic cancers.139 This diagnostic test of cfDNA methylation could dispense with invasive procedures to establish diagnosis, especially when cytoreduction is not necessary or the risks of invasive surgery outweigh benefits.139
Compared with plasma, the sequencing of ctDNA in cerebrospinal fluid (CSF) may more comprehensively characterize genomic and epigenetic alterations of brain tumors.138 Although cfDNA in CSF has a potential to supplement current diagnosis approaches, the use of CSF is limited owing to the invasive and morbid procedure.141 Novel biological, technological, and computational approaches are needed to increase the detection sensitivity for intracranial cancers, and there is still a long way to go.
Other cancers
The cfDNA-based liquid biopsy has also shown the potential to identify those cancers without any recommended screening tests, such as pancreatic, ovarian, nasopharyngeal, and esophageal cancer. A landmark research reported that Epstein-Barr virus (EBV) DNA in plasma identified early nasopharyngeal carcinoma in asymptomatic individuals with a sensitivity of 97.1% at a specificity of 98.6%,91 opening a new era of early non-invasive screening for this malignancy. A combination of KRAS mutation and protein biomarkers (CA19-9, TIMP1, and LRG1) could identify early pancreatic cancer from healthy controls with a sensitivity of 64.0% at a specificity of 94.0%.142 An interrogated 5-hmC markers of cfDNA could differentiate pancreatic ductal adenocarcinoma from healthy controls with an AUC of 0.74‒0.97 in two external validation cohorts.143 The methylation patterns in cfDNA discriminated patients with high-grade serous ovarian carcinoma from both healthy controls and those with a benign pelvic mass with a sensitivity of 41.4% at a specificity of 90.7%.144 For esophageal cancer, an early detection model including five methylated cfDNA markers distinguished cancer from healthy controls with sensitivities of 43.0%, 64.0%, 77.0%, and 92.0% for stages I‒IV at the specificity of 91.0%, respectively.145
To be noted, the lack of recommended screening tools for the above-mentioned cancers is probably owing to the low incidence, absence of specific high-risk factors, lack of simple diagnostic procedures, or rapid progression. It is more difficult to develop cfDNA-based screening methods with cost-effectiveness for these cancers without recommended screening tools. Based on early detection of these cancers, the improvement of the performance of cfDNA is expected.
Multi-cancer early detection and localization
In contrast with traditional screening methods aiming at a single cancer type,146, 147, 148, 149 detecting multiple cancers simultaneously with accurate TOO localization may steer clinicians onto a different path for early detection of cancers. The early detection of multi-cancers by plasma has several advantages. First, multiple cancers have a higher prevalence than single cancers in a general screening population. Hence, the early detection of multi-cancers may detect more patients with cancer than for single cancers, especially for those cancers with low prevalence or without specific high-risk factors. Second, the use of combined tests for early detection of single cancers generally cumulates the false-positive rate, potentially increasing unnecessary diagnostic workup. On the contrary, the early detection of multi-cancers with accurate TOO has the potential to prevent a diagnostic odyssey. Third, detecting multiple cancers simultaneously is simpler and time saving for the early detection of cancers in most cases.
One major obstacle to early detection of multi-cancers is the accurate TOO. The genomic and epigenomic alteration pattern of a specific cancer may be unique. In this study, t-stochastic neighbor embedding (t-SNE) was used to cluster cancers based on molecular similarities of methylation, CNVs and SNVs using the data of 11,117 tumor samples from 33 types of cancers in The Cancer Genome Atlas (TCGA) (Figure 3). Those cancers were clearly separated and clustered by the specific methylation features of the tissue origins. Additionally, the heatmap showed that either hyper- or hypo-methylation feature could accurately define each cancer type. However, tissue-specific clusters could not be defined based on CNVs or mutations. As plasma contains the DNA released from multiple tissues, it is possible to unveil the perturbed proportional contributions of different tissues into plasma by de-convoluting the sequencing data on the profiles of different tissues, providing information on TOO. Take lung cancer as an example; the cfDNA released from lung tissues is increased in the plasma owing to lung injury caused by tumor compression and invasion. This increase can be detected by mapping the sequencing data to the profiles of different tissues, and then lung cancer is suspected. A subsequent LDCT is subjected for further diagnosis (Figure 3). Among genomic and epigenomic alterations, the methylation of CpG sites provides not only tissue-specific information, but also cancer-specific information,58 making it a promising approach to the TOO of early detection of multi-cancers. Consistent results have demonstrated in a previous study that methylation patterns outperformed mutations and chromosomal alterations in multi-cancer detection and TOO.150
Figure 3.

Comparison of the tissue-of-origin efficacy of methylation, mutation, and CNV features
(A) Take lung cancer as an example. The cfDNA released from lung tissue is increased in the plasma owing to the lung injury caused by tumor compression and invasion, and this increase could be detected by mapping the sequencing data to the profiles of different tissues. Then lung cancer is suspected, and a subsequent LDCT is subjected to the patient for further diagnosis. (B–D) We performed t-SNE to decrease the dimensionality of methylation, mutation, and copy number data of the 33 cancer types in TCGA database. Particularly, a heatmap of methylation data shows the cancer subtype-specific hypomethylated and hypermethylated regions. ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; CNV, copy number variation; COAD, colon adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B-cell lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukemia; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; t-SNE, t-distributed stochastic neighbor embedding.; TGCT, testicular germ cell tumors; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma.
A recent blood test for early detection of multi-cancers based on cfDNA methylation detected more than 50 types of cancers across all stages, whose sensitivity of stages I–III was 43.9% and TOO accuracy was 93.0%.93 These results were further confirmed in another independent cohort with 5,309 participants (patients with cancer and healthy controls).151 Subsequently, a prospective interventional study, PATHFINDER (Assessment of the Implementation of an Investigational Multi-Early detection of cancers Test Into Clinical Practice; NCT04241796) was launched to assess the extent and types of diagnostic testing required to achieve diagnostic resolution after receiving a predicted positive result. The interim results showed that cancer signals were detected in 1.4% of all analyzable participants, and nearly half with diagnostic resolution had confirmed cancer, with an estimated 45% positive predictive value (PPV).152 Another multi-cancer detection based on cfDNA methylation yielded a sensitivity of 80.6% in six types of cancers at specificity of 98.3% in the validation group, and the TOO accuracy was 87.0% for top-two organ localization.94
In addition to cfDNA methylation, other attempts at multi-parametric analysis for cancer detection and TOO have been focusing on cfDNA mutations and protein biomarkers. CancerSEEK, a blood test assessing mutations in defined regions of 16 genes and 8 protein biomarkers, distinguished patients with cancers, including ovary, liver, stomach, pancreatic, esophagus, colorectal, lung, and breast cancers, from healthy controls. The sensitivities ranged from 33.0% to 98.0% at a specificity of 99.0%, and the median of TOO accuracy was 63.0%.28 Based on CancerSEEK, an exploratory prospective interventional study was conducted, named Detecting cancers Earlier through Elective mutation-based blood Collection and Testing (DETECT-A).92 The positive results of the CancerSEEK test at baseline were confirmed by a second test; e.g., imaging or positron emission tomography (PET)-CT scanning was used for further validation and localization. The screening procedure achieved a sensitivity of 15.6% at a specificity of 99% when combined with the baseline and second confirmation tests.92
The cfDNA fragmentation patterns have also been investigated in early detection of multi-cancers. A previous study reported that DELFI incorporated genome-wide fragmentation patterns in cfDNA sequenced by low-coverage WGS.77 The diagnostic sensitivities ranged from 57.0% to 99.0% in the seven cancer types at a specificity of 95.0%. In addition, DELFI combined with cfDNA mutations detected 91.0% of patients with cancer, with a TOO accuracy of 75.0%.77
Although multi-cancer detection has been promising in clinical applications, there are some problems that should be addressed. The unbalanced sensitivity among cancer types needs to be noted. In general, the sensitivity for breast, kidney, lung, esophagus, prostate, and uterine cancers at early stages was poor, probably owing to the low amount of ctDNA released into blood from these cancers, while the sensitivity for liver, colorectal, head and neck, and pancreatic cancers and lymphoma was relatively high. In addition, there are odds that cancer is detected with a misleading TOO. In this case, PET-CT as an indefinite diagnostic method needs to be carefully explored and discussed. Another concern is the relationship between the new early detection tests of multi-cancers and the current standard of care (SOC) screening tests. Presently, the sensitivity of cfDNA assays for lung and breast cancers with SOC screening tests is relatively lower than for other cancers. Thus, current early detection tests of multi-cancers cannot fully replace SOC screening tests; instead, they can be a complement. In PATHFINDER and DETECT-A studies,92,152 participants are all encouraged to stick to SOC screening tests, even with a negative test result to rule out the possibility of false-negative results.
Clinical phases of biomarker development for early detection of cancers
To provide a general perspective for the development of early detection of cancers, we describe the necessary phases of clinical trials of new diagnostic biomarkers before being approved by regulations. A guideline is proposed to describe the phases, which serves as an important principle, although not all the biomarkers need to consecutively go through each of the phases of development.153 Despite the unavailability of standardized criteria for clinical validity, benefit-risk, and clinical use relative to multi-cancer screening,154 three key clinical phases should be taken into account regarding cfDNA-based liquid biopsy tests: (1) the development and validation of assays, including analytical validation and clinical validation in case-control cohorts; (2) a prospective feasibility study in intended-use population with a small sample size to evaluate the feasibility of the test, involving sensitivity, specificity, PPV, and negative predictive value (NPV); and (3) a prospective pivotal study in intended-use population with a large sample size to evaluate the efficacy, safety, and eventual cost effectiveness.
In the first phase, analytical validation is needed to ensure that the assay is reliable and reproducible. The standardization and quality control of all analytical and preanalytical steps is crucial to implementation into patient care. Particularly external quality assessment schemes are important to ensure the laboratory-to-laboratory comparability. Additionally, how well the early detection model distinguishes cases from controls should be answered. Diagnostic accuracy, including sensitivity and specificity, needs to be evaluated as the primary outcome. Samples should be collected and processed under uniform standard operating procedures to minimize bias. It is also important to select appropriate risk-matched controls to mitigate false discovery rates and the overfitting of biomarker performance. Ideally, populations of cases and control should represent the targeted screening population, and selection bias is to be avoided. The cfDNA-based liquid biopsy model generated in this phase is going to be tested in the next prospective screening trials.
In the second phase, a prospective feasibility study needs to be conducted in the intended-use population, with a small sample size to further evaluate the sensitivity, specificity, PPV, and NPV. Sensitivity and specificity need to be balanced when the optimal cutoff value for the cfDNA-based liquid biopsy model is chosen in the intended-use population before the second phase (Figure 4). For example, high specificity is of great importance to multi-cancer screening in average-risk asymptomatic individuals. Otherwise, a considerable number of healthy individuals will be diagnosed as false positive, which may cause unnecessary invasive diagnostic procedures and anxieties. Assuming that the prevalence for multiple cancers in the general population is 1.0% at 90.0% specificity and a 90.0% sensitivity, the PPV is 8.3%, which means that one real patient with cancer has been detected with 11 cases of false positivity. If the specificity is increased from 90.0% to 99.0%, the PPV is dramatically increased to 47.6%. Comparatively, if the sensitivity increases from 90.0% to 99.0%, the PPV is mildly increased to 9.1%. Altogether, these indicate that specificity is more crucial when PPV is taken into consideration in multi-cancer screening assay.
Figure 4.

Balancing sensitivity and specificity in the cfDNA-based liquid biopsy model
The optimal cutoff value for the cfDNA-based liquid biopsy model should be determined based on different clinical sensorias ahead of its application in the intended-use population. For example, specificity is more important when PPV is taking into consideration in screening assay with cancer of a low prevalence. Assuming that the prevalence for a certain cancer in the general population is 1%, at 90% specificity and 90% sensitivity, the PPV is 8.3%, indicating that a true patient with cancer has been detected, with 11 false positives. If the specificity is increased from 90% to 99%, the PPV is dramatically increased to 47.6%. Comparatively, if the sensitivity increases from 90% to 99%, the PPV is mildly increased to 9.1%. However, under several scenarios, such as early detection in cancers that have specific high-risk factors or convenient diagnostic procedures, cfDNA test with high sensitivity and mild specificity is more acceptable. We propose a simplified formula for consideration to minimize the extra socioeconomic burden. Sen, sensitivity; Spe, specificity.
However, specificity is not always a priority. Under several scenarios, such as early detection in cancers that have specific high-risk factors or simple diagnostic procedures, cfDNA test with high sensitivity and mild specificity is more acceptable. Here, we propose a simplified formula to minimize the extra socio-economic burden (Figure 4). We define the extra cost for tumor omission caused by false negatives as A = Population × Prevalence × (1 − Sensitivity) × Cost of delayed diagnosis, and the additional cost for unnecessary diagnosis owing to false positives is B = Population × (1 − Prevalence) × (1 − Specificity) × Cost of over-diagnosis. Assuming that total extra cost is the sum of A and B, when the total extra cost is increased with decreasing sensitivity, wherein a cfDNA assay with higher sensitivity is preferred; conversely, when ) the total extra cost is increased with decreasing specificity, wherein a cfDNA test with higher specificity is preferred. It is important to note that this simplified criterion is only considered from the perspective of cost, regardless of effectiveness. How to balance specificity and sensitivity needs to be answered by cost-effectiveness studies.
In the third phase, a prospective pivotal study should be conducted in the intended-use population with a large sample size to evaluate the efficacy, safety, and eventual cost effectiveness. The evaluation of efficacy involves (1) the determination of detection rate and false referral rate based on positive results, (2) the practical efficacy of implementing the screening scheme, (3) the compliance of screen-positive populations with workup and treatment recommendations, and (4) the preliminary assessments of the effects of screening on costs and mortality associated with cancer. The assessment of safety includes (1) whether the screening will result in serious adverse events, (2) whether the screening will discourage participants from SOC screening, or (3) whether the screening will induce overwhelming anxiety and futility, as well as unnecessary invasive follow-up, especially in false positivity. The ultimate aim of screening is to decrease cancer mortality. Ideally, participants are randomized into two arms: one receiving the SOC screening and the other receiving the new cfDNA-based screening methods. Differences in cancer mortality and cost-effectiveness between the two arms will be studied.
Currently, many early detection models of cfDNA-based liquid biopsy have been built and validated in case-control studies, and several prospective cancer screening trials are ongoing. We review the ongoing clinical trials of cfDNA-based early detection posted on clinicltrials.gov (searches are up to January 2022), which are summarized in Table S1.
Further directions for early detection of cancers based on liquid biopsy
Improvement in technology and model development
The cfDNA-based liquid biopsy for early detection is mainly limited by the low signals shed from early-stage cancer, which directly affect the sensitivity of sequencing methods. Ultra-deep sequencing methods have been developed across different cancer types155, 156, 157 to increase sensitivity, by which LOD is established as low as 0.001%. In addition, the size selection of cfDNA fragments and the single-strand DNA library for NGS are also effective solutions.158 False-positive results are also a concern when multiple alterations are sequenced by NGS.46 Error-suppression strategies, such as the application of molecular barcodes and bioinformatic analysis pipelines of the data, minimize the risk of introducing errors during library preparation and subsequent sequencing steps.46 When ultrasensitive cfDNA mutation sequencing is used, equivalent depth sequencing of WBC DNA is essential to elimination of the influence of clonal hematopoiesis.55 Apart from analytical methods, some critical preanalytical processes, including the collection, conservation, centrifugation, and extraction protocols for cfDNA, are expected to be optimized to ensure the quality of cfDNA.
With the development of machine learning, more signals and integration of multiple parameters are exploited to boost the discovery and detection of cancer-specific signatures.159 The methods of machine learning range from univariable approaches such as logistic regression, to complex artificial neural networks containing multi-dimensional data,160 and help to build a diagnostic classifier based on the selected features.161 These methods have already been leveraged to improve the performance of different liquid biopsy assays and will facilitate their future integration into the clinical workflow.
Biomarkers beyond genomic or epigenomic alterations in cfDNA
The early detection of genomic or epigenetic alterations in blood relies heavily on the release of cfDNA by cancer cells and sensitivity is poor for those cancers that are cfDNA barren.49 Thus, new biomarkers that do not rely on cfDNA release are essential supplements. A different approach that tracks the body’s antitumor immune response was proposed based on detecting cancer-associated T cell receptors, which discriminated cancers from non-cancers with an AUC of more than 0.95.162 The microbiome is also involved in oncogenesis.163, 164, 165, 166 Based on the whole-genome and whole-transcriptome sequencing data in TCGA, a systematic characterization of cancer microbiome in 33 types of cancers was able to distinguished cancer samples from healthy controls.167 Extracellular vesicle surface proteins carry the molecular signatures of their parental cancer cells.168,169 In a small-sized sample, this method exhibited robust discrimination for early-stage cancer (stage I, n = 19) with a sensitivity of 95.0% at a specificity of 100.0%, while the accuracy of TOO in six cancer types was as low as 68.0%.168 In addition to cfDNA and proteins, miRNAs also have shown potentials for detecting a wide range of cancers including lung,170,171 liver,172 triple-negative breast,173 cervical,174 and gastric cancers.174 These novel biomarkers may collectively increase sensitivity for early detection of cancers, but large prospective clinical trials are still warranted.
Risk-stratified strategy for early detection of cancers
The Cancer Control Joint Action European Guide on Quality Improvement in Comprehensive Cancer Control has recommended that the benefit-harm trade-offs and cost effectiveness of screening programs should be estimated to guide decisions on implementation.175 A screening method suitable for a high-risk population or an average-risk population in application scenarios is a significant reference for government policymakers, insurance companies, and other payers.175 Identifying individuals at high risk for cancer is an ideal way to improve the cost effectiveness of new cfDNA tests. According to a recent study, breast cancer screening triennially in women ages 50 to 69 had better cost effectiveness, a higher benefit-to-harm ratio, and a lower incidence of over-diagnosis than in those with lower risk for breast cancer.176 Smoking status was involved as risk stratification to evaluate the cost-effectiveness of non-cardia gastric adenocarcinoma (NCGA) screening by serum pepsinogen, and the result showed that targeting high-risk smokers was a cost-effective strategy to decrease the mortality of intestinal-type NCGA.177 Although economic impacts have been evaluated for several traditional screening modalities,176,178, 179, 180, 181 thus far, few liquid biopsy-based early-detection studies have explored the cost effectiveness under risk stratification. One recent research used a serum miRNA biomarker panel to detect gastric cancer and demonstrated that the 12-miRNA panel was a cost-effective method relative to World Health Organization practice in symptomatic participants who were scheduled to undergo gastroscopy.182 This study provided a paradigm for evaluating the economic efficacy of cfDNA tests based on risk stratification.
There are two approaches to evaluation of the cost effectiveness of screening programs based on risk stratification,183 including clinical trial-based and model-based approaches. With clinical trial-based approaches, the efficacy of a new screening assay is evaluated by a well-designed retrospective or prospective case-control study, even by randomized controlled trials. In a model-based approach, natural history models184 and decision analysis models185 are used to study the long-term benefits and harm as well as the cost effectiveness of various screening strategies. Admittedly, trail-based data can provide robust evidence of efficacy for screening assays.186 However, it requires a large sample size, a long follow-up period, and even a prohibitively large research budget. Therefore, evidence derived from the model-based studies serves as an alternative to evaluate the effectiveness of risk-stratified screening. To date, much evidence on the effectiveness of risk-stratified screening is from model-based studies.176,178,181,187 However, the accuracy of the results of modeling mainly depends on model construction and input parameters, which is a challenge, especially in the early detection of multi-cancers as the definition of risk factors tends to be complicated (eg, carriers of high-risk mutations, carcinogenic exposures, intra-epithelial or pre-neoplastic traits, behavioral factors, characteristics of the individual including body habitus, polygenic risk scores, demographic, residence, and other non-biological factors). Moreover, it remains challenging to estimate over-diagnosis caused by the absence of data on disease progression rates for different risk groups.
Despite all odds, a risk-stratified strategy for early detection of cancers may have the potential to improve overall cost effectiveness and benefit-harm ratios in a population-based screening program.
Integration into the current clinical workflow
The cfDNA test is expected to be integrated into the clinical workflow after technological refinement and clinical trial validation. For single cancers with SOC screening methods, the new cfDNA-based test can be used before, with, or after SOC screening methods. In contrast, for single cancers without SOC screening methods, it is significant to balance the sensitivity and specificity of new cfDNA tests for cost effectiveness. In terms of multi-cancer screening, which is most likely used in general asymptomatic individuals, extremely high specificity and accurate TOO are top priorities. Participants with negative results should not be discouraged from SOC screening. Likewise, for those with positive screening results, the subsequent diagnosis is imperative. In many cases, a positive result triggers subsequent imaging or even an invasive diagnostic workup. A medical review committee including a number of disciplines (pathology department, surgical department, imaging department, etc.) and subjects (urology department, alimentary canal, gynecology, etc.) is needed to confirm the path of diagnosis according to baseline physical examination and family history (Figure 5). With the development of liquid biopsy in early detection of cancers, the establishment of a medical review committee shows the potential to revolutionize the healthcare prevention system in the future.
Figure 5.

Future direction for early detection of cancers based on cfDNA tests.
Conclusions and perspectives
cfDNA-based early detection has shown potentials in the early detection of both single- and multi-cancers. However, before it can be applied in clinic, several questions need to be answered. First, can multi-omics biomarkers increase sensitivity without sacrificing specificity? cfDNA-based detection technology needs to be further improved to increase the sensitivity for minute amounts of cfDNA, especially for early-stage cancer. Second, how do we integrate new early detection technologies into the current SOC screening to achieve a better screening performance? Even though it has been extensively studied and discussed in many published studies, validated clinical trials are in urgent need to demonstrate the extent of the feasibility and effectiveness of the new technologies combined with SOC screening modalities. Third, will the new cfDNA-based early detection test bring a survival benefit? Finally, how do we increase the public awareness of early detection of cancers? Governments, academic institutions, and nonprofit institutions are encouraged to organize health-promoting activities and facilitate social cancer screening cohesion.
With the great efforts in the past decade, liquid biopsy has revolutionized and is revolutionizing early detection of cancers not only by enabling minimally invasive molecular tests but also by boosting enormous enthusiasm for public health care. Although the development of liquid biopsy in cancer detection is facing technical difficulties, a lack of validated clinical trials, and other obstacles as discussed above, there is increasing evidence for the efficacy, safety, and cost effectiveness of liquid biopsy in early detection of cancers, which will increase the chance that liquid biopsy can be used as a routine of preventive medicine.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81961128025); Program of Shanghai Academic Research Leader (No. 19XD1420700); and Shanghai Municipal Key Clinical Specialty. We thank Yuzi Zhang, Yuchen Liao, Chenyang Wang, and Shaokun Chuai (Burning Rock Biotech) for their contributions to the manuscript.
Author contributions
J.F., J.W., and S.C. conceptualized the review and drafted the initial manuscript. Q.G., Q.Z., Z.W., and C.L. were responsible for writing and polishing the manuscript. Y.X., X.Z, H.L., and G.W. contributed to the discussion of content and preparation of the manuscript. P.C. prepared the analytic content based on TCGA datasets. All authors approve the final version of the article.
Declaration of interests
C.L., Y.X., P.C., X.Z., H.L., G.W., and S.C. are employees of Burning Rock Biotech. The other authors declare no competing interests.
Published Online: May 6, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xinn.2022.100259.
Contributor Information
Shangli Cai, Email: shangli.cai@brbiotech.com.
Jie Wang, Email: zlhuxi@163.com.
Jia Fan, Email: fan.jia@zs-hospital.sh.cn.
Lead contact website
https://www.zs-hospital.sh.cn/zsyy/n33/n35/n48/n123/index.html.
Supplemental information
References
- 1.Zeng H., Chen W., Zheng R., et al. Changing cancer survival in China during 2003–15: a pooled analysis of 17 population-based cancer registries. Lancet Glob. Health. 2018;6:e555–e567. doi: 10.1016/s2214-109x(18)30127-x. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer statistics, 2021. CA Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 3.Tanoue L.T., Tanner N.T., Gould M.K., Silvestri G.A. Lung cancer screening. Am. J. Respir. Crit. Care Med. 2015;191:19–33. doi: 10.1164/rccm.201410-1777ci. [DOI] [PubMed] [Google Scholar]
- 4.Smith R.A., Andrews K.S., Brooks D., et al. Cancer screening in the United States, 2018: a review of current American Cancer Society guidelines and current issues in cancer screening. CA Cancer J. Clin. 2018;68:297–316. doi: 10.3322/caac.21446. [DOI] [PubMed] [Google Scholar]
- 5.Rafferty E.A., Durand M.A., Conant E.F., et al. Breast cancer screening using tomosynthesis and digital mammography in dense and nondense breasts. JAMA. 2016;315:1784–1786. doi: 10.1001/jama.2016.1708. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y., Zhang L., Zhao G., et al. The clinical research of Thinprep Cytology Test (TCT) combined with HPV-DNA detection in screening cervical cancer. Cell Mol. Biol. 2017;63:92–95. doi: 10.14715/cmb/2017.63.2.14. [DOI] [PubMed] [Google Scholar]
- 7.Chen H., Li N., Ren J., et al. Participation and yield of a population-based colorectal cancer screening programme in China. Gut. 2019;68:1450–1457. doi: 10.1136/gutjnl-2018-317124. [DOI] [PubMed] [Google Scholar]
- 8.Tierney W.M. Cost-effectiveness of CT screening in the national lung screening trial. N. Engl. J. Med. 2015;372:387–388. doi: 10.1056/NEJMc1414726. [DOI] [PubMed] [Google Scholar]
- 9.Miller K.D., Nogueira L., Mariotto A.B., et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019;69:363–385. doi: 10.3322/caac.21565. [DOI] [PubMed] [Google Scholar]
- 10.Pantel K., Alix-Panabières C. Liquid biopsy and minimal residual disease — latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019;16:409–424. doi: 10.1038/s41571-019-0187-3. [DOI] [PubMed] [Google Scholar]
- 11.Heitzer E., Haque I.S., Roberts C.E.S., Speicher M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2018;20:71–88. doi: 10.1038/s41576-018-0071-5. [DOI] [PubMed] [Google Scholar]
- 12.Ignatiadis M., Sledge G.W., Jeffrey S.S. Liquid biopsy enters the clinic - implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021;18:297–312. doi: 10.1038/s41571-020-00457-x. [DOI] [PubMed] [Google Scholar]
- 13.Kahlert C., Melo S.A., Protopopov A., et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J. Biol. Chem. 2014;289:3869–3875. doi: 10.1074/jbc.c113.532267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thakur B.K., Zhang H., Becker A., et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014;24:766–769. doi: 10.1038/cr.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jahr S., Hentze H., Englisch S., et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659–1665. [PubMed] [Google Scholar]
- 16.Diehl F., Schmidt K., Choti M.A., et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008;14:985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.To E.W.H., Chan K.C.A., Leung S.F., et al. Rapid clearance of plasma Epstein-Barr virus DNA after surgical treatment of nasopharyngeal carcinoma. Clin. Cancer Res. 2003;9:3254–3259. [PubMed] [Google Scholar]
- 18.Yao W., Mei C., Nan X., Hui L. Evaluation and comparison of in vitro degradation kinetics of DNA in serum, urine and saliva: a qualitative study. Gene. 2016;590:142–148. doi: 10.1016/j.gene.2016.06.033. [DOI] [PubMed] [Google Scholar]
- 19.Lo Y.M.D., Zhang J., Leung T.N., et al. Rapid clearance of fetal DNA from maternal plasma. Am. J. Hum. Genet. 1999;64:218–224. doi: 10.1086/302205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamkovich S.N., Cherepanova A.V., Kolesnikova E.V., et al. Circulating DNA and DNase activity in human blood. Ann. N. Y. Acad. Sci. 2006;1075:191–196. doi: 10.1196/annals.1368.026. [DOI] [PubMed] [Google Scholar]
- 21.Botezatu I., Serdyuk O., Potapova G., et al. Genetic analysis of DNA excreted in urine: a new approach for detecting specific genomic DNA sequences from cells dying in an organism. Clin. Chem. 2000;46:1078–1084. doi: 10.1093/clinchem/46.8.1078. [DOI] [PubMed] [Google Scholar]
- 22.Reckamp K.L., Melnikova V.O., Karlovich C., et al. A highly sensitive and quantitative test platform for detection of NSCLC EGFR mutations in urine and plasma. J. Thorac. Oncol. 2016;11:1690–1700. doi: 10.1016/j.jtho.2016.05.035. [DOI] [PubMed] [Google Scholar]
- 23.Diehl F., Li M., Dressman D., et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. U S A. 2005;102:16368–16373. doi: 10.1073/pnas.0507904102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chused T.M., Steinberg A.D., Talal N. The clearance and localization of nucleic acids by New Zealand and normal mice. Clin. Exp. Immunol. 1972;12:465–476. [PMC free article] [PubMed] [Google Scholar]
- 25.Greytak S.R., Engel K.B., Parpart-Li S., et al. Harmonizing cell-free DNA collection and processing practices through evidence-based guidance. Clin. Cancer Res. 2020;26:3104–3109. doi: 10.1158/1078-0432.ccr-19-3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thierry A.R., El Messaoudi S., Gahan P.B., et al. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016;35:347–376. doi: 10.1007/s10555-016-9629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kustanovich A., Schwartz R., Peretz T., Grinshpun A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019;20:1057–1067. doi: 10.1080/15384047.2019.1598759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen J.D., Li L., Wang Y., et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359:926–930. doi: 10.1126/science.aar3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bettegowda C., Sausen M., Leary R.J., et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014;16:iii7. doi: 10.1093/neuonc/nou206.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun K., Jiang P., Chan K.C.A., et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. U S A. 2015;112:E5503–E5512. doi: 10.1073/pnas.1508736112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hestand M.S., Bessem M., van Rijn P., et al. Fetal fraction evaluation in non-invasive prenatal screening (NIPS) Eur. J. Hum. Genet. 2019;27:198–202. doi: 10.1038/s41431-018-0271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lo Y.M.D., Chan K.C.A., Sun H., et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2010;2:61ra91. doi: 10.1126/scitranslmed.3001720. [DOI] [PubMed] [Google Scholar]
- 33.Thierry A.R., Mouliere F., Gongora C., et al. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 2010;38:6159–6175. doi: 10.1093/nar/gkq421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Snyder M.W., Kircher M., Hill A.J., et al. Cell-free DNA comprises an in vivo nucleosome footprint that Informs its tissues-of-origin. Cell. 2016;164:57–68. doi: 10.1016/j.cell.2015.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagata S. Apoptotic DNA fragmentation. Exp. Cell Res. 2000;256:12–18. doi: 10.1006/excr.2000.4834. [DOI] [PubMed] [Google Scholar]
- 36.Jiang P., Chan C.W.M., Chan K.C.A., et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. U S A. 2015;112:E1317–E1325. doi: 10.1073/pnas.1500076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Underhill H.R., Kitzman J.O., Hellwig S., et al. Fragment length of circulating tumor DNA. PLoS Genet. 2016;12:e1006162. doi: 10.1371/journal.pgen.1006162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mouliere F., Chandrananda D., Piskorz A.M., et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018;10:eaat4921. doi: 10.1126/scitranslmed.aat4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gansauge M.-T., Meyer M. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat. Protoc. 2013;8:737–748. doi: 10.1038/nprot.2013.038. [DOI] [PubMed] [Google Scholar]
- 40.Fiala C., Diamandis E.P. Utility of circulating tumor DNA in cancer diagnostics with emphasis on early detection. BMC Med. 2018;16:166. doi: 10.1186/s12916-018-1157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abbosh C., Birkbak N.J., Wilson G.A., et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545:446–451. doi: 10.1038/nature22364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park G., Park J.K., Shin S.-H., et al. Characterization of background noise in capture-based targeted sequencing data. Genome Biol. 2017;18:136. doi: 10.1186/s13059-017-1275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Forshew T., Murtaza M., Parkinson C., et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012;4:136ra168. doi: 10.1126/scitranslmed.3003726. [DOI] [PubMed] [Google Scholar]
- 44.Kinde I., Wu J., Papadopoulos N., et al. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. U S A. 2011;108:9530–9535. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith T., Heger A., Sudbery I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017;27:491–499. doi: 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newman A.M., Bratman S.V., To J., et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014;20:548–554. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newman A.M., Lovejoy A.F., Klass D.M., et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016;34:547–555. doi: 10.1038/nbt.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phallen J., Sausen M., Adleff V., et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017;9:eaan2415. doi: 10.1126/scitranslmed.aan2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van der Pol Y., Mouliere F. Toward the early detection of cancer by decoding the epigenetic and environmental fingerprints of cell-free DNA. Cancer Cell. 2019;36:350–368. doi: 10.1016/j.ccell.2019.09.003. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y., Yao Y., Xu Y., et al. Pan-cancer circulating tumor DNA detection in over 10,000 Chinese patients. Nat. Commun. 2021;12:11. doi: 10.1038/s41467-020-20162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rolnik D.L., Yong Y., Lee T.J., et al. Influence of body mass index on fetal fraction increase with gestation and cell-free DNA test failure. Obstet. Gynecol. 2018;132:436–443. doi: 10.1097/aog.0000000000002752. [DOI] [PubMed] [Google Scholar]
- 52.Kolarova T.R., Gammill H.S., Nelson J.L., et al. At preeclampsia diagnosis, total cell-free DNA concentration is elevated and correlates with disease severity. J. Am. Heart Assoc. 2021;10:e021477. doi: 10.1161/jaha.121.021477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jaiswal S., Ebert B.L. Clonal hematopoiesis in human aging and disease. Science. 2019;366:eaan4673. doi: 10.1126/science.aan4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Genovese G., Kähler A.K., Handsaker R.E., et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014;371:2477–2487. doi: 10.1056/nejmoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chabon J.J., Hamilton E.G., Kurtz D.M., et al. Integrating genomic features for non-invasive early lung cancer detection. Nature. 2020;580:245–251. doi: 10.1038/s41586-020-2140-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Köhler F., Rodríguez-Paredes M. DNA methylation in epidermal differentiation, aging, and cancer. J. Invest. Dermatol. 2020;140:38–47. doi: 10.1016/j.jid.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 57.Chatterjee R., Vinson C. CpG methylation recruits sequence specific transcription factors essential for tissue specific gene expression. Biochim. Biophys. Acta. 2012;1819:763–770. doi: 10.1016/j.bbagrm.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dor Y., Cedar H. Principles of DNA methylation and their implications for biology and medicine. Lancet. 2018;392:777–786. doi: 10.1016/s0140-6736(18)31268-6. [DOI] [PubMed] [Google Scholar]
- 59.Chen X., Gole J., Gore A., et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat. Commun. 2020;11:3475. doi: 10.1038/s41467-020-17316-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ziller M.J., Gu H., Müller F., et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–481. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Q., Gu L., Adey A., et al. Tagmentation-based whole-genome bisulfite sequencing. Nat. Protoc. 2013;8:2022–2032. doi: 10.1038/nprot.2013.118. [DOI] [PubMed] [Google Scholar]
- 62.Meissner A., Gnirke A., Bell G.W., et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gu H., Smith Z.D., Bock C., et al. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc. 2011;6:468–481. doi: 10.1038/nprot.2010.190. [DOI] [PubMed] [Google Scholar]
- 64.Maunakea A.K., Nagarajan R.P., Bilenky M., et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao M.-T., Whyte J.J., Hopkins G.M., et al. Methylated DNA immunoprecipitation and high-throughput sequencing (MeDIP-seq) using low amounts of genomic DNA. Cell. Reprogram. 2014;16:175–184. doi: 10.1089/cell.2014.0002. [DOI] [PubMed] [Google Scholar]
- 66.Rauch T., Pfeifer G.P. Methylated-CpG island recovery assay: a new technique for the rapid detection of methylated-CpG islands in cancer. Lab. Invest. 2005;85:1172–1180. doi: 10.1038/labinvest.3700311. [DOI] [PubMed] [Google Scholar]
- 67.Shen S.Y., Burgener J.M., Bratman S.V., De Carvalho D.D. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat. Protoc. 2019;14:2749–2780. doi: 10.1038/s41596-019-0202-2. [DOI] [PubMed] [Google Scholar]
- 68.Shen S.Y., Singhania R., Fehringer G., et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018;563:579–583. doi: 10.1038/s41586-018-0703-0. [DOI] [PubMed] [Google Scholar]
- 69.Li M., Chen W.D., Papadopoulos N., et al. Sensitive digital quantification of DNA methylation in clinical samples. Nat. Biotechnol. 2009;27:858–863. doi: 10.1038/nbt.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guo S., Diep D., Plongthongkum N., et al. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat. Genet. 2017;49:635–642. doi: 10.1038/ng.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guo H., Zhu P., Guo F., et al. Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing. Nat. Protoc. 2015;10:645–659. doi: 10.1038/nprot.2015.039. [DOI] [PubMed] [Google Scholar]
- 72.Liang N., Li B., Jia Z., et al. Ultrasensitive detection of circulating tumour DNA via deep methylation sequencing aided by machine learning. Nat. Biomed. Eng. 2021;5:586–599. doi: 10.1038/s41551-021-00746-5. [DOI] [PubMed] [Google Scholar]
- 73.Diaz L.A., Jr., Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J. Clin. Oncol. 2014;32:579–586. doi: 10.1200/jco.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jammula S., Katz-Summercorn A.C., Li X., et al. Identification of subtypes of Barrett's esophagus and esophageal adenocarcinoma based on DNA methylation profiles and integration of transcriptome and genome data. Gastroenterology. 2020;158:1682–1697. doi: 10.1053/j.gastro.2020.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hahn O., Grönke S., Stubbs T.M., et al. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol. 2017;18:56. doi: 10.1186/s13059-017-1187-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang S.R., Schultheis A.M., Yu H., et al. Precision medicine in non-small cell lung cancer: current applications and future directions. Semin. Cancer Biol. 2020 doi: 10.1016/j.semcancer.2020.07.009. [DOI] [PubMed] [Google Scholar]
- 77.Cristiano S., Leal A., Phallen J., et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570:385–389. doi: 10.1038/s41586-019-1272-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ivanov M., Chernenko P., Breder V., et al. Utility of cfDNA fragmentation patterns in designing the liquid biopsy profiling panels to improve their sensitivity. Front. Genet. 2019;10:194. doi: 10.3389/fgene.2019.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Corces M.R., Granja J.M., Shams S., et al. The chromatin accessibility landscape of primary human cancers. Science. 2018;362:eaav1898. doi: 10.1126/science.aav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Polak P., Karlić R., Koren A., et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015;518:360–364. doi: 10.1038/nature14221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Whalley J.P., Buchhalter I., Rheinbay E., et al. Framework for quality assessment of whole genome cancer sequences. Nature communications. 2020;11:5040. doi: 10.1038/s41467-020-18688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu R.H., Wei W., Krawczyk M., et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat. Mater. 2017;16:1155–1161. doi: 10.1038/nmat4997. [DOI] [PubMed] [Google Scholar]
- 83.Cai J., Chen L., Zhang Z., et al. Genome-wide mapping of 5-hydroxymethylcytosines in circulating cell-free DNA as a non-invasive approach for early detection of hepatocellular carcinoma. Gut. 2019;68:2195–2205. doi: 10.1136/gutjnl-2019-318882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Qu C., Wang Y., Wang P., et al. Detection of early-stage hepatocellular carcinoma in asymptomatic HBsAg-seropositive individuals by liquid biopsy. Proc. Natl. Acad. Sci. U S A. 2019;116:6308–6312. doi: 10.1073/pnas.1819799116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen L., Abou-Alfa G.K., Zheng B., et al. Genome-scale profiling of circulating cell-free DNA signatures for early detection of hepatocellular carcinoma in cirrhotic patients. Cell Res. 2021;31:589–592. doi: 10.1038/s41422-020-00457-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang X., Wang Z., Tang W., et al. Ultra-sensitive and affordable assay for early detection of primary liver cancer using plasma cfDNA fragmentomics. Hepatology. 2021 doi: 10.1002/hep.32308. [DOI] [PubMed] [Google Scholar]
- 87.Luo H., Zhao Q., Wei W., et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci. Transl. Med. 2020;12:eaax7533. doi: 10.1126/scitranslmed.aax7533. [DOI] [PubMed] [Google Scholar]
- 88.Imperiale T.F., Ransohoff D.F., Itzkowitz S.H., et al. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014;370:1287–1297. doi: 10.1056/nejmoa1311194. [DOI] [PubMed] [Google Scholar]
- 89.Liang W., Chen Z., Li C., et al. Accurate diagnosis of pulmonary nodules using a non-invasive DNA methylation test. J. Clin. Invest. 2021;131:10. doi: 10.1172/jci145973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nuzzo P.V., Berchuck J.E., Korthauer K., et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat. Med. 2020;26:1041–1043. doi: 10.1038/s41591-020-0933-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chan K.C.A., Woo J.K.S., King A., et al. Analysis of plasma epstein-Barr virus DNA to screen for nasopharyngeal cancer. N. Engl. J. Med. 2017;377:513–522. doi: 10.1056/NEJMoa1701717. [DOI] [PubMed] [Google Scholar]
- 92.Lennon A.M., Buchanan A.H., Kinde I., et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science. 2020;369:eabb9601. doi: 10.1126/science.abb9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu M.C., Oxnard G.R., Klein E.A., et al. CCGA Consortium Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020;31:745–759. doi: 10.1016/j.annonc.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gao Q., Li B., Cai S., et al. Early detection and localization of multiple cancers using a blood-based methylation assay (ELSA-seq) J. Clin. Oncol. 2021;39:459. doi: 10.1200/jco.2021.39.3_suppl.459. [DOI] [Google Scholar]
- 95.Sung H., Ferlay J., Siegel R.L., et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 96.Church T.R., Black W.C., Aberle D.R., et al. Results of initial low-dose computed tomographic screening for lung cancer. N. Engl. J. Med. 2013;368:1980–1991. doi: 10.1056/nejmoa1209120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jemal A., Fedewa S.A. Lung cancer screening with low-dose computed tomography in the United States-2010 to 2015. JAMA Oncol. 2017;3:1278–1281. doi: 10.1001/jamaoncol.2016.6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Doria-Rose V.P., White M.C., Klabunde C.N., et al. Use of lung cancer screening tests in the United States: results from the 2010 National Health Interview Survey. Cancer Epidemiol. Biomarkers Prev. 2012;21:1049–1059. doi: 10.1158/1055-9965.EPI-12-0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mathios D., Johansen J.S., Cristiano S., et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes. Nat. Commun. 2021;12:5060. doi: 10.1038/s41467-021-24994-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hulbert A., Jusue-Torres I., Stark A., et al. Early detection of lung cancer using DNA promoter hypermethylation in plasma and sputum. Clin. Cancer Res. 2017;23:1998–2005. doi: 10.1158/1078-0432.ccr-16-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mcwilliams A., Tammemagi M.C., Mayo J.R., et al. Probability of cancer in pulmonary nodules detected on first screening CT. N. Engl. J. Med. 2013;369:910–919. doi: 10.1056/NEJMoa1214726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wolf A.M.D., Fontham E.T.H., Church T.R., et al. Colorectal cancer screening for average-risk adults: 2018 guideline update from the American Cancer Society. CA Cancer J. Clin. 2018;68:250–281. doi: 10.3322/caac.21457. [DOI] [PubMed] [Google Scholar]
- 103.Hewitson P., Glasziou P.P., Irwig L., et al. Screening for colorectal cancer using the faecal occult blood test, Hemoccult. Cochrane Database Syst. Rev. 2007;2011 doi: 10.1002/14651858.cd001216.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hewitson P., Glasziou P., Watson E., et al. Cochrane systematic review of colorectal cancer screening using the fecal occult blood test (hemoccult): an update. Am. J. Gastroenterol. 2008;103:1541–1549. doi: 10.1111/j.1572-0241.2008.01875.x. [DOI] [PubMed] [Google Scholar]
- 105.Zorzi M., Fedeli U., Schievano E., et al. Impact on colorectal cancer mortality of screening programmes based on the faecal immunochemical test. Gut. 2015;64:784–790. doi: 10.1136/gutjnl-2014-307508. [DOI] [PubMed] [Google Scholar]
- 106.Sokoro A., Singh H. Fecal occult blood test for evaluation of symptoms or for diagnostic testing. Am. J. Gastroenterol. 2020;115:679–680. doi: 10.14309/ajg.0000000000000560. [DOI] [PubMed] [Google Scholar]
- 107.Buskermolen M., Cenin D.R., Helsingen L.M., et al. Colorectal cancer screening with faecal immunochemical testing, sigmoidoscopy or colonoscopy: a microsimulation modelling study. BMJ. 2019;367:l5383. doi: 10.1136/bmj.l5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Regula J., Rupinski M., Kraszewska E., et al. Colonoscopy in colorectal-cancer screening for detection of advanced neoplasia. N. Engl. J. Med. 2006;355:1863–1872. doi: 10.1056/nejmoa054967. [DOI] [PubMed] [Google Scholar]
- 109.Imperiale T.F., Ransohoff D.F., Itzkowitz S.H., et al. Fecal DNA versus fecal occult blood for colorectal-cancer screening in an average-risk population. N. Engl. J. Med. 2004;351:2704–2714. doi: 10.1056/nejmoa033403. [DOI] [PubMed] [Google Scholar]
- 110.Ahlquist D.A. Multi-target stool DNA test: a new high bar for noninvasive screening. Dig. Dis. Sci. 2015;60:623–633. doi: 10.1007/s10620-014-3451-5. [DOI] [PubMed] [Google Scholar]
- 111.Piscitello A., Saoud L., Fendrick A.M., et al. Estimating the impact of differential adherence on the comparative effectiveness of stool-based colorectal cancer screening using the CRC-AIM microsimulation model. PLoS One. 2020;15:e0244431. doi: 10.1371/journal.pone.0244431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Song L., Jia J., Peng X., et al. The performance of the SEPT9 gene methylation assay and a comparison with other CRC screening tests: a meta-analysis. Sci. Rep. 2017;7:3032. doi: 10.1038/s41598-017-03321-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sun J., Fei F., Zhang M., et al. The role of m SEPT9 in screening, diagnosis, and recurrence monitoring of colorectal cancer. BMC Cancer. 2019;19:450. doi: 10.1186/s12885-019-5663-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lehmann-Werman R., Neiman D., Zemmour H., et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. U S A. 2016;113:E1826–E1834. doi: 10.1073/pnas.1519286113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Church T.R., Wandell M., Lofton-Day C., et al. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut. 2014;63:317–325. doi: 10.1136/gutjnl-2012-304149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Barták B.K., Kalmár A., Péterfia B., et al. Colorectal adenoma and cancer detection based on altered methylation pattern of SFRP1, SFRP2, SDC2, and PRIMA1 in plasma samples. Epigenetics. 2017;12:751–763. doi: 10.1080/15592294.2017.1356957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sui J., Wu X., Wang C., et al. Discovery and validation of methylation signatures in blood-based circulating tumor cell-free DNA in early detection of colorectal carcinoma: a case-control study. Clin. Epigenetics. 2021;13:26. doi: 10.1186/s13148-020-00985-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cai G., Cai M., Feng Z., et al. A multilocus blood-based assay targeting circulating tumor DNA methylation enables early detection and early relapse prediction of colorectal cancer. Gastroenterology. 2021;161:2053–2056.e2. doi: 10.1053/j.gastro.2021.08.054. [DOI] [PubMed] [Google Scholar]
- 119.Attwa M.H., El-Etreby S.A. Guide for diagnosis and treatment of hepatocellular carcinoma. World J. Hepatol. 2015;7:1632. doi: 10.4254/wjh.v7.i12.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Forner A., Llovet J.M., Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–1255. doi: 10.1016/s0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 121.Zhou J., Sun H., Wang Z., et al. Guidelines for the diagnosis and treatment of hepatocellular carcinoma (2019 edition) Liver Cancer. 2020;9:682–720. doi: 10.1159/000509424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Omata M., Cheng A.-L., Kokudo N., et al. Asia–Pacific clinical practice guidelines on the management of hepatocellular carcinoma: a 2017 update. Hepatol. Int. 2017;11:317–370. doi: 10.1007/s12072-017-9799-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Singal A., Volk M.L., Waljee A., et al. Meta-analysis: surveillance with ultrasound for early-stage hepatocellular carcinoma in patients with cirrhosis. Aliment. Pharmacol. Ther. 2009;30:37–47. doi: 10.1111/j.1365-2036.2009.04014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Moss J., Magenheim J., Neiman D., et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nature communications. 2018;9:5068. doi: 10.1038/s41467-018-07466-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kanwal F., Singal A.G. Surveillance for hepatocellular carcinoma: current best practice and future direction. Gastroenterology. 2019;157:54–64. doi: 10.1053/j.gastro.2019.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang Y.-J., Wu H.-C., Shen J., et al. Predicting hepatocellular carcinoma by detection of aberrant promoter methylation in serum DNA. Clin. Cancer Res. 2007;13:2378–2384. doi: 10.1158/1078-0432.ccr-06-1900. [DOI] [PubMed] [Google Scholar]
- 127.Kisiel J.B., Dukek B.A., VSR Kanipakam R., et al. Hepatocellular carcinoma detection by plasma methylated DNA: discovery, phase I pilot, and phase II clinical validation. Hepatology. 2019;69:hep.30244. doi: 10.1002/hep.30244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Luo B., Ma F., Liu H., et al. Cell-free DNA methylation markers for differential diagnosis of hepatocellular carcinoma. BMC Med. 2022;20:8. doi: 10.1186/s12916-021-02201-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Branco M.R., Ficz G., Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 2011;13:7–13. doi: 10.1038/nrg3080. [DOI] [PubMed] [Google Scholar]
- 130.Song M.A., Kwee S.A., Tiirikainen M., et al. Comparison of genome-scale DNA methylation profiles in hepatocellular carcinoma by viral status. Epigenetics. 2016;11:464–474. doi: 10.1080/15592294.2016.1151586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Okamoto Y., Shinjo K., Shimizu Y., et al. Hepatitis virus infection affects DNA methylation in mice with humanized livers. Gastroenterology. 2014;146:562–572. doi: 10.1053/j.gastro.2013.10.056. [DOI] [PubMed] [Google Scholar]
- 132.Moch H. An overview of renal cell cancer: pathology and genetics. Semin. Cancer Biol. 2013;23:3–9. doi: 10.1016/j.semcancer.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 133.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2019. CA Cancer J. Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 134.Schoots I.G., Zaccai K., Hunink M.G., Verhagen P.C. Bosniak classification for complex renal cysts reevaluated: a systematic review. J. Urol. 2017;198:12–21. doi: 10.1016/j.juro.2016.09.160. [DOI] [PubMed] [Google Scholar]
- 135.de Martino M., Klatte T., Haitel A., Marberger M. Serum cell-free DNA in renal cell carcinoma. Cancer. 2012;118:82–90. doi: 10.1002/cncr.26254. [DOI] [PubMed] [Google Scholar]
- 136.Skrypkina I., Tsyba L., Onyshchenko K., et al. Concentration and methylation of cell-free DNA from blood plasma as diagnostic markers of renal cancer. Dis. Markers. 2016;2016:1–10. doi: 10.1155/2016/3693096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zill O.A., Banks K.C., Fairclough S.R., et al. The landscape of actionable genomic alterations in cell-free circulating tumor DNA from 21,807 advanced cancer patients. Clin. Cancer Res. 2018;24:3528–3538. doi: 10.1158/1078-0432.ccr-17-3837. [DOI] [PubMed] [Google Scholar]
- 138.De Mattos-Arruda L., Mayor R., Ng C.K.Y., et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015;6:8839. doi: 10.1038/ncomms9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nassiri F., Chakravarthy A., Feng S., et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat. Med. 2020;26:1044–1047. doi: 10.1038/s41591-020-0932-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hoang-Xuan K., Bessell E., Bromberg J., et al. Diagnosis and treatment of primary CNS lymphoma in immunocompetent patients: guidelines from the European Association for Neuro-Oncology. Lancet Oncol. 2015;16:322–332. doi: 10.1016/s1470-2045(15)00076-5. [DOI] [PubMed] [Google Scholar]
- 141.Miller A.M., Shah R.H., Pentsova E.I., et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature. 2019;565:654–658. doi: 10.1038/s41586-019-0882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cohen J.D., Javed A.A., Thoburn C., et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc. Natl. Acad. Sci. U S A. 2017;114:10202–10207. doi: 10.1073/pnas.1704961114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Guler G.D., Ning Y., Ku C.-J., et al. Detection of early stage pancreatic cancer using 5-hydroxymethylcytosine signatures in circulating cell free DNA. Nat. Commun. 2020;11:5270. doi: 10.1038/s41467-020-18965-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Widschwendter M., Zikan M., Wahl B., et al. The potential of circulating tumor DNA methylation analysis for the early detection and management of ovarian cancer. Genome Med. 2017;9:116. doi: 10.1186/s13073-017-0500-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Qin Y., Wu C.W., Taylor W.R., et al. Discovery, validation, and application of novel methylated DNA markers for detection of esophageal cancer in plasma. Clin. Cancer Res. 2019;25:7396–7404. doi: 10.1158/1078-0432.ccr-19-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Provenzale D., Ness R.M., Llor X., et al. NCCN guidelines insights: colorectal cancer screening, version 2.2020: featured updates to the NCCN guidelines. J. Natl. Compr. Cancer Netw. 2020;18:1312–1320. doi: 10.6004/jnccn.2020.0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Telli M.L., Gradishar W.J., Ward J.H. NCCN guidelines updates: breast cancer. J. Natl. Compr. Cancer Netw. 2019;17:552–555. doi: 10.6004/jnccn.2019.5006. [DOI] [PubMed] [Google Scholar]
- 148.Partridge E.E., Abu-Rustum N., Giuliano A., et al. Cervical cancer screening. J. Natl. Compr. Cancer Netw. 2014;12:333–341. doi: 10.6004/jnccn.2014.0035. [DOI] [PubMed] [Google Scholar]
- 149.Wood D.E., Kazerooni E., Baum S.L., et al. Lung cancer screening, version 1.2015. J. Natl. Compr. Cancer Netw. 2015;13:23–34. doi: 10.6004/jnccn.2015.0006. [DOI] [PubMed] [Google Scholar]
- 150.Liu M.C., Jamshidi A., Venn O., et al. American Society of Clinical Oncology; 2019. Genome-wide Cell-free DNA (cfDNA) Methylation Signatures and Effect on Tissue of Origin (TOO) Performance. [Google Scholar]
- 151.Klein E.A. Clinical validation of a targeted MethylationBased multi-cancer early detection test. AACR. 2021:1167–1177. doi: 10.1016/j.annonc.2021.05.806. [DOI] [PubMed] [Google Scholar]
- 152.Beer T.M., McDonnell C.H., Nadauld L., et al. Interim results of PATHFINDER, a clinical use study using a methylation-based multi-cancer early detection test. J. Clin. Oncol. 2021;39:3010. doi: 10.1200/jco.2021.39.15_suppl.3010. [DOI] [Google Scholar]
- 153.Pepe M.S., Etzioni R., Feng Z., et al. Phases of biomarker development for early detection of cancer. J. Natl. Cancer Inst. 2001;93:1054–1061. doi: 10.1093/jnci/93.14.1054. [DOI] [PubMed] [Google Scholar]
- 154.Liu M.C. Transforming the landscape of early cancer detection using blood tests—commentary on current methodologies and future prospects. Br. J. Cancer. 2021;124:1475–1477. doi: 10.1038/s41416-020-01223-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Reinert T., Henriksen T.V., Christensen E., et al. Analysis of plasma cell-free DNA by ultradeep sequencing in patients with stages I to III colorectal cancer. JAMA Oncol. 2019;5:1124–1131. doi: 10.1001/jamaoncol.2019.0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Christensen E., Birkenkamp-Demtröder K., Sethi H., et al. Early detection of metastatic relapse and monitoring of therapeutic efficacy by ultra-deep sequencing of plasma cell-free DNA in patients with urothelial bladder carcinoma. J. Clin. Oncol. 2019;37:1547–1557. doi: 10.1200/jco.18.02052. [DOI] [PubMed] [Google Scholar]
- 157.Razavi P., Li B.T., Brown D.N., et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019;25:1928–1937. doi: 10.1038/s41591-019-0652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sanchez C., Snyder M.W., Tanos R., et al. New insights into structural features and optimal detection of circulating tumor DNA determined by single-strand DNA analysis. NPJ Genomic Med. 2018;3:31. doi: 10.1038/s41525-018-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Eraslan G., Avsec Ž., Gagneur J., Theis F.J. Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet. 2019;20:389–403. doi: 10.1038/s41576-019-0122-6. [DOI] [PubMed] [Google Scholar]
- 160.Ko J., Baldassano S.N., Loh P.-L., et al. Machine learning to detect signatures of disease in liquid biopsies – a user's guide. Lab. Chip. 2018;18:395–405. doi: 10.1039/c7lc00955k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sharif M.I., Li J.P., Naz J., Rashid I. A comprehensive review on multi-organs tumor detection based on machine learning. Pattern Recognit. Lett. 2020;131:30–37. doi: 10.1016/j.patrec.2019.12.006. [DOI] [Google Scholar]
- 162.Beshnova D., Ye J., Onabolu O., et al. De novo prediction of cancer-associated T cell receptors for noninvasive cancer detection. Sci. Transl. Med. 2020;12:eaaz3738. doi: 10.1126/scitranslmed.aaz3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Dejea C.M., Fathi P., Craig J.M., et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science. 2018;359:592–597. doi: 10.1126/science.aah3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Morgillo F., Dallio M., Della Corte C.M., et al. Carcinogenesis as a result of multiple inflammatory and oxidative hits: a comprehensive review from tumor microenvironment to gut microbiota. Neoplasia. 2018;20:721–733. doi: 10.1016/j.neo.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Thomas R.M., Jobin C. Microbiota in pancreatic health and disease: the next Frontier in microbiome research. Nat. Rev. Gastroenterol. Hepatol. 2020;17:53–64. doi: 10.1038/s41575-019-0242-7. [DOI] [PubMed] [Google Scholar]
- 166.Janney A., Powrie F., Mann E.H. Host-microbiota maladaptation in colorectal cancer. Nature. 2020;585:509–517. doi: 10.1038/s41586-020-2729-3. [DOI] [PubMed] [Google Scholar]
- 167.Poore G.D., Kopylova E., Zhu Q., et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature. 2020;579:567–574. doi: 10.1038/s41586-020-2095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Liu C., Zhao J., Tian F., et al. Low-cost thermophoretic profiling of extracellular-vesicle surface proteins for the early detection and classification of cancers. Nat. Biomed. Eng. 2019;3:183–193. doi: 10.1038/s41551-018-0343-6. [DOI] [PubMed] [Google Scholar]
- 169.Shurtleff M.J., Temoche-Diaz M.M., Schekman R. Extracellular vesicles and cancer: caveat lector. Annu. Rev. Cancer Biol. 2018;2:395–411. doi: 10.1146/annurev-cancerbio-030617-050519. [DOI] [Google Scholar]
- 170.Fehlmann T., Kahraman M., Ludwig N., et al. Evaluating the use of circulating MicroRNA profiles for lung cancer detection in symptomatic patients. JAMA Oncol. 2020;6:714–723. doi: 10.1001/jamaoncol.2020.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ying L., Du L., Zou R., et al. Development of a serum miRNA panel for detection of early stage non-small cell lung cancer. Proc. Natl. Acad. Sci. U S A. 2020;117:25036–25042. doi: 10.1073/pnas.2006212117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhou J., Yu L., Gao X., et al. Plasma microRNA panel to diagnose hepatitis B virus-related hepatocellular carcinoma. J. Clin. Oncol. 2011;29:4781–4788. doi: 10.1200/jco.2011.38.2697. [DOI] [PubMed] [Google Scholar]
- 173.Kahraman M., Röske A., Laufer T., et al. MicroRNA in diagnosis and therapy monitoring of early-stage triple-negative breast cancer. Sci. Rep. 2018;8:11584. doi: 10.1038/s41598-018-29917-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Snoek B.C., Verlaat W., Babion I., et al. Genome-wide microRNA analysis of HPV-positive self-samples yields novel triage markers for early detection of cervical cancer. Int. J. Cancer. 2019;144:372–379. doi: 10.1002/ijc.31855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tit A., Camilla A., Angela A., et al. 2017. European Guide on Quality Improvement in Comprehensive Cancer Control. [Google Scholar]
- 176.Pashayan N., Morris S., Gilbert F.J., Pharoah P.D.P. Cost-effectiveness and benefit-to-harm ratio of risk-stratified screening for breast cancer: a life-table aaamodel. JAMA Oncol. 2018;4:1504–1510. doi: 10.1001/jamaoncol.2018.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yeh J.M., Hur C., Ward Z., et al. Gastric adenocarcinoma screening and prevention in the era of new biomarker and endoscopic technologies: a cost-effectiveness analysis. Gut. 2016;65:563–574. doi: 10.1136/gutjnl-2014-308588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ten Haaf K., Jeon J., Tammemägi M.C., et al. Risk prediction models for selection of lung cancer screening candidates: a retrospective validation study. PLoS Med. 2017;14:e1002277. doi: 10.1371/journal.pmed.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pashayan N., Pharoah P.D.P. Overestimation of the benefit-to-harm ratio of risk-based mammography screening in the United Kingdom-reply. JAMA Oncol. 2019;5:428–429. doi: 10.1001/jamaoncol.2018.6504. [DOI] [PubMed] [Google Scholar]
- 180.Esserman L.J., LaCroix A.Z. Precision risk-based screening might maximize benefit and minimize harm. Nat. Rev. Clin. Oncol. 2018;15:661–662. doi: 10.1038/s41571-018-0093-0. [DOI] [PubMed] [Google Scholar]
- 181.Callender T., Emberton M., Morris S., et al. Polygenic risk-tailored screening for prostate cancer: a benefit-harm and cost-effectiveness modelling study. PLoS Med. 2019;16:e1002998. doi: 10.1371/journal.pmed.1002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.So J.B.Y., Kapoor R., Zhu F., et al. Development and validation of a serum microRNA biomarker panel for detecting gastric cancer in a high-risk population. Gut. 2021;70:829–837. doi: 10.1136/gutjnl-2020-322065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Payne K., Gavan S.P., Wright S.J., Thompson A.J. Cost-effectiveness analyses of genetic and genomic diagnostic tests. Nat. Rev. Genet. 2018;19:235–246. doi: 10.1038/nrg.2017.108. [DOI] [PubMed] [Google Scholar]
- 184.Dillon M., Flander L., Buchanan D.D., et al. Family history-based colorectal cancer screening in Australia: a modelling study of the costs, benefits, and harms of different participation scenarios. PLoS Med. 2018;15:e1002630. doi: 10.1371/journal.pmed.1002630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kastelein F., van Olphen S., Steyerberg E.W., et al. Surveillance in patients with long-segment Barrett's oesophagus: a cost-effectiveness analysis. Gut. 2015;64:864–871. doi: 10.1136/gutjnl-2014-307197. [DOI] [PubMed] [Google Scholar]
- 186.Cadier B., Bulsei J., Nahon P., et al. Early detection and curative treatment of hepatocellular carcinoma: a cost-effectiveness analysis in France and in the United States. Hepatology. 2017;65:1237–1248. doi: 10.1002/hep.28961. [DOI] [PubMed] [Google Scholar]
- 187.Kim J.J., Burger E.A., Sy S., Campos N.G. Optimal cervical cancer screening in women vaccinated against human papillomavirus. J. Natl. Cancer Inst. 2017;109:djw216. doi: 10.1093/jnci/djw216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.