

Abstract
Correlations between the biomass of phytoplankton and the biomass of bacteria and between the biomass of bacteria and the biomass of protozoans suggest that there is coupling between these compartments of the “microbial loop.” To investigate this coupling on the species level, bacteria and protozoans from untreated lake water inocula were allowed to grow on detritus of the green alga Ankistrodesmus falcatus or the cyanobacterium Oscillatoria limnetica in continuous-flow systems for 1 month. Denaturing gradient gel electrophoresis (DGGE) of the 16S and 18S rRNA genes was used to monitor the development of the bacterial community structure and the eukaryotic community structure, respectively. Nonmetric multidimensional scaling of the DGGE profiles revealed the changes in the microbial community structure. This analysis showed that significantly different bacterial communities developed on the green algal detritus and on the cyanobacterial detritus. Although similar results were obtained for the eukaryotic communities, the differences were not significant. Hence, our findings indicate that the origin of detritus can affect the structure of at least the bacterial community. A phylogenetic analysis of 20 18S ribosomal DNA clones that were isolated from the continuous cultures revealed that many sequences were related to the sequences of bacterivorous protozoans (members of the Ciliophora, Rhizopoda, Amoeba, and Kinetoplastida). One clone grouped in a recently established clade whose previously described members are all parasites. The affiliations of about 20% of the clones could not be determined.
In aquatic environments, photosynthetically produced organic carbon is decomposed by heterotrophic bacteria, which in turn are consumed by heterotrophic protozoans. Correlations between phytoplankton biomass and heterotrophic bacterial biomass (10, 12, 41) and between heterotrophic bacterial biomass and protozoan biomass (8, 9, 11, 28) suggest that there is coupling between the compartments of the so-called microbial loop (4). However, most descriptions of such relationships have been based on bulk measurements of the compartments, and whether there are correlations at the species level is an intriguing question. For example, is organic carbon derived from different phytoplankton species decomposed by different species of specialized bacteria? Or is protozoan species composition governed by bacterial species composition or just by the concentration of edible food particles? Although numerous studies have shown that bacteria have different rates of growth on natural carbon sources (6, 7, 16, 21, 32, 33) and that heterotrophic protozoans selectively graze on larger bacteria (15, 29, 31), little is known about whether the development of aquatic microbial communities depends on various organic carbon sources. This lack of information is attributable to the difficulty of determining the presence of microbial species due to the fact that the majority of these microorganisms cannot be grown with the current cultivation methods. Molecular techniques can be used to assess genetic composition without culturing all members of a community. To investigate the potential dependence of microbial community structure on the source of detritus in lakes, we used denaturing gradient gel electrophoresis (DGGE). We determined the bacterial community structure and the eukaryotic community structure (22, 35) for communities that developed on detritus derived from the green alga Ankistrodesmus falcatus or on detritus derived from the cyanobacterium Oscillatoria limnetica in specially designed two-stage continuous-flow systems (34). Since DGGE does not provide quantitative results (38), we converted the band patterns to reflect whether particular sequence types were present or absent. Our qualitative measures of microbial community structure were analyzed by nonmetric multidimensional scaling (NMDS) and a statistical method used for comparisons of the DGGE patterns. Special attention was paid to the eukaryotic microbial community. Since most heterotrophic protozoans are bacterivorous, we wanted to determine whether the carbon source used by the bacteria also influenced the community structure of the protozoans.
MATERIALS AND METHODS
Experimental setup.
Nonaxenic A. falcatus (Centre for Limnology strain E01) and Oscillatoria cf. limnetica (Centre for Limnology strain MR1) were grown under light-limiting conditions in modified Guillard medium (34) in the first stages (volume, 2 liters) of two continuous-flow systems (designated the Alga system and the Cyano system) until the steady state was reached (dilution rate, 0.36 day−1). The two-stage continuous-flow system has been described previously (34). The cultures were grown at a constant temperature (20°C) at pH 8.0 ± 0.2 with illumination by type TLE 32W/33 circular fluorescent lamps (Philips). The inner sides of the lamps facing the culture vessels were shielded with aluminum foil to decrease the light intensity. The light intensity for the Ankistrodesmus cultures (Alga system) was manipulated so that the chlorophyll a (Chla) concentrations in the two systems were approximately the same. Samples (10 ml per assay) were removed every 24 h and used to determine the concentrations of Chla and total suspended solids (TSS).
After 30 days the cultures were considered to be steady-state cultures based on the TSS and Chla concentrations. The two second stages (volume, 1 liter) (the replicates were designated Alga 1, Alga 2, Cyano 1, and Cyano 2) of each flow system were then filled with water from Lake Ketelmeer (The Netherlands) and connected to the first stages. Lake Ketelmeer is a small turbid lake (surface area, 35 km−2; mean depth, 3 m) with a short water retention time (∼1 week). This lake water was used as an inoculum for bacteria and heterotrophic eukaryotes and was chosen because both cyanobacteria and green algae are present in Lake Ketelmeer (36). If the origin of detritus governs microbial community structure, the lake water inoculum should have contained microorganisms that were specialized for decomposing these carbon sources. The second stages received UV-C-killed phytoplankton from the first stages at a dilution rate of 0.30 day−1. The UV-C intensities needed to kill O. limnetica and A. falcatus were 5 and 18 W · m−2, respectively. The procedures used to assess lethal UV-C intensities have been described previously (34). For 1 month, samples were removed and used for Chla, TSS, and community structure analyses (DGGE and 18S rRNA sequence determination).
Chla and TSS.
The Chla and TSS assays used have been described previously (34). Duplicate measurements were obtained with each assay.
DNA extraction, PCR, and DGGE.
DNA was released from cells by mechanical force (bead beating), phenol extraction, and ethanol precipitation (42). PCR primers for the V2 region were used for amplification of the 16S rRNA gene. The PCR primers used were F357GC (5′-CGCCCGCCGCGCCCCGCGCCCGGCCCGCCGCCCCCGCCCCCCTACGGGAGGCAGCAG-3′), which contains a GC-rich clamp and is specific for most members of the Bacteria, and R518 (5′-ATTACCGCGGCTGCTGG-3′), which is specific for most members of the Bacteria, Archaea, and Eucarya (22). Each PCR amplification was performed in a 50-μl reaction mixture containing approximately 100 ng of template DNA, 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 0.01% (wt/vol) gelatin, 1.5 mM MgCl2, each primer at a concentration of 0.5 μM, each deoxynucleotide at a concentration of 200 μM, 400 ng of bovine serum albumin, and 2.5 U of Taq DNA polymerase (Boehringer, Mannheim, Germany). PCR cycling was performed with a Perkin-Elmer model 480 thermocycler. The temperature cycling conditions were as follows: preincubation at 94°C for 5 min, followed by 25 cycles consisting of 94°C for 1 min, the annealing temperature (TA) for 1 min, and 72°C for 1 min. In the first 20 cycles the TA was decreased by 1°C stepwise each two cycles, from 65°C in the first cycle to 56°C in the 20th cycle. In the last five cycles the TA was 55°C. The 25 cycles were followed by 5 min of incubation at 72°C. The primers used for amplification of the 18S rRNA gene were F1427GC (5′-CGCCCGCCGCGCCCCGCGCCCGGCCCGCCGCCCCCGCCCCTCTGTGATGCCCTTAGATGTTCTGGG-3′) and R1616 (5′-GCGGTGTGTACAAAGGGCAGGG-3′). Both of these primers are specific for eukaryotic aquatic microorganisms (35). The temperature cycling conditions were as follows: preincubation at 94°C for 5 min, followed by 25 cycles consisting of 94°C for 1 min, 52°C for 1 min, and 72°C for 1 min and then a final extension step consisting of 72°C for 5 min. The reaction conditions were the same as described above. DGGE was performed as described previously (35, 42). Briefly, PCR products of similar sizes were separated on a 1.5-mm-thick vertical gel containing 8% (wt/vol) polyacrylamide (acrylamide/bisacrylamide ratio, 37.5:1) and a linear gradient consisting of the denaturants urea and formamide; the concentration of the denaturants increased from 35% at the top of the gel to 55% at the bottom for separation of the 16S ribosomal DNA (rDNA) fragment and from 30 to 55% for separation of the 18S rDNA fragment (100% denaturant was defined as 7 M urea and 40% [vol/vol] formamide). Equal amounts of PCR products were applied to the DGGE gel. The concentrations of PCR products were estimated by separating the products on 2.0% agarose gels, staining them with ethidium bromide (see below), and analyzing digitized images with ImageQuant software (Molecular Dynamics Ltd., Kemsing, England). A 50-μl portion of the sample containing the smallest amount of PCR product was loaded onto the DGGE gel; all other samples were loaded in amounts relative to this sample. Electrophoresis was performed at 60°C with a buffer containing 40 mM Tris, 40 mM acetic acid, and 1 mM EDTA (pH 7.6) (0.5× TAE buffer), and 75 V was applied to the submerged gel for 16 h. Nucleic acids were visualized by staining the gel for 1 h in 0.5× TAE buffer containing 0.5 mg of ethidium bromide per liter and then destaining it for 5 min in demineralized water and photographing it with a charge-coupled device camera (The Imager; Appligene, Illkirch, France). Digitized images were inverted by using the Photostyler software (Aldus Corporation, Seattle, Wash.). The contrast and gray balance of the entire image were adjusted to reduce the background.
Clone library construction.
To estimate the eukaryotic species present in the continuous-flow systems, the DNA extracted from three samples (Alga 1 stage on day 23, Alga 2 stage on day 23, and Cyano 1 stage on day 29) were mixed and used for clone library construction. 18S rDNA clones that were nearly full length were generated by using the Eucarya-specific primers E4 (5′-CTGGTTGATTCTGCCAGT-3′) and E1628 (5′-CGACGGGCGGTGTGTA-3′) (the numbers in the designations indicate Saccharomyces cerevisiae sequence positions). Each PCR amplification was performed in a 50-μl reaction mixture containing approximately 100 ng of template DNA, 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 0.01% (wt/vol) gelatin, 1.5 mM MgCl2, each primer at a concentration of 0.5 μM, each deoxynucleotide at a concentration of 200 μM, 400 ng of bovine serum albumin, and 2.5 U of Taq DNA polymerase (Boehringer). PCR cycling was performed with the Perkin-Elmer model 480 thermocycler programmed as follows: denaturation at 94°C for 5 min, followed by 25 cycles consisting of 94°C for 1 min, 54°C for 1 min, and 72°C for 1 min and a final extension step consisting of 68°C for 10 min. The cloning and sequencing procedures used have been described previously (42). A total of 120 clones were examined by DGGE. Only clones whose DGGE band positions were different were sequenced. The sequences were determined in two directions by using the following Texas red-labeled primers: M13 forward (5′-TGTAAAACGACGGCCAGT-3′), M13 reverse (5′-GAAACAGCTATGACCATG-3′), E382 forward (5′-CGGAGAGGGAGCCTGAG-3′), E565 reverse (5′-ATTACCGCGGCTGCTGG-3′), E1128 forward (5′-AAACTTAAAGGAATTGACG-3′), and E1179 reverse (5′-CCCGTGTTGAGTCAAATT-3′), (the numbers in the designations indicate S. cerevisiae sequence positions).
Phylogenetic analysis.
The 18S rDNA sequences obtained were compared with sequences obtained from GenBank/EMBL by using BLAST (1). The sequences with the highest levels of similarity were used as reference sequences for alignment. The alignment was based on secondary structure and was constructed by using the Dedicated Comparative Sequence Editor (13). Phylogenetic trees were constructed by using only unambiguously aligned sequence positions and maximum-likelihood analysis (PAUP*, test version 4.0d59; David L. Swofford, Laboratory of Molecular Systematics, Smithsonian Institution, Washington, D.C.). Each analysis was performed five times. Nucleotide frequencies and transition-to-transversion ratios were estimated from the data. Nucleotide substitution rates were assumed to follow a gamma distribution with a shape parameter of 0.5 and setting according to the HKY model (18). “Tree-bisection-reconnection” was used as a swapping algorithm. Maximum-parsimony bootstrap analysis (1,000 replicates) was used to assess the robustness of the trees.
Data analysis.
The DGGE patterns were converted to a binary (01) matrix (35). A Nei-Li distance matrix was calculated from the binary data (23). This distance matrix was analyzed by using the NMDS package from the Statistica software package (StatSoft, Inc., Tulsa, Okla.). This procedure presented the data in a Euclidean plane such that very similar values were plotted close together. The resulting graphical representation (NMDS map) was much easier to interpret than the original table of distances was. When it was applied to DGGE data, the NMDS map showed every band pattern (a reflection of the community structure at a particular point in time) as one point, and relative changes in community structure could be visualized and interpreted by connecting consecutive points (37, 38). The statistical significance of the differences in community structure due to the source of detritus was determined by using the Nei-Li distances and comparing the communities only at the same moment in time. Two communities fed with different types of detritus were considered to be different at a given moment in time if their Nei-Li distance was significantly greater than the Nei-Li distances between replicate communities. For each of the two sources of detritus we used two replicates and made six observations at different times. On the first sampling day (day 3) the distances between replicates were exceptionally small compared to the distances on later days, indicating that there was a lack of independence from the shared inoculum. Therefore, these distances were not included in the calculations. Thus, a total of 10 distances between replicates were used for the bacterial and eukaryotic community to calculate an average distance between replicates (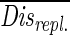 ). Each distance between communities growing on different sources of detritus was tested against this distribution and was considered significant if Disalga-cyano,t >
). Each distance between communities growing on different sources of detritus was tested against this distribution and was considered significant if Disalga-cyano,t > 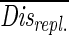 + (T · Stddis-repl.), where Disalga-cyano,t is the distance between two communities fed with algal or cyanobacterial detritus at time t, T is the Student t value (n = 9; P = 0.05), and Stddis-repl. is the standard deviation of
+ (T · Stddis-repl.), where Disalga-cyano,t is the distance between two communities fed with algal or cyanobacterial detritus at time t, T is the Student t value (n = 9; P = 0.05), and Stddis-repl. is the standard deviation of 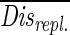 .
.
Nucleotide sequence accession numbers.
The eukaryotic clone sequences have been deposited in the EMBL database under accession no. AJ130849 to AJ130869.
RESULTS
Decomposition of detritus.
The Chla and TSS concentrations in both first stages (Alga system and Cyano system) are shown in Fig. 1. Although the first stages of both flow systems were at a steady state at the beginning of the experiment, in the Alga system the Chla concentration increased after 3 days, while the TSS concentration was constant for the first 15 days. In the Cyano system the Chla level was constant throughout the experiment, while the TSS concentration decreased during the first 15 days. As a result, the two systems had different Chla concentrations after day 15, but their TSS concentrations were about the same. On a dry weight basis, the second stages of the two flow systems received approximately the same amounts of UV-killed biomass after 2 weeks.
FIG. 1.

Concentrations of Chla and TSS in the two continuous-flow systems (Alga and Cyano systems). The asterisks indicate increases in the Chla and TSS concentrations that were caused by a pump defect. l, liter; d, day.
The patterns of decomposition in all of the second stages were similar. Theoretically, if the decomposition rate is zero, the concentrations of Chla and TSS in the second stages should increase until the levels in the first stage are reached. As soon as the Chla concentration started to decrease, the second stages became yellowish instead of green. In the Alga system, the decomposition rate exceeded the inflow of UV-killed cells after approximately 10 days, while in the Cyano system this situation occurred after 5 days. The sharp increases in the Chla and TSS concentrations in the Alga 2 stage were caused by a defective pump. The increases in the Chla and TSS concentrations in the Cyano 2 stage were apparently due to changes in the decomposition rate, and the green color reappeared in this stage.
Microbial community structure.
In the 16S rDNA DGGE analysis (Fig. 2A) 54 different bands were detected, while 58 different bands were detected in the 18S rDNA DGGE analysis (Fig. 2B). The relative changes in the structures of the bacterial and eukaryotic communities, as visualized by NMDS, are shown in Fig. 2C and D. Three days after the experiment started, the bacterial communities (Fig. 2C) growing on different types of detritus were still quite similar. A similar observation was made for the eukaryotic communities (Fig. 2D). A clear divergence between the bacterial communities grown on different types of detritus occurred on day 7. This trend persisted, and on day 17 the differences became significantly greater than the differences between the replicates (P < 0.05). Although the structures of the eukaryotic communities also changed, the trend was not as clear as the trend in the bacterial communities, and the differences between the replicates were as great as the differences between the treatments (green algal detritus versus cyanobacterial detritus).
FIG. 2.

Structures of the microbial communities depending on the source of detritus (green algae or cyanobacteria). (A) 16S rDNA-defined (bacterial) community, as revealed by DGGE. (B) 18S rDNA-defined (eukaryotic) community, as revealed by DGGE. (C) NMDS map of the bacterial communities grown on different types of detritus. (D) NMDS map of the eukaryotic communities grown on different types of detritus. Symbols: ○, Alga 1 stage; □, Alga 2 stage; ●, Cyano 1 stage; ■, Cyano 2 stage. The lines connect the community structures of replicate stages. The numbers indicate the numbers of days from the beginning of the experiment. Boldface numbers indicate significant differences (P < 0.05) in community structures between different sources of detritus. The white bands in panel B, lane c, indicate the band positions for the 18S rDNA clones (Table 1). For each of block of lanes, data for the six times when samples were obtained are shown in consecutive lanes. The arrow in panel A indicates the band belonging to O. limnetica; this band was not included in the Nei-Li distance calculation.
Eukaryotic phylogeny.
Of the 120 clones analyzed, 20 were found to have different DGGE bands. The sequences of these 20 clones were determined and phylogenetically analyzed by maximum-likelihood and parsimony methods (Fig. 3), and the frequency with which a band was found at the same position as the clones in the different stages of the two systems is shown in Table 1. Since 58 different band positions were identified on the 18S rDNA DGGE gels, our clone library accounted for at most one-third of the total eukaryotic diversity in the two systems. Of the 20 clones, 5 belonged to the Ciliophora (Fig. 3A), 3 belonged to the Rhizopoda (Fig. 3C), 3 belonged to the Metazoa (Fig. 3D), 1 belonged to the Drips (Fig. 3D), 1 belonged to the Amoeba (Fig. 3B), 1 belonged to the fungi (Fig. 3B), and 1 belonged to the Kinetoplastida (Fig. 3E), and the affiliations of 4 clones were not determined. Three of the sequences formed a unique terminal cluster with the fungi as the closest relatives (Fig. 3B).
FIG. 3.

Maximum-likelihood trees showing the phylogenetic positions of the 18S rDNA clones. The trees are rooted. The numbers indicate the levels of bootstrap support (percentages of 1,000 replicates) obtained in a parsimony analysis. (A) Tree constructed by using S. cerevisiae sequence positions 22 to 641 and 744 to 1643. (B) Tree constructed by using S. cerevisiae sequence positions 50 to 641 and 744 to 1643. (C) Tree constructed by using S. cerevisiae sequence positions 25 to 641 and 744 to 1643. (D) Tree constructed by using S. cerevisiae sequence positions 50 to 641 and 744 to 1643. (E) Tree constructed by using Leishmania donovani sequence positions 25 to 993, 1102 to 1300, and 1399 to 2067. Within the order Kinetoplastida, Bodo caudatus can be used as an outgroup (14). The EMBL accession numbers for the sequences are as follows: Gymnodinium beii, U37365; Alexandrium tamarense, X54946; Sarcocystis muris, M64244; Theileria parva, L02366; Glaucoma chattoni, X56533; Tetrahymena nanneyi, X56169; Opisthonecta henneguyi, X56531; Colpoda inflata, M97908; Blepharisma americanum, M97909; Hartmanella vermiformis, M95168; Acanthamoeba castellanii, M13435; Balamuthia mandrillaris, AF019071; Chytridium confervae, M59758; Saccharomyces cerevisiae, Z75578; Tilletia caries, U00972; Chlorarachnion reptans, X70809; Chlorarachnion sp. 1, U02075; Thaumatomonas sp., U42446; Paulinella chromatophora, X81811; Euglypha rotunda, X77692; Cercomonas 3, U42449; Cercomonas, U42450; Cercomonas 2, U42451; Heteromita globosa, U42447; Ichthyophonus hoferi, U25637; Psorospermium haeckelii, U33180; “Prototheca richardsi,” AF07445; Unknown L29455, L29455; Dermocystidium salmonis, U21337; Dermocystidium sp., U21336; Chaetonotus sp., AJ001735; Artemia salina, X01723; Brachionus plicatilis, U49911; Bodo caudatus, X53910; Trypanoplasma borreli, L14840; Blastocrithidia culicis, L29265; Phytomonas sp., L35076; Leptomonas sp., X53914; Crithidia fasciculata, X03450; Endotrypanum monterogeii, X53911; Leishmania donovani, X07773; Trypanosoma brucei, M12676; and Trypanosoma cruzi, X53917.
TABLE 1.
Identities and frequencies of occurrence (on six sampling dates) of the 18S rDNA clones in the second stages of our systems
| Clone(s)a | Taxon | Panel in Fig. 3 | Frequency (no. of dates)
|
|||
|---|---|---|---|---|---|---|
| Alga 1 | Alga 2 | Cyano 1 | Cyano 2 | |||
| LKM67 | Ciliophora | A | 5 | 4 | 4 | 1 |
| LKM48 | Rhizopoda | C | 1 | 0 | 0 | 0 |
| LKM62, LKM63b | Ciliophora | A | 1 | 0 | 0 | 2 |
| LKM43 | Ciliophora | A | 5 | 4 | 5 | 6 |
| LKM8 | Ciliophora | A | 3 | 3 | 4 | 4 |
| LKM45 | Rhizopoda | C | 2 | 4 | 4 | 6 |
| LKM88 | Metazoa | D | 3 | 0 | 3 | 4 |
| LKM74 | Amoebae | B | 3 | 4 | 1 | 1 |
| LKM30 | Rhizopoda | C | 2 | 4 | 0 | 3 |
| LKM36 | Ciliophora | A | 4 | 2 | 0 | 0 |
| LKM15 | LKM11c | B | 2 | 2 | 1 | 1 |
| LKM51 | DRIPsd | D | 0 | 3 | 1 | 3 |
| LKM33 | Fungi | D | 3 | 0 | 1 | 3 |
| LKM11 | LKM11c | B | 2 | 3 | 2 | 0 |
| LKM46 | LKM11c | B | 3 | 0 | 1 | 0 |
| LKM80 | Metazoa | D | 1 | 0 | 0 | 1 |
| LKM85 | Metazoae | 1 | 2 | 3 | 4 | |
| LKM101 | Kinetoplastida | E | 1 | 3 | 2 | 3 |
| LKM118 | Unknown | 0 | 1 | 0 | 0 | |
The clones are arranged in order of their melting positions on the DGGE gel, beginning at the top of the gel (Fig. 2).
Clones LKM62 and LKM63 produced the same DGGE band in a 30 to 55% denaturing gradient.
LKM11, LKM15, and LKM46 form a unique terminal clade whose affiliation is not known; this clade was provisionally named after clone LKM11.
The DRIPs phylogenetic clade currently comprises the following taxa: Dermocystidium salmonis, Dermocystidium sp., rosette agent (unknown L29455 sequence), Ichtypphonus hoferi, Psorospermium haeckelii (26), and a sequence from a protozoan formerly classified as the unpigmented unicellular alga “P. richardsi” (5).
The sequence of clone LKM85 was incomplete (S. cerevisiae sequence positions 4 to 558 and 630 to 1643) and therefore was not included in the phylogenetic analysis shown in Fig. 3D. The overall level of similarity of LKM85 to Brachionus plicatilis was 99.4%.
Most bands that were produced by the clones sequenced were not restricted to one of the two systems. Clone LKM36, a clone whose sequence was related to Ciliophora sequences, was the only clone that was frequently found only in the Alga system. Clones LKM48 and LKM118 were also found only in the Alga system. However, these clones were detected only once (Table 1).
DISCUSSION
We performed an experiment in which we investigated whether the origin of detritus can be a factor that governs microbial community structure. Although we found differences between replicates of the bacterial community growing on the same type of detritus, the effect of the carbon source (green algal detritus versus cyanobacterial detritus) was significantly greater (Fig. 2). In contrast to the bacterial community, the eukaryotic community developed rather chaotically; i.e., the differences between detritus sources were obscured by differences between replicates. This might be interpreted as due to poor reproducibility of the microcosm experiments and to the fact that development of microbial communities at the physical scale which we used is not determined solely by culture conditions, such as light, temperature, and nutrients. However, it is too soon for general conclusions to be made.
The abundance of a sequence was not included in our measurements of community structure since we used only the presence or absence of a DGGE band. Therefore, the changes in the community structure which we measured reflected only changes in species composition, not changes in species abundance. In addition, changes in the community structure, as detected by NMDS analysis, were influenced by the total number of bands in a DGGE pattern and by the number of bands shared by DGGE patterns. Differences in community structure could, therefore, have originated from differences in species diversity (the number of different DGGE bands obtained for two communities) or from differences in species richness (the total number of DGGE bands per community). In general, the bacterial richness of the Alga systems was somewhat greater than the bacterial richness of the Cyano systems. However, the differences in community structure that depended on the source of detritus were largely due to differences in bacterial diversity. The eukaryotic richness fluctuated greatly between sampling days and in all second stages. Thus, differences in the eukaryotic community structure between duplicates and between sources of detritus originated from variations in both eukaryotic richness and diversity.
Our results show that the origin of detritus can affect the structure of the microbial community, at least the bacterial community in our systems. Variations in the chemical composition of phytoplankton (25, 27, 30) and in bacterial substrate utilization may explain the effects on the bacterial community. Although some authors have described chemosensory feeding of bacterivorous protozoans (40), there is very strong evidence that size-selective feeding by protozoans occurs (15, 29, 31). If protozoan prey selection is based solely on size, one might not expect that detritus would have a strong effect on eukaryotic community structure unless the size spectrum of the bacterial community also changes due to the origin of the detritus.
Although we observed no significant differences between the eukaryotic communities growing on the different types of detritus, it was interesting to gain insight into the phylogenetic positions of the protozoan species developing in the flow systems, especially since eukaryotic microorganisms are largely neglected in molecular ecology. Since we constructed our clone library from three samples obtained on two sampling dates, this library does not represent the complete eukaryotic community. We obtained 20 different clones, while eukaryotic DGGE produced 58 different bands. Thus, our library accounts for one-third of the DGGE-detected eukaryotic diversity. Surprisingly, the clone library did not include A. falcatus. Apparently, the high UV-C intensity (18 W · m−2) needed to kill this alga destroyed its DNA. The phylogenetic position of the 18S rDNA clones was inferred by maximum-likelihood analysis (Fig. 3). Replicate analyses (n = 5) always resulted in the same phylogenetic tree. Since the second stages were kept in the dark, there was no algal and or cyanobacterial growth, and bacterivory or uptake of detritus or dissolved organic matter was the only possible mode of feeding. Not surprisingly, most of the sequences found were related to sequences of bacterivorous eukaryotes (members of the Ciliophora, Amoeba Rhizopoda, and Kinetoplastida). Microscopic observations confirmed that these bacterivorous eukaryotes were present. However, we did not identify species. Two sequences (LKM80 and LKM85) were closely related to the sequence of Brachionus plicatilis, a rotifer species capable of feeding on all members of the microbial loop (3). One sequence (LKM88) was closely related to the sequence of a Chaetonotus sp. (Gastrotricha). Since we did not prefilter the lake water inoculum, the metazoan grazers were not eliminated and could develop in the second stages of the flow systems. Three sequences (LKM11, LKM15, and LKM46) were not related to any of the eukaryotic sequences in the EMBL database and formed a unique terminal clade supported by a moderately strong parsimony bootstrap value. The closest relatives of these sequences were the fungi (Fig. 3B). We provisionally designated this cluster LKM11 after the first clone. Another sequence, LKM118, was not related to any previously described eukaryotic sequence. Phylogenetic analysis placed the LKM118 sequence just below the “crown” of the eukaryotic tree, although this position was very unstable (data not shown).
One sequence (LKM51) is particularly interesting. This sequence grouped in a recently established phylogenetic clade, the DRIPs clade, which was provisionally named after its first members (26). All of the sequences currently in this cluster are from protistan parasites of fish, crustaceans, and amphibians, and LKM51 exhibited a very high level of similarity (97.6%) with the “Prototheca richardsi” sequence. The latter sequence is from an agent that inhibits growth of amphibian larvae and was previously classified as a unicellular unpigmented alga (5). “P. richardsi” is the only DRIPs species whose free-living stage has been observed. None of the other DRIPs members have been cultured in vivo, and therefore the complete life histories of these parasites remain unclear. Apparently, our culture method supported growth of a free-living DRIPs species and may be used for further investigations of the life histories of these protistan parasites. Since we did not identify the eukaryotic microorganisms by microscopy, we have no information concerning the morphology of the species represented by sequence LKM51.
The resistance of most microbial species to cultivation has hampered the study of microbial community structure for many years. Recently introduced molecular techniques circumvent the problems and have been used increasingly to solve problems in microbial ecology. rDNA sequence information is now starting to reveal patterns and governing forces in natural microbial community structure (24). We used DGGE of the 16S and 18S rRNA genes to show that the origin of detritus can be a governing force in microbial community structure. Although many workers have used detritus as a natural carbon source to investigate bacterial production (17, 19, 39), to our knowledge we are the first researchers to describe detritus-dependent development of the microbial loop. Even though our study should be considered a survey and was carried out in small-scaled continuous-flow systems, the occurrence and subsequent decline of single-species phytoplankton blooms (2, 20) may result in organic carbon releases comparable to carbon releases in our flow system. The effects described above can be expected to take place in natural systems.
ACKNOWLEDGMENTS
We thank P. Schouten, H. Uittenhout, and G. M. van Hannen for construction of continuous culture boxes and UV-C devices and T. Beebe and M. Ragan for sharing the “P. richardsi” sequence before publication.
Footnotes
Publication no. 2525 of the Centre for Limnology, Netherlands Institute of Ecology, Maarssen, The Netherlands.
REFERENCES
- 1.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 2.Amon R M W, Benner R. Rapid cycling of high-molecular weight dissolved organic matter in the ocean. Nature. 1994;369:549–552. [Google Scholar]
- 3.Arndt H. Rotifers as predators on components of the microbial web (bacteria, heterotrophic flagellates, ciliates)—a review. Hydrobiologia. 1993;255/256:231–246. [Google Scholar]
- 4.Azam F, Fenchel T, Field J G, Gray J S, Meyer-Reil L-A, Thingstad F. The ecological role of water column microbes in the sea. Mar Ecol Prog Ser. 1983;10:257–263. [Google Scholar]
- 5.Beebee, T. Personal communication.
- 6.Bell W H. Bacterial utilization of algal extracellular products. 1. The kinetic approach. Limnol Oceanogr. 1980;25:1007–1020. [Google Scholar]
- 7.Bell W H, Sakshaug E. Bacterial utilization of algal extracellular products. 2. A kinetic study of natural populations. Limnol Oceanogr. 1980;25:1021–1033. [Google Scholar]
- 8.Berninger U-G, Finlay B J, Kuuppo-Leinikki P. Protozoan control of bacterial abundances in freshwater. Limnol Oceanogr. 1991;36:139–147. [Google Scholar]
- 9.Berninger U-G, Wickham S A, Finlay B J. Trophic coupling within the microbial food web: a study with fine temporal resolution in a eutrophic freshwater ecosystem. Freshwater Biol. 1993;30:419–432. [Google Scholar]
- 10.Bird D F, Kallf J. Empirical relationships between bacterial abundance and chlorophyll concentrations in fresh and marine waters. Can J Fish Aquat Sci. 1984;41:1015–1023. [Google Scholar]
- 11.Bjørnsen P K, Riemann B, Horsted S J, Nielsen T G, Pock-Sten J. Trophic interactions between heterotrophic nanoflagellates and bacterioplankton in manipulated seawater enclosures. Limnol Oceanogr. 1988;33:409–420. [Google Scholar]
- 12.Cole J, Findlay S, Pace M. Bacterial production in fresh and salt water ecosystems: a cross system overview. Mar Ecol Prog Ser. 1988;43:1–10. [Google Scholar]
- 13.De Rijk P, De Wachter R. DCSE, an interactive tool for sequence alignment and secondary structure research. Comput Appl Biosci. 1993;9:735–740. doi: 10.1093/bioinformatics/9.6.735. [DOI] [PubMed] [Google Scholar]
- 14.Fernandes A P, Nelson K, Beverley S M. Evolution of nuclear ribosomal RNA in kinetoplastid protozoa: perspectives on the age and origin of parasitism. Proc Natl Acad Sci USA. 1993;90:11608–11612. doi: 10.1073/pnas.90.24.11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.González J M, Iriberrri J, Egea L, Barcina I. Differential rates of digestion of bacteria by freshwater and marine phagotrophic protozoa. Appl Environ Microbiol. 1990;56:1851–1857. doi: 10.1128/aem.56.6.1851-1857.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gunnison D, Alexander M. Resistance and susceptibility of alga to decomposition by natural microbial communities. Limnol Oceanogr. 1974;20:64–70. [Google Scholar]
- 17.Hansen L, Krog G F, Søndergaard M. Decomposition of lake phytoplankton. 1. Dynamics of short-term decomposition. Oikos. 1986;46:37–44. [Google Scholar]
- 18.Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;4:406–425. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- 19.Jewel W J, McCarty P L. Aerobic decomposition of algae. Environ Sci Technol. 1971;5:1023–1031. [Google Scholar]
- 20.Kirchman D L, Suzuki Y, Garside C, Ducklow H W. High turnover rates of dissolved organic carbon during a spring phytoplankton bloom. Nature. 1991;352:612–614. [Google Scholar]
- 21.Mudryk Z, Donderski W. The occurrence of heterotrophic bacteria decomposing some macromolecular compounds in shallow estuarine lakes. Hydrobiologia. 1997;342/343:71–78. [Google Scholar]
- 22.Muyzer G, Dewaal E C, Uitterlinden A G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol. 1993;59:695–700. doi: 10.1128/aem.59.3.695-700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nei M, Li W H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nold S C, Zwart G. Patterns and governing forces in aquatic microbial communities. Aquat Ecol. 1998;32:17–35. [Google Scholar]
- 25.Oró J, Tornabene T G, Nooner D W, Gelpi E. Aliphatic hydrocarbons and fatty acids of some marine and freshwater microorganisms. J Bacteriol. 1967;93:1811–1818. doi: 10.1128/jb.93.6.1811-1818.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ragan M A, Goggin C L, Cawthorn R J, Cerenius L, Jamieson A V C, Plourde S M, Rand T G, Söderhäll K. A novel clade of protistan parasites near the animal-fungal divergence. Proc Natl Acad Sci USA. 1996;93:11907–11912. doi: 10.1073/pnas.93.21.11907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roessler P G. Environmental control of glycerolipid metabolism in microalgae: commercial implications and future research directions. J Phycol. 1990;26:393–399. [Google Scholar]
- 28.Sanders R W, Caron D A, Berninger U-G. Relationships between bacteria and heterotrophic nanoplankton in marine and fresh waters: an inter-ecosystem comparison. Mar Ecol Prog Ser. 1992;86:1–14. [Google Scholar]
- 29.Sherr B F, Sherr E B, McDaniel J. Effect of protistan grazing on the frequency of dividing cells in bacterioplankton assemblages. Appl Environ Microbiol. 1992;58:2381–2385. doi: 10.1128/aem.58.8.2381-2385.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shifrin N S, Chisholm S W. Phytoplankton lipids: interspecific differences and effects on nitrate, silicate and light-dark cycles. J Phycol. 1981;17:374–384. [Google Scholar]
- 31.Ŝimek K, Chraznowski T H. Direct and indirect evidence of size-selective grazing on pelagic bacteria by freshwater nanoflagellates. Appl Environ Microbiol. 1992;58:3715–3720. doi: 10.1128/aem.58.11.3715-3720.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tranvik L J, Höfle M G. Bacterial growth in mixed cultures on dissolved organic carbon from humic and clear waters. Appl Environ Microbiol. 1987;53:482–488. doi: 10.1128/aem.53.3.482-488.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Upton A C, Nedwell D B. Nutritional flexibility of oligotrophic and copiotrophic Antarctic bacteria with respect to organic substrates. FEMS Microbiol Ecol. 1989;62:1–6. [Google Scholar]
- 34.van Hannen E J, Gons H J. UVC-induced lysis and detritus production of Oscillatoria limnetica in a two-stage continuous-flow system. J Plankton Res. 1997;19:723–733. [Google Scholar]
- 35.van Hannen E J, van Agterveld M P, Gons H J, Laanbroek H J. Revealing eukaryotic genetic diversity in aquatic environments by denaturing gradient gel electrophoresis. J Phycol. 1998;34:206–213. [Google Scholar]
- 36.van Hannen, E. J., H. J. Gons, J. Ebert, H. L. Hoogveld, M. P. van Agterveld, G. Zwart, and H. J. Laanbroek. The microbial community structure in a large fresh water lagoon: an analysis using both flow cytometry and denaturing gradient gel electrophoresis. Submitted for publication.
- 37.van Hannen, E. J., M. Veninga, J. Bloem, H. J. Gons, and H. J. Laanbroek. Genetic changes in the bacterial community structure associated with protistan grazers. Arch. Hydrobiol., in press.
- 38.van Hannen E J, Zwart G, van Agterveld M P, Gons H J, Ebert J, Laanbroek H J. Changes in the bacterial and eukaryotic community structure after mass lysis of filamentous cyanobacteria associated with viruses. Appl Environ Microbiol. 1999;65:795–801. doi: 10.1128/aem.65.2.795-801.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Wambeke F. Influence of phytoplankton lysis or grazing on bacterial metabolism and trophic relationships. Microb Ecol. 1994;27:143–158. doi: 10.1007/BF00165814. [DOI] [PubMed] [Google Scholar]
- 40.Verity P G. Feeding in planktonic protozoans: evidence for non-random acquisition of prey. J Protozool. 1991;38:69–76. [Google Scholar]
- 41.White P A, Kalff J, Rasmussen J B, Gasol J M. The effect of temperature and algal biomass on bacterial production and specific growth rate in freshwater and marine habitats. Microb Ecol. 1991;21:99–118. doi: 10.1007/BF02539147. [DOI] [PubMed] [Google Scholar]
- 42.Zwart G, Huismans R, van Agterveld M P, Vande Peer Y, De Rijk P, Eenhoorn H, Muyzer G, van Hannen E J, Gons H J, Laanbroek H J. Divergent members of the bacterial division Verrucomicrobiales in a temperate freshwater lake. FEMS Microbiol Ecol. 1998;25:159–169. [Google Scholar]