

Abstract
Neuroblastoma (NB) is an enigmatic and deadliest pediatric cancer to treat. The major obstacles to the effective immunotherapy treatments in NB are defective immune cells and the immune evasion tactics deployed by the tumor cells and the stromal microenvironment. Nervous system development during embryonic and pediatric stages is critically mediated by non-coding RNAs such as micro RNAs (miR). Hence, we explored the role of miRs in anti-tumor immune response via a range of data-driven workflows and in vitro & in vivo experiments. Using the TARGET, NB patient dataset (n=249), we applied the robust bioinformatic workflows incorporating differential expression, co-expression, survival, heatmaps, and box plots. We initially demonstrated the role of miR-15a-5p (miR-15a) and miR-15b-5p (miR-15b) as tumor suppressors, followed by their negative association with stromal cell percentages and a statistically significant negative regulation of T and natural killer (NK) cell signature genes, especially CD274 (PD-L1) in stromal-low patient subsets. The NB phase-specific expression of the miR-15a/miR-15b-PD-L1 axis was further corroborated using the PDX (n=24) dataset. We demonstrated miR-15a/miR-15b mediated degradation of PD-L1 mRNA through its interaction with the 3'-untranslated region and the RNA-induced silencing complex using sequence-specific luciferase activity and Ago2 RNA immunoprecipitation assays. In addition, we established miR-15a/miR-15b induced CD8+T and NK cell activation and cytotoxicity against NB in vitro. Moreover, injection of murine cells expressing miR-15a reduced tumor size, tumor vasculature and enhanced the activation and infiltration of CD8+T and NK cells into the tumors in vivo. We further established that blocking the surface PD-L1 using an anti-PD-L1 antibody rescued miR-15a/miR-15b induced CD8+T and NK cell-mediated anti-tumor responses. These findings demonstrate that miR-15a and miR-15b induce an anti-tumor immune response by targeting PD-L1 in NB.
Keywords: neuroblastoma, miRNAs, PD-L1, natural killer cells, CD8+T cells, anti-tumor immune response, chemotherapy, patient-derived xenografts, survival
Graphical abstract
Neuroblastoma is one of the deadliest pediatric cancers and shows resistance to therapy due to a diminished anti-tumor immune response. Patients with higher PD-L1, reduced miR-15a, and miR-15b are further associated with poor survival. Our findings demonstrate that miR-15a and miR-15b induce an anti-tumor immune response by targeting PD-L1 in neuroblastoma.
Introduction
Neuroblastoma (NB) is the most common pediatric cancer affecting children younger than 5 years.1, 2, 3 NB develops from immature nerve cells, most commonly in adrenal glands situated above the kidney. NB accounts for 15% of childhood cancer-related mortality, and approximately 50% of children treated for high-risk NB have more aggressive tumor relapse with less than 20% 5-year overall survival.4, 5, 6, 7 During embryogenesis, multipotent neural crest cells differentiate into multiple cell types, including sympathetic neurons, adrenal medullary cells, and modified postganglionic sympathetic neurons.8,9 The deregulation of signaling pathways involved in the differentiation of neural crest cells leads to NB development in the sympathetic nervous system or medullar region of the adrenal glands. NBs are highly heterogeneous tumors consisting of various sub-cell types, and they display various genomic alterations, including MYCN amplification,10 different DNA ploidy patterns,11 deletion in the short arm of chromosome 1,12 a gain of chromosome 17q,13 and chromosome 11q deletion.14 These genomic alterations, along with International NB Staging System, stage histopathology and age are used to classify NB patients into risk groups.15,16 The high-risk NB patients are challenging to treat and require high doses of chemotherapy and radiotherapy in the clinic.17, 18, 19 The chances of tumor regression (>50%) and death rate (41.7% compared with 0% in low-risk and 5.2% in intermediate-risk patients) in high-risk patients are higher than other groups.19,20 Moreover, the use of intense treatment regimens has significant adverse effects on patients’ quality of life.
Several investigators have tried different therapeutic approaches like high-dose chemotherapy, surgery, radiation therapy, stem cell transplantation, retinoid therapy, and immunotherapy to improve clinical outcomes in aggressive high-risk NB patients.17,19,21,22 Immunotherapy has recently gained traction in high-risk NB treatment and improved survival rates in patients.22,23 Immunotherapy treatment harnesses the CD8+T and natural killer (NK) cells to recognize, target, and eradicate malignant cells.24, 25, 26 The immunotherapy approach of using chimeric monoclonal anti-disialoganglioside (GD2) antibody dinutuximab has shown some success in NB clinical trials but is associated with severe side effects including neuropathic pain, infection, infusion-related reactions, capillary leak syndrome, decreased sensation, and paresthesia.23,27 In NB, the levels of infiltrating CD8+T and NK cells correlate with therapy response.28,29 Elevated levels of programmed death-ligand 1 (PD-L1 or CD274) have been found in NB tumors.28,30,31 Two recent reports suggested that higher PD-L1 expression positively correlates with worse outcomes in NB patients.30,31 Patients with high PD-L1 expression had an increased risk of NB relapse and less overall survival compared with patients with low PD-L1 expression.30, 31, 32, 33 Therefore, therapies targeting PD-L1 could strengthen anti-tumor immunity and offer a promising strategy to treat high-risk NB.
Checkpoint inhibitor drugs that inhibit the PD-L1/PD-1 pathway have shown promising results in boosting immune responses and improving clinical outcomes in cancer patients. Food and Drug Administration (FDA) approval of PD-L1/PD-1 targeting drugs to treat cancer in recent years has attracted considerable interest in the discovery and development of improved PD-L1/PD-1 signaling pathway inhibitors. However, many patients do not respond well to these therapies and develop resistance and relapse.30,31,34 Therefore, there is a need for novel approaches that target PD-L1 upstream signaling pathways to overcome these challenges.
Micro RNAs (miRs) are key upstream regulators in NB.35, 36, 37 Several miRs have been reported to have roles in NB progression recently.38, 39, 40 For example, suppression of MYCN targeting tumor-suppressor miRNA let-7 by LIN28B upregulates MYCN and promotes proliferation in non-malignant neuroblasts.41 Amplification and overexpression of LIN28B or chromosomal loss of let-7 drives NB development and growth.42 Moreover, downregulation of miR-34a,43 miR-34b,44 miR-124b,45 miR-145,46 miR-542,47 miR-204,48 and miR-27b,49 is strongly associated with NB progression and therapy resistance. We recently showed that exosomal miR-155 induces chemotherapy resistance by targeting TERF1, an inhibitor of telomerase complex activation within the tumor microenvironment.50 Several investigators have reported that a distinct set of miRs activate/inhibit the immune system at multiple levels, including regulation of the checkpoint molecule PD-L1.50, 51, 52 For instance, miR-424(322) inhibits PD-L1 and CD80 expression, which promotes CD8+ T cells activation and reverses chemoresistance. The suppression of miR-21 in tumor-associated macrophages (TAMs) reprograms TAMs to proinflammatory anti-tumor type, improves CD8+T cell tumor infiltration and inhibits angiogenesis. Our previous study shows that NB cells transfer miR-21 in exosomes to monocytes, which upregulates oncogenic miR-155 expression in monocytes. Monocytes transfer this miR-155 to NB cells via exosomes and induce chemotherapy resistance.50,53,54
Several miR-based therapies, for example, tumor-suppressor miRs, miR-34 and miR-122, are in clinical development to treat cancer or other diseases.55 Nervous system development during embryonic and pediatric stages requires precise epigenetic regulation of proliferation and differentiation pathways that are critically mediated by non-coding RNAs (ncRNAs). These ncRNAs, primarily miRs, also contribute to several tumor suppressive and oncogenic pathways within the tumor microenvironment. However, how miRs influence T and NK cell function in NB remains poorly understood. In this study, we found that infiltration of fewer T and NK cells, diminished miR-15a-5p, miR-15b-5p (hereafter miR-15a, miR-15b) levels, and enhanced PD-L1 expression were positively associated with poor survival in NB patients and patient-derived xenograft (PDX) tumors. The miR-15a and miR-15b activate CD8+T and NK cell mediated anti-tumor immune response against NB by PD-L1 mRNA degradation through in vitro and in vivo approaches.
Results
Higher PD-L1 and reduced miR-15a and miR-15b associate with higher tumor-infiltrating lymphocyte exhaustion signatures in NB patients and PDX tumors
To prioritize tumor suppressor like miRs, we identified statistically significant downregulated miR (precursor miRs) genes using the survival status in the TARGET (Therapeutically Applicable Research to Generate Effective Treatments), NB patient (n=249) dataset as depicted in the Volcano plot Figure 1A. MiR-15a and miR-15b were selected to investigate further their tumor suppression mechanisms, especially their role in immune evasion. Immune- (NK cells, T lymphocytes), and stromal- [extracellular matrix (ECM), cancer-associated fibroblasts (CAFs), and mesenchymal stromal cells (MSCs)] cells are an area of current interest in the context of immune evasion mediated by tumor microenvironment in NB. The stromal cells comprising ECM, CAFs, and MSCs suppress effector immune cell activation and infiltration at the tumor site leading to immune evasion. Towards this, the 249 TARGET patient samples were classified into six categories using a multi-attribute criterion based on (1) overall survival (OS)- (2) event-free survival (EFS)- status (deceased/living), (3) INSS stage, (4) histology of tumor samples (favorable/unfavorable), (5) risk group (high/low), and (6) stroma cell percentages (low/high) as depicted in the heatmap top annotation (Figure 1B). Furthermore, to investigate the regulatory accepts of miR-15a and miR-15b against immune effectors like T- & NK-cells, we performed differential- and co-expression analyses against the Tumor Inflammation Signatures (TIS) like T-cell exhaustion and T- & NK- cell abundance, which was presented as heatmaps (Figure 1B-D). The TIS signature derived using 9000 tumor samples in TCGA comprised an 18-gene signature associated with four immune response processes108,33. TIS serves as an immune-phenotyping tool and can be a proxy for critical immune mediators. T cell/NK cell abundance (NKG7, CD8A & HLA-E) and T cell exhaustion (CD274, CD276, TIGIT, PDCD1LG2 & LAG3), which constitute two of the four immune processes, were deemed particularly relevant for investigation of the mechanistic aspects of miR-15a and miR-15b mediated antitumor immune responses. Our preliminary observations from the TARGET dataset (n=249) differential- and co-expression analyses demonstrated significant downregulation of tumor suppressor miR-15a and miR-15b (mean scaled expression = -0.28 & -0.669) among the two stromal-high cohorts. The immune evasion functions were further delineated by assessing the correlations with the eight TIS T-cell and NK-cell function signature genes across the six tumor categories based their aggressiveness (Figure 1B). Among the six clinical attributes used for patient stratification, the stromal cell percentage measure is the most relevant and conceptually related to immune evasion. It has demonstrated that tumors with lower stromal cell percentages (primary tumors) have potent antitumor immune responses. Higher stromal cell percentages (aggressive tumors) were associated with pro-tumor immune evasion phenotypes. Further, we determined the correlation between the mean expression values of miR-15a, miR-15b, and T- and NK- cell gene signatures in TARGET, NB patient cohorts. The expression of miR-15a and miR-15b is lower in stromal-high & aggressive tumors, as shown in Figure 1C. The co-expression analyses further corroborated these findings with stromal-low & primary tumors demonstrating high miR-15a and miR-15b expression and high mRNA inhibitory activity observed as a significant negative correlation (mean correlation z-value: -1.943) with CD274, the TIS signature genes with the most significant differential co-expression and a clear signal. Stromal-high & aggressive tumors with little or no miR-15a, and miR-15b expression/activity demonstrated relatively insignificant negative correlations with CD274 (mean correlation z-value: -1.461) (Figure 1C). We further performed a similar parallel analysis using PDX tumor data available from the dataset of genomic profiling of childhood tumor PDX models109. The 24 sample PDX dataset classified into six categories with varying aggressiveness using clinical attributes Phase and Age (Figure 1D). The miR-15a demonstrated high expression for the two least aggressive categories (mean: 0.5679 & 0.463), while its most significant target, CD274, showed suppressed expressions (mean: -0.399 & -0.3206) in the same two least aggressive categories. These observations from the TARGET dataset further corroborated with the PDX dataset reinforce our hypotheses that miR-15a/miR-15b-CD274 axis might contribute to immune evasion and tumor aggressiveness.
Figure 1.

High-risk NB patients and PDX tumors show higher tumor-infiltrating lymphocyte exhaustion signatures together with reduced miR-15a and miR-15b
(A) The Volcano plot depicting top-ranked up- (red) and down-regulated (blue) miRs (miR genes) in deceased versus living cohorts. The log-transformed TARGET dataset (n=249) was analyzed to prioritize miRs with statistically significant fold changes. The vertical red lines depict the fold change threshold of 1.77, corresponding to the log 2-fold change of 0.25. The horizontal red line indicates the P-value threshold of 0.03. The study prioritized miR-15a and miR-15b as tumor suppressor miRs for further characterization and validation. (B) The heatmap depicting the differential expression of miR-15a, miR-15b, and the T- and NK- cell signature genes across the TARGET dataset patient cohorts. The 249 TARGET dataset samples were classified into six categories with different aggressiveness based on a multi-attribute criterion depicted in the top annotation. The mean miR-15a and miR-15b log-transformed & scaled expression values for stromal-high and stromal-less tumor categories were -0.28 & -0.669. Two of the six categories were significantly enriched with stromal-high tumors. The values in stromal-high tumors were significantly lower than the corresponding mean values across the four other categories. This indicates potential antitumor immune response functions of miR-15a, and miR-15b. (C) Heat map of Pearson correlation Z-values between the mean miR-15a & miR-15b expression values and the eight genes of T- and NK- cell signatures. CD274 showed a strong signal with significant negative correlation profiles in stromal-low tumors versus the stromal-high tumors. This is consistent with the previous observations that miR-15a, miR-15b are tumor suppressors whose expression is higher in stromal-low tumors leading to better mR inhibitory functions captured as the significant negative correlations with target mRNA in these cohorts. (D) Heatmap depicting the differential expression of miR-15a, miR-15b, and the T- and NK- cell signature genes across the PDX dataset patient cohorts. The 24 PDX dataset samples were classified into six categories with different aggressiveness based on the Phase and Age depicted in the heatmap’s top annotation. The mean miR-15a scaled expression values for the two less aggressive categories were high (0.5679 & 0.463), while its target mRNA CD274 demonstrated suppressed expression (-0.399 & -0.3206), corroborating our hypotheses that miR-15a/miR-15b-CD274 axis might contribute to immune evasion and tumor aggressiveness. (E) Kaplan-Meier curves showing event-free and overall survival probability rates with different levels of PD-L1 in NB patients. (F) Immunofluorescence images of PD-L1 on NB patient tumor and normal tissue microarrays photographed at 5X. (G) Western blotting analysis of PD-L1 in whole lysates of PDX tumors of NB patients collected at the diagnosis and progression stages. Images were captured at low and higher exposures (exp). (H) IHC staining of PD-L1 in PDX tumors of NB patients at the diagnosis and progression stages.
From the TIS signature genes, we observed a remarkable, highly statistically significant correlation between increased PD-L1 expression and poor survival in the TARGET, NB patients (Figures 1E and S1A). We further screened the relationship between miR-15a and survival status from other NB patient datasets. We observed that the expression of DLEU2, a host gene of miR-15a (GSE16476, n=88), is associated with relapse-free and overall survival in NB patients (GSE16476, n = 88), (Figure S1B). Similarly, high miR-15b expression is significantly associated with better progression-free survival (p=0.048) and overall survival (p=0.056) in NB patients (Tumor NB ALT-Westermann−144-tpm-gencode19 in R2 database) (Figure S1C). Further, as given in Figure 1D-G, higher levels of miR-15a found in low-risk vs. high-risk. Later, we performed a tissue microarray (TMA) analysis to examine the PD-L1 protein expression level in NB and normal tissues. As shown in Figures 1F and S1J, PD-L1 protein expressed much higher in NB tumor tissues than in non-neoplastic normal tissues. Moreover, we checked PD-L1 expression in NB PDX models (derived from patient tumor cells collected at the diagnosis and relapse stages) that we established by expansion in nude mice, as illustrated in Figure S1K. In western blotting analysis, we saw a trend of PD-L1 upregulation in relapse-specific PDX tumors versus those taken at diagnosis (Figure 1G, lanes 3, 6, 7 versus 1, 2, 4, 5). Next, immunohistochemical (IHC) staining of PD-L1 showed similar results in PDX tumors (Figures 1H and S1L). Furthermore, high levels of PD-L1 expression were detected on the surface of GD2+ tumor cells isolated from PDX tumor tissues from NB patients (Figures S1M and S1N). These observations demonstrate that higher PD-L1 and diminished miR-15a and miR-15b are associated with adverse clinical features and poor survival in NB patients.
MiR-15a and miR-15b target PD-L1 in NB cells
To evaluate the role of miR-15a and miR-15b in PD-L1 regulation if any, we first transfected SK-N-AS, NB-19, and SK-N-BE(2) cell lines with precursors of miR-15a, miR-15b, and non-targeting control miRs for 48 h. The quantification of miRs is shown in Figure S2A. Cells showed a significant reduction of PD-L1 mRNA upon receiving miR mimics (Figure 2A). A similar decrease was also seen in PD-L1 total protein levels in miRs transfected cells (Figures 2B and S2B), and the downregulation was more pronounced in SK-N-AS and NB-19 cells than SK-N-BE(2). Hence, we chose these two cell lines for further studies. To explore the mechanistic aspects of PD-L1 regulation by miRs, we checked if any complementary sequence exists between PD-L1 mRNA and miRs using in silico analysis tools including TargetScan. Figure S2C shows the presence of complementary binding sites between miR-15a, miR-15b, and 3′UTR of PD-L1 mRNA. To investigate whether these miRs regulate PD-L1 through 3′UTR, we cloned PD-L1-3′UTR (NM_014,143.4) wild-type region and seven base mutations at nucleotides 567–573 in the miR binding sites of the 3′UTR region referred to as 3′UTR mutant into a commercially available luciferase reporter vector pEZX-MT06 (Cat. #HmiT117860-MT06, Cat. #CS-HmiT117860-MT06-01; GeneCopoeia, Rockville, MD). SK-N-AS cells were transfected with precursors of miR-15a, miR-15b, or control miR oligonucleotides together with PD-L1-3′UTR wild-type or mutatant constructs for 48h. MiR-15a and miR-15b showed a reduction in luciferase activity with PD-L1-3′UTR wild type but not with PD-L1 mutant (Figure 2C). These observations indicate that miR-15a and miR-15b target PD-L1 through 3′UTR (Figure 2A-C). Further, we evaluated whether surface PD-L1 expression is also regulated by these miRs. We analysed surface PD-L1 expression in miR-15a and miR-15b transfected SK-N-AS, NB-19 cells, and an SK-N-B(E)2 cell line stably expressing miR-15a by flow cytometry. As shown in Figures 2D–2F, reduced surface PD-L1 expression was found, suggesting that miR-15a and miR-15b target PD-L1 in NB. The representative flow cytometric plots showing fluorescence minus one (FMO) control to omit background signals are shown in Figure S2D. The quantification of miR-15a in stable miR-15a expressing SK-N-B(E)2 cell line is shown in Figure S2E. To further confirm that the effect is not off-target, we silenced endogenous miR-15a and miR-15b in NB cells by transfecting antisense oligonucleotides (miR inhibitors). Conversely, with the introduction of miR inhibitors, cells exhibited increased PD-L1 expression (Figures 2G and S2F). The quantification of miR-15a and miR-15b in NB cells after transfection with miR inhibitors is shown in Figure S2G. Next, we performed Ago2 immunoprecipitation to see whether these miRs bind to PD-L1 mRNA in NB. A schematic presentation of Ago2-RNA-IP is depicted in Figure S2H. As shown in Figure 2H, anti-Ago2 antibodies successfully immunoprecipitated PD-L1 mRNA but not GAPDH mRNA. Further, Ago2 also immunoprecipitated miR-15a and miR-15b but not U6, a small nucleolar RNA, suggesting that miR-15a and miR-15b regulate PD-L1 via direct interaction with PD-L1 mRNA in NB cells.
Figure 2.

MiR-15a and miR-15b target PD-L1 in NB cells
(A,B) A RT-qPCR quantification graph for PD-L1 mRNA (A), western blotting for PD-L1 total protein (B) in NB cells transfected with miR-15a, miR-15b, or control (ctrl) miRs for 48 h. (C) Luciferase reporter assay in SK-N-AS cells cotransfected with either luciferase reporter vector containing the PD-L1-3’UTR wild-type or mutant in the presence or absence of miR-15a and miR-15b for 48 h, followed by measuring the luciferase activity. (D-F) Representative flow cytometric plots for PD-L1 surface expression analyzed by using anti-PD-L1 phycoerythrin (PE) conjugated antibody in SK-N-AS and NB-19 cells transfected with miR-15a, miR-15b or ctrl miRs for 48 h (D and E) or miR-15a expressing stable SK-N-B(E)2 cells (F). (G) Western blotting for PD-L1 total protein in NB cells transfected with inhibitors of miRs such as α-miR-15a, α-miR-15b or α- ctrl miRs for 48 h. (H) A RT-qPCR quantification graph for Ago2-occupied PD-L1 mRNA (first panel), Ago2-occupied miR-15a (second panel), and Ago2-occupied miR-15b (third panel). SK-N-BE(2) cells were transfected with miR-15a, miR-15b, or ctrl miRs for 48 h followed by immunoprecipitation (IP) with α-Ago2 antibody. Ago2 bound RNA complexes were eluted, purified, and quantified for Ago2 bound mRNA and miRs by RT-qPCR using TaqMan assays. GAPDH mRNA and U6, a non-coding small nuclear RNA, were used as non-specific controls. The data were compared with the control IgG-bound mRNA or miR and set to one for normalization. Data represent the mean ± standard error of three to four independent biological experiments. Statistical analyses were performed using a two-sided unpaired t-test. *p < 0.001.
MiR-15a and miR-15b promote CD8+T cell proliferation and induce CD8+T cell-mediated NB cytotoxicity
We tested the effect of miR-15a and miR-15b on CD8+T cell-mediated cytotoxicity against NB cells. NB cells were treated with miR-15a and miR-15b mimics for 24 h and cocultured with activated CD8+T cells for an additional 24 h. As shown in Figures 3A and 3B, a higher percentage of CD8+T cells showed intracellular granzyme B and perforin expression after coculture (E:T=1:1) with miR-15a and miR-15b expressing SK-N-AS (Figures 3A, 3B top panels, and S3G), and NB-19 (Figures 3A, 3B, bottom panels, and S3H) cells. Next, we checked proliferation capacity and Ki-67 (a nuclear protein associated with cell proliferation) staining in cytotoxic CD8+T cells after coculture (E:T=1:1) with miR-15a and miR-15b expressing NB cells. Interestingly, CD3+CD8+T cells showed more proliferation (61% in miR-15a and miR-15b versus 33% control miRs, Figures 3C, S3I1, and S3I2) and higher Ki-67 expression (Figures 3D and S3J) in the presence of miR expressing SK-N-AS (Figures 3C and 3D, top panels), and NB-19 (Figures 3C and 3D, bottom panels) cells than control group cells. These results showed that NB cells expressing miR-15a and miR-15b induce proliferation in CD8+T cells. In the same experiments, we further checked the functional effect of activated CD8+ T cells on NB cytotoxicity by measuring cleaved caspase-3, an apoptosis marker in target NB cells. As shown in Figures 3E and S3K, caspase-3 cleavage increased from 6% to 16.2% and 22.8% (control miR versus miR-15a and miR-15b) in SK-N-AS cells (Figure 3E, top panel), and 19% to 24.7% and 25.5% (control versus miR-15a and miR-15b) in NB-19 cells (Figure 3E, bottom panel). These results suggest that miRs expressing NB cells were more prone to CD8+T cell-mediated apoptosis. Further, to confirm these findings, we cocultured activated CD8+T cells (E:T=5:1) with stable luciferase gene expressing miR-15a and miR15b treated SK-N-BE(2) cells for 24 h and performed the luciferase reporter assay as described in the materials and methods section. We found significantly reduced luciferase activity in miR expressing cells than in their control group, suggesting that miR-15a and miR-15b induce CD8+T-cell-mediated NB cytotoxicity (Figure 3F).
Figure 3.

MiR-15a and miR-15b promote CD8+T cell activation,proliferation and induce CD8+T cell-mediated NB cytotoxicity
(A–D) Representative flow cytometric plots of human peripheral CD8+ T cells showing the expression of intracellular Granzyme B (A), intracellular Perforin (B), surface CD3 and CD8 (C), and intracellular Ki-67 (D) after coculture (E:T=1:1) with miR-15a and miR-15b expressing SK-N-AS (A–D, top panels), and NB-19 (A–D, bottom panels) cells for 24 h. (E) Representative flow cytometric plots of miR-15a and miR-15b expressing SK-N-AS (E, top panel), and NB-19 (E, bottom panel) showing the expression of intracellular active caspase-3 upon coculture (E:T=1:1) with activated human CD8+ T cells for 24 h. (F) A quantification graph showing normalized luciferase activity in SK-N-BE(2) cells expressing miR-15a and miR-15b upon coculture (E:T=5:1) with activated CD8+ T cells for 24 h. Untouched CD8+ T cells were isolated from PBMCs of healthy human blood donors by negative selection using the MojoSort human CD8+ T cell Isolation Kit (BioLegend). For 7 days, CD8+ T cells were activated and expanded using human T-activator CD3/CD28 Dynabeads and used for coculture experiments with NB cells. NB cells were transfected with miRs for 24 h and used for coculture experiments with activated CD8+T cells for an additional 24 h. Non-targeting control (ctrl) miR cells served as a control group. Cells were permeabilized (intracellular), fixed, and stained for respective antibodies described in the materials and methods section and analyzed by flow cytometry. The percentage of Granzyme B+ (A), Perforin+ (B), CD3/CD8+ (C), ki67+ (D), and cleaved caspase-3+ cells (E) are shown in each of their respective plots. Data represent the mean ± standard error of three to five independent biological experiments. Statistical analyses were performed using a two-sided unpaired t-test.
MiR-15a and miR-15b promote NK cell activation and induce NK cell-mediated NB cytotoxicity
We tested the effect of miR-15a and miR-15b on NK cell-mediated anti-tumor immune response against NB cells. Representative flow cytometric pseudo color plots showing human NK cells purity before and after expansion ex vivo using irradiated K562-mbIL21 feeder cells are depicted in Figures S4A and S4B. The FMOs related to the experiments are shown in Figures S4C–S4E. First, NB cells were treated with miR-15a and miR-15b mimics for 48 h and cocultured with activated NK cells for an additional 5 h. NK cells displayed remarkably higher perforin expression upon coculture with miR expressing NB cells than control-miRs (Figures 4A, 4B, S4F, and S4G). Further, fluorescence intensity analysis showed increased accumulation of CD107a molecules on the NK cells placed on NB cells expressing miRs (17% to27.2% and 42.5%, SK-N-AS; 21.8% to31.3% and 37.7%, NB-19) (Figures 4C and S4H). These results show that miR-15a and miR-15b in NB cells might enhance the activation of NK cells.
Figure 4.

MiR-15a and miR-15b promote NK cell activation and NK cell-mediated NB cytotoxicity
(A–C) Representative flow cytometric plots of human NK cells showing the expression of intracellular perforin (A and B) and surface CD107a (C) after coculture with miR-15a and miR-15b expressing SK-N-AS (E:T=1:1) (A and C, top panels), NB-19 (E:T=0.25:1) (A and C, bottom panels), and miR-15a expressing stable SK-N-B(E)2 (E:T=1:1) (B) cells for 5 h. (D–H) Representative flow cytometric plots of miR-15a and miR-15b expressing SK-N-AS (D and F), NB-19 (E and G), and miR-15a expressing stable SK-N-B(E)2 cells (H) showing the expression of intracellular active caspase-3 (D and E) and the % of dead cells (F–H) upon coculture with or without activated human NK Cells for 5 h. (I) Phase-contrast microscope images of miR-15a expressing stable SK-N-B(E)2 cells showing dead cells upon coculture (E:T=1:1) with or without activated NK cells for 5 h. Untouched NK cells were isolated from PBMCs of healthy human blood donors by negative selection using the MojoSort human NK Cell Isolation Kit. NK cells were propagated and activated ex vivo using irradiated K562-mbIL21 feeder cells and IL-2 for 14 days and used for coculture experiments with NB cells. NB cells stably expressing miRs or overexpressing them by transfection were used for coculture experiments with NK cells. Non-targeting control (ctrl) miR cells served as a control group. Cells were permeabilized (intracellular), fixed, and stained for respective antibodies described in the materials and methods section and analyzed by the Attune Acoustic Focusing Flow Cytometer. The percentage of Perforin+ (A and B), CD107+ (C), cleaved caspase-3+ (D and E), and dead cells (F–H) are shown in each of their respective plots. Data represent the mean ± standard error of three to five independent biological experiments. Statistical analyses were performed using a two-sided unpaired t-test.
To understand the functional consequences of NK cell activation on tumor cell cytotoxicity, we assayed for cleaved caspase-3, a marker for cell death, and the number of dead cells in miR-15a and miR-15b expressing SK-N-AS and NB-19 cells after coculture (E:T=1:1 for SK-N-AS and E:T=0.25:1 for NB-19) with or without NK cells for 5 h. NK cells significantly induced caspase-3 cleavage from 27.1% to 46.9% and 44.6% in SK-N-AS and 49% to 62% and 61% in NB-19 cells in the presence of miRs compared with control-miRs (Figures 4D and 4E). Quantification of cleaved caspase-3 is shown in Figures S4I and S4J. Next, we detected dead NB cells using Zombie Aqua, an amine-reactive, a water-soluble fluorescent dye that stains dead cells due to their compromised membranes. As shown in Figure 4F, NK cells coculture with SK-N-AS cells induced a dose-dependent increase of dead NB cells from 3.2% to 25.4% and 47.1% at an E&T ratio of 0.5: 1 and 1:1. The overexpression of miR-15a and miR-15b further sensitized SK-N-AS cells to NK cell-mediated cytotoxicity. A similar trend of NK cell-mediated cytotoxicity was further noticed in miR expressing NB-19 (E:T=0.25:1 and 0.5:1) and SK-N-B(E)2 (E:T=1:1) cells (Figures 4G and 4H). Quantified NK cell-mediated NB cytotoxicity levels are given in Figures S4K–S4M. These results were further confirmed in phase-contrast images of miR-15a stably expressing SK-N-B(E)2 cells showing higher frequency of dead cells upon coculture with activated NK cells (Figures 4I, S4N, and S4O). These results collectively show that miR-15a and miR-15b promote NK cell-mediated release of cytotoxic granule perforin and cleaved caspase-3 activation, enhancing the efficacy of NK cell cytotoxicity against NB cells.
PD-L1 blockade rescues miR-15a and miR-15b mediated immune activation and cytokine secretion
We determined to test the role of PD-L1 in miR-15a and miR-15b mediated regulation of CD8+T and NK cell functions in NB. First, SK-N-AS cells were blocked with anti-PD-L1 or IgG control antibody for 24 h. followed by transfection with miR-15a and miR-15b mimics for an additional 24 h. As shown in Figure 5A, miR-15a and miR-15b effectively silenced (47% to 26% and 28.4%) surface PD-L1 in cells treated with control antibody but not PD-L1 antibody. These results confirmed an effective blockage of surface PD-L1 in SK-N-AS cells. These cells were cocultured (E:T=1:1) with CD8+T cells for 24 h, NK cells for 5 h and analyzed for activation markers granzyme B on CD8+T cells and CD107a on NK cells. As shown in Figures 5B, 5C, S5A, and S5B, blocking cell surface PD-L1 significantly enhanced granzyme B secretion from 40% to 54% in CD8+T cells and degranulation marker CD107a from 34.2% to 63.7% in NK cells. Consistent with previous results, SK-N-AS cells treated with miR-15a and miR-15b significantly increased granzyme B secretion (40% to46.7% and 49.3%, p=0.0034 and p=0.0008) in cocultured CD8+T cells. Further, we noticed a similar trend with CD107a (34.2% to 49.7% and 56%, p=0.0025 and p=0.0006) in cocultured NK cells in the control IgG antibody treatment group, but no significant difference in PD-L1 antibody treated group.
Figure 5.

Targeting PD-L1 is required for miR-15a- and miR-15b-mediated immune cell activation, and cytokine production
(A and D) Representative flow cytometric plots showing the surface PD-L1 expression in SK-N-AS cells that were blocked with anti-PD-L1 or IgG control antibody for 24 h followed by transfection with miR-15a and miR-15b mimics for an additional 24 h (A), and NB-19 cells treated with miR-15a, miR-15b mimics and PD-L1 siRNA for 48 h (D). (B, C, and E) Representative flow cytometric plots showing the expression of intracellular granzyme B in CD8+ T cells (B), and surface CD107a in NK cells (C and E) cocultured (24 h for CD8+ T cells, 5 h for NK cells) with miR-15a and miR-15b expressing SK-N-AS cells (E:T=1:1) treated with anti-PD-L1 antibody for 24 h (B, C) or NB-19 cells (E:T=0.25:1) treated with PD-L1 siRNA for 48 h (E). (F) Representative ELISA assay-based quantification graphs of IFN-γ in the culture medium of NK cells after coculture (E:T=1:1) with miR-15a and miR-15b expressing SK-N-AS cells pretreated with anti-PD-L1 antibody for 24 h. NB cells were treated with anti-PD-L1 antibody (15 μg/mL) for 24 h followed by miRs transfection for another 24 h (SK-N-AS) or PD-L1 siRNA (NB-19) for 48 h and used for coculture experiments with activated CD8+T (SK-N-AS) or NK (NB-19) cells. Control (ctrl) miR or IgG antibody treated cells served as a control group. Untouched CD8+ T cells and NK cells were isolated, expanded, and activated as previously described, and were used for coculture experiments with NB cells. Cells were permeabilized (intracellular), fixed, stained with antibodies as described in the materials and methods section, and analyzed by the Attune Acoustic Focusing Flow Cytometer. The percentage of surface PD-L1+ (A and D), intracellular granzyme B+ (B), and surface CD107a+ (C and E) are shown in each of their respective plots. Data represent the mean ± standard error of three to five independent biological experiments. Statistical analyses were performed using a two-sided unpaired t-test. ∗p < 0.05, ∗∗p < 0.001.
Next, to confirm the role of PD-L1 in miR-15a and miR-15b mediated anti-tumor response, we followed another approach of silencing PD-L1 using small interfering RNAs (siRNA). As shown in Figure 5D, PD-L1 siRNAs effectively silenced (47.3% to31.5%, p<0.05) surface PD-L1 in NB-19 cells compared with control siRNAs. These cells were used for coculture experiments with NK cells. NK cells showed a significant increase of CD107a expression upon coculture with miR-15a and miR-15b expressing NB-19 cells that are treated with control siRNAs but no significant difference in cells treated with PD-L1 siRNA (Figures 5E and S5C).These results further confirmed that PD-L1 suppression plays an important role in miR-15a and miR-15b-mediated immune cell activation. In addition, we analyzed the secretion of soluble factor interferon (IFN)-γ by ELISA in cell culture supernatants of effector NK cells coincubated (E:T=1:1) with miR-15a and miR-15b expressing target SK-N-AS cells and treated with control IgG or anti-PD-L1 antibody. Interestingly, we did not see a significant difference in INF-γ secretion in control miR versus miR-15a, miR-15b treated SK-N-AS cells and cocultured with NK cells. However, anti-PD-L1 antibody treatment increased INF-γ concentration in the media upon coculture with NK cells, which was further decreasing after miR-15a and miR-15b overexpression, implying that these miRs suppressed INF-γ secretion from NK cells (Figure 5F). Altogether, these results indicate that suppression of PD-L1 by miR-15a and miR-15b mediate immune cell activation and cytokine secretion response in NB.
MiR-15a regulates CD8+ T and NK cell-mediated anti-tumor immune response in NB in vivo
Based on these in vitro findings, we next sought to investigate the role of miR-15a and miR-15b in modulating immune response using an immune-competent C57BL/6 mice model. We chose miR-15a for our in vivo experiments as both miR-15a and miR-15b share a common complementary sequence with PD-L1 mRNA (Figure S2C). We subcutaneously injected C57/BL6 mice with murine stable NB975 cells expressing either GFP-control miR or GFP-miR-15a for 30 days and evaluated tumor growth. A significant (p<0.05) repression of tumor growth, weight, and volume were observed in the mice that received miR-15a cells compared with control miR cells (Figures 6A, 6B, and S6A). Tumor tissues were harvested, freshly prepared single-cell suspensions, and analyzed PD-L1 on GFP+ve tumor cells by flow cytometry. As shown in Figures 6C and 6D, tumor cells of the mice that received miR-15a cells showed a significant (p<0.05) reduction of surface PD-L1 (64.9%–33.4%) compared to control miR cells, suggesting that miR-15a targets PD-L1 in vivo. Next, CD8+T and NK cells infiltration was evaluated using a flow cytometer. As depicted in Figures 6E, 6F, S6B, and S6C, miR-15a tumors showed a significant increase in CD8+T cell infiltration (30.8% versus 45.8%, p=0.002), and Ki-67, a CD8+T cell proliferation marker from 6.73% to 9.48% (p=0.018) when compared with the control miR group. Further, the infiltrated CD8+T cells in miR-15a-specific tumors produced higher levels of INF-γ (0.72% versus 1.72%, p=0.038) (Figures 6G and S6D) than the control miR group. These results suggest that miR-15a promoted T cell priming and infiltration into NB tumors.
Figure 6.

MiR-15a activates anti-tumor immune response against NB in vivo
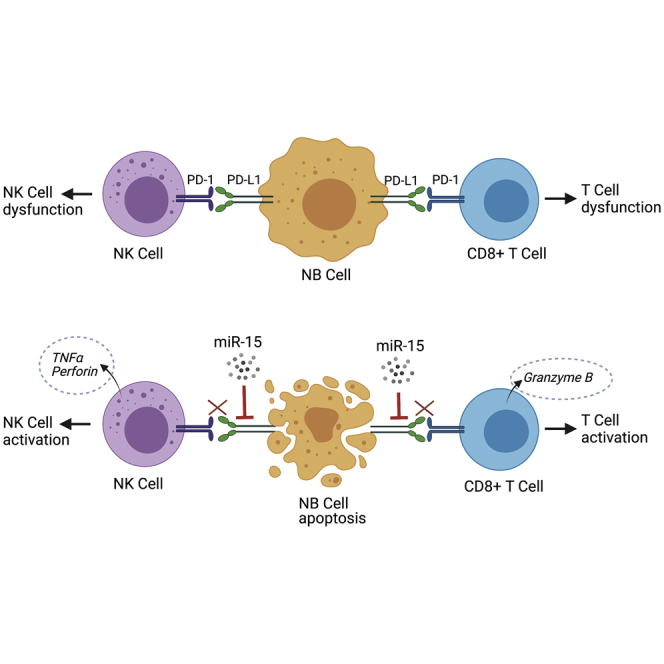
(A) Photographs showing tumor pictures, and (B) summary graph showing tumor weight of the C57BL/6 mice that received subcutaneous murine NB-975 cells stably expressing GFP-miR-15a or GFP-control (ctrl) miR for 30 days. (C) Representative flow cytometric plots showing the percentage of surface PD-L1 expression on GFP+ tumor cells, and (D) quantification, (E) percentage of infiltrated CD8 cells, (F and G) Ki-67, IFN-γ positive infiltrated CD8+ T cells, and (H) perforin positive CD3−NK1.1+ mouse NK cells analyzed from the single-cell suspension of tumors tissues from C57BL/6 mice that received (subcutaneous) murine NB-975 cells stably expressing miR-15a or Ctrl miR for 30 days. (I) Representative IHC images of CD34 (murine endothelial cells) stained microvessels at 20× magnification. Murine NB975 cells stably expressing GFP-miR-15a or GFP-ctrl miR were injected (subcutaneous) into the left flank of immune-competent C57BL/6 mice. After 30 days, mice were euthanized, tumor tissues were harvested, photographed, and weighed. Tumor tissues were excised, prepared single-cell suspension, and stained for PD-L1 (α-mouse PE-PD-L1), infiltrated CD8+T cells (α-mouse PE-CD8a), Ki-67 (α-mouse BV421-Ki67), IFN-γ (α-mouse BV42-IFN-γ), CD3−NK1.1+ mouse NK cells (α-mouse AF700-CD3, and α-mouse APC/FireTM 750-NK1.1), perforin (α-mouse PE-perforin) by flow cytometry using their respective antibodies. (C, and E–H) Representative flow cytometric plots showing an FMO control of cells stained with all fluorochromes except one used to set the background signal for the analysis were given. Bar graphs are shown as the mean ± standard error (n=4 mice per group). Statistical analyses were performed using a two-sided unpaired t-test. (I) A schematic model showing the modulation of anti-tumor immune response in the absence (top panel) or presence (bottom panel) of miR-15 (miR-15a, miR-15b) in NB.
Tumor-infiltrating CD25+CD4+ regulatory T cells (Tregs) with Foxp3 expression play a key role in suppressing anti-tumor activity and promoting tumor growth.56 Thus, we examined the effect of miR-15a on CD25+CD4+Foxp3+ Tregs in NB tumors. As shown in Figures S6E and S6F, tumors of mice that received miR-15a cells had a significantly lower (p=0.0016) Treg population than the control group, indicating that miR-15a repressed Tregs in the tumor microenvironment to prevent immune escape. We next evaluated the activation status of tumor-reactive NK cells in tumors by analyzing perforin on CD3− (to exclude T cells), NK1.1+ (a marker for mouse NK cells) gated population from the single-cell suspension of tumor tissues. We found higher perforin expression (16.5% versus 27.1%, p=0.013) in the miR-15 group tumors than the control miR group tumors (Figures 6H and S6G). Last, we examined microvessel infiltration by IHC staining using an anti-mouse CD34 antibody, which stains endothelial cells. As shown in Figures 6I and S6H, tumors of mice that received control miR cells showed intense neovascularization as indicated by high CD34 staining, which was significantly decreased in tumors of mice that received miR-15a cells . The mean microvessel count/field in control miR group tumors was 233±44 (SD) and 101±52 (SD) in miR-15a group tumors (Figure S6I). These observations indicate that miR-15a decreases tumor microvessel density in vivo. Overall, our data strongly support the role of miR-15a and miR-15b in anti-tumor immune response through PD-L1 degradation.
Discussion
Tumor-immune interactions are intricate, and efforts to capture this complexity via a single marker such as PD-L1 expression as measured by IHC, or tumor mutation load as a surrogate of potential tumor antigenicity, yields limited and incomplete information. This is a significant concern for the analyses of pediatric tumors’ immunogenicity since their tumor mutation burden is negligible. The complex and dynamic nature of the tumor-immune microenvironment also involves stromal cells consisting of extracellular matrix, cancer-associated fibroblasts, and mesenchymal stromal cells. These cells prevent the infiltration of immune mediators while enabling drug resistance and metastasis via mesenchymal cancer cell permeation. This data-driven study represents one of the earliest attempts to use stromal cell percentages to interrogate non-coding RNA molecules with tumor antigenic function. Using differential- and co-expression studies on TARGET (n=249) and PDX (n=24) datasets, the stromal-cell specific expression and co-expression of miR-15a, miR-15b, and its target gene PD-L1 were established. These findings were experimentally validated in great detail using in vitro and in vivo approaches. Various cell surface and other immunophenotyping markers were quantified, reinforcing the crucial role of the miR-15a/miR-15b & PD-L1 axis in regulating various immune mediators involved in NB tumor immunogenicity.
PD-L1 surface expression on cancer cells is primarily involved in immune cell dysfunctions, and many studies have reported its prognostic significance in several cancer types.57, 58, 59, 60 PD-L1 expression in tumor cells attenuate T cell activation and functions.60,61 Currently, the FDA has approved the use of three PD-L1 inhibitors: (1) atezolizumab for non-small cell lung cancer (NSCLC),62 (2) durvalumab for NSCLC and urothelial carcinoma (UC),63 and (3) avelumab for UC and renal cell carcinoma.64,65 Moreover, several other inhibitors are in different phases of clinical trials for various cancer types.66 However, one of the challenges limiting the effectiveness of anti-PD-L1 therapy is that checkpoint molecules acquire several mutations during tumor progression, 67 suggesting the importance of targeting upstream regulators of checkpoint molecules.68,69
MiRs have been shown to regulate various oncogenes in NB.70,71 Hence miR-mediated targeting of PD-L1 could have great translational potential to stage 4 NB patients. Hence, we have developed workflows for elucidating regulatory networks of immune evasion to prioritize novel miRs upstream of PD-L1 regulation. The present study explored a miR-based approach to target immune checkpoint molecule PD-L1, which sensitized NB cells to CD8+T and NK cell-mediated cytotoxicity. Multiple studies have demonstrated the association of poor survival with fewer tumor-infiltrating lymphocytes, including T cells and NK cells in NB.28,33,72, 73, 74, 75 Moreover, NB tumors employ several immune evasion strategies, including the expression of immune checkpoint molecules.76, 77, 78, 79, 80 Our bioinformatic co-expression analysis from the TARGET data highlighted the presence of exhaustive immune signatures, including PD-L1, in NB patients and PDX tumors. We further found that lower expression of PD-L1 is associated with better progression-free and overall survival in NB patients. Moreover, PD-L1 mRNA levels are higher in patients with tumor relapse. In contrast, high expression levels of miR-15a were positively correlated with good progression-free and overall survival of NB patients. Patients who had a high risk or died from the disease expressed lower miR-15a levels. Besides, miR-15a expressed differentially across different NB stages, with the lowest expression found in most aggressive and therapy-resistant stage 4 patients. Further, we found an inverse correlation between PD-L1 and miR-15a expression in NB patients. These analyses implied that higher PD-L1 and lower miR-15a expressions are associated with poor clinical features in NB.
Our experimental results showed that miR-15 family in particular miR-15a and miR-15b degraded PD-L1 mRNA and protein in several NB cell lines, including SK-N-AS, NB-19, and SK-N-BE(2). Further, enrichment of miR-15a, miR-15b, and PD-L1 mRNA with Ago2, a core component of RNA-induced silencing complex (RISC), revealed a specific interaction between PD-L1, miR-15a, miR-15b within the RISC complex. RISC plays a major role in regulating gene expression through Argonaute RISC Catalytic Component2 (Ago2)-mediated interaction with miRs and the complementary sequence of the target mRNA, thus serving as an indicator of target gene regulation.81 PD-L1, a membrane-anchored protein, binds to its receptor protein PD-1 on the surface of immune cells. Thus, surface expression of PD-L1 on cancer cells is a critical component of determining the anti-tumor immune response. CD8+T cells are the main components of an adaptive immune response against tumor cells.82,83 NK cells are the main effector immune cells that play essential roles in tumor surveillance and demonstrate anti-tumor cell cytotoxicity without the requirement of the major histocompatibility complex.84 Both CD8+T and NK cells express PD-1 receptors, making them more susceptible to PD-L1-mediated dys-function by tumor cells.85,86 MiR-15a treatment significantly reduced cell surface PD-L1 in NB cell lines suggesting that miR-15a could promote CD8+T and NK cell-mediated anti-tumor immune responses in NB cells.52
NB cells upon receiving miR-15a and miR-15b induced CD8+T and NK cell activation markers such as granzyme B, perforin, and CD107A, as well as CD8+T cell clonal expansion. CD107A, also known as lysosomal-associated membrane protein-1 (LAMP-1), is a marker for NK cell activation and protects NK cells from degranulation-associated damage.87,88 Cytotoxicity experimental results revealed that NK cells were more efficient in lysing NB cells upon miRs treatment. Enhanced granzyme B and perforin secretions and activation of CD8+T and NK cells by miR-15a and miR-15b are associated with a more prominent cleavage of caspase-3 in target NB cells. Caspase-3 is one of the endpoint executioner caspases, which cleaves various target proteins, including PARP-1, leading to an apoptotic breakdown of the cell,89 suggesting that miR-15a and miR-15b play an essential role in executing immune cell-mediated NB cell apoptosis.
Further, mechanistic experiments interfering PD-L1 by siRNAs or blocking the surface PD-L1 using anti-PD-L1 antibody revealed efficient CD8+T and NK cell-mediated anti-tumor immune responses against NB cells. Studies have also shown that tumor necrosis factor (TNF)-α is critical for NK cells in tumor response as it promotes cell recruitment at the tumor site.90 TNF-α binds to TNF receptor-2 (TNFR2) on the surface of NK cells and promotes INF-γ production in NK cells. NK cells control tumor growth by releasing TNF-α and IFN-γ.90, 91, 92 IFN-γ and TNF-α synergistically enhance NK cell-induced tumor cytotoxicity through the NF-κB-dependent pathway.91 However, we did not see a significant difference in the induction of IFN-γ in NK cells upon treatment with miRs. Interestingly, NB cells treatment with miR-15a, miR-15b in the presence of anti-PD-L1 antibody significantly decreased IFN-γ secretion from cocultured NK cells. MiR-15a, miR-15b have complementary binding sequences to 3′UTR of IFN-γ mRNA, and studies have shown that miR-15a, miR-15b inhibit IFN-γ production in NK cells.93 Since miR-15a, miR-15b are not good for NB cells, we believe NB cells can secrete out these miRs in the medium. The uptake of miR-15a, miR-15b by NK cells could be responsible for decreased IFN-γ production in NK cells after coculture with miRs overexpressing NB cells. IFN-γ is known to induce surface PD-L1 expression on cancer cells, protecting cancer cells from anti-tumor immunity and playing a role in chemoresistance.94, 95, 96, 97 MiR-15 mediated inhibition of IFN-γ secretion might act as a double-edged sword that prevents IFN-γ-induced PD-L1 upregulation and promotes CD8+T and NK cell functions by directly targeting PD-L1. In vivo experiments provided evidence that miR-15a promotes the activation and infiltration of CD8+T and NK cells and reduces tumor vasculature, tumor size, and tumor-promoting CD4+CD25+FoxP3+ Tregs in tumors. These results further suggest that miR-15a mediates immune regulation and, therefore, should be considered in treating patients together with approved drugs .
In summary, we have provided evidence that diminished miR-15a, miR-15b, and higher PD-L1 expression are associated with immune cell exhaustion signatures and tumor relapse in NB patients and PDX tumors. PD-L1 mediates immune evasion, whereas targeting PD-L1 using miR-15a and miR-15b oligonucleotides leads to immune cell activation, cytokine secretion, and immune cell-mediated NB cytotoxicity and anti-tumor immune response in vitro. Injection of miR-15a expressing cells in immune-competent C57/BL6 mouse reduced tumor growth and tumor vessel formation while increasing the activation and infiltration of immune cells, suggesting that degradation of PD-L1 by miR-15a and miR-15b activate anti-tumor immune responses in high-risk, low immunogenic NB patients.
Materials and methods
Patient datasets, PDX datasets, missing value imputation, data mining, and survival analysis
The NB TARGET (n=249) dataset was downloaded from the web of the National Cancer Institute (https://ocg.cancer.gov/programs/target/projects/neuroblastoma). According to the International NB Staging System,102 and the Risk Group (INRG) systems,103 disease stages and risk status were categorized. The clinical characteristics of NB patients such as sex, MYCN amplification, risk, survival, stage, and progression were also available. The R2, a web-based genomics analysis and visualization application platform (http://r2.amc.nl) was used for survival analysis using (i) TARGET (n=249), (ii) Versteeg, GSE16476 (n=88),100 and (iii) Tumor NB ALT-Westermann–144-tpm-gencode19-R2 dataset (n=139) datasets. Moreover, the following datasets were also used for the box plot analysis of miR-15a, miR-15b, and PD-L1, (i) Versteeg, GSE16476 (n=88),100 (ii) Saadati, GSE73515 (n=105),101 (iii) Fischer, GSE120572 (n=394),98 and (iv) Asgharzadeh, GSE3446 (n=234).99 These datasets were downloaded from the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/). The NB PDX (n=24) dataset was downloaded from the supplementary information of the Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design.109 The clinical characteristics of PDX tumors derived from NB patients such as diagnosis, progression relapse, age, overall, and event-free survival time were also available.109 Patient/PDX RNA-sequencing and microarray-based gene/miRNA expression matrices and the corresponding clinical attributes were processed and analyzed (box-, line-, scatter- plots & heatmaps) through Bioconductor and CRAN libraries using the R software (https://www.r-project.org/).111
Patient stratification using clinical attributes and tumor-stromal cell percentages
A systematic meta-analysis of tumor stroma quantified in 16 solid cancer types from 2732 patients revealed that the stroma fraction was highly variable within and across the tumor types. Kidney cancer showed the lowest while pancreato-biliary type periampullary cancer demonstrated the highest stroma proportion (median 19% and 73% respectively)110. Despite the wide variation, no significant association between stromal abundance and prognosis was observed. Given this context, we have chosen a stromal cell percent cut-off that yielded the approximately 50-50 stratification of the 249 patients in concert with survival status.
Missing value imputation using multivariate imputation by chained equations
Multivariate imputation by chained equations (MICE) is an imputation method based on Fully Conditional Specification, where different models impute incomplete attributes. Hence, MICE can impute missing values in datasets with continuous, binary, and categorical attributes by using either logistic or linear regression for each attribute. While expression/count data mostly had continuous data, the tumor sample attribute file had continuous and categorical variables. The integrity of the variable types was verified before imputation was attempted. The number of multiple imputations (m) and the number of iterations (maxit) varied from their default value of 5. The best parametric settings were determined after preliminary explorations and assessments of the imputation quality. Both TARGET and PDX datasets had multiple missing values in the expression and tumor sample attribute files.
Representative probe prioritization
Both TARGET and PDX datasets had multiple probes per gene. Furthermore, both these datasets were derived from different array platforms. Any meaningful meta-analyses and corroboration assessments require a standardized approach for prioritizing the best representative probe of the gene. Prioritization of the best representative probe prior to the application of statistical tests can, in some instances, be detrimental to the consistency between the obtained results and the proposed hypotheses. However, such prioritization represents a best practices protocol with an unbiased, objective, and data-driven approach with robust statistical significance and predictive utility.
Human tissue samples and peripheral blood mononuclear cells
Deidentified formalin-fixed and embedded paraffin blocks of NB patient tissue samples were obtained as a tissue microarray (#NB642a) from US Biomax Inc (26 patient samples, in duplicates). All procedures with NB patient tissue microarray followed our institutional ethical standards and were approved by the human ethics committee. These blocks were used for the Immunofluorescence (IF) analysis. Freshly obtained human peripheral blood mononuclear cells (PBMCs) from the Elutriation Core Facility of our institute were used for the isolation of CD8+T and NK cells.
PDX tumors
We have established NB-specific PDX tumors in collaboration with C. Patrick Reynolds, MD, PhD, from Texas Tech University Health Sciences Center through the Alex’s Lemonade Stand Foundation (ALSF)/Children’s Oncology Group (COG) Childhood Cancer Repository (https://www.cccells.org) and the Childhood Solid Tumor Network at St. Jude Children’s Research Hospital.70 In brief, patient tumor samples were surgically removed, cut into multiple pieces in sterile Hanks’ balanced salt solution supplemented with antibiotics. The deidentified patient tumor cells were subcutaneously injected into 4- to 6-week-old nude mice. Mice were observed for palpable tumors, and the tumor tissues were harvested once the tumor reached end volume, then a single-cell suspension was prepared for re-implantation in the next set of mice, with multiple passages for expansion. These tumors were used for the western blotting analysis, P0 = Passage 0, P1 = Passage 1 (Figure S1K). Our institutional ethics committee approved the study methodologies. The PDX tumor tissues COG-N-564x, COG-N-453x, COG-N-496x, COG-N-603x, COG-N-470x, COG-N-424x, and SJNBL013763_x1 were used for the Western blotting and IHC analysis.
Cell lines and cell culture conditions
Human NB cell lines SK-N-AS, NB-19, and SK-N-BE(2) were obtained from American Type Culture Collection (ATCC) or the COG Cell Culture and Xenograft Repository (www.cogcell.org). Feeder K562.mbIL21.4-1BBL (K562 cells genetically modified to express membrane-bound interleukin-21 [mbIL21] and 4-1 BB [CD137] Ligand [CD137L]) is a generous gift from Siddappa Byrareddy, PhD, from the University of Nebraska Medical Center (UNMC). The murine NB975 cell line was kindly provided by Leonid Metelitsa, MD, PhD, from Texas Children’s Hospital. SK-N-BE(2), NB-19, K562, and NB975 cells were cultured in RPMI and SK-N-AS cells in DMEM media, each containing 10% heat-inactivated fetal bovine serum (FBS) supplemented with penicillin/streptomycin (50 U/mL), L-glutamine, sodium pyruvate, and non-essential amino acids, as described earlier. Cell lines were cultured in a 5% CO2 humidified atmosphere at 37°C. Cell lines were authenticated by short tandem repeats analysis and were frequently tested to ensure they were free from mycoplasma.
CD8+T cell isolation, activation, and culture
PBMCs from healthy human donors were processed using Histopaque-1077 reagent and CD8+T cells were isolated using MojoSort Human CD8+T cell isolation kit (Biolegend, #480012) according to the manufacturer’s instructions. The CD8+T cells were stimulated by Dynabeads Human T-Activator CD3/CD28 (Thermo Fisher Scientific, #11131D) at a 1:1 ratio of T cells to beads in the growth medium of 10% FBS in RPMI-1640 for 48 h. The activated CD8+T cells were harvested, and the purity was analyzed via flow cytometry using anti-CD3, anti-CD8, and anti-CD4 antibodies. The CD8+T cells used in the study were CD3+CD8+CD4−. The gating strategy is given in Figure S3A through staining with CD3-AF700, CD8-PE, and CD4-BV785 antibodies. The FMOs related to the experiments are shown in Figures S3B–S3F. CD8+T cells isolated with >90% purity were used for coculture experiments with NB cells. The activated CD8+T cells were cocultured with SK-N-AS, NB-19, and stable luciferase gene expressing SK-N-B(E)2 cells for 24 h at an Effector [E] CD8+T, and Target [T] NB cell ratio of 1:1 (SK-N-AS and NB-19) and 5:1 (SK-N-B(E)2) respectively.
NK cell isolation, expansion, and culture
Untouched NK cells were isolated from PBMCs using the MojoSort Human NK Cell isolation kit (BioLegend, #480054) per the manufacturer’s instructions. In brief, PBMCs (1×107/100 μL MojoSort buffer) were incubated with Biotin-Antibody Cocktail (10 μL) against lineage-specific targets for 15 min on ice. The cells were washed with MojoSort buffer, followed by the addition of streptavidin magnetic nanobeads (10 μL) for 15 min on ice, and resuspended in MojoSort buffer (2.5 mL). The cells were placed in the magnet for 5 min at room temperature (RT), and the unbound NK cells were separated by pouring the liquid into a fresh tube. The cells were cultured in RPMI-1640 supplemented with 10% FBS and 1% penicillin/streptomycin together with recombinant (r) human (h) IL-2 (20 ng/mL), and rhIL-15 (50 ng/mL) for 4 days and were used for expansion. The obtained NK cells were cultured with irradiated K562 CSTX002 mbIL21.41bbL cells at a ratio of 1:1 in the presence of rhIL-2 (20 ng/mL) and expanded for 2 weeks. NK cells were replenished with fresh K562 cells once a week and rhIL-2 twice a week. The purity of NK cells was determined by staining with anti-CD56-APC/Fire750, and CD16-PE antibodies, whereas contaminating T cells were labeled by anti-CD3-AF700 antibodies, using flow cytometry. The NK cells used in the study were CD56+CD16+CD3− (Figures S4A and S4B) and were used for coculture experiments with NB cells or frozen in liquid nitrogen in a freezing medium containing 95% FBS for future use. These activated NK cells were used in coculture experiments with SK-N-AS cells (E:T=1:1), NB-19 cells (E:T=0.25:1), and SK-N-B(E)2 cells (E:T=1:1) with stable miR-15a expression for 5 h.
PD-L1 cell surface blocking
Surface PD-L1 on NB cells was blocked by treatment with control immunoglobulin (Ig) G or anti-PD-L1 antibody (15 μg/mL, BioXCell, Lebanon, NH) for 24 h followed by transfection with miR-15a and miR-15b oligonucleotides for an additional 24 h. After wash out the unbound antibody, these cells were used for coculture experiments with CD8+T and NK cells.
MiRNAs, siRNAs, and transfections
We purchased hsa-miR-15a-5p mimics (Life Technologies, #4464066, ID: MC10235), hsa-miR-15b-5p mimics (Life Technologies, #4464066, ID: MC10904), hsa-anti-miR-15a inhibitors (Life Technologies, 4464084, ID: MH10235), hsa-anti-miR-15b inhibitors (Life Technologies, #4464084, ID: MH10904), and PD-L1 specific siRNAs (Life Technologies, #AM16706, ID:134191). The corresponding non-targeting control miRs, anti-miRs, and siRNAs were also purchased simultaneously. NB cells (1.9×105/well) were seeded in a 12-well plate in a regular growth medium for 24 to 48 h. Cells were transfected with 50 to 60 nM concentrations of miRs, siRNAs, or their corresponding control miRs in Opti-MEM reduced serum medium (Life Technologies, #31985070) using RNAiMAX (Life Technologies, #13778150) transfection reagent for 7 to 8 h. This was followed by replacing a regular growth medium for 24h at 37°C in 5% CO2 and used for the experiments. For SK-N-BE (2) cells transient transfection, cells were serum-starved for overnight in RPMI media containing 2% FBS before transfection.
Lentiviral transductions
Lentiviral vectors carrying human precursor (pre) miR-15a (#PMIRH15aPA-1), non-functional human GFP control miR (#PMIRH000-PA-1), mouse pre-miR-15a (#MMIR-15a+16-1-PA-CL), and non-functional mouse GFP control miR (#MMIR-000-PA-1) constructs were purchased from System Biosciences. Lentiviral particles were generated in HEK293T cells (3×106/8 mL 10% FCS DMEM medium) that were co-transfected with lentiviral miRNA vector (10 μg), and packaging plasmids (pVSV-G [5 μg], pMDL [10 μg], pREV [5 μg]) using polyethyleneimine (PEI) transfection reagent at a ratio of 1:4 for DNA and PEI for 24 h followed by fresh DMEM (10% FCS) media replacement. The lentiviral supernatants were collected at 48 and 96 h, centrifuged (200 g/5 min), and filtrated through a 0.45-μm syringe filter. The lentivirus was concentrated using a PEG-it virus precipitation solution (System Biosciences, LV825A-1). For viral infections, NB cells were seeded in a 6-well plate (1×105/well) and were infected with lentivirus at 40% confluence for 24 h followed by fresh regular growth medium replacement, and the cells were allowed to grow for 3 to 5 days. The transduced cells were selected in the presence of puromycin (5 μg/mL). Further, to keep a pure population, GFP-positive cells were sorted by flow cytometry.
Western blotting and antibodies
Cells or tumor tissue samples were lysed in radioimmunoprecipitation assay buffer containing freshly added EDTA-free protease and phosphatase inhibitor tablets for 30 min on ice. The protein samples were resolved on 10% gradient SDS-PAGE and transferred to a nitrocellulose membrane, followed by blocking with 5% BSA for 1 h at RT. The membranes were incubated with primary antibodies, including monoclonal mouse anti-human PD-L1/CD274 (Proteintech, #66248-1-Ig, 1:8,000, overnight at 4°C), horseradish peroxidase (HRP)-conjugated anti-β-actin (Proteintech, #HRP60008, 1:12000, for 2 h at RT). After secondary antibody incubation at RT for 1 h, membranes were developed using an enhanced chemiluminescent reagent. The images were captured on X-ray film and quantified using ImageJ analysis software.104
RNA isolation and reverse transcription quantitative real-time PCR
Total RNA was isolated from the cells using the PureLinkTM RNA Mini Kit (#12183025) followed by reverse transcription for miRNAs using TaqMan MicroRNA Reverse Transcription Kit (Life Technologies, #4366597) and mRNA using iScript cDNA synthesis kit (Bio-Rad, #1708891). A reverse transcription quantitative real-time PCR (RT-qPCR) analysis was performed in triplicates on an ABI StepOne RT-qPCR system (Applied Biosystems) with SYBR Green Master Mix (Bio-Rad) for mRNA expression or TaqMan Universal Master Mix II (Life Technologies, #4440041) for miRNAs, as described earlier. Calculations were performed using the ΔΔCτ method, normalized with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA or U6 small nucleolar RNA, and the quantifications were presented as fold changes.105 RT-qPCR primers for PD-L1 and GAPDH were purchased from Integrated DNA Technologies, Inc., and the sequences are as follows. PD-L1 Forward: 5′-TGGCATTTGCTGAACGCATTT-3′, Reverse: 5′-TGCAGCCAGGTCTAATTGTTTT-3′; GAPDH Forward: 5′-GATTCCACCCATGGCAAATTC-3′, Reverse: 5′-AGCATCGCCCCACTTGATT-3′. The miR-15a (ID: 000389), miR-15b (ID: 000390) and U6 snRNA (ID: 001973) primers were purchased from Life Technologies (ID:4427975).
Flow cytometry analysis
The assessment of PD-L1, CD8+T, and NK cell surface markers, cleaved caspase-3, perforin, granzyme B, Ki-67, Foxp3, INF-γ and cell survival analysis was performed by immunofluorescence staining followed by flow cytometric analysis. In brief, cells were collected at 350 × g for 5 min, washed with, and suspended in PBS (1.0 × 105/100 μL), and all steps were performed in the dark unless stated. The dead cells were excluded by incubation with Zombie Aqua fixable viability dye (BioLegend, # 423102) at RT for 30 min. Before immunostaining, cells were incubated with Fc receptor blocking solution (TruStain FcX for human, #422302 or TruStain FcX PLUS for mouse #156604, BioLegend) diluted in ice-cold cell staining buffer (BD Biosciences) on ice for 15 min to reduce non-specific antibody binding. For the analysis of surface GD2, PD-L1, CD3, CD4, CD8, CD16, CD25, CD56, CD107a, and NK1.1 proteins, the cells were incubated with fluorochrome-conjugated anti-GD2, anti-PD-L1, anti-CD3, anti-CD4, anti-CD8, anti-CD16, anti-CD25, anti-CD56, anti-CD107a, and anti-NK1.1 antibodies on ice for 20 min. For measurement of intracellular cleaved caspase-3, perforin, granzyme B, Ki-67, Foxp3, and INF-γ, cells were washed with staining buffer (2x), fixed and permeabilized with Cyto-Fast Fix/Perm Buffer (BioLegend) at RT for 20 min followed by staining with the fluorochrome-conjugated anti-perforin, anti-granzyme B, anti-Ki67, anti-Foxp3, anti-INF-γ, or unconjugated anti-cleaved caspase-3 antibodies at RT for 25 min. The cells were further incubated with the fluorochrome-conjugated secondary antibody at RT for another 40 min if stained with an unconjugated primary antibody. After incubation, cells were washed with Cyto-Fast Perm buffer (2x) and cell staining buffer (1x), then suspended in cell staining buffer. The specificity of the antibodies was verified by staining with respective isotype control antibodies and FMOs. The samples were analyzed freshly or within 24 h using the Attune Acoustic Focusing Flow Cytometer (Thermo Fischer Scientific) or LSRII (Becton Dickinson). The data evaluation was performed using the FlowJo v10.8.1 program.
Antibodies used for flow cytometry
The following fluorescent dye-conjugated antibodies were purchased from BioLegend, CA and were used for flow cytometry analysis. PE anti-human CD274 (clone 29E.2A3, #329706), PE/Cyanine 7 anti-human CD274 (clone MIH3, #374506), PE anti-human Ganglioside GD2 (clone 14G2a, #357304), APC anti-human/mouse granzyme B (clone QA16A02, #372204), PE/Dazzle 594 anti-human perforin (clone dG9, #308132), PE anti-human CD8 (clone SK1, #344706), Alexa Fluor 700 anti-human CD3 (clone OKT3, #317340), Brilliant Violet 785 anti-human CD4 (clone OKT4, #317442), APC anti-human CD107a (clone H4A3, #328620), PE anti-human CD16 (clone 3G8, #302008), APC/FireTM 750 anti-human CD56 (NCAM) (clone 5.1H11, #362554), anti-mouse CD34 (clone MEC14.7, #119302), Brilliant Violet 421 anti-mouse IFN-γ (clone XMG1.2, #505830), Brilliant Violet 421 anti-mouse/human ki-67 (clone 11F6, #151208), Brilliant Violet 785 anti-mouse CD4 (clone GK1.5, #100453), Pacific Blue anti-mouse CD25 (clone PC61, #102022), Alexa Fluor 647 anti-mouse/rat/human Foxp3 (clone150D, #320014), PE/Cyanine7 anti-mouse F4/80 (clone BM8, #123114), anti-mouse PE-CD274 (10F.9G2, #124308), APC/FireTM 750 anti-mouse NK1.1 (clone PK136, #108752), PE anti-mouse perforin (clone S16009A, #154306), Alexa Fluor 647 donkey anti-rabbit (clone Poly4064) secondary antibody and Zombie Aqua Fixable Viability Kit (#423101). The unconjugated cleaved caspase-3 was purchased from Cell Signaling Technology (#9661).
Enzyme-linked immunosorbent assay
The concentration of INF-γ in the culture supernatants was assayed by Enzyme-Linked ImmunoSorbent Assay (ELISA) using LEGEND MAX Human IFN-γ ELISA (BioLegend, #430107) kit per the manufacturer’s instructions. In brief, the cell culture supernatants were collected and human INF-γ standards at 500, 250, 125, 62.5, 31.3 and 15.6 pg/mL concentrations were freshly prepared. The ELISA plates were washed (4x) with PBS containing 0.05% Tween 20, and blocked (200 μL/well) with PBS containing 10% FBS at RT on a shaker for 1 h. The cell culture supernatant samples (3x diluted), or human IFN-γ standards (100 μL/well) were added at RT for 2 h on a shaker. The plates were washed (4x), then the human INF-γ antibody was added (100 μL/well) and incubated for 1 h at RT with shaking. The plates were washed (5x), and the avidin-HRP solution was added (100 μL/well) and incubated at RT for 30 min with shaking. The plates were washed (5x) before adding tetramethylbenzidine substrate solution (100 μL/well) at RT for 5 min in the dark or until the development of blue color. The reaction was stopped by a stop solution (100 μL/well), and the optical density was measured at 450 nm and 570 nm on a plate reader. The final readings were obtained by subtracting 570 nm absorbance from 450 nm and compared with IFN-γ standard curve. Each sample was assayed in triplicate and repeated more than three times.
Direct tumor cell killing quantification by luminescence assay
Luciferase gene expressing stable SK-N-B(E)2 cells were used in CD8+T cell-mediated direct tumor cell killing assay. The cells were seeded (0.9 × 105/well) into a 12-well plate for 48 h, followed by transfection with miR-15a, miR-15b, or non-targeting control mimics for 9 h. Next, activated CD8+T cells (effector [E]) were cocultured with SK-N-B(E)2 (target [T]) cells at a 5:1 E and T ratio for 48 h. The supernatants containing the effector CD8+T cells were removed, and the remaining dead detached target cancer cells were carefully washed away by rinsing twice with PBS (1 mL). The target cancer cells were collected, washed with PBS, and lysed (100 μL) in reporter lysis buffer (Promega Corporation, Madison, WI; #E3971) with a freeze-thaw cycle to achieve complete cell lysis. The cleared cell lysates (80 μL) were transferred to a Corning/Costar white, flat-bottom 96-well plates, followed by the addition of substrate (80 μL/well) D-luciferin (Promega Corporation, #E151A). The luminescence was measured using a luminescence plate reader (Infinite 200 PRO, Tecan). The luminescence units were normalized to control (tumor target cells without any effector CD8+T cells) set to 100. The results were reported as the percentage of relative luminescence units.
PD-L1 3′UTR constructs and luciferase reporter assays
Publicly available online bioinformatics databases such as TargetScan (www.targetscan.org), miRNA Target Prediction Database (mirdb.org), and microRNA target prediction (www.microrna.orgg) were used to predict the potential miR-15a, miR-15b binding sites in the 3′UTR of PD-L1 mRNA. The predicted miR binding sites in the 3′UTR region of PD-L1 mRNA referred to as 3′ UTR wild type (WT) and mutations in the miRNA binding sites (seven bases) of the 3′UTR region referred to as 3′UTR mutant were cloned into a luciferase vector pEZX-MT06 (Cat. #HmiT117860-MT06, Cat. #CS-HmiT117860-MT06-01; GeneCopoeia, Rockville, MD). NB cells were transfected with these luciferase plasmid constructs (1 μg) with miR-15a, miR-15b, or control miR oligonucleotides (100 nM), using lipofectamine reagent. After 48 h, the cells were lysed and the luciferase activity was measured by luciferase assay reagent (Promega) using the luminometer following manufacturer's instructions. The luciferase activity was expressed relative to control miR, which was set to 100.
Ago2 protein-associated RNA immunoprecipitation
Immunoprecipitation of Ago2 protein-bound RNA-protein complexes was performed as described previously.70 NB cells were transfected with miR-15a, miR-15b, and control miRs for 48 h. After transfection, cells were collected at 300×g, washed with PBS, and lysed in polysome lysis buffer (5 mM MgCl2, 100 mM KCl, 10 mM HEPES [pH 7.0], 0.5% Nonidet P-40, 1 mM DTT, 100 U of RNase inhibitor/mL) supplemented with 20 mM EDTA and protease inhibitors on ice for 20 min. The lysates were centrifuged to remove cell debris, and clear lysates were collected. The lysates were diluted to 1:10 in freshly made NT2 buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1 mM MgCl2, 0.05% Nonidet P-40, 1 mM DTT, 100 U of RNase inhibitor/mL) supplemented with 20 mM EDTA and protease inhibitors and were incubated with α-Ago2 primary antibody at 4°C for 6 h, followed by incubation with protein A beads for an additional 2 h. The beads were then washed with NT2 buffer five times and treated with DNase I and proteinase K. The beads were eluted with NT2 buffer containing 0.1% SDS, and RNAs were extracted using phenol-chloroform and ethanol precipitation. The Ago2 bound immunoprecipitated RNA was used for the analysis of PD-L1 mRNA and miR-15a, miR-15b by RT-qPCR assay and normalized to IgG-immunoprecipitations as described.50
Immunohistochemistry and immunofluorescence
IHC and IF staining for PD-L1 and CD34 proteins was performed through our institutional core facility on a Discovery Ultra staining system (Roche Tissue Diagnostics, Ventana Medical Systems, Inc.). In brief, tissue samples were processed by fixing (70% ethanol), embedded in paraffin, and cut into 5-μm sections. The sections were deparaffinized in EZ prep, a mild detergent solution at 69°C for 24 min (Roche Diagnostics, #950-102), and antigen retrieval was achieved by incubation with cell-conditioning solution (Roche Diagnostics, #950-124), a Tris EDTA buffer (pH 8.2), for 32 min at 95°C. To prevent unspecific signal, we blocked endogenous peroxidase activity by treating with Inhibitor CM, available within the DISCOVERY ChromoMap DAB Kit (Roche Diagnostics, #760-159) for 8 min at RT. The sections were incubated with either rabbit monoclonal PD-L1 antibodies (clone 28-8, Abcam, #ab205921) or anti-mouse CD34 for endothelial cells (clone MEC14.7, BioLegend, #119302) at 37°C for 30 min using the Discovery Benchmark ULTRA advanced staining system. The HRP-conjugated secondary antibody (Roche Diagnostics, #760-4815) incubation was performed at 37°C for 16 min followed by diaminobenzidine (DAB) signal detection using DISCOVERY ChromoMap DAB Kit. The sections were counterstained with hematoxylin II for microscopic examination. The images were acquired using a fluorescence microscope (Leica Microsystems Inc, USA). Tumor microvessel density was assessed according to the criteria described by Weidner and colleagues.106,107 First, the entire tumor section was scanned at low magnification to find areas having high vessel density. Then, images of four such fields were digitally acquired at high magnification (20×). Microvessel density was quantified by manual counting using Photoshop (version 22.0.1), and an average of four fields from each tumor was determined. Four mice from each group were included in the analysis.
NB mouse xenograft model
All animal studies were conducted in accordance with and approved by the UNMC’s Institutional Animal Care and Use Committee. In vivo immune response was evaluated using NB cell line-derived xenografts. We purchased 5- to 6-week-old C57/BL6 mice from The Jackson Laboratory (Bar Harbor, ME). We used only female mice for in vivo experiments to avoid biological variables. NB975, a murine NB cell line that stably expresses murine GFP-miR-15a, or non-targeting GFP-control miR, suspended in Matrigel (Corning Corporation, #CB-40230C), were subcutaneously inoculated into the right flank (3×106/100 μL) of C57/BL6 mice within the animal care facility at UNMC. Mice were monitored daily to ensure that the injection sites were healthy. On day 30, the mice were sacrificed, and the tumors were excised, photographed, and the final tumor volumes were measured using the equation: V=L×W2/2 (L: tumor length; W: tumor width). Tumor tissues were lysed using standard protocols, prepared single-cell suspensions and then processed for immunophenotyping by flow cytometry using the methods described above.
Statistical analysis
All the statistical analyses were performed using GraphPad Prism software. The data were presented as the mean ± standard error or individual values derived from independent experiments and/or independent biological replicates. The Kruskal-Wallis test was used for multiple comparisons between groups. The survival analysis was calculated by the Kaplan-Meier method and the log-rank test. Statistical significance was calculated using a two-sided unpaired t-test. P values smaller than 0.05 were considered statistically significant.
Data and code availability
Data were generated by the authors and are available upon request from the corresponding author.
Acknowledgments
Dr. Challagundla’s laboratory was supported by the NIH/NCI grant CA197074; the Buffett Cancer Center, which is supported by the National Cancer Institute under award number CA036727, in collaboration with the UNMC/Children’s Hospital & Medical Center Child Health Research Institute Pediatric Cancer Research Group; and the Department of Biochemistry & Molecular Biology start-up funds. The results published here are in whole or part based upon data generated by the Therapeutically Applicable Research to Generate Effective Treatments (https://ocg.cancer.gov/programs/target) initiative, phs000218. The data used for this analysis are available at https://portal.gdc.cancer.gov/projects. We are grateful to Jonas Nance and Kristyn Mccoy at the Childhood Cancer Repository (supported by Alex's Lemonade Stand Foundation), Åsa Karlström, PhD, at the Childhood Solid Tumor Network of St. Jude Children’s Research Hospital for arranging NB PDX tumor cells; Myriam Gorospe, PhD, at NIH for sharing RNA-IP protocol, Vikas Pathania for data mining using R program and Sita Devi for data mining from the R2 database, and the tissue sciences core facility of UNMC for performing IF and IHC staining. The authors thank Matthew Sandbulte, PhD, of the Child Health Research Institute at Children’s Hospital & Medical Center and the UNMC for editorial assistance.
Author contributions
A.S.P., P.P., and K.B.C. conceptualization, designed studies, performed experiments, expanded patient-derived xenografts, performed patient data mining, survival, correlation analyses, analyzed data, and wrote the manuscript. S.C., O.A.O., and T.S.O. assisted in western blotting, flow cytometry experiments, samples preparation, acquisition, and analysis. S.C.G. and D.W.C. provided suggestions to the study design and assisted in manuscript preparation, review, and edits. N.K.C. assisted in isolating PBMCs, provided NB cell lines, reviewed, and edited the manuscript. S.N.B. assisted in flow cytometry experiments, provided K562 feeder cell line, reviewed and edited the manuscript. K.B.C. acquired funding, coordinated with all coauthors, and supervised the project.
Declaration of interests
The authors have declared that no competing interest exists.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.03.010.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer statistics, 2021. CA Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Mahapatra S., Challagundla K.B. StatPearls; 2020. Cancer, Neuroblastoma. [PubMed] [Google Scholar]
- 3.Prathipati A., Pathania A.S., Chaturvedi N.K., Gupta S.C., Byrareddy S.N., Coulter D.W., Challagundla K.B. SAP30, an Oncogenic Driver of Progression, Survival, and Drug Resistance in High-Risk Neuroblastoma. Mol. Ther. Nucleic Acids. 2022 doi: 10.1016/j.omtn.2022.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mueller S., Matthay K.K. Neuroblastoma: biology and staging. Curr. Oncol. Rep. 2009;11:431–438. doi: 10.1007/s11912-009-0059-6. [DOI] [PubMed] [Google Scholar]
- 5.London W.B., Castel V., Monclair T., Ambros P.F., Pearson A.D., Cohn S.L., Berthold F., Nakagawara A., Ladenstein R.L., Iehara T., Matthay K.K. Clinical and biologic features predictive of survival after relapse of neuroblastoma: a report from the International Neuroblastoma Risk Group project. J. Clin. Oncol. 2011;29:3286–3292. doi: 10.1200/jco.2010.34.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith M.A., Seibel N.L., Altekruse S.F., Ries L.A., Melbert D.L., O'Leary M., Smith F.O., Reaman G.H. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J. Clin. Oncol. 2010;28:2625–2634. doi: 10.1200/jco.2009.27.0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spix C., Pastore G., Sankila R., Stiller C.A., Steliarova-Foucher E. Neuroblastoma incidence and survival in European children (1978-1997): report from the automated childhood cancer information system project. Eur. J. Cancer. 2006;42:2081–2091. doi: 10.1016/j.ejca.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 8.Yamamoto M., Yanai R., Arishima K. Study of migration of neural crest cells to adrenal medulla by three-dimensional reconstruction. J. Vet. Med. Sci. 2004;66:635–641. doi: 10.1292/jvms.66.635. [DOI] [PubMed] [Google Scholar]
- 9.Chung K.F., Sicard F., Vukicevic V., Hermann A., Storch A., Huttner W.B., Bornstein S.R., Ehrhart-Bornstein M. Isolation of neural crest derived chromaffin progenitors from adult adrenal medulla. Stem Cell. 2009;27:2602–2613. doi: 10.1002/stem.180. [DOI] [PubMed] [Google Scholar]
- 10.Huang M., Weiss W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013;3:a014415. doi: 10.1101/cshperspect.a014415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kusafuka T., Fukuzawa M., Oue T., Yoneda A., Okada A., Satani M. DNA flow cytometric analysis of neuroblastoma: distinction of tetraploidy subset. J. Pediatr. Surg. 1994;29:543–547. doi: 10.1016/0022-3468(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 12.Ritke M.K., Shah R., Valentine M., Douglass E.C., Tereba A. Molecular analysis of chromosome 1 abnormalities in neuroblastoma. Cytogenet. Cell Genet. 1989;50:84–90. doi: 10.1159/000132729. [DOI] [PubMed] [Google Scholar]
- 13.Plantaz D., Mohapatra G., Matthay K.K., Pellarin M., Seeger R.C., Feuerstein B.G. Gain of chromosome 17 is the most frequent abnormality detected in neuroblastoma by comparative genomic hybridization. Am. J. Pathol. 1997;150:81–89. [PMC free article] [PubMed] [Google Scholar]
- 14.Mlakar V., Jurkovic Mlakar S., Lopez G., Maris J.M., Ansari M., Gumy-Pause F. 11q deletion in neuroblastoma: a review of biological and clinical implications. Mol. Cancer. 2017;16:114. doi: 10.1186/s12943-017-0686-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brisse H.J., McCarville M.B., Granata C., Krug K.B., Wootton-Gorges S.L., Kanegawa K., Giammarile F., Schmidt M., Shulkin B.L., Matthay K.K., et al. Guidelines for imaging and staging of neuroblastic tumors: consensus report from the International Neuroblastoma Risk Group Project. Radiology. 2011;261:243–257. doi: 10.1148/radiol.11101352. [DOI] [PubMed] [Google Scholar]
- 16.Davidoff A.M. Neuroblastoma. Semin. Pediatr. Surg. 2012;21:2–14. doi: 10.1053/j.sempedsurg.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith V., Foster J. High-risk neuroblastoma treatment review. Children (Basel) 2018;5:114. doi: 10.3390/children5090114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matthay K.K., Villablanca J.G., Seeger R.C., Stram D.O., Harris R.E., Ramsay N.K., Swift P., Shimada H., Black C.T., Brodeur G.M., et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children's Cancer Group. N. Engl. J. Med. 1999;341:1165–1173. doi: 10.1056/nejm199910143411601. [DOI] [PubMed] [Google Scholar]
- 19.Coughlan D., Gianferante M., Lynch C.F., Stevens J.L., Harlan L.C. Treatment and survival of childhood neuroblastoma: evidence from a population-based study in the United States. Pediatr. Hematol. Oncol. 2017;34:320–330. doi: 10.1080/08880018.2017.1373315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herd F., Basta N.O., McNally R.J.Q., Tweddle D.A. A systematic review of re-induction chemotherapy for children with relapsed high-risk neuroblastoma. Eur. J. Cancer. 2019;111:50–58. doi: 10.1016/j.ejca.2018.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer J., Pohl A., Volland R., Hero B., Dubbers M., Cernaianu G., Berthold F., von Schweinitz D., Simon T. Complete surgical resection improves outcome in INRG high-risk patients with localized neuroblastoma older than 18 months. BMC Cancer. 2017;17:520. doi: 10.1186/s12885-017-3493-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armideo E., Callahan C., Madonia L. Immunotherapy for high-risk neuroblastoma: management of side effects and complications. J. Adv. Pract. Oncol. 2017;8:44–55. doi: 10.6004/jadpro.2017.8.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu A.L., Gilman A.L., Ozkaynak M.F., London W.B., Kreissman S.G., Chen H.X., Smith M., Anderson B., Villablanca J.G., Matthay K.K., et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. New Engl. J. Med. 2010;363:1324–1334. doi: 10.1056/nejmoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehlert K., Hansjuergens I., Zinke A., Otto S., Siebert N., Henze G., Lode H. Nivolumab and dinutuximab beta in two patients with refractory neuroblastoma. J. Immunother. Cancer. 2020;8:e000540. doi: 10.1136/jitc-2020-000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020;11:3801. doi: 10.1038/s41467-020-17670-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shirinbak S., Chan R.Y., Shahani S., Muthugounder S., Kennedy R., Hung L.T., Fernandez G.E., Hadjidaniel M.D., Moghimi B., Sheard M.A., et al. Combined immune checkpoint blockade increases CD8+CD28+PD-1+ effector T cells and provides a therapeutic strategy for patients with neuroblastoma. Oncoimmunology. 2021;10:1838140. doi: 10.1080/2162402x.2020.1838140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morandi F., Frassoni F., Ponzoni M., Brignole C. Novel immunotherapeutic approaches for neuroblastoma and malignant melanoma. J. Immunol. Res. 2018;2018:1–12. doi: 10.1155/2018/8097398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mina M., Boldrini R., Citti A., Romania P., D'Alicandro V., De Ioris M., Castellano A., Furlanello C., Locatelli F., Fruci D. Tumor-infiltrating T lymphocytes improve clinical outcome of therapy-resistant neuroblastoma. Oncoimmunology. 2015;4:e1019981. doi: 10.1080/2162402x.2015.1019981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu J., Hodgins J.J., Marathe M., Nicolai C.J., Bourgeois-Daigneault M.C., Trevino T.N., Azimi C.S., Scheer A.K., Randolph H.E., Thompson T.W., et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Invest. 2018;128:4654–4668. doi: 10.1172/jci99317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zuo S., Sho M., Sawai T., Kanehiro H., Maeda K., Yoshida M., Tsukada R., Nomura M., Okuyama H. Potential role of the PD-L1 expression and tumor-infiltrating lymphocytes on neuroblastoma. Pediatr. Surg. Int. 2020;36:137–143. doi: 10.1007/s00383-019-04616-9. [DOI] [PubMed] [Google Scholar]
- 31.Saletta F., Vilain R.E., Gupta A.K., Nagabushan S., Yuksel A., Catchpoole D., Scolyer R.A., Byrne J.A., McCowage G. Programmed death-ligand 1 expression in a large cohort of pediatric patients with Solid tumor and association with clinicopathologic features in neuroblastoma. JCO Precision Oncol. 2017;1:1–12. doi: 10.1200/po.16.00049. [DOI] [PubMed] [Google Scholar]
- 32.Nallasamy P., Chava S., Verma S.S., Mishra S., Gorantla S., Coulter D.W., Byrareddy S.N., Batra S.K., Gupta S.C., Challagundla K.B. PD-L1, inflammation, non-coding RNAs, and neuroblastoma: Immuno-oncology perspective. Semin. Cancer Biol. 2018;52:53–65. doi: 10.1016/j.semcancer.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei J.S., Kuznetsov I.B., Zhang S., Song Y.K., Asgharzadeh S., Sindiri S., Wen X., Patidar R., Najaraj S., Walton A., et al. Clinically relevant cytotoxic immune cell signatures and clonal expansion of T-cell receptors in high-risk MYCN-not-amplified human neuroblastoma. Clin. Cancer Res. 2018;24:5673–5684. doi: 10.1158/1078-0432.ccr-18-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kluger H.M., Tawbi H.A., Ascierto M.L., Bowden M., Callahan M.K., Cha E., Chen H.X., Drake C.G., Feltquate D.M., Ferris R.L., et al. Defining tumor resistance to PD-1 pathway blockade: recommendations from the first meeting of the SITC Immunotherapy Resistance Taskforce. J. Immunother. Cancer. 2020;8:e000398. doi: 10.1136/jitc-2019-000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Misiak D., Hagemann S., Bell J.L., Busch B., Lederer M., Bley N., Schulte J.H., Huttelmaier S. The MicroRNA landscape of MYCN-amplified neuroblastoma. Front. Oncol. 2021;11:647737. doi: 10.3389/fonc.2021.647737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ooi C.Y., Carter D.R., Liu B., Mayoh C., Beckers A., Lalwani A., Nagy Z., De Brouwer S., Decaesteker B., Hung T.T., et al. Network modeling of microRNA-mRNA interactions in neuroblastoma tumorigenesis identifies miR-204 as a direct inhibitor of MYCN. Cancer Res. 2018;78:3122–3134. doi: 10.1158/0008-5472.can-17-3034. [DOI] [PubMed] [Google Scholar]
- 37.Perri P., Ponzoni M., Corrias M.V., Ceccherini I., Candiani S., Bachetti T. A focus on regulatory networks linking MicroRNAs, transcription factors and target genes in neuroblastoma. Cancers (Basel) 2021;13:5528. doi: 10.3390/cancers13215528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aravindan N., Subramanian K., Somasundaram D.B., Herman T.S., Aravindan S. MicroRNAs in neuroblastoma tumorigenesis, therapy resistance, and disease evolution. Cancer Drug Resist. 2019;2:1086–1105. doi: 10.20517/cdr.2019.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pathania A.S., Prathipati P., Pandey M.K., Byrareddy S.N., Coulter D.W., Gupta S.C., Challagundla K.B. The emerging role of non-coding RNAs in the epigenetic regulation of pediatric cancers. Semin. Cancer Biol. 2021;S1044-579X:00116-4. doi: 10.1016/j.semcancer.2021.04.015. [DOI] [PubMed] [Google Scholar]
- 40.Mudgapalli N., Shaw B.P., Chava S., Challagundla K.B. The transcribed-ultra conserved regions: novel non-coding RNA players in neuroblastoma progression. Noncoding RNA. 2019;5:39. doi: 10.3390/ncrna5020039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molenaar J.J., Domingo-Fernandez R., Ebus M.E., Lindner S., Koster J., Drabek K., Mestdagh P., van Sluis P., Valentijn L.J., van Nes J., et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat. Genet. 2012;44:1199–1206. doi: 10.1038/ng.2436. [DOI] [PubMed] [Google Scholar]
- 42.Powers J.T., Tsanov K.M., Pearson D.S., Roels F., Spina C.S., Ebright R., Seligson M., de Soysa Y., Cahan P., TheiBen J., et al. Multiple mechanisms disrupt the let-7 microRNA family in neuroblastoma. Nature. 2016;535:246–251. doi: 10.1038/nature18632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cole K.A., Attiyeh E.F., Mosse Y.P., Laquaglia M.J., Diskin S.J., Brodeur G.M., Maris J.M. A functional screen identifies miR-34a as a candidate neuroblastoma tumor suppressor gene. Mol. Cancer Res. 2008;6:735–742. doi: 10.1158/1541-7786.mcr-07-2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Antonellis P., Carotenuto M., Vandenbussche J., De Vita G., Ferrucci V., Medaglia C., Boffa I., Galiero A., Di Somma S., Magliulo D., et al. Early targets of miR-34a in neuroblastoma. Mol. Cell. Proteomics. 2014;13:2114–2131. doi: 10.1074/mcp.m113.035808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nolan J.C., Salvucci M., Carberry S., Barat A., Segura M.F., Fenn J., Prehn J.H.M., Stallings R.L., Piskareva O. A context-dependent role for MiR-124-3p on cell phenotype, viability and chemosensitivity in neuroblastoma in vitro. Front. Cell Dev Biol. 2020;8:559553. doi: 10.3389/fcell.2020.559553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang H., Pu J., Qi T., Qi M., Yang C., Li S., Huang K., Zheng L., Tong Q. MicroRNA-145 inhibits the growth, invasion, metastasis and angiogenesis of neuroblastoma cells through targeting hypoxia-inducible factor 2 alpha. Oncogene. 2014;33:387–397. doi: 10.1038/onc.2012.574. [DOI] [PubMed] [Google Scholar]
- 47.Althoff K., Lindner S., Odersky A., Mestdagh P., Beckers A., Karczewski S., Molenaar J.J., Bohrer A., Knauer S., Speleman F., et al. miR-542-3p exerts tumor suppressive functions in neuroblastoma by downregulating Survivin. Int. J. Cancer. 2015;136:1308–1320. doi: 10.1002/ijc.29091. [DOI] [PubMed] [Google Scholar]
- 48.Ryan J., Tivnan A., Fay J., Bryan K., Meehan M., Creevey L., Lynch J., Bray I.M., O'Meara A., Davidoff A.M., Stallings R.L. MicroRNA-204 increases sensitivity of neuroblastoma cells to cisplatin and is associated with a favourable clinical outcome. Br. J. Cancer. 2012;107:967–976. doi: 10.1038/bjc.2012.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee J.J., Drakaki A., Iliopoulos D., Struhl K. MiR-27b targets PPARgamma to inhibit growth, tumor progression and the inflammatory response in neuroblastoma cells. Oncogene. 2012;31:3818–3825. doi: 10.1038/onc.2011.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Challagundla K.B., Wise P.M., Neviani P., Chava H., Murtadha M., Xu T., Kennedy R., Ivan C., Zhang X., Vannini I., et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J. Natl. Cancer Inst. 2015;107:djv135. doi: 10.1093/jnci/djv135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mehta A., Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat. Rev. Immunol. 2016;16:279–294. doi: 10.1038/nri.2016.40. [DOI] [PubMed] [Google Scholar]
- 52.Xu S., Tao Z., Hai B., Liang H., Shi Y., Wang T., Song W., Chen Y., OuYang J., Chen J., et al. miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat. Commun. 2016;7:11406. doi: 10.1038/ncomms11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Richard H., Pokhrel A., Chava S., Pathania A., Katta S.S., Challagundla K.B. Exosomes: novel players of therapy resistance in neuroblastoma. Adv. Exp. Med. Biol. 2020;1277:75–85. doi: 10.1007/978-3-030-50224-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gunda V., Pathania A.S., Chava S., Prathipati P., Chaturvedi N.K., Coulter D.W., Pandey M.K., Durden D.L., Challagundla K.B. Amino acids regulate cisplatin insensitivity in neuroblastoma. Cancers (Basel) 2020;12:E2576. doi: 10.3390/cancers12092576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rupaimoole R., Slack F.J. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 56.Togashi Y., Shitara K., Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019;16:356–371. doi: 10.1038/s41571-019-0175-7. [DOI] [PubMed] [Google Scholar]
- 57.Patel S.P., Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 2015;14:847–856. doi: 10.1158/1535-7163.mct-14-0983. [DOI] [PubMed] [Google Scholar]
- 58.Davis A.A., Patel V.G. The role of PD-L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J. Immunother. Cancer. 2019;7:278. doi: 10.1186/s40425-019-0768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McClanahan F., Hanna B., Miller S., Clear A.J., Lichter P., Gribben J.G., Seiffert M. PD-L1 checkpoint blockade prevents immune dysfunction and leukemia development in a mouse model of chronic lymphocytic leukemia. Blood. 2015;126:203–211. doi: 10.1182/blood-2015-01-622936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen G., Huang A.C., Zhang W., Zhang G., Wu M., Xu W., Yu Z., Yang J., Wang B., Sun H., et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–386. doi: 10.1038/s41586-018-0392-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Juneja V.R., McGuire K.A., Manguso R.T., LaFleur M.W., Collins N., Haining W.N., Freeman G.J., Sharpe A.H. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 2017;214:895–904. doi: 10.1084/jem.20160801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.v18/05/2020. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-first-line-treatment-metastatic-nsclc-high-pd-l1-expression; 2020. [Accessed 17 April 2022].
- 63.v16/02/2018. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-durvalumab-after-chemoradiation-unresectable-stage-iii-nsclc; 2018. [Accessed 17 April 2022].
- 64.v30/06/2020. https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-avelumab-urothelial-carcinoma-maintenance-treatment; 2020. [Accessed 17 April 2022].
- 65.v14/05/2019. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-avelumab-plus-axitinib-renal-cell-carcinoma; 2019. [Accessed 17 April 2022].
- 66.Akinleye A., Rasool Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019;12:92. doi: 10.1186/s13045-019-0779-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Puccini A., Battaglin F., Iaia M.L., Lenz H.J., Salem M.E. Overcoming resistance to anti-PD1 and anti-PD-L1 treatment in gastrointestinal malignancies. J. Immunother. Cancer. 2020;8:e000404. doi: 10.1136/jitc-2019-000404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu B., Tang L., Chen S., Yin C., Peng S., Li X., Liu T., Liu W., Han C., Stawski L., et al. Targeting the upstream transcriptional activator of PD-L1 as an alternative strategy in melanoma therapy. Oncogene. 2018;37:4941–4954. doi: 10.1038/s41388-018-0314-0. [DOI] [PubMed] [Google Scholar]
- 69.Arens R., Scheeren F.A. Genetic screening for novel regulators of immune checkpoint molecules. Trends Immunol. 2020;41:692–705. doi: 10.1016/j.it.2020.06.005. [DOI] [PubMed] [Google Scholar]
- 70.Chava S., Reynolds C.P., Pathania A.S., Gorantla S., Poluektova L.Y., Coulter D.W., Gupta S.C., Pandey M.K., Challagundla K.B. miR-15a-5p, miR-15b-5p, and miR-16-5p inhibit tumor progression by directly targeting MYCN in neuroblastoma. Mol. Oncol. 2020;14:180–196. doi: 10.1002/1878-0261.12588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Neviani P., Wise P.M., Murtadha M., Liu C.W., Wu C.H., Jong A.Y., Seeger R.C., Fabbri M. Natural Killer-derived exosomal miR-186 inhibits neuroblastoma growth and immune escape mechanisms. Cancer Res. 2018;79:1151–1164. doi: 10.1158/0008-5472.can-18-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Camisaschi C., Renne S.L., Beretta V., Rini F., Spagnuolo R.D., Tuccitto A., Podda M.G., Parmiani G., Rivoltini L., Collini P., et al. Immune landscape and in vivo immunogenicity of NY-ESO-1 tumor antigen in advanced neuroblastoma patients. BMC Cancer. 2018;18:983. doi: 10.1186/s12885-018-4910-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Facchetti P., Prigione I., Ghiotto F., Tasso P., Garaventa A., Pistoia V. Functional and molecular characterization of tumour-infiltrating lymphocytes and clones thereof from a major-histocompatibility-complex-negative human tumour: neuroblastoma. Cancer Immunol. Immunother. 1996;42:170–178. doi: 10.1007/s002620050267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Layer J.P., Kronmuller M.T., Quast T., van den Boorn-Konijnenberg D., Effern M., Hinze D., Althoff K., Schramm A., Westermann F., Peifer M., et al. Amplification of N-Myc is associated with a T-cell-poor microenvironment in metastatic neuroblastoma restraining interferon pathway activity and chemokine expression. Oncoimmunology. 2017;6:e1320626. doi: 10.1080/2162402x.2017.1320626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Olle Hurtado M., Wolbert J., Fisher J., Flutter B., Stafford S., Barton J., Jain N., Barone G., Majani Y., Anderson J. Tumor infiltrating lymphocytes expanded from pediatric neuroblastoma display heterogeneity of phenotype and function. PLoS One. 2019;14:e0216373. doi: 10.1371/journal.pone.0216373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dondero A., Pastorino F., Della Chiesa M., Corrias M.V., Morandi F., Pistoia V., Olive D., Bellora F., Locatelli F., Castellano A., et al. PD-L1 expression in metastatic neuroblastoma as an additional mechanism for limiting immune surveillance. Oncoimmunology. 2016;5:e1064578. doi: 10.1080/2162402x.2015.1064578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moghimi B., Muthugounder S., Jambon S., Tibbetts R., Hung L., Bassiri H., Hogarty M.D., Barrett D.M., Shimada H., Asgharzadeh S. Preclinical assessment of the efficacy and specificity of GD2-B7H3 SynNotch CAR-T in metastatic neuroblastoma. Nat. Commun. 2021;12:511. doi: 10.1038/s41467-020-20785-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pistoia V., Morandi F., Bianchi G., Pezzolo A., Prigione I., Raffaghello L. Immunosuppressive microenvironment in neuroblastoma. Front. Oncol. 2013;3:167. doi: 10.3389/fonc.2013.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Raffaghello L., Prigione I., Airoldi I., Camoriano M., Morandi F., Bocca P., Gambini C., Ferrone S., Pistoia V. Mechanisms of immune evasion of human neuroblastoma. Cancer Lett. 2005;228:155–161. doi: 10.1016/j.canlet.2004.11.064. [DOI] [PubMed] [Google Scholar]
- 80.Wienke J., Dierselhuis M.P., Tytgat G.A.M., Kunkele A., Nierkens S., Molenaar J.J. The immune landscape of neuroblastoma: challenges and opportunities for novel therapeutic strategies in pediatric oncology. Eur. J. Cancer. 2021;144:123–150. doi: 10.1016/j.ejca.2020.11.014. [DOI] [PubMed] [Google Scholar]
- 81.Peters L., Meister G. Argonaute proteins: mediators of RNA silencing. Mol. Cell. 2007;26:611–623. doi: 10.1016/j.molcel.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 82.Martinez-Lostao L., Anel A., Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res. 2015;21:5047–5056. doi: 10.1158/1078-0432.ccr-15-0685. [DOI] [PubMed] [Google Scholar]
- 83.Menares E., Galvez-Cancino F., Caceres-Morgado P., Ghorani E., Lopez E., Diaz X., Saavedra-Almarza J., Figueroa D.A., Roa E., Quezada S.A., Lladser A. Tissue-resident memory CD8(+) T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat. Commun. 2019;10:4401. doi: 10.1038/s41467-019-12319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Anfossi N., Andre P., Guia S., Falk C.S., Roetynck S., Stewart C.A., Breso V., Frassati C., Reviron D., Middleton D., et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25:331–342. doi: 10.1016/j.immuni.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 85.Petrovas C., Casazza J.P., Brenchley J.M., Price D.A., Gostick E., Adams W.C., Precopio M.L., Schacker T., Roederer M., Douek D.C., Koup R.A. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 2006;203:2281–2292. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mariotti F.R., Petrini S., Ingegnere T., Tumino N., Besi F., Scordamaglia F., Munari E., Pesce S., Marcenaro E., Moretta A., et al. PD-1 in human NK cells: evidence of cytoplasmic mRNA and protein expression. Oncoimmunology. 2019;8:1557030. doi: 10.1080/2162402x.2018.1557030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cohnen A., Chiang S.C., Stojanovic A., Schmidt H., Claus M., Saftig P., JanBen O., Cerwenka A., Bryceson Y.T., Watzl C. Surface CD107a/LAMP-1 protects natural killer cells from degranulation-associated damage. Blood. 2013;122:1411–1418. doi: 10.1182/blood-2012-07-441832. [DOI] [PubMed] [Google Scholar]
- 88.Alter G., Malenfant J.M., Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 89.Porter A.G., Janicke R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 90.Smyth M.J., Kelly J.M., Baxter A.G., Korner H., Sedgwick J.D. An essential role for tumor necrosis factor in natural killer cell-mediated tumor rejection in the peritoneum. J. Exp. Med. 1998;188:1611–1619. doi: 10.1084/jem.188.9.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang R., Jaw J.J., Stutzman N.C., Zou Z., Sun P.D. Natural killer cell-produced IFN-gamma and TNF-alpha induce target cell cytolysis through up-regulation of ICAM-1. J. Leukoc. Biol. 2012;91:299–309. doi: 10.1189/jlb.0611308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jewett A., Gan X.H., Lebow L.T., Bonavida B. Differential secretion of TNF-alpha and IFN-gamma by human peripheral blood-derived NK subsets and association with functional maturation. J. Clin. Immunol. 1996;16:46–54. doi: 10.1007/bf01540972. [DOI] [PubMed] [Google Scholar]
- 93.Sullivan R.P., Leong J.W., Schneider S.E., Keppel C.R., Germino E., French A.R., Fehniger T.A. MicroRNA-deficient NK cells exhibit decreased survival but enhanced function. J. Immunol. 2012;188:3019–3030. doi: 10.4049/jimmunol.1102294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Garcia-Diaz A., Shin D.S., Moreno B.H., Saco J., Escuin-Ordinas H., Rodriguez G.A., Zaretsky J.M., Sun L., Hugo W., Wang X., et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–1201. doi: 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen J., Feng Y., Lu L., Wang H., Dai L., Li Y., Zhang P. Interferon-gamma-induced PD-L1 surface expression on human oral squamous carcinoma via PKD2 signal pathway. Immunobiology. 2012;217:385–393. doi: 10.1016/j.imbio.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 96.Abiko K., Matsumura N., Hamanishi J., Horikawa N., Murakami R., Yamaguchi K., Yoshioka Y., Baba T., Konishi I., Mandai M. IFN-gamma from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br. J. Cancer. 2015;112:1501–1509. doi: 10.1038/bjc.2015.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yu M., Peng Z., Qin M., Liu Y., Wang J., Zhang C., Lin J., Dong T., Wang L., Li S., et al. Interferon-gamma induces tumor resistance to anti-PD-1 immunotherapy by promoting YAP phase separation. Mol. Cell. 2021;81:1216–1230.e9. doi: 10.1016/j.molcel.2021.01.010. [DOI] [PubMed] [Google Scholar]
- 98.Ackermann S., Cartolano M., Hero B., Welte A., Kahlert Y., Roderwieser A., Bartenhagen C., Walter E., Gecht J., Kerschke L., et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science. 2018;362:1165–1170. doi: 10.1126/science.aat6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Asgharzadeh S., Pique-Regi R., Sposto R., Wang H., Yang Y., Shimada H., Matthay K., Buckley J., Ortega A., Seeger R.C. Prognostic significance of gene expression profiles of metastatic neuroblastomas lacking MYCN gene amplification. J. Natl. Cancer Inst. 2006;98:1193–1203. doi: 10.1093/jnci/djj330. [DOI] [PubMed] [Google Scholar]
- 100.Molenaar J.J., Koster J., Zwijnenburg D.A., van Sluis P., Valentijn L.J., van der Ploeg I., Hamdi M., van Nes J., Westerman B.A., van Arkel J., et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483:589–593. doi: 10.1038/nature10910. [DOI] [PubMed] [Google Scholar]
- 101.Henrich K.O., Bender S., Saadati M., Dreidax D., Gartlgruber M., Shao C., Herrmann C., Wiesenfarth M., Parzonka M., Wehrmann L., et al. Integrative genome-scale Analysis identifies epigenetic mechanisms of transcriptional deregulation in unfavorable neuroblastomas. Cancer Res. 2016;76:5523–5537. doi: 10.1158/0008-5472.can-15-2507. [DOI] [PubMed] [Google Scholar]
- 102.Brodeur G.M., Pritchard J., Berthold F., Carlsen N.L., Castel V., Castelberry R.P., De Bernardi B., Evans A.E., Favrot M., Hedborg F. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. 1993;11:1466–1477. doi: 10.1200/jco.1993.11.8.1466. [DOI] [PubMed] [Google Scholar]
- 103.Monclair T., Brodeur G.M., Ambros P.F., Brisse H.J., Cecchetto G., Holmes K., Kaneko M., London W.B., Matthay K.K., Nuchtern J.G., et al. The international neuroblastoma risk group (INRG) staging system: an INRG task force report. J. Clin. Oncol. 2009;27:298–303. doi: 10.1200/jco.2008.16.6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pathania A.S., Wani Z.A., Guru S.K., Kumar S., Bhushan S., Korkaya H., Seals D.F., Kumar A., Mondhe D.M., Ahmed Z., et al. The anti-angiogenic and cytotoxic effects of the boswellic acid analog BA145 are potentiated by autophagy inhibitors. Mol. Cancer. 2015;14:6. doi: 10.1186/1476-4598-14-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 106.Weidner N., Semple J.P., Welch W.R., Folkman J. Tumor angiogenesis and metastasis--correlation in invasive breast carcinoma. N. Engl. J. Med. 1991;324:1–8. doi: 10.1056/nejm199101033240101. [DOI] [PubMed] [Google Scholar]
- 107.Weidner N., Folkman J., Pozza F., Bevilacqua P., Allred E.N., Moore D.H., Meli S., Gasparini G. Tumor angiogenesis: a new significant and independent prognostic indicator in early-stage breast carcinoma. J. Natl. Cancer Inst. 1992;84:1875–1887. doi: 10.1093/jnci/84.24.1875. [DOI] [PubMed] [Google Scholar]
- 108.Danaher P., Warren S., Lu R., Samayoa J., Sullivan A., Pekker I., Wallden B., Marincola F.M., Cesano A. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): results from The Cancer Genome Atlas (TCGA) J Immunother Cancer. 2018;6:63. doi: 10.1186/s40425-018-0367-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rokita J.L., Rathi K.S., Cardenas M.F., Upton K.A., Jayaseelan J., Cross K.L., Pfeil J., Egolf L.E., Way G.P., Farrel A., Kendsersky N.M., et al. Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design. Cell Rep. 2019;29:1675–1689.e1679. doi: 10.1016/j.celrep.2019.09.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Micke P., Strell C., Mattsson J., Martin-Bernabe A., Brunnstrom H., Huvila J., Sund M., Warnberg F., Ponten F., Glimelius B., Hrynchyk I., et al. The prognostic impact of the tumour stroma fraction: A machine learning-based analysis in 16 human solid tumour types. EBioMedicine. 2021;65:103269. doi: 10.1016/j.ebiom.2021.103269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data were generated by the authors and are available upon request from the corresponding author.