

Abstract
Disturbance of macrophage-associated lipid metabolism plays a key role in atherosclerosis. Crosstalk between autophagy deficiency and inflammation response in foam cells (FCs) through epigenetic regulation is still poorly understood. Here, we demonstrate that in macrophages, oxidized low-density lipoprotein (ox-LDL) leads to abnormal crosstalk between autophagy and inflammation, thereby causing aberrant lipid metabolism mediated through a dysfunctional transcription factor EB (TFEB)–P300–bromodomain-containing protein 4 (BRD4) axis. ox-LDL led to macrophage autophagy deficiency along with TFEB cytoplasmic accumulation and increased reactive oxygen species generation. This activated P300 promoted BRD4 binding on the promoter regions of inflammatory genes, consequently contributing to inflammation with atherogenesis. Particularly, ox-LDL activated BRD4-dependent super-enhancer associated with liquid–liquid phase separation (LLPS) on the regulatory regions of inflammatory genes. Curcumin (Cur) prominently restored FCs autophagy by promoting TFEB nuclear translocation, optimizing lipid catabolism, and reducing inflammation. The consequences of P300 and BRD4 on super-enhancer formation and inflammatory response in FCs could be prevented by Cur. Furthermore, the anti-atherogenesis effect of Cur was inhibited by macrophage-specific Brd4 overexpression or Tfeb knock-out in Apoe knock-out mice via bone marrow transplantation. The findings identify a novel TFEB-P300-BRD4 axis and establish a new epigenetic paradigm by which Cur regulates autophagy, inhibits inflammation, and decreases lipid content.
Key words: Macrophage, Autophagy, TFEB, P300, BRD4, Inflammation, Atherosclerosis, Curcumin
Keywords: Acetyl-H3, acetyl-histone 3; ATG5, autophagy-related 5; BET, bromodomain and extra-terminal; BRD4, bromodomain protein 4; ChIP, chromatin immunoprecipitation; CQ, chloroquine; Cur, curcumin; CVDs, cardiovascular diseases; Dil-ox-LDL, 1,1′-dioctadecyl-3,3,3′,3′-tetramethy-lindocarbocyanine perchlorate labeled oxidized low-density lipoprotein; FCs, foam cells; HFD, high-fat diet; IL-1β, interleukin 1β; LIR, LC3-interacting region; MCP-1, monocyte chemotactic protein 1; mTORC1, mammalian target of rapamycin complex 1; NAC, N-acetyl-l-cysteine; ORO, Oil red O; ox-LDL, oxidized low-density lipoprotein; qRT-PCR, quantitative real-time polymerase chain reaction; Re-ChIP, re-chromatin immunoprecipitation; ROS, reactive oxygen species; SE, super-enhancer; siRNAs, small interference RNAs; TFEB, transcription factor EB; TNF-α, tumor necrosis factor α
Graphical abstract
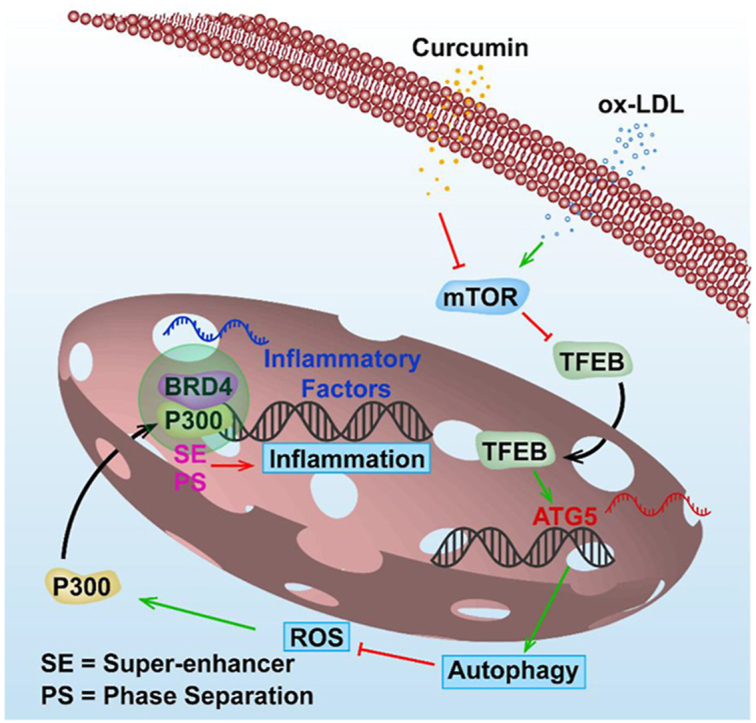
Through promoting TFEB nuclear translocation, curcumin enhances foam cells autophagy to inhibit P300 activity and the subsequent phase separation protein BRD4-dependent super-enhancer formation of the inflammatory genes, finally ameliorating atherogenesis.
1. Introduction
As an underlying pathological process of life-threatening cardiovascular diseases (CVDs), the incidence of atherosclerosis is rising globally. Atherosclerosis has become a costly public health issue1. Dysregulated of lipid catabolism and a maladaptive inflammatory response driven by the intramural retention of lipid-laden macrophages in susceptible areas of the arterial wall are the major contributors to atherosclerosis2. Initially, plasma-derived lipoproteins that are sequestered in the subendothelial space following their modification are often engulfed by macrophages and balanced by reverse cholesterol transporters. Excess lipid forces the formation of foam cells (FCs) that contribute to the atherosclerotic lesion. Secretion of proinflammatory cytokines by FCs causes the plaque inflammatory state and defective efferocytosis. FCs also exacerbate the formation of the vulnerable advanced lesion with a necrotic lipid core, whose rupture results in serious clinical complications. Thus, macrophages as the key integrators of inflammatory and metabolic signals have a pivotal role in atherogenesis. Efficiently ameliorating dysfunctional lipid catabolism and FCs inflammation may be a promising strategy for the regression of atherosclerosis.
Autophagy is an evolutionarily conserved cellular self-renewal mechanism that is critical in bulk degradation of aging or damaged cytoplasmic material via double-membrane autophagosomes with ensuing lysosomal fusion3. Accumulating evidence indicates that adequate induction of autophagy delays atherogenesis through multiple mechanisms, including promoting lipid breakdown, inhibiting inflammation and oxidative stress4,5. Impairment of autophagy usually occurs in advanced plaque. Injury of the lysosome by the lethal accumulation of cholesterol crystals contributes to plaque development and vulnerability. The resulting release of lysosomal hydrolases aggravates apoptosis, leading to a vicious cycle6,7. So far, in medications against atherosclerosis, only rapamycin or everolimus are working as the promoters of autophagy. Unfortunately, they have many side effects including cytokine production, dyslipidemia, and hyperglycemia8. Transcription factor EB (TFEB) is an emerging regulator of the autophagy–lysosome pathway via binding to E-box sequences (CANNTG)9. The protective effects of TFEB have been widely confirmed on a variety of diseases, such as Alzheimer's, diabetes, ischemic injury, as well as atherosclerosis from in vivo and in vitro models9,10. No effective drug targeting TFEB in CVDs has been described.
Many natural products with excellent pharmacological properties are reported to be good candidates for the prevention of atherosclerosis. Curcumin (Cur) is a natural polyphenol extracted from turmeric curry spice. Cur has multiple biological properties and health benefits without corresponding toxicity. Numerous studies have confirmed the favorable therapeutic effects of Cur on a broad array of diseases11. Although its role in the prevention of atherosclerosis has been shown, Cur's anti-atherogenic mechanism is still unclear. Recently, Cur is reported to regulate autophagy by inhibiting the mammalian target of rapamycin (mTOR) activity in endothelial cells undergoing oxidative stress12. Phosphorylation that is dependent on mammalian target of rapamycin complex 1 (mTORC1) controls TFEB nuclear export and inhibits autophagy in normal conditions13. Whether Cur can delay atherosclerosis by activating autophagy via TFEB and overcome the side effects of traditional autophagy modulating medicines has yet not been investigated.
Atherosclerosis is characterized by abnormal autophagy and chronic inflammation of the arterial wall. However, the mechanism involved in the crosstalk between autophagy and inflammation is still unclear. An imbalance between the two processes may trigger pathologic atherogenesis. Bromodomain protein 4 (BRD4), a member of the bromodomain and extra-terminal (BET) family proteins, usually localizes at gene promoters and enhancers. BRD4 acts as a scaffold for transcription factors. BRD4 recognizing acetylated histones through bromodomains, regulates the chromatin landscape, and promotes the transactivation of pathologic genes14. Interestingly, BRD4 has also been implicated in CVDs, including pulmonary arterial hypertension, heart failure and cardiac hypertrophy, due to the epigenetic activation of proinflammatory genes15, 16, 17. Whether BRD4 is involved in the atherogenesis and whether Cur can delay atherosclerosis by a BRD4-dependent epigenetic regulatory mechanism has not been reported. Given the inhibitory effect of autophagy on inflammation, whether there is a link between the crucial autophagy regulator TFEB and the inflammation regulator BRD4 is also unclear.
The present study examined two closely related issues. First, the novel epigenetic mechanism was investigated to understand the abnormal crosstalk between autophagy and inflammation, which will cause aberrant lipid metabolism in FCs. Second, we sought to identify an effective drug to optimize the crosstalk between autophagy and inflammation during anthogenesis. We demonstrate that Cur, a natural P300-specific histone acetyltransferases inhibitor, regulates the novel epigenetic TFEB–P300–BRD4 axis. The findings explain the pro-autophagy-induced anti-inflammation behavior of Cur in the formation of FCs.
2. Materials and methods
2.1. Cell culture
Human monocytic THP-1 cells (American Type Culture Collection, Manassas, VA, USA) were cultured in RPMI 1640 medium with 10% fetal bovine serum supplemented with 20 μg/mL penicillin–streptomycin at 37 °C in a 5% CO2 humidified atmosphere. To establish the FCs models, THP-1 cells were firstly differentiated into macrophages by 100 ng/mL phorbol-12-myristate-13-acetate (La Jolla, CA, USA) for 72 h at 1.0 × 106 cells/mL, which were subsequently transformed to FCs induced by 50 μg/mL oxidized low-density lipoprotein (ox-LDL, Peking Union-Biology Co., Ltd., China) for 24 h. For the autophagy inhibition experiment, the cells were pre-incubated with 1 mmol/L 3-MA in serum-free medium for 2 h and then incubated with ox-LDL.
2.2. Atherosclerosis model
Male Apoe knock-out mice (Apoe–/–, 8 weeks) on the C57BL/6 background were purchased from Sippr-BK Laboratory Animal Co., Ltd. (Shanghai, China). The mice were housed in a specific pathogen-free facility under controlled conditions (temperature, 22 ± 2 °C; relative humidity, 55 ± 15%; noise, < 60 dB; light/dark cycle, 12/12 h). Male Apoe–/– mice were fed with a high-fat diet (HFD, 21% fat and 0.15% cholesterol; Xietong Organism Inc., China) for 16 weeks to induce atherosclerosis. Mice in the Cur group received daily Cur (20 mg/kg body weight) by gastric gavages for 16 weeks together with HFD. All of the Apoe–/– mice consumed the HFD throughout the experiment. All experimental procedures in animals were conducted by the National Institute of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee of Nanjing Medical University (IACUC-2103042), Nanjing, China.
2.3. Construct generation and lentiviral particle production
The recombinant lentiviral construct specifically expressing Brd4 or deleting Tfeb in macrophages was produced by molecular cloning methods. Firstly, the full-length Brd4 and Tfeb cDNA was obtained from murine peripheral blood mononuclear cells and amplified by reverse transcription-polymerase chain reaction (RT-PCR). Then, the PCR product was inserted into the self-inactivating lentiviral vector pLVX-Puro (Clontech, Mountain View, CA, USA). The macrophage-specific human CD68 promoter (2.9 kb) along with an 89 bp intronic enhancer were amplified from human genomic DNA by PCR. After deleting the cytomegalovirus promoter from the vector, the CD68 promoter was incorporated upstream of the coding sequence to generate the final Brd4-expressing lentiviral construct pLVCD68-Brd4 overexpression and Tfeb-deleting lentiviral construct pLVCD68-Tfeb knock-out (Tfeb KO). Then, the viruses were harvested from the medium of 293T cells transient co-transfected with pLVCD68-Brd4, pLVCD68-Tfeb or Lenti-X HTX Packaging Mix (Clontech, USA) according to the manufacturer's instructions. Similarly, a recombinant lentiviral vector pLVCD68 that contains the CD68 promoter and lacks the Brd4 coding sequence or the Tfeb coding sequence was also generated and used as a control.
2.4. Bone marrow transplantation
Bone marrow cells (BMCs) from the femurs and tibias of donor male Apoe–/– mice were filtered through 30 μm nylon gauze to obtain the single-cell suspensions. Then, they were transfected by recombinant lentiviral pLVCD68-Brd4 oe or pLVCD68-Tfeb KO supernatant for 24 h at an MOI of 20 with 1 × 108 TU/mL titers containing 4 μg/mL polybrene and 2000 U/mL recombinant human macrophage-colony stimulating factor (rhM-CSF, R&D Systems, Minneapolis, MN, USA). Next, the cells were cultured for another 4 days in fresh medium with 2000 U/mL rhM-CSF and harvested before injection. Lethally irradiated (9 Gy) the male recipient Apoe–/– mice transplanted with 5 × 106 lentiviral transduced BMCs via tail vein injection within 6 h of irradiation. After 4 weeks, mice were fed with a HFD for an additional 16 weeks. The mice received BMCs transduced with pLVCD68 lentivirus used as control.
2.5. Atherosclerotic lesion analysis
The aortas were perfused with PBS and excised from the proximal aortic arch to the common iliac artery. Then, the connective and adipose tissues were removed from the aorta and the section of aortas was cut longitudinally stained with Oil red O (ORO) solution. Finally, the specimens were digitally photographed.
For histological analysis, the section of aortas was fixed in 4% paraformaldehyde at 4 °C for 24 h, dehydrated in a graded series of alcohol (70%, 80%, 90%, 95% and 100%, each 90 min), embedded in 5 μm-thick paraffin. Then, they were stained with hematoxylin and eosin, ORO, Sirius red and Masson. Images were captured using a light microscope (Olympus BX53, Olympus Corp.) at magnification, 10 ×, 20 × and 40 ×.
2.6. Cell viability analysis
The cell viability was evaluated by the cell counting kit 8 (CCK-8) assay (Beyotime, Beijing, China) according to the procedure. After different treatments, 100 μL 1640 medium containing CCK-8 at 10:1 dilution ratio was added to the cells incubating for 1 h in the dark. The survival cells were reflected by the absorbance of each well at 450 nm obtained via SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, USA).
2.7. Cell transfection
Small interference RNAs (siRNAs) used to knock down human TFEB, autophagy-related 5 (ATG5) and BRD4 (GenePharma, Shanghai, China), as well as the plasmid used to overexpress BRD4, was imported into the macrophages in 35 mm cultures. Firstly, lipofectamineTM 3000 (Thermo Fisher Scientific, Madison, USA) were diluted in Opti-MEM medium (Thermo Fisher Scientific, Madison, USA). Then, the master mix of siRNA/DNA was prepared by diluting siRNAs/DNA in the Opti-MEM medium (Supporting Information Table S1). After that, the diluted siRNAs/DNA was added into diluted lipofectamineTM 3000 at 1:1 ratio and incubated for 15 min at room temperature (RT). Finally, the siRNA/DNA–lipid complex was added into cells incubating for 1–3 days. Immunoblotting was used to analyze the transfection efficiency of different siRNAs and plasmids.
2.8. Immunofluorescence
After different treatments, the cells seeded in glass-bottom cell culture dishes were rinsed in PBS followed by fixed in 4% paraformaldehyde and soaked in 0.1% Triton X-100 for 30 min, respectively. After blocked with 3% bovine serum albumin for 1 h, the cells were incubated with specific primary antibodies overnight at 4 °C. The cells were then incubated with appropriate TRITC/FITC-conjugated secondary antibodies (diluted in 1% bovine serum albumin) or Bodipy (Invitrogen, Carlsbad, CA, USA) for neutral lipid staining for 1 h at 37 °C. For the detection of phase separation, macrophages seeded in glass-bottom cell culture dishes were treated with 3% hexanediol for 15 s after ox-LDL stimulation for 24 h, then incubated with BRD4 and MED1 primary antibodies. The cell nuclei were counterstained with DAPI. The sections were viewed under a confocal laser scanning microscope (LSM800, Zeiss, Oberkochen, Germany).
2.9. Immunoblot analysis
The whole-cell extracts were obtained in RIPA buffer containing protease and phosphatase inhibitors on ice. The nuclear and cytosolic fractions were obtained by the nuclear/cytosol fractionation kit (Beyotime, Beijing, China). Equal amounts of protein samples were separated in 6%–15% SDS-PAGE and transferred onto PVDF membranes (Millipore, Billerica, MA, USA) which was blocked in 5% skimmed milk for 2 h at RT. Then, the membranes were immunoblotted with specific primary antibodies at 4 °C overnight with slight agitation followed by HRP-conjugated secondary antibodies (Jackson Labs, USA) for 1 h at RT (Supporting Information Table S2). Finally, the immunoreactive bands were detected by the enhanced chemiluminescence method (Amersham Imager 600, GE, USA). According to the Guidelines for the use and interpretation of assays for monitoring autophagy18, we evaluated the expression of autophagy by comparing LC3 II with actin.
2.10. CO-immunoprecipitation
After different treatments, the cells were lysed in lysis buffer (Beyotime, Beijing, China) and placed on a low-speed rotating shaker for 30 min at 4 °C. After centrifuged at 12,000 × g for 10 min, 50 μL of the whole cell lysates was loaded as input sample and the remaining cell lysates were incubated with the indicated antibody with protein A/G agarose beads at 4 °C overnight. The next day, beads were washed with lysis buffer for 5 times and boiled at 95 °C for 10 min. All samples were subjected to SDS-PAGE separation and immunoblotted with the indicated antibodies.
2.11. Quantitative real-time PCR (qRT-PCR)
Total cellular RNAs were isolated using Trizol (Takara, Japan) according to the manufacturer's instructions and quantified by NanodropTM OneC (Thermo Fisher Scientific, Madison, USA), followed by reverse-transcribed into cDNA using HiScript® II Q RT SuperMix for qPCR (Vazyme, Jiangsu, China). qRT-PCR was carried out with cDNA in triplicate using AceQ® qPCR SYBR® Green Master Mix (Vazyme, China) on QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific, USA). Gene expression values were normalized against that of Actin. Data were analyzed with the 2ΔΔCt method. Structures of all primers used are listed in Supporting Information Table S3.
2.12. Oil Red O lipid staining
The lipid accumulation in cells was analyzed by ORO (Sigma, St. Louis, MO, USA) staining. Firstly, the FCs were fixed in 4% paraformaldehyde followed by dehydrated with 60% isopropanol for 2 min at RT. Next, filtered 0.3% ORO solution was used to stain the lipid in FCs for 10 min at RT. Finally, DAPI was used to counterstain cell nuclei. After washing, they were photographed under an optical microscope and quantified by Image-pro plus.
2.13. Dil-ox-LDL uptake assay
The uptake capacity of ox-LDL in FCs after different treatments were tracked by 1,1′-dioctadecyl-3,3,3′,3′-tetramethy-lindocarbocyanine perchlorate labeled ox-LDL (Dil-ox-LDL, Yiyuan Biotech, Guangzhou, China). The FCs were loaded with 20 μg/mL Dil-ox-LDL for 6 h at 37 °C in the dark and then counterstained with DAPI to track the nucleus. Finally, the FCs were detected under a fluorescence microscope at 514/565 nm.
2.14. Reactive oxygen species (ROS) assay
The generation of intracellular ROS was detected by DCFH-DA staining. Briefly, the cells after different treatments were firstly washed with RPMI 1640 and then incubated with DCFH-DA (Beyotime, Beijing, China) at 1:1000 ratio for 30 min at 37 °C in the dark. After washing, the fluorescent signal was measured by fluorescence microscope at 488/525 nm.
2.15. Chromatin immunoprecipitation (ChIP) and re-chromatin immunoprecipitation (Re-ChIP)
ChIP was performed as previously described19. Briefly, the cells grown in 15-cm dishes were cross-linked in 1% formaldehyde at RT for 10 min after different treatments, quenched in 0.125 mol/L glycine for 5 min and collected by centrifugation followed by sonicated in SDS lysis buffer to generate ∼500 bp chromatin fragments. The lysates were centrifugated at 4 °C for 10 min and 20 μL supernatant was used as input for quantitation. The left supernatant was diluted 10 times with dilution buffer and precleared in 20 μL protein A–agarose beads at 4 °C for 3 h. Subsequently, the supernatant was incubated with indicated antibodies at 4 °C with rotation overnight followed by incubating with 40 μL protein A–agarose beads at 4 °C for 2 h. The Txn stop buffer with 0.4 mg/mL glycogen and proteinase K (0.45 mg/mL, Roche, USA) was used to digest protein in immunoprecipitated DNA at 37 °C for 1 h. After that, DNA was isolated in phenol/chloroform/isoamyl alcohol (25:24:1) and precipitated in ethanol. For Re-ChIP, immunoprecipitated complexes were eluted with the elution buffer (1% SDS, 100 mmol/L NaCO3), diluted with the Re-ChIP buffer (1% Triton X-100, 2 mmol/L EDTA, 150 mmol/L NaCl, 20 mmol/L Tris pH 8.1), which was subjected to immunoprecipitation with BRD4 and MED1 antibody. Finally, the enrichment of immunoprecipitated material relative to input with gene-specific primers to the specified regions was determined by qRT-PCR (Supporting Information Table S4).
2.16. Chromatin conformation capture assay-qPCR (3C-qPCR)
After different treatments, the cells were cross-linked in 1% formaldehyde at RT for 10 min, quenched in 0.125 mol/L glycine for 5 min and centrifuged at 1000 × g for 10 min at 4 °C. Then, the pellets were lysed in lysis buffer for 10 min on ice and centrifuged to get nuclei, which were digested in restriction enzyme buffer containing 0.3% SDS for 1 h at 37 °C. Triton X-100 was added to the buffer for 1 h at 37 °C followed by 400 U XbaI incubation overnight to digest DNA. Next, the nuclei were incubated in 1.6% SDS for 25 min at 65 °C, suspended in ligation buffer containing 1% Triton X-100 for 1 h at 37 °C, then ligated by 100 U ligase for 4 h at 16 °C followed by 30 min at RT. Reaction was terminated, and de-crosslinking was performed with PK buffer (5 mmol/L EDTA, pH 8.0; 10 mmol/L Tris–HCl, pH 8.0; 0.5% SDS) at 65 °C overnight. After digested in 300 μg RNase for 45 min at 37 °C, DNA was isolated in phenol/chloroform/isoamyl alcohol (25:24:1) and precipitated in ethanol. 3C-DNA was amplified by qPCR. BAC 3C DNA was used to determine the PCR efficiency for each set of primers. The actin promoter was used as a negative control.
2.17. 1,6-Hexanediol disrupts BRD4 and MED1 phase separation
The experiments were performed as previously descried20. THP-1 cells seeded in glass-bottom cell culture dishes and treated with 3% hexanediol for 15 s after ox-LDL stimulation for 24 h. Immunofluorescence staining with BRD4 and MED1 and the cell nuclei were counterstained with DAPI. The sections were viewed under a confocal laser scanning microscope (LSM800, Zeiss, Oberkochen, Germany).
2.18. Statistical analysis
Results obtained are presented as mean ± standard error of mean (SEM) based on five independent experiments. Differences among the treatment groups were assessed with one-way ANOVA followed by Tukey's test for post hoc comparison when appropriate. Unpaired two-tailed Student's t test was used for comparisons between two groups. Statistical significance was defined as P value < 0.05. Statistical analyses were performed with GraphPad Prism 8.0 software.
3. Results
3.1. Cur restores FCs autophagy and attenuates lipid metabolic dysfunction by promoting TFEB nuclear translocation
To determine whether Cur protects macrophage-derived FCs against ox-LDL injury, we initially used the CCK-8 assay to detect cell viability. As shown in Fig. 1A, the cell viability of macrophages decreased significantly in the process of differentiation into FCs and was restored by Cur in a dose-dependent manner. At the same time, there was no obvious difference in the cell viability of FCs treated with 20 μmol/L Cur for 0, 6, 12, or 24 h (Fig. 1B). The cell viability of FCs decreased when the cells were treated with 20 μmol/L Cur for 48 h, which was consistent with previous report21. Therefore, FCs treated with 20 μmol/L Cur for 24 h were subsequently used.
Figure 1.

Curcumin (Cur) restores foam cells (FCs) autophagy and attenuates lipid metabolic dysfunction by promoting TFEB nuclear translocation. (A, B) Cell viability of FCs was measured by the cell counting kit 8 (CCK-8) assay. (C–E) Immunoblot analysis of LC3 II and P62 in FCs with different treatments. (F) Immunofluorescence analysis of LC3 (green) puncta formation in macrophages treated by oxidized low-density lipoprotein (ox-LDL) combined with Cur. Scale bar = 20 μm. (G) Immunoblot analysis of the autophagic flux in Cur-treated FCs pretreated with chloroquine (CQ). (H) Immunoblot analysis of TFEB subcellular distribution in FCs. (I) Immunoblot of mTOR/p-mTOR, AKT/p-AKT, calcineurin, and TFEB/p-TFEB in Cur-treated FCs. (J) Immunoblot analysis of the effect of mTORC1-specific agonist l-leucine (0.5 mmol/L) on the activity of mTOR and TFEB in Cur-treated FCs. (K, L) Immunoblot (K) and immunofluorescence (L) analysis of autophagy related proteins in Cur-treated FCs after siTFEB. Scale bar = 20 μm. (M, N) The subcellular distribution of TFEB in Cur-treated FCs combined with 3-MA and siATG5 was detected by immunoblot (M) and immunofluorescence (N). Scale bar = 20 μm. (O, P) The lipid accumulation (O) and the lipid uptake capacity (P) of Cur-treated FCs combined with siTFEB or siATG5 was evaluated by Oil red O (ORO) staining and 1,1′-dioctadecyl-3,3,3′,3′-tetramethy-lindocarbocyanine perchlorate labeled oxidized low-density lipoprotein (Dil-ox-LDL), separately. Scale bar = 100 μm. (Q) Immunofluorescence analysis of lipid catabolism in FCs through double fluorescence labeling with Bodipy (red) and LC3 (green). Scale bar = 20 μm. (R) Immunofluorescence analysis of lipid catabolism in Cur-treated FCs combined with siTFEB or siATG5 through double fluorescence labeling with Bodipy (red) and LC3 (green) at 24 h. Scale bar = 20 μm. Data are expressed as mean ± SEM, n = 5; ∗P < 0.05, ∗∗∗P < 0.001; ###P < 0.001.
Overloaded lipid accumulation usually causes autophagy deficiency, which exacerbates the pathogenesis of atherosclerosis3. To evaluate the effect of Cur on the autophagy of FCs, immunofluorescence and immunoblot analyses were performed to detect the expression levels of autophagy-specific markers. Upon autophagy induction, microtubule-associated protein 1 light chain 3-I (MAP1LC3/LC3 I) is converted to LC3 II through lipidation, promoting the formation of autophagosomes22. Meanwhile, sequestosome-1 (SQSTM1)/P62 targets damaged cytoplasmic material for lysosomal degradation. The results reveal that macrophage autophagy was downregulated by ox-LDL as evidenced by the reduced expression of LC3 II along with the increased P62 expression (Fig. 1E and Supporting Information Fig. S1C). However, they were reversed by Cur in concentration- and time-dependent manners (Fig. 1C and Fig. S1A, Fig. 1D and S1B). Similarly, compared with non-treated cells, immunofluorescence examination revealed that the number of LC3 puncta was decreased in FCs which was restored by Cur (Fig. 1F and Fig. S1D). Therefore, FCs incubated with 20 μmol/L Cur for 24 h is chosen as the optimal condition.
During the process of autophagy, the encapsulated contents as well as LC3 II and P62 in autophagosomes are usually degraded by fusing with lysosomes, which is called autophagic flux3. To examine the Cur-induced autophagy, FCs were pretreated with a lysosomal inhibitor (chloroquine, CQ, 20 μmol/L, 1 h) to prevent the fusion and subsequent proteolysis. Compared with Cur-treated FCs, the expression of LC3 II and P62 were further increased in Cur combined with CQ (Fig. 1G and Fig. S1E). There was no further significant increase of LC3 II and P62 in FCs in response to CQ, suggesting a defect in autophagic flux in FCs. The collective findings indicate that Cur restores the autophagy level of FCs by promoting the formation of both autophagosomes and autophagic flux.
To detect whether TFEB was involved in Cur-induced autophagy in FCs, we assessed the cellular distribution of TFEB. Cur did not affect total TFEB expression. While the nuclear levels of TFEB gradually increased and reached the most at 24 h after Cur, the cytosol levels of TFEB presented the opposite trend (Fig. 1H and Fig. S1F). In most cases, mTORC1-dependent phosphorylation controls TFEB nuclear export and inhibits autophagy in the normal condition13. TFEB stability can also be regulated by other kinases, such as AKT and calcineurin23,24. We confirmed that Cur did not affect the expression of calcineurin and phosphorylated AKT (p-AKT) in FCs. Nevertheless, phosphorylated mTOR (p-mTOR; Ser2448), as the indicator of mTORC1 activation, and its downstream effector p-S6K1 were downregulated in Cur-treated FCs along with p-TFEB (Fig. 1I and Fig. S1G, Fig. 1K and Fig. S1I). The decreased expression of p-TFEB caused by Cur was largely recovered when co-treated with the mTORC1 specific activator l-leucine (Fig. 1J and Fig. S1H), indicating that Cur effectively promoted TFEB nuclear translocation in FCs by inhibiting mTOR activity. Furthermore, TFEB deficiency resulting from treatment with small interfering RNA (siRNA) markedly inhibited the autophagy activation in Cur-treated FCs (Fig. 1K, Fig. S1I and Supporting Information Fig. S2A, Fig. 1L and Fig. S1J). However, autophagy inhibitor 3-methyladenine (3-MA) or autophagy deficiency via siATG5 had no effect on Cur-induced TFEB nuclear translocation in immunoblot analysis (Fig. 1M, Fig. S1K and Fig. S2B) and immunofluorescence (Fig. 1N). These data demonstrate that Cur restores FCs autophagy mainly by activating TFEB nuclear translocation.
Cur could inhibit the decrease of ATG5 mRNA in macrophages caused by ox-LDL, which was reversed by siTFEB (Fig. S2C). Moreover, Cur increased the binding of TFEB to the ATG5 promoter regions as observed by ChIP (Fig. S2D). However, there was no protein interaction between TFEB and ATG5 (Fig. S2E). To further confirm whether TFEB influences the open state of chromosomes, ChIP analysis was performed to detect the open (active) and condensed chromatin landmarks. Open chromatin is generally marked by H3K4me1 and H3K27ac25. Transcriptionally repressed (condensed) chromatin is generally marked by H3K9me3 and H3K27me326. Cur inhibited the binding of H3K9me3 and H3K27me3 to the ATG5 promoter regions and increased the binding of H3K27ac and H3K4me1 to the ATG5 promoter regions, which could be reversed by siTFEB (Fig. S2F). These results suggest that Cur could promote the open state of ATG5 gene located chromatin by increasing TFEB nuclear translocation.
Cur reportedly enhances lipid degradation in FCs. The underlying mechanism is unclear. Whether the effect of Cur on lipid degradation depends on TFEB-related autophagy was assessed. As shown in Fig. 1O and Fig. S1L, Cur apparently eliminated the lipid accumulation in FCs as stained by ORO. A binding assay using Dil-ox-LDL shows that Cur diminished the lipid uptake of FCs as evidenced by decreased red fluorescence intensity (Fig. 1P and Fig. S1M). At 24 h of treatment, Cur also resulted in an increased number of LC3-positive autophagosomes along with a decreased number of Bodipy-positive lipid. The findings suggest that Cur promoted the lipid breakdown of FCs (Fig. 1Q). All of the above changes induced by Cur in FCs were inhibited by siTFEB and siATG5 (Fig. 1O, P and R). Therefore, Cur decreased the lipid accumulation in FCs by decreasing lipid uptake and increasing lipid catabolism.
3.2. Cur reduces inflammation in FCs by inhibiting P300 and BRD4 to alter the chromatin microenvironment for inflammatory gene expression
Proinflammatory cytokines participate in immune-mediated plaque formation and vulnerability. As shown in Fig. 2A and B, the increased mRNA levels of inflammatory cytokines, including interleukin 1β (IL-1β), interleukin 6 (IL-6), interleukin 8 (IL-8), tumor necrosis factor α (TNF-α), and monocyte chemotactic protein 1 (MCP-1) induced by ox-LDL were markedly suppressed by Cur. The anti-inflammatory effects of Cur on FCs were also eliminated by TFEB and autophagy deficiency. These results indicate that Cur attenuated inflammation in FCs by enhancing TFEB-related autophagy.
Figure 2.

Cur reduces inflammation in FCs by inhibiting P300 and BRD4 to alter the chromatin environment for inflammatory gene expression. (A, B) Relative mRNA levels of inflammatory genes, including IL-1β, IL-6, IL-8, TNF-α, and MCP-1 in ox-LDL-treated macrophages combined with Cur in the presence of siTFEB, siATG5, 3-MA, or CQ. (C, D) The expressions of BRD2, BRD3, BRD4, and actin in ox-LDL-treated macrophages combined with Cur were measured by immunoblot (C) and immunofluorescence (D). Scale bar = 100 μm. (E, F) Relative mRNA levels of inflammatory genes in ox-LDL-treated macrophages combined with BRD4 inhibitor JQ-1 (1 μmol/L) or siBRD4 (E), BRD4 overexpression (oeBRD4) plasmid or P300 activator CTB (F). (G) Chromatin immunoprecipitation (ChIP) analysis of the enrichment of no-antibody (NA), P300, BRD2, BRD3, BRD4 and H3 at −1 kb from the promoter, at the promoter, and at +1 kb from the promoter of inflammatory genes (IL-8, IL-1β, TNF-α) in Cur-treated FCs. Data are expressed as mean ± SEM, n = 5. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001; ##P < 0.01, ###P < 0.001; $P < 0.05, $$P < 0.01, $$$P < 0.001.
The epigenetic reader protein BRD4 functions as a critical coactivator of the transactivation of inflammatory-genes during aeroallergen-induced inflammation and heart failure27,28. To detect whether BRD4 is involved in the activation of inflammation in FCs, we first examined the expressions of the BET family genes, including BRD2, BRD3, and BRD4. Immunoblot and immunofluorescence analyses revealed that BRD4 was increased in FCs, which could be markedly inhibited by Cur, while BRD2 and BRD3 did not change (Fig. 2C, D and Supporting Information Fig. S3A). Furthermore, ox-LDL-induced inflammation in macrophages was substantially diminished when BRD4 was suppressed by siRNA or the inhibitor JQ-1 (Fig. 2E and Fig. S2G). The findings indicate the involvement of BRD4 in the activation of inflammation in FCs. The histone acetyltransferases CBP/P300 can facilitate the recruitment of BRD4 to activate enhancers and to control cell identity gene induction in adipogenesis and myogenesis29. In the present study, the activity of P300 reflected by the expression of acetyl-histone 3 (acetyl-H3) was also increased by ox-LDL, which was inhibited by Cur (Fig. 2C). ox-LDL also effectively increased the acetylated levels of H3K9 and H3K27, rather than those of H3K14, H3K18, and H3K23. The increases were reversed by the P300 inhibitor C646 (Fig. S3B). These results suggest that the specific site of P300 acetylation on FC histones is H3K9 and H3K27. Furthermore, plasmid-mediated enforced overexpression of BRD4 (oeBRD4, Fig. S2H) and the P300 activator CTB markedly repressed the anti-inflammatory effect of Cur on FCs (Fig. 2F). The collective results indicate that BRD4 and P300 are positive regulators involved in the inflammation of FCs, whose activities could be inhibited by Cur.
ChIP of the promoter elements (−1 to +1 kb) of inflammatory genes was used to confirm the mechanism underlying the regulation of P300 and BRD4 on FC-mediated inflammation. We systematically used ChIP to probe for BRD2, BRD3, BRD4, P300, and H3 (positive control) and no-antibody control (NA). No significant changes were evident in NA, BRD2, and BRD3 concerning IL-8, IL-1β, TNF-α, IL-6, and MCP-1 in each treatment group (Fig. 2G and Fig. S3C). Strikingly, the promoter regions of inflammatory genes were enriched for BRD4 and P300 in ox-LDL-treated macrophages, which was inhibited by Cur. The control marker H3 was pulled down evenly. The positive effects of Cur on lipid catabolism in vitro were all reversed by BRD4 overexpression (Fig. S3D–S3F). The collective results implicate BRD4 and P300 were involved in the control of the chromatin status to transactivate inflammatory genes, thereby promoting lipid accumulation. The decreased expression of LC3 II in FCs was rescued by siBRD4, consistent with the previous observation that BRD4 is also a transcriptional repressor of autophagy (Fig. S3G).
3.3. Epigenetic landscape of P300–BRD4-dependent inflammatory genes in Cur treated FCs
We next analyzed whether the effect of BRD4 on inflammation in Cur-treated FCs relies on P300 activity. BRD4 deficiency in FCs reduced the enrichment of BRD4 at the promoter regions of these inflammatory genes, while having no effect on P300 (Fig. 3A and Supporting Information Fig. S4A). ChIP data of IL-8, IL-1β, IL-6, TNF-α and MCP-1 revealed that the inhibitory effect of Cur on the binding of BRD4 and P300 to the promoter regions of inflammatory genes in FCs was reversed by oeBRD4 and the P300 activator CTB (Fig. 3B and Fig. S4B, Fig. 3C and S4C). However, CTB and oeBRD4 did not affect the expression or the activity of each other (Fig. 3D). CTB promoted increased binding of BRD4 to the promoter regions of the inflammatory genes in the presence of ox-LDL (Fig. 3C). In contrast, oeBRD4 had no effect on the binding of P300 (Fig. 3B). Consistently, P300 cooperates with the downstream histone acetylation reader BRD4 to achieve transcriptional activation30. Thus, Cur could reduce inflammation in FCs by inhibiting the P300–BRD4 pathway.
Figure 3.

Epigenetic landscape of P300-BRD4 dependent inflammatory genes in Cur treated FCs. (A) ChIP analysis of the enrichment of P300, BRD2, BRD3, BRD4 and H3 at −1 kb from the promoter, at the promoter and at +1 kb from the promoter of inflammatory genes (IL-8, IL-1β) in ox-LDL-treated macrophages combined with JQ-1 or siBRD4. (B, C) ChIP analysis of the enrichment of P300, BRD2, BRD3, BRD4 and H3 at −1 kb from the promoter, at the promoter and at +1 kb from the promoter of inflammatory genes (IL-8, IL-1β) in ox-LDL-treated macrophages combined with Cur in the presence of oeBRD4 (B) or CTB (C). (D) Immunoblot analysis of the expressions of P300, BRD4, actin, acetyl-H3 and H3 in Cur-treated FCs in the presence of oeBRD4 and CTB. Data are expressed as mean ± SEM, n = 5. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001; #P < 0.05, ###P < 0.001; ns, no significance.
3.4. Cur inhibits the P300–BRD4 pathway by attenuating ROS generation through TFEB-activated autophagy
Cur is reportedly an inhibitor of P300. However, the detailed mechanism is unclear. Interestingly, we observed that Cur reduced BRD4 and P300 enrichment in TFEB oriented chromatin, which was abrogated by TFEB deficiency (Fig. 4A and Supporting Information Fig. S5A), as were the expressions of acetyl-H3 and BRD4 (Fig. 4B). The observations prompted us to consider how Cur decreases BRD4 expression in FCs and whether the regulation of TFEB on P300 activity relies on the activation of autophagy.
Figure 4.

Cur inhibits the P300–BRD4 pathway by attenuating ROS generation through TFEB-activated autophagy. (A) ChIP analysis of the enrichment of P300, BRD2, BRD3, BRD4 and H3 at −1 kb from the promoter, at the promoter and at +1 kb from the promoter regions of inflammatory genes (IL-8, IL-1β, TNF-α) in Cur-treated FCs combined with siTFEB. (B) Immunoblot analysis of the expressions of P300, BRD2, BRD3, BRD4, Actin, acetyl-H3, and H3 in Cur-treated FCs combined with siTFEB. (C, D) Immunoblot of acetyl-H3, H3, BRD4 and actin in FCs treated with siATG5 and Cur (C) or the reactive oxygen species (ROS) scavenger NAC (D). (E) The ROS level in FCs treated with Cur in the presence of siTFEB or siATG5 was detected using the DCFH-DA probe. Scale bar = 50 μm. Data are expressed as mean ± SEM, n = 5. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001; #P < 0.05, ##P < 0.01, ###P < 0.001.
Two protein degradation systems, including ubiquitin–proteasome and autophagy systems, have been described31. We first used the proteasome inhibitor MG132 to explore the degradation pathway of BRD4 in Cur-treated FCs. The decreased expression of BRD4 in Cur-treated FCs was not changed by MG132 (Fig. S5B). This result prompted us to explore whether Cur decreased BRD4 expression via autophagy. Scrutiny of the iLIR autophagy database revealed an LC3-interacting region (LIR) motif in BRD4, which ensures targeting of autophagy receptors to LC3 anchored in the phagophore membrane (Fig. S5C). The LIR sequence is presented in Fig. S5D. Use of the autophagy flux inhibitor CQ revealed that the decrease of BRD4 in Cur-treated FCs was restored by CQ (Fig. S5E), suggesting that the BRD4 was degraded by autophagy in Cur-treated FCs. The interaction between BRD4 and LC3 in Cur-treated FCs, which was further increased in the presence of CQ (Fig. S5F). Collectively, these findings confirm that Cur could degrade BRD4 in FCs by autophagy activation.
The inhibitory effect of Cur on P300 activity was attenuated in the presence of siATG5 (Fig. 4C), indicating that Cur can decrease P300 activity via TFEB-mediated autophagy activation. ROS could upregulate P300 activity-dependent CD36 expression and promote FCs formation32. Increased activation of autophagy can effectively decrease ROS generation through mitophagy33. We observed that the increased P300 activity mediated by ox-LDL was blocked by the ROS scavenger N-acetyl-l-cysteine (NAC) (Fig. 4D). This finding suggests the involvement of ROS in P300 activation during FC formation, which is consistent with previous studies. Moreover, Cur substantially inhibited the generation of ROS as indicated by DCFH-DA fluorescence staining, which was reversed by siTFEB or siATG5 (Fig. 4E). Over the course of exploring the basis of the ROS-mediated increase of P300 activity in FCs, we observed that phosphorylation of non-receptor tyrosine kinase BTK increased in ox-LDL-treated macrophages, which was inhibited by the ROS scavenger NAC (Fig. S5G). Furthermore, the increased acetylation of total histone in ox-LDL-treated macrophages was inhibited by BTK antisense oligonucleotides (ASO, Fig. S5H). These results confirm that ox-LDL resulted in ROS production, which in turn activated BTK, followed by increased P300 activity in macrophages.
The collective data suggest that ROS is an activator of P300 that promotes the binding of BRD4 to the promoters of inflammatory genes. This binding can be inhibited by TFEB-induced autophagy upon treatment with Cur.
3.5. Cur regulates chromatin open state in FCs in a BRD4-dependent manner
For the well-known proinflammatory cytokines IL-8 and IL-1β, the putative super-enhancer (SE) in blood predicted by the comprehensive human SE database was denoted as a horizontal line in blue in Fig. 5A and Supporting Information Fig. S6A34. In this putative SE regions, strong signals of active SE marked by high levels of H3K27ac were found to be located in proximal to the IL-8 and IL-1β promoters. The corresponding ChIP-seq data from three human blood cell lines (K56235, CD14-positive monocyte36 and THP-137) were displayed using the Integrative Genomics Viewer38 (Fig. 5A and Fig. S6A). To confirm the interaction between the promoters of IL-8 and these potential enhancer clusters as well as IL-1β, we performed a quantitative analysis of chromosome conformation capture assays (3C-qPCR). The primers for promoter and enhancer locus 2 in IL-8 and locus 4 in IL-1β showed the greatest interaction strength and strongest response to Cur, JQ-1, siBRD4, and P300 inhibitor C646 (Fig. 5B and Fig. S6B). An SE that strongly enriches for the H3K27 acetylation (H3K27ac), BRD4, and the mediator complex 1 (MED1) is comprised of clusters of transcriptional enhancers39. The IL-8 and IL-1β promoters were strongly enriched for BRD4, MED1, and H3K27ac in FCs, which were inhibited by JQ-1, siBRD4, or Cur (Fig. 5C and Fig. S6C). Re-ChIP assay results demonstrate that BRD4 and MED1 occupied the same DNA element of IL-8 and IL-1β in FCs, which was inhibited by Cur or siBRD4 (Fig. 5D and Fig. S6D). ox-LDL upregulated the expressions of BRD4, MED1, H3K27ac and H3K4me1 and decreased H3K27me3 expression. The findings suggest that ox-LDL contributed to the opening state of chromatin, which was reversed by Cur or siBRD4 (Fig. 5E). The collective findings indicate that ox-LDL promotes the enrichments of BRD4 and MED1 in the promoter regions of inflammation genes to form SE and thereby enhance the transactivation of inflammation, which can be inhibited by Cur.
Figure 5.

Cur regulates the chromatin open state in FCs in a BRD4-dependent manner. (A) ChIP-seq peaks (H3K27ac levels) and putative super-enhancer (SE) location (highlighted by the red box) around IL-8. Cell lines are marked with different colors, with K562 in blue, CD14-positive monocytes in light blue, and THP-1 in green. The putative SE region is marked by the horizontal blue line. (B) 3C-qPCR analysis of the long-distance interactions between the IL-8 promoter and seven enhancer loci in FCs after different treatments (x-axis means seven enhancer loci of IL-8 promoter). (C) ChIP analysis of the enrichments of BRD4, MED1, and H3K27ac at the SE regions of IL-8 in Cur-treated FCs combined with JQ-1, siBRD4, as well as H3. (D) Re-ChIP was performed with the 1st round pull-down of MED1 antibody and 2nd round pull-down of the BRD4 antibody. (E) Immunoblot analysis of the expressions of BRD4, MED1, H3, and chromatin active markers (H3K27ac, H3K4me1 and H3K27me3) in Cur-treated FCs combined with JQ-1 or siBRD4. Data are expressed as mean ± SEM, n = 5. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001; ns, no significance.
3.6. Cur regulates BRD4-dependent SE associated with phase separation on inflammatory genes
Based on the latest research on phase separation published in Science20, we investigated the sensitivity of BRD4 and MED1 to 1,6-hexanediol, a compound known to disrupt liquid-like condensates. The architectural features of BRD4 and MED1 that are prone to phase separation are shown in Fig. 6A. Compared with macrophages, a dramatic increase was evident in the number of BRD4 and MED1 puncta in the nuclei of FCs. The increase was reversed by 1,6-hexanediol (Fig. 6B). 1,6-Hexanediol inhibited the greatest interaction strength between promoter and enhancer locus 2 in IL-8 and locus 4 in IL-1β (Fig. 6C and Fig. S6E), the binding of BRD4 and MED1 to the SE region of IL-8 and IL-1β (Fig. 6D and Fig. S6F), and the upregulation of IL-8 and IL-1β mRNA (Fig. 6E and Fig. S6G) in FCs. The increased BRD4 and MED1 puncta on macrophages treated with the phase separation activator PEG were also inhibited by Cur or the P300 inhibitor C646 (Fig. 6F and J). PEG increased the interaction between the IL-8 promoter and the enhancer locus 2, as well as the IL-1β promoter and the enhancer locus 4 (Fig. 6G and Fig. S6H). PEG also increased BRD4 and MED1 binding to the SE region of IL-8 and IL-1β (Fig. 6H and K, Fig. S6I and S6K). These reactions were inhibited by both Cur and C646, which were evident at the mRNA level for IL-8 and IL-1β (Fig. 6I and L, Fig. S6J and S6L). The increased BRD4 and MED1 puncta induced by ox-LDL was significantly inhibited by Cur (Fig. 6M). The collective findings indicate that Cur regulates BRD4-dependent SE associated with phase separation on inflammatory genes.
Figure 6.

Cur regulates BRD4-dependent SE associated with phase separation on inflammatory genes. (A) A color-coded schematic representation of the aligned amino acid sequences and corresponding prion-like domain disorder propensity plots for BRD4 and MED1 generated using the prion-like amino acid composition pool. The gray line indicates the low complexity region. (B) Immunofluorescence analysis of the puncta of BRD4 (green) and MED1 (green) in ox-LDL-treated macrophages exposed to 3% 1,6-hexanediol for 15 s. Scale bar = 20 μm. (C) 3C-qPCR analysis of the long–distance interactions between the IL-8 promoter and seven enhancer loci (the positions of the different loci are described in Fig. 5A) in FCs treated with 1,6-hexanediol (x-axis means seven enhancer loci of IL-8 promoter). (D) ChIP analysis of the enrichment of BRD4 and MED1 at the SE regions of IL-8 in ox-LDL-treated macrophages combined with 3% 1,6-hexanediol, as well as H3. (E) Relative mRNA level of IL-8 in ox-LDL-treated macrophages exposed to 3% 1,6-hexanediol. (F–L) Macrophages were treated with PEG along with Cur or C646. We detected the puncta of BRD4 (green) and MED1 (green) by immunofluorescence (F, J), the long–distance interactions between IL-8 promoter and seven enhancer loci in macrophages with different treatments by 3C-qPCR (x-axis means seven enhancer loci of IL-8 promoter) (G), the enrichment of BRD4 and MED1 at the SE regions of IL-8 by ChIP assay (H, K), and the mRNA level of IL-8 by qRT-PCR (I, L). Scale bar = 20 μm. (M) Immunofluorescence analysis of the puncta of BRD4 (green) and MED1 (green) in ox-LDL-treated macrophages combined with Cur. Scale bar = 20 μm. Data are expressed as mean ± SEM, n = 5. ∗∗P < 0.01, ∗∗∗P < 0.001.
3.7. Specific ectopic expression of BRD4 and TFEB in macrophages abolishes the protective effect of Cur in Apoe–/– mice
Compared with Apoe–/– mice fed with normal chow (NC), the expression of BRD4 of aortas was increased in Apoe–/– mice fed with a HFD (Fig. 7A). Macrophage-specific Brd4 overexpressing Apoe–/– mice were constructed through bone marrow transplantation. C57BL/6 bone marrow cells transduced with either pLVCD68-Brd4 overexpression (pLVCD68-Brd4 oe) or pLVCD68 vector were transplanted into the Apoe–/– mice. After consuming the HFD for 16 weeks, atherosclerotic plaques were assessed. There was no difference in survival rate among different groups of mice (Supporting Information Fig. S7A). We confirmed that the mRNA level of Brd4 increased markedly in mice transplanted with pLVCD68-Brd4 oe-transduced BMCs and was partially decreased in the presence of Cur (Fig. 7B). Compared with the mice that received pLVCD68 + DMSO, the lesion area and lipid content of aortas from the mice that received pLVCD68 + Cur significantly decreased as assessed by morphometric analyses of lesion area in the aorta en face (Fig. 7C, Fig. S7B) and cross-sections of the aortic arch (Fig. 7D, Fig. S7C). Furthermore, compared with mice treated with pLVCD68 + DMSO, there was a trend toward increased plaque collagen content measured by Sirius red and Masson staining in plaques (Fig. 7D, Fig. S7C), and decreased inflammation of arteries from mice treated with pLVCD68 + Cur (Fig. 7E). No difference in blood pressure was evident in all groups of mice (Fig. S7D). However, the protective effect of Cur on atherosclerosis was significantly inhibited in mice transplanted with pLVCD68-Brd4 oe-transduced BMCs (Fig. 7C–E). Cur effectively decreased the plasma lipids in HFD-fed Apoe–/– mice that had received a bone marrow transplant from pLVCD68 macrophages. The decrease was reversed by macrophage-specific Brd4 overexpression (Fig. S7E).
Figure 7.

Specific ectopic expression of BRD4 and TFEB in macrophages reduces the protective effect of Cur in Apoe–/– mice. (A) Immunoblot analysis of BRD4 in aortas from Apoe–/– mice fed with the normal chow (NC) and high-fat diet (HFD). All of the Apoe–/– mice were fed with the HFD throughout the experiment. (B) Relative Brd4 mRNA level in Apoe–/– mice bone marrow cells (BMCs), which was transplanted with pLVCD68-Brd4 overexpression transduced BMCs combined with Cur. (C) Representative images of ORO-stained aortic lesion in Apoe–/– mice treated as in (B). (D) Representative images of ORO, hematoxylin and eosin, Sirius red and Masson's trichrome staining of cross-sections of the aorta from mice treated as in (B). Scale bar = 50 μm. (E) Relative mRNA levels of inflammatory genes of aortas from Apoe–/– mice treated as in (B). (F) Immunoblot analysis of TFEB in Apoe–/– mice fed with a HFD and treated with Cur. (G) Relative Tfeb mRNA level in Apoe–/– mice BMCs, which was transplanted with pLVCD68-Tfeb knock-out (TFEB KO) transduced BMCs combined with Cur. (H) Representative images of ORO-stained aortic lesion in Apoe–/– mice treated as in (G). (I) Representative images of ORO, hematoxylin and eosin, Sirius red and Masson's trichrome staining of cross-sections of the aorta from mice treated as in (G). Scale bar = 50 μm. (J) Relative mRNA levels of inflammatory genes of the aorta from mice treated as in (G). (K) Dihydroethidium (DHE) staining was used to detect the ROS content of aorta from mice treated as in (G). Scale bar = 20 μm. (L) Immunoblot analysis of LC3 II, P62, BRD4, and Acetyl-H3 in aorta from mice treated as in (G). Data are expressed as mean ± SEM, n = 5. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001; ###P < 0.001.
To further demonstrate the role of Tfeb in the protective effect of Cur on atherosclerosis, we first assessed the Tfeb activity. In aortas of HFD-fed Apoe–/– mice, the phosphorylation level of TFEB was decreased by Cur treatment, while the total TFEB protein expression did not change between groups (Fig. 7F and Fig. S7F). We constructed the macrophage-specific Tfeb knock-out (KO) Apoe–/– mice by bone marrow transplantation. qRT-PCR confirmed the marked decrease in the mRNA level of Tfeb in mice transplanted with pLVCD68-Tfeb-KO-transduced BMCs (Fig. 7G). There was no difference in survival rate in all groups (Fig. S7G). The protective effects of Cur on the lesion area, lipid content, plaque collagen, inflammation, and plasma lipids were all inhibited in mice transplanted with pLVCD68-Tfeb-KO-transduced BMCs (Fig. 7H–J, Fig. S7H, S7I and S7K). No difference was evident in blood pressure in all groups of mice (Fig. S7J). The increased ROS generation and LC3 expression, and decreased expression of BRD4 in Cur-treated Apoe–/– mice were also reversed in macrophage-specific Tfeb KO mice (Fig. 7K and L, Fig. S7L). We also confirmed that the decreased P300 activity in the presence of Cur was also reversed in mice with macrophage-specific Tfeb KO (Fig. 7L and Fig. S7L). The collective findings demonstrate that Cur could effectively enhance autophagy in FCs via TFEB and decrease P300 activity and BRD4 expression.
4. Discussion
In this study, we observed that ox-LDL led to abnormal crosstalk between autophagy and inflammation through the TFEB–P300–BRD4 axis. A proposed model is presented in Fig. 8. We also shed light on the mechanism of how Cur benefits atherosclerosis by promoting autophagy and thereby inhibiting inflammation in the FCs. These findings implicate Cur as a potential therapeutic agent for atherosclerosis with several novel drug targets identified.
Figure 8.

Proposed model of the autophagy enhanced by Cur ameliorates inflammation in atherogenesis via the TFEB–P300–BRD4 axis. ox-LDL leads to the autophagy deficiency and ROS production in macrophages. This resulted in the activation of P300 which promotes increased binding of BRD4 to the promoter regions of inflammatory genes. This consequently contributes to inflammation which leads to the formation of FCs. Cur can significantly increase autophagy activity in FCs by promoting TFEB nuclear translocation through the inhibition of mTOR. This optimizes lipid catabolism. Autophagy can effectively attenuate the ROS level and P300 activity. The decreased acetylated histone resulting from the suppression of P300 lessens the attraction of BRD4 to the promoter regions of inflammatory genes in FCs and finally attenuates the inflammation. Especially, Cur inhibits the formation of BRD4-dependent SE of the inflammatory genes associated with phase separation.
Our results show that Cur markedly ameliorated autophagy deficiency in FCs and increased the autophagic flux. Therefore, Cur has a protective role in atherosclerosis. TFEB is a novel master molecule of the autophagy–lysosome pathway. Normally, TFEB is localized on the lysosome membrane in an inactivated form via phosphorylation by mTORC113. Interestingly, Cur has been reported to ameliorate intestinal barrier injury via AMPK–TFEB-dependent mitophagy40. The Cur derivative Cur20 attenuates cerebral ischemic injury via HIF-1alpha/VEGF/TFEB-dependent angiogenesis41. Another Cur analog could induce autophagy by directly binding to and activating TFEB in an mTOR-independent manner to treat neurodegenerative diseases42. However, the TFEB response upon Cur treatment in FCs has never been reported. Here, we demonstrate that Cur induced the time-dependent redistribution of TFEB in the nuclei of FCs and corresponding activation of autophagy. These events were completely inhibited by silencing TFEB. Pharmacological inhibition of autophagy had little effect on TFEB nuclear translocation, implicating TFEB was the upstream regulator of autophagy. The expression of p-mTOR (Ser2448) in FCs was downregulated by Cur and was partly reversed by TFEB silence, while other previously reported TFEB activity regulators like AKT and calcineurin were not affected in FCs24. The collective findings support the viewpoint that Cur significantly improves the autophagy of FCs by promoting mTORC1-dependent TFEB nuclear translocation. Furthermore, Cur could inhibit the binding of H3K9me3 and H3K27me3 to the ATG5 promoter region and increase the H3K4me1 and H3K27ac binding of to the ATG5 promoter region. These events can be reversed by siTFEB. These results suggest that Cur could influence the open state of chromosomes by increasing TFEB nuclear translocation. In addition, Cur led to an increase in the content of autophagosomes and decreased Bodipy+ lipid droplets in FCs, as well as the reduced lipid uptake and accumulation. All of the above events were reversed by TFEB or ATG5 deficiency, suggesting that TFEB nuclear translocation and subsequently autophagy activation are crucial in Cur-mediated promotion of lipid degradation.
Inflammation is causally related to atherogenesis43. TFEB overexpression has been reported to delay atherosclerotic plaques by an anti-inflammatory effect through the activation of autophagy in macrophages and endothelial cells by an underlying mechanism that is still unclear5,44. We observed that the inhibitory effect of Cur on the elevated mRNA levels of inflammatory cytokines mRNA levels (IL-1β, IL-6, IL-8, TNF-α and MCP-1) in FCs were partially reversed by the silence of TFEB or ATG5. As a potential natural anti-inflammation product, Cur can inhibit the inflammation by fine-tuning the TFEB- and ATG5-related inflammatory responses.
Recently, epigenetic regulation has been found to play a critical role in gene expression via post-translational modification of core histones in the genome, resulting in alternative chromatin configuration and altered gene transcription45. The expression of inflammatory genes is tightly controlled by chromatin “readers” that specifically bind acetylated histones and provide a scaffold for sequence-specific transcription factors that comprise the transcriptional activation complex46. Moreover, BET proteins as “readers” of histone acetylation have been demonstrated to mediate the inflammatory response by remodeling the communication between enhancer and RNA polymerase in heart failure and pulmonary artery hypertension15,27. Whether BET families take part in the epigenetic regulation of inflammation in FCs has never been reported. Amazingly, we found that the expression of BRD4, but not BRD2 and BRD3, is dramatically increased in FCs. These events can be prevented by Cur. Consistent with the report that BRD4 acts as a transcriptional repressor of autophagy, the inhibition of autophagy in FCs was partially rescued by siBRD447. The anti-inflammation effect of Cur in FCs could be reversed by BRD4 overexpression. Additionally, we confirmed that either the overexpression of Brd4 or the knock-out of Tfeb in macrophage exacerbated atherosclerosis in Apoe–/– mice and abolished the anti-atherosclerotic effects of Cur. The decreased ROS generation, increased expression of LC3, and decreased H3 activation upon Cur treatment were all inhibited by macrophage-specific Tfeb KO. Mechanically, ChIP analysis showed that under ox-LDL stimulation, the aberrant occupancy of the promoters of inflammatory genes by BRD4 was reversed by Cur and attenuated by TFEB silencing. The collective findings indicate that activated TFEB is a negative regulator in the upstream of BRD4 in Cur-treated FCs. In assessing how Cur decreases BRD4 expression in FCs, we found that the Cur mediated decreases of BRD4 was reversed by inhibition of autophagic flux, rather than by a proteasome inhibitor. The finding was supported by the prediction that BRD4 harbors the LC3-interacting region (LIR) motif based on the iLIR autophagy database. Interaction between BRD4 and LC3 was demonstrated in Cur-treated FCs. The interaction was further increased in the presence of CQ. The collective findings confirmed that Cur can degrade BRD4 in FCs by activating autophagy.
Histone acetylation is usually achieved by histone acetyltransferases including P300. The latter is a major enzyme that mediates the function of BRD4. The role of P300 in atherosclerosis is controversial. One study reported that P300 promotes the atherogenesis by activating vascular smooth muscle cells proinflammatory genes48. However, another study showed that P300 rescues the base excision repair enzyme activity to delay the plaque development by decreasing oxidative damage49. The results of our present study show that P300 activity was upregulated in FCs as evidenced by the increase of acetylated histone 3. This event was reversed by Cur, which is consistent with the reported that Cur is an inhibitor of P30050. Cur was reported to inhibit the binding efficiency of histones and acetyl CoA to P300 via binding with P300/CBP at a specific site51. However, we proposed that Cur could inhibit P300 in another way. Here, P300 activity in FCs was downregulated in the presence of Cur, the same as the effect of the ROS broad scavenger NAC, in accordance with the report that ROS can increase P300 activity32. This reminds us that ROS is the upstream activator of P300. In exploring whether TFEB activation caused by Cur regulated P300 through ROS, we observed that ROS generation in FCs was inhibited by Cur. This event was reversed by TFEB or ATG5 deficiency. These findings indicate that Cur can inhibit ROS generation by activating autophagy due to TFEB nuclear translocation, which in turn reduces P300 activity. We confirmed that ROS increases the P300 activity by increasing BTK phosphorylation. Although P300 is widely reported to be a negative regulator of autophagy, we confirmed that autophagy can also negatively regulate the activity of P300. This finding broadens the understanding of the interplay between P300 and autophagy.
The SE is composed of clusters of transcriptional enhancers, which are strongly enriched for the binding of MED1, BRD4, H3K27ac and other transcriptional coactivators to genes essential for certain cellular function20. Data from bioinformation databases reveal an SE region at the promoter of inflammatory genes, which was furtherly enhanced with ox-LDL treatment and inhibited by Cur. Thus, SE could be a new target for Cur. Phase separation is a physicochemical process by which a homogeneous liquid solution of macromolecular components separates into a dense phase and a dilute phase52. Biomolecular condensates produced by LLPS allow rapid movement of components into and within the dense phase and exhibit properties of liquid droplets, such as fusion and fission. The aberrant forms of phase separation are now thought to mediate many complex human diseases, such as neurodegenerative diseases, cancers, and infectious diseases53. Recently, BRD4 has been reported to be involved in the formation of LLPS at SEs with coactivator MED1 to control gene expression20. In the present study, BRD4 was upregulated in macrophages by treatment with the classical stimulator ox-LDL, which promoted the expression of inflammatory genes. Notably, LLPS in FCs was associated with BRD4. All these findings implicate FCs as a new model to develop new drugs targeting phase separation and BRD4, with the aim of curing atherosclerosis. Lipoamide and its related compound lipoic acid as the first identified drug could inhibit the phase transition of FUS in amyotrophic lateral sclerosis54. Therefore, Cur could be a potential drug targeting phase separation due to the similar antioxidative effect with lipoic acid.
The discovery of novel functions of natural compounds by screening new drug targets for various diseases has become a popular approach. Compounds including trehalose, resveratrol and adipoRon are protective for CVDs based on the activation of TFEB10,55,56. The present findings confirm that Cur can restore the autophagy and autophagic flux in FCs damaged by ox-LDL by regulating mTORC1-dependent TFEB nuclear translocation. Thus, TFEB could be a potentially valuable drug target for CVDs. Cur decreases histone acetylation mediated by P300 by inhibiting ROS generation by autophagy activation, in turn reducing the occupancy of BRD4 on the promotors of inflammatory genes via various epigenetic mechanisms. This results in the downregulation of inflammation. Our collective findings suggest a novel mechanism of the protective effect of Cur on FCs, which may be a potential therapeutic approach for the treatment of atherosclerosis related CVDs.
5. Conclusions
Cur significantly increased the activation of autophagy in FCs by promoting TFEB nuclear translocation. This occurred due to the inhibition of mTOR and optimized lipid catabolism. Autophagy can effectively attenuate the generation of ROS. The decreased acetylated histone as the result of the suppression of ROS on P300 activity would attract less BRD4 to the promoter regions of inflammatory genes in FCs. Ultimately, inflammation would be attenuated. Especially, Cur regulated BRD4-dependent SE was associated with LLPS on regulatory regions of inflammatory genes. The collective evidence in the present study implicates that Cur as a potential therapeutic agent for atherosclerosis. Several novel drug targets were identified.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (31771334, 81970428 and 81800385); the Key Research Plan of the National Natural Science Foundation of China (81820108002); the Major Research Plan of the National Natural Science Foundation of China (91649125 and 91639204); University Natural Science Research of Jiangsu Province (18KJB310008, China); Jiangsu Province Health and Family Planning Commission Scientific Research Project (H2017011, China); Jiangsu Provincial Medical Youth Talent (QNRC2016432, China); Top Talents Project Foundation of “Six-One Project” for High-level Health Talents of Jiangsu Province (LGY2020055, China); and Technology Development Foundation of Nanjing Medical University (2017NJMUZD020, China). This research was also supported by the program of special professor of Jiangsu Province, the program of the special medical experts of Jiangsu Province and the program of innovation and entrepreneurship team plan of Jiangsu Province, China.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2021.12.014.
Contributor Information
Bingjian Wang, Email: wofcardio96@126.com.
Yong Ji, Email: yongji@njmu.edu.cn.
Hongshan Chen, Email: hongshanchen@njmu.edu.cn.
Author contributions
Hongshan Chen, Yong Ji, Bingjian Wang, Xuesong Li and Ruigong Zhu, conceived and designed the study. Xuesong Li, Ruigong Zhu and Hong Jiang and Xian Ji performed the in vitro experiments. Xuan Wu, Jiajing Chen, Quanwen Yin and Jiaming Gu performed the in vivo experiments. Bioinformatics analysis was performed by Hong Jiang. All authors analyzed the data. Xuesong Li, Ruigong Zhu, Hong Jiang and Hongshan Chen wrote the manuscript. Yong Ji modified the language of this article.
Conflicts of interest
The authors have no conflicts of interest to declare.
Appendix A. Supporting information
The following is the Supplementary data to this article:
References
- 1.Benjamin E.J., Virani S.S., Callaway C.W., Chamberlain A.M., Chang A.R., Cheng S., et al. Heart disease and stroke statistics—2018 update: a report from the American Heart Association. Circulation. 2018;137:e67–492. doi: 10.1161/CIR.0000000000000558. [DOI] [PubMed] [Google Scholar]
- 2.Khoury M.K., Yang H., Liu B. Macrophage biology in cardiovascular diseases. Arterioscler Thromb Vasc Biol. 2021;41:e77–81. doi: 10.1161/ATVBAHA.120.313584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bravo-San Pedro J.M., Kroemer G., Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res. 2017;120:1812–1824. doi: 10.1161/CIRCRESAHA.117.311082. [DOI] [PubMed] [Google Scholar]
- 4.Brophy M.L., Dong Y., Wu H., Rahman H.N., Song K., Chen H. Eating the dead to keep atherosclerosis at bay. Front Cardiovasc Med. 2017;4:2. doi: 10.3389/fcvm.2017.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Evans T.D., Jeong S.J., Zhang X., Sergin I., Razani B. TFEB and trehalose drive the macrophage autophagy–lysosome system to protect against atherosclerosis. Autophagy. 2018;14:724–726. doi: 10.1080/15548627.2018.1434373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu X., Tang Y., Cui Y., Zhang H., Zhang D. Autophagy is associated with cell fate in the process of macrophage-derived foam cells formation and progress. J Biomed Sci. 2016;23:57. doi: 10.1186/s12929-016-0274-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan X.M., Li W., Brunk U.T., Dalen H., Chang Y.H., Sevanian A. Lysosomal destabilization during macrophage damage induced by cholesterol oxidation products. Free Radic Biol Med. 2000;28:208–218. doi: 10.1016/s0891-5849(99)00220-8. [DOI] [PubMed] [Google Scholar]
- 8.Martinet W., De Loof H., De Meyer G.R. mTOR inhibition: a promising strategy for stabilization of atherosclerotic plaques. Atherosclerosis. 2014;233:601–607. doi: 10.1016/j.atherosclerosis.2014.01.040. [DOI] [PubMed] [Google Scholar]
- 9.Napolitano G., Ballabio A. TFEB at a glance. J Cell Sci. 2016;129:2475–2481. doi: 10.1242/jcs.146365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sergin I., Evans T.D., Zhang X., Bhattacharya S., Stokes C.J., Song E., et al. Exploiting macrophage autophagy–lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun. 2017;8:15750. doi: 10.1038/ncomms15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel S.S., Acharya A., Ray R.S., Agrawal R., Raghuwanshi R., Jain P. Cellular and molecular mechanisms of curcumin in prevention and treatment of disease. Crit Rev Food Sci Nutr. 2020;60:887–939. doi: 10.1080/10408398.2018.1552244. [DOI] [PubMed] [Google Scholar]
- 12.Han J., Pan X.Y., Xu Y., Xiao Y., An Y., Tie L., et al. Curcumin induces autophagy to protect vascular endothelial cell survival from oxidative stress damage. Autophagy. 2012;8:812–825. doi: 10.4161/auto.19471. [DOI] [PubMed] [Google Scholar]
- 13.Napolitano G., Esposito A., Choi H., Matarese M., Benedetti V., Di Malta C., et al. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat Commun. 2018;9:3312. doi: 10.1038/s41467-018-05862-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang N., Wu R., Tang D., Kang R. The BET family in immunity and disease. Signal Transduct Target Ther. 2021;6:23. doi: 10.1038/s41392-020-00384-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meloche J., Lampron M.C., Nadeau V., Maltais M., Potus F., Lambert C., et al. Implication of inflammation and epigenetic readers in coronary artery remodeling in patients with pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol. 2017;37:1513–1523. doi: 10.1161/ATVBAHA.117.309156. [DOI] [PubMed] [Google Scholar]
- 16.Cully M. Cardiovascular disease: BET inhibitor attenuates heart failure. Nat Rev Drug Discov. 2017;16:453. doi: 10.1038/nrd.2017.125. [DOI] [PubMed] [Google Scholar]
- 17.Wang Q., Sun Y., Li T., Liu L., Zhao Y., Li L., et al. Function of BRD4 in the pathogenesis of high glucoseinduced cardiac hypertrophy. Mol Med Rep. 2019;19:499–507. doi: 10.3892/mmr.2018.9681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J., Abeliovich H., Acevedo Arozena A., et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H., Ruiz P.D., Novikov L., Casill A.D., Park J.W., Gamble M.J. MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation. Nat Struct Mol Biol. 2014;21:981–989. doi: 10.1038/nsmb.2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sabari B.R., Dall'Agnese A., Boija A., Klein I.A., Coffey E.L., Shrinivas K., et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018;361:eaar3958. doi: 10.1126/science.aar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin X.L., Liu M.H., Hu H.J., Feng H.R., Fan X.J., Zou W.W., et al. Curcumin enhanced cholesterol efflux by upregulating ABCA1 expression through AMPK–SIRT1–LXRalpha signaling in THP-1 macrophage-derived foam cells. DNA Cell Biol. 2015;34:561–572. doi: 10.1089/dna.2015.2866. [DOI] [PubMed] [Google Scholar]
- 22.Dikic I., Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–364. doi: 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 23.Pi H., Li M., Zou L., Yang M., Deng P., Fan T., et al. AKT inhibition-mediated dephosphorylation of TFE3 promotes overactive autophagy independent of MTORC1 in cadmium-exposed bone mesenchymal stem cells. Autophagy. 2019;15:565–582. doi: 10.1080/15548627.2018.1531198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medina D.L., Di Paola S., Peluso I., Armani A., De Stefani D., Venditti R., et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–299. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bysani M., Agren R., Davegardh C., Volkov P., Ronn T., Unneberg P., et al. ATAC-seq reveals alterations in open chromatin in pancreatic islets from subjects with type 2 diabetes. Sci Rep. 2019;9:7785. doi: 10.1038/s41598-019-44076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Risca V.I., Denny S.K., Straight A.F., Greenleaf W.J. Variable chromatin structure revealed by in situ spatially correlated DNA cleavage mapping. Nature. 2017;541:237–241. doi: 10.1038/nature20781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duan Q., McMahon S., Anand P., Shah H., Thomas S., Salunga H.T., et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aah5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian B., Hosoki K., Liu Z., Yang J., Zhao Y., Sun H., et al. Mucosal bromodomain-containing protein 4 mediates aeroallergen-induced inflammation and remodeling. J Allergy Clin Immunol. 2019;143:1380–1394.e9. doi: 10.1016/j.jaci.2018.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J.E., Park Y.K., Park S., Jang Y., Waring N., Dey A., et al. BRD4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat Commun. 2017;8:2217. doi: 10.1038/s41467-017-02403-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan S.H., Tang Y., Miao L., Darwich-Codore H., Vejnar C.E., Beaudoin J.D., et al. BRD4 and P300 confer transcriptional competency during zygotic genome activation. Dev Cell. 2019;49:867–881.e8. doi: 10.1016/j.devcel.2019.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dikic I. Proteasomal and autophagic degradation systems. Annu Rev Biochem. 2017;86:193–224. doi: 10.1146/annurev-biochem-061516-044908. [DOI] [PubMed] [Google Scholar]
- 32.Kotla S., Rao G.N. Reactive oxygen species (ROS) mediate P300-dependent STAT1 protein interaction with peroxisome proliferator-activated receptor (PPAR)-gamma in CD36 protein expression and foam cell formation. J Biol Chem. 2015;290:30306–30320. doi: 10.1074/jbc.M115.686865. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Lin Q., Li S., Jiang N., Shao X., Zhang M., Jin H., et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019;26:101254. doi: 10.1016/j.redox.2019.101254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang Y., Qian F., Bai X., Liu Y., Wang Q., Ai B., et al. SEdb: a comprehensive human super-enhancer database. Nucleic Acids Res. 2019;47:D235–D243. doi: 10.1093/nar/gky1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pope B.D., Ryba T., Dileep V., Yue F., Wu W., Denas O., et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515:402–405. doi: 10.1038/nature13986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohaghegh N., Bray D., Keenan J., Penvose A., Andrilenas K.K., Ramlall V., et al. NextPBM: a platform to study cell-specific transcription factor binding and cooperativity. Nucleic Acids Res. 2019;47:e31. doi: 10.1093/nar/gkz020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pott S., Lieb J.D. What are super-enhancers?. Nat Genet. 2015;47:8–12. doi: 10.1038/ng.3167. [DOI] [PubMed] [Google Scholar]
- 40.Cao S., Wang C., Yan J., Li X., Wen J., Hu C. Curcumin ameliorates oxidative stress-induced intestinal barrier injury and mitochondrial damage by promoting Parkin dependent mitophagy through AMPK–TFEB signal pathway. Free Radic Biol Med. 2020;147:8–22. doi: 10.1016/j.freeradbiomed.2019.12.004. [DOI] [PubMed] [Google Scholar]
- 41.Zhang R., Zhao T., Zheng B., Zhang Y., Li X., Zhang F., et al. Curcumin derivative Cur20 attenuated cerebral ischemic injury by antioxidant effect and HIF-1alpha/VEGF/TFEB-activated angiogenesis. Front Pharmacol. 2021;12:648107. doi: 10.3389/fphar.2021.648107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song J.X., Sun Y.R., Peluso I., Zeng Y., Yu X., Lu J.H., et al. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy. 2016;12:1372–1389. doi: 10.1080/15548627.2016.1179404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adao R., Ait-Oufella H. Scientists on the spot: inflammation in atherosclerosis. Cardiovasc Res. 2021;117:e7–8. doi: 10.1093/cvr/cvaa314. [DOI] [PubMed] [Google Scholar]
- 44.Lu H., Fan Y., Qiao C., Liang W., Hu W., Zhu T., et al. TFEB inhibits endothelial cell inflammation and reduces atherosclerosis. Sci Signal. 2017;10:1–13. doi: 10.1126/scisignal.aah4214. [DOI] [PubMed] [Google Scholar]
- 45.Pons D., de Vries F.R., van den Elsen P.J., Heijmans B.T., Quax P.H., Jukema J.W. Epigenetic histone acetylation modifiers in vascular remodelling: new targets for therapy in cardiovascular disease. Eur Heart J. 2009;30:266–277. doi: 10.1093/eurheartj/ehn603. [DOI] [PubMed] [Google Scholar]
- 46.Clapier C.R., Cairns B.R. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 47.Sakamaki J.I., Wilkinson S., Hahn M., Tasdemir N., O'Prey J., Clark W., et al. Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol Cell. 2017;66:517–532.e9. doi: 10.1016/j.molcel.2017.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu Y., Zhang L., Liao X., Sangwung P., Prosdocimo D.A., Zhou G., et al. Kruppel-like factor 15 is critical for vascular inflammation. J Clin Invest. 2013;123:4232–4241. doi: 10.1172/JCI68552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shah A., Gray K., Figg N., Finigan A., Starks L., Bennett M. Defective base excision repair of oxidative DNA damage in vascular smooth muscle cells promotes atherosclerosis. Circulation. 2018;138:1446–1462. doi: 10.1161/CIRCULATIONAHA.117.033249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu L., Fu Y., Zheng Y., Ma M., Wang C. Curcumin inhibits proteasome activity in triple-negative breast cancer cells through regulating P300/miR-142-3p/PSMB5 axis. Phytomedicine. 2020;78:153312. doi: 10.1016/j.phymed.2020.153312. 1-9. [DOI] [PubMed] [Google Scholar]
- 51.Balasubramanyam K., Varier R.A., Altaf M., Swaminathan V., Siddappa N.B., Ranga U., et al. Curcumin, a novel P300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem. 2004;279:51163–51171. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- 52.Alberti S., Gladfelter A., Mittag T. Considerations and challenges in studying liquid–liquid phase separation and biomolecular condensates. Cell. 2019;176:419–434. doi: 10.1016/j.cell.2018.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alberti S., Dormann D. Liquid–liquid phase separation in disease. Annu Rev Genet. 2019;53:171–194. doi: 10.1146/annurev-genet-112618-043527. [DOI] [PubMed] [Google Scholar]
- 54.Sevgiler Y., Karaytug S., Karayakar F. Antioxidative effects of N-acetylcysteine, lipoic acid, taurine, and curcumin in the muscle of Cyprinus carpio L. exposed to cadmium. Arh Hig Rada Toksikol. 2011;62:1–9. doi: 10.2478/10004-1254-62-2011-2082. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y.T., Chen J., Li X., Umetani M., Chen Y., Li P.L., et al. Contribution of transcription factor EB to adipoRon-induced inhibition of arterial smooth muscle cell proliferation and migration. Am J Physiol Cell Physiol. 2019;317:C1034–C1047. doi: 10.1152/ajpcell.00294.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou X., Yang J., Zhou M., Zhang Y., Liu Y., Hou P., et al. Resveratrol attenuates endothelial oxidative injury by inducing autophagy via the activation of transcription factor EB. Nutr Metab (Lond) 2019;16:42. doi: 10.1186/s12986-019-0371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.