

Abstract
To find new potential tyrosinase inhibitors, a diverse range of 2-arylchromone-4-thione derivatives (2a–2p) were designed and synthesized by employing a multistep strategy, and the newly synthesized compounds, for the first time, were screened in vitro for their tyrosinase inhibitory activity. In this context, the newly synthesized compounds (2a–2p) were characterized using a combination of several spectroscopic techniques including Fourier transform infrared, UV–vis, 1H NMR, and 13C NMR spectroscopies and electron ionization–mass spectrometry. All the target compounds were potent against tyrosinase as compared to the standard inhibitor kojic acid (half-maximal inhibitory concentration (IC50) = 12.6 ± 0.6 μM). The compounds (2a–2p) produced IC50 values in the range from 1.12 ± 0.04 to 5.68 ± 0.13 μM. Among the synthesized 4-thioflavones and 4-thioflavonols, the compound 2n exhibited excellent tyrosinase inhibitory activity with the lowest IC50 of 1.12 ± 0.04 μM that could be recommended as potential lead candidates to cure tyrosinase-mediated hyperpigmentation in the future. A kinetic study of compound 2n revealed that compound 2n inhibited tyrosinase in a competitive mode. Furthermore, the nontoxic performance of the most beneficial compounds ranging from 1 to 25 g/mL was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide test method for A375 human melanoma cells for the highly efficient target compounds (2m, 2n, 2o, and 2p). Moreover, a molecular modeling study was performed against tyrosinase enzyme (2Y9X) to check the binding interactions of the synthesized compounds (2a–2p) against the target protein. Furthermore, quantitative structure-activity relationship studies were conducted based on an antityrosinase assay. The value of the correlation coefficient (R2) 0.9997 shows that there was a good correlation between (2a–2p) structures and selected properties. The geometry optimization of all complexes was performed by using Gaussian 09. Additionally, a drug-likeness research was used to establish the potent analogues’ positive action as a new antityrosinase agent (2n, 2o, and 2p).
1. Introduction
Tyrosinase (EC 1.14.18.1) is a dinuclear copper-containing metalloenzyme that is found abundantly in nature and plays an influential role in the biosynthesis pathway of melanin pigment (Raper-Mason pathway).1 The melanogenesis process is catalyzed by the tyrosinase enzyme, and it is thus comprised of two steps. Initially, o-diphenol conversion takes place from monophenol by hydroxylation (monophenolase activity), and subsequently, o-diphenol gets converted into o-quinone (o-diphenolase activity) by an oxidation process; finally, the formed quinone product is the key species for the synthesis of the melanin pigment.2 Melanin as the main natural biological pigment has a high determining role in skin and hair pigmentation,3 which results in a photoprotective function in humans.4 However, the accumulation of an abnormal amount of melanin causes dermatological disorders such as freckles, melasma, lentigo, etc.5 and increases the risk of cancer and other skin diseases.6 Also, recent studies reported that problems associated with melanogenesis are linked to the neurodegenerative diseases including Alzheimer’s, Parkinson’s, and Huntington’s diseases. Although melanogenesis is a complicated process, it is highly accepted that the tyrosinase enzyme regulates the biosynthetic pathway for melanin formation.7 Moreover, tyrosinase is responsible for enzymatic reactions that brown perished fruits and vegetables. Since tyrosinase catalyzes rate-limiting steps of melanogenesis, it has been recognized as a therapeutic target to control melanin synthesis. Therefore, tyrosinase inhibition provides a promising therapeutic potential in pigmentation disorders, skin cancers, and neurodegenerative disorders. Furthermore, tyrosinase inhibitors have become increasingly important in the food industry as well as in the medicinal and cosmetic products due to decreasing the excessive accumulation of pigmentation resulting from the enzyme action.8−12 Over the past decades, many tyrosinase inhibitors have been reported, but only a few possess enough potency and safety in the application of food and pharmaceutical industries.13 Moreover, the undesirable phenomena related to melanin have persuaded researchers to search for new potent tyrosinase inhibitors used in skin whitening and antibrowning of foods.14
Moreover, hyperpigmentation is one of the world’s most serious health issues. Despite multiple studies on anti-hyperpigmentation drug synthesis, there is still a high need for the design, synthesis, and discovery of effective anti-hyperpigmentation medications to address the difficulties associated with existing commercial treatments, such as toxicity and drug resistance.15−17 Therefore, controlling a tyrosinase-dependent mechanism of melanogenesis might be the basis for a potential antimelanoma therapy, and the inhibition of melanogenesis is one reasonable method to cure hyperpigmentation. A large number of tyrosinase inhibitors have been discovered and reported; however, because of their side effects only a few of these inhibitors have been commercially applied.18−21,22a Therefore, the above-mentioned problems prompted us to develop new safer, more effective, and cheaper tyrosinase inhibitors. Some substances have been well-described as tyrosinase inhibitors such as kojic acid and arbutin; however, these compounds possess undesirable side effects, including carcinogenicity and cytotoxicity.23
In this context, in order to treat the ailments related to overpigmentation in human bodies, there is a strong urge to design and develop new, stable, safe, and efficient tyrosinase inhibitors with high efficacy. Also, it appears as an important objective in the food and medicinal chemistry.
Moreover, numerous sulfur-containing tyrosinase inhibitors have been reported in the literature. It is well-known that oxidation caused by reactive oxygen species (ROS) is a major cause of cellular aging, melanogenesis, mutagenesis, and carcinogenesis.24 Sulfur-containing compounds usually possess good antioxidant and antimelanogenic activities, which can be speculated by their ability to scavenge free radicals or by their capacity to affect key redox enzymes and chelate metal ions.25 Interestingly, recent studies have highlighted that sulfur-containing small molecules played a key role in a wide range of fundamental biological functions, which intensely attracted many researchers to develop new thio-compounds in disease therapy and prevention.26 The sulfur atom of the compound irreversibly binds/complexes to binuclear copper ions in the active site of tyrosinase and inhibits the activity.27 Most of those compounds have an inherent function in chelating metal ions, which plays a critical role in the inhibition of tyrosinase.
Encouraged by these findings, we decided to extend the structural diversity by the replacement of oxygen of the chromone scaffold with sulfur at position 4. The scientists recently investigated the structural effects of replacing an oxygen atom with a sulfur atom.22a−22c,28 This issue may become crucial in drug design, since the H-bond acceptor (HNA) capacity of the O and S atoms are different. Furthermore, because the X–C bond length is significantly longer in the case of X=S than with X=O, the increased ring size may affect the fit of the ligand to the receptor.
During recent years, compounds containing a chromone moiety have become potential targets for many medicinal chemists. The significance of chromone derivatives lies in their varied biological activities and a unique position in heterocyclic chemistry.29−35 Among various chromones, the 4H-chromone core has acquired a superior position owing to its wide-ranging variety of bioactivity.36 The chromone nucleus is the essential part of many therapeutically important agents. Despite a wide range of activities, thiochromones have been less attended.37 In this context, 4-thioflavones and 4-thioflavonols (2-arylchromones) (Figure 1) are interesting targets because of their diverse biological properties and easy access from the corresponding oxygen analogues;38 thus, they have been reported to possess potent biological properties such as anticancer, antimicrobial, green pesticides, etc.39−41
Figure 1.

Representative structures of (a) thioflavone and (b) thioflavonol.
Several research groups are now pursuing various methods for flavone and flavonol syntheses and structural modification of the chromone ring in order to better understand the various roles of thiochromones.41−49 As a result, a naturally obtained flavone moiety with a wide range of biological activities can be used as a starting point for the synthesis of purely synthetic flavone derivatives with various functional groups at various positions on their skeleton.50 However, even though the conversion of the carbonyl (C=O) group in the 2-phenylchromone scaffold into the thiocarbonyl (C=S) group has been typically described as a viable approach,31,33,51−54 the inhibitory potential against the tyrosinase enzyme of sulfur-containing flavones and flavonols has been not reported yet.
The potential pharmacological significance and the reduced amount of synthetic work on these compounds have motivated us to synthesize these new compounds and also assess their role as a mushroom tyrosinase enzyme inhibitor. Furthermore, because of the noteworthy importance of flavone and flavonol derivatives in medicinal chemistry and our continuous effort to develop effective tyrosinase inhibitors, we intended to discover the tyrosinase inhibition potential of 4-thioflavones and 4-thioflavonols, which has not been reported. Likewise, it is also required to study the effect of oxygen replacement with sulfur and various substituents on the tyrosinase inhibitory potential of these scaffolds. The biological results have also been validated by computational studies.
2. Results and Discussion
2.1. Chemistry
The complete synthesis of target compounds is depicted in Scheme 1. The first step involves the base-catalyzed condensation of substituted 2′-hydroxyacetophenone with different aryl aldehydes in methanol solvent to produce intermediate substituted 2′-hydroxychalcones. From these synthesized chalcone derivatives, varyingly substituted flavone and flavonol derivatives were synthesized as follows. In order to prepare flavone derivatives, 2′-hydroxychalcones were treated with an I2–dimethyl sulfoxide (DMSO) mixture as the oxidizing system at 140 °C to get the title compounds (1a–1g) in moderate to good yields. In the case of flavonols, substituted 2-hydroxychalcones were produced in situ from aromatic aldehyde and 2′-hydroxyacetophenone and then transformed into the required flavonols (1h–1p) via a one-pot synthesis using a hydrogen peroxide (H2O2, 35%) solution. Before we proceeded to the next step, both flavones (1a–1g) and flavonols (1h–1p) were purified through recrystallization in ethanol and characterized by UV, Fourier transform infrared (FTIR), and NMR spectroscopic techniques. Eventually, 2-arylchromes (flavones and flavonols) were treated individually with Lawesson’s reagent in the presence of anhydrous toluene. The reaction mixture was refluxed for 20–24 h under an inert atmosphere to produce the desired 2-arylchromone-4-thiones (4-thioflavones (2a–2g) and 4-thioflavonols (2h–2p)) in good to excellent yields. These target compounds were purified through recrystallization in ethanol and afterward were characterized by UV–vis, FTIR, and NMR spectroscopic techniques. The molecular masses of the final 2-arylchromone-4-thiones were determined by Electron Ionization (EI) mass spectrometry. Overall, all the obtained spectroscopic data unequivocally corroborate the structures of newly synthesized 4-thioflavones (2a–2g) and 4-thioflavonols (2h–2p).
Scheme 1. Synthesis of Flavones (1a–1g), Flavonols (1h–1p), Thioflavones (2a–2g), and Thioflavonols (2h–2p).
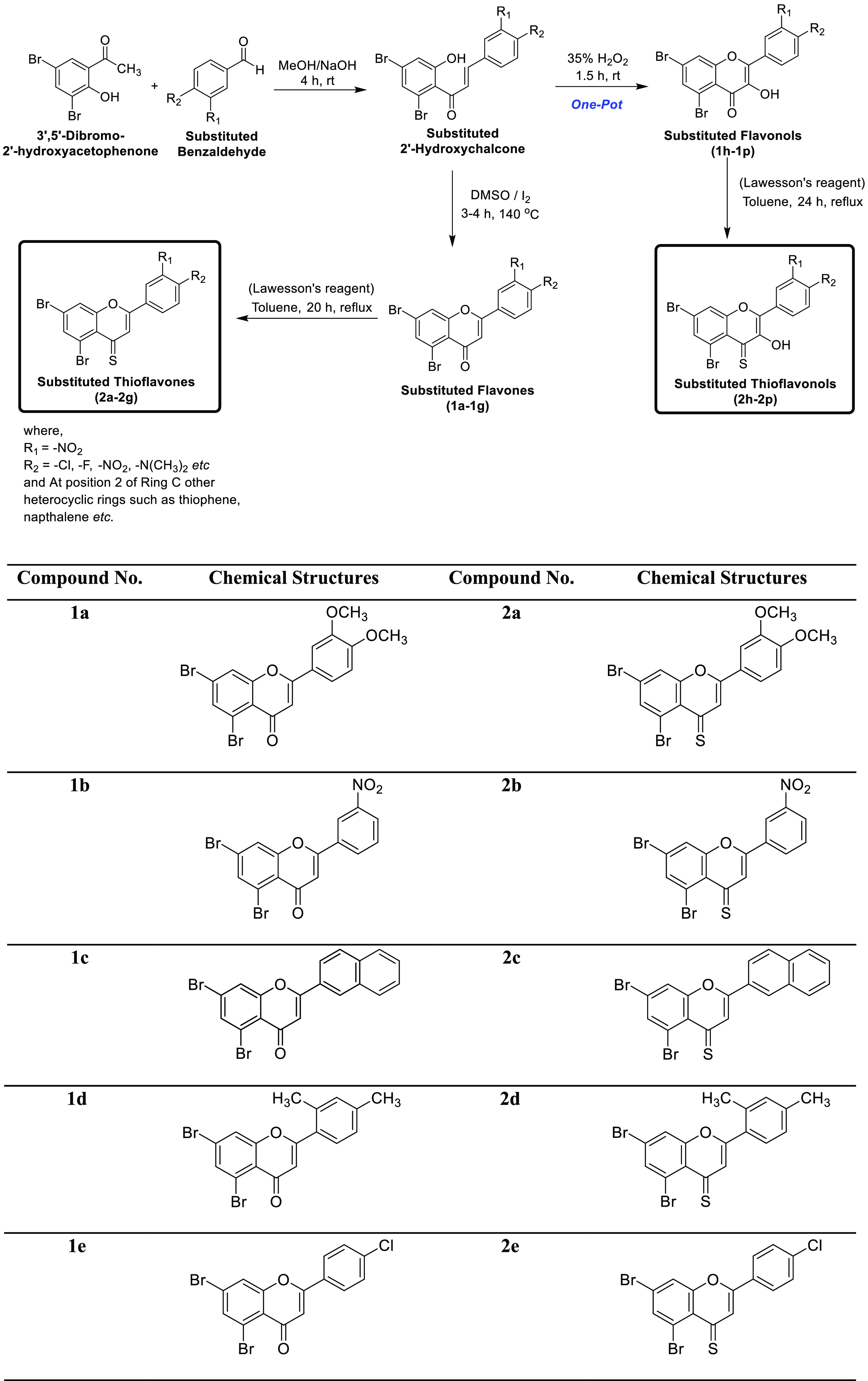
The spectral data of already known substituted flavones (1a–1g) and flavonols (1h–1p) have been reported in the literature.22a,35 However, the spectral data of the newly synthesized 4-thioflavones (2a–2g) and 4-thioflavonols (2h–2p) are given below.
5,7-Dibromo-2-(3,4-dimethoxyphenyl)-4H-chromene-4-thione (2a)
Yellow crystalline solid; yield: 86%; mp 238–240 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.9; UV–vis λmax (CH2Cl2) = 379 nm; IR (KBr, cm–1) vmax: 3057, 1573, 1541, 1447, 1263, 1181, 781, 676; 1H NMR (400 MHz, DMSO-d6): δ 8.47 (d, J = 4.0 Hz, 1H, Ar–H), 8.13 (d, J = 4.0 Hz, 1H, Ar–H), 7.92 (dd, J = 4.0, 8.0 Hz, 1H, Ar–H), 7.76 (d, J = 4.0 Hz, 1H, Ar–H), 7.24 (d, J = 8.0 Hz, 1H, Ar–H), 6.57 (s, 1H, −C=CH), 3.93 (s, 3H, OMe), 3.91 (s, 3H, OMe); 13C NMR (101 MHz, DMSO-d6): δ 188.2, 148.4, 146.2, 144.8, 140.0, 138.2, 137.8, 134.5, 131.5, 130.5, 128.0, 127.8, 126.5, 120.0, 114.2, 56.5, 56.3; accurate mass (EI-MS) of [M]·+: Calcd. for C17H1279Br2O3S 453.8874; found 453.8865.
5,7-Dibromo-2-(3-nitrophenyl)-4H-chromene-4-thione (2b)
Green amorphous solid; yield: 78%; mp 155–157 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.7; UV–vis λmax (CH2Cl2) = 361 nm; IR (KBr, cm–1) vmax: 3042, 1645, 1595, 1440, 1304, 1193, 774, 685; 1H NMR (400 MHz, DMSO-d6): δ 9.00 (d, J = 4.0 Hz, 1H, Ar–H), 8.93 (d, J = 4.0 Hz, 1H, Ar–H), 8.48 (d, J = 4.0 Hz, 1H, Ar–H), 8.15 (d, J = 4.0 Hz, 1H, Ar–H), 7.96–7.92 (m, 2H, Ar–H), 7.46 (s, 1H, −C=CH); 13C NMR (101 MHz, DMSO-d6): δ 186.7, 151.6, 148.7, 147.6. 145.8, 141.0, 138.4, 137.1, 132.5, 131.0, 130.5, 128.5, 127.0, 125.6, 119.1, 115.0; accurate mass (EI-MS) of [M]+·: Calcd. for C15H779Br2NO3S 438.8513; found 438.8517.
5,7-Dibromo-2-(naphthalen-2-yl)-4H-chromene-4-thione (2c)
Green solid; yield: 88%; mp 165–167 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.6; UV–vis λmax (CH2Cl2) = 332 nm; IR (KBr, cm–1) vmax: 3067, 1662, 1585, 1440, 1262, 1196, 798, 685; 1H NMR (400 MHz, DMSO-d6): δ 8.51 (d, J = 4.0 Hz, 1H, Ar–H), 8.18 (d, J = 4.0 Hz, 1H, Ar–H), 7.68–7.55 (m, 4H, Ar–H), 7.28–7.20 (m, 3H, Ar–H), 6.58 (s, 1H, −C=CH); 13C NMR (101 MHz, DMSO-d6): δ 188.1, 148.6, 147.0, 141.5, 139.2, 138.4, 133.3, 132.0, 130.6, 130.0, 128.5, 128.0, 127.6, 127.5, 126.6, 125.2, 125.1, 118.8, 114.5; accurate mass (EI-MS) of [M]·+: Calcd. for C19H1079Br2OS 443.8819; found 443.8804.
5,7-Dibromo-2-(2,4-dimethylphenyl)-4H-chromene-4-thione (2d)
Yellow solid; yield: 82%; mp 215–217 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.9; UV–vis λmax (CH2Cl2) = 383 nm; IR (KBr, cm–1) vmax: 3068, 1646, 1589, 1495, 1302, 1193, 763, 669; 1H NMR (400 MHz, DMSO-d6): δ 8.43 (d, J = 4.0 Hz, 1H, Ar–H), 8.17 (d, J = 4.0 Hz, 1H, Ar–H), 7.65 (d, J = 4.0 Hz, 1H, Ar–H), 7.61 (d, J = 8.0 Hz, 1H, Ar–H), 6.90 (dd, J = 4.0, 8.0 Hz, 1H, Ar–H), 6.64 (s, 1H, −C=CH), 2.54 (s, 3H, CH3), 2.51 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6): δ 185.8, 149.2, 147.8, 145.6, 141.7, 138.5, 137.0, 132.6, 131.1, 129.1, 128.5, 127.2, 126.1, 119.3, 113.8, 21.8, 21.6; accurate mass (EI-MS) of [M]·+: Calcd. for C17H1279Br2OS 421.8976; found 421.8961.
5,7-Dibromo-2-(4-chlorophenyl)-4H-chromene-4-thione (2e)
Green solid; yield: 74%; mp 195–197 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.6; UV–vis λmax (CH2Cl2) = 391 nm; IR (KBr, cm–1) vmax: 3067, 1646, 1527, 1475, 1274, 1170, 780, 701; 1H NMR (400 MHz, DMSO-d6): δ 8.47 (d, J = 4.0 Hz, 1H, Ar–H), 8.29 (d, J = 8.0 Hz, 2H, Ar–H), 8.12 (d, J = 4.0 Hz, 1H, Ar–H), 7.76 (d, J = 8.0 Hz, 2H, Ar–H), 6.59 (s, 1H, −C=CH); 13C NMR (101 MHz, DMSO-d6): δ 188.1, 150.2, 148.5, 145.2, 141.7, 138.5, 132.0, 130.7, 128.4, 127.6, 126.0, 118.8, 114.6; accurate mass (EI-MS) of [M]·+: Calcd. for C15H779Br235ClOS 427.8273; found 427.8260.
5,7-Dibromo-2-(thiophen-2-yl)-4H-chromene-4-thione (2f)
Green amorphous solid; yield: 72%; mp 236–238 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.7; UV–vis λmax (CH2Cl2) = 355 nm; IR (KBr, cm–1) vmax: 3076, 1645, 1599, 1487, 1305, 1186, 791, 682; 1H NMR (400 MHz, DMSO-d6): δ 8.42 (d, J = 4.0 Hz, 1H, Ar–H), 8.22 (dd, J = 4.0, 8.0 Hz, 1H, Ar–H), 8.15 (d, J = 4.0 Hz, 1H, Ar–H), 7.46–7.43 (m, 2H, Ar–H), 6.59 (s, 1H, −C=CH); 13C NMR (101 MHz, DMSO-d6): δ 183.5, 149.6, 147.8, 144.3, 139.5, 133.0, 131.2, 130.0, 129.5, 127.0, 126.7, 119.0, 114.2; accurate mass (EI-MS) of [M]·+: Calcd. for C13H679Br2OS2 399.8227; found 399.8224.
5,7-Dibromo-2-(1-tosyl-1H-indol-6-yl)-4H-chromene-4-thione (2g)
Red solid; yield: 80%; mp 147–149 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.8; UV–vis λmax (CH2Cl2) = 331 nm; IR (KBr, cm–1) vmax: 2922, 1659, 1595, 1439, 1298, 1168, 787, 652; 1H NMR (400 MHz, DMSO-d6): δ 8.60 (d, J = 4.0 Hz, 1H, Ar–H), 8.47 (d, J = 4.0 Hz, 1H, Ar–H), 8.30 (d, J = 8.0 Hz, 2H, Ar–H), 8.14 (d, J = 4.0 Hz, 1H, Ar–H), 8.00 (d, J = 8.0 Hz, 2H, Ar–H), 7.75 (s, 1H, Ar–H), 7.65–7.58 (m, 2H, Ar–H), 6.60 (s, 1H, −C=CH), (2.37 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6): δ 189.0, 149.8, 148.5, 146.4, 141.2, 138.4, 137.4, 132.7, 131.2, 130.6, 128.7, 128.0, 127.5, 126.7, 125.4, 125.0, 124.2, 121.6, 119.8, 118.4, 116.4, 114.6, 112.4, 21.3; accurate mass (EI-MS) of [M]·+: Calcd. for C24H1579Br2NO3S2; 586.8860; found 586.8865.
5,7-Dibromo-3-hydroxy-2-(p-tolyl)-4H-chromene-4-thione (2h)
Yellow solid; yield: 89%; mp 245–247 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.9; UV–vis λmax (CH2Cl2) = 375 nm; IR (KBr, cm–1) vmax: 3278, 3064, 2851, 1625, 1583, 1381, 1297, 692; 1H NMR (400 MHz, DMSO-d6): δ 9.37 (s, 1H, OH), 8.49 (d, J = 8.0 Hz, 1H, Ar–H), 8.13 (d, J = 4.0 Hz, 1H, Ar–H), 7.58 (d, J = 8.0 Hz, 2H, Ar–H), 7.37 (d, J = 8.0 Hz, 2H, Ar–H), 2.35 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6): δ 187.2, 150.0, 148.4, 146.3, 141.4, 138.3, 137.4, 132.4, 129.7, 129.0, 127.5, 127.1, 126.0, 118.8, 114.5, 21.7; accurate mass (EI-MS) of [M]·+: Calcd. for C16H1079Br2O2S 423.8768; found 423.8760.
5,7-Dibromo-3-hydroxy-2-(naphthalen-2-yl)-4H-chromene-4-thione (2i)
Red solid; yield: 72%; mp 240–242 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.5; UV–vis λmax (CH2Cl2) = 338 nm; IR (KBr, cm–1) vmax: 3054, 3002, 2970, 1578, 1508, 1386, 1257, 745; 1H NMR (400 MHz, DMSO-d6): δ 8.46 (s, 1H, OH), 8.23 (d, J = 4.0 Hz, 1H, Ar–H), 8.14 (d, J = 4.0 Hz, 1H, Ar–H), 7.52–7.45 (m, 2H, Ar–H), 7.41–7.38 (m, 2H, Ar–H), 7.30–7.23 (m, 3H, Ar–H); 13C NMR (101 MHz, DMSO-d6): δ 187.8, 151.0, 149.8, 148.3, 146.2, 141.3, 138.7, 136.6, 131.0, 130.7, 129.8, 129.0, 128.2, 127.7, 127.3, 126.6, 125.0, 119.5, 114.7; accurate mass (EI-MS) of [M]·+: Calcd. for C19H1079Br2O2S 459.8768; found 459.8750.
5,7-Dibromo-3-hydroxy-2-(1-tosyl-1H-indol-6-yl)-4H-chromene-4-thione (2j)
Light-brown amorphous solid; yield: 75%; mp 135–137 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.7; UV–vis λmax (CH2Cl2) = 350 nm; IR (KBr, cm–1) vmax: 3059, 2920, 1741, 1646, 1598, 1359, 1297, 681; 1H NMR (400 MHz, DMSO-d6): δ 8.91 (s, 1H, OH), 8.62 (d, J = 4.0 Hz, 1H, Ar–H), 8.49 (d, J = 4.0 Hz, 1H, Ar–H), 8.26 (d, J = 8.0 Hz, 2H, Ar–H), 8.13 (d, J = 4.0 Hz, 1H, Ar–H), 8.03 (d, J = 8.0 Hz, 2H, Ar–H), 7.78–7.73 (m, 1H, Ar–H), 7.68–7.60 (m, 3H, Ar–H), 2.33 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6): δ 189.1, 151.3, 149.6, 148.2, 146.7, 142.0, 140.2, 138.3, 138.2, 132.5, 131.8, 130.2, 128.5, 128.0, 127.8, 126.0, 125.3, 123.7, 120.7, 119.3, 118.1, 116.5, 114.8, 21.6; accurate mass (EI-MS) of [M]·+: Calcd. for C24H1579Br2NO4S2 602.8809; found 602.8812.
5,7-Dibromo-2-(2,4-dimethylphenyl)-3-hydroxy-4H-chromene-4-thione (2k)
Bright yellow solid; yield: 80%; mp 217–219 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.6; UV–vis λmax (CH2Cl2) = 367 nm; IR (KBr, cm–1) vmax: 3065, 3051, 2917, 1613, 1587, 1387, 1258, 688; 1H NMR (600 MHz, DMSO-d6): δ 8.78 (s, 1H, OH), 8.54 (d, J = 6.0 Hz, 1H, Ar–H), 8.43 (d, J = 6.0 Hz, 1H, Ar–H), 7.61 (d, J = 12.0 Hz, 1H, Ar–H), 7.26 (s, 1H, Ar–H), 7.22 (d, J = 12.0 Hz, 1H, Ar–H), 2.38 (s, 6H, 2 × CH3); 13C NMR (151 MHz, DMSO-d6): δ 187.2, 148.0, 146.7, 145.4, 141.4, 138.3, 138.0, 132.0, 130.8, 130.7, 129.8, 127.0, 126.8, 119.1, 114.2, 21.4, 20.4; accurate mass (EI-MS) of [M]·+: Calcd. for C17H1279Br2O2S 437.8925; found 437.8913.
5,7-Dibromo-2-(4-(dimethylamino)phenyl)-3-hydroxy-4H-chromene-4-thione (2l)
Dark-red amorphous solid; yield: 70%; mp 282–284 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.5; UV–vis λmax (CH2Cl2) = 353 nm; IR (KBr, cm–1) vmax: 3058, 2918, 1604, 1565, 1505, 1396, 1254, 1195; 1H NMR (400 MHz, DMSO-d6): δ 8.91 (s, 1H, OH), 8.51 (d, J = 4.0 Hz, 1H, Ar–H), 8.14 (d, J = 4.0 Hz, 1H, Ar–H), 8.05 (d, J = 8.0 Hz, 2H, Ar–H), 7.88 (d, J = 8.0 Hz, 2H, Ar–H), 2.95 (s, 6H, −N(CH3); 13C NMR (75 MHz, DMSO-d6): δ 188.5, 151.3, 149.0, 147.6, 141.2, 138.4, 132.2, 131.0, 130.1, 129.0, 128.4, 127.6, 126.0, 118.8, 113.6, 40.0; accurate mass (EI-MS) of [M]·+: Calcd. for C17H1379Br2NO2S 452.9034; found 452.9038.
5,7-Dibromo-2-(4-chlorophenyl)-3-hydroxy-4H-chromene-4-thione (2m)
Orange powder; yield: 72%; mp 230–232 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.7; UV–vis λmax (CH2Cl2) = 350 nm; IR (KBr, cm–1) vmax: 3061, 2849, 1912, 1763, 1589, 1447, 1326, 1252, 659; 1H NMR (400 MHz, DMSO-d6): δ 8.81 (s, 1H, OH), 8.46 (d, J = 4.0 Hz, 1H, Ar–H), 8.30 (d, J = 8.0 Hz, 2H, Ar–H), 8.13 (d, J = 4.0 Hz, 1H, Ar–H), 7.80 (d, J = 8.0 Hz, 2H, Ar–H); 13C NMR (101 MHz, DMSO-d6): δ 186.6, 150.3, 149.0, 148.7, 146.3, 139.5, 132.6, 130.8, 129.8, 129.3, 128.0, 126.4, 125.8, 119.2, 113.4; accurate mass (EI-MS) of [M]·+: Calcd. for C15H779Br235ClO2S 443.8222; found 443.8210.
5,7-Dibromo-3-hydroxy-2-(3-nitrophenyl)-4H-chromene-4-thione (2n)
Orange solid; yield: 82%; mp 231–233 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.8; UV–vis λmax (CH2Cl2) = 355 nm; IR (KBr, cm–1) vmax: 3062, 2987, 1590, 1521, 1488, 1383, 1207, 692; 1H NMR (400 MHz, DMSO-d6): δ 9.40 (s, 1H, OH), 8.95 (d, J = 4.0 Hz, 1H, Ar–H), 8.89 (dd, J = 4.0, 8.0 Hz, 1H, Ar–H), 8.47 (d, J = 4.0 Hz, 1H, Ar–H), 8.14 (d, J = 4.0 Hz, 1H, Ar–H), 7.87–7.80 (m, 2H, Ar–H); 13C NMR (101 MHz, DMSO-d6): δ 186.8, 149.8, 148.0, 146.3, 141.4, 138.6, 133.2, 131.1, 130.0, 128.4, 127.2, 126.6, 125.3, 119.5, 114.8; accurate mass (EI-MS) of [M]·+: Calcd. for C15H779Br2NO4S 454.8463; found 454.8445.
5,7-Dibromo-3-hydroxy-2-(4-nitrophenyl)-4H-chromene-4-thione (2o)
Orange-red solid; yield: 75%; mp 131–133 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.7; UV–vis λmax (CH2Cl2) = 354 nm; IR (KBr, cm–1) vmax: 3060, 2920, 1651, 1599, 1429, 1334, 1299, 682; 1H NMR (400 MHz, DMSO-d6): δ 9.38 (s, 1H, OH), 8.90 (d, J = 8.0 Hz, 2H, Ar–H), 8.49 (d, J = 4.0 Hz, 1H, Ar–H), 8.15 (d, J = 4.0 Hz, 1H, Ar–H), 8.04 (d, J = 8.0 Hz, 2H, Ar–H); 13C NMR (101 MHz, DMSO-d6): δ 186.5, 150.0, 148.3, 146.7, 141.5, 133.8, 131.8, 130.5, 127.5, 126.3, 125.7, 119.2, 114.4; accurate mass (EI-MS) of [M]·+: Calcd. for C15H779Br2NO4S 454.8463; found 454.8450.
5,7-Dibromo-2-(3,4-dimethoxyphenyl)-3-hydroxy-4H-chromene-4-thione (2p)
Orange-red solid; yield: 87%; mp 241–243 °C; Rf (ethyl acetate/n-hexane 1:3) = 0.9; UV–vis λmax (CH2Cl2) = 353 nm; IR (KBr, cm–1) vmax: 3061, 3009, 2836, 1599, 1539, 1390, 1297, 667; 1H NMR (400 MHz, DMSO-d6): δ 8.87 (s, 1H, OH), 8.48 (d, J = 4.0 Hz, 1H, Ar–H), 8.12 (d, J = 4.0 Hz, 1H, Ar–H), 7.93 (dd, J = 4.0, 8.0 Hz, 1H, Ar–H), 7.78 (d, J = 4.0 Hz, 1H, Ar–H), 7.27 (d, J = 8.0 Hz, 1H, Ar–H), 3.95 (s, 3H, OMe), 3.93 (s, 3H, OMe); 13C NMR (101 MHz, DMSO-d6): δ 187.2, 150.1, 149.0, 146.5, 141.1, 138.5, 132.3, 130.7, 128.6, 128.0, 127.4, 126.0, 125.7, 118.8, 114.5, 56.3, 56.0; accurate mass (EI-MS) of [M]·+: Calcd. for C17H1279Br2O4S 469.8823; found 469.8807.
2.2. FTIR Study
The structures of the synthesized brominated 4-thioflavones (2a–2g) and 4-thioflavonols (2h–2p) were confirmed by the absorption peaks found at their corresponding frequencies. Peaks indicating absorption for v(O–H), v(C=O), and v(C=C) in the ranges of 3391–2990, 1565–1678, and 1450–1550 cm–1, respectively, individually established the presence of carbonyl linkage in the oxyflavones (1a–1g) and oxyflavonols (1h–1p). However, IR spectra of the corresponding 4-thioflavones (2a–2g) and 4-thioflavonols (2h–2p) appear slightly different. For instance, there is an additional peak in the region of 1200–1360 cm–1 indicating v(C=S) linkage. Additionally, in the IR spectrum, the disappearance of the C=O signal (1610–1665 cm–1) and the appearance of a C=S new signal around 1200–1300 cm–1 explicitly endorses the replacement of oxygen by a sulfur atom in the target molecules. The remaining peaks correspond to other functional groups like OH (3274–3374 cm–1) and C=C (1400–1510 cm–1). The functionalities present in the compound were identified, and a satisfactory vibrational band assignment was made for the fundamental modes of vibration by observing the position, shape, and intensity of the bands (Figure S1 in the Supporting Information).
2.3. NMR Study
For further structural support, 1H and 13C NMR spectra of the oxyflavonols and 4-thioflavonols were recorded in DMSO-d6 (Figure S2 in the Supporting Information). The NMR spectroscopic technique plays an essential role in explaining the replacement of oxygen by a sulfur atom in target compounds. The 1H and 13C NMR spectra of compound 1k (Figure 2) give two characteristics signals; one peak around δ = 9.47–10.0 ppm belongs to the hydroxyl group (−OH) at position 3, and the other two doublets in the range of δ = 8.20–8.37 ppm are linked to two protons (Hb & Hc) present on ring A of the flavonol skeleton. The proton (Ha) of compound 1k is more deshielded due to the presence of a highly electronegative O″ atom in the vicinity, and thus it is accountable to give a signal. Remarkably, all NMR peaks in the 1H NMR spectrum of 1k are broader in shape and shifted toward downfield. This phenomenon is attributed to the intra- and intermolecular hydrogen bonding and the electron-withdrawing nature of the carbonyl group present in the simple flavonols because the electronegative oxygen results in a decrease of the electron density on the hydroxyl proton as well as other protons present in the molecule.
Figure 2.

Stacked 1H NMR spectra of compounds 1k and 2k (magnified aromatic region).
However, the 1H NMR spectrum of compound 2k (corresponding to 4-thioflavonol) shows a characteristic singlet in the range of δ = 8.0–9.0 ppm due to a hydroxyl (−OH) present at position 3 of the 2-arylchromone-4-thione scaffold. Likewise, the 1H NMR spectrum exhibits two characteristic doublets around δ = 8.40–8.55 ppm with J = 4.0 Hz by virtue of two protons (Hb & Hc) present on ring A of the 4-thioflavonol skeleton. Noteworthy, all signals present in the spectrum of 2k look sharper and are considerably upfield as compared to the NMR signals of the compound 1k. This is because of less effective hydrogen bonding and less electronegativity of sulfur atom. For example, the proton (Ha) of compound 2k is relatively less deshielded due to the presence of a less electronegative S″ atom in the neighborhood, and thus it is liable to give an upfield and sharp signal as compared to compound 1k.
Similarly, in compounds 1k and 2k, there are other characteristic doublets around δ = 7.47–7.50 ppm and δ = 7.61–7.58 ppm for the aromatic protons labeled as He and Hf, respectively (Figure 2). The Hf protons are less shielded and slightly downfield in the 1H NMR spectrum of 4-thioflavonol 2k and more shielded and upfield in flavonol 1k. Furthermore, the proton NMR spectrum of compound 2k displays a sharp singlet at δ = 7.26 ppm for the Hd of 4-thioflavonol, whereas it exhibits a broader and merged singlet at δ = 7.21 ppm for the Hd of the flavonol motif (Figure 2).
The replacement of oxygen with sulfur in 2-arylchromone-4-thione derivatives was further confirmed from the 13C NMR spectra, which were again taken in DMSO-d6 (Figure 3). In 13C NMR spectra of compounds 1k and 2k, the peak of a carbonyl carbon (C=O) and thiocarbonyl carbon (C=S) appears around δ = 172 and 186 ppm, respectively. In the in latter, the concerned peak moved downfield, which indicated that the electron density decreased around the carbon atom on the thiocarbonyl group because of the larger size of S than O. In compound 1k, the carbonyl carbon peak moved upfield, which indicated that the electron density increased around the carbon atom of carbonyl group, probably because of the compact nature and high electron density on the O atom. Additionally, conjugation between the α,β-unsaturated double bond and keto group is considerably more effective due to the (-I)-effect of oxygen in simple 2-arylchromones. However, their corresponding thio analogues are lacking or have less effective conjugation due to the low electronegativity of S in comparison to that of O. So, all these factors make the carbon of the thioketo group resonate at high chemical shift values. Interestingly, the nature and position of other carbon peaks in both precursors (2-arylchmromones) and target compounds (2-arylchromone-4-thiones) are mostly identical. The peaks at δ = 20.4 and 21.6 ppm represent the aliphatic methyl groups present on ring B in compounds 1k and 2k.
Figure 3.

Stacked 13C NMR spectra of compounds 1k and 2k (whole spectra).
2.4. Mushroom Tyrosinase Inhibition Assay
In a continuation of our prior investigations on the subject of mushroom tyrosinase inhibition,22,24,55 brominated thioflavones (2a–2g) and thioflavonols (2h–2p) were examined for their antityrosinase activity as described in the Experimental section. All the target compounds (2a–2p) showed moderate-to-good inhibition against mushroom tyrosinase enzyme (Figure 4 and Table 1). The half-maximal inhibitory concentration (IC50) values of the investigated compounds and the reference kojic acid (IC50 = 12.6 ± 0.6 μM) are shown in the table below. The IC50 values for the compounds (2a–2p) ranged from 1.12 ± 0.04 to 5.68 ± 0.13 μM.
Figure 4.
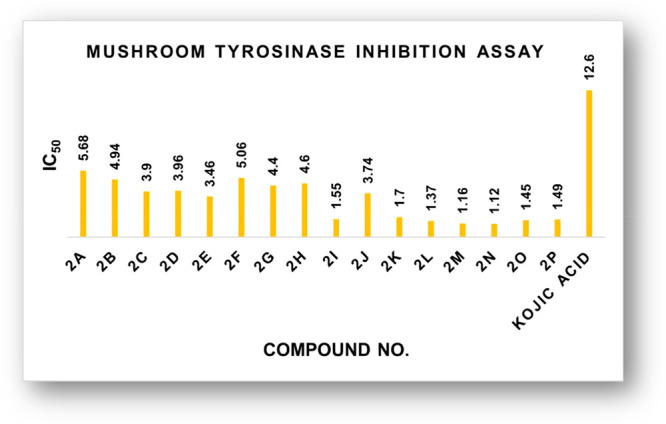
Comparative tyrosinase inhibitory study graph of 4-thioflavones and 4-thioflavonols.
Table 1. Tyrosinase Inhibitory Efficiency of Synthesized Derivatives (2a–2p).
| compound No. | mushroom tyrosinase inhibitionaIC50 ± SEM (μM) |
|---|---|
| 2a | 5.68 ± 0.13 |
| 2b | 4.94 ± 0.16 |
| 2c | 3.90 ± 0.17 |
| 2d | 3.96 ± 0.14 |
| 2e | 3.46 ± 0.07 |
| 2f | 5.06 ± 1.00 |
| 2g | 4.40 ± 0.13 |
| 2h | 4.60 ± 0.15 |
| 2i | 1.55 ± 0.10 |
| 2j | 3.74 ± 0.28 |
| 2k | 1.70 ± 0.07 |
| 2l | 1.37 ± 0.01 |
| 2m | 1.16 ± 0.07 |
| 2n | 1.12 ± 0.04 |
| 2o | 1.45 ± 0.08 |
| 2p | 1.49 ± 0.8 |
| kojic acidb | 12.6 ± 0.6 |
IC50 values (mean ± standard error of the mean).
Standard inhibitor for mushroom tyrosinase.
Among all the synthesized 2-arylchromone-4-thiones, the compound (2n) exhibited excellent tyrosinase inhibitory activity with the lowest IC50 = 1.12 ± 0.04 μM. Therefore, compounds 2i (IC50 = 1.55 ± 0.10 μM), 2k (IC50 = 1.70 ± 0.07 μM), 2l (IC50 = 1.37 ± 0.01 μM), 2m (IC50 = 1.16 ± 0.07 μM), 2o (IC50 = 1.45 ± 0.08 μM), and 2p (IC50 = 1.49 ± 0.8 μM) could be recommended as potential lead candidates to cure tyrosinase-mediated hyperpigmentation in the future.
2.5. Kinetic Study
A kinetic study of the most active compound 2n was evaluated by a Lineweaver–Burk plot analysis to determine the mode of inhibition mechanism of this class of compounds. Different concentrations of compound 2n and substrate were employed to determine the enzyme’s reaction rate in the enzyme kinetic experiments. Plotting 1/V versus 1/[S] yielded a series of lines with varying slopes intersecting at the Y-axis (Figure 5a). According to the analysis, the Km values increased with increasing concentrations of 2n, but Vmax remained constant. These findings suggest that 2n is a competitive inhibitor of tyrosinase. The enzyme inhibitor dissociation constant (Ki) was calculated by a replot of slopes (Lineweaver–Burk plot) against various concentrations of 2n. As illustrated in Figure 5b, the Ki value of 2n is 1.98 μM. The kinetic study results showed that 2n is a competitive tyrosinase inhibitor with a Ki value of 1.98 μM.
Figure 5.
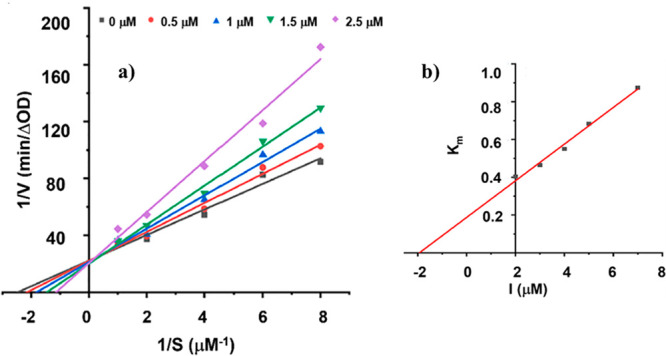
(a) Lineweaver–Burk plot for compound 2n. (b) Secondary replot of slopes (Lineweaver–Burk plot) vs various concentrations of compound 2n.
2.6. Cytotoxicity Study
There are several skin-lightening chemicals in the market, such as kojic acid, phenylthiourea (PTU), hydroquinone arbutin, and others, that are widely used as skin-whitening agents.51b,51c However, to meet the criteria, the potent skin-whitening agent must be trustworthy and free of cytotoxic side effects. Herein, we synthesized 2a–2p tyrosinase inhibitors in this study, and they all showed good effectiveness against a mushroom tyrosinase inhibitor when compared to ordinary kojic acid. However, for the cytotoxicity test, we chose four extremely powerful inhibitors (2m, 2n, 2o, and 2p). The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test for A375 human melanoma cells was used to conduct the cell toxicity investigation. The cells were cultured for 24 h at different concentrations (1, 5, 10, 15, 20, and 25 μg/mL) of the most powerful compounds 2m, 2n, 2o, and 2p, with the results of the cell viability assay shown in Figure 6. In the cell viability test, all of the compounds have a cell viability of ∼80% when compared to nontreated cells. Thus, in the concentration range of 0–25 μg/mL, none of the powerful compounds are harmful to the A375 human melanoma cells (p < 0.05).
Figure 6.

Cell viability measurement using the MTT assay. Cells were untreated (control) or treated with the most potent compounds 2m, 2n, 2o, and 2p at different concentrations (1, 5, 10, 15, and 25 μg/mL) in DMSO for 24 h.
Nonetheless, after a comparison of the compounds in the series, the most potent compounds 2m, 2n, 2o, and 2p were shown to have the best results in cell cytotoxicity tests. As a result, in the realm of medicinal chemistry, the target compound 2n might be used as an efficient tyrosinase inhibitor for the development of a novel drug.
2.7. Structure–Activity Relationship
All synthetic analogues (2a–2p) were subjected to their in vitro tyrosinase inhibitory activity. The inhibitory activity data presented in Table 1 led us to generate an initial structure–activity relationship (SAR) model in order to investigate the effect of substituent on the aryl ring as well as the type of substituents on the activity profile that could be explored (Figure 7).
Figure 7.
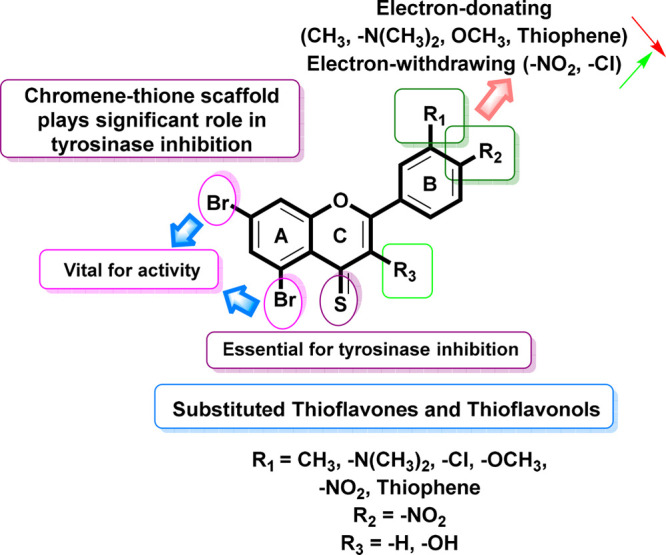
SAR of 4-thioflavone and 4-thioflavonol analogues.
Interestingly, the compounds 2n (IC50 = 1.12 ± 0.04 μM) and 2o (IC50 = 1.45 ± 0.08 μM) have a −Br group at the fifth and seventh positions in ring A and an electron-withdrawing group (−NO2) at the third and fourth positions, respectively, on ring B of the 3-hydroxythioflavone carbon skeleton, distinctly resulting in increased inhibitory activity, and thus it was found to be the most potent tyrosinase inhibitor among all the synthetic brominated 4-thioflavone and 3-hydroxy-4-thioflavone derivatives. Henceforth, the meta −NO2-substituted compound 2n disclosed increased activity relative to its para-analogue 2o.
Furthermore, the third most-potent compound of this series is compound 2m (IC50 = 1.16 ± 0.07 μM), which has bromo groups present on Ring A and an electron-withdrawing group (−Cl) group at the para position on ring B (Figure 7). Probably, this group and its position are appropriate to agreeably interact with the active pocket of the tyrosinase.
Additionally, compounds 2a (IC50 = 5.68 ± 0.13 μM), 2b (IC50 = 4.94 ± 0.16 μM), 2c (IC50 = 3.90 ± 0.17 μM), 2e (IC50 = 3.46 ± 0.07 μM), 2g (IC50 = 4.40 ± 0.13 μM), 2h (IC50 = 4.60 ± 0.15 μM), 2i (IC50 = 1.55 ± 0.10 μM), 2k (IC50 = 1.70 ± 0.07 μM), 2l (IC50 = 1.37 ± 0.01 μM), and 2p (IC50 = 1.96 ± 0.8 μM) were found to be even more active than the standard kojic acid (IC50 = 12.6 ± 0.6 μM). It is notable that all these derivatives demonstrated exceptional inhibitory activity against the tyrosinase enzyme.
However, compounds 2d (IC50 = 3.96 ± 0.14 μM), 2f (IC50 = 5.06 ± 1.00 μM), and 2j (IC50 = 3.74 ± 0.28 μM) were found to be less active among the 4-thioflavone and 3-hydroxy-4-thioflavone series against the envisioned enzyme. These findings reflect that highly hydrophilic, that is, thiophene, −(CH3)2 & sulfonamide-like groups on both aryl rings (B) are accountable for their low activities because of their diminished interaction with the tyrosinase. A detailed SAR relation was established and depicted in Figure 7.
2.8. Molecular Docking Assay
The glide-dock module was implemented for the docking studies of selected ligands 2n and 2o against the crystal structure of the target protein mushroom tyrosinase (Protein Data Bank (PDB) identifier (ID) 2Y9X) (Figure 8). The docking scores/energies were calculated (Table 2), and images of docked complexes were also generated (Figure 9a,b). The predicted binding interactions by all docking processes suggested the docking scores (kcal/mol) in the range from −4.2620 to −8.5064 represent the good-to-best interactions. The ligation mode was mainly focused on an OH site. The main binding-receptor backbones were histidine, phenylalanine, alanine, and active-site copper ions. The displayed images of docking complexes (Figures 10a,b and 11a,b), especially with the interacting image, clarify the formerly concluded data.1,7,14 The surface maps were built over the receptor atoms, leading to a good view of the electrostatic interactions inside the docking complexes. Good inhibition of compounds 2n and 2o exhibit high occupancy of the inside surface grooves pointing to the best blocking of active-site amino acids. The meta nitro-substituted phenyl ring of best docked score (−8.5064 kcal/mol) inhibitor 2n is predicted to form a π–π stacking with active-site residues His259 and His263, whereas the dibromo-substituted phenyl ring is predicted if this inhibitor is picking up a π-interaction with the side-chain residue His244. The para nitrophenyl moiety of the second-highest docked score (−7.7652 kcal/mol) compound 2o is interacting with amino acids His85 and His259 through π–π stacking, and the dibromo-substituted phenyl ring of this inhibitor is further stabilized by another π-interaction with active-site residue His244.
Figure 8.
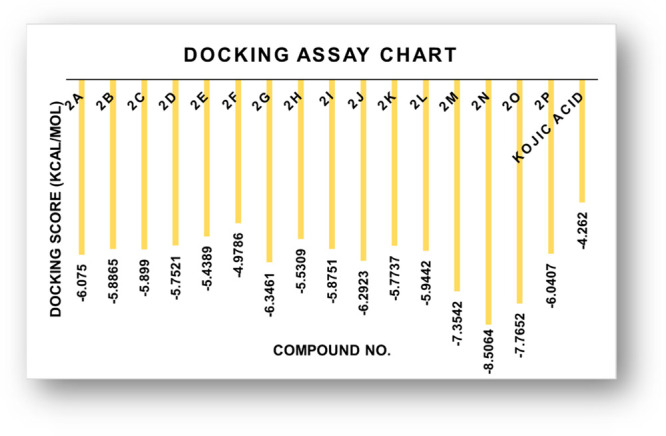
Graphical depiction of docking energy values of thioflavone and thioflavonol analogues (2a–2p).
Table 2. Binding Energies of the Compounds (2a–2p) against Various Modes of Tyrosinase Enzyme.
| compound No. | docking score PDB ID 2Y9X (kcal/mol) | compound No. | docking score PDB ID 2Y9X (kcal/mol) |
|---|---|---|---|
| 2a | –6.0750 | 2i | –5.8751 |
| 2b | –5.8865 | 2j | –6.2923 |
| 2c | –5.8990 | 2k | –5.7737 |
| 2d | –5.7521 | 2l | –5.9442 |
| 2e | –5.4389 | 2m | –7.3542 |
| 2f | –4.9786 | 2n | –8.5064 |
| 2g | –6.3461 | 2o | –7.7652 |
| 2h | –5.5309 | 2p | –6.0407 |
| kojic acid (standard) | –4.2620 (kcal/mol) |
Figure 9.

(a) 2D ligand-interaction diagram of compound 2n. (b) 2D ligand-interaction diagram of compound 2o.
Figure 10.
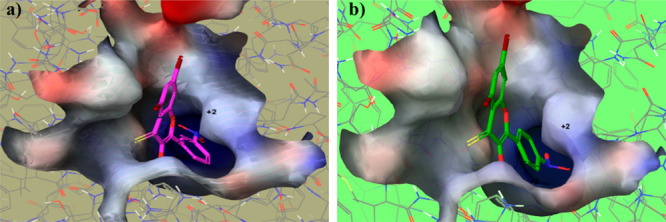
(a) Ball-and-surface diagram of compound 2n. (b) Ball-and-surface diagram of compound 2o.
Figure 11.

(a) Ball-and-stick diagram of compound 2n. (b) Ball-and-stick diagram of compound 2o.
2.9. Quantitative Structure–Activity Relationship Model
In the present study, the antityrosinase activity of synthesized ligands was further analyzed for cross-correlation with chemical descriptors to examine common structure–activity relationship with chemical descriptors. A correlation was found between the observed parameters with a correlation coefficient (R2) value of 0.9996. The atomic models of each compound (2a–2p) with its biological activity were constructed keeping in view the patterns of hydrophilic/phobic interactions that best express the binding nature of each compound as accessed from pMIC (logarithmic minimum inhibitory concentration) values.56e The pMICs were separated as training and test sets. Since there are no previous data for the tyrosinase inhibitory activity of 2a–2p, pMIC values were used for both training and test sets. The test set and training set values clearly show a positive correlation between the pMIC and biological activity (Figure 12a,b).
Figure 12.

QSAR model. (a) Training set. (b) Test set.
The various parameters and pMIC correlation coefficient value obtained are shown here. SD: standard deviation of regression = 0.0364; F: variance ratio (ratio of the model variance to the observed activity variance) = 4721.88; P: significance level of variance ratio = 1.076 × 10–10; RMSE: the RMS error in the test group predictions = 0.031 47; R2: regression coefficient = 0.9997; Q2: correlation coefficient = 0.9996; Pearson-r: correlation among the predicted and observed activity for the test group = 0.9999.
2.10. Density Functional Theory (DFT) Study
2.10.1. HOMO–LUMO Analysis
The highest occupied molecular orbital (HOMO) (donor) and lowest unoccupied molecular orbital (LUMO) (acceptor) parameters are very important in many disciplines of chemistry to specify the formation of charge. Electrons are directly involved in many chemical reactions, and therefore the HOMO and LUMO affect the chemical action of the molecule. Here, the HOMO and LUMO analyses of 5,7-dibromo-3-hydroxy-2-(3-nitrophenyl)-4H-chromene-4-thione (2n) and 5,7-dibromo-3-hydroxy-2-(4-nitrophenyl)-4H-chromene-4-thione (2o) derivatives were accomplished with the B3LYP function and 3-21G basic set. The quantum calculations of compounds 2n and 2o were performed in Gaussian by using the GaussView interface. The resulting HOMO/LUMO, ionization potential, energy gap, and electron affinity values and other descriptors were calculated (Table 3). The energy gap is used to indicate the structure stability, and the molecule with a higher energy gap is more stable than the molecule with a smaller gap.53 The energy gap for selected compounds presents the order 2o < 2p < 2m < 2n, indicating that the compound 2n is potentially more stable than the others. Moreover, the HOMO/LUMO distributions of the selected compounds were calculated (Figure 13). In addition, electron affinity, ionization potential, chemical hardness, electronic chemical potential, and chemical softness are calculated by using HOMO/LUMO energy values.
Table 3. HOMO and LUMO Energy Values and Other Related Parameters of 2m, 2n, 2o, and 2p.
| compounds |
||||
|---|---|---|---|---|
| parameters (eV) | 2m | 2n | 2o | 2p |
| ELUMO | –0.114 34 | –0.117 32 | –0.126 83 | –0.107 87 |
| EHOMO | –0.224 92 | –0.228 57 | –0.230 21 | –0.211 53 |
| energy gap |EHOMO – ELUMO| | 0.110 58 | 0.111 25 | 0.103 38 | 0.103 66 |
| ionization potential (I = −EHOMO) | 0.224 92 | 0.228 57 | 0.230 21 | 0.211 53 |
| electron affinity (A = −ELUMO) | 0.114 34 | 0.117 32 | 0.126 83 | 0.107 87 |
| chemical hardness (η = (I – A)/2) | 0.055 29 | 0.055 62 | 0.051 69 | 0.051 83 |
| chemical softness (ζ = 1/2η) | 9.043 22 | 8.988 76 | 9.673 05 | 9.646 92 |
| electronegativity (χ = (I + A)/2) | 0.169 63 | 0.172 94 | 0.178 52 | 0.1597 |
| chemical potential (μ = −(I + A)/2) | –0.169 63 | –0.172 94 | –0.178 52 | –0.1597 |
| electrophilicity index (ω = μ2/2η) | 3.068 00 | 3.109 40 | 3.453 66 | 3.081 22 |
Figure 13.

HOMO and LUMO plots of compounds 2m, 2n, 2o, and 2p in B3LYP/3-21 G.
2.10.2. Molecular Electrostatic Potential
The molecular electrostatic potential (MEP) could be an important approach to confirm the evidence for interaction of these molecules as inhibitors. MEP defines the size, shape, and positive, negative, and neutral regions in a molecule with the help of color grading. The increasing order of potential is red < orange < yellow < green < blue. It is very easy to find favorable sites for an attack of a nucleophile and electrophile by following the color order. The blue color suggests the area with maximum electrostatic capability, which indicates the absence of the electrons in that region and therefore the favored site for a nucleophilic attack, while the red color indicates the areas with low electrostatic potential, which suggests the abundance of electrons and therefore a favored site for an electrophilic attack.58 According to the MEP analysis, the areas of least potential are positioned at the oxygen atoms of the nitro group (red coded region) in each system as shown in Figure 14. The hydrogen of a benzene ring seems to show that a positive (blue) potential in each system appears to have increased potential. The degree to which drug-like molecules bind to receptors at the active sites of the targeted receptor is primarily determined by the difference in mapping this electrostatic potential around them.
Figure 14.
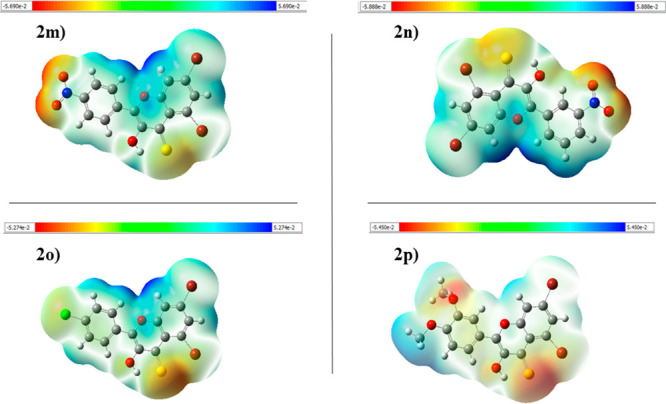
Molecular electrostatic potential of compounds 2m, 2n, 2o, and 2p.
2.10.3. Chemo-Informatics and Lipinski’s Rule
The expected chemo-informatics properties like polarizability, solubility, and polar surface area (PSA) were computed. On the basis of a literature review, a standard value for molar molecular weight (160–480) and number of atoms (20–70) was determined.34,35 The anticipated values of 2n, 2o, and 2p are substantially better than standard values and all other produced compounds, according to the findings. The computational results predicted that 2n, 2o, and 2p possesses 5, 5, and 5 HBA (≤10), 1, 1, and 1 hydrogen-bond donors (HBD) (≤5) and 4.09, 4.11, and 4.79 LogP (<5), 17.51, 12.78, and 7.14 mg/L LogS and PSA 60.46, 60.46, and 37.46 A2 (≤120A2) values, respectively, which significantly justified its drug-like behavior with drug-like model scores of −0.58, −0.44, and −0.02 (Table 4 and Figure 15). The findings reveal that a number of analogues have drug-like effects while not breaking any of the constraints that regulate their therapeutic potential.
Table 4. Pharmacokinetic Assessment of Synthesized Thioflavone and Thioflavonol Analogues (2a–2p)a.
| compound No. | molecular formula | molecular weight (g/mol) | No. HBAb | No. HBDc | molecular LogPd | molecular LogSe (mg/L) | molecular PSAf A2 | drug-likeness model score | Lipinski’s rule of 5 |
|---|---|---|---|---|---|---|---|---|---|
| 2a | C17H1279Br2O3S | 453.8874 | 4 | 0 | 5.86 | 0.64 | 23.80 | –0.15 | No |
| 2b | C15H779Br2NO3S | 438.8513 | 4 | 0 | 5.16 | 1.43 | 46.80 | –0.67 | No |
| 2c | C19H1079Br2OS | 443.8819 | 2 | 0 | 7.48 | 0.03 | 8.27 | –0.46 | No |
| 2d | C17H1279Br2OS | 421.8976 | 2 | 0 | 6.90 | 0.41 | 8.45 | –0.55 | No |
| 2e | C15H779Br2ClOS | 427.8273 | 2 | 0 | 6.92 | 0.04 | 8.54 | –0.20 | No |
| 2f | C13H679Br2OS2 | 399.8227 | 3 | 0 | 5.78 | 0.30 | 9.56 | –0.57 | No |
| 2g | C24H1579Br2NO3S2 | 586.8860 | 4 | 0 | 8.15 | 0.30 | 37.64 | –0.40 | No |
| 2h | C16H1079Br2O2S | 423.8768 | 3 | 1 | 5.69 | 0.88 | 22.20 | –0.29 | No |
| 2i | C19H1079Br2O2S | 459.8768 | 3 | 1 | 6.41 | 0.06 | 21.93 | –0.31 | No |
| 2j | C24H1579Br2NO4S2 | 602.8809 | 5 | 1 | 7.08 | 0.52 | 51.30 | –0.27 | No |
| 2k | C17H1279Br2O2S | 437.8925 | 3 | 1 | 6.06 | 1.14 | 22.20 | –0.42 | No |
| 2l | C17H1379Br2NO2S | 452.9034 | 3 | 1 | 5.31 | 3.38 | 25.01 | –0.36 | No |
| 2m | C15H779Br235ClO2S | 443.8222 | 3 | 1 | 5.84 | 0.23 | 22.20 | –0.08 | No |
| 2n | C15H779Br2NO4S | 454.8463 | 5 | 1 | 4.09 | 17.51 | 60.46 | –0.58 | Yes |
| 2o | C15H779Br2NO4S | 454.8463 | 5 | 1 | 4.11 | 12.78 | 60.46 | –0.44 | Yes |
| 2p | C17H1279Br2O4S | 469.8823 | 5 | 1 | 4.79 | 7.14 | 37.46 | –0.02 | Yes |
The table above depicts all of Lipinski’s RO5 components.
Number of hydrogen-bond acceptors.
Number of hydrogen-bond donors.
Octanol–Water partition coefficient.
Measured solubility.
Total polar surface area.
Figure 15.
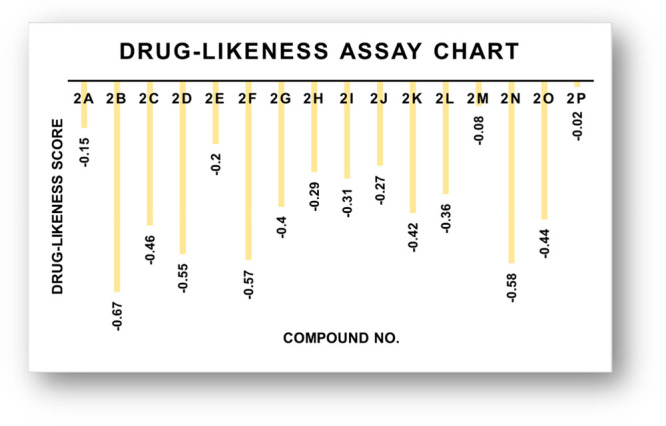
Drug-likeness score bar chart.
3. Conclusions
In summary, a series of new 4-thioflavones (2a–2g) and 4-thioflavonols (2h–2p) were synthesized using precedent methodologies starting from substituted 2′-hydroxyacetophenone and various substituted aromatic aldehydes over two steps and characterized by various spectral techniques (UV–vis, FTIR, NMR, mass spectrometry, etc.). All the synthetic compounds (2a–2p) were evaluated in vitro against the mushroom tyrosinase enzyme. In order to explore activities of synthesized thioflavones and thioflavonols in terms of potent tyrosinase inhibitors, we herein, for the first time, report that the 4-thioflavones and 3-hydroxy-4-thioflavones are potential new tyrosinase inhibitors. The in vitro assay revealed that all the synthesized target analogues (2a–2p) show excellent activity against mushroom tyrosinase compared with standard kojic acid (IC50 = 12.6 ± 0.6 μM). The compounds (2a–2p) exhibited IC50 values in the range from 1.12 ± 0.04 to 5.68 ± 0.13 μM. Among the synthesized 4-thioflavones and 4-thioflavonols, the compound (2n) exhibited excellent tyrosinase inhibition with the lowest IC50 value of 1.12 ± 0.04 μM. Furthermore, compounds 2i (IC50 = 1.55 ± 0.10 μM), 2k (IC50 = 1.70 ± 0.07 μM), 2l (IC50 = 1.37 ± 0.01 μM), 2m (IC50 = 1.16 ± 0.07 μM), 2o (IC50 = 1.45 ± 0.08 μM), and 2p (IC50 = 1.49 ± 0.8 μM) may prove to be effective inhibitors of tyrosinase. The inhibitory potential of the compounds (2a–2p) was also studied using molecular docking method. Moreover, kinetic studies revealed that compound 2n has a competitive inhibition mechanism and that its Ki value is 1.98 μM. The cell viability of the potential compounds (2m, 2n, 2o, and 2p) has been calculated at various concentrations ranging from 0 to 25 μg/mL using the MTT assay method for A375 human melanoma. The computational evaluation concludes that many of the tested molecules, theoretically, will not have complications with oral bioavailability. Because of its low toxicity and its high inhibition activity, it could represent a milestone on the path toward new valuable agents in medical fields, where it was recently suggested that tyrosinase could play key role.
4. Experimental Section
4.1. Materials and Methods
The required chemicals were purchased from Sigma-Aldrich and Merck and used as received. Melting points were determined on an Electrothermal melting point apparatus and are uncorrected. IR spectra were recorded on a Bio-Rad spectrophotometer. The IR values are expressed in υ units. NMR spectra were measured on a Bruker DRX 400 instrument (1H, 400 MHz; 13C, 101 MHz). The UV absorption spectra have been recorded on the Jasco UV–vis V-670 instrument using a QUARTZ cell in very dilute solutions prepared in different solvents. Accurate mass measurements were performed with the Fisons VG sector-field instrument (EI) and an FT-ICR mass spectrometer.
4.2. General Procedures for the Syntheses of Flavone (1a–1g), Flavonol (1h–1p), Thioflavone (2a–2g), and Thioflavonol (2h–2p) Derivatives35,36,38
A mixture of 3′,5′-dibromo-2′-hydroxyacetophenone (294 mg, 1.0 mmol) and 5.0 mL of an aqueous sodium hydroxide solution (30%) was dissolved in 15.0 mL of distilled methanol and stirred for 30 min at room temperature followed by the addition of substituted aromatic aldehyde (1.0 mmol) dropwise, and the reaction mixture was further stirred for 3–4 h at the same temperature. The progress of the intermediate chalcone formation was monitored by comparative thin-layer chromatography (TLC) using ethyl acetate/n-hexane (1:3) as a mobile phase. After the completion of the reaction (indicated by TLC), the contents of the reaction mixture were then acidified by dilute HCl (10%) and poured onto ice-cold water. The solid obtained was filtered, washed several times with water, and eventually recrystallized from ethanol to afford the pure product. In the first route, the substituted chalcone (1.0 mmol) was dissolved in DMSO (10 mL) and oxidatively cyclized in the presence of I2 (254 mg, 1.0 mmol). The mixture was refluxed at 130 °C for 3–5 h. Upon reaction completion, as indicated by TLC, the solution was cooled to room temperature and poured onto crushed ice, and sodium thiosulfate was added to the reaction mixture followed by an excessive amount of water to allow precipitation. The product was filtered and allowed to dry at room temperature, and the solid was recrystallized from ethanol to furnish the pure flavone derivatives (1a–1g). In the second route, the resultant chalcones (in situ) so formed were converted into corresponding substituted 3-hydroxyflavone derivatives (1h–1p) by conventional Algar-Flynn-Oyamada (AFO) conditions upon oxidative cyclization using H2O2 (35%) in methanol and allowing the reaction mixture to stir for a further 1 h at the same temperature. After the oxidative cyclization of flavone and flavonols, the synthesized substituted flavone and flavonols (1.0 mmol) were treated with Lawesson’s reagent (526 mg, 1.3 mmol) in refluxing anhydrous toluene (15 mL) for 24 h. Subsequently, the excess solvent was evaporated under vacuum to furnish the dark colored residue, which was recrystallized by ethanol to obtain the pure 4-thioflavones (2a–2g) and 4-thioflavonols (2h–2p) in excellent yields.
4.3. Enzyme Inhibition Assay
4.3.1. Mushroom Antityrosinase Assay
To evaluate the antityrosinase inhibitory activity of target compounds, an assay was performed as previously reported with slight modifications.2,6,55 Briefly, the wells of a 96-well plate were filled with 140 μL of phosphate buffer (20 mM, pH 6.8). Then 20 μL of mushroom tyrosinase (250 U/mL) and 20 μL of test compound were added, and the mixture was incubated at room temperature for 10 min. Later, 20 μL (0.85 mM) of 3,4-dihydroxyphenylalanine (L-DOPA) was added to the mixture. The stock solution of the examined compounds was prepared using DMSO. The plate was incubated in the dark for 20 min at 25 °C. Finally, using a plate reader, the absorbance of dopachrome was measured at 400 nm (Biotek ELX 808). Each assay was conducted as three separate replicates. The following formula was used to compute percentage inhibition
where X = absorbance of the enzyme with a test compound, and Y = absorbance of the enzyme without a test compound. Later, the concentration of the test compound necessary to achieve 50% tyrosinase inhibition (IC50) was determined by the data analysis and graphing software, Origin.
4.4. Kinetic Analysis
The experimental approach was identical to that used in the tyrosinase inhibition assay, and a series of experiments was performed to establish the mode of inhibition. The concentrations of 2n were 2.0, 1.5, 1.0, and 0.5 μM. Lineweaver–Burk plots were used to assess the Km and Vmax of tyrosinase.29,51a
4.5. Cytotoxicity
4.5.1. Cell Culture and Treatment of 2m, 2n, 2o, and 2p
Korean Cell Line Bank provided human malignant melanoma A375 cells (KCLB No. 80003). The cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco/Invitrogen) with 10% fetal bovine serum (FBS, Gibco/Invitrogen) at 37 °C, 5% CO2, and 95% air in an incubator.51b−51d
4.5.2. Cell Proliferation Assay
Cells were seeded into 96-well plates at a density of 0.4 × 105 cells per well in 100 μL of medium and cultured for 24 h. Following an overnight incubation, the cell medium was withdrawn, and the cells were treated for 24 h with concentration gradients of 2m, 2n, 2o, and 2p (1, 5, 10, 15, 20, and 25 μg/mL). Three duplicate wells were included in each treatment. The cells treated with DMSO served as a negative control. The medium was removed, 0.5 mg/mL MTT was added to each well, and the cells were incubated for 6 h at 37 °C in an incubator. To stop the MTT reaction, the medium was withdrawn, and 100 μL of solubilization buffer (10% sodium dodecyl sulfate (SDS), 0.01 N HCl) was added to each well. The plate was covered with foil and agitated on an orbital shaker for 1 h to dissolve formazan crystals for the determination of formazan crystal quantity, which was measured using a microplate reader and the absorbance at 595 nm (Molecular Devices). The treatment was repeated three times, and the findings were computed as percentages of growth inhibition based on the mean of two independent measurements (standard error of the mean (SEM)). The percentage of viable cells was estimated using the equation below.51b−51d
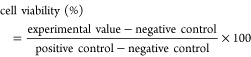 |
4.6. Molecular Modeling Study
The two-dimensional (2D) structures of the ligands (2a–2p) were sketched using ChemDraw Professional (v15.2), prepared in their neutral form and configured by adding hydrogens and minimizing the energy in the applied OPLS3 force field in the Ligprep module of Maestro Schrodinger. The prepared ligands were further employed for the docking process. The X-ray crystal structure of the target protein (PDB ID 2Y9X) was downloaded from the Research Collaboratory for Structural Bioinformatics (RCSB) protein data bank and configured by the addition of hydrogen and by the removal of the water molecules beyond 5 Å. The receptor grid box (20 Å) was generated by selecting the cocrystallized ligand tropolone in the active site. The default docking setup parameters were employed for Glide-XP-docking experiment. The inhibitors were docked against a prepared selected protein with the default settings until the most stable docking complexes were reached, and the top 15 postures for each ligand are reported. The scoring energies, which were the mean values of trials using the London dG scoring function, were upgraded by two unrelated refinements by the triangular Matcher methods. The interacting complexes as well as electrostatic maps for the interacting surfaces were extracted, in addition to essential interaction parameters. The binding mode was predicted based on the extracted parameters such as ligand, receptor backbones (amino acids), interaction type, bond lengths, and internal and scoring energies. The visual inspection and three-dimensional (3D) graphical images of the best scored docking complex were also generated using Maestro.22a
4.7. Quantitative Structure Activity Relationship (QSAR) Studies
The Maestro Schrodinger Suite (v2017-2) was used to perform Quantitative Structure Activity Relationship (QSAR) calculations. The 3D structure of compounds (2a–2p) was drawn and prepared by Ligprep as discussed in the molecular modeling study. Then, the prepared structures were used for QSAR investigations. All the docked molecules were used as a training set, whereas the QSAR model correlates the activities with inherent properties of each molecule in a test set. Various molecular descriptors were employed to determine these properties. In the first step of QSAR studies, descriptors were generated that encode structural information. In the second step, a statistical regression analysis was used to correlate the structural variation encoded by the descriptors with the variation in the biological activity of protein. A multiple linear regression analysis was performed to test the reliability of results using inhibitory activity as dependent and descriptor as predictor variables. The compounds incorporated in the collection of data sets have inhibitory potencies with IC50 values varying from 1.12 to 5.68 μM and converted to pMICs according to the following equation.51e−51h
The QSAR models with p < 0.05 were derived with reasonable correlation of inhibition activity and the individual descriptor to ensure the statistical reliability.51i
4.8. Computational (DFT) Calculations
Density functional theory (DFT) is based on the molecule’s energy and is calculated by using electron density rather than the wave function. The electron correlations make the findings more consistent with the experimental results. In this work, the 3D geometries of the chromone derivatives (2m, 2n, 2o, and 2p) are drawn by the GaussView (5.0.16) interface and calculated in the Gaussian (v.09) program. DFT calculations were performed using basic set 3-21G, B3LYP.56−60 A Frontier Molecular Orbitals analysis of selected compounds was obtained after an optimization in DMSO solution. MEP surfaces were drawn to calculate the charge distributions in a molecule to understand the interaction of molecules.
4.9. Drug-Likeness Study
The Molinspiration tool was used to evaluate the drug-like properties of the aforementioned compounds. Lipinski’s rule of five (RO5) provides the basis for estimating the bioavailability and pharmacokinetics of a molecule when administered orally. Computer-aided drug design (CADD) is a fantastic tool for researchers searching for new chemical entities for biomedical applications to save time and effort.29,30
Acknowledgments
The authors acknowledge the Deanship of Scientific Research at Umm Al-Qura University for supporting this work by Grant No. 22UQU4320545DSR12. The authors extend their sincere appreciation to Taif University Researchers Supporting Project No. TURSP-2020/312, Taif University, Taif, Saudi Arabia. The authors gratefully acknowledge the financial support by Higher Education Commission of Pakistan HEC under research project NRPU No. 15800.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c01841.
(PDF)
Author Contributions
E.U.M.: Conceptualization, supervision, funding acquisition, investigation, writing—review and editing; J.A.: Data analysis and collection, first-draft preparation; E.M.H.: Data analysis and collection, first-draft preparation. Y.N.: Performed molecular docking, QSAR and DFT studies; A.S.A.: Data analysis and collection, first-draft preparation, formal analysis; N.N.: Data analysis and collection, first-draft preparation, formal analysis; A.S.: Co-supervision, manuscript writing; R.A.A.: Data analysis and collection, first-draft preparation. S.A.A.: Main idea, supervision, investigation, data analysis, funding acquisition, final writing, and editing the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Rafiq M.; Nazir Y.; Ashraf Z.; Rafique H.; Afzal S.; Mumtaz A.; Hassan M.; Ali A.; Afzal K.; Yousuf M. R.; et al. Synthesis, computational studies, tyrosinase inhibitory kinetics and antimelanogenic activity of hydroxy substituted 2-[(4-acetylphenyl) amino]-2-oxoethyl derivatives. J. Enzyme Inhib. Med. Chem. 2019, 34 (1), 1562–1572. 10.1080/14756366.2019.1654468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo S.-Y.; Sharma V. K.; Sharma N. Mushroom tyrosinase: recent prospects. J. Agric. Food Chem. 2003, 51 (10), 2837–2853. 10.1021/jf020826f. [DOI] [PubMed] [Google Scholar]
- Tsuji-Naito K.; Hatani T.; Okada T.; Tehara T. Modulating effects of a novel skin-lightening agent, α-lipoic acid derivative, on melanin production by the formation of DOPA conjugate products. Bioorg. Med. Chem. 2007, 15 (5), 1967–1975. 10.1016/j.bmc.2006.12.042. [DOI] [PubMed] [Google Scholar]
- Ismail T.; Shafi S.; Srinivas J.; Sarkar D.; Qurishi Y.; Khazir J.; Alam M. S.; Kumar H. M. S. Synthesis and tyrosinase inhibition activity of trans-stilbene derivatives. Bioorg. Chem. 2016, 64, 97–102. 10.1016/j.bioorg.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Khan M. T. H.Heterocyclic compounds against the enzyme tyrosinase essential for melanin production: biochemical features of inhibition. In Bioactive Heterocycles III ;Springer, 2007; pp 119–138. [Google Scholar]
- Ali A.; Ashraf Z.; Rafiq M.; Kumar A.; Jabeen F.; Lee G. J.; Nazir F.; Ahmed M.; Rhee M.; Choi E. H. Novel amide derivatives as potent tyrosinase inhibitors; in-vitro, in-vivo antimelanogenic activity and computational studies. Med. Chem. 2019, 15 (7), 715–728. 10.2174/1573406415666190319101329. [DOI] [PubMed] [Google Scholar]
- Nazir Y.; Saeed A.; Rafiq M.; Afzal S.; Ali A.; Latif M.; Zuegg J.; Hussein W. M.; Fercher C.; Barnard R. T.; et al. Hydroxyl substituted benzoic acid/cinnamic acid derivatives: Tyrosinase inhibitory kinetics, anti-melanogenic activity and molecular docking studies. Bioorg. Med. Chem. Lett. 2020, 30 (1), 126722–126731. 10.1016/j.bmcl.2019.126722. [DOI] [PubMed] [Google Scholar]
- Friedman M. Food browning and its prevention: an overview. J. Agric. Food Chem. 1996, 44 (3), 631–653. 10.1021/jf950394r. [DOI] [Google Scholar]
- Matsuura R.; Ukeda H.; Sawamura M. Tyrosinase inhibitory activity of citrus essential oils. J. Agric. Food Chem. 2006, 54 (6), 2309–2313. 10.1021/jf051682i. [DOI] [PubMed] [Google Scholar]
- Mayer A. M.; Harel E. Polyphenol oxidases in plants. Phytochemistry 1979, 18 (2), 193–215. 10.1016/0031-9422(79)80057-6. [DOI] [Google Scholar]
- Nihei K.-i.; Kubo I. Identification of oxidation product of arbutin in mushroom tyrosinase assay system. Bioorg. Med. Chem. Lett. 2003, 13 (14), 2409–2412. 10.1016/S0960-894X(03)00395-0. [DOI] [PubMed] [Google Scholar]
- Uiterkamp A. S.; Mason H. Magnetic dipole-dipole coupled Cu (II) pairs in nitric oxide-treated tyrosinase: A structural relationship between the active sites of tyrosinase and hemocyanin. Proc. Natl. Acad. Sci. U. S. A. 1973, 70 (4), 993–996. 10.1073/pnas.70.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gelder C. W.; Flurkey W. H.; Wichers H. J. Sequence and structural features of plant and fungal tyrosinases. Phytochemistry 1997, 45 (7), 1309–1323. 10.1016/S0031-9422(97)00186-6. [DOI] [PubMed] [Google Scholar]
- Nazir Y.; Rafique H.; Roshan S.; Shamas S.; Ashraf Z.; Rafiq M.; Tahir T.; Qureshi Z.-U.-R.; Aslam A.; Asad M. H. H. B. Molecular Docking, synthesis, and tyrosinase inhibition activity of acetophenone amide: Potential inhibitor of melanogenesis. BioMed. Res. Int. 2022, 2022, 1040693–1040705. 10.1155/2022/1040693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan K. M.; Mughal U. R.; Khan M. T. H.; Zia-Ullah; Perveen S.; Iqbal Choudhary M. Oxazolones: new tyrosinase inhibitors; synthesis and their structure–activity relationships. Bioorg. Med. Chem. 2006, 14 (17), 6027–6033. 10.1016/j.bmc.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Butt A. R. S.; Abbasi M. A.; Aziz-ur-Rehman; Siddiqui S. Z.; Raza H.; Hassan M.; Shah S. A. A.; Shahid M.; Seo S.-Y. Synthesis and structure-activity relationship of tyrosinase inhibiting novel bi-heterocyclic acetamides: Mechanistic insights through enzyme inhibition, kinetics and computational studies. Bioorg. Chem. 2019, 86, 459–472. 10.1016/j.bioorg.2019.01.036. [DOI] [PubMed] [Google Scholar]
- Gawande S. S.; Warangkar S. C.; Bandgar B. P.; Khobragade C. N. Synthesis of new heterocyclic hybrids based on pyrazole and thiazolidinone scaffolds as potent inhibitors of tyrosinase. Bioorg. Med. Chem. 2013, 21 (10), 2772–2777. 10.1016/j.bmc.2012.12.053. [DOI] [PubMed] [Google Scholar]
- Nazir Y.; Rafique H.; Kausar N.; Abbas Q.; Ashraf Z.; Rachtanapun P.; Jantanasakulwong K.; Ruksiriwanich W. Methoxy-substituted tyramine derivatives synthesis, computational studies and tyrosinase inhibitory kinetics. Molecules 2021, 26 (9), 2477–2482. 10.3390/molecules26092477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hałdys K.; Goldeman W.; Jewgiński M.; Wolińska E.; Anger-Góra N.; Rossowska J.; Latajka R. Halogenated aromatic thiosemicarbazones as potent inhibitors of tyrosinase and melanogenesis. Bioorg. Chem. 2020, 94, 103419–103428. 10.1016/j.bioorg.2019.103419. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Zheng L.; Li Y.; Su F.; Yue X.; Tang W.; Ma X.; Nie J.; Li H. Synthesis and structure–activity relationships and effects of phenylpropanoid amides of octopamine and dopamine on tyrosinase inhibition and antioxidation. Food Chem. 2012, 134 (2), 1128–1131. 10.1016/j.foodchem.2012.02.152. [DOI] [PubMed] [Google Scholar]
- Ullah S.; Park C.; Ikram M.; Kang D.; Lee S.; Yang J.; Park Y.; Yoon S.; Chun P.; Moon H. R. Tyrosinase inhibition and anti-melanin generation effect of cinnamamide analogues. Bioorg. Chem. 2019, 87, 43–55. 10.1016/j.bioorg.2019.03.001. [DOI] [PubMed] [Google Scholar]
- a Ashraf J.; Mughal E. U.; Alsantali R. I.; Obaid R. J.; Sadiq A.; Naeem N.; Ali A.; Massadaq A.; Javed Q.; Javid A.; et al. Structure-based designing and synthesis of 2-phenylchromone derivatives as potent tyrosinase inhibitors: In vitro and in silico studies. Bioorg. Med. Chem. 2021, 35, 116057–116071. 10.1016/j.bmc.2021.116057. [DOI] [PubMed] [Google Scholar]; b Nagy P. I. Replacement of oxygen by sulfur in small organic molecules. 3. Theoretical studies on the Tautomeric Equilibria of the 2OH and 4OH-substituted oxazole and thiazole and the 3OH and 4OH-substituted isoxazole and isothiazole in the isolated state and in solution. Int. J. Mol. Sci. 2016, 17 (7), 1094–1102. 10.3390/ijms17071094. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Nagy P. I. In-solution conformational analysis of the XCYCH3 moiety for small esters and ethers with all combinations of X, Y= O, S. Molecules 2013, 18 (7), 8063–8082. 10.3390/molecules18078063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obaid R. J.; Mughal E. U.; Naeem N.; Sadiq A.; Alsantali R. I.; Jassas R. S.; Moussa Z.; Ahmed S. A. Natural and synthetic flavonoid derivatives as new potential tyrosinase inhibitors: A systematic review. RSC Adv. 2021, 11 (36), 22159–22198. 10.1039/D1RA03196A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Shu C.; Liu L.; Huang Q.; Peng Y. Design and synthesis of thiourea derivatives with sulfur-containing heterocyclic scaffolds as potential tyrosinase inhibitors. Bioorg. Med. Chem. 2016, 24 (8), 1866–1871. 10.1016/j.bmc.2016.03.013. [DOI] [PubMed] [Google Scholar]
- Liu J.; Yi W.; Wan Y.; Ma L.; Song H. 1-(1-Arylethylidene) thiosemicarbazide derivatives: A new class of tyrosinase inhibitors. Bioorg. Med. Chem. 2008, 16 (3), 1096–1102. 10.1016/j.bmc.2007.10.102. [DOI] [PubMed] [Google Scholar]
- Lam K. W.; Syahida A.; Ul-Haq Z.; Rahman M. B. A.; Lajis N. H. Synthesis and biological activity of oxadiazole and triazolothiadiazole derivatives as tyrosinase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20 (12), 3755–3759. 10.1016/j.bmcl.2010.04.067. [DOI] [PubMed] [Google Scholar]
- Havasi M. H.; Ressler A. J.; Parks E. L.; Cocolas A. H.; Weaver A.; Seeram N. P.; Henry G. E. Antioxidant and tyrosinase docking studies of heterocyclic sulfide derivatives containing a thymol moiety. Inorg. Chim. Acta 2020, 505, 119495–119509. 10.1016/j.ica.2020.119495. [DOI] [Google Scholar]
- Ha Y. M.; Park Y. J.; Kim J.-A.; Park D.; Park J. Y.; Lee H. J.; Lee J. Y.; Moon H. R.; Chung H. Y. Design and synthesis of 5-(substituted benzylidene) thiazolidine-2, 4-dione derivatives as novel tyrosinase inhibitors. Eur. J. Med. Chem. 2012, 49, 245–252. 10.1016/j.ejmech.2012.01.019. [DOI] [PubMed] [Google Scholar]
- Battin E. E.; Brumaghim J. L. Antioxidant activity of sulfur and selenium: a review of reactive oxygen species scavenging, glutathione peroxidase, and metal-binding antioxidant mechanisms. Cell Biochem. Biophy. 2009, 55 (1), 1–23. 10.1007/s12013-009-9054-7. [DOI] [PubMed] [Google Scholar]
- Kessler D. Enzymatic activation of sulfur for incorporation into biomolecules in prokaryotes. FEMS Microbiol. Rev. 2006, 30 (6), 825–840. 10.1111/j.1574-6976.2006.00036.x. [DOI] [PubMed] [Google Scholar]
- Chang T.-S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10 (6), 2440–2475. 10.3390/ijms10062440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- N Masum M.; Yamauchi K.; Mitsunaga T. Tyrosinase inhibitors from natural and synthetic sources as skin-lightening agents. Rev. Agric. Sci. 2019, 7, 41–58. 10.7831/ras.7.41. [DOI] [Google Scholar]
- Zolghadri S.; Bahrami A.; Hassan Khan M. T.; Munoz-Munoz J.; Garcia-Molina F.; Garcia-Canovas F.; Saboury A. A. A comprehensive review on tyrosinase inhibitors. J. Enzyme Inhib. Med. Chem. 2019, 34 (1), 279–309. 10.1080/14756366.2018.1545767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsantali R. I.; Mughal E. U.; Naeem N.; Alsharif M. A.; Sadiq A.; Ali A.; Jassas R. S.; Javed Q.; Javid A.; Sumrra S. H. Flavone-based hydrazones as new tyrosinase inhibitors: Synthetic imines with emerging biological potential, SAR, molecular docking and drug-likeness studies. J. Mol. Struct. 2022, 1251, 131933–131951. 10.1016/j.molstruc.2021.131933. [DOI] [Google Scholar]
- Ashraf J.; Mughal E. U.; Sadiq A.; Naeem N.; Muhammad S. A.; Qousain T.; Zafar M. N.; Khan B. A.; Anees M. Design and synthesis of new flavonols as dual α-amylase and α-glucosidase inhibitors: Structure-activity relationship, drug-likeness, in vitro and in silico studies. J. Mol. Struct. 2020, 1218, 128458–128471. 10.1016/j.molstruc.2020.128458. [DOI] [Google Scholar]
- Ullah Mughal E.; Ayaz M.; Hussain Z.; Hasan A.; Sadiq A.; Riaz M.; Malik A.; Hussain S.; Choudhary M. I. Synthesis and antibacterial activity of substituted flavones, 4-thioflavones and 4-iminoflavones. Bioorg. Med. Chem. 2006, 14 (14), 4704–4711. 10.1016/j.bmc.2006.03.031. [DOI] [PubMed] [Google Scholar]
- Mughal E. U.; Javid A.; Sadiq A.; Murtaza S.; Zafar M. N.; Khan B. A.; Sumrra S. H.; Tahir M. N.; Kanwal; Khan K. M. Synthesis, structure-activity relationship and molecular docking studies of 3-O-flavonol glycosides as cholinesterase inhibitors. Bioorg. Med. Chem. 2018, 26 (12), 3696–3706. 10.1016/j.bmc.2018.05.050. [DOI] [PubMed] [Google Scholar]
- Mughal E. U.; Sadiq A.; Ashraf J.; Zafar M. N.; Sumrra S. H.; Tariq R.; Mumtaz A.; Javid A.; Khan B. A.; Ali A.; et al. Flavonols and 4-thioflavonols as potential acetylcholinesterase and butyrylcholinesterase inhibitors: Synthesis, structure-activity relationship and molecular docking studies. Bioorg. Chem. 2019, 91, 103124–103135. 10.1016/j.bioorg.2019.103124. [DOI] [PubMed] [Google Scholar]
- Mughal E. U.; Sadiq A.; Ayub M.; Naeem N.; Javid A.; Sumrra S. H.; Zafar M. N.; Khan B. A.; Malik F. P.; Ahmed I. Exploring 3-Benzyloxyflavones as new lead cholinesterase inhibitors: synthesis, structure–activity relationship and molecular modelling simulations. J. Biomol. Struct. Dyn. 2021, 39, 1–14. 10.1080/07391102.2020.1803136. [DOI] [PubMed] [Google Scholar]
- Alsharif M. A.; Naeem N.; Mughal E. U.; Sadiq A.; Jassas R. S.; Kausar S.; Altaf A. A.; Zafar M. N.; Mumtaz A.; Obaid R. J.; et al. Experimental and theoretical insights into the photophysical and electrochemical properties of flavone-based hydrazones. J. Mol. Struct. 2021, 1244, 130965–130981. 10.1016/j.molstruc.2021.130965. [DOI] [Google Scholar]
- Keri R. S.; Budagumpi S.; Pai R. K.; Balakrishna R. G. Chromones as a privileged scaffold in drug discovery: A review. Eur. J. Med. Chem. 2014, 78, 340–374. 10.1016/j.ejmech.2014.03.047. [DOI] [PubMed] [Google Scholar]
- Ravishankar D.; Corona G.; Hogan S. M.; Spencer J. P.; Greco F.; Osborn H. M. Thioflavones as novel neuroprotective agents. Bioorg. Med. Chem. 2016, 24 (21), 5513–5520. 10.1016/j.bmc.2016.09.006. [DOI] [PubMed] [Google Scholar]
- Pingaew R.; Saekee A.; Mandi P.; Nantasenamat C.; Prachayasittikul S.; Ruchirawat S.; Prachayasittikul V. Synthesis, biological evaluation and molecular docking of novel chalcone–coumarin hybrids as anticancer and antimalarial agents. Eur. J. Med. Chem. 2014, 85, 65–76. 10.1016/j.ejmech.2014.07.087. [DOI] [PubMed] [Google Scholar]
- Martins I. s. L.; Charneira C.; Gandin V.; Ferreira da Silva J. L.; Justino G. a. C.; Telo J. o. P.; Vieira A. J.; Marzano C.; Antunes A. M. Selenium-containing chrysin and quercetin derivatives: Attractive scaffolds for cancer therapy. J. Med. Chem. 2015, 58 (10), 4250–4265. 10.1021/acs.jmedchem.5b00230. [DOI] [PubMed] [Google Scholar]
- Anderson S. N.; Larson M. T.; Berreau L. M. Solution or solid–it doesn’t matter: visible light-induced CO release reactivity of zinc flavonolato complexes. Dalton Trans 2016, 45 (37), 14570–14580. 10.1039/C6DT01709F. [DOI] [PubMed] [Google Scholar]
- Dong J.; Zhang Q.; Meng Q.; Wang Z.; Li S.; Cui J. The chemistry and biological effects of thioflavones. Mini Rev. Med. Chem. 2018, 18 (20), 1714–1732. 10.2174/1389557518666180515145633. [DOI] [PubMed] [Google Scholar]
- Singh M.; Kaur M.; Silakari O. Flavones: An important scaffold for medicinal chemistry. Eur. J. Med. Chem. 2014, 84, 206–239. 10.1016/j.ejmech.2014.07.013. [DOI] [PubMed] [Google Scholar]
- Arroo R. R.; Sari S.; Barut B.; Özel A.; Ruparelia K. C.; Şöhretoğlu D. Flavones as tyrosinase inhibitors: kinetic studies in vitro and in silico. Phytochem. Anal. 2020, 31 (3), 314–321. 10.1002/pca.2897. [DOI] [PubMed] [Google Scholar]
- Falcone Ferreyra M. L.; Rius S.; Casati P. Flavonoids: biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 2012, 3, 222–234. 10.3389/fpls.2012.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierziak J.; Kostyn K.; Kulma A. Flavonoids as important molecules of plant interactions with the environment. Molecules 2014, 19 (10), 16240–16265. 10.3390/molecules191016240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dinya Z.; Sztaricskai F.; Lévai A.; Litkei G. Photoelectron (Hel) spectroscopy of flavonoids and thioflavonoids, ii. photoelectron spectra of chromone and 1-thiochromone derivatives. Croat. Chem. Acta 1993, 66 (2), 265–278. [Google Scholar]; b Saeed A.; Ahmed A.; Ali Channar P.; Shabir G.; Hassan A.; Zia-ur-Rehman; Abbas Q.; Hassan M.; Raza H.; Seo S.-Y.; et al. Identification of novel C-2 symmetric Bis-Azo-Azamethine molecules as competitive inhibitors of mushroom tyrosinase and free radical scavengers: synthesis, kinetics, and molecular docking studies. J. Biomol. Struct. Dyn. 2020, 1–10. 10.1080/07391102.2020.1857841. [DOI] [PubMed] [Google Scholar]; c Kuo S.-C.; Ibuka T.; Huang L.-J.; Lien J.-C.; Yean S.-R.; Huang S.-C.; Lednicer D.; Morris-Natschke S.; Lee K.-H. Synthesis and cytotoxicity of 1, 2-disubstituted naphth [2, 3-d] imidazole-4, 9-diones and related compounds. J. Med. Chem. 1996, 39 (7), 1447–1451. 10.1021/jm950247k. [DOI] [PubMed] [Google Scholar]; d Mehmood R.; Sadiq A.; Alsantali R. I.; Mughal E. U.; Alsharif M. A.; Naeem N.; Javid A.; Al-Rooqi M. M.; Chaudhry G.-e.-S.; Ahmed S. A. Synthesis and evaluation of 1, 3, 5-triaryl-2-pyrazoline derivatives as potent dual inhibitors of urease and α-glucosidase together with their cytotoxic, molecular modeling and drug-likeness studies. ACS Omega 2022, 7 (4), 3775–3795. 10.1021/acsomega.1c06694. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Adeniji S. E.; Uba S.; Uzairu A. Future Journal of Pharmaceutical sciences. Future 2018, 4, 284–295. 10.1016/j.fjps.2018.10.003. [DOI] [Google Scholar]; f Khatkar A.; Nanda A.; Kumar P.; Narasimhan B. Synthesis, antimicrobial evaluation and QSAR studies of p-coumaric acid derivatives. Arab. J. Chem. 2017, 10, S3804–S3815. 10.1016/j.arabjc.2014.05.018. [DOI] [Google Scholar]; g van Zanden J. J.; Wortelboer H. M.; Bijlsma S.; Punt A.; Usta M.; van Bladeren P. J.; Rietjens I. M.; Cnubben N. H. Quantitative structure activity relationship studies on the flavonoid mediated inhibition of multidrug resistance proteins 1 and 2. Biochem. Pharmacol. 2005, 69 (4), 699–708. 10.1016/j.bcp.2004.11.002. [DOI] [PubMed] [Google Scholar]; h Hevener K. E.; Ball D. M.; Buolamwini J. K.; Lee R. E. Quantitative structure–activity relationship studies on nitrofuranyl anti-tubercular agents. Bioorg. Med. Chem. 2008, 16 (17), 8042–8053. 10.1016/j.bmc.2008.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Sarwar M. W.; Riaz A.; Dilshad S. M. R.; Al-Qahtani A.; Nawaz-Ul-Rehman M. S.; Mubin M. Structure activity relationship (SAR) and quantitative structure activity relationship (QSAR) studies showed plant flavonoids as potential inhibitors of dengue NS2B-NS3 protease. BMC Struc. Bio. 2018, 18 (1), 1–10. 10.1186/s12900-018-0084-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai T. The construction and application of C= S bonds. Top. Curr. Chem. 2018, 376 (4), 1–21. 10.1007/s41061-018-0209-0. [DOI] [PubMed] [Google Scholar]
- Tran B. L.; Cohen S. M. Flavothionato metal complexes: implications for the use of hydroxyflavothiones as green pesticides. Chem. Commun. 2006, 2, 203–205. 10.1039/B512185J. [DOI] [PubMed] [Google Scholar]
- Xiao X.; Wang X.; Gui X.; Chen L.; Huang B. Natural flavonoids as promising analgesic candidates: a systematic review. Chem. Biodivers. 2016, 13 (11), 1427–1440. 10.1002/cbdv.201600060. [DOI] [PubMed] [Google Scholar]
- Ali A.; Ashraf Z.; Kumar N.; Rafiq M.; Jabeen F.; Park J. H.; Choi K. H.; Lee S.; Seo S.-Y.; Choi E. H.; et al. Influence of plasma-activated compounds on melanogenesis and tyrosinase activity. Sci. Rep. 2016, 6 (1), 1–20. 10.1038/srep21779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf J.; Mughal E. U.; Alsantali R. I.; Sadiq A.; Jassas R. S.; Naeem N.; Ashraf Z.; Nazir Y.; Zafar M. N.; Mumtaz A.; et al. 2-Benzylidenebenzofuran-3 (2 H)-ones as a new class of alkaline phosphatase inhibitors: synthesis, SAR analysis, enzyme inhibitory kinetics and computational studies. RSC Adv. 2021, 11 (56), 35077–35092. 10.1039/D1RA07379F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.Gaussian09. http://www.gaussian.com/2009. (accessed March 11, 2022)
- Hayani S.; Sert Y.; Baba Y. F.; Benhiba F.; Chahdi F. O.; Laraqui F.-Z.; Mague J. T.; El Ibrahimi B.; Sebbar N. K.; Rodi Y. K.; et al. New alkyl (cyclohexyl) 2-oxo-1-(prop-2-yn-1-yl)-1, 2-dihydroquinoline-4-carboxylates: Synthesis, crystal structure, spectroscopic characterization, hirshfeld surface analysis, molecular docking studies and DFT calculations. J. Mol. Struct. 2021, 1227, 129520–129534. 10.1016/j.molstruc.2020.129520. [DOI] [Google Scholar]
- Mendoza-Huizar L. H.; Rios-Reyes C. H.; Zuñiga-Trejo H. A computational study of the chemical reactivity of isoxaflutole herbicide and its active metabolite using global and local descriptors. J. Serb. Chem. Soc. 2020, 85 (9), 1163–1174. 10.2298/JSC191105024M. [DOI] [Google Scholar]
- Ashraf J.; Mughal E. U.; Sadiq A.; Bibi M.; Naeem N.; Ali A.; Massadaq A.; Fatima N.; Javid A.; Zafar M. N.; et al. Exploring 3-hydroxyflavone scaffolds as mushroom tyrosinase inhibitors: synthesis, X-ray crystallography, antimicrobial, fluorescence behaviour, structure-activity relationship and molecular modelling studies. J. Biomol. Struct. Dyn. 2021, 39 (18), 7107–7122. 10.1080/07391102.2020.1805364. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.