

Abstract
Glycosite-specific antibody‒drug conjugatess (gsADCs), harnessing Asn297 N-glycan of IgG Fc as the conjugation site for drug payloads, usually require multi-step glycoengineering with two or more enzymes, which limits the substrate diversification and complicates the preparation process. Herein, we report a series of novel disaccharide-based substrates, which reprogram the IgG glycoengineering to one-step synthesis of gsADCs, catalyzed by an endo-N-acetylglucosaminidase (ENGase) of Endo-S2. IgG glycoengineering via ENGases usually has two steps: deglycosylation by wild-type (WT) ENGases and transglycosylation by mutated ENGases. But in the current method, we have found that disaccharide LacNAc oxazoline can be efficiently assembled onto IgG by WT Endo-S2 without hydrolysis of the product, which enables the one-step glycoengineering directly from native antibodies. Further studies on substrate specificity revealed that this approach has excellent tolerance on various modification of 6-Gal motif of LacNAc. Within 1 h, one-step synthesis of gsADC was achieved using the LacNAc-toxin substrates including structures free of bioorthogonal groups. These gsADCs demonstrated good homogeneity, buffer stability, in vitro and in vivo anti-tumor activity. This work presents a novel strategy using LacNAc-based substrates to reprogram the multi-step IgG glycoengineering to a one-step manner for highly efficient synthesis of gsADCs.
KEY WORDS: Site-specific ADCs, ENGase, LacNAc, One-step assembly, Potent in vivo efficacy
Graphical abstract
A LacNAc-based substrate reprograms the multi-step IgG glycoengineering and enables the efficient one-step synthesis of glycosite-specific antibody–drug conjugates (gsADCs), catalyzed by a wild-type endo-glycosidase (Endo-S2). This method also facilitates diverse modification of biotinylation, fluorescent labeling, extra glycan elongation, etc. for gain of functions.

1. Introduction
Antibody–drug conjugates (ADCs), covalently linking a potent cytotoxin onto the antibody, attract great and broad research interests as novel biotherapeutics1,2. To date, total 12 ADCs have been approved by US Food and Drug Administration (FDA). These approved ADCs are synthesized majorly by random conjugation of the payloads on lysine or cysteine residues, resulting in heterogeneity in conjugation sites and drug‒antibody ratios (DARs). In the past decade, site-specific conjugation has been proved as a feasible strategy to improve the therapeutic index of ADCs1,3,4, such as the THIOMAB technology3, unnatural-amino-acids incorporation5, 6, 7, as well as chemoenzymatic ligations with sortase A8,9 or transglutaminase10,11.
Harnessing the conserved N-glycosylation site on Fc Asn297 as the payload loading site12,13, the concept of glycosite-specific ADCs (gsADCs) has been developed by our and other research groups. Normally, synthesis of gsADCs requires antibody glycoengineering with an unnatural sugar either by a glycosyltransferase or an endo-β-N-acetyl-glucosaminidase (ENGase), followed by a bioorthogonal reaction to assemble the toxin payload. Boons14, Zhu15, Zhou16, van Delft17, and Thompson18 groups, utilized various glycosyltransferases to develop gsADCs, while Davis19, as well as our group20,21, synthesized gsADCs via ENGase-catalyzed transglycosylation. High homogeneity is the advantage of gsADCs, however, it's still a challenge to achieve gsADCs efficiently from native antibodies, due to the spiny glycoengineering steps. Multiple steps, multiple enzymes, functionalized sugars, modified drugs, and biorthogonal reactions, are usually indispensable in IgG glycoremodeling and payload conjugation, which limited the application of gsADCs. A more robust synthetic process for gsADCs bearing diversified linkages is required for gain of functions.
As summarized in Fig. 1, the previous synthesis of gsADCs (Fig. 1A), either by glycosyltransferase or ENGase, required 3–4 steps with at least two enzymes for IgG glycoengineering and payload conjugation. And, these methods all rely on the biorthogonal reactions. Moreover, the complicated synthesis and limited substrate specificity of UDP (uridine diphosphate)/CMP (cytidine monophosphate)-monosaccharides also hindered the efficient preparation and structural diversification of gsADCs for further evaluations. On the contrary, herein, we reported a facile and high-efficient method for synthesis of gsADCs in one step with a disaccharide‒drug substrate catalysed by one enzyme. The disaccharide of N-acetyl-lactosamine (LacNAc) was found as a perfect substrate of wild-type (WT) endoglycosidase from Streptococcus pyogenes of serotype M49 (Endo-S2) for “one-step” IgG glycan-remodeling. With the LacNAc derivatives, we prepared diverse functionalized antibodies directly from native antibody, including gsADCs, in a single one-step transglycosylation process (Fig. 1C). These LacNAc-based gsADCs showed high potent antitumor activity against both in vitro and in vivo. This is the first time, to the best of our knowledge, that glycosite-specific ADCs are prepared efficiently from native IgGs in a single one-step process.
Figure 1.

Strategies for synthesis of glycosite-specific ADCs.
2. Materials and methods
2.1. Chemicals and reagents
Endo-M22, Endo-S and Endo-S D233Q23, Endo-S2 and Endo-S2 D184M24, Endo-F3 and Endo-F3 D165A25, Endo-D and Endo-D Q431A and Endo-D N322Q26, Endo-A27, and Alfc28 were expressed in E. coli following the reported procedure. Sialylglycopeptide (SGP) was isolated and purified from egg yolk powder following previously reported procedures21,29. 2-Chloro-1,3-dimethyl-1H-benzimidazol-3-ium chloride (CDMBI)30, O-(2-azidoethyl)-hydroxylamine hydrochloride, O-(2-propynylethyl)-hydroxylamine hydrochloride, biotin-ONH2, NH2-VC-PAB-MMAE, DBCO-FITC were synthesized following previously reported procedures20. N-Acetylglucosamine (GlcNAc), N-acetylgalactosamine (GalNAc), LacNAc, N,N′-diacetylchitobiose, N,N′,N″-triacetylchitotriose, maltotriose were purchased from Acmec (Shanghai, China). 2-Chloro-1,3-dimethylimidazolidinium chloride (DMC), 7-(diethylamino) coumarin-3-carboxylic acid, 2-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU), mono-Fmoc ethylene diamine hydrochloride, sodium borohydride (NaBH4), NiCl2·6H2O, 3-azido-1-propanamine, sodium cyanoborohydride (NaCNBH3), 2-propynylamine, N-benzylthiourea, hydroxylamine hydrochloride, Fmoc-lysine-OH were purchased from Bidepharm (Shanghai, China). DBCO-CONHS, DBCO-Cy3, DBCO-PEG4-Biotin, BCN-O-PNP were purchased from Flechem (Shanghai, China) and Confluore (Xi'an, China). MMAE was purchased from Resuperpharmtech (Shanghai, China). Trastuzumab was purchased from Roche (Shanghai, China). Galactose oxidase, HRP and catalase were purchased from Sangon Biotech (Shanghai, China). Tween-20, protein-A, Hoechst 33258 and cell fix solution were purchased from Yeasen (China, Shanghai). Bio-gel P2 was purchased from BIO-RAD. Waters Sep-Pak Vac column was purchased from Waters, RPMI 1640, cell grade PBS were purchased from Hyclone (GE Health). 8-well (155411) plates, Matrigel were purchased from Thermo Scientific. Centrifugal filter was purchased from Millipore Corporation. Other chemical reagents and solvents were purchased from Sinopharm Chemical Reagent Co. (Shanghai, China). Nuclear magnetic resonance (NMR) spectra were measured on a Varian-MERCURY Plus-400 or 500 instrument. Antibody internalization was measured on a Leica TCS-SP8 STED instrument.
2.2. Equipment and conditions
2.2.1. High performance liquid chromatography (HPLC)
Method A: Analytical RP-HPLC was performed on a Thermo ultimate 3000 instrument with a C18 column (Thermo, Acclaim™ 120, 5 μm, 4.6 mm × 250 mm) at 40 °C. The column was eluted with a linear gradient of 2%–90% acetonitrile containing 0.1% TFA in 30 min at a flow rate of 1 mL/min. Method B: Analytical RP-HPLC was performed on a Thermo ultimate 3000 instrument with an Agilent InfinityLab Poroshell 120 column. The column was eluted with a linear gradient of 2%–90% acetonitrile containing 0.02% NH3 in 30 min at a flow rate of 1 mL/min. Method C: Semi-preparative HPLC was performed on a Beijing ChuangXinTongHeng LC3000 (preparative) instrument with a preparative C-18 column (5 μm, 10 mm × 250 mm). The column was eluted with a suitable gradient of aqueous acetonitrile containing 0.1% TFA (or 0.1% NH3 for oxazoline purification) at a flow rate of 8 mL/min.
2.2.2. Electron spray ionization mass spectrometry (ESI-MS) and liquid chromatography mass spectrometry (LC‒MS)
The ESI-MS spectra were measured on a Waters Xevo G2-XS Q-TOF. The small molecules were analyzed using a Waters C18 column (ACQUITY UPLC BEH C18, 1.7 μm, 2.1 mm × 50 mm) and eluted with a linear gradient of 10%–70% acetonitrile containing 0.1% formic acid in 1.3 min at a flow rate of 0.3 mL/min. Mass spectra of small molecule were recorded in the mass range of 50–3000 under high resolution mass-spec mode (HRMS, standard 3200 m/z, 4 GHz). Key source parameters: Cone Gas of 50 L/h; Desolvation Gas of 800 L/h; source temperature of 120 °C; Desolvation temperature of 200 °C; Capillary voltage of 3000 V; Collision Cell RF Offset of 150 V; Collision Cell RF Gain of 10. The proteins samples were measured with a Waters C4 column (ACQUITY UPLC Protein BEH C4, 1.7 μm, 2.1 mm × 50 mm) at 80 °C. The column was eluted with a linear gradient of 5%–90% acetonitrile containing 0.1% formic acid in 4 min at a flow rate of 0.3 mL/min. The mass spectra of proteins were performed under the extended mass range mode (High 20,000 m/z, 1 GHz) and the data were collected in the mass range of 500–3500. Key source parameters: Cone Gas of 50 L/h; Desolvation Gas of 800 L/h; source temperature of 120 °C; Desolvation temperature of 600 °C; Capillary voltage of 3000 V; Collision Cell RF Offset of 600 V; Collision Cell RF Gain of 0.
2.3. General procedures for the synthesis of oxazoline
To a solution of various sugar substrates (43 μmoL) in D2O (1.56 mL) was added 2-chloro-1,3-dimethyl-1H-benzimidazol-3-ium chloride (CDMBI, 46.44 mg, 215 μmoL) and K3PO4 (137 mg, 0.6445 mmoL), The reaction mixture was incubated at 0 °C for 2 h. After that, the mixture was purified by Bio-gel P2 column or directly subjected to NMR analysis.
2.4. Transglycosylation test of ENGase and oxazoline substrates
2.4.1. Deglycosylation of wild-type trastuzumab to give (Fucα1,6)GlcNAc-trastuzumab 1a and GlcNAc-trastuzumab 1b
A solution of wild-type trastuzumab (33 mg) in a phosphate buffer (50 mmol/L, pH 7.4) was incubated with Endo-S (100 ng) at 37 °C overnight. LC‒MS monitoring indicated the complete hydrolysis of the heterogeneous glycoforms at glycosylation sites. The antibody was subjected to protein A-agarose resin (1 mL) that was pre-equilibrated with a phosphate buffer (0.2 mol/L, pH 7.4). The column was washed with phosphate buffer (0.2 mol/L, pH 7.4, 3 mL), phosphate buffer (0.2 mol/L, pH 5.0, 0.1% Tween-20, 3 mL) and phosphate buffer (0.2 mol/L, pH 5.0, 3 mL) respectively to remove the impurities. The bound IgG was released with glycine-HCl (100 mmol/L, pH 2.7, 0.8 mL) and the elution fractions were immediately neutralized with 10 mol/L NaOH. The fractions containing the antibody component were combined and concentrated by centrifugal filtration (Millipore) to give the corresponding (Fucα1,6)GlcNAc-trastuzumab. For synthesis of GlcNAc-trastuzumab 1b, Endo-S (100 ng) and Alfc (3.3 mg) were co-incubated with wild-type trastuzumab at 37 °C overnight, and other procedures were as the same as above mentioned.
2.4.2. General procedures for transglycosylation activity determination of sugar oxazolines and ENGases with (Fucα1,6)GlcNAc-trastuzumab 1a
A solution of (Fucα1,6)GlcNAc-trastuzumab (50 μg) and various sugar oxazolines (15 nmol) in a Tris buffer (50 mmol/L, pH 7.2, 10 μL) was incubated with different endoglycosidases (2 μg) at 30 °C for 3 h and the transglycosylation of each sugar and ENGase were determined with LC‒MS.
2.5. Synthesis of LacNAc analogues
The detailed procedures for the synthesis of LacNAc analogues 2f‒h, 4a‒i, 11 could be found in the Supporting Information.
2.6. Glycoengineering of trastuzumab with LacNAc derivatives and Endo-S2
A solution of wild-type trastuzumab or GlcNAc-trastuzumab 1b (1 mg) and derived LacNAc oxazolines (0.1 μmol) in a phosphate buffered saline (PBS) buffer (50 mmol/L, pH 7.0, 200 μL) was incubated with Endo-S2 (80 μg) at 30 °C for 0.5–12 h. LC‒MS monitoring indicated the complete reaction of wild-type trastuzumab to give the transglycosylation product carrying LacNAc derivatives. The reaction mixture was immediately subjected to affinity chromatography via protein A resin following the above procedure to give the corresponding glycoengineered antibodies 3g and 6a‒i with core-fucosylation and 3g′, 7a and 7c without core-fucosylation.
2.7. Affinity determination of glycoengineered trastuzumab and Fc receptors
Homogeneous time-resolved fluorescence resonance energy transfer based CD16a(V176)/CD32a(H167)/CD64/FcRn binding assay. The total volume of the reaction system was 20 μL. When measuring antibody's binding to human CD16a(V176)/CD32a(H167)/CD64(CDA-H82E9/CDA-H82E6/FCA-H82E8, ACRO Biosystems), the concentrations of the d2-labeled native human IgG1 protein (IgG1-d2) and biotinylated human CD16a(V176)/CD32a(H167)/CD64 were each 10 nmol/L, and the concentration of streptavidin-Eu cryptate (610SAKLA, PerkinElmer) was 2.5 nmol/L and a test sample was diluted with the PBS pH 7.2 (1×) (20012-027, Life Technologies). When measuring antibody's binding to human FcRn, the concentrations of the IgG1-d2 and biotinylated human FcRn (FCM-H82W4, ACRO Biosystems) were each 3 nmol/L, and the concentration of streptavidin-Eu cryptate was 0.75 nmol/L and a test sample was diluted with the PPI-Europium detection buffer (pH 6.0) (61DB9RDF, PerkinElmer). The IgG1-d2, streptavidin-Eu cryptate, receptor and the diluted test sample were then added to a HTRF 96-well-low-volume plate (#66PL96025, PerkinElmer) and well mixed, the plate was sealed with a sealing film and incubated at 25 ± 1 °C for 2 h ± 5 min for CD16a(V176)/CD32a(H167)/FcRn binding measurement or incubated for 4 h ± 5 min for CD64 binding measurement without shaking. After incubation, the fluorescent signals were obtained on the Spark 10M (Tecan) with excitation wavelength at 320 nm and emission wavelength at 620 and 665 nm, respectively. The fluorescent signals of the sample were plotted against log-transformed concentrations using a four-parameter logistic model for curve-fitting (GraphPad Prism 8).
2.8. One-step synthesis of LacNAc based glycosite-specific ADCs
A solution of wild-type trastuzumab (1 mg) and drug-linker 11 (0.1 mmol) in a PB buffer (50 mmol/L, pH 7.0, 200 μL) was incubated with Endo-S2 (80 μg) at 30 °C for 1 h. LC‒MS monitoring indicated the complete transfer of drug-linker to the transglycosylation sites of trastuzumab. The reaction mixture was immediately subjected to affinity chromatography via protein A resin following the above procedure to give the corresponding gsADC-2.
2.9. Two-step synthesis of LacNAc based biotinylated/Cy3-labled trastuzumab and glycosite-specific ADCs
2.9.1. Synthesis of biotinylated/Cy3-labled trastuzumab
A solution of Azido-LacNAc-trastuzumab (1 mg) in a phosphate buffer (50 mmol/L, pH 7.4, 200 μL) was incubated with DBCO-PEG4-biotin 8a or DBCO-Cy3 8b (80 nmoL) at 37 °C for 2 h at dark. LC‒MS monitoring indicated the complete reaction of azido-LacNAc-trastuzumab to give biotin/Cy3-tagged LacNAc-trastuzumab. The reaction mixture was concentrated by centrifugal filtration (Millipore) to give the corresponding Biotin or Cy3-tagged LacNAc-trastuzumab (9a or 9b).
2.9.2. Synthesis of gsADC-1
A solution of 6a (1 mg) and drug-linker 10 (80 nmol) in a PB buffer (50 mmol/L, pH 7.0, 200 μL) was incubated at 37 °C for 4 h. LC‒MS monitoring indicated the complete conjugation of 10 on glycosylation sites. The reaction mixture was concentrated by centrifugal filtration (Millipore) to give the corresponding gsADC-1.
2.10. Cell culture
SK-Br-3, NCI-N87 and MDA-MB-231 were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 100 U/mL penicillin and streptomycin (Invitrogen, Carlsbad, CA, USA) at 37 °C with 5% CO2.
2.11. In vitro activity determination of gsADCs
The MTT assay was used to measure the in vitro efficiency of gsADCs. HER2 positive tumor cells SK-Br-3, NCI-N87 and HER2 negative tumor cells MDA-MB-231 were cultured in a 10% FBS-containing RPMI 1640 medium and were planted into 96-well plates with 6000 cells per well. The plates were incubated overnight at 37 °C in a 5% CO2 cell incubator. gsADC-1‒gsADC-3 were diluted 5-fold with RPMI 1640 medium from an initial concentration of 100 nmol/L, totally nine concentration gradients. The ADC samples were added to three wells with every single concentration and the 96-well plates were cultured at 37 °C in a 5% CO2 cell incubator for three days. Then MTT solution was added and incubated at 37 °C for 4 h, 10% SDS solution was added to dissolve the formazan. Optical density (OD) value was measured at 570 nm using an BioTek Epoch. EC50 values and the cell viability curve were calculated by GraphPad software.
2.12. Hydrophobic interaction chromatography of gsADCs
ADC samples (20 μg) were dissolved in 1 × PBS (20 μL). HIC analysis was performed with a butyl-NPR column, 4.6 cm × 10 cm, 2.5 μm particle size (TSKgel) with a linear gradient of 100% mobile phase A (2 mol/L (NH4)2SO4 in 25 mmol/L phosphate buffer, pH 7.0) to 100% mobile phase B (25 mmol/L phosphate buffer, pH 7.0) in 15 min.
2.13. Stability analysis of gsADCs
ADC samples (200 μg) were dissolved in 1 × PBS (200 μL) and heated at 60 °C for 36 h. Aliquots were taken at intervals and were analyzed by Zenix-C SEC-300 column, 7.8 mm × 300 mm, 3 μm particle size with a gradient of 100% mobile phase (150 mmol/L Sodium phosphate, pH 7.0) in 15 min.
2.14. In vivo assay of gsADCs
Five-weeks old female athymic BALB/c nu/nu mice were purchased from Shanghai Institute of Materia Medica and randomly divided into groups of five mice each. Animal handling and procedures were approved and performed according to the requirements of the Institutional Animal Care and Use Committee (IACUC) of Shanghai Institute of Materia Medica, CAS (license: SYXK (Shanghai) 2015-0027 and 2019-0032). All xenograft models were established by s.c. inoculation in the flanks of the mice. NCI-N87 cells were established by injecting 3 × 106 cells suspended in a Matrigel matrix. After tumor volume reached about 150 mm3, tumor-bearing mice were randomized based on tumor volumes. The xenograft mice were injected i.p. with each ADC sample or PBS at a dose of 3 mg/kg, dosing on Days 1, 4, and 7. The tumor volume was calculated by formula: length × width2 × 1/2. The in vivo activity was calculated by GraphPad software.
3. Results and discussion
3.1. Screening of new saccharide substrates and ENGases for one-step IgG glycoengineering
ENGases hydrolyze the glycosidic bond between GlcNAc-β1,4-GlcNAc of N-glycans, then transfer a glycan oxazoline onto the GlcNAc-containing acceptor, therefore fulfilling the two-step glycoengineering22, 23, 24, 25, 26,31, 32, 33, 34. To find a suitable enzyme and new saccharide substrates for potential one-step transglycosylation, we tested 10 ENGases on their catalytic activity to assemble the mono-saccharide GlcNAc oxazoline (2a) onto the deglycosylated trastuzumab (Fucα1,6GlcNAc-Tras, also named GN(F)-Tras, 1a), a therapeutic mAb against HER2 positive breast cancer (Fig. 2A). The data revealed that GlcNAc-ox 2a could be transglycosylated onto GN(F)-Tras by Endo-S2 (Fig. 2B, Supporting Information Fig. S1) but not other 9 ENGases. Although the initial yield is quite low (about 5%), it is an exciting and promising start point since Endo-S2 exhibited the potential activity for very simple substrate. Thereafter, we determined the transglycosylation of Endo-S2 with various sugar substrates, including GalNAc-ox 2b, LacNAc-ox 2c, (GlcNAc)2-ox 2d, (GlcNAc)3-ox 2e, NeuNAcα2,3-LacNAc-ox 2f, NeuNAcα2,6-LacNAc-ox 2g, and Galβ1,4-(Fucα1,3)GlcNAc-ox 2h. Interestingly, the transglycosylation with LacNAc oxazoline 2c and NeuNAcα2,6-LacNAc 2g achieved about 70% yields, even without optimization on the reaction conditions (Fig. 2C, Supporting Information Fig. S2). Meanwhile, the sialylation on 6-position of galactose (2g) showed barely no influence on Endo-S2-catalyzed transglycosylation, compared to LacNAc 2c, on the contrary, the 3-sialylation of Gal (2f) or 3-fucosylation of GlcNAc (2h) dramatically reduce the yield (<10%). These data demonstrated that the modifications on 6-position of Gal is excellently tolerated by Endo-S2 catalysis. After the optimization of reaction conditions, encouragingly, LacNAc-ox 2c could be quantitatively transferred onto the N-glycosylation site of both the deglycosylated and native trastuzumab in one step (Supporting Information Fig. S3). Endo-S2 hydrolyzes native glycoforms of IgG and simultaneously assembles LacNAc back onto the deglycosylated IgG without hydrolysis of the newly glyco-remodeled product, that reprograms the multi-step IgG engineering to a robust one-step manner.
Figure 2.

Screening of ENGases and new sugar oxazolines for one-step IgG glycoengineering. (A) Transglycosylation reaction and substrate structures of sugar oxazolines. (B) Transglycosylation yields of GlcNAc(Fuc)-Tras (1a) with GlcNAc-ox (2a) catalyzed by various ENGases. (C) Endo-S2-catalyzed transglycosylation yields of GlcNAc(Fuc)-Tras (1a) with various sugar oxazolines (2a‒h).
3.2. LacNAc derivatization on the 6-hydroxyl of Gal
Since modification of Gal 6-position of LacNAc is tolerated for Endo-S recognition, we next determined antibody glycoengineering with various LacNAc derivatives. The synthesis of these derivatives is summarized in Scheme 1. Using LacNAc 2c as the starting material, we firstly oxidized the 6-position of Gal by a galactose oxidase to give the 6-aldehyde group, a useful functional group for next functionalization. Then, we introduced azido, alkyne, biotin onto LacNAc with hydroxylamine-containing molecules and gave LacNAc derivatives 4a, 4c, 4h. Meanwhile, CHO-LacNAc was reacted with amine-containing molecules by reductive amination reaction to give LacNAc derivatives 4b, 4d, 4i. We also turned aldehyde group into amino group for further modification, including coupling with isothiocyanate and carboxylic acid to give LacNAc derivatives 4f and 4g. In another aspect, LacNAc was easily elongated by fucosyltransferase and sialyltransferase to give fucosylated and sialylated LacNAc 2f‒h.
Scheme 1.

Synthesis of LacNAc derivatives. Reagents and conditions: (a) galactose oxidase, HRP, catalase, O2, 50 mmol/L PB, pH 7.0, 30 °C; (b) O-(2-azidoethyl)-hydroxylamine hydrochloride, O-(2-propynylethyl)-hydroxylamine hydrochloride, or biotin-ONH2, pH 7.2, rt; (c) 3-azido-1-propanamine, 2-propynylamine, or coumarin-NH2, NaCNBH3, pH 6.0, 0 °C; (d) NH2OH·HCl, Na2CO3, CH3OH/H2O, rt; NaBH4, NiCl2·6H2O, 0 °C; (e) N-benzylthiourea, pH 8.0, rt; (f) DBCO-CONHS, pH 7.4, rt; (g) GDP-Fuc, α(1,3)-fucosyltransferase; (h) α(2,3)-sialyltransfererase, CMP-sialic acid, Tris-HCl buffer (100 mmol/L, pH 8.0); or α(2,6)-sialyltransferase (Pd2,6ST), CMP-sialic acid, Tris-HCl buffer (100 mmol/L, pH 8.0).
3.3. One-step IgG glycoengineering with LacNAc derivatives for gain of functions
With these LacNAc derivatives in hand, we performed the transglycosylation under optimized conditions and monitored the process by LC‒MS. As showed in Fig. 3, all these LacNAc-based compounds can serve as substrates for Endo-S2-catalysed transglycosylation and gave the glycoengineered antibodies, 3c and 6a‒i, rapidly in high yields (see Supporting Information Fig. S4 for detailed LC‒MS profiles). Endo-S2 has been reported to hydrolysis the heterogeneous N-glycans from IgG N-glycosylation site, including complex type, high mannose type, and hybrid type35. Additionally, the Endo-S2 mutants have been found to be capable of transferring N-glycan oxazoline to both fucosylated and non-fucosylated IgGs24. Hence, besides the native trastuzumab, the defucosylated trastuzumab (GN-Tras, 1b), obtained by deglycosylation with Endo-S and defucosylation with Alfc (α-fucosidase from Lactobacillus casei)28, also exhibited equal efficiency of glycoengineering via LacNAc-Endo-S2 approach, giving glycoengineered trastuzumab 3g′, 7a and 7c.
Figure 3.

One-step IgG glycoengineering with various LacNAc derivatives.
Then we tested the binding affinity of these new LacNAc-based glycoengineered trastuzumabs with four different Fc receptors, CD16a, CD32a, CD64 and FcRn. As shown in Table 1, defucosylated LacNAc-trastuzumab exhibited a 10-time folds enhanced affinity against CD16a than core-fucosylated and deglycosylated samples (7a/7c vs 6a/6c or 1a/1b), implicating that both fucosylation and glycan length contribute to CD16a binding. As to CD32a and CD64, the IgG glycan length but not fucosylation is important. All the glycoengineered trastuzumabs indicated comparable binding affinities with FcRn and fucosylation slightly enhance the FcRn interaction. These data predict that LacNAc-based IgGs may maintain antibody-dependent cell-mediate cytotoxicity (ADCC) (related to CD16a) by defucosylation and keep a good pharmacokinetic profile (related to FcRn).
Table 1.
Binding affinity (EC50, nmol/L) of glycoengineered trastuzumab with various Fc receptors.
| Entry | mAb | Glycoform | CD16a (V176) | CD32a (H167) | CD64 | FcRn |
|---|---|---|---|---|---|---|
| 1 | Tras | 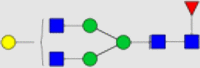 |
92.04 | 301.7 | 12.02 | 11.48 |
| 2 | 1a | 3342 | 7946 | 75.08 | 24.3 | |
| 3 | 1b | 441.4 | 9797 | 57.67 | 20.91 | |
| 4 | 6a | 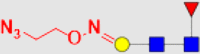 |
635.5 | 2014 | 40.03 | 13.48 |
| 5 | 6c | 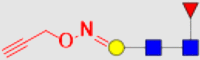 |
691.6 | 1996 | 36.14 | 14.59 |
| 6 | 7a | 68.47 | 1829 | 32.57 | 17.41 | |
| 7 | 7c | 65.2 | 1972 | 29.65 | 15.68 | |
| 8 | S2G2(F)-Tras | 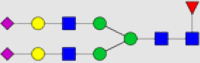 |
81.69 | 132.4 | 10.78 | 5.149 |
| 9 | S2G2-Tras | 15.45 | 135.8 | 9.612 | 20.73 |
With azido- or DBCO-tagged IgG (6a/6g), further functionalization with biotin, Cy3, or a trisaccharide can be readily achieved via click chemistry (Fig. 4). The resulted Cy3-Tras (9b) was employed in tracing the internalization of HER2 receptor on SK-Br-3 cells during 4–8 h incubation (Fig. 4F). Moreover, the efficient assembly of 1-azido-maltotriose onto 6g demonstrated that further glycan-elongation on the LacNAc motif is facile and practical. This approach opens a new avenue for IgG glycoengineering with various non-native glycoforms for gain of functions.
Figure 4.

Further functionalization of glycoengineered trastuzumab via click chemistry. (A) Assembly of biotin, Cy3, and maltotriose onto glycoengineered trastuzumab. (B) LC‒MS of Az-LacNAc-Tras 6a. (C) LC‒MS of DBCO-LacNAc-Tras 6g. (D) LC‒MS of Biotin-LacNAc-Tras 9a. (E) LC‒MS of Cy3-LacNAc-Tras 9b. (F) Confocal imaging of cell-surface HER2 internalization using 9b.
3.4. LacNAc enables one-step efficient synthesis of glycosite-specific ADCs
The robust IgG glycoengineering using LacNAc-based substrates encouraged us to apply this method in one-step or two-step synthesis of glycosite-specific ADCs (Fig. 5A). As expected, the Az-LacNAc-Tras (6a) bearing an azido tag can readily assemble a payload drug via click chemistry, using a cyclooctyne-tagged toxin (BCN-(PEG24)-VC-PAB-MMAE, 10), giving the glycosite-specific ADC (gsADC-1) (Fig. 5C and D).
Figure 5.

Synthesis of glycosite-specific ADCs using LacNAc-based substrates. (A) General procedure of one-step or two-step synthesis of gsADCs. (B) Small molecules used in gsADCs assembly. (C–E), LC‒MS profiles of Az-LacNAc-Tras 6a (C), gsADC-1 (D) and gsADC-2 (E). (F) SDS-PAGE of one-step synthesis of gsADC-2. Lane 0: protein marker; Lane 1: deglycosylated Tras (1a); Lane 2: gsADC-2. (G) Conjugation-site analysis of gsADC-2 via PNGase F digestion. (H) Antibody molecular weight of gsADC-2 after digestion with PNGase F. (I) HRMS profile of glycan–drug complex released from gsADC-2.
More importantly, a one-step synthesis of gsADCs can be achieved using a LacNAc-payload substrate, which is free of bioorthogonal reaction. As shown in Fig. 5A and B, we synthesized a MMAE-LacNAc oxazoline without any bioorthogonal tag (11) to perform the one-step IgG glycoengineering catalyzed by WT Endo-S2. LC‒MS and SDS-PAGE showed that the native Trastuzumab was totally consumed within 1 h and a gsADC with DAR of 2 (gsADC-2) was obtained in >95% yield (Fig. 5E and F). To confirm the site-specific conjugation on IgG glycosite, we digested gsADC-2 with PNGase F to release the N-glycosylation of Trastuzumab or Ides to separate the Fab and Fc domain (Fig. 5G). The LC‒MS characterization of antibody moiety demonstrated a 3767 molecular weight decrease (Fig. 5H), which is equals to 2 mol of Fuc-α1,6-GlcNAc-β1,4-GlcNAc-b1,4-Gal-VC-PAB-MMAE complex 12, of gsADC-2 after PNGase F digestion. Meanwhile, the compound 12 released from gsADC-2 by PNGase F was also determined by HRMS and was found exactly the expected structure (Fig. 5I). Both protein analysis and glycan–drug complex analysis indicated the correct structure of gsADCs that LacNAc-VC-PAB-MMAE was assembled onto the trastuzumab N-glycosylation site by WT Endo-S2 (Fig. 5G and Supporting Information Fig. S5).
3.5. LacNAc-based gsADCs showed high homogeneous and stability
For comparison to these LacNAc-based gsADCs, we also prepared a full-glycan gsADCs with DAR of 4 (gsADC-3) by conjugating a BCN-VC-PAB-MMAE to Az-S2G2F-trastuzumab as our previously reported work (Supporting Information Fig. S6)13. Hydrophobic Interaction Chromatography (HIC) analysis was carried out and the data showed the high purity and homogeneity of these gsADCs (Fig. 6B). Moreover, these new gsADCs exhibited good performance in buffer stability, with only 10.2% and 14.6% of aggregations after incubated in 1 × PBS at 60 °C for 36 h (Fig. 6C).
Figure 6.

HIC and SEC analysis of gsADCs. (A) Detailed structures of 3 gsADCs. (B) HIC analysis of 3 gsADCs. (C) SEC analysis of gsADC-1 and gsADC-2.
3.6. LacNAc-based gsADCs showed potent in vitro activity selectively on HER2-positive cell lines
The in vitro activities of these three gsADCs were evaluated against two HER2-positive tumor cells, SK-Br-3 and NCI-N87, and one HER2-negative tumor cell, MDA-MB-231. All gsADCs possessed excellent in vitro activities against HER2 positive tumor cells and non-cytotoxicity against HER2-negative tumor cells, indicating a targeted cell killing (Fig. 7, Table 2).
Figure 7.

In vitro activity determination of 3 gsADCs against two HER2 positive tumor cell lines, SK-Br-3 (A) and NCI-N87 (B), and HER2 negative tumor cell line MDA-MB-231 (C).
Table 2.
IC50 values of gsADCs against SK-Br-3, NCI-N87 and MDA-MB-231a.
| Entry | Cell line | gsADC-1 | gsADC-2 | gsADC-3 |
|---|---|---|---|---|
| 1 | SK-Br-3 | 0.045 | 0.044 | 0.071 |
| 2 | NCI-N87 | 0.152 | 0.163 | 0.19 |
| 3 | MDA-MB-231 | >100 | >100 | >100 |
All data were shown in nmol/L.
3.7. LacNAc-based gsADCs exhibited excellent efficacy in vivo antitumor activity
We next evaluated the in vivo activities of these three gsADCs and a cysteine conjugated ADC (Supporting Information Fig. S7) with NCI–N87 xenograft mice model. As shown in Fig. 8, gsADC-3 and Cys-ADC maintained its tumor inhibiting activity till Day-15 and the tumor volumes began to rebound in the following days. However, for LacNAc-based gsADC-1 and gsADC-2 (DAR = 2), both exhibited unremitting tumor inhibition, showing a much better in vivo efficacy than full-glycan gsADC-3 (DAR = 4) (Fig. 8A and C). Since LacNAc is shorter than full-length N-glycans, these LacNAc-based gsADCs can hide the hydrophobic MMAE more inside the Fc domain, therefore leading to better stability and efficacy in vivo. These data clearly demonstrated that LacNAc-based gsADCs via one-step synthesis are advantageous and attractive in both synthetic procedure and biological functions.
Figure 8.

In vivo efficacy assay of 4 ADCs on the NCI-N87 xenograft mice model. (A) Tumor volume curve. Red arrows indicate the time-points of ADC administration. Two-tailed t test was used to assess statistical significance between treatment and control groups. ∗∗∗∗P < 0.0001, comparing all ADCs with vehicle; ▹▹▹▹P < 0.0001, comparing gsADC-2 with Cys-conjugated ADC (n = 5 per group). Data = mean ± SEM. (B) Body weight curve. (C) Tumor images after dissection.
4. Conclusions
In conclusion, efficient one-step IgG glycoengineering and glycosite-specific ADCs synthesis were achieved. The LacNAc-based substrates and WT Endo-S2 demonstrated a reprogrammed procedure for IgG glycoengineering, which avoided the multi-enzymatic treatment and payload conjugation in previous approaches. Various modifications on 6-Gal of LacNAc were well tolerated by Endo-S2, allowing structural diversification in IgG glycoengineering including the glycan elongation. In addition, the one-step synthesis of gsADCs does not rely on the bioorthogonal reactions for payload assembly, which may reduce the risk of triazole-induced immunogenicity. These novel LacNAc-based gsADCs demonstrated excellent homogeneity, stability, in vitro and in vivo anti-tumor activity, in comparison with a full-glycan gsADC and a Cys-based ADC. This method is also compatible with other therapeutic antibodies, such as Rituximab, bispecific antibodies and Fc-fused nanobodies (data not shown). For the first time, we report the one-step synthesis of gsADCs from native IgG antibodies, which exhibited great potentials in IgG glycoengineering for structural diversification and gain of functions.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (NSFC, No., 2187116 and 82003574), Shanghai Municipal Science and Technology Major Project, the Shanghai Sail Program (No. 19YF1457100), the Special Research Assistant Program (Chinese Academy of Sciences, CAS), and Natural Science Foundation of Shandong Province (ZR2017BC062).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2021.12.013.
Contributor Information
Feng Tang, Email: tangfeng2013@simm.ac.cn.
Wei Huang, Email: huangwei@simm.ac.cn.
Author contributions
Wei Shi performed the screening of sugars and ENGases, LacNAc modifications, trastuzumab glycoengineering, linker-drug synthesis, gsADCs synthesis, LC‒MS analysis, HIC analysis, buffer stability analysis, in vitro and in vivo cytotoxicity determination. Wanzhen Li and Jianxin Zhang co-prepared the linker-drug complex, LC‒MS analysis, HIC analysis and in vivo cytotoxicity determination. Tiehai Li synthesized the trisaccharides, NeuNAα2,3LacNAc, NeuNAcα2,6LacNAc, Galβ1,4(Fucα1,3)GlcNAc. Yakai Song conducted the Fc binding affinity to Fc receptors. Yue Zeng helped in in vivo cytotoxicity determination. Shuquan Fan expressed and purified the fucosidase Alfc and helped in the analysis of defucosylation. Zeng Lin and Qian Dong helped in the compounds NMR analysis. Likun Gong helped in the discussion of Fc receptor affinity determination. Feng Tang and Wei Huang conceived the idea, supervised experimental design, analyzed the data, and wrote the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Beck A., Goetsch L., Dumontet C., Corvaia N. Strategies and challenges for the next generation of antibody‒drug conjugatess. Nat Rev Drug Discov. 2017;16:315–337. doi: 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- 2.Walsh S.J., Bargh J.D., Dannheim F.M., Hanby A.R., Seki H., Counsell A.J., et al. Site-selective modification strategies in antibody‒drug conjugatess. Chem Soc Rev. 2021;50:1305–1353. doi: 10.1039/d0cs00310g. [DOI] [PubMed] [Google Scholar]
- 3.Junutula J.R., Raab H., Clark S., Bhakta S., Leipold D.D., Weir S., et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925–932. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 4.Lhospice F., Bregeon D., Belmant C., Dennler P., Chiotellis A., Fischer E., et al. Site-specific conjugation of monomethyl auristatin E to anti-CD30 antibodies improves their pharmacokinetics and therapeutic index in rodent models. Mol Pharm. 2015;12:1863–1871. doi: 10.1021/mp500666j. [DOI] [PubMed] [Google Scholar]
- 5.Axup J.Y., Bajjuri K.M., Ritland M., Hutchins B.M., Kim C.H., Kazane S.A., et al. Synthesis of site-specific antibody‒drug conjugatess using unnatural amino acids. Proc Natl Acad Sci U S A. 2012;109:16101–16106. doi: 10.1073/pnas.1211023109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zimmerman E.S., Heibeck T.H., Gill A., Li X., Murray C.J., Madlansacay M.R., et al. Production of site-specific antibody‒drug conjugatess using optimized non-natural amino acids in a cell-free expression system. Bioconjug Chem. 2014;25:351–361. doi: 10.1021/bc400490z. [DOI] [PubMed] [Google Scholar]
- 7.Oller-Salvia B., Kym G., Chin J.W. Rapid and efficient generation of stable antibody‒drug conjugatess via an encoded cyclopropene and an inverse-electron-demand Diels-Alder reaction. Angew Chem Int Ed. 2018;57:2831–2834. doi: 10.1002/anie.201712370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beerli R.R., Hell T., Merkel A.S., Grawunder U. Sortase enzyme-mediated generation of site-specifically conjugated antibody drug conjugates with high in vitro and in vivo potency. PLoS One. 2015;10:e0131177. doi: 10.1371/journal.pone.0131177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harmand T.J., Bousbaine D., Chan A., Zhang X., Liu D.R., Tam J.P., et al. One-pot dual labeling of IgG 1 and preparation of C-to-C fusion proteins through a combination of sortase A and butelase 1. Bioconjug Chem. 2018;29:3245–3249. doi: 10.1021/acs.bioconjchem.8b00563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schneider H., Deweid L., Avrutina O., Kolmar H. Recent progress in transglutaminase-mediated assembly of antibody‒drug conjugatess. Anal Biochem. 2020;595:113615. doi: 10.1016/j.ab.2020.113615. [DOI] [PubMed] [Google Scholar]
- 11.Jeger S., Zimmermann K., Blanc A., Grunberg J., Honer M., Hunziker P., et al. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew Chem Int Ed. 2010;49:9995–9997. doi: 10.1002/anie.201004243. [DOI] [PubMed] [Google Scholar]
- 12.Zhou Q. Site-specific antibody conjugation for ADC and beyond. Biomedicines. 2017;5:64. doi: 10.3390/biomedicines5040064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L.X., Tong X., Li C., Giddens J.P., Li T. Glycoengineering of antibodies for modulating functions. Annu Rev Biochem. 2019;88:433–459. doi: 10.1146/annurev-biochem-062917-012911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X., Fang T., Boons G.J. Preparation of well-defined antibody‒drug conjugatess through glycan remodeling and strain-promoted azide-alkyne cycloadditions. Angew Chem Int Ed. 2014;53:7179–7182. doi: 10.1002/anie.201402606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu Z., Ramakrishnan B., Li J., Wang Y., Feng Y., Prabakaran P., et al. Site-specific antibody‒drug conjugation through an engineered glycotransferase and a chemically reactive sugar. MAbs. 2014;6:1190–1200. doi: 10.4161/mabs.29889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Q., Stefano J.E., Manning C., Kyazike J., Chen B., Gianolio D.A., et al. Site-specific antibody‒drug conjugation through glycoengineering. Bioconjug Chem. 2014;25:510–520. doi: 10.1021/bc400505q. [DOI] [PubMed] [Google Scholar]
- 17.van Geel R., Wijdeven M.A., Heesbeen R., Verkade J.M., Wasiel A.A., van Berkel S.S., et al. Chemoenzymatic conjugation of toxic payloads to the globally conserved N-glycan of native mAbs provides homogeneous and highly efficacious antibody‒drug conjugatess. Bioconjug Chem. 2015;26:2233–2242. doi: 10.1021/acs.bioconjchem.5b00224. [DOI] [PubMed] [Google Scholar]
- 18.Thompson P., Ezeadi E., Hutchinson I., Fleming R., Bezabeh B., Lin J., et al. Straightforward glycoengineering approach to site-specific antibody-pyrrolobenzodiazepine conjugates. ACS Med Chem Lett. 2016;7:1005–1008. doi: 10.1021/acsmedchemlett.6b00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parsons T.B., Struwe W.B., Gault J., Yamamoto K., Taylor T.A., Raj R., et al. Optimal synthetic glycosylation of a therapeutic antibody. Angew Chem Int Ed. 2016;55:2361–2367. doi: 10.1002/anie.201508723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang F., Yang Y., Tang Y., Tang S., Yang L., Sun B., et al. One-pot N-glycosylation remodeling of IgG with non-natural sialylglycopeptides enables glycosite-specific and dual-payload antibody‒drug conjugatess. Org Biomol Chem. 2016;14:9501–9518. doi: 10.1039/c6ob01751g. [DOI] [PubMed] [Google Scholar]
- 21.Tang F., Wang L.X., Huang W. Chemoenzymatic synthesis of glycoengineered IgG antibodies and glycosite-specific antibody‒drug conjugatess. Nat Protoc. 2017;12:1702–1721. doi: 10.1038/nprot.2017.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Umekawa M., Huang W., Li B., Fujita K., Ashida H., Wang L.X., et al. Mutants of mucor hiemalis endo-beta-N-acetylglucosaminidase show enhanced transglycosylation and glycosynthase-like activities. J Biol Chem. 2008;283:4469–4479. doi: 10.1074/jbc.M707137200. [DOI] [PubMed] [Google Scholar]
- 23.Huang W., Giddens J., Fan S.Q., Toonstra C., Wang L.X. Chemoenzymatic glycoengineering of intact IgG antibodies for gain of functions. J Am Chem Soc. 2012;134:12308–12318. doi: 10.1021/ja3051266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li T., Tong X., Yang Q., Giddens J.P., Wang L.X. Glycosynthase mutants of endoglycosidase S2 Show potent transglycosylation activity and remarkably relaxed substrate specificity for antibody glycosylation remodeling. J Biol Chem. 2016;291:16508–16518. doi: 10.1074/jbc.M116.738765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giddens J.P., Lomino J.V., Amin M.N., Wang L.X. Endo-F3 glycosynthase mutants enable chemoenzymatic synthesis of core-fucosylated triantennarycomplex type glycopeptides and glycoproteins. J Biol Chem. 2016;291:9356–9370. doi: 10.1074/jbc.M116.721597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan S.Q., Huang W., Wang L.X. Remarkable transglycosylation activity of glycosynthase mutants of endo-D, an endo-beta-N-acetylglucosaminidase from Streptococcus pneumoniae. J Biol Chem. 2012;287:11272–11281. doi: 10.1074/jbc.M112.340497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujita K., Tanaka N., Sano M., Kato I., Asada Y., Takegawa K. Synthesis of neoglycoenzymes with homogeneous N-linked oligosaccharides using immobilized endo-beta-N-acetylglucosaminidase A. Biochem Biophys Res Commun. 2000;267:134–138. doi: 10.1006/bbrc.1999.1963. [DOI] [PubMed] [Google Scholar]
- 28.Li T., DiLillo D.J., Bournazos S., Giddens J.P., Ravetch J.V., Wang L.X. Modulating IgG effector function by Fc glycan engineering. Proc Natl Acad Sci U S A. 2017;114:3485–3490. doi: 10.1073/pnas.1702173114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun B., Bao W., Tian X., Li M., Liu H., Dong J., et al. A simplified procedure for gram-scale production of sialylglycopeptide (SGP) from egg yolks and subsequent semi-synthesis of Man3GlcNAc oxazoline. Carbohydr Res. 2014;396:62–69. doi: 10.1016/j.carres.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 30.Noguchi M., Fujieda T., Huang W.C., Ishihara M., Kobayashi A., Shoda S.I. A practical one-step synthesis of 1,2-oxazoline derivatives from unprotected sugars and its application to chemoenzymatic β-N-acetylglucosaminidation of disialo-oligosaccharide. Helv Chim Acta. 2012;95:1928–1936. [Google Scholar]
- 31.Huang W., Li C., Li B., Umekawa M., Yamamoto K., Zhang X., et al. Glycosynthases enable a highly efficient chemoenzymatic synthesis of N-glycoproteins carrying intact natural N-glycans. J Am Chem Soc. 2009;131:2214–2223. doi: 10.1021/ja8074677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L.X., Huang W. Enzymatic transglycosylation for glycoconjugate synthesis. Curr Opin Chem Biol. 2009;13:592–600. doi: 10.1016/j.cbpa.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang W., Yang Q., Umekawa M., Yamamoto K., Wang L.X. Arthrobacter endo-beta-N-acetylglucosaminidase shows transglycosylation activity on complex-type N-glycan oxazolines: one-pot conversion of ribonuclease B to sialylated ribonuclease C. Chembiochem. 2010;11:1350–1355. doi: 10.1002/cbic.201000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodfellow J.J., Baruah K., Yamamoto K., Bonomelli C., Krishna B., Harvey D.J., et al. An endoglycosidase with alternative glycan specificity allows broadened glycoprotein remodelling. J Am Chem Soc. 2012;134:8030–8033. doi: 10.1021/ja301334b. [DOI] [PubMed] [Google Scholar]
- 35.Sjogren J., Cosgrave E.F., Allhorn M., Nordgren M., Bjork S., Olsson F., et al. EndoS and EndoS2 hydrolyze Fc-glycans on therapeutic antibodies with different glycoform selectivity and can be used for rapid quantification of high-mannose glycans. Glycobiology. 2015;25:1053–1063. doi: 10.1093/glycob/cwv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.