

Abstract
Nevoid basal cell carcinoma syndrome (NBCCS) is an autosomal dominant disorder with an increased incidence of tumors, such as basal cell carcinomas and medulloblastomas. The PTCH1 gene, responsible for NBCCS, suppresses the hedgehog signaling pathway, which is recognized as one of the important pathways in tumorigenesis and, thus, is a therapeutic target in cancer. In the present study, we generated PTCH1−/− induced pluripotent stem cells (iPSCs) from NBCCS patient-derived iPSCs (PTCH1+/−) by gene editing. The proliferation of PTCH1−/− iPSCs was accelerated due to the activation of the hedgehog signaling pathway. When PTCH1−/− iPSCs were subcutaneously injected into immunodeficient mice, the resulting teratomas almost exclusively contained immature ectodermal lineage cells expressing medulloblastoma markers, and the percentages of the area occupied by medulloblastoma-like tissue were larger in PTCH1−/− teratomas than in PTCH1+/− teratomas. In contrast, in PTCH1+/+ teratomas, medulloblastoma-like tissue positive for all of these medulloblastoma markers was not observed. The present results indicate the importance of PTCH1 in medulloblastoma formation and the suitability of these gene-edited iPSCs and PTCH1−/− teratomas as models for the formation of tumors, such as medulloblastomas and Hh-related tumors.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12672-022-00498-x.
Keywords: Nevoid basal cell carcinoma syndrome, PTCH1, Medulloblastoma, iPSCs
Introduction
Nevoid basal cell carcinoma syndrome (NBCCS; OMIM 109400), also known as Gorlin syndrome, is an autosomal dominant disorder that is characterized by a variety of developmental abnormalities and an increased incidence of tumors, such as medulloblastomas, basal cell carcinomas (BCC), and keratocystic odontogenic tumors [1, 2]. The gene responsible for NBCCS is PTCH1, the human homolog of the Drosophila patched gene, located on 9q21.2 [3, 4]. This gene encodes a protein of 1,447 amino acid residues containing two large extracellular loops and 12-pass transmembrane domains that serves as a receptor for hedgehog (Hh) ligands, namely Sonic hedgehog, Desert hedgehog and Indian hedgehog. The Hh signaling pathway plays an important role in embryonic development, cell proliferation, and tumorigenesis [5, 6]. In the absence of Hh binding, PTCH1 suppresses 7-pass transmembrane protein smoothened (SMO), another component of the Hh signaling pathway. After the binding of ligands to PTCH1, the inhibitory effects of SMO are released, resulting in the activation of GLI transcription factors and alterations in target gene expression. Therefore, genetic mutations in PTCH1 lead to the constitutive activation of signaling that results in the development of NBCCS [7–10].
The Hh signaling pathway was recently recognized as one of the important pathways for carcinogenesis and as a therapeutic target in cancer. It remains active and plays important roles in the regulation of tissue homeostasis and stem cell maintenance in adults [11]. Besides NBCCS, the up-regulation of Hh signaling has been implicated in many sporadic malignancies, including breast cancer, pancreatic cancer, lung cancer, prostate cancer, BCC, and medulloblastomas [12–15]. Vismodegib, an inhibitor of SMO, is the first oral therapeutic agent to be tested for the treatment for BCC and Sonic hedgehog subgroup medulloblastomas in adults [16, 17]. Despite positive outcomes in Phase II clinical trials, safer and less toxic drugs are required due to side effects and the acquisition of resistance after the mutation of the SMO gene [17, 18]. Therefore, the establishment of Hh-related tumor models is expected for the effective screening of therapeutic small chemicals.
We previously reported the formation of medulloblastoma-like tissue in teratomas generated from NBCCS patient-derived induced pluripotent stem cells (iPSCs) and the loss of heterozygosity or a second mutation in the PTCH1 gene in the medulloblastoma, but not in the non-medulloblastoma portion of the teratoma [19]. In the present study, we disrupted a wild-type PTCH1 allele remaining in NBCCS-iPSCs using the CRISPR/Cas9 system and injected PTCH1−/− iPSCs into immunodeficient mice to form teratomas. The resulting tumors almost exclusively contained immature ectodermal lineage cells expressing medulloblastoma markers and may be a promising tool for the screening of small molecule drugs against Hh-related tumors.
Materials and methods
Ethical considerations
All experiments described below were reviewed and approved by the Institutional Review Board of Kitasato University School of Medicine, Chiba University Graduate School of Medicine and the National Center for Child Health and Development. Written informed consent was obtained from the patients. All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of Kitasato University School of Medicine (approval number: 2021-099). In all experiments, the size of the tumors did not exceed 10% of body weight, which is the tumor burden permitted by the committee. All experiments were performed in accordance with the guidelines of the National Institutes of Health, the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, and ARRIVE guidelines.
Plasmid construction
In order to alter the PTCH1 gene sequence in human iPSCs, a pair of phosphorylated and annealed oligonucleotides listed in supplementary Table S1 were cloned into the BbsI sites of the chimeric guide RNA and human codon-optimized Cas9 expression vector plasmids, pX330 or pX260, obtained from AddGene (https://www.addgene.org/) according to the manufacturer’s instructions.
Antibodies
Antibodies to SSEA4 (Merck Millipore), glial fibrillary acidic protein (GFAP) (R&D Systems), α-smooth muscle actin (α-SMA) (R&D Systems), alpha fetoprotein (AFP) (R&D Systems), β-III tubulin (Promega), synaptophysin (Agilent), Ki67 (Abcam), SHH (SantaCruz), and cleaved caspase-3 (Cell Signaling Technology) were used in the present study. An Alexa488-conjugated anti-mouse goat antibody (ThermoFisher) was used as a secondary antibody for immunocytochemistry.
Generation of iPSCs from NBCCS patients
The generation of NBCCS-specific iPSCs (NBCCS-iPSCs) from three cases, G11 (c.3130_3131dupGC), G36 (approximately 1.1-Mb deletion including the entire PTCH1 gene), and G72 (c.274delT), was previously described [20–22]. In brief, fibroblasts were obtained from non-cancerous tissues adjacent to the cancer during surgery and infected with Sendai viruses expressing the human transcription factors OCT3/4, SOX2, KLF4, and C-MYC. The emerging colonies were picked up and expanded.
Cell culture
Human iPSCs were maintained in StemFit AK02N human iPSC culture medium (Takara-bio) on a dish coated with iMatrix-511 matrix (Takara-bio) at 37 °C in humidified air with 5% CO2. Medium was changed every other day. Human iPSC colonies comprising closely packed cells were dissociated with a 1:1 solution of TrypLE reagent (ThermoFisher) and DPBS (ThermoFisher) containing 0.5 mM EDTA after a 2 h treatment with 10 μM Y-27632 (WAKO chemical), and then scraped. Dissociated cells were seeded at a density of 1.3 × 103 cells/cm2 and cultured in medium supplemented with 10 μM Y-27632 until the next day.
Editing of the PTCH1 gene in NBCCS-iPSCs
In total, 2 × 105 dissociated NBCCS-iPSCs were transfected with 1 μg of the CRISPR/Cas9 vector plasmids, pX330-G11mt or pX330-G11wt for G11-iPSC, pX330-G36wt1 or pX330-G36wt2 for G36-iPSC, and pX330-G72wt or pX260-G72mt for G72-iPSC, and 1 μg of the puromycin expression vector, pENTR-lox-Puro [23], using the 4D Nucleofector device (program CB-150) and P4 Primary Cell 4D-Nucleofector solution (both from Lonza) in the 20-μl format, and then plated on a 100-mm dish coated with iMatrix-511 matrix. Forty-eight hours after transfection, iPSCs were treated with 0.75 μg/ml puromycin for 16 h and cultured for 2 weeks. Each colony was split in two, with one half being maintained in culture. The other half was used for genotyping. Cells were lysed in lysis buffer (65 mM Tris–HCl pH8.0, 15.3 mM ammonium sulfate, 1 mM 2-melcaptoethanol, 1 mM EDTA, 0.5% Triton-X100, and 3 μg/ml proteinase K) at 55 °C for 2 h, and then incubated at 95 °C for 12 min to inactivate proteinase K. Extracted genomic DNA was used for PCR direct sequencing as a template. The primers used for PCR direct sequencing are listed in supplementary Table S2.
Immunocytochemistry of iPSCs
In total, 2 × 103 dissociated iPSCs were plated on the Nunc Lab-Tek Permanox 4-well slide chamber (Merck Millipore) coated with iMatrix-511. Ninety-six hours later, cells were fixed with PBS containing 4% paraformaldehyde at 4 °C for 1 h and permeabilized with PBS containing 0.3% Triton-X100 at room temperature for 1 h. After a treatment with the blocking solution, Block ACE (Bio-Rad), at room temperature for 1 h, cells were stained with the primary antibody followed by the Alexa488-conjugated secondary antibody. Cells were observed under the confocal microscope, LSM710 (Zeiss).
Cell proliferation assay
A cell proliferation assay was performed using the BrdU cell proliferation ELISA kit (Abcam) according to the manufacturer’s instructions. In brief, dissociated 400 iPSCs were plated on a 96-well plate coated with iMatrix-511. After 24, 48, 72, 96, 120 or 144 h of culture, cell were labeled with BrdU for 24 h, and fixed. Then, cells were treated with anti-BrdU antibody, peroxidase-conjugated secondary antibody, and TMB peroxidase substrate. Rates of BrdU incorporation were measured by reading the plate using the SpectraMax M2 spectrophotometer (Molecular Devices) at a wavelength of 450 nm.
Teratoma formation
Confluent human iPSCs cultured in a 60-mm dish were dissociated, suspended in 400 μl of a 1:1 solution of DPBS and Matrigel hESC-Qualified Matrix (Corning), and then subcutaneously injected into the dorsal flanks of CB17/IcrJcl-Prkdcscid immunodeficient mice (CLEA Japan, Inc.). Eight to twelve weeks after the injection, tumors were dissected and fixed in Maskedform (Japan Tanner). Paraffin-embedded tissue was sliced and stained with hematoxylin and eosin.
Histological analysis of teratoma
Hematoxylin and eosin staining was conventionally performed. Immunohistochemistry was conducted using a combination of the microwave oven heating and Histofine Simple Stain MAX-PO (Nichirei Bioscience) methods. In brief, slices were treated by microwave oven heating in 10 mM citrate buffer (pH6.0) or 10 mM Tris–EDTA buffer (pH9.0) for 15 min for antigen retrieval after deparaffinization, and incubated at 4 °C overnight with optimized dilutions of primary antibodies. Slice images were obtained using the microscope, BZ-9000 (Keyence), and combined using the software, BZ-H4XD (Keyence), to create composite images. In order to analyze the rate of apoptosis, the images of neuroepithelium including neural tubes were obtained using × 20 objective lens and counted cleaved caspase-3-positive cells in each image.
Quantitative RT-PCR
Quantitative RT-PCR was performed as described previously [23]. In brief, 5 µg of total RNA was reverse-transcribed and the resulting cDNA was used as a template for quantitative PCR. Primers used in the analysis are listed in supplementary Table S2. GAPDH served as the endogenous control. The expression levels of the Hh target genes were quantified using the Pfaffl analysis method [24].
Statistical analysis
Data are represented as means ± standard errors of the mean (SEM), and were compared using the Student’s t-test (Fig. 2) or Welch’s t-test (Fig. 4c).
Fig. 2.

Hh signaling was accelerated in PTCH1−/− iPSC lines. a Cell proliferation assay of PTCH1-edited iPSCs. Eight hundred cells were plated on a 96-well plate and cell numbers were measured every 24 h using the BrdU incorporation assay. b The expression levels of the Hh-signaling target genes, PTCH1, GLI1, and HHIP1 were normalized by GAPDH mRNA levels. Data are presented as means ± SEM (n = 3). *: p < 0.05, **: p < 0.01, ***: p < 0.001
Fig. 4.

Immunohistological analysis of marker proteins in teratomas. a The expression of the germ layer marker proteins, GFAP, αSMA, and AFP, was immunohistochemically analyzed in a PTCH1+/+ teratoma (G11 PTCH1+/+) (upper column) and PTCH1−/− teratoma (G72 PTCH1−/−) (lower column). Scale bar: 100 μm. b Expression of the marker proteins for medulloblastoma. A series of sections of a PTCH1−/− teratoma (G36 PTCH1−/−) were stained with anti-synaptophysin, anti-βIII tubulin, and anti-Ki67 antibodies. Scale bar: 60 μm. c Medulloblastomas occupied a larger area in PTCH1−/− teratomas than in PTCH1+/− teratomas. The percentage of areas of medulloblastoma-like tissues positive for βIII tubulin, synaptophysin, and Ki67 was calculated in PTCH1+/− and PTCH1−/− teratomas. Closed circles and error bars represent the mean ± SEM (PTCH1+/−; n = 5, PTCH1−/−; n = 9). Open circles represent each value
Results
Generation of PTCH1−/− iPSCs from NBCCS-iPSCs
In order to establish the Hh signaling-related tumor model, we disrupted the wild-type PTCH1 allele remaining in three NBCCS-derived iPSC lines, G11-iPSC, G36-iPSC, and G72-iPSC [21, 22], by introducing the CRISPR/Cas9 expression vector, which targets the wild-type PTCH1 sequence. We obtained two clones from each parental iPSC, named G72 PTCH1−/−, G36 PTCH1−/−, and G11 PTCH1−/− (Fig. 1, Table 1).
Fig. 1.

Schematic presentation of PTCH1-edited NBCCS iPSCs. a, b The disruption of the wild-type allele a and the correction of the mutated allele to the wild type b in G11-iPSC. The red and green arrowheads in the top panel indicate the locations of the nearest up- and downstream SNPs, respectively. The red and green arrows indicate the positions of the primer pairs used for PCR direct sequencing. c The disruption of the wild-type PTCH1 allele in G36-iPSC. Note that the entire PTCH1 on allele 2 was deleted in G36. d, e The disruption of the wild-type allele d and the correction of the mutated allele e in G72-iPSC. CRISPR/Cas9 target sequences and the protospacer adjacent motifs (PAM) were indicated by underlines and dotted underlines, respectively
Table 1.
PTCH1 mutations of gene-edited and parental iPSCs
| Name of iPSCs | Allele 1 | Allele 2 |
|---|---|---|
| G11 | wt | c.3130_3131dupGC |
| G11 PTCH1−/−#1 | c.3130_3131dupGC | c.3130_3131dupGC |
| G11 PTCH1−/−#2 | c.3130_3131dupGC | c.3130_3131dupGC |
| G11 PTCH1+/+#1 | wt | wt |
| G11 PTCH1+/+#2 | wt | wt |
| G36 | wt | large deletion |
| G36 PTCH1−/−#1 | c.570_571insA | large deletion |
| G36 PTCH1−/−#2 | c.1207delT | large deletion |
| G72 | wt | c.274delT |
| G72 PTCH1−/−#1 | c.364_365delGA | c.274delT |
| G72 PTCH1−/−#2 | c.364_365delGA | c.274delT |
| G72 PTCH1+/+ | wt | wt |
In G11-PTCH1−/− iPSCs, the wild-type PTCH1 sequence was eliminated, whereas the heterozygosity of the nearest SNPs was maintained (Fig. 1a, lower panel). Deletions between these SNP sites were not detected by PCR direct sequencing (data not shown). These results indicated that the wild-type PTCH1 allele (allele 1) was edited into an identical sequence with a germline mutation via homologous recombination using allele 2.
To confirm the disruption of the wild-type PTCH1 allele in G72-iPSC, we performed TA cloning to select clones in which only the wild-type PTCH1 allele was mutated because the CRISPR/Cas9 target sequence may edit wild-type and mutated alleles. Two G72-PTCH1−/− clones coincidently obtained the same mutation, c.364_365delGA, on allele 1 (Fig. 1d, lower panel).
In G36-iPSC, the wild-type allele (allele 1) was selectively detected by PCR due to the entire deletion of the PTCH1 allele in allele 2. The remaining allele was efficiently disrupted by c.570_571insA or c.1207delT in clones 1 and 2, respectively (Fig. 1c).
To produce PTCH1+/+ clones from G11-iPSC, mutated PTCH1 allele-specific CRISPR/Cas9 vectors were transfected into G11-iPSC. Two clones were obtained in which the mutated allele (allele 1) was converted into the wild-type sequence. The possibility of the deletion of allele 2 was excluded by PCR direct sequencing and the heterozygosity of nearby SNPs (Fig. 1b, lower panel).
To correct the mutated PTCH1 allele in G11-iPSC, we employed the pX260 vector, which recognizes a target sequence of 30 bp in length [25] instead of pX330 because we were unable to identify a protospacer adjacent motif within 20 bp around the germline mutation. The possibility of the deletion of allele 2 was excluded by PCR direct sequencing and the heterozygosity of the nearby PNP of the in/del type, rs71366292 (Fig. 1e, lower panel). The genotypes of the edited clones are listed in Table 1. The expression of pluripotent markers, such as SSEA4, OCT3/4, NANOG, and SOX2, were confirmed by immunofluorescence and RT-PCR after gene editing (Fig. S1).
Characterization of PTCH1−/− iPSCs
We investigated whether Hh signaling was accelerated in PTCH1-edited iPSCs by measuring BrdU incorporation into newly synthesized DNA and by RT-qPCR of Hh target genes. All PTCH1−/− iPSC clones proliferated more rapidly than parental NBCCS-iPSCs. In contrast, PTCH1+/+ clones proliferated more slowly than parental cells. These results are consistent with activated and inhibited Hh signaling in PTCH1−/− iPSCs and PTCH1+/+ iPSCs, respectively (Fig. 2a). The expression levels of Hh-signaling target genes, such as PTCH1, GLI1, and HHIP1, were also investigated in these iPSCs (Fig. 2b). The expression of PTCH1 and HHIP1 was significantly stronger and weaker, respectively, in PTCH1−/− clones and PTCH1+/+ clones than in parental iPSCs. Changes in the expression of GLI1 were less evident than those of other target genes.
Teratoma formation
We subcutaneously transplanted these iPSCs into the dorsal flanks of immunodeficient mice to generate teratomas. As we reported previously [19], paraffin-embedded sections of the resulting tumors contained the components of the three germ layers, such as the neural tube, cartilage, and the intestine, in teratomas generated from parental NBCCS-iPSCs as well as gene-edited PTCH1+/+ iPSCs (Fig. 3a–c, f–h). In addition, we confirmed the presence of melanocytes as a marker for highly differentiated ectodermal cells [26] in these teratomas (Fig. 3d, i). On the other hand, PTCH1−/− teratomas frequently lacked mesodermal and/or endodermal components. Among the 10 PTCH1−/− teratomas investigated, only one had all 3 germ layers, whereas three had ectodermal and endodermal tissues, and the remaining 6 teratomas only contained the ectodermal component (Fig. 3k, Table 2). In addition, no melanocytes were found in PTCH1−/− teratomas.
Fig. 3.
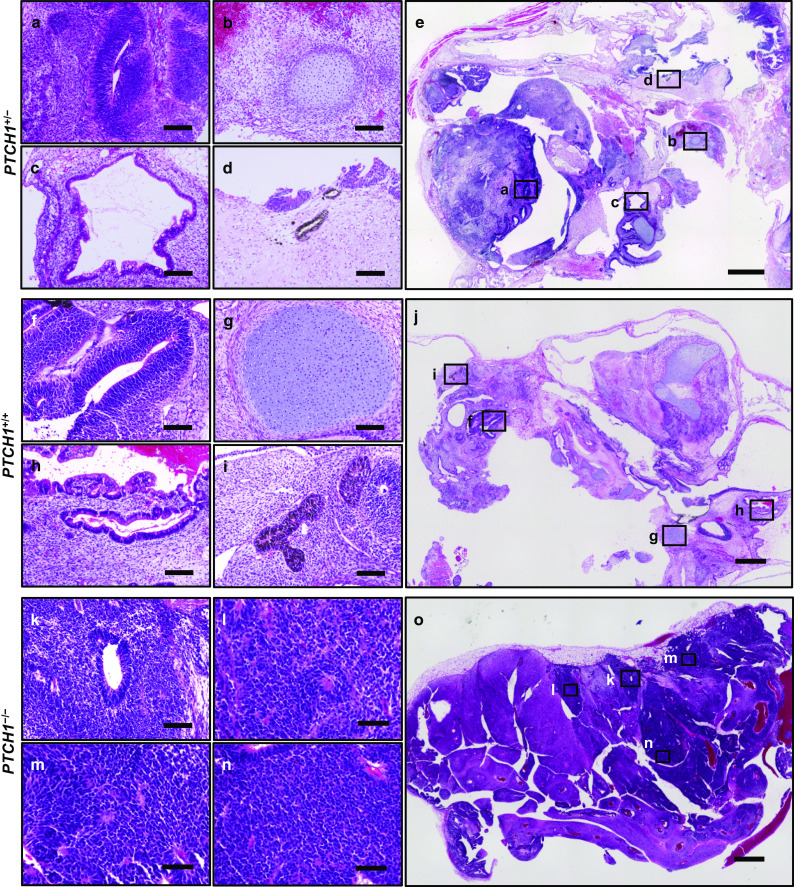
HE staining of PTCH1+/−, PTCH1+/+, and PTCH1−/− teratomas. HE staining of a PTCH1+/− teratoma (G11-NBCCS) (a-e), PTCH1+/+ teratoma (G11 PTCH1+/+) (f–j), and PTCH1−/− teratoma (G36 PTCH1−/−) (k–o). Neural tubes (a, f, k), cartilage (b, g), the intestine (c, h), and melanocytes d, i are shown. Whole-slice images of teratomas are shown in e, j, and o. Higher magnifications of the areas indicated by rectangles in e, j, and o are shown in a-d, f-i, and k-n. Scale bars: 100 μm in a–d, f–i, and k, 1 mm in e, j, and o, 60 μm in l–n
Table 2.
Numbers of teratomas with 3 germ layer components and melanocytes
| PTCH1+/+ | PTCH1+/− | PTCH1−/− | |
|---|---|---|---|
| Neural tube | 5/5 | 6/6 | 10/10 |
| Cartilage | 5/5 | 5/6 | 1/10 |
| The intestine | 5/5 | 6/6 | 3/10 |
| Melanocytes | 4/5 | 4/6 | 0/10 |
We then confirmed these results by immunohistochemistry. We stained the marker proteins of the three germ layers: GFAP for the ectoderm, α-SMA for the mesoderm, and AFP for the endoderm (Fig. 4a). The PTCH1+/+ and PTCH1+/− teratomas were positive for all of these marker proteins. In contrast, in PTCH1−/− teratomas, GFAP was positive in most areas, whereas α-SMA-positive signals were only observed in perivascular cells, and AFP was negative. These results indicate that PTCH1−/− iPSCs predominantly differentiate into ectodermal cells and remain in an undifferentiated state in vivo.
In addition, medulloblastoma-like tissues were frequently observed in PTCH1−/− teratomas (Fig. 3l–n). These medulloblastoma-like tissues were confirmed to be strongly positive for medulloblastoma markers, such as βIII tubulin (Tuj1), synaptophysin, and Ki67 (Fig. 4b). The percentages of the area occupied by medulloblastomas were significantly larger in PTCH1−/− teratomas than in PTCH1+/− teratomas (Fig. 4c, Fig. S2). These results support the suitability of PTCH1−/− teratomas as a model for Hh-related tumors, including medulloblastomas. In contrast, no medulloblastoma-like tissue positive for medulloblastoma markers was observed in teratomas generated from gene-edited PTCH1+/+ iPSCs (Fig. 3f–j), demonstrating that the rescuing a mutated allele in PTCH1+/− iPSCs inhibited medulloblastoma formation.
PTCH1 has not only been described as an Hh receptor and inhibitor of SMO, but also other functions have been unveiled (e.g. the pro-apoptotic signaling coming from the PTCH1 receptor in the absence of the Hh ligand) [27]. We investigated the expression levels of SHH in the iPSC lines by RT-PCR, and explored the expression of SHH protein and the rate of apoptosis in teratomas using anti-SHH and anti-cleaved caspase-3 antibodies, respectively. Only two G72-PTCH1−/−-derived iPSC lines expressed SHH, in which the expression levels were very low (SHH Ct > 28 vs. GAPDH Ct < 18) (Fig. S3). Regardless of SHH expressions in limited lines of original iPSCs, the SHH protein was weakly, but widely expressed at various tissues such as intestinal epitheliums, gland-like tissues, and neural tissues in PTCH1+/− teratomas and in PTCH1−/− teratomas (Fig. 5a–f). Apoptotic cells were found in PTCH1+/− and in PTCH1−/− teratomas, but there was no statistical difference of the apoptotic rate between two genotypes (Fig. 5g–i), suggesting the apoptosis in these teratoma was induced by ligand-free PTCH1-independent fashion.
Fig. 5.
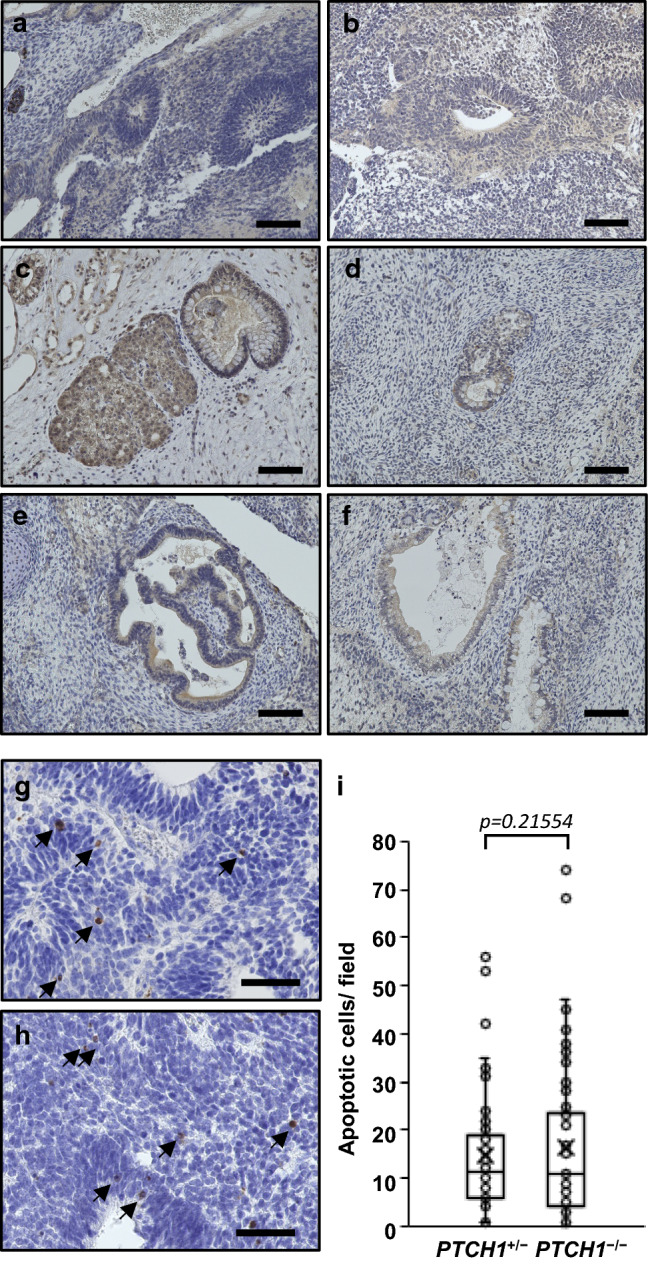
The SHH expressions and apoptotic cells in teratomas. a–f Immunohistological staining of SHH. SHH was expressed in neural tissues (a, b), glands c, d and intestinal epithelium e, f in PTCH1+/− a, c and e and PTCH1−/− teratomas (b, d and f). g–i Apoptosis in PTCH1+/− g and PTCH1−/− teratoma (h). Cleaved caspase-3 positive apoptotic cells were indicated by allows. Numbers of apoptotic cells counted in each field were displayed in the boxplot (i). Open circles and cross signs indicate each value of apoptotic cells and the averages, respectively (PTCH1+/−; n = 55, PTCH1−/−; n = 85). Scale bars: 100 μm in a–f, 50 μm in g and h
Discussion
Disease-specific iPSCs from patients with germline mutations are useful for understanding the initiation and progression of genetic disorders and examining relevant therapeutic targets and reagents. We previously demonstrated that when NBCCS-iPSCs were injected into immunodeficient mice, they developed teratomas containing small fractions of medulloblastoma-like tissues. These tissues carried secondary mutations in PTCH1, such as LOH and small insertions/deletions [19]. Previous studies reported the formation of the Hh subgroup of medulloblastomas by injecting neuroepithelial stem cells derived from NBCCS-iPSCs into the cerebellum [28, 29]. These findings prompted us to generate iPSCs in which both of the PTCH1 alleles were mutated and test their ability to form tumors.
Although teratomas from parental NBCCS-iPSCs (PTCH1+/− iPSCs), as well as gene-edited PTCH1+/+ iPSCs, contained all components of the three germ layers, those from gene-edited PTCH1−/− iPSCs predominantly differentiated into ectodermal tissues. Moreover, highly differentiated ectodermal cells, such as melanocytes, were rarely observed in PTCH1−/− iPSC-derived teratomas.
As described above, we previously reported that teratomas derived from NBCCS-iPSCs (PTCH1+/−) generated medulloblastoma-like tissues [19]. The major difference between teratomas from NBCCS-iPSCs and gene-edited PTCH1−/− teratomas is that the latter mostly comprise ectodermal tissues and markedly larger areas were occupied by medulloblastoma-like tissues in the latter than in the former.
Hh signaling is reported to be involved in the differentiation of pluripotent stem cells into the neuroectodermal lineage in addition to the differentiation and proliferation of neural stem cells in mice and humans. Mouse and human pluripotent stem cells were previously induced to motor neuron progenitors or midbrain-specific neurons in the presence of Hh proteins [30, 31]. On the other hand, Smo−/− or Ihh−/− mouse ES cells failed to generate embryoid bodies containing neuroectodermal cells and nestin-positive neural stem cells [32]. Our results showing that PTCH1−/− teratomas predominantly consisted of neuroectodermal cells are consistent with these findings. Importantly, the absence of medulloblastoma in teratomas generated from gene-edited PTCH1+/+ iPSCs confirmed the idea that PTCH1 is indeed a driver gene in the formation of medulloblastomas observed in our study.
Apoptosis has a profound effect on tumor formation. Whereas PTCH1 was reported to induce apoptosis in the absence of its ligands, such as SHH, there was no statistical difference of the apoptotic rate between PTCH1+/− and PTCH1−/− teratomas in this study. The extensive expression of SHH protein in PTCH1+/− teratoma and presence of apoptotic cells in PTCH1−/− teratoma also support the idea that apoptosis both in PTCH1+/− and PTCH1−/− teratomas are induced by a PTCH1-independent manner. Since several groups have been reported that proapoptotic protein, BAX, expressed frequently in immature teratomas [33, 34], BAX expression possibly accounts for the induction of apoptosis in PTCH1+/− and PTCH1−/− teratomas.
BCC, the most frequent neoplasia in NBCCS, is also accompanied by secondary mutations in the wild-type PTCH1 gene [12, 35]. However, the pathogenic characteristics of BCC, e.g. peripheral palisading and stromal retraction, were not observed in PTCH1−/− teratomas in the present study. Since the onset of BCC is markedly later than that of medulloblastoma (the mean onset ages of BCC and medulloblastoma are 37.4 and 1.8 years, respectively, in the Japanese population [36]), the in vitro differentiation of iPSCs into keratinocytes, the primary component of the epidermis, before a subcutaneous injection may be required to generate BCC.
Since PTCH1−/− iPSCs are one step closer to tumor formation according to the Knudson’s two-hit hypothesis [37], these gene-edited iPSCs may be a good model for the formation of medulloblastoma in NBCCS and may also be useful for drug screening to identify personalized treatments for this tumor type.
Supplementary Information
Supplementary file1 Table S1. Oligonucleotides used for CRISPR/Cas9 vector construction. (XLS 33 KB)
Supplementary file2 Table S2. Oligonucleotides used for PCR. (XLS 36 KB)
Supplementary file3 Fig. S1. PTCH1-edited NBCCS iPSCs expressed pluripotency markers. (PDF 1341 KB)
Supplementary file4 Fig. S2. Whole-slice images and portions of medulloblastomas in PTCH1+/− and PTCH1−/− teratomas. (PDF 1070 KB)
Supplementary file5 Fig. S3. The expression of SHH in iPSCs. (PDF 783 KB)
Acknowledgements
We thank Hiromi Hatsuse for technical assistance. The present study was supported by JSPS KAKENHI Grants (JP15K19041 and JP18K07205) to K. N.
Author contributions
KN and TMi designed experiments. CK and KN performed experiments and analyzed data. TMo, YI, KF, and AU contributed materials. CK and KN prepared all figures. CK, KN, KF, AU, and TMi discussed the data and wrote the manuscript. All authors read and approved the final manuscript.
Funding
The present study was supported by JSPS KAKENHI Grant Numbers JP15K19041 and JP18K07205 to K. N.
Data availability
The data that support the findings of this study are available from the corresponding author, KN, upon reasonable request.
Code availability
Not applicable.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gorlin RJ. Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore) 1987;66:98–113. doi: 10.1097/00005792-198703000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Gorlin RJ. Goltz RW Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N Engl J Med. 1960;262:908–912. doi: 10.1056/nejm196005052621803. [DOI] [PubMed] [Google Scholar]
- 3.Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 4.Johnson RL, Rothman AL, Xie J, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 5.Rubin LL, de Sauvage FJ. Targeting the Hedgehog pathway in cancer. Nat Rev Drug Discov. 2006;5:1026–1033. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]
- 6.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22:2454–2472. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 7.Fan Z, Li J, Du J, et al. A missense mutation in PTCH2 underlies dominantly inherited NBCCS in a Chinese family. J Med Genet. 2008;45:303–308. doi: 10.1136/jmg.2007.055343. [DOI] [PubMed] [Google Scholar]
- 8.Fujii K, Ohashi H, Suzuki M, et al. Frameshift mutation in the PTCH2 gene can cause nevoid basal cell carcinoma syndrome. Fam Cancer. 2013;12:611–614. doi: 10.1007/s10689-013-9623-1. [DOI] [PubMed] [Google Scholar]
- 9.Kijima C, Miyashita T, Suzuki M, Oka H, Fujii K. Two cases of nevoid basal cell carcinoma syndrome associated with meningioma caused by a PTCH1 or SUFU germline mutation. Fam Cancer. 2012;11:565–570. doi: 10.1007/s10689-012-9548-010. [DOI] [PubMed] [Google Scholar]
- 10.Pastorino L, Ghiorzo P, Nasti S, et al. Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A. 2009;149a:1539–1543. doi: 10.1002/ajmg.a.32944. [DOI] [PubMed] [Google Scholar]
- 11.Hooper JE, Scott MP. Communicating with Hedgehogs. Nat Rev Mol Cell Biol. 2005;6:306–317. doi: 10.1038/nrm1622. [DOI] [PubMed] [Google Scholar]
- 12.Bonilla X, Parmentier L, King B, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet. 2016;48:398–406. doi: 10.1038/ng.3525. [DOI] [PubMed] [Google Scholar]
- 13.Yang ZJ, Ellis T, Markant SL, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14:135–145. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bailey JM, Mohr AM, Hollingsworth MA. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene. 2009;28:3513–3525. doi: 10.1038/onc.2009.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karhadkar SS, Bova GS, Abdallah N, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 16.Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164–1172. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 17.Robinson GW, Orr BA, Wu G, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase ii pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J Clin Oncol. 2015;33:2646–2654. doi: 10.1200/jco.2014.60.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basset-Seguin N, Sharpe HJ, de Sauvage FJ. Efficacy of Hedgehog pathway inhibitors in Basal cell carcinoma. Mol Cancer Ther. 2015;14:633–641. doi: 10.1158/1535-7163.Mct-14-0703. [DOI] [PubMed] [Google Scholar]
- 19.Ikemoto Y, Miyashita T, Nasu M, et al. Gorlin syndrome-induced pluripotent stem cells form medulloblastoma with loss of heterozygosity in PTCH1. Aging (Albany NY) 2020;12:9935–9947. doi: 10.18632/aging.103258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagao K, Fujii K, Saito K, et al. Entire PTCH1 deletion is a common event in point mutation-negative cases with nevoid basal cell carcinoma syndrome in Japan. Clin Genet. 2011;79:196–198. doi: 10.1111/j.1399-0004.2010.01527.x. [DOI] [PubMed] [Google Scholar]
- 21.Ikemoto Y, Takayama Y, Fujii K, et al. Somatic mosaicism containing double mutations in PTCH1 revealed by generation of induced pluripotent stem cells from nevoid basal cell carcinoma syndrome. J Med Genet. 2017;54:579–584. doi: 10.1136/jmedgenet-2016-104490. [DOI] [PubMed] [Google Scholar]
- 22.Ikehara H, Fujii K, Miyashita T, et al. Establishment of a Gorlin syndrome model from induced neural progenitor cells exhibiting constitutive GLI1 expression and high sensitivity to inhibition by smoothened (SMO) Lab Invest. 2020;100:657–664. doi: 10.1038/s41374-019-0346-2. [DOI] [PubMed] [Google Scholar]
- 23.Nagao K, Iwai Y. Miyashita T RCAN1 is an important mediator of glucocorticoid-induced apoptosis in human leukemic cells. PLoS ONE. 2012;7:e49926. doi: 10.1371/journal.pone.0049926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirobe T. How are proliferation and differentiation of melanocytes regulated? Pigment Cell Melanoma Res. 2011;24:462–478. doi: 10.1111/j.1755-148X.2011.00845.x. [DOI] [PubMed] [Google Scholar]
- 27.Thibert C, Teillet MA, Lapointe F, Mazelin L, Le Douarin NM. Mehlen P Inhibition of neuroepithelial patched-induced apoptosis by sonic hedgehog. Science. 2003;301:843–846. doi: 10.1126/science.1085405. [DOI] [PubMed] [Google Scholar]
- 28.Huang M, Tailor J, Zhen Q, et al. Engineering genetic predisposition in human neuroepithelial stem cells recapitulates medulloblastoma tumorigenesis. Cell Stem Cell. 2019;25:433–46.e7. doi: 10.1016/j.stem.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Susanto E, Marin Navarro A, Zhou L, et al. Modeling SHH-driven medulloblastoma with patient iPS cell-derived neural stem cells. Proc Natl Acad Sci U S A. 2020;117:20127–20138. doi: 10.1073/pnas.1920521117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu BY, Zhang SC. Differentiation of spinal motor neurons from pluripotent human stem cells. Nat Protoc. 2009;4:1295–1304. doi: 10.1038/nprot.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaeger I, Arber C, Risner-Janiczek JR, et al. Temporally controlled modulation of FGF/ERK signaling directs midbrain dopaminergic neural progenitor fate in mouse and human pluripotent stem cells. Development. 2011;138:4363–4374. doi: 10.1242/dev.066746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maye P, Becker S, Siemen H, et al. Hedgehog signaling is required for the differentiation of ES cells into neurectoderm. Dev Biol. 2004;265:276–290. doi: 10.1016/j.ydbio.2003.09.027. [DOI] [PubMed] [Google Scholar]
- 33.Addeo R, Crisci S, D'Angelo V, et al. Bax mutation and overexpression inversely correlate with immature phenotype and prognosis of childhood germ cell tumors. Oncol Rep. 2007;17:1155–1161. [PubMed] [Google Scholar]
- 34.Hiroshima K, Toyozaki T, Iyoda A, Yusa T, Fujisawa T, Ohwada H. Apoptosis and proliferative activity in mature and immature teratomas of the mediastinum. Cancer. 2001;92:1798–1806. doi: 10.1002/1097-0142(20011001)92:7<1798::AID-CNCR1696>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 35.Unden AB, Holmberg E, Lundh-Rozell B, et al. Mutations in the human homologue of Drosophila patched (PTCH) in basal cell carcinomas and the Gorlin syndrome: different in vivo mechanisms of PTCH inactivation. Cancer Res. 1996;56:4562–4565. [PubMed] [Google Scholar]
- 36.Endo M, Fujii K, Sugita K, Saito K, Kohno Y, Miyashita T. Nationwide survey of nevoid basal cell carcinoma syndrome in Japan revealing the low frequency of basal cell carcinoma. Am J Med Genet A. 2012;158a:351–357. doi: 10.1002/ajmg.a.34421. [DOI] [PubMed] [Google Scholar]
- 37.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file1 Table S1. Oligonucleotides used for CRISPR/Cas9 vector construction. (XLS 33 KB)
Supplementary file2 Table S2. Oligonucleotides used for PCR. (XLS 36 KB)
Supplementary file3 Fig. S1. PTCH1-edited NBCCS iPSCs expressed pluripotency markers. (PDF 1341 KB)
Supplementary file4 Fig. S2. Whole-slice images and portions of medulloblastomas in PTCH1+/− and PTCH1−/− teratomas. (PDF 1070 KB)
Supplementary file5 Fig. S3. The expression of SHH in iPSCs. (PDF 783 KB)
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, KN, upon reasonable request.
Not applicable.