

Abstract
Vascular endothelial growth factor-A (VEGF) is the predominant angiogenic factor that is expressed in solid tumors. Besides its critical function in mediating tumor angiogenesis, multiple studies have demonstrated that VEGF also contributes to tumor immunosuppression. VEGF interferes with immune cell trafficking indirectly by promoting a vascular immune barrier through VEGF receptor (VEGFR) activity on endothelial cells. However, VEGFRs are also expressed on multiple immune cell types, including T cells (effector T cells, Tregs) and myeloid cells (DCs, TAMs, MDSCs), where VEGF can have direct effects on immune cell phenotype and function. Thus, it is not surprising that strategies targeting VEGF/VEGFRs have shown efficacy in alleviating tumor-associated immunosuppression and have been combined with immunotherapies, especially immune checkpoint blockade. In this review, we discuss the direct and indirect effects of VEGF on the immunosuppressive tumor microenvironment with particular focus on the direct regulation of immune cells through VEGFR2 activity. We also summarize preclinical and clinical observations of combining antiangiogenesis agents with immunotherapies for the treatment of solid tumors.
Keywords: angiogenesis, immune regulation, tumor microenvironment, VEGF
1 |. INTRODUCTION
Tumors exploit conserved immune regulatory pathways to evade immune-mediated elimination. Tumor cells and stromal cells in the tumor microenvironment (TME) engage immune checkpoints, including cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed cell death protein 1 (PD-1) expressed on T cells to suppress the function of cytotoxic T lymphocytes (CTLs) and prevent antitumor immune activity. The advent of immune checkpoint blockade therapy has revolutionized the treatment of many tumor types and has become a central therapeutic strategy for subsets of patients with advanced malignances. This strategy was first demonstrated using antibodies that block CTLA-4 function and resulted in inhibition of tumor growth and durable antitumor memory in mice.1 CTLA-4 expressed on T cells binds to B7 molecules such as CD80 and CD86 on APCs and subsequently blocks T-cell priming and activation.2 The blockade of PD-1 on T cells or its ligands, PD-L1 and PD-L2, expressed by cancer cells and host myeloid cells, also enhances the antitumor activity of tumor antigen-primed CTLs.3 Under physiologic conditions, the activation of CTLA-4 or PD-1 pathways on T cells contributes to the maintenance of tolerance to self-antigens and prevents autoimmunity. However, tumors up-regulate the expression of CLTA-4 and PD-1 ligands to abrogate the downstream effects of T-cell activation.4 In essence, immune checkpoint blockade removes the brake on T-cell activation and triggers adaptive immune responses in appropriately primed CTLs.3
Immune checkpoint strategies are approved for first-line therapy for multiple indications, such as melanoma, lung cancer, and metastatic colorectal cancer, and when combined with other treatments has elicited remarkable antitumor responses in patients with a variety of solid tumors.5–7 Although immune checkpoint blockade leads to durable responses in melanoma patients, only a fraction of cancer patients benefit from immune checkpoint blockade, and the rate of complete response to anti-PD-1 or anti-CTLA-4 antibodies remains low.5,8 Therefore, multiple strategies are currently under investigation to improve the therapeutic efficacy of immunotherapies. Importantly, tumors with preexisting tumor-infiltrating lymphocytes (TILs) and a less immunosuppressive microenvironment, which are considered as immunologically “hot,” tend to have better response to immune checkpoint inhibitors (ICIs).9 Abnormal tumor angiogenesis has been described as a major component among various factors in the immunosuppressive TME that limit the therapeutic benefit of ICIs in patients.10 The abnormal vascular network that results from tumor angiogenesis restricts efficient lymphocyte infiltration into the tumor site, which compromises the efficacy of immunotherapies.6 Angiogenesis, the process of generating a new vascular network through the sprouting of an existing vessel in response to proangiogenic factors, is crucial for the progression and metastasis of solid tumors.11 However, angiogenesis is also associated with immunosuppression; thus angiogenesis and immunosuppression likely occur in parallel during tumor formation and progression.12 Indeed, a variety of proangiogenic factors, especially vascular endothelial growth factor-A (hereafter referred to as VEGF), a primary stimulant of angiogenesis, have immunosuppressive functions. Thus, targeting angiogenic pathways has been exploited in an attempt to restore antitumor immune responses.6,13 In this review, we discuss the effects of VEGF on regulating the tumor immune microenvironment, including the modulatory effects of VEGF on tumor endothelium and different immune cell types. As VEGF receptors and their functions on myeloid cells have been under investigation recently, we focus on the direct effects of VEGF on regulating different myeloid cells and review the latest preclinical and clinical observations on the immunostimulatory outcomes of antiangiogenic agents. The strategies of antiangiogenic therapies discussed in this review have been widely explored and their effects on modulating tumor immune microenvironment are summarized in Table 1.
TABLE 1.
Summarized regulation of tumor immune microenvironment by antiangiogenic therapy
| Agent | Target | Effects of antiangiogenic therapy | References |
|---|---|---|---|
| Smallmolecule tyrosine kinase inhibitor | |||
| Sunitinib | VEGFRs, c-Kit, PDGFR, Flt-3, RET, CSF-1R | Up-regulates CXCL10, CXCL11 in tumor vasculature; reduces Tregs; increases blood myeloid DCs; decreases MDSCs; reduces Treg and type 1 T-cell suppression | [34][76][78][106][131][132][136] |
| Sorafenib | VEGFRs, c-Kit, PDGFR, RAF-kinases, RET | Inhibits DCs function; promotes DCs differentiation and activation. | [76][77] |
| Axitinib | VEGFRs, c-Kit, PDGFR | Suppresses TIM-3 expression on T cells; decreases suppressive capacity of MDSCs; leads to the dysfunction of DCs | [53][135][79] |
| Apatinib | VEGFR2, c-Kit, c-SRC | Limits the recruitment of TAMs | [109] |
| Monoclonal antibody | |||
| Bevacizumab | VEGF-A | Increases the infiltration of proliferating CD8+ T cells; reduces Treg recruitment and decreases the proportion of Ki67+ Tregs; reduces MDSCs; promotes differentiation of DCs | [54][59][61][62][80][106] |
| A2V | VEGF-A, ANGPT2 | Up-regulates CXCL10, VCAM-1, and PD-L1 on blood vessels; reprograms TAMs; suppresses tumor growth | [35][115][116] |
| DC101 | VEGFR2 | Promotes T cells infiltration; polarizes TAMs from M2 to M1; decreases suppressive capacity of MDSCs | [40][134] |
| r84/mcr84 | VEGF-A | Decreases FasL in tumor vasculature; increases CD8+ T cells infiltration; decreases MDSCs; decreases PD-L1 expression on MDSCs. | [39][106][131] |
| B20.4.1.1 | VEGF-A | Increases antigen-specific response of CD8+ T cells | [51] |
| Ramucirumab | VEGFR2 | Increases CD8+ T-cell infiltration; reduces PD-1 expression on CD8+ T cells; reduces Tregs | [55] |
Abbreviations: DCs, dendritic cells; M1, proinflammatory macrophage; M2, anti-inflammatory macrophage; MDSC, myeloid-derived suppressor cell; TAM, tumor-associated macrophage; Treg, regulatory T cell.
2 |. THE VEGF FAMILY AND VEGFRS
The VEGF family is comprised of VEGFA-D and placenta growth factor (PIGF). These growth factors bind to receptor tyrosine kinases, namely VEGF receptors VEGFR1–3, with different affinities and many of them also interact with neuropilins and heparan sulfate proteoglycans as coreceptors.14 Among these receptors, VEGFR2 is the dominant receptor mediating VEGF proangiogenic activity in endothelial cells.15 VEGF binding leads to receptor homodimerization, resulting in the phosphorylation of tyrosine residues and activation of the kinase domain, which recruits adaptor molecules and mediates intracellular signaling pathways that regulate endothelial cell survival, migration, and proliferation.16,17
VEGF is a potent angiogenic factor that exists as 4 different isoforms, VEGF121, VEGF165, VEGF189, and VEGF206, due to alternative splicing.18 These isoforms differ in their interaction with the coreceptors and extracellular matrix while maintaining similar affinities for VEGFR2.19 VEGF has 2 main receptors: VEGFR1 and VEGFR2, which are expressed widely in vascular endothelial cells. Although VEGF has at least a 10-fold higher affinity for VEGFR1 compared with VEGFR2, VEGFR1 phosphorylation induced by VEGF is weak and VEGFR1 signaling remains poorly understood.14,20 Furthermore, convincing genetic data utilizing mice homozygous for VEGFR1-targeted mutation indicates that VEGFR1 functions during development as a negative regulator of VEGF-induced VEGFR2 function.21 Furthermore, a soluble VEGFR1 variant has been reported to form a heterodimer with VEGFR2 and reduce VEGFR2 signaling.20 Therefore, VEGFR1 is typically considered as a decoy receptor that controls the amount of available VEGF, thus negatively regulating VEGFR2 signaling. VEGFR2 is essential for vasculature formation during embryonic development. Mice deficient in VEGFR2 die in utero as a result of a failure to develop organized blood vessels and due to disrupted hematopoietic precursors.22 Although VEGFRs were considered to be expressed exclusively on endothelial cells, it is now well established that VEGFRs are also expressed on other cell types, including some tumor cells, T cells, dendritic cells (DCs), tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs).18,23–25
3 |. VEGF INDUCES AN IMMUNE VASCULAR BARRIER
The success of immunotherapy requires the infiltration of effector immune cells, their extravasation into the tumor stroma, and their direct interaction with tumor cells. However, tumor vasculature is characterized as leaky, irregular, lacking pericyte support, and inefficient in perfusion.11 Consequently, tumor vasculature is impaired in its function of nutrient and oxygen delivery as well as facilitating immune cell infiltration. The process of immune cell infiltration consists of homing of immune cells, adhesion to the endothelium, and cytokine-mediated diapedesis.2 Due to the activity of angiogenic factors, including VEGF, tumor-associated endothelial cells have a suppressed expression of adhesion molecules, such as VCAM-1 and ICAM-1. Reduced endothelial expression of adhesion molecules limits T-cell attachment to the vessel and decreases efficient T-cell infiltration.26,27 However, it has been demonstrated that VEGF signals to 2 parallel downstream pathways that have opposite function to finely regulate adhesion molecules expression. In vitro stimulation of normal endothelial cells with VEGF results in the activation of NF-κB through the PLCγ-sphingosine kinase-PKC pathway, which stimulates the transcription of adhesion molecules, including ICAM-1 and VCAM-1. At the same time, VEGF-mediated activation of the PI3K pathway actively suppresses the expression of ICAM-1 and VCAM-1.28 In addition, on tumor-associated endothelial cells, VEGF inhibits TNF-α-induced VCAM-1 and ICAM-1 expression.29 These effects are reversed by VEGF blockade.29 In addition to VCAM-1 and ICAM-1, CD34 that has been widely used as a stem-cell marker has also been identified as an adhesion molecule required for immune cell adhesion.30 The expression of CD34 is also suppressed on tumor endothelial cells in response to VEGF and fibroblast growth factors.31
Besides the regulatory function of adhesion molecules, the infiltration of immune cells also requires inflammatory cytokines, such as TNF-α to mediate leukocyte capture, rolling, and transmigration.32,33 VEGF is also reported to interfere with proinflammatory TNF-α signaling in endothelial cells.34 VEGF treatment inhibits TNF-α-mediated regulation of CXCL10 and CXCL11, which contributes to decreased T-cell adhesion to endothelial cells. VEGFR2 inhibition by small molecule inhibitors up-regulated the expression of CXCL10 and CXCL11 in tumor vasculature in a B16 melanoma model, thus improving T-cell recruitment.34 The dual inhibition of angiopoietin-2 and VEGF also results in the activation of proinflammatory signaling pathways, including the TNF-α, type I and II IFNs, and NF-κB pathways, in endothelial cells sorted from murine breast tumors. Consistently, the blockade of angiogenic factors results in the up-regulation of CXCL10, VCAM-1, and PD-L1 expression on CD31+ blood vessels.35 VEGF and TNF-α produced by tumor cells also enable human proangiogenic monocyte extravasation into tumors through the GATA3-induced suppression of CX3CL1 on vessels.36
Furthermore, tumor endothelial cells create a selective immune barrier by up-regulating the death mediator Fas ligand (FasL).37 A prior study found no correlation between FasL expression and VEGF level in acute myeloid leukemia patient samples38; however, recent studies illustrate that VEGF, IL-10, and prostaglandin E2 (PGE2)induce FasL expression on tumor endothelium cooperatively.37 The interaction between Fas expressed on effector CD8+ T cells and FasL mediates cell death and limits cytotoxic CD8+ T-cell infiltration but does not affect the accumulation of regulatory T cells (Tregs).37 The blockade of VEGF together with the pharmacologic inhibition of PGE2 or COX-2 (the enzyme responsible for the formation of PGE2) results in decreased FasL expression by tumor vasculature and a significant increase in cytotoxic CD8+ T-cell infiltration into the tumor niche in multiple models.37,39 It is worth noting that, compared with high dose anti-VEGFR2 treatment, lower doses were shown to promote T cell infiltration more potently in preclinical breast cancer models, since high-dose antiangiogenic therapy tends to excessively prune tumor vessels, creating a hypoxic and immunosuppressive TME. Thus, optimized doses of antiangiogenic therapies can normalize the tumor vasculature in multiple aspects and support immune cell trafficking.40,41
4 |. DIRECT EFFECTS OF VEGF ON T CELLS
CD4+ and CD8+ T cells are the main lymphocytes in cell-mediated immunity and are central to antitumor immunity.42 CD8+ T cells recognize tumor-specific antigens bound to MHC class I expressed on tumor cells. This recognition leads to direct killing of tumor cells through T cell release of cytotoxins such as perforin and granzymes.43 CD4+ T cells, on the other hand, are highly versatile and can differentiate into several subtypes responding to different cytokine stimuli, which allow them to provide help and regulation to effector immune cells and function as coordinators of adaptive immunity. CD4+ T cells are mainly classified as T-helper cell and Treg, with Tregs predominantly mediating immunosuppression.44 VEGF has direct effects on T cells. VEGF at concentrations found in advanced-stage cancer patients disrupts the development of T cells from hematopoietic progenitor cells (HPCs) and leads to thymic atrophy.45 Additionally, VEGF infusion into tumor-free mice decreases T-cell fraction and the T cell/B cell ratios in lymph nodes and spleen.46
4.1 |. VEGF directly reduces effector T cell activity
Among VEGFRs, VEGFR2 is expressed on T cells. Studies evaluating the expression of different types of VEGFRs on T lymphocytes from healthy donors show that VEGFR1 and VEGFR3 are difficult to detect, whereas VEGFR2 is detected on the surface of CD3+ T cells. Moreover, after anti-CD3 activation, VEGFR2 is up-regulated.47 Similarly, purified CD8+ T cells from tumor-free mice have a low level of VEGFR1 and VEGFR2 expression, but stimulation with anti-CD3 antibodies increases VEGFR expression.48 Further, the same study found that tumor infiltrating CD8+ T cells have a higher expression of VEGFR1 and VEGFR2 compared with splenic T cells.48 Hypoxia also induces VEGF and VEGFR2 expression in murine T-cell lines in a time-dependent manner, and VEGF secreted by activated T cells leads to Th1 polarization.49 For T lymphocytes from human PBMCs and T cells isolated from the ascites of ovarian cancer patients, VEGF has been shown to directly suppress T cell proliferation and reduce the cytotoxic activity of T cells through VEGFR2.47,50 These effects can be attenuated by administration of anti-VEGFR2 antibodies.50
In addition, VEGF contributes to the regulation of expression of inhibitory checkpoint molecules on CD8+ T cells in tumors through the activation of the VEGFR2-PLCγ-calcineurin-NFAT pathway.48 By up-regulating the immune checkpoint molecules PD-1, CTLA-4, T-cell immunoglobulin mucin-3 (TIM-3), and lymphocyte activation gene-3 (LAG-3), VEGF directly enhances T-cell exhaustion, which contributes to immune escape. A recent study has revealed that a T-cell exhaustion-specific transcriptional program, including the up-regulation of immune inhibitory molecules induced by VEGF, is dependent on the transcription factor TOX in microsatellite stable colorectal cancers.23 A VEGFR2-specific knockout in T cells, produced by crossing VEGFR2flx/flx mice with LCK-Cre mice, resulted in improved overall survival in a syngenic MC38 model. TILs from VEGFR2 conditional knockout mice displayed higher proliferation and cytokine production capacity with down-regulated TOX and immune inhibitory molecule expression.23
Given the importance of CD8+ T cells in the antitumor immune response, and the direct modulation of CD8+ T cells by VEGF, multiple strategies for VEGF blockade have been shown to decrease immune checkpoint molecule expression on T cells and restore antigen-specific CD8+ T-cell effector function in preclinical models.23,48,51,52 A recent study demonstrated that the inhibition of VEGFR2 but not VEGFR1 enhances cytokine production and the antigen-specific response of CD8+ effector T cells.51 In response to anti-VEGF therapy, which leads to a hypoxic environment, CD8+ T cells in CT26 tumors possess stabilized HIF-1α and up-regulated HIF-1α target genes that support CD8+ effector T-cell function.51 In addition, in PBMCs from patients with recurrent glioblastoma, treatment with the VEGFR inhibitor axitinib suppressed TIM-3 expression on CD4+ and CD8+ T cells.53 Further, therapy with the VEGF-blocking antibody bevacizumab results in elevated expression of genes related to Th1 chemokines and further enhances the infiltration of proliferating CD8+ T cells in metastatic renal cell carcinoma.54 Recently, an immune profile analysis of PBMCs and TILs from pre- and post-ramucirumab (anti-VEGFR2) therapy in advanced gastric cancer patients confirmed an enhanced CD8+ T-cell infiltration and a reduced PD-1 expression by CD8+ T cells.55
4.2 |. VEGF effect on Tregs
Tregs characterized as CD4+CD25+FoxP3+ are critical for maintaining peripheral tolerance under normal conditions and contribute to tumor immune suppression.56 Tregs inhibit the antitumor activity of CD8+ T cells and NK cells with various mechanisms including secretion of inhibitory cytokines, such as IL-10, IL-35, TGF-β, and adenosine nucleosides that suppress effector T cell function.56 In addition, Tregs have also been shown to inhibit the cytotoxicity of CD8+ T cells and NK cells by killing these effector cells in granzyme B and perforin dependent manner.57 Thus, high intratumoral Tregs correlate with poor disease outcomes in multiple tumor types, including pancreatic, ovarian, and liver cancer.58 The expression pattern of VEGFRs on Tregs is similar to that of effector T cells: VEGFR1 and VEGFR2 are expressed on a small population of Tregs in naïve mice, but the percentages of VEGFR1+ and VEGFR2+ Tregs are elevated in tumor-bearing mice.59 The inhibition of VEGF in CT26 tumor-bearing mice reduced Treg accumulation by directly inhibiting Treg proliferation through VEGFR2 but did not affect the suppressive capacity of Tregs.59 Consistently, the selective blockade of VEGF from binding to VEGFR2 decreases the number of Tregs in genetically engineered mouse models of pancreatic cancer.40 Similarly, silencing tumor-derived VEGF limits Treg infiltration and proliferation in tumor-draining lymph nodes in B16 melanoma.60
Consistent with preclinical studies, VEGF blockade by bevacizumab reduces Treg recruitment and decreases the proportion of Ki67+ Tregs in peripheral blood of patients with metastatic colorectal cancer and glioblastoma.59,61,62 Primary TILs from advanced gastric cancer patients treated with the VEGFR2-blocking antibody ramucirumab also exhibit reduced Tregs, and in vitro assays confirmed VEGF promotes VEGFR2+ Treg proliferation, which could be reversed by ramucirumab.55 In addition, studies also indicate that the up-regulation of Treg signature genes or an increased number of Tregs contributes to immunologic resistance to anti-VEGF therapy in glioblastoma.53,63
5 |. EFFECTS OF VEGF ON MYELOID CELLS
5.1 |. DCs
DCs are professional APCs that are critical for the initiation of an antigen-specific antitumor immune response. Immature DCs derive from bone-marrow HPCs and have a low expression of costimulatory molecules and MHC class molecules, and thus have limited capacity for antigen processing and presenting.64,65 DCs can be activated to undergo maturation by a variety of environmental inflammatory stimuli, including soluble factors secreted from tumor cells.66 The maturation of DCs results in efficient antigen presenting and reduced antigen uptake.65,67 Activated DCs are characterized by up-regulated MHC and costimulatory molecule (CD80 and CD86) expression. The activation of DCs also alters the expression of chemokine receptors and cytokines.64
Multiple factors in the TME can lead to the dysfunction of DCs. Immature DCs in PBMCs from cancer patients of different cancer types correlate with an increased level of VEGF in plasma.68 VEGF was initially reported to directly inhibit DC maturation from CD34+ precursors.46,66 The mechanism was illustrated later by evaluating the binding of VEGF to HPCs. VEGF binding to VEGFR1 on HPCs inhibits the activation of NF-κB signaling in HPCs, resulting in the defective maturation of DCs.69 Embryonic stem cells from VEGFR1- or VEGFR2-deficient mice were exposed to VEGF or PIGF, which revealed that VEGFR1 is the dominant mediator for the inhibitory effect of VEGF on DC maturation, whereas the tyrosine kinase activity of VEGFR2 contributes to early hematopoietic differentiation.24 Recent studies confirmed in peripheral blood from chronic myeloid leukemia and prostate cancer patients that VEGF inhibits the maturation and function of DCs, and this inhibitory effect is associated with high plasma levels of VEGF.70,71 Further, neuropilin-1, a coreceptor for VEGF, is necessary to suppress the maturation of murine bone marrow-derived DCs that are induced by LPS.72 Although VEGF has no significant effect on mature DCs with regard to phenotype, cytokine production, and the induction of apoptosis, VEGF disrupts mature DC stimulation of allogeneic T cells, an effect mediated by VEGFR2, indicating different functions of VEGF receptors in the maturation process of DCs.73 Furthermore, VEGFR2 expression is abundant on plasmacytoid DCs from human and mouse tissues, but not on those isolated from blood. VEGFR2 also contributes to the homeostasis of plasmacytoid DCs and their response to IFN-α.74 In addition, an investigation of blood monocyte-derived myeloid DCs (MDCs) from ovarian cancer patients demonstrated that VEGF can suppress MDC maturation from progenitor cells and up-regulate PD-L1 expression on MDCs, which can be reversed by blocking VEGF activity.75 Elevated PD-L1 expression on MDCs impairs MDC-mediated T-cell activation. However, VEGF does not alter PD-L1, CD80, or CD86 expression levels on LPS-stimulated mature MDCs.
The modification of DC function by targeting the VEGF axis has been explored widely. Small molecule tyrosine kinase inhibitors, such as sunitinib, sorafenib, and axitinib, have been approved for therapy of multiple malignant diseases. However, the effects of these inhibitors on DC function remain controversial, likely due to the diverse targets of these inhibitors as they also inhibit other tyrosine kinases besides VEGFRs. Sorafenib, but not sunitinib, was reported to inhibit DC function by reducing cytokine secretion and suppressing costimulatory molecule expression as well as the induction of antigen-specific T cells.76 In contrast, another study demonstrated that sorafenib reversed the inhibitory effect of VEGF on DC differentiation and produced an enhanced expression of HLA-DR and CD86.77 The main difference between these studies was that the latter one focused on the differentiation process of DCs. Sunitinib was shown to increase the level of blood myeloid DCs in patients with advanced renal cell cancer experiencing regression.78 Treatment with axitinib, on the other hand, leads to the dysfunction of DCs with inhibited expression of activation markers and costimulatory receptors and an impaired induction of T-cell proliferation.79 These complex and sometimes opposite results reinforce the importance of understanding the effect of multitarget small molecule inhibitors when combining with immunotherapeutic approaches. In contrast, bevacizumab treatment has shown more consistent results, where it can restore the differentiation of human monocytes to DCs.77 Consistently, elevated DCs and reduced immature progenitor cells in peripheral blood have been detected in cancer patients after bevacizumab administration.80 In vitro studies also suggest that supernatant from breast cancer cell lines with VEGF expression ablated by shRNA induces PBMC-derived DCs to up-regulate CD80, CD86, and HLA-DR expression and enhances DC-mediated T-cell cytotoxicity.81
5.2 |. TAMs
Macrophages are professional phagocytes of the innate immune system that contribute to maintaining tissue homeostasis.82 They respond to danger signals and endogenous molecules and are capable of inducing an inflammatory response and triggering adaptive T-cell responses together with other immune cells.82,83 Macrophages respond differently to various microenvironment stimuli and traditionally have been divided into 2 general phenotypes based on their functions: proinflammatory macrophages (M1) in response to Th1-associated cytokines or LPS and anti-inflammatory macrophages (M2) activated by IL-4 or IL-13 from Th2 cells. Although the M1 and M2 categorization of macrophages is convenient, in reality, macrophages in tissues and tumors exist within a spectrum of phenotypes and protein expression.84,85 Generally, M1 macrophages are considered to have antitumorigenic activity, whereas M2 macrophages are considered to have protumorigenic activities.83,86 Macrophages are recruited early to tumor sites, and these TAMs mostly resemble M2-type macrophages and can generally promote metastasis, stimulate tumor angiogenesis, and lead to immunosuppression.87–89 Clinical studies have shown that the infiltration of M2-like macrophages into tumors confers a poor clinical prognosis in many types of cancers, such as pancreatic, prostate, and breast, and in Hodgkin’s lymphoma.88,90–92
The infiltration of macrophages into tumors is mediated by a variety of cytokines, chemokines, and growth factors, including VEGF.93 VEGFR1 is known to be expressed on macrophages and functions as a chemotactic receptor.94,95 VEGFR1 was initially found to mediate VEGF-induced human monocyte migration.96 Qian et al.97 identified a subset of TAMs expressing VEGFR1 in breast cancer (metastasis-associated macrophages), which were remarkably enriched in metastatic sites. By utilizing a macrophage-specific VEGFR1-deleted genetic model, they further suggested that VEGFR1 signaling on metastasis-associated macrophages is essential for breast cancer metastasis.97 Relatedly, a recent study showed that VEGFR1+ metastasis-associated macrophages are highly angiogenic in murine cancer models and colorectal cancer patients with liver metastasis. Additionally, high VEGFR1+ monocytes in liver metastasis or in circulating blood correlate with a worse prognosis in colorectal cancer patients.98 VEGFR1 signaling on bone marrow-derived macrophages inhibits IL-4-induced arginase-1 expression.99 In contrast to VEGFR1, which has been widely acknowledged to be expressed on macrophages, macrophages under normal conditions express low to no VEGFR2.25 However, multiple studies have demonstrated that a subset of TAMs express VEGFR2, which is the dominant receptor mediating VEGF-induced TAM infiltration into tumors.25 Furthermore, selectively inhibiting VEGF binding to VEGFR2 decreases the recruitment of VEGFR2+ TAMs.25,100 Consistent with preclinical studies, the population of VEGFR2+/CD45bright/CD14+ monocytes is prominent in circulating blood from cancer patients compared with healthy donors, which might be a potential marker for the efficacy of antiangiogenic treatment.101 Huang et al.102 recently evaluated the function of VEGFR2 in the myeloid cell lineage and confirmed an elevated expression level of VEGFR2 on myeloid cells is associated with murine glioma grade and progression-free survival (PFS) in high-grade glioma patients. Mechanistically, VEGFR2 expression on bone marrow-derived cells contributes to the differentiation of myeloid lineages and proangiogenic function, a process potentially driven by the inhibitor of DNA binding protein 2, which was found to be an upstream regulator of VEGFR2 expression.102
TAMs also participate in angiogenesis by recruiting endothelial cells through secretion of proangiogenic factors and chemokines, including VEGF, bFGF, CXCL8, and CXCL12.83 In addition, TAMs express MMP9, which mediates extracellular matrix degradation releasing matrix-associated VEGF.103 VEGF produced by TAMs induces a high-density vessel network, infiltration of macrophages, and acceleration of tumor progression in the PyMT tumor model.104 Additional studies validated that VEGF derived from myeloid-lineage cells (mainly macrophages and neutrophils) is critical for the characteristics of tumor vasculature, as a myeloid-specific deletion of Vegf attenuated the formation of tumor vasculature. However, myeloid depletion of Vegf resulted in accelerated tumor progression but sensitization to chemotherapy in multiple breast cancer models.105
Preclinical evidence suggests that VEGF blockade significantly reduces the recruitment of immunosuppressive macrophages in breast cancer models.100,106 Other studies demonstrated that low doses of an anti-mouse VEGFR2 antibody, DC101, can polarize TAMs from the immunosuppressive M2-like macrophages toward an immunostimulatory M1-like phenotype that show an elevated secretion of chemokines (such as CXCL9 and CXCL11) that facilitate T-cell recruitment.40 Similarly, apatinib, a tyrosine kinase inhibitor selectively targeting VEGFR2, when used at low doses also limits the recruitment of TAMs and modulates the immunosuppressive TME to benefit immune checkpoint blockade in lung cancer.107
Interestingly, resistance to antiangiogenic therapies is commonly associated with TAMs, as hypoxia caused by antiangiogenic agents promotes a compensatory recruitment of angiogenic TAMs and other myeloid cells.108,109 The depletion of macrophages after adaptive resistance to VEGF blockade improved the survival of ovarian cancer tumor-bearing animals.110 Microarray analysis identified macrophage migration inhibitory factor (MIF) as another mediator for increased macrophage recruitment.111 Reduced MIF contributes to bevacizumab resistance in glioblastoma patients and xenograft models by promoting the expansion of M2-like macrophages.111 Tie2-expressing monocytes/macrophages (TEMs) with high proangiogenic capacity are also involved in the escape of malignant gliomas from antiangiogenic treatment.112,113 Angiopoietin-2, the ligand of Tie2, contributes to the homing of TEMs and promotes the proangiogenic activity of TEMs.114 The dual targeting of angiopoietin-2/VEGF in multiple preclinical models has been shown to suppress tumor growth efficiently and reprogram TAMs toward a proinflammatory M1-like phenotype.35,115,116
5.3 |. MDSCs
MDSCs are a population of highly immunosuppressive immature myeloid cells that coexpress CD11b and Gr1. MDSCs were initially identified in the context of the TME, whereas in healthy individuals, immature myeloid cells differentiate into mature myeloid cells, such as macrophages, DCs, and granulocytes.117 There are 2 main populations of MDSCs that increase significantly in tumors: monocytic MDSCs (M-MDSCs) and polymorphonuclear MDSCs (PMN-MDSCs).118 PMN-MDSCs are the dominant population in tumor-bearing animals and are able to inhibit antigen-specific CD8+ T cells but are less immunosuppressive than M-MDSCs in suppressing T-cell activation in vitro.117,119 Multiple mechanisms are exploited by MDSCs to modulate innate and adaptive immune responses, including the expression of arginase-1, which depletes arginase, a lymphocyte nutrient; the production of reactive oxygen species; the reduction of effector cell trafficking; and the expansion of Tregs.117,120,121 Tumor-derived soluble factors contribute to myeloid cell recruitment and function. For example, proinflammatory factors such as IL-1β, IL-6, IFN-γ, and IL-4 trigger the rapid generation of MDSCs from precursors and induce MDSC-induced immunosuppression.122,123 Gabrilovich et al.46 demonstrated that the in vivo infusion of VEGF leads to dysfunction of DCs and increases the production of immature myeloid cells. Immature myeloid cells, especially MDSCs that accumulate in cancer patients, correlate with serum VEGF and disease progression.68,124,125 Additionally, increased levels of all MDSC subpopulations in circulation are associated with disease progression.126 A pronounced accumulation of MDSCs has been detected in murine pancreatic tumors, and their levels are associated with elevated intratumoral VEGF levels during tumor progression. Further, depletion of MDSCs improves the survival of tumor-bearing animals.127
VEGFR1 and VEGFR2 are reported to make distinct contributions to cancer-associated hematopoiesis. Huang et al.128 dissected the functions of VEGFR1 and VEGFR2 by VEGF infusion and demonstrated that VEGFR2 is the main mediator for VEGF-induced accumulation of CD11b+Gr1+ cells, whereas the effect of VEGFR1 is limited. The accumulation of immature myeloid cells and their inability to differentiate into mature DCs caused by VEGF is associated with the constitutive activation of Jak2/STAT3 signaling.129 A recent study confirmed VEGFR1 and VEGFR2 expression on tumor infiltrating MDSCs in an ovarian tumor model.14 However, different from VEGFR1+ MDSCs, whose proportion remains unchanged across organs, the frequency of VEGFR2+ MDSCs increases significantly in tumors, indicating that VEGF-mediated recruitment of MDSCs into tumors is mainly dependent on VEGFR2 signaling.14 Further evaluation showed that VEGFR2+ MDSCs are arginase-1+, suggesting that VEGFR2+ MDSCs possess a high immunosuppressive capacity.14 Our recent study also demonstrated elevated expression level of VEGFR2 on MDSCs from tumor-bearing animals, which contributes to the immunosuppressive myeloid cell phenotype. We exploited the use of a myeloid-specific Vegfr2-deleted mouse model130 and found that loss of Vegfr2 specifically in myeloid cells reduced the immune suppressive function of tumor associated MDSCs.
Therefore, it is reasonable to restrain MDSC accumulation and reverse the immunosuppressive function by disrupting the VEGF-VEGFR2 axis. We and others have demonstrated that multiple anti-VEGF strategies, including the use of antibodies and sunitinib, can decrease the number of MDSCs and result in less T cell suppressive capacity in inflammatory 4T1 breast cancer model and MC38 colon cancer model.106,130,131 Sunitinib inhibited MDSC recruitment via VEGFR inhibition and/or the inhibition of STAT3 in MDSCs.132 Sunitinib treatment or selective inhibition of VEGF binding to VEGFR2 also significantly decreased PD-L1 expression on MDSCs and other myeloid cells.130,131 The depletion of MDSCs caused by sunitinib treatment further benefits vaccine efficacy and leads to an enhanced antigen-specific T-cell response in preclinical tumor models.133 Further studies suggested that the anti-VEGFR2 antibody DC101 does not effect MDSC mobilization but that DC101 attenuates the ability of M-MDSCs to inhibit T-cell proliferation.134 Consistently, M-MDSCs from axitinib-treated mice also exhibit a reduced suppressive capacity on T cells in a melanoma model.135 Clinical studies also reveal that sunitinib treatment in renal cell carcinoma patients results in a reduction in peripheral MDSCs, which correlates with a reduction in Tregs and type 1 T-cell suppression.136 Similarly, bevacizumab significantly reduces the percentage of PMN-MDSCs in the peripheral blood of patients with unresectable non-small cell lung cancer (NSCLC).126
Myeloid cells, including MDSCs, have been thought to be involved in the resistance of tumors to anti-VEGF treatments. Compared with anti-VEGF-sensitive tumors, refractory tumors are often associated with an increase in tumor-infiltrating CD11b+Gr1+ MDSCs and bone marrow MDSCs, which produce high levels of MMP9 and can acquire an endothelial phenotype, resulting in tumor vascularization.137,138 Gene expression analysis of bone marrow MDSCs from refractory tumors reveals an enrichment of inflammatory cytokines, which are markers of myeloid cell differentiation as well as proangiogenic factors.137 STAT3 signaling was also found to be essential for MDSC-mediated tumor angiogenesis.139 In ovarian cancer murine models and clinical samples that are resistant to anti-VEGF therapy, there is a significant increase in Gr1+ MDSCs in hypoxic regions. This elevated infiltration is thought to be mediated by GM-CSF.140 Thus, strategies targeting MDSCs are being explored to overcome the resistance to antiangiogenic therapy.141 The vitamin A metabolite all-trans retinoic acid has been found to induce the differentiation of MDSCs into ature myeloid cells and potentiate anti-VEGFR2 therapy in breast cancer models.142 Additionally, MDSC depletion with anti-Ly6G antibodies combined with the small molecule kinase inhibitor sorafenib suppresses MDSC infiltration and improves the therapeutic efficacy of sorafenib in syngeneic orthotopic liver tumors.143 Similarly, combination therapy with anti-Gr1 antibodies and anti-VEGF neutralizing antibodies also shows significant activity in controlling the growth of refractory tumors.137
6 |. ANTIANGIOGENIC THERAPY IN COMBINATION WITH IMMUNOTHERAPY
As discussed above, VEGF has direct and indirect effects on the immune system and contributes to tumor immune evasion, which is summarized in Figure 1. As a result, strategies targeting VEGF or the VEGF–VEGFR axis can promote an immunostimulatory microenvironment. However, for patients with advanced disease, single-agent antiangiogenic therapy is likely not sufficient to generate a robust and durable immune response. Therefore, strategies that combine antiangiogenic therapy with immunotherapy are being pursued vigorously.
FIGURE 1.
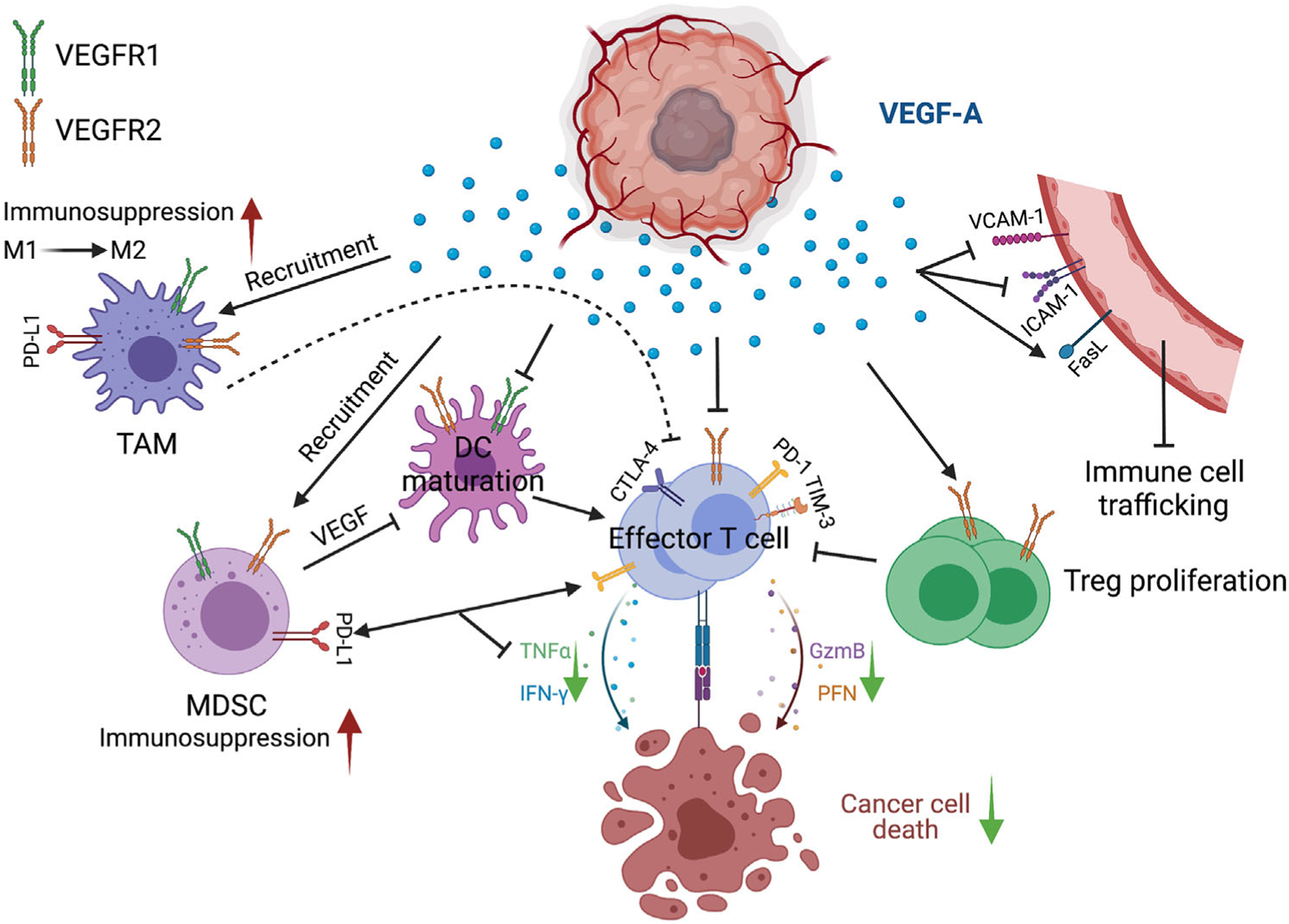
Direct and indirect effects of tumor-secreted VEGF in promoting tumor immunosuppression. By regulating adhesion molecules or FasL expression, VEGF leads to a tumor vasculature that limits immune cell trafficking and infiltration. VEGF directly functions on effector T cells and Tregs through VEGFR2. VEGF enhances the exhaustion status and inhibits the proliferation of effector T cells, while promoting Treg proliferation. VEGF also directly modulates the phenotype of myeloid cells through both VEGFR1 and VEGFR2 and contributes to the cells’ immunosuppressive function on effector T cells, thus reducing T-cell cytotoxic capacity toward cancer cells. DC, dendritic cell; MDSC, myeloid-derived suppressor cell; TAM, tumor-associated macrophage; Treg, regulatory T cell; GzmB, granzyme B; PFN, perforin. Green arrow pointing down, decreased; red arrow pointing up, increased
6.1 |. Immune checkpoint blockade
The combination of ICIs with certain antiangiogenic agents has been investigated in preclinical models and in cancer patients. In preclinical models, including pancreatic neuroendocrine tumor and mammary carcinoma models that are poorly responsive to anti-VEGFR2, PD-L1 was found to be up-regulated due to IFN-γ produced by T cells. Antibody-mediated blockade of PD-L1 and VEGFR2 prolonged antitumor responses, reduced tumor burden, and increased animal survival. Efficacy was associated with the formation of high endothelial venules that mediated lymphocyte infiltration.144 Similarly, a combination of anti-PD-1 antibodies with DC101 was found to enhance antigen-specific T-cell response and improve animal survival in a microsatellite-stable colorectal cancer model.23 A recent study also demonstrated the efficacy of the dual inhibition of VEGF and angiopoietin-2 together with PD-1 blockade in a murine model of glioblastoma. Response was associated with elevated tumor-associated CD8+ T cells and vessel normalization.145 Additionally, the small molecule inhibitor axitinib in combination with anti-PD-1 or anti-TIM-3 antibodies resulted in enhanced therapeutic efficacy in syngeneic murine models.146 Furthermore, blocking CD47, which is the ligand of another immune checkpoint regulator, SIRPα, in NSCLC that is resistant to antiangiogenic therapy potentiates the benefits of VEGFR blockade.147
The pretreatment level of serum VEGF has been reported to negatively correlate with clinical response in patients with metastatic melanoma treated with anti-CTLA-4 antibodies and in patients with advanced NSCLC receiving anti-PD-1 antibodies. These studies indicate that VEGF may be a potential biomarker for therapy and they highlight the rationale of targeting VEGF in these patients.148,149 Indeed, clinical trials have shown that the combination of a selective VEGF inhibitor, axitinib, with pembrolizumab (an anti-PD-1 antibody) in treatment-naïve advanced renal cell cancer patients is well tolerated.150 A randomized phase 2 clinical trial of atezolizumab (anti-PD-L1) alone or atezolizumab combined with bevacizumab versus sunitinib was completed in previously untreated patients with metastatic renal cell carcinoma. Median PFS with combination therapy was 11.7 months (95% CI, 8.4–17.3) compared with 8.4 months (95% CI, 7.0–14.0) in those treated with sunitinib alone, and 6.1 months (95% CI, 5.4–13.6) in those receiving atezolizumab monotherapy.151 The efficacy of combination therapy was superior in PD-L1+ patients, who had a median PFS of 14.7 months (95% CI, 8.2–25.1).151 Biomarker analysis of patient tissue from this trial demonstrated that a high expression of an effector T-cell gene signature is associated with improved overall objective response rates and PFS in the combination arm.151 A phase 3 clinical trial is currently ongoing to confirm these results.2
In patients with metastatic renal carcinoma who had already received first-line ICIs, the use of second-line VEGFR tyrosine kinase inhibitors led to a partial response in 39.7% of the patients and stable disease in 52.9%.152 Besides renal cell carcinoma, axitinib together with pembrolizumab (anti-PD-1 antibody) has been applied to patients with advanced sarcomas in a phase 2 trial, indicating manageable toxicity and preliminary efficacy. This was particularly so for patients with alveolar soft-part sarcoma, 72.7% of whom reached 3-month PFS (95% CI 37.1–90.3).153 The combination of bevacizumab and nivolumab (anti-PD-1 antibody) was also found to have activity in patients with relapsed ovarian cancer in a phase 2 clinical trial, while interestingly, the majority of responses occurred in patients with lower tumor PD-L1 expression.154
In conclusion, multiple preclinical studies have demonstrated promising results from combining antiangiogenic agents with ICIs. Clinical trials have demonstrated manageable toxicity with some indications of improved efficacy. A large number of clinical trials are currently under way to assess the application of combination regimens in various cancer types.
6.2 |. Tumor vaccines
Tumor vaccines, such as DC vaccines and vaccines targeting tumor-associated antigens, are designed to induce an immune response toward one or multiple self-antigens or tumor-specific antigens. However, challenges in the development of cancer vaccines still remain due to immune self-tolerance.155 In addition, the compromised immune system in most patients in clinical trials likely contributes to disappointing results.156,157 As discussed before, antiangiogenic agents promote effector cell infiltration and can reverse the immunosuppressive microenvironment. Therefore, it might be beneficial to utilize antiangiogenic treatment to boost vaccine-mediated immune responses. In preclinical studies, vaccines targeting proangiogenic factors or tumor vasculature have shown the potential of antivascular effects and the control of tumor growth by simultaneously targeting angiogenesis and stimulating an antitumor immune response.158
Sunitinib treatment combined with OVA peptide-pulsed DC was shown to exhibit superior antitumor effects in B16-OVA melanomas by facilitating antigen-specific T-cell response while reducing MDSCs and Tregs in tumors and tumor-draining lymph nodes.159 The antitumor effect was more potent when sunitinib was administrated at the time of initial vaccine boost.159 A HER-2 peptide vaccine in combination with DC101 was investigated in a genetic murine model of Her2+ breast cancer, which resulted in inhibited tumor growth.160 Similarly, low-dose DC101 treatment combined with a whole cancer cell vaccine enhanced vaccine therapy in an orthotopic breast cancer model and improved animal survival in an MMTV-PyMT model.40
A preliminary clinical study reported that a vaccine (ERC-1671) generated from cellular and tumor lysate components of glioblastoma multiforme (GBM) showed signs of efficacy in patients with progressive gliomas after bevacizumab failure.161 A larger cohort of GBM patients who failed bevacizumab therapy were treated with ERC-1671, resulting in minimal toxicity and a 100% 6-month overall survival and 77% 40-week survival compared with 33% and 10% in patients in the control group.162 A phase 2 clinical trial is ongoing to verify the efficacy of ERC-1671 in combination with bevacizumab in recurrent GBM.163 Recently, a personalized cancer vaccine developed by using autologous DCs pulsed with whole-tumor cell lysates was tested in recurrent ovarian cancer patients, in order to address the safety and feasibility of combination therapy with bevacizumab and cyclophosphamide.164 Besides bevacizumab, sunitinib has also been combined with a multipeptide cancer vaccine containing 10 tumor-associated peptides for advanced renal cell carcinoma. A phase 3 trial of this strategy was disappointing, as the combination did not improve overall survival compared with first-line therapy,165 highlighting the complexity of combining small molecule inhibitors with tumor vaccines.
6.3 |. Adoptive cell transfer
A therapy involving the adoptive transfer of immune cells has been developed based on an antiangiogenic strategy. This means introducing engineered cytotoxic T lymphocytes with chimeric T-cell receptors that target VEGFR2.166 After adoptive transfer into tumor-bearing mice, these engineered T cells are designed to efficiently target cells expressing VEGFRs, and they have led to the inhibition of tumor growth in multiple syngeneic murine models and human xenografts. Combining this approach with an angiogenesis inhibitor, TNP-470, further enhanced this effect.166 Investigators used the adoptive transfer of mouse and human T cells expressing chimeric antigen receptors (CARs) that target VEGFR2. They consistently found that this generated antigen-specific immune responses significantly suppressed tumor growth and improved survival in 5 different types of vascularized syngeneic tumors.167 The CAR T cells that target VEGFR2 also reduce VEGFR2+ MDSCs and reverse the immunosuppressive microenvironment.168 The simultaneous transfer of engineered syngeneic CAR T cells that target VEGFR2 and T cells that are specific for tumor antigens induces tumor regression in B16 melanomas compared with either of these T-cell treatments alone.169 Researchers also observed a remarkable expansion and durability in the transferred tumor antigen-specific T cells.169 Similarly, anti-VEGF antibody treatment enhanced the efficacy of adoptive cell transfer therapy with infused T cells targeting B16 melanoma cell-specific markers even in established tumors (~200 mm2) that do not respond well to anti-VEGF therapy alone. The enhancement was likely due to increases in tumorinfiltrating T cells caused by VEGF blockade.170 In addition, bevacizumab in combination with the adoptive transfer of cytokine-induced killer cells, which were derived from PBMCs, had synergistic effects on controlling tumor growth in NSCLC murine models.171 However, there are still limitations of anti-VEGFR2 CAR T cells as a monotherapy since immune barriers within the tumor vasculature still exists, such as FasL and adhesion molecules expression which attenuate T cell function. A recent study revealed that after anti-VEGFR2 CAR T cell transfer, VEGF was up-regulated, which further competed for receptor binding and compromised the efficacy of anti-VEGFR2 CAR T cells.172 Furthermore, the efficacy of anti-VEGFR2 CAR T cells is limited unless the CAR T cells coexpressed IL-12 or IL-15 to augment tumor control,.168,173
Clinically, bevacizumab together with a tumor cell lysate-pulsed DC vaccine followed by the adoptive transfer of autologous vaccine-primed T cells were used to treat a small cohort of recurrent ovarian cancer patients.174 This combination strategy turned out to be well-tolerated and elicited durable immune responses, including reduced circulating Tregs and elevated CD8+ T cells.174 Further investigation of such combinations, including how they might be modified, are warranted in clinical studies in larger cohorts.
6.4 |. Type I IFN and STING agonists
The IFNs are a family of cytokines that modulate the immune response or have a direct effect on targeted cells. In the TME, IFNs can be produced by multiple cell types, where they directly target tumor cells or stimulate T cells and activate immune responses.175 There are 3 major types of IFNs, type I, type II, and type III IFNs, which signal through different pathways. Type I IFNs, including IFN-α, IFN-β, and other subtypes, are essential for antiviral immunity and can be rapidly produced by fibroblasts and monocytes after the stimulation of pattern-recognition receptors.176 Type I IFNs are important in cancer as type I IFNs regulate cell proliferation, apoptosis, migration, and differentiation.175 Type I IFNs can also contribute to the activation and maturation of DCs, further benefiting antigen presentation and T cell priming.177 In addition, type I IFNs are essential for expansion of stem-like T cells.178 One of the type I IFNs, IFN-α, has been widely explored in the clinic for treating different types of cancer, such as hematologic cancers, melanoma, and other advanced metastatic diseases.175,179,180 Type II IFN, known as IFN-γ, also promotes antiviral immunity and is critical in coordinating the innate and adaptive immune response.176 IFN-γ is mainly produced by NK cells in antiviral innate immune response, whereas CD4+ and CD8+ T cells are the major sources of IFN-γ in adaptive immune response. In the TME, IFN-γ functions as a cytotoxic cytokine to initiate apoptosis in tumor cells, while it also mediates ICI molecules expression, such as PD-L1, which in turn stimulates immunosuppression.181
A decade ago, a phase 3 clinical trial was performed by combining bevacizumab with IFN-α, which was the historic standard treatment for renal cell carcinoma in patients with previously untreated metastatic disease. Although there were benefits in overall survival, they did not reach the criteria for significance, likely due to toxicity.182 Similar results were observed with bevacizumab combined with IFNα-2α.183
The STING signaling pathway triggered by cytosolic DNA is essential in host defense and can induce antitumor immune responses. The activation of the STING pathway in APCs drives the production of type I IFN and enhances T-cell priming.184 Accumulating evidence shows the importance of STING expression in endothelial cells, and Yang et al.185 recently confirmed the correlation of endothelial STING expression and intratumoral CD8+ T-cell infiltration. They further reported that a STING agonist delivered by intratumoral injection normalizes the vasculature in Lewis lung carcinoma tumors, and they demonstrated that the combination of a STING agonist with VEGFR2 blockade results in complete tumor regression. A triple combination of a STING agonist, immune checkpoint blockade (anti-PD-1 or anti-CTLA-4), and anti-VEGFR2 antibodies also leads to dramatic tumor control, with mice that received the triple therapy exhibiting long-lasting tumor-specific immune memory.185 Table 2 summarizes selected preclinical studies evaluating the combination efficacy of antiangiogenesis therapies and immunotherapies in multiple models.
TABLE 2.
Selected preclinical studies of antiangiogenic reagents in combination with immunotherapies
| Antiangiogenic therapy | Immunotherapy | Tumormodel | Key results (arrows indicate increase/decrease) | References |
|---|---|---|---|---|
| Immune checkpoint blockade | ||||
| DC101 (Anti-VEGFR2) | 10F.9G2 (Anti-PD-L1) | Pancreatic cancer; breast cancer | Tumor burden;↓ animal survival;↑ lymphocyte infiltration↑ |
[144] |
| DC101 (Anti-VEGFR2) | RPM1–14 (Anti-PD-1) | Microsatellite-stable colorectal cancer | Animal survival;↑ antigen-specific T-cell response↑ |
[23] |
| Trebananib (Ang-1/Ang-2 peptibody); Aflibercept (VEGF-trap) | RPM1–14 (Anti-PD-1) | Glioblastoma | CD8+ T cells; ↑ vessel normalization↑ |
[145] |
| Axitinib | Anti-PD-1; Anti-TIM-3 | Colon cancer; Lewis lung carcinoma |
Mast cells;↓ TAMs;↓ T-cell response↑ |
[146] |
| B20-4.1.1 (Anti-VEGF) | RPM1–14 (Anti-PD-1) | Colon cancer | PD-1+CD8+ T cells; ↓ antitumor effect↑ |
[48] |
| VEGFR1-Fc | SIRPα-Fc | NSCLC | Resistance to antiangiogenic therapy;↓ macrophage phagocytosis;↑ animal survival↑ |
[147] |
| Tumor vaccines | ||||
| Sunitinib | OVA peptide-pulsed DC (VAC) | Melanoma | Antigen-specific T cells;↑ MDSCs;↓ Tregs↓ |
[159] |
| DC101 (Anti-VEGFR2) | HER-2 peptide vaccine | Breast cancer | Tumor growth;↓ lytic activity of CD8+ T cells ↑ |
[16] |
| DC101 (Anti-VEGFR2) | Whole cancer cell vaccine | Breast cancer | Animal survival↑ | [40] |
| Adoptive cell transfer | ||||
| NA | DC101-CAR T cells | Melanoma; colon cancer; fibrosarcoma | Tumor growth;↓ animal survival;↑ antigen-specific T cells;↑ MDSCs↓ |
[166] [167] [168] |
| DC101-CAR T cells | Tumor antigen-specific CAR T cells | Melanoma | Tumor regression;↑ infiltration and persistence of tumor-specific T cells↑ |
[169] |
| B20-4.1.1-PHAGE (Anti-VEGF); DC101 (Anti-VEGFR2) | Tumor antigen-specific CAR T cells | Melanoma | Antitumor effect;↑ infiltration of tumor-specific T cells↑ |
[170] |
| Bevacizumab (Anti-VEGF) | Cytokine-induced killer cells | NSCLC | Antitumor effect;↑ vessel normalization;↑ tumor hypoxia↓ |
[171] |
| STING agonist | ||||
| DC101 (Anti-VEGFR2) | cGAMP or ADU-S100; J43 (anti-PD-1); 9D9 (anti-CTLA-4) | Lewis lung carcinoma; Colon cancer; Breast cancer |
Tumor regression;↑ animal survival;↑ vessel normalization↑ |
[185] |
Abbreviations: DC, dendritic cell; MDSC, myeloid-derived suppressor cell; NSCLC, non-small cell lung cancer; TAM, tumor-associated macrophage; Treg, regulatory T cell. NA, not applicable.
7 |. CONCLUSIONS
Compelling evidence indicates that the effect of blocking VEGF activity in solid tumors extends beyond the inhibition of angiogenesis and can modulate the immune system. In preclinical and clinical settings, dozens of studies have demonstrated the benefit of combining VEGF blockade with anticancer immunotherapy. However, increased therapy-induced toxicity is a concern, especially in the context of small molecule tyrosine kinase inhibitors that have a significant toxicity profile as single agents. Thus, antiangiogenic agents with higher specificity might be more manageable regarding toxicity and be the preferred combinatorial agent with immunotherapy. For example, the selective blockade of VEGF–VEGFR2 signaling results in potent tumor control with limited toxicity as well as an improved immune microenvironment compared with broader spectrum inhibitors.106,186 Combination with immunotherapies might exacerbate the risk of toxicity and adverse events associated with antiangiogenic strategies. Ongoing and future clinical studies combining antiangiogenetic agents with immunotherapies will likely address these questions and could result in therapies that enhance the activity of ICIs in a wide range of cancer patients.
Anti-VEGF therapies have shown the potential to increase lymphocyte infiltration and switch the immunosuppressive microenvironment to a more immune-stimulatory one, but the elimination of cancer cells by a T cell-mediated immune response is a multistep process.43 Thus, other obstacles that limit the efficacy of immunotherapies likely exist. For example, low immunogenicity in tumors could represent a persistent challenge resulting in low levels of tumor antigen-specific T cells. Another challenge is represented by tumors with abundant stroma, such as pancreatic ductal adenocarcinoma. The stroma may represent a barrier to a productive antitumor immune response that is not overcome by antiangiogenic therapy.187 Furthermore, therapy-induced resistance might still develop. Alternative proangiogenic pathways can be elevated after anti-VEGF therapy, and resistance to combined therapy might arise from the dependence of tumors on other immune checkpoint pathways. For example, a recent study has pointed out THA in genetically engineered mouse models of lung cancer progressing after anti-PD-1 therapy, TIM-3 is up-regulated on T cells.188
In addition, optimization is required for antiangiogenesis agents in combination with immunotherapy. Although the applications for anti-PD-1/anti-PD-L1 antibodies together with bevacizumab or other kinase inhibitors are shown in the majority in clinical trials, our recent data have suggested that the specific inhibition of VEGF binding to VEGFR2 leads to the down-regulation of PD-L1 on myeloid cells.130 Considering that PD-L1 expression on host bone marrow-derived cells is essential for the response to PD-L1 blockade,189 our data suggest that anti-VEGF therapy might benefit from combination with immune checkpoint molecules other than PD-1/PD-L1. Indeed, we found that the efficacy of combining VEGF-specific blockade with anti-CTLA-4 antibodies is superior to combination with PD-1 blockade in a breast cancer syngeneic model. Meanwhile, the dose of agents targeting angiogenesis and the sequence of administrating antiangiogenesis agents and ICIs are also key considerations.
In summary, angiogenic factors, especially VEGF, are significant contributors to the tumor immune microenvironment. VEGF impacts the development, recruitment, and phenotype of many types of immune cells through VEGFR2 expression that are relevant to tumor progression. Thus, the rationale for combining antiangiogenic therapy with immunotherapy for the treatment of multiple types of cancer is robust. Multiple addressable challenges remain, but overall, this combination strategy has significant potential to provide benefit to cancer patients.
Summary sentence.
We review the function of vascular endothelial growth factor-A (VEGF) in regulating immune cells in the tumor microenvironment and how the use of anti-VEGF strategies might influence the efficacy of cancer immunotherapies.
ACKNOWLEDGMENT
The authors would like to thank Dave Primm of the UT Southwestern Medical Center for help in editing this article.
Funding information
Effie Marie Cain Fellowship in Angiogenesis Research and R01 CA243577 to RAB
Abbreviations:
- CAR
chimeric antigen receptor
- CTL
cytotoxic T lymphocyte
- CTLA-4
cytotoxic T lymphocyte antigen 4
- DC
dendritic cell
- FasL
Fas ligand
- GBM
glioblastoma multiforme
- HPC
hematopoietic progenitor cell
- ICAM-1
intercellular adhesion molecule 1
- ICI
immune checkpoint inhibitor
- IFN
interferon
- LAG-3
lymphocyte activation gene-3
- LPS
lipopolysaccharide
- M1
proinflammatory macrophage
- M2
anti-inflammatory macrophage
- MDC
myeloid DC
- MDSC
myeloid-derived suppressor cell
- MHC
major histocompatibility complex
- MIF
migration inhibitory factor
- M-MDSC
monocytic MDSC
- NK
natural killer
- NSCLC
non-small cell lung cancer
- PBMC
peripheral blood mononuclear cell
- PD-1
programmed cell death protein 1
- PD-L1
programmed cell death 1-ligand 1
- PD-L2
programmed cell death 1-ligand 2
- PFS
progression-free survival
- PGE2
prostaglandin E2
- PIGF
placenta growth factor
- PMN-MDSC
polymorphonuclear MDSC
- RTK
receptor tyrosine kinase
- TAM
tumor-associated macrophage
- TEM
Tie2-expressing monocyte/macrophage
- TIL
tumor-infiltrating lymphocyte
- TME
tumor microenvironment
- Treg
regulatory T cell
- VAC
OVA peptide-pulsed DC
- VCAM-1
vascular cell adhesion protein 1
- VEGF
vascular endothelial growth factor-A
- VEGFR
VEGF receptor
Footnotes
DISCLOSURE
The authors declare no conflict of interest.
REFERENCES
- 1.Leach DR, Krummer MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (80-). 1996;271:1734–1736. 10.1126/science.271.5256.1734 [DOI] [PubMed] [Google Scholar]
- 2.Khan KA, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat Rev Clin Oncol. 2018;15:310–324. 10.1038/nrclinonc.2018.9. [DOI] [PubMed] [Google Scholar]
- 3.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sznol M, Chen L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin Cancer Res. 2013;19:1021–1034. 10.1158/1078-0432.CCR-12-2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. 2018;15:325–340. 10.1038/nrclinonc.2018.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, Song W, Shao C, Shi Y, Han W. Emerging predictors of the response to the blockade of immune checkpoints in cancer therapy. Cell Mol Immunol. 2019;16:28–39. 10.1038/s41423-018-0086-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ribas A, Hamid O, Daud A et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315:1600–1609. 10.1001/jama.2016.4059. [DOI] [PubMed] [Google Scholar]
- 9.Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res. 2016;22:1865–1874. 10.1158/1078-0432.CCR-15-1507. [DOI] [PubMed] [Google Scholar]
- 10.Munn LL, Jain RK, Vascular regulation of antitumor immunity. Science (80-). 2019;365:544–545. 10.1126/science.aaw7875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 12.Motz GT, Coukos G. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat Rev Immunol. 2011;11:702–711. 10.1038/nri3064. [DOI] [PubMed] [Google Scholar]
- 13.Horikawa N, Abiko K, Mastsumara N, et al. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin Cancer Res. 2017;23:587–599. 10.1158/1078-0432CCR-16-0387.. [DOI] [PubMed] [Google Scholar]
- 14.Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol. 2016;17:611–625. 10.1038/nrm.2016.87. [DOI] [PubMed] [Google Scholar]
- 15.Veikkola T, Karkkainen M, Claesson-Welsh L, Alitalo K. Regulation of angiogenesis via vascular endothelial growth factor receptors 1. Cancer Res.2000;Jan 15;60(2):203–12. [PubMed] [Google Scholar]
- 16.Koch S, Claesson-Welsh L Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437:169–183. 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- 17.Shibuya MJB Review Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases. J Biochem. 2013. Jan;153(1):13–9. 10.1093/jb/mvs136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kowanetz M, Ferrara N. Vascular endothelial growth factor signaling pathways: therapeutic perspective. Clin Cancer Res. 2006;12:5018–5022. 10.1158/1078-0432.CCR-06-1520. [DOI] [PubMed] [Google Scholar]
- 19.Chen TT, Alfonso Luque, Sunyoung Lee, Anderson Sean M., Tatiana Segura, Iruela-Arispe M. Luisa. Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J Cell Biol. 2010;188:595–609. 10.1083/jcb.200906044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25. 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- 21.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 22.Shalaby F, Rossant J, Yamaguchi TP, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 23.Kim CG, Jang M, Kim Y, et al. VEGF-A drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci Immunol. 2019;4. 10.1126/sciimmunol.aay0555. [DOI] [PubMed] [Google Scholar]
- 24.Dikov MM, Ohm JE, Ray N, et al. Differential roles of vascular endothelial growth factor receptors 1 and 2 in dendritic cell differentiation. J Immunol. 2005;174:215–222. 10.4049/JIMMUNOL.174.1.215. [DOI] [PubMed] [Google Scholar]
- 25.Dineen SP, Lynn KD, Holloway SE, et al. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008;68:4340–4346. 10.1158/0008-5472.CAN-07-6705. [DOI] [PubMed] [Google Scholar]
- 26.Piali L, Fichtel A, Terpe HJ, Imhof BA, Gisler RH Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. J Exp Med. 1995;181:811–816. 10.1084/jem.181.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riegler J, Gill H, Ogasawara A, et al. VCAM-1 density and tumor perfusion predict T-cell infiltration and treatment response in preclinical models. Neoplasia (United States). 2019;21:1036–1050. 10.1016/j.neo.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim I, Moon SO, Kim SH, Kim HJ, Koh YS, Koh GY. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J Biol Chem. 2001;276:7614–7620. 10.1074/jbc.M009705200. [DOI] [PubMed] [Google Scholar]
- 29.Wu X, Giobbie-Hurder A, Liao X, et al. VEGF neutralization plus CTLA-4 blockade alters soluble and cellular factors associated with enhancing lymphocyte infiltration and humoral recognition in Melanoma. Cancer Immunol Res. 2016;4:858–868. 10.1158/2326-6066.CIR-16-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nielsen JS, McNagny KM. Novel functions of the CD34 family. J Cell Sci. 2008;121:3683–3692. 10.1242/JCS.037507. [DOI] [PubMed] [Google Scholar]
- 31.Dirkx AEM, oude Egbrink MG, Castermans K, et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J. 2006;20:621–630. 10.1096/fj.05-4493com. [DOI] [PubMed] [Google Scholar]
- 32.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. 10.1038/NRI2156. [DOI] [PubMed] [Google Scholar]
- 33.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–815. 10.1038/NRI2171. [DOI] [PubMed] [Google Scholar]
- 34.Huang H, Langenkamp E, Georganaki M, et al. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-κ B-induced endothelial activation. FASEB J. 2015;29:227–238. 10.1096/fj.14-250985. [DOI] [PubMed] [Google Scholar]
- 35.Schmittnaegel M, Rigamonti N, Kadioglu E et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med. 2017;12;9(385):eaak9670. 10.1126/scitranslmed.aak9670. Published online 2017. [DOI] [PubMed] [Google Scholar]
- 36.Sidibe A, Ropraz P, Jemelin S, et al. Angiogenic factor-driven inflammation promotes extravasation of human proangiogenic monocytes to tumours. Nat Commun. 2018;9. 10.1038/s41467-017-02610-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Motz GT, Santoro SP, Wang LP, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20:607–615. 10.1038/nm.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JJ, Chung IJ, Park MR, Ryang DW, Park CS, Kim HJ. Increased angiogenesis and Fas-ligand expression are independent processes in acute myeloid leukemia. Leuk Res. 2001;25:1067–1073. 10.1016/S0145-2126(01)00082-0. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Kirane A, Huang H et al. Cyclooxygenase-2 inhibition potentiates the efficacy of vascular endothelial growth factor blockade and promotes an immune stimulatory microenvironment in preclinical models of pancreatic cancer. Mol Cancer Res. 2019;17:348–355. 10.1158/1541-7786.MCR-18-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang Y, Yuan J, Righi E et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci USA. 2012;109:17561–17566. 10.1073/pnas.1215397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang Y, Kim BYS, Chan CK, Hahn SM, Weissman IL, Jiang W. Improving immune–vascular crosstalk for cancer immunotherapy. Nat Rev Immunol. 2018;18:195–203. 10.1038/nri.2017.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol 2020 2011. 2020;20:651–668. 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 44.Tay RE, Richardson EK, Toh HC. Revisiting the role of CD4+ T cells in cancer immunotherapy—new insights into old paradigms. Cancer Gene Ther. 2020;28:5–17. 10.1038/s41417-020-0183-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohm JE, Gabrilovich DI, Sempowski GD et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood. 2003;101:4878–4886. 10.1182/blood-2002-07-1956. [DOI] [PubMed] [Google Scholar]
- 46.Gabrilovich D, Ishida T, Oyama T et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–4166. 10.1182/blood.v92.11.4150.423k45_4150_4166. [DOI] [PubMed] [Google Scholar]
- 47.Ziogas AC, Gavalas NG, Tsiatas M, et al. VEGF directly suppresses activation of T cells from ovarian cancer patients and healthy individuals via VEGF receptor Type 2. Int J Cancer. 2012;130:857–864. 10.1002/ijc.26094. [DOI] [PubMed] [Google Scholar]
- 48.Voron T, Colussi O, Marcheteau E, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212:139–148. 10.1084/jem.20140559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mor F, Quintana FJ, Cohen IR. Angiogenesis-inflammation cross-talk: vascular endothelial growth factor is secreted by activated T cells and induces Th1 polarization. J Immunol. 2004;172:4618–4623. 10.4049/JIMMUNOL.172.7.4618. [DOI] [PubMed] [Google Scholar]
- 50.Gavalas NG, Tsiatas M, Tsitsilonis O et al. VEGF directly suppresses activation of T cells from ascites secondary to ovarian cancer via VEGF receptor type 2. Br J Cancer. 2012;107:1869–1875. 10.1038/bjc.2012.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Almeida PE, Mak J, Hernandez G et al. Anti-VEGF treatment enhances CD8 + T-cell antitumor activity by amplifying hypoxia. Cancer Immunol Res.2020. Jun;8(6):806–818. Published online 2020. 10.1158/2326-6066.CIR-19-0360 [DOI] [PubMed] [Google Scholar]
- 52.Malo CS, Khadka RH, Ayasoufi K, et al. Immunomodulation mediated by anti-angiogenic therapy improves CD8 T cell immunity against experimental glioma. Front Oncol. 2018;8. 10.3389/fonc.2018.00320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Du Four S, Maenhout SK, Benteyn D et al. Disease progression in recurrent glioblastoma patients treated with the VEGFR inhibitor axitinib is associated with increased regulatory T cell numbers and T cell exhaustion. Cancer Immunol Immunother. 2016;65:727–740. 10.1007/s00262-016-1836-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wallin JJ, Bendell JC, Funke R et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624. 10.1038/ncomms12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tada Y, Togashi Y, Kotani D, et al. Targeting VEGFR2 with Ramucirumab strongly impacts effector/activated regulatory T cells and CD8+ T cells in the tumor microenvironment. J Immunother Cancer. 2018;6. 10.1186/s40425-018-0403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cao X, Cai SF, Fehniger TA et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27:635–646. 10.1016/J.IMMUNI.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 58.Lapeyre-Prost A, et al. Immunomodulatory activity of VEGF in cancer. Int Rev Cell Mol Biol. 2017; 330:295–342. 10.1016/bs.ircmb.2016.09.007 [DOI] [PubMed] [Google Scholar]
- 59.Terme M, Pernot S, Marcheteau E et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013;73:539–549. 10.1158/0008-5472.CAN-12-2325. [DOI] [PubMed] [Google Scholar]
- 60.Courau T, Nehar-Belaid D, Florez L et al. TGF-β and VEGF cooperatively control the immunotolerant tumor environment and the efficacy of cancer immunotherapies. JCI insight. 2016;1:e85974. 10.1172/jci.insight.85974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wada J, Suzuki H, Fuchino R, et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res. 2009; 29:881–888. [PubMed] [Google Scholar]
- 62.Thomas AA, Fisher JL, Hampton TH et al. Immune modulation associated with vascular endothelial growth factor (VEGF) blockade in patients with glioblastoma. Cancer Immunol Immunother. 2017;66:379–389. 10.1007/s00262-016-1941-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Long Y, Tao H, Karachi A et al. Dysregulation of glutamate transport enhances Treg function that promotes VEGF blockade resistance in glioblastoma. Cancer Res. 2020;80:499–509. 10.1158/0008-5472.CAN-19-1577. [DOI] [PubMed] [Google Scholar]
- 64.Tan JKH, O’Neill HC. Maturation requirements for dendritic cells in T cell stimulation leading to tolerance versus immunity. J Leukoc Biol. 2005;78:319–324. 10.1189/jlb.1104664. [DOI] [PubMed] [Google Scholar]
- 65.Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol. 2018;9. 10.3389/fimmu.2018.00978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gabrilovich DI, Chen HL, Girgis KR et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2:1096–1103. 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- 67.Wilson NS, El-Sukkari D, Villadangos JA. Dendritic cells constitutively present self antigens in their immature state in vivo and regulate antigen presentation by controlling the rates of MHC class II synthesis and endocytosis. Blood. 2004;103:2187–2195. 10.1182/blood-2003-08-2729. [DOI] [PubMed] [Google Scholar]
- 68.Almand B, Resser JR, Lindman B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6:1755–66. [PubMed] [Google Scholar]
- 69.Oyama T, Ran S, Ishida T et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J Immunol. 1998;160:1224–1232. http://www.ncbi.nlm.nih.gov/pubmed/9570538. Accessed October 20, 2019. [PubMed] [Google Scholar]
- 70.Boissel N, Rousselot P, Raffoux E et al. Defective blood dendritic cells in chronic myeloid leukemia correlate with high plasmatic VEGF and are not normalized by imatinib mesylate. Leukemia. 2004;18:1656–1661. 10.1038/sj.leu.2403474. [DOI] [PubMed] [Google Scholar]
- 71.Bai WK, Zhang W, Hu B. Vascular endothelial growth factor suppresses dendritic cells function of human prostate cancer. Onco Targets Ther. 2018;11:1267–1274. 10.2147/OTT.S161302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oussa NAE, Dahmani A, Gomis M et al. VEGF requires the receptor NRP-1 to inhibit lipopolysaccharide-dependent dendritic cell maturation. J Immunol. 2016;197:3927–3935. 10.4049/jimmunol.1601116. [DOI] [PubMed] [Google Scholar]
- 73.Mimura K, Kono K, Takahashi A, Kawaguchi Y, Fujii H. Vascular endothelial growth factor inhibits the function of human mature dendritic cells mediated by VEGF receptor-2. Cancer Immunol Immunother. 2007;56:761–770. 10.1007/s00262-006-0234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Agudo J, Ruzo A, Tung N et al. The miR-126-VEGFR2 axis controls the innate response to pathogen-associated nucleic acids. Nat Immunol. 2014;15:54–62. 10.1038/ni.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Orleans N, Clinic M, Blockade of B7-H1 improves myeloid dendritic cell – mediated antitumor immunity. 2003;9. 10.1038/nm [DOI] [PubMed] [Google Scholar]
- 76.Hipp MM, Hilf N, Walter S et al. Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune responses. Blood. 2008;111:5610–5620. 10.1182/blood-2007-02-075945. [DOI] [PubMed] [Google Scholar]
- 77.Alfaro C, Suarez N, Gonzalez A et al. Influence of bevacizumab, sunitinib and sorafenib as single agents or in combination on the inhibitory effects of VEGF on human dendritic cell differentiation from monocytes. Br J Cancer. 2009;100:1111–1119. 10.1038/sj.bjc.6604965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Van Cruijsen H, van derVeldt AA, Vroling L et al. Sunitinib-induced myeloid lineage redistribution in renal cell cancer patients: cD1c+ dendritic cell frequency predicts progression-free survival. Clin Cancer Res. 2008;14:5884–5892. 10.1158/1078-0432.CCR-08-0656. [DOI] [PubMed] [Google Scholar]
- 79.Heine A, et al. The VEGF-receptor inhibitor axitinib impairs dendritic cell phenotype and function. PLoS One. 2015;10. 10.1371/journal.pone.0128897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Osada T, et al. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol Immunother. 2008;57:1115–1124. 10.1007/s00262-007-0441-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang H, et al. Inhibition of vascular endothelial growth factor by small interfering RNA upregulates differentiation, Maturation and function of dendritic cells. Exp Ther Med. 2015;9:120–124. 10.3892/etm.2014.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Glass CK, Natoli G. Molecular control of activation and priming in macrophages. Nat Immunol. 2016;17:26–33. 10.1038/ni.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Albini A, et al. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: implications for immunotherapy. Front Immunol. 2018;9. 10.3389/fimmu.2018.00527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6. 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Krzyszczyk P, et al. The role of macrophages in acute and chronic wound healing and interventions to promote pro-wound healing phenotypes. Front Physiol. 2018;9:419. 10.3389/fphys.2018.00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Allavena P, Mantovani A. Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol. 2012;167:195–205. 10.1111/j.1365-2249.2011.04515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mantovani A, et al. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416. 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155–1166. 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hu H, et al. The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumor Biol. 2016;37:8657–8664. 10.1007/s13277-015-4741-z. [DOI] [PubMed] [Google Scholar]
- 91.Xu J, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73:2782–2794. 10.1158/0008-5472.CAN-12-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Steidl C, Lee T, Shah SP et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med. 2010;362:875–885. 10.1056/NEJMoa0905680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 94.Murakami M, Zheng Y, Hirashima M et al. VEGFR1 tyrosine kinase signaling promotes lymphangiogenesis as well as angiogenesis indirectly via macrophage recruitment. Arterioscler Thromb Vasc Biol. 2008;28:658–664. 10.1161/ATVBAHA.107.150433. [DOI] [PubMed] [Google Scholar]
- 95.Casazza A, Laoui D, Wenes M et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013;24:695–709. 10.1016/j.ccr.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 96.Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marmé D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87:3336–3343. 10.1182/blood.v87.8.3336.bloodjournal8783336. [DOI] [PubMed] [Google Scholar]
- 97.Qian B-Z, Zhang H, Li J, et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J Exp Med. 2015;212:1433–1448. 10.1084/jem.20141555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Valls AF, Knipper K, Giannakouri E et al. VEGFR1+ metastasis-associated macrophages contribute to metastatic angiogenesis and influence colorectal cancer patient outcome. Clin Cancer Res. 2019;25:5674–5685. 10.1158/1078-0432.CCR-18-2123. [DOI] [PubMed] [Google Scholar]
- 99.Zou Y, Chen Q, Ye Z, Li X, Ju R. VEGFR1 signaling regulates IL-4-mediated arginase 1 expression in macrophages. Curr Mol Med. 2017;17: 304–311. 10.2174/1566524017666171106114537. [DOI] [PubMed] [Google Scholar]
- 100.Roland CL, Dineen SP, Lynn KD et al. Inhibition of vascular endothelial growth factor reduces angiogenesis and modulates immune cell infiltration of orthotopic breast cancer xenografts. Mol Cancer Ther. 2009. Jul;8(7):1761–71. 10.1158/1535-7163.MCT-09-0280. Published online 2009. [DOI] [PubMed] [Google Scholar]
- 101.Vroling L, Yuana Y, Schuurhuis GJ et al. VEGFR2 expressing circulating (progenitor) cell populations in volunteers and cancer patients. Thromb Haemost. 2007;98:440–450. 10.1160/TH07-03-0225. [DOI] [PubMed] [Google Scholar]
- 102.Huang Y, Rajappa P, Hu W et al. A proangiogenic signaling axis in myeloid cells promotes malignant progression of glioma. J Clin Invest. 2017;127:1826–1838. 10.1172/JCI86443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol. 2014. 10.3389/fphys.2014.00075. 5 MAR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lin EY, Li JF, Bricard G et al. Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Mol Oncol. 2007;1:288–302. 10.1016/j.molonc.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stockmann C, Doedens A, Weidemann A et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008;456:814–819. 10.1038/nature07445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Roland CL, Lynn KD, Toombs JE, et al. Cytokine levels correlate with immune cell infiltration after anti-VEGF therapy in preclinical mouse models of breast cancer. PLoS One. 10.1371/journal.pone.0007669. Published online 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhao S, Ren S, Jiang T et al. Low-dose apatinib optimizes tumor microenvironment and potentiates antitumor effect of PD-1/PD-L1 blockade in lung cancer. Cancer Immunol Res. 2019;7:630–643. 10.1158/2326-6066.CIR-17-0640. [DOI] [PubMed] [Google Scholar]
- 108.De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23:277–286. 10.1016/j.ccr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 109.Wroblewski M, Bauer R, Cubas Córdova M et al. Mast cells decrease efficacy of anti-angiogenic therapy by secreting matrix-degrading granzyme B. Nat Commun. 2017;8. 10.1038/s41467-017-00327-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dalton HJ, Pradeep S, McGuire M, et al. Macrophages facilitate resistance to anti-VEGF therapy by altered VEGFR expression. Clin Cancer Res. 2017;23:7034–7046. 10.1158/1078-0432.CCR-17-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Castro BA, Flanigan P, Jahangiri A et al. Macrophage migration inhibitory factor downregulation: a novel mechanism of resistance to anti-angiogenic therapy. Oncogene. 2017;36:3749–3759. 10.1038/onc.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gabrusiewicz K, Liu D, Cortes-Santiago N et al. Anti-vascular endothelial growth factor therapy-induced glioma invasion is associated with accumulation of Tie2-expressing monocytes. Oncotarget. 2014;5:2208–2220. 10.18632/oncotarget.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.De Palma M, Venneri MA, Galli R et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–226. 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 114.Coffelt SB, Tal AO, Scholz A et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 2010;70:5270–5280. 10.1158/0008-5472.CAN-10-0012. [DOI] [PubMed] [Google Scholar]
- 115.Peterson TE, Kirkpatrick ND, Huang Y et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc Natl Acad Sci USA. 2016;113:4470–4475. 10.1073/pnas.1525349113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kloepper J, Riedemann L, Amoozgar Z et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc Natl Acad Sci USA. 2016;113:4476–4481. 10.1073/pnas.1525360113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V et al. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Youn J-I, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802. 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Movahedi K, Guilliams M, Van den Bossche J et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell suppressive activity. Blood. 2008;111:4233–4244. 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 120.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg SMyeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70:68–77. 10.1158/0008-5472.CAN-09-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pan PY, Ma G, Weber KJ et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010;70:99–108. 10.1158/0008-5472.CAN-09-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Marigo I, Bosio E, Solito S et al. Tumor-induced tolerance and immune suppression depend on the C/EBPβ transcription factor. Immunity. 2010;32:790–802. 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 123.Gallina G, Dolcetti L, Serafini P et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. 2006;116:2777–2790. 10.1172/JCI28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Arihara F, Mizukoshi E, Kitahara M et al. Increase in CD14+HLA-DR-/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol Immunother. 2013;62:1421–1430. 10.1007/s00262-013-1447-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shen P, Wang A, He M, Wang Q, Zheng S. Increased circulating Lin-/lowCD33+HLA-DR- myeloid-derived suppressor cells in hepatocellular carcinoma patients. Hepatol Res. 2014;44:639–650. 10.1111/hepr.12167. [DOI] [PubMed] [Google Scholar]
- 126.Koinis F, Vetsika EK, Aggouraki D et al. Effect of first-line treatment on myeloid-derived suppressor cells’ subpopulations in the peripheral blood of patients with non-small cell lung cancer. J Thorac Oncol. 2016;11:1263–1272. 10.1016/j.jtho.2016.04.026. [DOI] [PubMed] [Google Scholar]
- 127.Karakhanova S, et al. Characterization of myeloid leukocytes and soluble mediators in pancreatic cancer: importance of myeloid-derived suppressor cells. Oncoimmunology. 2015;4. 10.1080/2162402X.2014.998519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huang Y, et al. Distinct roles of VEGFR-1 and VEGFR-2 in the aberrant hematopoiesis associated with elevated levels of VEGF. Blood. 2007;110:624–631. 10.1182/blood-2007-01-065714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nefedova Y, et al. Hyperactivation of STAT3 Is involved in abnormal differentiation of dendritic cells in cancer. J Immunol. 2004;172:464–474. 10.4049/jimmunol.172.1.464. [DOI] [PubMed] [Google Scholar]
- 130.Zhang Y, et al. VEGFR2 activity on myeloid cells mediates immune suppression in the tumor microenvironment. JCI Insight. 2021. 10.1172/JCI.INSIGHT.150735. Published online October 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ozao-Choy J, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–2522. 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xin H, et al. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69:2506–2513. 10.1158/0008-5472.CAN-08-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Draghiciu O, et al. Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. Oncoimmunology. 2015;4:1–11. 10.4161/2162402X.2014.989764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Secondini C, et al. Arginase inhibition suppresses lung metastasis in the 4T1 breast cancer model independently of the immunomodulatory and anti-metastatic effects of VEGFR-2 blockade. Oncoimmunology. 2017;6. 10.1080/2162402X.2017.1316437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Du Four S, et al. Axitinib increases the infiltration of immune cells and reduces the suppressive capacity of monocytic MDSCs in an intracranial mouse melanoma model. Oncoimmunology. 2015;4:e998107. 10.1080/2162402X.2014.998107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ko JS, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148–2157. 10.1158/1078-0432.CCR-08-1332. [DOI] [PubMed] [Google Scholar]
- 137.Shojaei F, Ferrara N. Refractoriness to antivascular endothelial growth factor treatment: role of myeloid cells. Cancer Res. 2008;68:5501–5504. 10.1158/0008-5472.CAN-08-0925. [DOI] [PubMed] [Google Scholar]
- 138.Yang L, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–421. 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 139.Kujawski M, et al. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest. 2008;118:3367–3377. 10.1172/JCI35213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Horikawa N, et al. Anti-VEGF therapy resistance in ovarian cancer is caused by GM-CSF-induced myeloid-derived suppressor cell recruitment. Br J Cancer. 2020;122:778–788. 10.1038/s41416-019-0725-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Iwai T, et al. Capecitabine reverses tumor escape from anti-VEGF through the eliminating CD11bhigh/Gr1high myeloid cells. Oncotarget. 2018;9:17620–17630. 10.18632/oncotarget.24811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bauer R, et al. Blockade of myeloid-derived suppressor cell expansion with all-trans retinoic acid increases the efficacy of antiangiogenic therapy. Cancer Res. 2018;78:3220–3232. 10.1158/0008-5472.CAN-17-3415. [DOI] [PubMed] [Google Scholar]
- 143.Chang CJ, et al. Targeting tumor-infiltrating Ly6G+ myeloid cells improves sorafenib efficacy in mouse orthotopic hepatocellular carcinoma. Int J Cancer. 2018;142:1878–1889. 10.1002/ijc.31216. [DOI] [PubMed] [Google Scholar]
- 144.Allen E, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med. 2017;9:eaak9679. 10.1126/scitranslmed.aak9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Di Tacchio M, et al. Tumor vessel normalization, immunostimulatory reprogramming, and improved survival in glioblastoma with combined inhibition of PD-1, angiopoietin-2, and VEGF. Cancer Immunol Res. 2019;7:1910–1927. 10.1158/2326-6066.CIR-18-0865. [DOI] [PubMed] [Google Scholar]
- 146.Läubli H, et al. The multi-receptor inhibitor axitinib reverses tumor-induced immunosuppression and potentiates treatment with immune-modulatory antibodies in preclinical murine models. Cancer Immunol Immunother. 2018;67:815–824. 10.1007/s00262-018-2136-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhang X, et al. Blocking CD47 efficiently potentiated therapeutic effects of anti-angiogenic therapy in non-small cell lung cancer. J Immunother Cancer. 2019;7. 10.1186/s40425-019-0812-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yuan J, et al. Pretreatment serum VEGF is associated with clinical response and overall survival in advanced melanoma patients treated with ipilimumab. Cancer Immunol Res. 2014;2:127–132. 10.1158/2326-6066.CIR-13-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shibaki R, et al. Predictive value of serum VEGF levels for elderly patients or for patients with poor performance status receiving anti-PD-1 antibody therapy for advanced non-small-cell lung cancer. Cancer Immunol Immunother. 10.1007/s00262-020-02539-2. Published online 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Atkins MB, et al. Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: a non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. Lancet Oncol. 2018;19:405–415. 10.1016/S1470-2045(18)30081-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.McDermott DF, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med. 2018;24:749–757. 10.1038/s41591-018-0053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shah AY, et al. Outcomes of patients with metastatic clear-cell renal cell carcinoma treated with second-line VEGFR-TKI after first-line immune checkpoint inhibitors. Eur J Cancer. 2019;114:67–75. 10.1016/j.ejca.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wilky BA, et al. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: a single-centre, single-arm, phase 2 trial. Lancet Oncol. 2019;20. 10.1016/S1470-2045(19)30153-6. [DOI] [PubMed] [Google Scholar]
- 154.Liu JF, et al. Assessment of combined nivolumab and bevacizumab in relapsed ovarian cancer: a phase 2 clinical trial. JAMA Oncol. 2019;5:1731–1738. 10.1001/jamaoncol.2019.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wentink MQ, Huijbers EJ, deGruijl TD, Verheul HM, Olsson AK, Griffioen AW. Vaccination approach to anti-angiogenic treatment of cancer. Biochim Biophys Acta - Rev Cancer. 2015;1855:155–171. 10.1016/j.bbcan.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 156.Vansteenkiste JF, Cho BC, Vanakesa T et al. Magrit, a double-blind, randomized, placebo-controlled phase III study to assess the efficacy of the Recmage-A3 + As15 cancer immunotherapeutic as adjuvant therapy in patients with resected mage-A3-positive non-small cell lung cancer (Nsclc). Ann Oncol. 2014;25:iv409. 10.1093/annonc/mdu347.1. [DOI] [PubMed] [Google Scholar]
- 157.Butts C, Socinski MA, Mitchell PL et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15:59–68. 10.1016/S1470-2045(13)70510-2. [DOI] [PubMed] [Google Scholar]
- 158.Facciponte JG, Ugel S, De Sanctis F et al. Tumor endothelial marker 1-specific DNA vaccination targets tumor vasculature. J Clin Invest. 2014;124:1497–1511. 10.1172/JCI67382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bose A, Taylor JL, Alber S et al. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int J Cancer. 2011;129:2158. 10.1002/IJC.25863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Manning EA, Ullman JG, Leatherman JM et al. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res. 2007;13:3951–3959. 10.1158/1078-0432.CCR-07-0374. [DOI] [PubMed] [Google Scholar]
- 161.Bota DA, Alexandru-Abrams D, Pretto C et al. Use of ERC-1671 vaccine in a patient with recurrent glioblastoma multiforme after progression during bevacizumab therapy: first published report. Perm J. 2015;19:41. 10.7812/TPP/14-042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Schijns VEJC, Pretto C, Devillers L, et al. First clinical results of a personalized immunotherapeutic vaccine against recurrent, incompletely resected, treatment-resistant glioblastoma multiforme (GBM) tumors, based on combined allo- and auto-immune tumor reactivity. Vaccine. 2015;33:2690–2696. 10.1016/j.vaccine.2015.03.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bota DA, Chung J, Dandekar M et al. Phase II study of ERC1671 plus bevacizumab versus bevacizumab plus placebo in recurrent glioblastoma: interim results and correlations with CD4+ T-lymphocyte counts. CNS Oncol. 2018;7:CNS22. 10.2217/cns-2018-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tanyi JL, Bobisse S, Ophir E et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci Transl Med. 2018;10. 10.1126/scitranslmed.aao5931. [DOI] [PubMed] [Google Scholar]
- 165.Rini BI, Stenzl A, Zdrojowy R et al. IMA901, a multipeptide cancer vaccine, plus sunitinib versus sunitinib alone, as first-line therapy for advanced or metastatic renal cell carcinoma (IMPRINT): a multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2016;17:1599–1611. 10.1016/S1470-2045(16)30408-9. [DOI] [PubMed] [Google Scholar]
- 166.Niederman TMJ, Ghogawala Z, Carter BS, Tompkins HS, Russell MM, Mulligan RC. Antitumor activity of cytotoxic T lymphocytes engineered to target vascular endothelial growth factor receptors. Proc Natl Acad Sci USA. 2002;99:7009–7014. 10.1073/pnas.092562399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chinnasamy D, Yu Z, Theoret MR et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest. 2010;120:3953–3968. 10.1172/JCI43490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chinnasamy D, Yu Z, Kerkar SP et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. 2012;18:1672–1683. 10.1158/1078-0432.CCR-11-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chinnasamy D, Tran E, Yu Z, Morgan RA, Restifo NP, Rosenberg SA. Simultaneous targeting of tumor antigens and the tumor vasculature using t lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res. 2013;73:3371–3380. 10.1158/0008-5472.CAN-12-3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010;70:6171–6180. 10.1158/0008-5472.CAN-10-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tao L, Huang G, Shi S, Chen L. Bevacizumab improves the antitumor efficacy of adoptive cytokine-induced killer cells therapy in non-small cell lung cancer models. Med Oncol. 2014;31. 10.1007/s12032-013-0777-3. [DOI] [PubMed] [Google Scholar]
- 172.Lanitis E, Kosti P, Ronet C et al. VEGFR-2 redirected CAR-T cells are functionally impaired by soluble VEGF-A competition for receptor binding. J Immunother Cancer. 2021;9:e002151. 10.1136/JITC-2020-002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lanitis E, Rota G, Kosti P et al. Optimized gene engineering of murine CAR-T cells reveals the beneficial effects of IL-15 coexpression. J Exp Med. 2021;218. 10.1084/JEM.20192203/211522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kandalaft LE, Powell DJJr, Chiang CL, et al. Autologous lysate-pulsed dendritic cell vaccination followed by adoptive transfer of vaccine-primed ex vivo co-stimulated t cells in recurrent ovarian cancer. Oncoimmunology. 2013;2. 10.4161/onci.22664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16:131–144. 10.1038/nrc.2016.14. [DOI] [PubMed] [Google Scholar]
- 176.Lee AJ, Ashkar AA. The dual nature of type I and type II interferons. Front Immunol. 2018;9:2061. 10.3389/FIMMU.2018.02061/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Vatner RE, Janssen E. DCs and the link between innate and adaptive tumor immunity. Mol Immunol. 2019;110:13–23. 10.1016/J.MOLIMM.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Li H, Liu Z, Liu L, et al. AXL targeting restores PD-1 blockade sensitivity of STK11/LKB1 mutant NSCLC through expansion of TCF1+ CD8 T cells. Cell Reports Med. 2022;3:100554. 10.1016/J.XCRM.2022.100554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Stein BL, Tiu RV. Biological rationale and clinical use of interferon in the classical BCR-ABL-negative myeloproliferative neoplasms. J Interf Cytokine Res. 2013;33:145–153. 10.1089/jir.2012.0120. [DOI] [PubMed] [Google Scholar]
- 180.Mocellin S, Pföhler C, Bewarder M, et al. Interferon alpha adjuvant therapy in patients with high-risk melanoma: a systematic review and meta-analysis. J Natl Cancer Inst. 2010;102:493–501. 10.1093/jnci/djq009. [DOI] [PubMed] [Google Scholar]
- 181.Minn AJ. Interferons and the immunogenic effects of cancer therapy. Trends Immunol. 2015;36:725–737. 10.1016/j.it.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Rini BI, Halabi S, Rosenberg JE, et al. Phase III trial of bevacizumab plus interferon alfa versus interferon alfa monotherapy in patients with metastatic renal cell carcinoma: final results of CALGB 90206. J Clin Oncol. 2010;28:2137–2143. 10.1200/JCO.2009.26.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Escudier B, Bellmunt J, Négrier S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol. 2010;28:2144–2150. 10.1200/JCO.2009.26.7849. [DOI] [PubMed] [Google Scholar]
- 184.Woo SR, Fuertes MB, Corrales L, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–842. 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Yang H, Lee WS, Kong SJ, et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest. 2019;129:4350–4364. 10.1172/JCI125413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sullivan LA, Carbon JG, Roland CL, et al. r84, a novel therapeutic antibody against mouse and human VEGF with potent antitumor activity and limited toxicity induction. Gartel AL, ed. PLoS One. 2010;5:e12031. 10.1371/journal.pone.0012031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18:4266–4276. 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7. 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tang H, Liang Y, Anders RA et al. PD-L1 on host cells is essential for PD-L1 blockade–mediated tumor regression. J Clin Invest. 2018;128:580–588. 10.1172/JCI96061. [DOI] [PMC free article] [PubMed] [Google Scholar]